

Abstract
Aims:
To assess the burden of transactive response DNA-binding protein of 43kDa (TDP-43) inclusions in a unique cohort of old-age patients with genetic-frontotemporal lobar degeneration (gFTLD-TDP) and compare these patients with sporadic old-age individuals with TDP-43, either in the presence of Alzheimer’s disease (AD-TDP) or in isolation (pure-TDP).
Methods:
The brain bank at Mayo Clinic-Jacksonville was searched for cases ≥75 years old at death with TDP-43 extending into middle frontal cortex. Cases were split into the following groups: 1) gFTLD-TDP (n=15) with progranulin (GRN)/C9ORF72 mutations; 2) AD-TDP (n=10)-cases with median Braak neurofibrillary tangle (NFT) stage VI, Thal phase V; 3) pure-TDP (n=10)-cases with median Braak NFT stage I, Thal phase I. Clinical data were abstracted; TDP-43 burden was calculated using digital pathology.
Results:
Amnestic Alzheimer’s dementia was the clinical diagnosis in ≥50% patients in each group. The distribution of TDP-43 burden in gFTLD-TDP and AD-TDP, but not pure-TDP, was limbic-predominant targeting CA1 and subiculum. Patients with gFTLD-TDP had higher burden in entorhinal cortex compared to AD-TDP. TDP-43 burden in middle frontal cortex did not differ between the three groups.
Conclusions:
In old age it is challenging to clinically and pathologically differentiate gFTLD-TDP from AD-TDP and pure-TDP-43 based on burden. Like AD-TDP, old age gFTLD-TDP have a limbic predominant TDP-43 distribution. Given that amnestic Alzheimer’s dementia was the most common clinical diagnosis regardless of group suggests that TDP-43 directly and indirectly targets limbic regions.
Keywords: TDP-43, mutation, frontotemporal lobar degeneration, Alzheimer’s disease, old-age FTLD-TDP, LATE-NC
INTRODUCTION
Clinico-pathologic correlates in neurodegenerative diseases are heterogeneous and complex as the same abnormally aggregated proteins are linked to a wide array of phenotypes and often demographically and genetically different susceptibility groups. Phenotypical heterogeneity has been described in Alzheimer’s disease[1], α-synucleinopathies [2], and tauopathies [3]. Phenotypical differences in the above-mentioned neurodegenerative disorders have been associated with differences in pathologic distribution, types of inclusion and burden of abnormal proteins [4–8]; however, these clinico-pathologic associations are not always consistent. In the past decade transactive response DNA-binding protein of 43kDa (TDP-43) proteinopathies [9] have joined the rank of phenotypically heterogeneous neurodegenerative disorders, however, it is unclear to what extent they are pathologically heterogeneous.
Phosphorylated TDP-43 inclusions were first identified in patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration (FTLD)[10], however, since then have been widely reported as co-pathology in other neurodegenerative disorders, particularly Alzheimer’s and Lewy body diseases[11–16]. Patients with mixed Alzheimer’s disease (AD) and TDP-43 pathologies tend to be older than those considered to have FTLD-TDP and those with pure AD (i.e. without TDP-43) and usually have an amnestic presentation[17]. Elderly patients with TDP-43 and none/low likelihood AD also phenotypically resemble those with typical Alzheimer’s dementia and have TDP-43 aggregation most often but not always limited to limbic structures[18, 19]. With-that-said, TDP-43 aggregation in neocortical regions (e.g., middle frontal cortex) is not uncommon in older adults [20–22]. Pathologically, TDP-43 proteinopathy detected in the brains of older patients is similar to type A FTLD-TDP or can be associated with neurofibrillary tangles (NFT) [20, 23].
Some clinical phenotypes are strongly suggestive of underlying TDP-43 pathology and even of its certain morphologic type[23]; additionally, the presence of certain mutations (progranulin (GRN)[24] and C9ORF72 [25]) is considered diagnostic for FTLD-TDP. Nevertheless, the diagnostic certainty diminishes in older patients as the phenotypes become non-specific [17, 19]. Moreover, it is unclear whether genetic and phenotypical differences translate into pathologic differences in distribution and burden of TDP-43 pathology, especially in a demographically homogeneous population. Unfortunately, quantitative data on TDP-43 burden are scarce. Existing studies investigating the spectrum of pathologic differences among TDP-43 proteinopathies are limited by semi-quantitative methods or confounded by differences in age and differences in TDP-43 stage across comparison groups[26]. It is well known that young-onset FTLD-TDP cases are associated with striking TDP-43 burden in the frontal and temporal cortices. However, little is known about old-age genetically confirmed FTLD-TDP and whether these cases differ from similarly old age-matched sporadic cases with TDP-43 that do not have an FTLD genetic mutation, and may or may not have co-existent AD.
Given these unknowns, we sought to investigate TDP-43 burden in old-age individuals (age at death ~ 80 years) with genetically confirmed FTLD-TDP (GRN or C9ORF72) (gFTLD-TDP) and determine whether these cases differ from cases in this age-range that also have TDP-43 pathology affecting the frontal lobe (stage 6, TDP-43 in the middle frontal cortex [19, 21, 22]) but who do not have an FTLD genetic mutation. The latter sporadic group was further divided into cases with mixed TDP-43 and high likelihood AD (AD-TDP), and cases with TDP-43 and none or low likelihood of AD (pure-TDP).
MATERIALS AND METHODS
Cases
We searched our neuropathologic brain bank database at Mayo Clinic, Jacksonville, FL to identify all cases who were ≥75 years old at death and were TDP-43 positive with available brain tissue to conduct immunohistochemistry and digital quantitative burden analysis. Cases were designated as TDP-43 positive if TDP-43 immunoreactive neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear or neurofibrillary tangle (NFT)-associated inclusions were identified in the amygdala and hippocampus. All TDP-43 positive cases were further evaluated in order to categorize each case into one of the six TDP-43 stages based on published criteria: stage 1, TDP-43 deposition is limited to the amygdala; stage 2, TDP-43 deposition extends into entorhinal cortex and/or subiculum; stage 3, TDP-43 deposition extends into hippocampus dentate granule cell layer and/or occipitotemporal cortex; stage 4, TDP-43 extends into the insula, ventral striatum, basal forebrain, and/or inferior temporal cortex; stage 5, TDP-43 extends into substantia nigra, inferior olive, and/or midbrain tectum; and stage 6, TDP-43 deposition extends into middle frontal cortex and/or basal ganglia[21, 22]. For this study, because we aimed to assess TDP-43 burden in the frontal lobe, only cases with TDP-43 stage 6 were chosen for further analysis. That is, it would not make sense to quantify TDP-43 in frontal neocortex in cases with TDP-43 < stage 6 where there is no TDP-43 in the frontal lobe. TDP-43 typing was also performed[20, 27].
Genetic analysis
All 35 cases were screened for mutations in progranulin and C9ORF72 genes. Exons 0–12 and the 3′-untranslated region of the GRN gene were amplified by polymerase chain reaction using previously published primers and protocol [24, 28]. The presence of an expanded GGGGCC hexanucleotide repeat in C9ORF72 was determined by the repeat primed polymerase chain reaction method[25].
Study group designation
Cases were assigned to one of the following three groups based on genetic status and likelihood/burden of AD neuropathologic changes: 1) gFTLD-TDP (n=15) – cases with genetically confirmed progranulin (GRN), n=6, or C9ORF72, n=9, mutations; 2) AD-TDP (n=10) – cases that were negative for GRN and C9ORF72 mutations with Braak NFT stage ≥V and Thal phase ≥4; 3) pure-TDP (n=10) – cases that were negative for GRN and C9ORF72 mutations with Braak NFT stage ≤III and Thal phase ≤2. All cases had to have available hippocampal and middle frontal paraffin blocks for sectioning and further analysis. Demographic and clinical information including sex, race/ethnicity, education (years), family history of neurodegenerative disorders, clinical phenotypic diagnosis, score of the last prior to death Mini-Mental Status Examination (MMSE)[29], age at onset and disease duration was abstracted for all 35 cases.
Pathologic evaluation
Autopsy was performed within 24 hours of death in all cases. Brain tissue in all cases had no areas of discolouration, softening or putrefaction. All cases were evaluated according to standard neuropathologic examination by a single neuropathologist (DWD) following cortical sampling according to the Consortium to Establish a Registry for Alzheimer’s disease (CERAD)[30] with thioflavin S fluorescent microscopy used to assign Braak neurofibrillary tangle (NFT) stage[31], Thal phase[32], CERAD neuritic plaque score and cerebral amyloid angiopathy (CAA) scores[33]. The presence of vascular lesions (micro-infarcts, lacunar infarcts [<1cm], large infarcts [≥ 1cm]) as well as arteriolosclerosis was recorded. A five-point vascular composite score (0 – 4) incorporating all four vascular lesions (CAA, arteriolosclerosis, micro-infarcts, and lacunar/large infarcts) was used to grade cerebrovascular disease: 0 = no vascular legions present; 1 = mild arteriolosclerosis or CAA only; 2 = moderate-severe arteriolosclerosis or CAA only; 3 = presence of cortical micro-infarcts without lacunar or large infarcts; 4 = presence of lacunar or large infarcts [34, 35].
The presence of Lewy bodies in amygdala, limbic, brainstem regions or neocortical regions was documented in accordance with published consensus report[5]. Presence of any Lewy bodies rendered a case to be Lewy body disease (LBD) positive. Likelihood of AD pathology was established according to consensus recommendations [36, 37].
Hippocampal sclerosis[18] was diagnosed in cases where there was neuronal loss in the CA1 and the subiculum of the hippocampus out of proportion to the degree of AD pathology in those regions[38]; argyrophilic grains disease (AGD) was diagnosed if silver and tau-positive spindle-shaped lesions in transentorhinal and entorhinal cortex, amygdala or temporal allocortex were identified[39].
TDP-43 burden quantitative digital analysis
In all 35 cases, serial sectioning of paraffin blocks with hippocampal and middle frontal regions was performed. TDP-43 immunohistochemistry was performed using phosphorylated TDP-43 (pTDP-43) antibody (pS409/410, 1:5000 mouse monoclonal, Cosmo Bio Co., LTD) that recognized TDP-43 with phosphorylated epitopes (phosphoserine 409 and 410). Immunohistochemistry was performed using a DAKO Autostainer (Universal Staining System Carpinteria, California). All slides were scanned at 40X magnification using Aperio AT2 slide scanner (Leica Biosystems Microsystems Inc.) and stored on a dedicated server. The following regions were manually traced in Aperio ImageScope software (version 12.4.3.5008, Leica Biosystems Microsystems Inc.): dentate fascia, cornu Ammonis 1 (CA1) and subiculum of the hippocampus, entorhinal cortex (ERC) (Figure 1A) and 2 similar in size regions in the sulci of middle frontal cortex where grey matter is parallel to the white matter (Figure 1B). Each traced region was inspected for the presence of artifacts and/or debris which were excised with a negative pen tool. The burden of TDP-43 inclusions was analysed using the Aperio Positive Pixel Count algorithm (version 9, Leica Biosystems Microsystems Inc.). Positive and strong positive pixels were considered to be TDP-43 positive inclusions whereas negative and weak positive pixels were considered to be TDP-43 negative (Figure 1D and F). The total area was standardized by calculating the average total pixel count for each region (dentate fascia, CA1, etc.) and dividing the total pixel count of the given region for a specific participant by the average total pixel count for a region. The acquired ratio was used to multiply each case’s positive and strong positive pixel count for that specific region representing adjusted positive and strong positive pixel count. TDP-43 burden in a specific region was calculated by dividing the sum of adjusted positive and strong positive pixel counts by the average total pixel count and expressed in per cent (%). TDP-43 burden in the middle frontal cortex represented the average burden of the two sampled regions.
Figure 1. Region selection and quantitative TDP-43 burden analysis methodology.
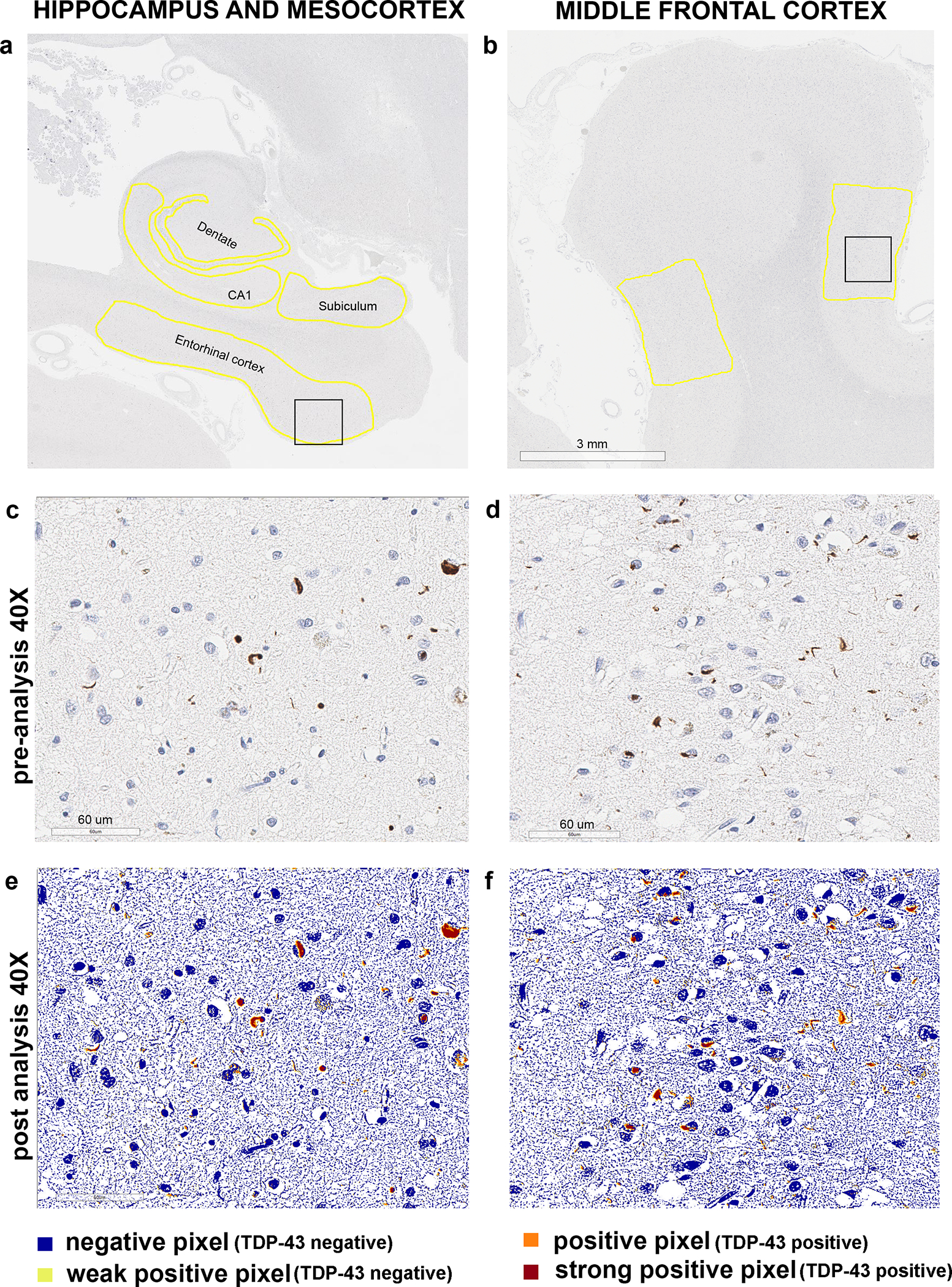
Panels a-b show how regions of interest were traced for each study sample. Black squares represent the regions which were magnified at 40X in panels c-f. Panels c and e show 40X magnified pre-analysis and post-analysis region of the entorhinal cortex; panels d and f show 40X magnified pre-analysis and post-analysis region of the middle frontal cortex. Positive and strong positive pixels were considered TDP-43 positive
Statistical analysis
All statistical analysis was performed in JMP Pro software (version 14.1.0, 2018 SAS Institute Inc.) Nonparametric Steel-Dwass test was used to compare continuous variables among three groups in order to control for multiple comparisons. Fisher’s exact test was used for the categorical variables. Significance was set at p <0.05.
Figures were generated in RStudio software (version 1.2.5042; RStudio Inc.). Adobe Photoshop CC 2018 (version 19.1.6; Adobe Systems Inc.) was used for figure assembly and labelling.
RESULTS
Representative images of TDP-43 inclusions in the entorhinal and middle frontal cortices for each group, and specifically for GRN and C9ORF72 genetic mutation carriers, are shown in Figure 2. Demographic and clinical characteristics of the cases by group are summarized in Table 1, and no statistically significant differences were identified. The most frequent ante mortem clinical diagnosis in all the groups was Alzheimer’s dementia, including 53% of gFTLD-TDP cases and 50% of pure-TDP cases, followed by an FTLD spectrum clinical diagnosis. The gFTLD-TDP cases had a lower frequency of high likelihood AD compared to AD-TDP but did not differ from pure-TDP (Table 2). The AD-TDP group had a higher frequency of Lewy body disease compared to gFTLD-TDP.
Figure 2. Representative images of TDP-43 positive inclusions for each study group and mutation type.
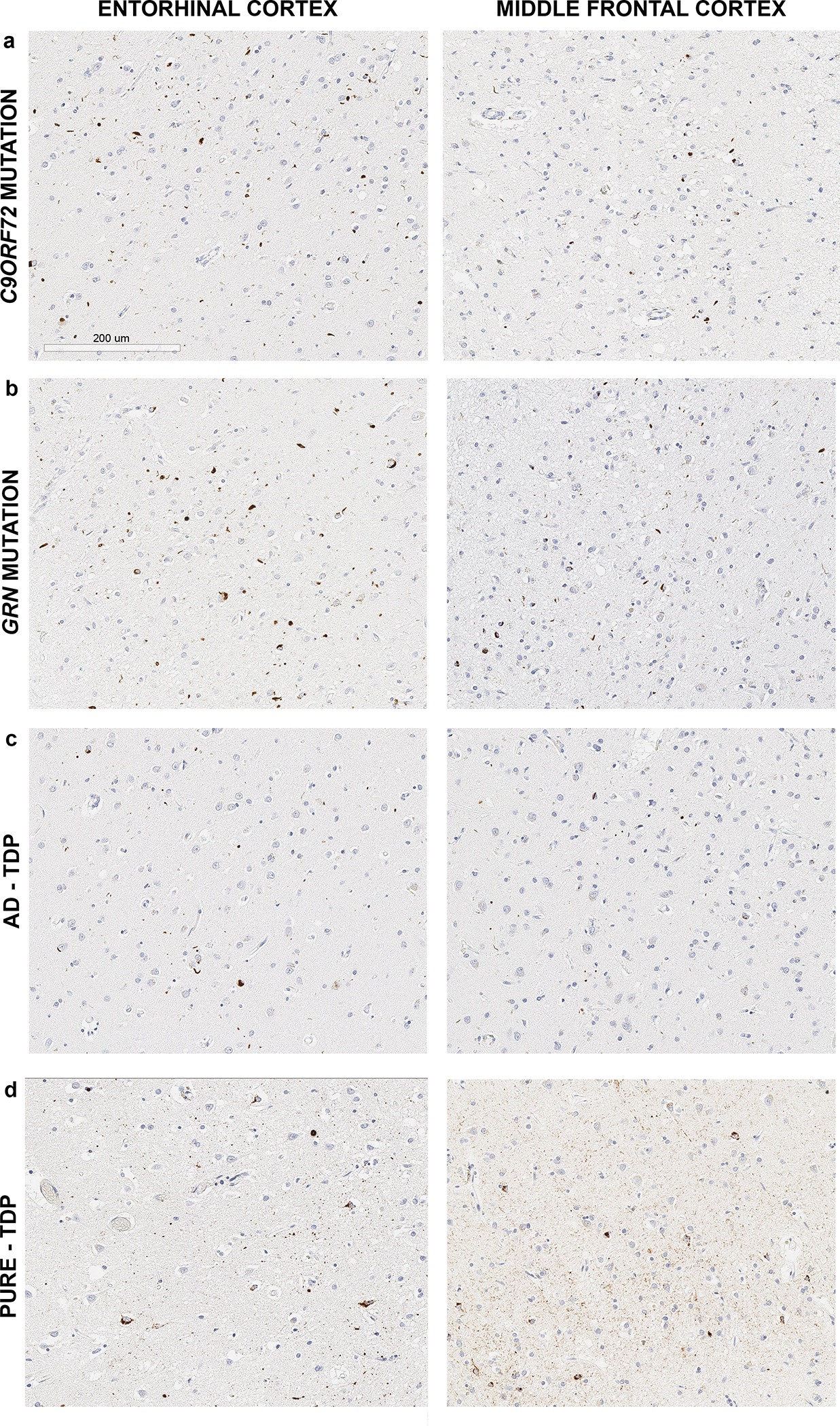
All images were taken at 20X magnification
Table 1.
Demographic and clinical characteristics of the three study groups
| Characteristic | No. or median (% or interquartile range) | |||
|---|---|---|---|---|
| gFTLD-TDP (n=15) | AD-TDP (n=10) | Pure-TDP (n=10) | p-value | |
| Female | 6 (40%) | 5 (50%) | 4 (40%) | 0.91 |
| White/Caucasian | 14 (93%) | 10 (100%) | 10 (100%) | >0.99 |
| Age at onset, years | 68 (66 – 70) | 69 (67 – 75) | 73 (67 – 77) | 0.20 |
| Disease duration, years | 10 (9 – 15) | 11 (10 – 18) | 9 (5 – 14) | 0.27 |
| Age at death, years | 79 (76 – 84) | 83 (78 – 87) | 78 (77 – 87) | 0.24 |
| Family history | 10 (67%) | 8 (80%) | 4 (40%) | 0.18 |
| Education, years | 15 (12 – 16) | 16 (12 – 17) | 16 (14 – 17) | 0.67 |
| Last MMSE/30 | 13 (5 – 25) | 17 (5 – 22) | 16 (2 – 22) | 0.85a |
| Clinical diagnosis | ||||
| FTLD clinical spectrum diagnosis | 5 (33%) | 3 (30%) | 3 (30%) | |
| Frontotemporal dementia (FTD) | 2 (13%) | 0 (0%) | 0 (0%) | 0.75 |
| FTD-Motor Neuron Disease | 0 (0%) | 0 (0%) | 1 (10%) | |
| Primary progressive aphasia | 2 (13%) | 1 (10%) | 1 (10%) | |
| Progressive supranuclear palsy | 1 (7%) | 0 (0%) | 1 (10%) | |
| Corticobasal syndrome | 0 (0%) | 2 (20%) | 0 (0%) | |
| Alzheimer’s dementia | 8 (53%) | 7 (70%) | 5 (50%) | |
| Dementia with Lewy bodies | 1 (7%) | 0 (0%) | 1 (10%) | |
| Vascular dementiab | 1 (7%) | 0 (0%) | 1 (10%) | |
Data are represented as median (interquartile range) for continuous variables or number (per cent) for categorical variables. P-values are from Kruskal-Wallis test for continuous variables or Fisher’s exact test for categorical variables
Adjusted for disease duration at the time of last MMSE
No or minimal evidence (vascular score 0/1) of cerebrovascular disease on brain autopsy in both cases
Table 2.
Neuropathologic characteristics of the three study groups
| No. or median (% or interquartile range) | ||||
|---|---|---|---|---|
| Characteristic | gFTLD-TDP (n=15) | AD-TDP (n=10) | Pure-TDP (n=10) | p-value |
| Time to autopsy, hours | 3 (3 – 5) | 18 (4 – 19) | 8.5 (3 – 14) | 0.13 |
| Brain weight, grams | 920 (740 – 1040) | 980 (818 – 1095) | 970 (900 – 1180) | 0.37 |
| Tau | ||||
| Median Braak NFT stage | 2.5 (1 – 3) | 5.5 (4 –6) | 1 (1 – 2) | <0.0001a,c |
| Braak 0 | 2 (13%) | 0 (0%) | 1 (10%) | |
| Braak I | 2 (13%) | 0 (0%) | 5 (50%) | |
| Braak II | 3 (20%) | 0 (0%) | 3 (30%) | |
| Braak III | 5 (33%) | 0 (0%) | 1 (10%) | |
| Braak IV | 1 (7%) | 2 (20%) | 0 (0%) | |
| Braak V | 2 (13%) | 3 (30%) | 0 (0%) | |
| Braak VI | 0 (0%) | 5 (50%) | 0 (0%) | |
| Amyloid-ß | ||||
| Median CERAD | 0 (0 – 0) | 3 (1 – 3) | 0 (0 – 0) | <0.0001a,c |
| Median Thal phase | 1 (0 – 2) | 5 (4 – 5) | 1 (0 – 1) | <0.0001a,c |
| 0 (no amyloid-β) | 5 (33%) | 0 (0%) | 4 (40%) | |
| 1 (neocortical) | 6 (40%) | 0 (0%) | 4 (40%) | |
| 2 (allocortex) | 2 (13%) | 0 (0%) | 2 (20%) | |
| 3 (thalamus, striatum) | 1 (7%) | 0 (0%) | 0 (0%) | |
| 4 (brainstem) | 1 (7%) | 4 (40%) | 0 (0%) | |
| 5 (cerebellum) | 0 (0%) | 6 (60%) | 0 (0%) | |
| TDP-43 type | ||||
| Type A/alpha | 13 (87%) | 10 (100%) | 9 (90%) | 0.77 |
| Type B | 2 (13%) | 0 (0%) | 1 (10%) | |
| Alzheimer’s disease neuropathologic changes | ||||
| High/intermediate likelihood | 3 (20%) | 10 (100%) | 0 (0%) | |
| Low likelihood/none | 12 (80%) | 0 (0%) | 10 (100%) | <0.0001a,c |
| Hippocampal sclerosis | 10 (67%) | 7 (70%) | 8 (80%) | 0.89 |
| moderate/severe | 9 (60%) | 7 (70%) | 8 (80%) | 0.60 |
| Lewy body disease | 0 (0%) | 4 (40%) | 1 (10%) | 0.015 a |
| Incidental | 0 (0%) | 0 (0%) | 1 (10%) | |
| Brainstem-predominant | 0 (0%) | 1 (10%) | 0 (0%) | |
| Amygdala-predominant | 0 (0%) | 0 (0%) | 0 (0%) | |
| Transitional/limbic | 0 (0%) | 2 (20%) | 0 (0%) | |
| Diffuse/neocortical | 0 (0%) | 1 (10%) | 0 (0%) | |
| Cerebrovascular disease | ||||
| Vascular score (0 – 4) | 1 (0 – 2) | 2 (1 – 3) | 1.5 (1 – 4) | 0.20 |
| 0 (no lesions) | 7 (47%) | 0 (0%) | 2 (20%) | |
| 1 (mild CAA/arteriolosclerosis) | 1 (7%) | 4 (40%) | 3 (30%) | |
| 2 (≥moderate CAA/arteriolosclerosis) | 4 (27%) | 2 (20%) | 1 (10%) | 0.12 |
| 3 (micro-infarcts, no large infarcts) | 2 (13%) | 2 (20%) | 1 (10%) | |
| 4 (lacunar/large infarcts) | 1 (7%) | 2 (20%) | 3 (30%) | |
| Other pathologies | ||||
| Argyrophilic grains disease | 2 (13%) | 0 (0%) | 0 (0%) | 0.50 |
Data are represented as median (interquartile range) for continuous variables or number (per cent) for categorical variables. P-values are from Kruskal-Wallis test for continuous variables or Fisher’s exact test for categorical variables
significant difference between gFTLD-TDP and AD-TDP, p<0.05
significant difference between gFTLD-TDP and pure-TDP, p<0.05
significant difference between AD-TDP and pure-TDP, p<0.05
TDP-43 burden in gFTLD-TDP did not differ from AD-TDP or pure-TDP in the frontal cortex, dentate fascia, or subiculum (Figure 3). The gFTLD-TDP cases had a higher TDP-43 burden in the entorhinal cortex compared to AD-TDP and showed a trend for a higher burden in the CA1 region compared to the pure-TDP cases (Figure 3). Overall, there was a limbic-predominant pattern of TDP-43 deposition in gFTLD-TDP, with the highest burdens of TDP-43 in CA1, the subiculum and entorhinal cortex compared to the frontal cortex. The AD-TDP cases similarly showed a limbic-predominant pattern with the highest burdens of TDP-43 occurring in CA1 and subiculum. This limbic-predominant pattern was not observed in pure-TDP where with highest burden in frontal neocortex.
Figure 3. Box plots showing regional TDP-43 burden differences among the three study groups.
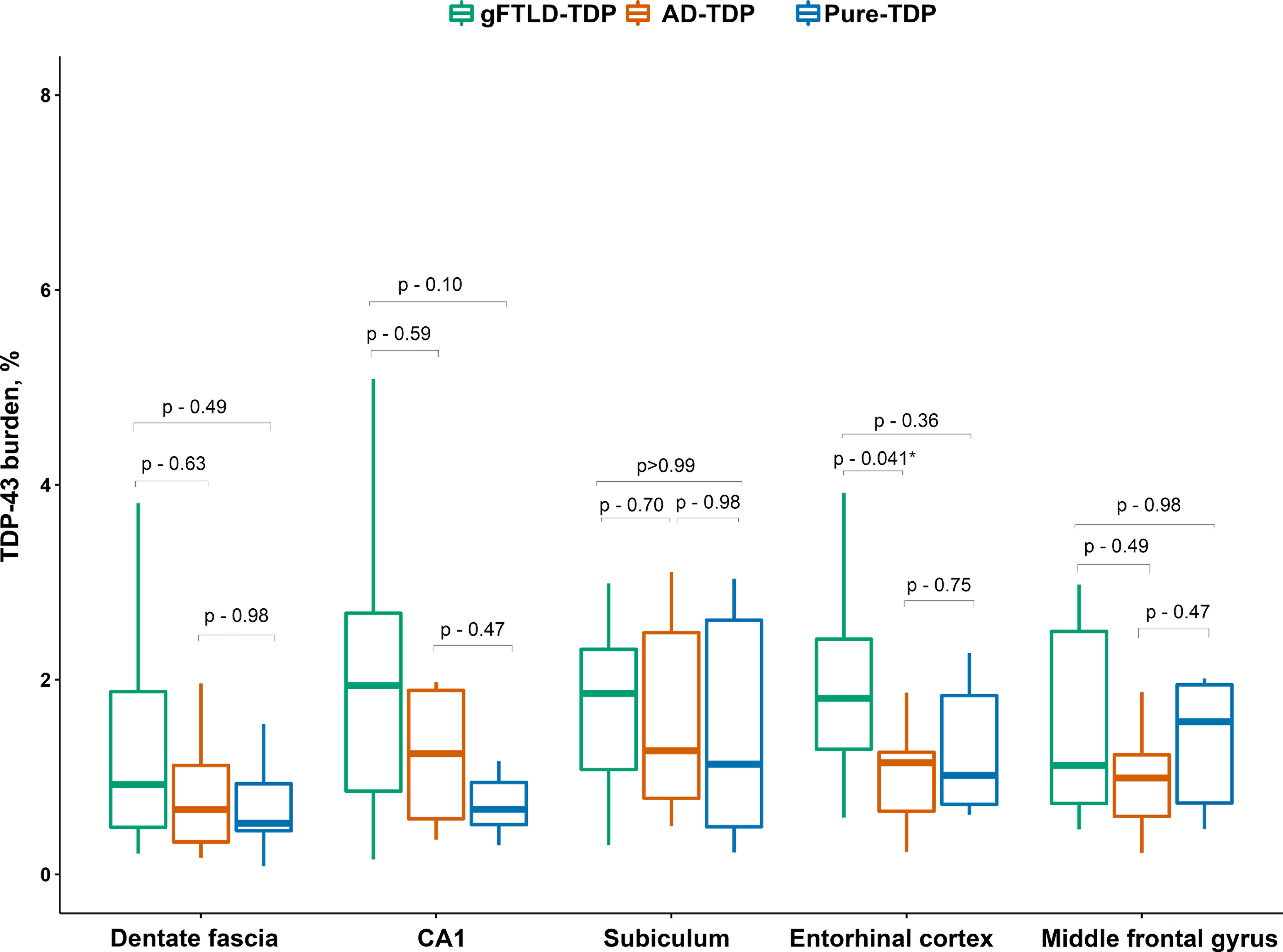
The line in box plots represents the median, the box represents interquartile range and the whiskers correspond to minimum and maximum values. P-values are from Steel-Dwass test
Within the gFTLD-TDP group, there were no differences in demographic variables between GRN and C9ORF72 positive cases, pathologically C9ORF72 cases had more tau NFT pathology (Supplemental Table 1). Cases with C9ORF72 mutations (n=9) tended to have higher TDP-43 burden in the middle frontal gyrus compared to the cases with GRN mutations (n=6), although difference did not reach significance (Supplemental Figure 1).
Three of fifteen gFTLD-TDP cases (20%) had concomitant intermediate/high likelihood AD pathology. We, therefore, compared regional TDP-43 burden and its distribution in gFTLD-TDP case with intermediate/high likelihood AD pathology to gFTLD-TDP cases with none/low likelihood AD pathology and found no statistically significant differences or trends.
Most of our cases were gFTLD-TDP type A or AD-TDP type α. We re-ran all our analyses with inclusion of only TDP-43 type A/α cases (gFTLD-TDP, n = 13; AD-TDP, n =10; pure-TDP, n=9). The results were unchanged as with the full cohort. Likewise, the results of analysis did not change when GRN and C9ORF72 positive cases with TDP-43 inclusions type A were compared.
DISCUSSION
In this study of old-age genetic FTLD-TDP, non-mutation associated mixed AD and TDP-43 proteinopathies and pure TDP-43 cases the three groups were not easily discernible from each other based on TDP-43 burden, although, some regional differences were identified. Most importantly, old-age genetic FTLD-TDP (gFTLD-TDP) shows a limbic predominant pattern of TDP-43 burden, similar to AD-TDP type-α stage 6, with both groups having a limbic-predominant TDP-43 burden, whereas pure TDP-43 stage 6 showed a relatively equally distributed or more neocortical TDP-43 burden.
The findings of our study are novel, thought provoking and challenge current concepts in the field. While GRN and C9ORF72 mutations always present with TDP-43 pathology, are considered to have high penetrance and usually present with an early-onset disease [24, 25], we report a series of mutation cases that had TDP-43 at autopsy, justifying that the mutations are the primary cause of the TDP-43 aggregation, but surprisingly lived until old age; most patients also had later than usual onset of cognitive or behavioural difficulties. These old at death gFTLD-TDP cases had more TDP-43 burden in limbic and mesocortical regions, particularly CA1 hippocampal subfield, subiculum and entorhinal cortex. Therefore, old gFTLD-TDP cases have a limbic-predominant distribution of TDP-43 burden with less pathology in the neocortex, which is different from genetic and sporadic young onset FTLD-TDP cases that die at a younger age[40]. This finding while challenging the current belief is in fact similar to that seen for tau and AD where a higher burden of tau pathology in cortical regions is characteristic of young early-onset Alzheimer’s disease[4, 41] but not old age AD. It is well known that age is related to the amount, distribution and speed of aggregation of abnormal protein, at least for tau, which appears to be similar for TDP-43. Therefore, the burden and distribution of TDP-43 related to mutations in GRN and C9ORF72 genes might represent a pathologic and possibly phenotypic continuum of mutation-associated disease rather than alternative age-related diagnoses.
When compared to other old non-mutation-associated/sporadic TDP-43 cases with or without concomitant AD pathology, it is important to emphasize that TDP-43 burden did not differ among the three groups in most regions analysed. Most importantly, TDP-43 burden in gFTLD-TDP cases did not differ in the middle frontal cortex, which is discordant with the findings reported from another study where patients with FTLD-TDP, often mutation-associated, had more TDP-43 pathology in the frontal regions compared to mixed AD-TDP/pure-TDP patients referred to as limbic-predominant age-related TDP-43 encephalopathy (LATE) [19, 26]. There are several explanations for these differences. First, in the above-mentioned study, the patients with FTLD-TDP were on average two decades younger than those with LATE. That is, they were not matched for age at death, and, as we mentioned before, age is a known factor that can affect the amount and distribution of abnormal protein deposition [4, 41]. Second, not all patients in the pure LATE group had TDP-43 inclusions that extended into neocortex, hence, it is not surprising that cortical burden would be less in those cases compared to the FTLD-TDP cases where in all instances TDP-43 did extended into frontal cortex [26]. Lastly, in contrast to our study that used digital quantitative methods for burden determination, the other study used a semi-quantitative approach to measure the amount of TDP-43 pathology. Our results, therefore, challenge the suggestion made previously that TDP-43 burden in frontal cortex can differentiate FTLD-TDP from non-FTLD TDP-43. Additionally, FTLD with motor neuron disease (FTLD-MND) cases, an FTLD-TDP variant, typically do not have much TDP-43 in neocortex [27, 42–44].
Whereas no difference in TDP-43 burden was found in the frontal cortex, that was not the case for limbic and mesocortical regions. Our finding of higher TDP-43 burden in the entorhinal cortex in gFTLD-TDP cases compared to AD-TDP, but not pure-TDP, is of interest. This finding provides evidence that there are regional differences in TDP-43 burden between mutation-associated vs. non-mutation cases in the patients over age of 75, though not in the regions one would expect.
One co-pathology that needs to be mentioned is hippocampal sclerosis. The CA1 was the region with the highest TDP-43 burden overall in the gFTLD-TDP cases. The relatively higher burden in CA1 could be related to the frequency of hippocampal sclerosis among groups. Though not statistically significant, most patients with pure-TDP had hippocampal sclerosis compared to the AD-TDP and gFTLD-TDP cases, with lowest frequency of severe hippocampal sclerosis among cases with C9ORF72 expansion. The lower burden of TDP-43 in CA1 of patients with pure-TDP may be related to the higher degree of gliosis and replacement of the neuronal population with scar tissue. This raises the question whether the lower burden of TDP-43 in the pure-TDP group is the sign of milder TDP-43 pathology or on the contrary the marker of higher burden of TDP-43 prior to death that led to atrophy and gliosis?
Another theory for higher TDP-43 burden in CA1 and other limbic or mesocortex can be the reflection of age-related predilection of protein aggregation as it is seen with tau in early onset vs. typical Alzheimer’s dementia[4]. In that context, it is relevant to point out the similarities in TDP-43 distribution between old-age gFTLD-TDP and AD-TDP observed in this study. Though there was a significant difference in TDP-43 burden in the entorhinal cortex between gFTLD-TDP and AD-TDP, is this difference sufficient to make a distinction between these two pathologic entities? An even bigger question, is there a true difference between gFTLD-TDP and AD-TDP, or do the latter cases in fact represent sporadic old-age FTLD with AD (oFTLD-AD).
Our results do not provide evidence of differences in TDP-43 burden depending on the type of FTLD mutation, however, certain trends have been observed. Both the C9ORF72 and GRN cases showed a limbic-predominant pattern of TDP-43 deposition. Though not statistically significant, cases with C9ORF72 mutation tended to have higher TDP-43 burden in middle frontal gyrus, as well as CA1 and entorhinal cortex. An earlier study showed that there might be potential differences in the burden of C-terminal, N-terminal and full length TDP-43 species associated with presence or absence of a GRN or C9ORF72 mutation, however, the sample size was small to compare the burden of above mentioned TDP-43 species between the mutations[45]. Differences in TDP-43 species, types of inclusions, as well as other factors might play a role in overall TDP-43 burden differences between patients with GRN or C9ORF72 mutations. This warrants a larger study aimed to investigate TDP-43 burden differences between cases with GRN and C9ORF72 mutations.
Another interesting finding from our study was the phenotypic presentation of our cases. Though it is expected that most cases with AD-TDP would have had clinical Alzheimer’s dementia, amnestic Alzheimer’s dementia was also the clinical diagnosis for half or more of cases in both the gFTLD-TDP and pure-TDP groups. One would certainly not predict the most frequent clinical diagnosis in any genetic FTLD-TDP group to not be a frontotemporal dementia, and instead be an amnestic Alzheimer’s dementia. This finding emphasizes that the majority of patients with GRN or C9ORF72 mutations who develop symptom at older age do not present with the typical features of frontotemporal dementia commonly encountered at younger age[46]. It is unclear, however, to what degree the clinical diagnosis in these cases is driven by the actual clinical phenotype, which is more typical for Alzheimer’s dementia, or whether it is biased by the old age; the finding of a limbic pattern of TDP-43 burden would be in support of the former. Regardless, based on pathologic data and the fact that amnestic Alzheimer’s dementia was the most common clinical diagnosis across all three groups suggests that TDP-43 must be targeting the hippocampus, either directly or indirectly in the elderly.
In terms of pure-TDP cases, where half of the patients received a clinical diagnosis of Alzheimer’s dementia without having any significant Alzheimer’s pathology on autopsy, this finding reaffirms that TDP-43 proteinopathy not only aggravates Alzheimer’s associated clinical phenotype [17, 47] but now provides evidence that TDP-43 in isolation is capable of causing cognitive impairment severe enough to render a dementia. The high frequency of hippocampal sclerosis in the pure-TDP cases might further be responsible for Alzheimer’s dementia phenotype in this group[48, 49].
Quantitative burden analysis is one of the biggest strengths of this study. Strict selection criteria allowing for distinct not overlapping groups add robustness to our analysis increasing the chance to detect differences if such exist. Importantly, our groups did not differ in age which is a strong risk factor for multiple neurodegenerative diseases and can be associated with differences in burden[4, 14]. The sample size, considering the rarity of pure-TDP and gFTLD-TDP cases that die at an older age, is quite large, however, larger sample size could have allowed detection of additional differences, we otherwise observed only at a trend level. We were unable to compare the burden between different types of inclusions, e.g., dystrophic neurites vs. intracytoplasmic inclusions. The gFTLD-TDP group in our study included patients only with mutations in GRN or C9ORF72 genes, which are the most frequent mutations that lead to FTLD-TDP; however, in rare instances mutations in other genes (i.e. VCP, OPTN)[50, 51] have been reported. Therefore, the results of our study would not generalize to those mutation-associated FTLD cases. In addition, our findings are specific to AD+TDP and Pure TDP were TDP-43 deposition has spread to frontal cortex, i.e. Josephs stage 6 or LATE_NC stage 3.
In conclusion, in patients of advanced age, it is challenging to distinguish neurodegenerative disorders not just phenotypically but also pathologically. Access to demographic, clinical and genetic data might be needed to help guide neuropathologic diagnosis, however, might also lead to additional biases. Nevertheless, there are some differences in TDP-43 burden in genetic FTLD-TDP vs. Alzheimer’s associated vs. pure TDP-43 cases (at stage 6) predominantly in limbic and mesocortical structures. Patients harbouring mutations have higher TDP-43 burden compared to non-mutation associated TDP-43 proteinopathies in mesocortex; however, mutations in patients who died at older age were not associated with higher TDP-43 burden in neocortical regions compared to non-mutation TDP-43 cases in contrast to younger patients with mutations [26]. Whether these differences are of sufficient magnitude to be clinically impactful is, however, unclear and further research is warranted. There were also similarities between old gFTLD-TDP and sporadic TDP-43 with and without AD that would be in support of the existence of old-age FTLD-TDP (oFTLD-TDP).
Supplementary Material
ACKNOWLEDGEMENTS
MB participated in study design, performed digital and statistical analysis of the data and interpreted the results, drafted the manuscript, all figures and tables. MCB and RR developed, performed and interpreted the genetic analysis, revised the manuscript for intellectual content. DWD performed pathologic evaluation of all the cases, revised the manuscript for intellectual content. KAJ conceptualized, designed and supervised the study, provided the funding, revised the manuscript for intellectual content. All authors read and approved the final manuscript.
This study was funded by National Institutes of Health grant R01 AG037491–11. This grant served for the design and conduct of the study, collection, management and analysis of the collected data. The sponsor had no role in study design; collection, analysis and interpretation of data; writing the report; or in the decision to submit the article for publication.
COMPETING INTERESTS
RR has patents US9486541B2 and US20090291444A1. KAJ is the recipient of the NIH grant that funded the present study. Other authors declare that they have no competing interests.
Footnotes
DATA SHARING AND DATA ACCESSIBILITY
Anonymized data are available from the corresponding author upon request from any qualified investigator for purposes of replicating procedures and results.
ETHICAL APPROVAL
This study was approved by the Mayo Clinic institutional review board; all participants and/or their proxies signed a written informed consent form before taking part in any research activities in accordance with the Declaration of Helsinki.
REFERENCES
- 1.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. The Lancet Neurology 2014; 13: 614–29 [DOI] [PubMed] [Google Scholar]
- 2.McCann H, Stevens CH, Cartwright H, Halliday GM. α-Synucleinopathy phenotypes. Parkinsonism & related disorders 2014; 20: S62–S7 [DOI] [PubMed] [Google Scholar]
- 3.Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism & related disorders 2016; 22: S29–S33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. The Lancet Neurology 2011; 10: 785–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017; 89: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80: 496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauw J-J, Daniel S, Dickson D, Horoupian D, Jellinger K, Lantos P, McKee A, Tabaton M, Litvan I. Preliminary NINDS neuropathologic criteria for Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy). Neurology 1994; 44: 2015– [DOI] [PubMed] [Google Scholar]
- 8.Litvan I, Hauw J, Bartko J, Lantos P, Daniel S, Horoupian D, McKee A, Dickson D, Bancher C, Tabaton M. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. Journal of Neuropathology & Experimental Neurology 1996; 55: 97–105 [DOI] [PubMed] [Google Scholar]
- 9.Tan RH, Kril JJ, Fatima M, McGeachie A, McCann H, Shepherd C, Forrest SL, Affleck A, Kwok JB, Hodges JR. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain 2015; 138: 3110–22 [DOI] [PubMed] [Google Scholar]
- 10.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130–3 [DOI] [PubMed] [Google Scholar]
- 11.Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM, Petrucelli L, Senjem ML, Ivnik RJ, Parisi JE. TAR DNA‐binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Annals of neurology 2015; 78: 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J. TDP‐43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain pathology 2017; 27: 472–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buciuc M, Whitwell JL, Boeve BF, Ferman TJ, Graff-Radford J, Savica R, Kantarci K, Fields JA, Knopman DS, Petersen RC. TDP-43 is associated with a reduced likelihood of rendering a clinical diagnosis of dementia with Lewy bodies in autopsy-confirmed cases of transitional/diffuse Lewy body disease. Journal of neurology 2020: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018; 141: 2181–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amador‐Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis. Handbook of clinical neurology 2008; 89: 569–72 [DOI] [PubMed] [Google Scholar]
- 16.Amador‐Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff‐Radford NR, Hutton ML, Dickson DW. TDP‐43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Annals of neurology 2007; 61: 435–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta neuropathologica 2014; 127: 811–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson DW, Davies P, Bevona C, Van Hoeven K, Factor S, Grober E, Aronson M, Crystal H. Hippocampal sclerosis: a common pathological feature of dementia in very old (≥ 80 years of age) humans. Acta neuropathologica 1994; 88: 212–21 [DOI] [PubMed] [Google Scholar]
- 19.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, Matchett BJ, Jack CR, Knopman DS, Petersen RC. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta neuropathologica 2019; 137: 227–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, Petersen RC, Dickson DW. Staging TDP-43 pathology in Alzheimer’s disease. Acta neuropathologica 2014; 127: 441–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta neuropathologica 2016; 131: 571–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta neuropathologica 2011; 122: 111–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916–9 [DOI] [PubMed] [Google Scholar]
- 25.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, Van Deerlin VM, Abner EL, Jicha GA, Barber JM. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 2020; 143: 2844–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta neuropathologica 2009; 118: 349–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Human molecular genetics 2006; 15: 2988–3001 [DOI] [PubMed] [Google Scholar]
- 29.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. Journal of psychiatric research 1975; 12: 189–98 [DOI] [PubMed] [Google Scholar]
- 30.Mirra SS, Heyman A, McKeel D, Sumi S, Crain BJ, Brownlee L, Vogel F, Hughes J, Van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41: 479– [DOI] [PubMed] [Google Scholar]
- 31.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta neuropathologica 2006; 112: 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800 [DOI] [PubMed] [Google Scholar]
- 33.Ellis R, Olichney JM, Thal L, Mirra S, Morris J, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 1996; 46: 1592–6 [DOI] [PubMed] [Google Scholar]
- 34.Josephs KA, Martin PR, Weigand SD, Tosakulwong N, Buciuc M, Murray ME, Petrucelli L, Senjem ML, Spychalla AJ, Knopman DS. Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 2020: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, Kalaria RN, King A, Lammie GA, Mann D, Neal J. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 2016; 139: 2957–69 [DOI] [PubMed] [Google Scholar]
- 36.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & dementia 2012; 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica 2012; 123: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rauramaa T, Pikkarainen M, Englund E, Ince PG, Jellinger K, Paetau A, Alafuzoff I. Consensus recommendations on pathologic changes in the hippocampus: a postmortem multicenter inter-rater study. Journal of Neuropathology & Experimental Neurology 2013; 72: 452–61 [DOI] [PubMed] [Google Scholar]
- 39.Jellinger KA. Dementia with grains (argyrophilic grain disease). Brain pathology 1998; 8: 377–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yousef A, Robinson JL, Irwin DJ, Byrne MD, Kwong LK, Lee EB, Xu Y, Xie SX, Rennert L, Suh E. Neuron loss and degeneration in the progression of TDP-43 in frontotemporal lobar degeneration. Acta neuropathologica communications 2017; 5: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gefen T, Gasho K, Rademaker A, Lalehzari M, Weintraub S, Rogalski E, Wieneke C, Bigio E, Geula C, Mesulam M-M. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 2012; 135: 1554–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta neuropathologica 2006; 112: 539–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandmeir NJ, Geser F, Kwong LK, Zimmerman E, Qian J, Lee VM-Y, Trojanowski JQ. Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta neuropathologica 2008; 115: 123–31 [DOI] [PubMed] [Google Scholar]
- 44.King A, Maekawa S, Bodi I, Troakes C, Al‐Sarraj S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP‐43 proteinopathies but are only rarely additionally immunopositive for phosphorylation‐dependent TDP‐43. Neuropathology 2011; 31: 239–49 [DOI] [PubMed] [Google Scholar]
- 45.Josephs KA, Zhang Y-J, Baker M, Rademakers R, Petrucelli L, Dickson DW. C-terminal and full length TDP-43 specie differ according to FTLD-TDP lesion type but not genetic mutation. Acta neuropathologica communications 2019; 7: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, Van Swieten JC, Seelaar H, Dopper EG, Onyike CU. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134: 2456–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, Rademakers R, Boeve BF, Parisi JE, Smith GE. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008; 70: 1850–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer disease and associated disorders 2011; 25: 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brenowitz WD, Monsell SE, Schmitt FA, Kukull WA, Nelson PT. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. Journal of Alzheimer’s Disease 2014; 39: 691–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. Journal of Neuropathology & Experimental Neurology 2007; 66: 152–7 [DOI] [PubMed] [Google Scholar]
- 51.Hortobágyi T, Troakes C, Nishimura AL, Vance C, Van Swieten JC, Seelaar H, King A, Al-Sarraj S, Rogelj B, Shaw CE. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta neuropathologica 2011; 121: 519–27 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.