

Abstract
Background
Integration of human papillomavirus (HPV) into the host genome is a dominant feature of invasive cervical cancer (ICC), yet the tumorigenicity of cis genomic changes at integration sites remains largely understudied.
Methods
Combining multi-omics data from The Cancer Genome Atlas with patient-matched long-read sequencing of HPV integration sites, we developed a strategy for using HPV integration events to identify and prioritise novel candidate ICC target genes (integration-detected genes (IDGs)). Four IDGs were then chosen for in vitro functional studies employing small interfering RNA-mediated knockdown in cell migration, proliferation and colony formation assays.
Results
PacBio data revealed 267 unique human–HPV breakpoints comprising 87 total integration events in eight tumours. Candidate IDGs were filtered based on the following criteria: (1) proximity to integration site, (2) clonal representation of integration event, (3) tumour-specific expression (Z-score) and (4) association with ICC survival. Four candidates prioritised based on their unknown function in ICC (BNC1, RSBN1, USP36 and TAOK3) exhibited oncogenic properties in cervical cancer cell lines. Further, annotation of integration events provided clues regarding potential mechanisms underlying altered IDG expression in both integrated and non-integrated ICC tumours.
Conclusions
HPV integration events can guide the identification of novel IDGs for further study in cervical carcinogenesis and as putative therapeutic targets.
Subject terms: Cervical cancer, Cancer genomics, Targeted resequencing, Data mining, Tumour virus infections
Background
Integration of the high-risk human papillomavirus (HPV) genome into the host genome associates with invasive cervical cancer (ICC) progression [1, 2], contributing to the malignant phenotype by modifying both viral and host gene expression. The chief effect of integration on the viral genome is upregulation of the HPV oncogenes, E6 and E7. E6 and E7 target important regulatory host factors such as p53 and retinoblastoma proteins [3], and their elevated expression in ICC causes nucleotide depletion, leading to stress on replication, double-strand breaks and preferential integration of the viral genome at regions sensitive to replication stress [4]. However, since not all ICC tumours exhibit elevated expression of E6 and/or E7 [5–7], other host-centric disease mechanisms likely exist and remain uncharacterised.
Evidence suggests that host genome alterations resulting from HPV integration are integral to the development of ICC [5, 8–12]. HPV integration can drive rearrangements and amplification of the host genome, leading to dysregulated transcription of adjacent cancer-associated genes. For example, HPV18 integrations in HeLa cervical cancer cells drive amplification of the MYC locus [8] and integration in patient samples target other known ICC oncogenes, including ERBB2 and RAD51B [11, 13, 14]. However, The Cancer Genome Atlas (TCGA) analysis of HPV integration from its RNA-sequencing (RNAseq) data demonstrates that <10% of HPV integration events are linked to established oncogenes [11], suggesting that most HPV integration sites are currently of unknown functional significance in ICC. Collectively, this affords a great opportunity to identify potential new ICC-specific vulnerabilities since oncogenic driver function is dependent on tissue-specific transcriptional and proteomic networks [15, 16], and anticancer drug response often depends on the anatomical cancer type [17].
The current study aims to leverage a large amount of existing data—including TCGA whole-genome sequencing (WGS), RNAseq, DNA methylation and updated outcome data—for the discovery of novel genes that may play a role in cervical carcinogenesis. We have termed these putative new ICC target genes integration-detected genes (IDGs), as their study was initiated by their proximity to an HPV integration event. Moreover, to precisely characterise the complex structure of integration events not discernible from short-read sequencing data, HPV-enriched tumour DNA from eight patient-matched samples was subjected to Pacific Biosciences (PacBio) long-read sequencing. Genes within 2 Mb of each integration site were recorded and, integrating PacBio long-read sequencing data with matching TCGA multi-omics data, subjected to a series of filtering measures to narrow the list of candidate IDGs. Four candidate IDGs from four unique integration events were then selected for functional study in cervical cancer cell lines. Taken together, our data provide unique insights into the genomic compartments surrounding viral–cellular DNA junctions and demonstrate the utility of in-depth characterisation of HPV integration structure/function for the identification of better-defined target genes for treating cervical disease.
Methods
Study population
Subjects included eight women with ICC (Supplementary Table S1) with matched multi-omics data from TCGA. All samples were obtained at the time of diagnosis and before administering any treatment. Procedures for processing and quality management of samples were as described by TCGA [11]. HPV typing was performed as previously described [11, 18, 19] and showed excellent concordance with TCGA and the Medical College of Wisconsin (MCW) typing when applicable. The study protocol was approved by MCW’s Institutional Review Board.
DNA isolation
Tumour (5–10 mg) DNA was extracted with the Gentra Puregene Tissue Kit (Qiagen, Germantown, MD), modified to include two 1× phosphate-buffered saline (PBS) washes to dissolve OCT prior to cell lysis and to exclude RNase A digestion. Quantity and quality of DNA were assessed using the Qubit dsDNA BR Assay (Invitrogen, Waltham, MA) and ultraviolet spectroscopy, respectively.
HPV-targeted PacBio long-read sequencing and viral integration analysis
SMRTbell® libraries for sequencing on the PacBio Sequel® System were constructed as outlined in the PacBio Procedure & Checklist–Multiplexed Genomic DNA Target Capture Using IDT xGen® Lockdown® Probes. Custom probes were generated against the HPV16 genomic sequence (see Supplementary Methods for more detail) and used to enrich tumour DNA for HPV prior to sequencing. Pooled libraries were sequenced on the PacBio Sequel II at the MCW GSPMC Sequencing Service Facility.
Sequencing data were demultiplexed using the PacBio SMRTLink analysis tool (v5.1.0). The PacBio bam2fastx tool (SMRTLink v5.1.0) was then used to produce raw sequencing data in FASTQ format. BWA-mem with ‘-x pacbio’ setting was used to align reads to a hybrid human–HPV reference genome. SAMTOOLS was used to sort alignments by genomic coordinates and bedtools was used to identify chimeric reads mapped to both human (GRCh37/hg19) and HPV16 or HPV70 (see Supplementary Methods).
Identification and prioritisation of candidate IDGs
Clonal analysis
TCGA WGS data from the eight tumours were analysed with ViFi [20] for detection of human–HPV chimeric reads. For each viral integration event, we calculated integration allele fraction (IAF) using published methodology [21]. Of note, for one sample lacking TCGA WGS data (TCGA-C5-A8XH), IAF was calculated with the same methodology but using whole-exome sequencing (WXS) data. Up to 50% of WXS data contains sequences outside targeted exons [22], thus WXS data were sufficient for the ViFi identification and clonal analysis of the USP36 (ubiquitin-specific peptidase 36) integration site in this tumour.
Integrated tumour-specific expression (Z-score)
Expression of each candidate IDG was converted to a standard score using Z-transformation. Analysis was conducted in R [23] and TCGA CESC (cervical squamous cell carcinoma and endocervical adenocarcinoma; used interchangeably with ICC hereafter) RNAseq expression data (n = 304 tumours) was obtained using the package TCGA2STAT. Sorted barplots were produced using ggplot2 [24].
Survival analysis
Survival association of candidate IDG expression was assessed with Kaplan–Meier plot and log-rank test. Using the R package survminer, tumours were split into high and low expressing groups according to the optimal cutoff points based on maximally ranked statistic methods (see Supplementary Methods for more details). Hazard ratios (HRs) of high vs low expression were calculated with the R package survival. MCW-ICC cervical tumours (n = 142) for survival analysis were obtained from participants enrolled in the Cervical Cancer Genetic Epidemiology (CerGE) study (Supplementary Table S1). Subjects in the CerGE study were recruited and consented to a trio study developed to investigate inherited genetic polymorphisms and HPV subtypes and variants contributing to cervical cancer [18, 25, 26]. The sample size of n = 142 tumours allowed for 80% power to detect HRs of 1.6 or greater.
Annotation of HPV integration sites for altered expression of candidate IDGs
Integration events associated with candidate IDGs were annotated using PacBio long-read sequencing data, sample-matched multi-omics data from TCGA (RNAseq, copy number variation (CNV) and methylation) and tracks from the UCSC Genome Browser (GRCh37/hg19; [27]). UCSC Genome Browser tracks used were as follows: RefSeq UCSC gene annotations, GeneHancer [28] for the location of gene-specific enhancer and promoter regions and HeLa-specific chromatin state (Roadmap Epigenomics Project [29] and Cistrome Analysis Hub [30]). Of note, the Cistrome Analysis Hub track was lifted over from GRCh38/hg38 and matched to the precise, corresponding location in GRCh37/hg19. TCGA data sources were uploaded for visualisation using the Integrative Genomics Viewer [31, 32]. PacBio reads were additionally visualised and presented using Ribbon [33]. MEXPRESS [34] and EDGE in TCGA [35] websites were used to probe TCGA CESC cohort data for gene-specific regulatory relationships.
Functional testing of candidate IDGs
Cell culture
SiHa and HeLa cervical cancer cells from the American Type Culture Collection (ATCC; Manassas, VA) were maintained in Eagle’s minimum essential medium supplemented with 10% foetal bovine serum (FBS) and 1% GlutaMAX™ (Gibco). MCW2 (HPV18+) cervical cancer cells were maintained in F medium. Cells were harvested using Trypsin-EDTA and counted with a hemocytometer using 0.4% Trypan Blue. Cell line authentication was performed by ATCC STR profiling (SiHa and HeLa) or RNAseq (MCW2) and each cell line was tested for mycoplasma contamination before beginning in vitro functional assays.
siRNA-mediated knockdown
SiHa, HeLa or MCW2 cells were seeded (2.0 × 105) into a 6-well dish to adhere overnight. The following day, cells were transfected with siRNAs (25 nM) targeting four human genes (siBNC1 (small interfering basonuclin 1), siRSBN1 (round spermatid basic protein 1), siUSP36, and siTAOK3 (TAO kinase 3)) or a negative control siRNA (siCONT), using DharmaFECT1 reagent (Horizon Discovery, Waterbeach, UK). Transfection media were replaced with fresh media 24 h later and functional assays were performed 48 h later. In addition to providing IDG expression data for survival association in the MCW-ICC cohort, quantitative reverse transcription-PCR (qRT-PCR) was used to determine siRNA-mediated knockdown efficiency [36]. RNA was isolated using mirVana miRNA Isolation Kit (Invitrogen) and complementary DNA (cDNA) was synthesised using High-Capacity RNA-to-cDNA Kit (Applied Biosystems). The expression was assessed using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) on a StepOnePlus Real-Time PCR System (Applied Biosystems). The thermal cycling programme included an enzyme activation step at 95 °C for 10 min, followed by 40 cycles of a 10 s denaturing step at 95 °C, and a 1 min annealing/extension step at 60 °C. Fluorescent intensity was measured at 60 or 62 °C at the end of each cycle. SiHa experiments were repeated using a second, unique siRNA targeting each of the four IDGs (labelled #2). siRNA and primer sequences are listed in Supplementary Table S2.
Western blotting
IDG protein expression was determined by western blotting using primary antibodies for BNC1 (NBP2-24721; Novus Biologicals, Centennial, CO), RSBN1 (NBP1-57724; Novus Biologicals), USP36 (NBP2-74806; Novus Biologicals) and TAOK3 (ab150388; Abcam, Cambridge, MA) and horseradish peroxidase-conjugated secondary antibodies. Chemiluminescence was visualised using Molecular Imager ChemiDoc XRS + (Bio-Rad) and visualised protein bands were quantified using Image Lab Software (Bio-Rad). See Supplementary Methods for additional details.
Transwell cell migration assays
After ~16 h of serum starvation, transfected cells were harvested using HyQTase and seeded (1 × 105 cells in 100 μl) into the top chamber of the transwell (Corning, NY), while Dulbecco’s modified Eagle’s medium + 20% FBS (600 μl) was added as a chemoattractant to the bottom chamber. After a 6 h incubation at 37 °C, cells that migrated through each transwell were fixed and stained with crystal violet. Four independent sections of each transwell were visualised using light microscopy for counting the number of migrating cervical cancer cells in each condition.
Cell growth assays
Cervical cancer cell proliferation was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) according to the manufacturer’s directions. Transfected cells were seeded onto 96-well plates (100 µl of 0.02 × 106 cells/ml per well). CellTiter 96® AQueous One Solution was added 2 h, 1 day, 3 days, or 5 days later and incubated for 30 min in a humidified, 5% CO2 atmosphere. Absorbance at 490 nm was recorded using an Infinite 200 PRO plate reader (Tecan, Switzerland). Similarly, cell colony formation was assessed by seeding transfected cells into 6-well plates (2000 cells/well) for incubation in a humidified, 5% CO2 atmosphere. Growth media were refreshed every 3–4 days. Two weeks later, colonies were fixed and stained with crystal violet (Sigma) and images were taken for ImageJ [37] analysis.
Cell-based assay statistical analysis
Statistical analyses for in vitro cell line experiments were performed using GraphPad Prism 9.0 (La Jolla, CA). One representative experiment of triplicate experiments is shown for cell proliferation, migration, and colony formation assays, while messenger RNA expression data represent the average across at least three unique experiments. Results were reported as percentages or means ± standard error of the mean (SEM), as appropriate. All datasets were assessed for normality using the Shapiro–Wilk normality test. Differences between means were analysed using the two-tailed Student’s t test or Welch’s t test (if Bartlett’s test suggested that dataset variances were not similar). For proliferation assay results, differences among means were analysed by two-way analysis of variance (ANOVA). When the ANOVA showed significant differences, pairwise comparisons between means were assessed using Dunnett’s multiple comparisons testing. P values <0.05 were considered significant.
Results
PacBio long-read sequencing of HPV-enriched ICC tumour DNA
HPV-enriched DNA from eight ICC tumours was subjected to long-read sequencing on the PacBio Sequel II. Compared with the ~240 bp resolution of reads covering HPV integration sites from TCGA short-read sequencing, PacBio captured up to ~10 kb of chimeric HPV-flanking host genomic DNA at an average sequencing depth of 69× (median = 13; range = 1–4551), providing higher spatial resolution of complex HPV integration events (discussed in more detail below). Further, higher spatial resolution allowed for the assignment of multiple chimeric human–HPV breakpoints to the same integration event, thus defined here as the collection of one or more chimeric breakpoints occurring within 1.5 Mb of each other.
PacBio long-read sequencing analysis revealed that the eight ICC tumours harboured a total of 267 unique HPV–human breakpoints comprising 87 total HPV integration events (Supplementary Table S3). The number of HPV integration events (1–72) and human–HPV chimeric breakpoints (1–233) varied greatly across samples (Supplementary Table S3). However, sample TCGA-C5-A2LX contributed most to this variability as it harboured significantly more integration events (n = 72) compared to the average number of events (n = 2) observed in the remaining seven samples. Supplementary Table S4 lists HPV and human genome coordinates for each of the 267 chimeric breakpoints. In agreement with other HPV integration studies, frequent breakpoints occurred in the E1 gene of HPV16 (Supplementary Fig. S1) and within intergenic regions across most of the human genome (Supplementary Fig. S2). PacBio integration analysis identified all but three integration events reported by TCGA RNAseq analysis, all of which were flagged as ‘discordant’, meaning only one of two TCGA analysis sites identified the integration event [11]. Further, PacBio sequencing of HPV-enriched tumour DNA uncovered novel integration events not reported in TCGA’s original analysis (n = 60; open circles in Supplementary Fig. S2).
Prioritisation of candidate IDGs for functional study
The main objective of the current study was to develop a workflow for identifying and filtering candidate IDGs from HPV integration signatures exhibited by each tumour. The rationale behind using HPV integrations to pinpoint important cancer genes comes from accumulating evidence that the virus appears to integrate into or near cancer-related genes [5, 7, 38–40]. Further, when the virus integrates into these important, cancer-promoting regions (i.e. those harbouring cancer-related genes and/or regulatory elements), it can result in clonal enrichment within the tumour. Thus, we used HPV integration events as a beacon, illuminating candidate genes that could play a vital role in ICC.
Our list of candidate IDGs began with all genes within 2 Mb on either side of each integration event, resulting in 3399 total genes representing all 87 events (Supplementary Fig. S3A; 265 of these were duplicate genes due to similar integration events found in more than one tumour; thus, n = 3134 unique genes). The 2 Mb cutoff was designed to capture potential integration-induced alterations in DNA that could disrupt nearby topographically associated domain (TAD) chromatin states and associated gene expression [41–44]. TAD disruptions can result in altered chromosomal contacts and rewiring of enhancer–promoter interactions, resulting in aberrant gene expression [41–44].
Next, to pare down the list of candidate IDGs, we implemented a series of filters for their prioritisation (Supplementary Fig. S3A). The first filter criterion was a prominent clonal representation of the integration event, as higher frequency insertions are more likely to be enriched within the tumour. Following published methodology [21] and employing ViFi analysis software [20], we calculated IAF for each event from the sample-matched TCGA data (WGS or WXS; last column of Supplementary Table S4). Of note, TCGA short-read sequencing data was used for clonal analysis since long-read sequencing samples were baited for HPV, thus affecting the relative proportion of integrated to wild-type reads. Employing a 10% cutoff for IAF values (at least 10% of reads covering the integration region harboured a human–viral chimeric breakpoint) reduced the number of potential IDGs to 1697 from 40 clonal events. Next, genes were filtered based on their expression level in the tumour with associated viral integration compared to expression across the rest of TCGA’s ICC cohort (Z-score) with the rationale that, if integration specifically affects the expression of a nearby gene, then an expression of that gene in the integrated patient should be an outlier in the cohort. After removing genes lacking TCGA RNAseq expression data (e.g. uncharacterised RNA genes and miRNAs; n = 238), we employed the Z-score filter (cutoff ≥1.5 or ≤−1.5), leaving 170 potential IDGs from 40 clonal events. The third filter required that a gene’s expression (TCGA RNAseq) be correlated with overall survival (OS) and/or recurrence-free ICC survival (RFS), with an HR in agreement with the expression in the integrated tumour (i.e. positive Z-score and HR > 1 or negative Z-score and HR < 1). Of note, we chose a less stringent p value cutoff for the survival association filter (p < 0.2) due to the limited survival information in TCGA’s ICC cohort. Employing the final survival filter resulted in 84 candidate IDGs from the 40 clonal integration events in eight samples (Supplementary Fig. S3A and Table S5). Ingenuity pathway analysis revealed that our list of 84 candidate IDGs contained 15 enzymes, 1 growth factor, 5 kinases, 4 peptidases, 1 phosphatase, 14 transcriptional regulators, 3 transmembrane receptors, 6 transporters and 35 listed as ‘other’ (Supplementary Table S5). Of note, 18 (21.4%) of the 84 candidate IDGs were associated with integration events not previously reported by TCGA, which used polyA-selected RNAseq data for integration analysis vs HPV-enriched tumour DNA used here.
After prioritisation using our filtering criteria above, we chose four IDGs for further validation. Figure 1a–d depict Z-score plots generated from TCGA cervical cancer cohort RNAseq data for each of the four IDGs passing our filtering criteria. BNC1 (Fig. 1a) is a zinc-finger protein highly expressed in epithelial cells, RSBN1 (Fig. 1b) is a histone demethylase, USP36 (Fig. 1c) is a multifunctional deubiquitinase and TAOK3 (Fig. 1d) is a serine/threonine kinase involved in the regulation of MAPK signalling. In addition, using TCGA RNAseq expression data, we next examined how the expression of each IDG was associated with OS in TCGA’s CESC cohort (Fig. 1e–h). Expression of all four IDGs met our cutoff for survival association (p < 0.2; less stringent cutoff applied here due to the limited scope of TCGA survival data), with USP36 (Fig. 1g) exhibiting the most significant survival association in TCGA’s CESC cohort. In comparison to known oncogenes previously shown to be affected by HPV integration (e.g. ERBB2, RAD51B and PVT1), a pan-cancer and ICC-specific PubMed literature search of all four putative IDGs resulted in significantly fewer results (Supplementary Fig. S3B), suggesting that little is known about their function in cancer and, more specifically, in ICC.
Fig. 1. Filtering and prioritisation results of four candidate IDGs chosen for validation and functional testing.

Clonality (IAF = integration allele fraction) values and Z-score plots for BNC1 (a), RSBN1 (b), USP36 (c), and TAOK3 (d) demonstrate how each IDG passed our first two filtering criteria. Z-score plots depict a black bar for each TCGA ICC sample (x-axis; CESC = cervical squamous cell carcinoma and endocervical adenocarcinoma; n = 304) and their corresponding IDG-specific expression levels (y-axis; TCGA RNAseq data). Red lines mark IDG expression in the integrated tumour. Numerical values in red (b and c) = RSEM values for the integrated tumour. Next, we filtered on the association of BNC1 (e), RSBN1 (f), USP36 (g), and TAOK3 (h) expression with overall survival in the TCGA ICC cohort (HR = hazard ratio). ICC-specific overall survival association of BNC1 (i), RSBN1 (j), USP36 (k), and TAOK3 (l) expression (measured via qRT-PCR) was validated in a second ICC cohort (MCW-ICC; n = 142).
Before moving to functional assessment, we next sought to validate the association of IDG expression with outcome in a second ICC cohort (MCW-ICC, n = 142; Fig. 1i–l). Expression of BNC1, RSBN1, USP36 and TAOK3 was measured via qRT-PCR to investigate the association with OS. Kaplan–Meier survival analysis showed that high expression of BNC1 (Fig. 1i) and RSBN1 (Fig. 1j) was significantly associated with poorer OS, while USP36 (Fig. 1k) and TAOK3 (Fig. 1l) did not reach significance, but exhibited an observable trend suggesting poorer survival with their increased expression.
Long-range sequencing data provide potential clues regarding the regulation of IDG expression in integrated and non-integrated ICC tumours
To be considered a valuable ICC target gene at the broader population level, the expression of any potential IDG should be altered in both integrated and non-integrated ICC tumours. Thus, before advancing the four candidate IDGs to functional assessment, our final objective leveraged existing sample-matched TCGA data, publicly available datasets from the UCSC Genome Browser (GRCh37/hg19; [27]) and integrated structural information from PacBio sequencing for in-depth annotation of integration sites to infer potential mechanisms underlying altered expression of candidate IDGs (Figs. 2–5). Of note, each of the integrated genomic regions depicted in Figs. 2–5 exhibited CNV gains according to TCGA CNV analysis (red bars below RNAseq data in each figure). While we expect the virus is responsible for driving the surrounding genome amplification, we cannot rule out the possibility that these regions were already amplified in the tumour prior to viral integration.
Fig. 2. Annotation of HPV integration affecting BNC1 expression in TCGA-C5-A2LV.

PacBio long-read sequencing, TCGA, and UCSC Genome Browser (http://genome.ucsc.edu) data were used to annotate the HPV integration site proximal to the candidate IDG, BNC1. TCGA-C5-A2LV long-read data (PacBio) and TCGA sequencing (RNAseq), CNV (blue = loss; red = gain), and methylation data (blue = hypomethylation; red = hypermethylation) covering the area of integration (a; red box). The Integrative Genomics Viewer (IGV) was used for the visualisation of PacBio and TCGA RNAseq read alignments. PacBio coverage displays the read depths at each locus with a grey bar chart. PacBio alignments show individual aligned reads, where grey lines represent reads aligning to the human reference genome. For RNAseq, the coverage and alignment tracks are the same, but in between the two is a splice junction track that provides a visualisation of reads spanning splice junctions. Blue lines in the RNAseq IGV image connect reads spanning splice junctions. UCSC Genome Browser GeneHancer track suggests that the integration site is adjacent to a BNC1-specific enhancer (EnH; grey bar) ~88 kb from its promoter (red bar). In addition, HeLa cell-specific cistrome analysis (bottom of a) suggests that these regulatory regions are indeed applicable to cervical cancer. The Ribbon programme was used to generate a schematic of a single PacBio read covering the area of integration, showing how the HPV genome is inserted (red) with human sequence flanking both sides (blue; b). Thick bars across the top (b) represent the HPV and human reference genomes that are connected by dashed lines to a single PacBio read covering the integration to show how it specifically mapped to each genome. Data from all PacBio long reads covering the integration event were used to schematically annotate the integration event (c). Breakpoints identified from TCGA short-read sequencing (SR) of tumour RNA are highlighted in the yellow boxes. The dashed line represents a portion of the human genome not covered by PacBio reads. Collectively, the data support potential HPV integration-induced upregulation of BNC1 enhancer RNA (eRNA), leading to increased BNC1 expression.
Fig. 5. Annotation of HPV integration affecting TAOK3 expression in TCGA-C5-A2LX.

PacBio long-read sequencing, TCGA, and UCSC Genome Browser (http://genome.ucsc.edu) data were used to annotate the HPV integration site within the candidate IDG, TAOK3. TCGA-C5-A2LX long-read data (PacBio) and TCGA sequencing (RNAseq), CNV (blue = loss; red = gain), and methylation data (blue = hypomethylation; red = hypermethylation) covering integration event is depicted in panel (a). The Ribbon programme was used to generate a schematic of two PacBio reads covering the area of integration. Thick bars across the top (b) represent the HPV and human reference genomes, which are connected by dashed lines to two unique PacBio reads covering the integration to show how they are specifically mapped to each genome. Data from all PacBio long reads covering the integration event were used to hand annotate the integration event (c). Breakpoints identified from TCGA short-read sequencing (SR) are highlighted in the yellow boxes. PacBio sequencing successfully captured the entirety of the HPV insertion, which comprised almost two full copies of the HPV16 genome (pink) flanked on both sides by intron 9 of TAOK3 (olive).
The integration event affecting BNC1 expression in TCGA-C5-A2LV is depicted in Fig. 2 and demonstrates how annotation of integration events can drive the development of mechanistic hypotheses regarding the altered expression of putative IDGs in both integrated and non-integrated tumours. Examining TCGA RNAseq data from TCGA-C5-A2LV, we noted robust expression of both genes flanking the integration, BNC1 and SH3GL3, in addition to an intergenic region immediately adjacent to the event (Fig. 2, red box). UCSC Genome Browser data revealed that the adjacent intergenic region of increased expression corresponds to a BNC1-specific enhancer (GeneHancer ID: GH15J083364; [28, 45]). Interestingly, TCGA copy number variation analysis of TCGA-C5-A2LV showed amplification of this enhancer region (Fig. 2; pink bar below RNAseq), yet shallow deletion of the BNC1 gene region (Fig. 2; light blue bar below RNAseq) despite its expression being very high in this tumour. Hence, our annotation of this integration event suggests that BNC1 expression in TCGA-C5-A2LV may be driven by HPV16 URR-dependent alteration of this BNC1-specific enhancer.
Sixteen tumours from the TCGA ICC cohort exhibited higher BNC1 expression than the integrated sample (see Fig. 1a, samples to the right of the red line), highlighting an interesting question: If BNC1 upregulation in TCGA-C5-A2LV (integrated sample) is HPV-driven, what is the mechanism of upregulation in non-integrated samples? Using online tools EDGE in TCGA [35] and MEXPRESS [34], we explored factors that could potentially explain elevated BNC1 expression in the rest of TCGA’s ICC cohort (non BNC1-integrated samples). The top factor correlated with higher BNC1 expression was hypomethylation of a specific site in its promoter (cg26429925; Pearson r = −0.78, p = 4.46e − 37; Supplementary Figure S4A). Using TCGA methylation and RNAseq data, we compared cg26429925 methylation in the top vs the bottom 25% of BNC1 expressers and found that BNC1 promotor methylation was significantly lower in the high BNC1-expressing ICC tumours (Supplementary Figure S4B). In fact, the integrated sample also displayed BNC1 promoter hypomethylation (Supplementary Fig. S4B; red data point), suggesting that BNC1 expression in this sample may be regulated by more than just HPV integration. Thus, while the HPV integration event specific to TCGA-C5-A2LV nominated BNC1 as a putative ICC gene target, analysis of ICC patient survival (see Fig. 1e, i) and genomic data (Supplementary Figs. 4A, B) support a broader impact of BNC1 on ICC.
Annotations of the other three integration events affecting the expression of RSBN1, USP36 and TAOK3 are depicted in Figs. 3, 4 and 5, respectively. HPV16 integration within RSBN1 in TCGA-C5-A3HD revealed complex rearrangement and high expression of multiple adjacent regions (Fig. 3). The regions of greatest amplification harbour an RSBN1-specific promoter and enhancer (GH01J113809 and GH01J113808; red box in Fig. 3) poised upstream of the inserted HPV16 genome, suggesting that the virus may trigger expression of these gene-specific regulatory elements. MEXPRESS analysis suggested that CNV was the top factor affecting RSBN1 expression across the TCGA ICC cohort (Pearson’s r = 0.481, p = 6.98e − 17; Supplementary Figs. S4C, D) and, in tumours with no CNV, the top factor associated with high RSBN1 expression was promoter hypomethylation (cg23078294; Pearson’s r = −0.19, p = 0.009; Supplementary Fig. S4E).
Fig. 3. Annotation of HPV integration affecting RSBN1 expression in TCGA-C5-A3HD.
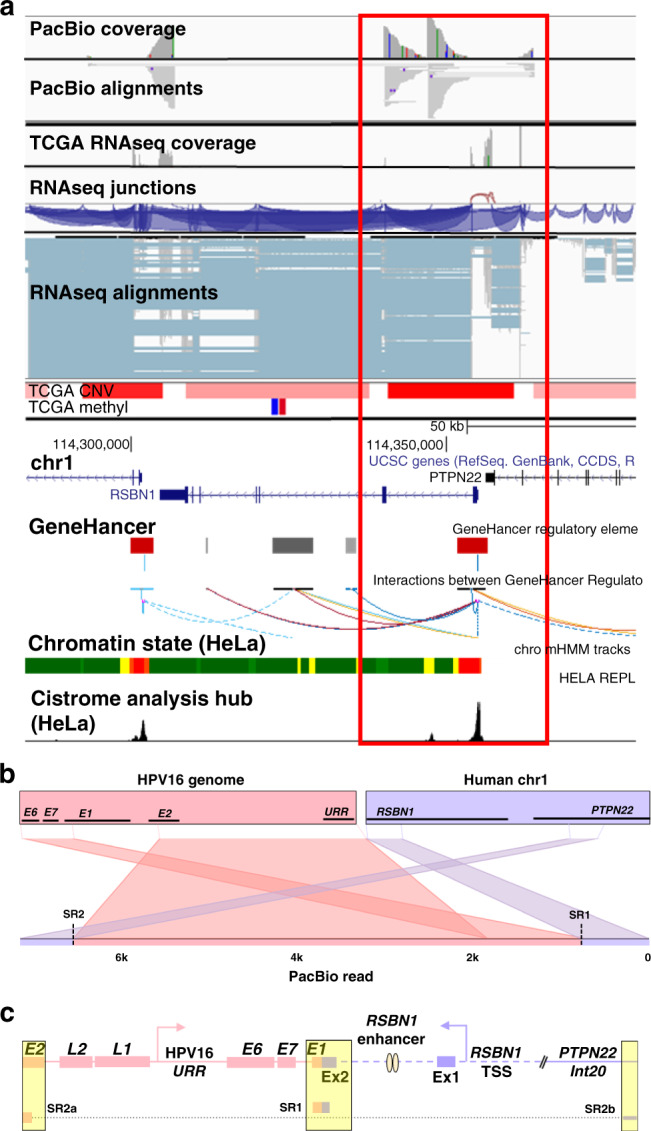
PacBio long-read sequencing, TCGA, and UCSC Genome Browser (http://genome.ucsc.edu) data were used to annotate the HPV integration site within the candidate IDG, RSBN1. TCGA-C5-A3HD long-read data (PacBio) and TCGA sequencing (RNAseq), CNV (blue = loss; red = gain), and methylation data (blue = hypomethylation; red = hypermethylation) covering the integration event, which spans ~50 kb of the human genome (a). The Ribbon programme was used to generate a schematic of a single PacBio read (b) covering the area of integration including the human–viral breakpoint with the highest IAF value (exon 2 of RSBN1; red box in a). Thick bars across the top (b) represent the HPV and human reference genomes, which are connected by dashed lines to a single PacBio read covering the integration to show how it specifically mapped to each genome. Data from all PacBio long reads covering the integration event were used to hand annotate the integration event (c). Breakpoints identified from TCGA short-read sequencing (SR) are highlighted in the yellow boxes. SRa and SRb are segments (connected by dotted line) of a single Illumina read spanning a breakpoint connecting two non-contiguous sequences of the human genome (represented as a diagonal double line in PacBio long read). The dashed purple line represents a portion of the human genome not covered by PacBio reads. The regions of greatest amplification harbour an RSBN1-specific promoter and enhancer (GeneHancer track; red and grey boxes, respectively) poised adjacent to the inserted HPV16 genome, possibly suggesting viral-driven expression of these gene-specific regulatory elements.
Fig. 4. Annotation of HPV integration affecting USP36 expression in TCGA-C5-A8XH.

PacBio long-read sequencing, TCGA, and UCSC Genome Browser (http://genome.ucsc.edu) data were used to annotate the HPV integration site within the candidate IDG, USP36. TCGA-C5-A8XH long-read data (PacBio) and TCGA sequencing (RNAseq), CNV (blue = loss; red = gain), and methylation data (blue = hypomethylation; red = hypermethylation) covering the integration event spanning ~200 kb of the human genome (a). The Ribbon programme was used to generate a schematic of two PacBio reads covering the area of integration. Thick bars across the top (b) represent the HPV and human reference genomes, which are connected by dashed lines to two unique PacBio reads covering the integration to show how they are specifically mapped to each genome. Data from all PacBio long reads covering the integration event were used to hand annotate the integration event (c). Breakpoints identified from TCGA short-read sequencing (SR) are highlighted in the yellow boxes. The diagonal double line represents a breakpoint connecting two non-contiguous sequences of the human genome. TCGA RNAseq data suggest the expression of the fused USP36-encoding DNA with upstream intergenic DNA located between the SCAT and CYTH1 genes and sharp upregulation of USP36 expression beginning at intron 4, potentially driven by the inserted viral URR.
PacBio analysis of TCGA-C5-A8XH revealed a multipart integration pattern spanning ~200 kb of chr17, resulting in fusion of USP36-encoding DNA with upstream intergenic DNA located between the SCAT and CYTH1 genes (Fig. 4). Although the impact of this DNA fusion is yet to be determined, TCGA RNAseq confirmed sharp upregulation of USP36 expression beginning at intron 4, which may be driven by the inserted viral URR (Fig. 4). Like RSBN1, the top factor associated with USP36 expression across the TCGA ICC cohort was CNV (Pearson’s r = 0.505, p = 2.66e − 18; Supplementary Figs. S5A, B). Further, in tumours without CNV in this region, higher USP36 expression was significantly correlated with hypomethylation of a CpG site within USP36 intron 17 (cg25288675; Pearson’s r = 0.300, p < 0.0001; Supplementary Fig. S5C).
In our final example, annotation of the TAOK3 integration site in TCGA-C5-A2LX revealed amplification of a discrete segment of TAOK3 DNA (Fig. 5). PacBio sequencing successfully captured the entirety of the HPV insertion, which comprised almost two full copies of the HPV16 genome flanked on both sides by intron 9 of TAOK3 (Fig. 5). Interestingly, this specific integration has been proposed to result in a circular extrachromosomal DNA (eccDNA) structure composed of the integrated HPV16 genome flanked by ~12.3 kb TAOK3 DNA (introns 9–11) on one side and ~26.1 Kb TAOK3 DNA (introns 4–9) on the other [20]. Further supporting the potential importance of TAOK3 in ICC, eccDNA occurs in nearly half of human cancers and most commonly involves driver oncogenes that are amplified in expression as a result [46]. Across the rest of the TCGA ICC cohort, hypomethylation of a CpG site within intron 1 of TAOK3 was significantly correlated with its higher expression (cg01431992; Pearson’s r = −0.456, p = 5.84e − 16; Supplementary Fig. S5D), with the bottom 25% of TAOK3 expressers exhibiting significantly greater methylation at this site compared to the top 25% (Supplementary Fig. S5E). In summary, our in-depth annotation of integration events using multiple data sources provided testable hypotheses regarding mechanisms underlying altered candidate IDG expression in both integrated and non-integrated ICC tumours.
Functional testing of candidate IDGs in cervical cancer cell lines
As discussed above, results from our filtering scheme and annotation of IDG-associated integration events provided sufficient support to investigate the potential functional role of BNC1, RSBN1, USP36 and TAOK3 in ICC. Thus, using siRNAs designed to target each of the four candidate IDGs, we knocked down their expression in cervical cancer cell lines (Fig. 6a and Supplementary Fig. S6A) to explore how their loss affected select oncogenic processes. Knockdown of all four IDGs resulted in significantly decreased SiHa and HeLa cell migration (Fig. 6b and Supplementary Fig. S6B). Further, knockdown of all four candidate IDGs resulted in significantly decreased SiHa cell proliferation, while HeLa cell proliferation was only significantly decreased following BNC1, RSBN1, and USP36 knockdown (Fig. 6c). Colony formation assays revealed knockdown of all four candidate IDGs significantly decreased SiHa colony number, while HeLa colony numbers were significantly decreased only after BNC1 and USP36 knockdown (Fig. 6d and Supplementary Fig. S6C). The effects of IDG knockdown on SiHa cell migration, proliferation and colony formation were further validated employing a second, unique siRNA (Fig. 6 and Supplementary Fig. S6; labelled #2 in SiHa panels). Finally, we performed IDG knockdown in a patient-derived cervical cancer cell line (MCW2; Supplementary Fig. S6D) and examined the effects on cell proliferation. Like HeLa, MCW2 proliferation was significantly decreased upon knockdown of BNC1 and USP36, while TAOK3 and RSBN1 knockdown had a minimal effect (Supplementary Fig. S6E). Collectively, our data provide proof of principle for identification and functional validation of candidate IDGs via in-depth characterisation and prioritisation of host genes affected by HPV integration.
Fig. 6. Functional testing of candidate IDGs in cervical cancer cell lines.

Two cervical cancer established cell lines (SiHa and HeLa) were subjected to siRNA-mediated knockdown (KD) of each candidate IDG (labelled #1) or scrambled negative control (siCONT) and tested in three functional assays. Of note, results were validated in SiHa cells using a second, unique siRNA targeting each IDG (labelled #2 in each SiHa graph). Knockdown of each IDG was first confirmed via qRT-PCR (a). KD of all four candidate IDGs significantly decreased SiHa and HeLa cell migration (b). KD of all four IDGs significantly decreased SiHa proliferation (c; day 5). In HeLa cells, TAOK3 KD did not significantly affect cell proliferation, while KD of BNC1 (3d and 5d), RSBN1 (3d and 5d) and USP36 (3d) significantly decreased cell proliferation (c). SiHa colony formation was significantly decreased following KD of all four IDGs, while HeLa colony formation was only significantly affected by KD of BNC1 and USP36 (d). Each experiment was run in triplicate and data are presented as mean ± standard error of the mean. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Discussion
Here, we provide details of a multi-omics approach designed to thoroughly annotate HPV integration events for the identification and prioritisation of putative novel ICC targets, which we term IDGs. The power in our approach is rooted in the combination of complementary long-read sequencing of HPV-enriched tumour DNA with TCGA short-read genome sequencing data for identification and analysis of HPV integration events. Combining these datasets, we identified 87 integration events in eight ICC samples (see Supplementary Table S4). Next, we generated a list of genes from the 87 events (all genes within 2 Mb on either side of the integration) and filtered them based on their integration event clonality, an altered expression associated with viral integration and association with ICC outcome. Implementation of our filtering methods successfully cut the number of candidate IDGs from 3134 to 84. Finally, four candidate IDGs were further prioritised based on their unknown function in ICC and preliminary in vitro functional testing results presented here (see Fig. 6) suggest that each may play a potential oncogenic role in ICC. Of note, we also examined if the expression of these four IDGs associated with OS in cancers originating from other tissues of Müllerian origin represented in TCGA. Data from the ovarian cancer cohort (OV) indicated an association of high expression with worse OS for BNC1 (Supplementary Fig. S7A) and TAOK3 (Supplementary Fig. S7D), but not RSBN1 (Supplementary Fig. S7B) or USP36 (Supplementary Fig. S7C), while high expression of BNC1 (Supplementary Fig. S8A), USP36 (Supplementary Fig. S8C) and TAOK3 (Supplementary Fig. S8D), but not RSBN1 (Supplementary Fig. S8B), associated with worse OS in endometrial cancer (UCEC). Consequently, the potential oncogenic effects exerted by these genes may extend to other gynaecological cancer cohorts.
Long-read sequencing data enabled the filling in of certain gaps arising from the sole analysis of the original TCGA data, providing better spatial and structural resolution of integration events (see Figs. 2–5). Specifically, our approach combining long-read sequencing data with matched TCGA ‘omics data enabled a more thorough annotation of each integration event associated with the four IDGs studied. For example, the integration events affecting BNC1 and TAOK3 exhibited a single insertion site of HPV with human DNA flanking both sides, a pattern previously described as a co-linear HPV integration architecture [47]. Interestingly, the TAOK3 integration site in TCGA-C5-A2LX has been proposed to result in a chimeric extrachromosomal circular DNA [20], a feature also supported by our combined analysis of long-read and TCGA ‘omics data. Alternatively, the more complex integrations affecting RSBN1 and USP36 appear to reflect non-linear insertions where human genomic DNA is flanked by HPV DNA [47]. Additionally, while annotating the integration event affecting BNC1 expression in TCGA-C5-A2LV, we noticed that the RNAseq data showed high expression in an intergenic region adjacent to the integration event. Further examination of this area in the UCSC Genome Browser revealed the presence of a BNC1 enhancer, thus we postulated that increased BNC1 expression in TCGA-C5-A2LV may be a result of the virus driving expression of the BNC1 enhancer RNA (eRNA). Indeed, eRNAs are RNA molecules transcribed from DNA at active enhancers, have been shown to be involved in multiple cancer-associated signalling pathways [48–50] and can potentiate oncogenesis when altered by genetic and/or epigenetic changes [51].
The idea to use HPV integration sites for IDG identification began with our original studies using Illumina short-read sequencing of HPV-enriched ICC tumour DNA [19]. Integration analysis from this study revealed multiple integrations in the lncRNA PVT1, a gene that at the time of our analysis (2013) was associated with frequent translocations in Burkitt’s lymphoma [52, 53] and amplification in breast and ovarian cancers [54]. This ultimately resulted in our characterisation of PVT1 function in ICC [36] and this lncRNA is now a widely accepted oncogene with over 450 cancer-related publications. The current study lends further support for using integration analysis as a mechanism for pinpointing genes that may be of functional significance in driving ICC. For example, the list of 84 candidate IDGs produced from our filtering/prioritisation scheme included known oncogenes, such as NRAS (NRAS proto-oncogene, GTPase) and PVT1, and other cancer-related genes such as TOP2A (DNA topoisomerase II alpha), SOCS3 (suppressor of cytokine signalling 3) and GADD45A (growth arrest and DNA damage-inducible alpha). Thus, our results strongly reinforce the idea that HPV integration frequently disrupts important cancer genes [5, 7, 38–40] and, since the cancer genome has many genes of unknown biological significance, the approach outlined here should guide the characterisation of critical genes and their associated pathways in ICC [55, 56].
We identified HPV integration events using PacBio long-read sequencing of HPV-enriched tumour DNA from eight ICC samples. By specifically targeting those regions of the genome that harbour HPV genome sequences, pre-sequencing enrichment of DNA libraries creates a more cost-effective way to employ long-range sequencing of viral integrations in multiple samples. Further, as stated above, long-read sequencing provides details of HPV integration events not possible with standard sequencing technologies. For example, our PacBio data conclusively shows that integration events are a collection of unique, chimeric breakpoints that can span extensive regions of the human genome, thus resulting in extensive genomic disruption like that described in the literature [57]. Much work has focused on deciphering the importance of the 3D genome and how alterations to its structure, specifically at topologically associating domains (TADs), may contribute to carcinogenesis [58]. In fact, a recent publication [59] provides evidence of HPV integration-induced alterations to local chromosome architecture and 3D genome remodelling, resulting in the viral hijacking of a host enhancer in the region of integration. Future avenues of research investigating how HPV integration alters the 3D genome could shed light on novel mechanisms underlying ICC progression.
The chief goal of the current study was to examine the potential of HPV integration sites to highlight putative genes affecting cervical carcinogenesis using a novel pipeline and filtering system. Although we highlight four excellent candidate IDGs here, it is important to note that much more must be done before they could be considered true ICC gene targets. Future steps for more in-depth functional characterisation of these and other IDGs include supplementary in vitro work to determine the main pathways and potential accessory molecules crucial to cancer-promoting properties of each IDG, which may be guided by the existing literature. For example, BNC1 knockdown in mammary epithelial cells results in elevated levels of E-cadherin [60], suggestive of a role for this transcription factor in epithelial-to-mesenchymal transition. RSBN1 expression is induced by hypoxia in breast cancer cells [61], USP36 promotes ERK and Akt signalling in HeLa cells [62] and TAOK3 promotes tumour initiation and metastasis in pancreatic cells [63] and regulates human T-cell receptor signalling [64, 65]. Importantly, these oncogenic processes and signalling pathways are well characterised and easily testable, thus providing a logical springboard for future experiments delving into the precise role of the four candidate IDGs in cervical carcinogenesis. Finally, once IDG function and pathway information are complete, in vivo IDG validation and targeting would be tested using siRNA-mediated knockdown and/or CRISPR-based gene editing in cervical cancer cell lines and orthotopic xenograft models.
Although we are generally pleased with the performance of our pipeline, it does have limitations. For instance, shortcomings of our filtering criteria are that (1) expression of true oncogenes does not necessarily need to associate with survival and (2) Z-score values only tell us how the expression of a candidate IDG in the integrated sample compares to its expression across the rest of the cohort. However, it is important to note that our use of these filters was an attempt at paring down a very large number of genes to a more manageable one for study. Further, we appreciate that not all integration events will be associated with clinically relevant targets. In fact, there is still much debate regarding whether HPV integration-induced alterations to the human genome are a key player in cervical carcinogenesis or if these host genomic alterations were already present in the tumour prior to viral integration. Despite the shortcomings, our data support the use of this pipeline as a first step in exploiting HPV integration events to drive the discovery of important ICC genes. Our future work will continue to prioritise and test the function of remaining candidate IDGs from these samples and others from a large ICC cohort, with the ultimate goals of (1) fine-tuning of our pipeline filters and (2) discovering similarities across tumours/integrations for improved stratification of ICC patients into clinically actionable cohorts based on their HPV integration signatures and other oncogenic driver events.
Supplementary information
Acknowledgements
We thank the Genomic Sciences and Precision Medicine Center (GSPMC) at the Medical College of Wisconsin for their assistance and technical support with PacBio sequencing.
Author contributions
MI, Y-WH, PL, MJF and JSR designed the study concept and experiments; MI, Y-WH and MX performed the experiments; MI, S-WT, Y-WH and PL acquired, analysed and interpreted the data; MI, MJF and JSR wrote the paper. All authors discussed the results and had final approval of the submitted manuscript.
Funding information
This work was supported by the NCI R21CA241013 (to JSR) and the Women’s Health Research Programme in the Department of Obstetrics and Gynecology, Medical College of Wisconsin.
Data availability
The PacBio long-read sequencing data generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA640649.
Code availability
All codes used to generate results presented are publicly available and cited with the first mention.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was approved by the Medical College of Wisconsin’s Institutional Review Board. All the cervical cell lines used in this study were purchased from the American Type Cell Collection (ATCC, Manassas, VA).
Consent to publish
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01545-0.
References
- 1.Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004;64:3878–84. doi: 10.1158/0008-5472.CAN-04-0009. [DOI] [PubMed] [Google Scholar]
- 2.Pett M, Coleman N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol. 2007;212:356–67. doi: 10.1002/path.2192. [DOI] [PubMed] [Google Scholar]
- 3.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 4.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–46. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci USA. 2014;111:15544–9. doi: 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banister CE, Liu C, Pirisi L, Creek KE, Buckhaults PJ. Identification and characterization of HPV-independent cervical cancers. Oncotarget. 2017;8:13375–86. doi: 10.18632/oncotarget.14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan H, Krawczyk E, Blancato J, Albanese C, Zhou D, Wang N, et al. HPV positive neuroendocrine cervical cancer cells are dependent on Myc but not E6/E7 viral oncogenes. Sci Rep. 2017;7:45617. doi: 10.1038/srep45617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–11. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warburton A, Redmond CJ, Dooley KE, Fu H, Gillison ML, Akagi K, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018;14:e1007179. doi: 10.1371/journal.pgen.1007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009;5:e1000397. doi: 10.1371/journal.ppat.1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research, N., Albert Einstein College of, M., Analytical Biological, S., Barretos Cancer, H., Baylor College of, M., Beckman Research Institute of City of, H. et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–84. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McBride AA, Warburton A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017;13:e1006211. doi: 10.1371/journal.ppat.1006211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–5. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513. doi: 10.1038/ncomms3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33:690–705. doi: 10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, et al. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell. 2018;173:499–514. doi: 10.1016/j.cell.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17:239–53. doi: 10.1038/nrc.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Borecki I, Nguyen L, Ma D, Smith K, Huettner PC, et al. CD83 gene polymorphisms increase susceptibility to human invasive cervical cancer. Cancer Res. 2007;67:11202–8. doi: 10.1158/0008-5472.CAN-07-2677. [DOI] [PubMed] [Google Scholar]
- 19.Liu P, Iden M, Fye S, Huang YW, Hopp E, Chu C, et al. Targeted, deep sequencing reveals full methylation profiles of multiple HPV types and potential biomarkers for cervical cancer progression. Cancer Epidemiol Biomark Prev. 2017;26:642–50. doi: 10.1158/1055-9965.EPI-16-0368. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen ND, Deshpande V, Luebeck J, Mischel PS, Bafna V. ViFi: accurate detection of viral integration and mRNA fusion reveals indiscriminate and unregulated transcription in proximal genomic regions in cervical cancer. Nucleic Acids Res. 2018;46:3309–25. doi: 10.1093/nar/gky180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Kost J, Sulovari A, Wong N, Liang WS, Cao J, et al. A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome Res. 2019;29:819–30. doi: 10.1101/gr.242529.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Y, Long J, He J, Li CI, Cai Q, Shu XO, et al. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:194. doi: 10.1186/1471-2164-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Foundation for Statistical Computing. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
- 24.Hadley W. Ggplot2. New York: Springer Science+Business Media, LLC; 2016.
- 25.Martin MP, Borecki IB, Zhang Z, Nguyen L, Ma D, Gao X, et al. HLA-Cw group 1 ligands for KIR increase susceptibility to invasive cervical cancer. Immunogenetics. 2010;62:761–5. doi: 10.1007/s00251-010-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu KJ, Rader JS, Borecki I, Zhang Z, Hildesheim A. CD83 polymorphisms and cervical cancer risk. Gynecol Oncol. 2009;114:319–22. doi: 10.1016/j.ygyno.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017; 2017: bax028. 10.1093/database/bax028. [DOI] [PMC free article] [PubMed]
- 29.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729–D735. doi: 10.1093/nar/gky1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nattestad M, Aboukhalil R, Chin CS, Schatz MC. Ribbon: intuitive visualization for complex genomic variation. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koch A, Jeschke J, Van Criekinge W, van Engeland M, De Meyer T. MEXPRESS update 2019. Nucleic Acids Res. 2019;47:W561–65. doi: 10.1093/nar/gkz445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rau A, Flister M, Rui H, Auer PL. Exploring drivers of gene expression in the Cancer Genome Atlas. Bioinformatics. 2018. 10.1093/bioinformatics/bty551. [DOI] [PMC free article] [PubMed]
- 36.Iden M, Fye S, Li K, Chowdhury T, Ramchandran R, Rader JS. The lncRNA PVT1 contributes to the cervical cancer phenotype and associates with poor patient prognosis. PLoS ONE. 2016;11:e0156274. doi: 10.1371/journal.pone.0156274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Z, Chattopadhyay N, Liu WJ, Chan C, Pignol JP, Reilly RM. Optimized digital counting colonies of clonogenic assays using ImageJ software and customized macros: comparison with manual counting. Int J Radiat Biol. 2011;87:1135–46. doi: 10.3109/09553002.2011.622033. [DOI] [PubMed] [Google Scholar]
- 38.Durst M, Croce CM, Gissmann L, Schwarz E, Huebner K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc Natl Acad Sci USA. 1987;84:1070–4. doi: 10.1073/pnas.84.4.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bodelon C, Untereiner ME, Machiela MJ, Vinokurova S, Wentzensen N. Genomic characterization of viral integration sites in HPV-related cancers. Int J Cancer. 2016;139:2001–11. doi: 10.1002/ijc.30243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannizzaro LA, Durst M, Mendez MJ, Hecht BK, Hecht F. Regional chromosome localization of human papillomavirus integration sites near fragile sites, oncogenes, and cancer chromosome breakpoints. Cancer Genet Cytogenet. 1988;33:93–98. doi: 10.1016/0165-4608(88)90054-4. [DOI] [PubMed] [Google Scholar]
- 41.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuijers J, Manteiga JC, Weintraub AS, Day DS, Zamudio AV, Hnisz D, et al. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 2018;23:349–60. doi: 10.1016/j.celrep.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belokopytova PS, Nuriddinov MA, Mozheiko EA, Fishman D, Fishman V. Quantitative prediction of enhancer-promoter interactions. Genome Res. 2020;30:72–84. doi: 10.1101/gr.249367.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. 2016;54:1.30.1–33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 46.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543:122–5. doi: 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmes A, Lameiras S, Jeannot E, Marie Y, Castera L, Sastre-Garau X, et al. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom Med. 2016;1:16004. doi: 10.1038/npjgenmed.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, et al. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol Cell. 2017;67:1049–58. doi: 10.1016/j.molcel.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ron G, Globerson Y, Moran D, Kaplan T. Promoter-enhancer interactions identified from Hi-C data using probabilistic models and hierarchical topological domains. Nat Commun. 2017;8:2237. doi: 10.1038/s41467-017-02386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–84. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kron KJ, Bailey SD, Lupien M. Enhancer alterations in cancer: a source for a cell identity crisis. Genome Med. 2014;6:77. doi: 10.1186/s13073-014-0077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Webb E, Adams JM, Cory S. Variant (6; 15) translocation in a murine plasmacytoma occurs near an immunoglobulin kappa gene but far from the myc oncogene. Nature. 1984;312:777–9. doi: 10.1038/312777a0. [DOI] [PubMed] [Google Scholar]
- 53.Graham M, Adams JM. Chromosome 8 breakpoint far 3’ of the c-myc oncogene in a Burkitt’s lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 1986;5:2845–51. doi: 10.1002/j.1460-2075.1986.tb04578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13:5745–55. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- 55.Haynes WA, Tomczak A, Khatri P. Gene annotation bias impedes biomedical research. Sci Rep. 2018;8:1362. doi: 10.1038/s41598-018-19333-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maertens A, Tran VH, Maertens M, Kleensang A, Luechtefeld TH, Hartung T, et al. Functionally enigmatic genes in cancer: using TCGA data to map the limitations of annotations. Sci Rep. 2020;10:4106. doi: 10.1038/s41598-020-60456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24:185–99. doi: 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kantidze OL, Gurova KV, Studitsky VM, Razin SV. The 3D genome as a target for anticancer therapy. Trends Mol Med. 2020;26:141–9. doi: 10.1016/j.molmed.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao C, Hong P, Huang X, Lin D, Cao G, Wang L, et al. HPV-CCDC106 integration alters local chromosome architecture and hijacks an enhancer by three-dimensional genome structure remodeling in cervical cancer. J Genet Genomics. 2020;47:437–50. doi: 10.1016/j.jgg.2020.05.006. [DOI] [PubMed] [Google Scholar]
- 60.Feuerborn A, Mathow D, Srivastava PK, Gretz N, Grone HJ. Basonuclin-1 modulates epithelial plasticity and TGF-beta1-induced loss of epithelial cell integrity. Oncogene. 2015;34:1185–95. doi: 10.1038/onc.2014.54. [DOI] [PubMed] [Google Scholar]
- 61.Abu-Jamous B, Buffa FM, Harris AL, Nandi AK. In vitro downregulated hypoxia transcriptome is associated with poor prognosis in breast cancer. Mol Cancer. 2017;16:105. doi: 10.1186/s12943-017-0673-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim SY, Choi J, Lee DH, Park JH, Hwang YJ, Baek KH. PME-1 is regulated by USP36 in ERK and Akt signaling pathways. FEBS Lett. 2018;592:1575–88. doi: 10.1002/1873-3468.13039. [DOI] [PubMed] [Google Scholar]
- 63.Bian Y, Teper Y, Mathews Griner LA, Aiken TJ, Shukla V, Guha R, et al. Target deconvolution of a multikinase inhibitor with antimetastatic properties identifies TAOK3 as a key contributor to a cancer stem cell-like phenotype. Mol Cancer Ther. 2019;18:2097–110. doi: 10.1158/1535-7163.MCT-18-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ormonde JVS, Li Z, Stegen C, Madrenas J. TAOK3 regulates canonical TCR signaling by preventing early SHP-1-mediated inactivation of LCK. J Immunol. 2018;201:3431–42. doi: 10.4049/jimmunol.1800284. [DOI] [PubMed] [Google Scholar]
- 65.Ormonde JVS, Nie Y, Madrenas J. TAOK3, a regulator of LCK-SHP-1 crosstalk during TCR signaling. Crit Rev Immunol. 2019;39:59–81. doi: 10.1615/CritRevImmunol.2019030480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PacBio long-read sequencing data generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA640649.
All codes used to generate results presented are publicly available and cited with the first mention.