

Abstract
Introduction:
There are more than two dozen bispecific antibodies (BsAb’s) in development with a variety of designs which are relevant to breast cancer. The field of BsAbs for breast cancer includes agents that co-direct immune recognition of the cancer cell, target unique cancer antigens, and target the microenvironment. BsAb’s are being developed for use as antibody-drug conjugates and as homing signals for engineered T-cells.
Areas covered:
This review covers potential targets for bispecific antibodies, agents in pre-clinical development, agents in clinical trials, combinatorial therapies and future directions.
Expert opinion:
There is no BsAb approval expected for breast cancer in the near term, but late stage trials are underway. Future BsAb roles in breast cancer are possible given unmet needs in estrogen receptor+ disease, residual disease and de-escalating chemotherapy use. The HER2+ space shows hints of success for BsAb’s, but is already crowded. Areas of unmet need still exist.
Keywords: bispecific antibody, breast cancer, immunotherapy, clinical trials, cellular therapy
1. Introduction and Background
A bispecific antibody (BsAb) is generally a protein construct that can simultaneously bind two or more different antigens and is based off naturally occurring mammalian antibody protein sequences (Figure 1). Usually, one end of the BsAb construct targets an antigen on the effector cell and the other end of the construct targets an antigen on tumor cells. There are currently many variations in design and application which are relevant to breast cancer therapy and research. Historically, the earliest description of antibody was made by Paul Ehrlich in 1891 and the “lock-and-key” specificity of antibodies were confirmed by Linus Pauling in the 1940s. When the antibody structure was published in 1972, it resulted in a Nobel prize, but full recognition of the natural bispecificity of an antibody molecule not well reported until 1983 by Aalberse et al.1 The first lab-made BsAb was reported in 1983 by Nisonoff et al.2 In 1984, a BsAb that could recruit T-cells in vivo was reported by Staerz et al3. In the 1990s with the advent of hybridoma technologies, there were several clinical trials using BsAbs, but the problem of anti-mouse antibodies and the difficulty in manufacturing large quantities of BsAbs limited the early development.
Figure 1:
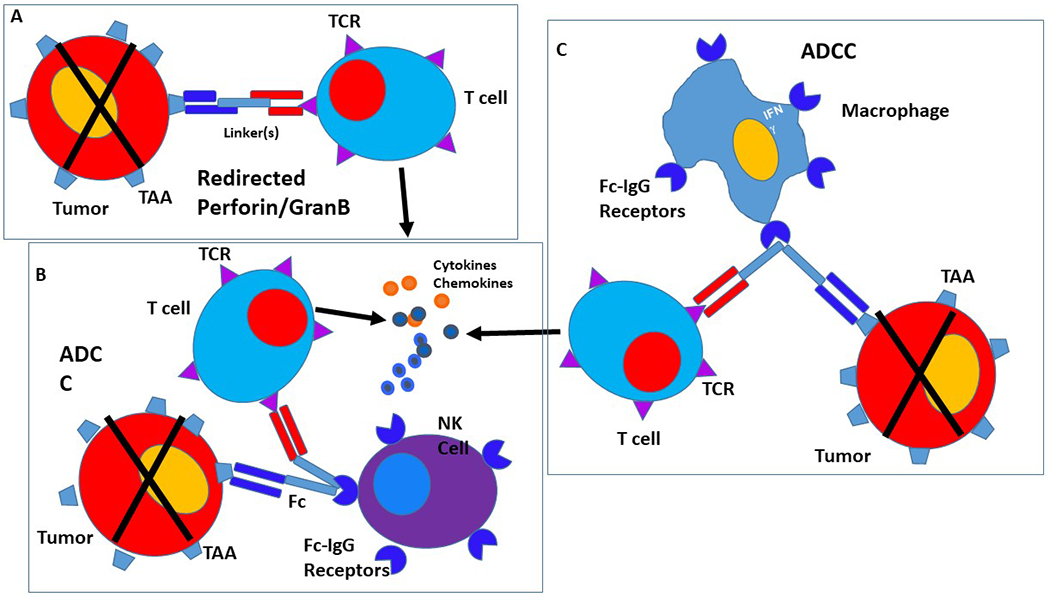
Mechanisms involved in bispecific antibody mediated cytotoxicity. A: Non-MHC restricted perforin/granzyme B mediated killing by redirected T cells. B: Antibody dependent cellular cytotoxicity mediated by Fc-recepter binding of bipecific antibody, NK cell and effector T cell. C: Antibody dependent cellular cytotoxicity mediated by T effector cell and monocyte.
With some improvements in manufacturing in the 1990’s, came clinical trials using bispecific antibodies chemically conjugated to HER2 or EGFR with bi-specificity against CD64 for recruitment of monocytes, macrophages and activated neutrophils. The studied agents were MDX-210 (a HER2 and CD64 BsAb), MDX-H210 (humanized version of MDX-210) and MDX-447 for EGFR and CD64 co-targeting.4,5 Unfortunately, trials of those three agents lacked clinical responses. Follow-up analysis revealed that effective tumor lysis required high BsAb concentrations and high effector to target cell ratios of 40:1. A solution to this problem was to replace the CD64 targeting with FcγRIII (CD16) targeting which would be expressed by macrophages and natural killer cells; but the resulting BsAb (HRS-3/A9) required four day continuous infusions and was not pursued for further clinical development.
Ultimately, the first BsAb to be approved by a regulatory body was Catumaxomab (Fresenius Biotech, Germany).6 Catumaxomab is a Murine IgG2a anti-CD3 hemi-antibody with rat IgG2b anti-epithelial cell adhesion molecule (EpCAM) which was approved in Europe in 2009 for palliation of malignant ascites. Catumaxomab is a Trifunctional antibody or Triomab (Fig 2) that recognizes Fcγ receptor types I and III, thus directing dendritic cells, macrophages and NK cells to the tumor cells. This BsAb successfully resulted in high levels of Th1 cytokine release and puncture-free ascites control when given by intraperitoneal infusion. Unfortunately, Catumaxomab was unable to be tolerated intravenously as there was nonspecific activation of T-cells manifested by cytokine release syndrome. The agent was withdrawn from the European Medicines Agency (EMA) in 2017 and from the US in 2013 for commercial reasons.
Figure 2:
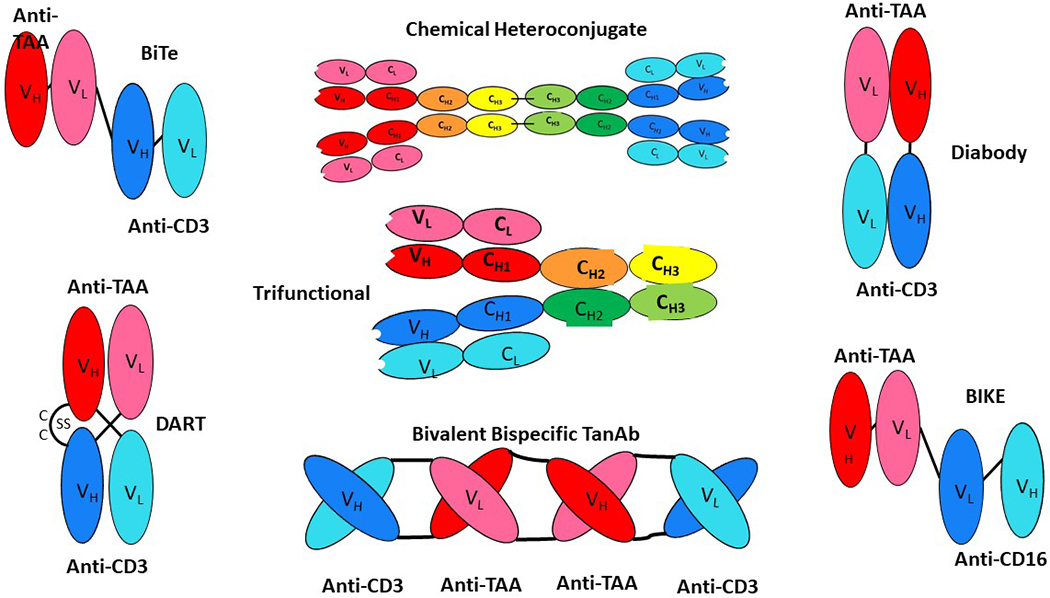
Various bispecific antibody formats are shown as whole antibody molecules or engineered antibody derivatives containing variable regions of heavy and light chains. The heavy chains targeting CD3 and TAA are depicted in dark blue and red respectively. The light chains targeting CD3 and TAA are depicted in light blue and pink, respectively BiTE = Bispecific T cell engager; Chemical heteroconjugate = two antibodies linked; DART= Dual-affinity retargeting bispecific antibody; Trifunctional Antibody: quadroma fusion with Fc-binding portion; TanB; bispecific tetravalent chimeric antibody construct; BIKE: Bispecific killer cell engagers; Diabody: basic construct of genetically fusing variable domains of the heavy and light chains with linkers. The heavy chains targeting CD3 and TAA are depicted in dark blue and red respectively. The light chains targeting CD3 and TAA are depicted in light blue and pink, respectively.
There are currently two FDA approved BsAbs on the market. The first is blinatumomab (Amgen, Thousand Oaks, CA) with dual binding of CD3 and CD19, and which was FDA approved for acute lymphoblastic leukemia (ALL) in 2017.7 The second BsAb on the market is emicizumab (Chugai/Roche, Basel Switzerland), which targets clotting factors IXa and X and is used for hemophilia A. Contextually, these approvals lay a foundation for future bispecific approvals for solid tumors. Blinatumumab notably produced a 44% complete response rate and an overall median survival of 7.7 months in a widely treated ALL population. It also achieved MRD negativity (minimal residual disease) after one cycle in 78% of ALL patients in the BLAST trial which led to added FDA label indications for blinatumumab. Studies are ongoing to add blinatumumab to initial treatment, post-transplant maintenance and in combination with tyrosine kinase inhibitors. Indeed in a review article from 2021, it was stated that blinotumumab and antibody-based treatment approaches may become a success story in ALL.8 Perhaps one lesson that should be learned here was that the CD19 target was often downregulated after BsAb treatment which rendered future CAR-T therapy difficult. This may be relevant as cellular therapies for breast cancer are rapidly developing. It is also notable that adherent breast cancers by definition will have lower drug exposure than hematologic malignancies, but that feature alone has not deterred antibody development in general or in the BsAb space.
The most critical need for development of BsAb’s in breast cancer may be that breast cancer remains the only major solid tumor for which checkpoint-based immunotherapies are rarely used; the exception being the PD-L1+ triple negative setting. We will therefore discuss successes, challenges and future directions for the development of BsAbs for breast cancer with an emphasis on the recent and ongoing clinical trials of BsAbs for breast cancer.
2. Potential Targets in Breast Cancer:
Breast cancer is generally considered to be poorly immunogenic, especially the estrogen and progesterone positive subtypes.9 Historically, immunotherapies and vaccines for breast cancer focused on known antigens such as HER2, MUC1, EGFR and a few cancer testis antigens.10,11 More recent screening on protein microarrays have allowed direct identification of novel cancer antigens in breast cancer samples and cell lines.12 Table 1 below is a summary of the growing list of targetable antigens currently being explored for bispecific technologies in breast cancer.
TABLE 1.
Cancer antigens relevant to breast cancer and examples of BsAbs reported to target those antigens.
| Cancer antigen | Example of BsAb | Ref |
|---|---|---|
| Trop2 | CD3 x Trop2 | 57 |
| EGFR | CD3 x EGFR EGFR x mPEG |
58
59 |
| EphA10 | CD3 x EphA10 | 60 |
| P-cadherin | CD3 x cadherin | 61 |
| EpCAM | CD3 x EpCAM | 6,30 |
| CEACAM5 | CD3 x CEACAM5 | 57 |
| HER2 | mPEG x HER2 | 62 |
| HER3 | HER3 x EGFR HER2 x HER3 |
45
63 |
| p95HER2 | CD3 x p95HER2 | 48 |
| Notch | EGFR x Notch | 46 |
| MAGE-A4 | MAGE-A4 x CD3 | 64 |
| Mesothelin | CD16 x mesothelin | 47 |
| Muc1 | CD16 x MUC1 | 65 |
As background, HER2 is a well validated target in breast cancer. At the current cut-point for clinical use, HER2 is only consider over-expressed in 15%-20% of breast cancers. Fortunately, for the non-amplified population, there is a rapidly growing body of literature to suggest that lower level expression of HER2 may be relevant for therapies including BsAb’s targeted at HER2.13 Also as discussed below, there are recently efforts to target the full length HER2, truncated versions (i.e. p95HER2), and HER2/HER3 heterodimers.
It is notable that the Trop2 receptor is recently fully validated as target in breast cancer as highlighted by the 2020 FDA approval of the Sacituzumab govitecan antibody (anti-Trop-2 ADC). In the development of that agent, it was learned that 80% of triple negative breast cancers express Trop-2 (average of 90,000 molecules of Trop-2 per cell).14 Not surprisingly, Trop-2 BsAb’s are recently described and studied.15,16 At least four additional Trop-2 ADC agents are in the pipeline in late stage studies.
Efforts to utilize EGFR as a target for breast cancer have been elusive for more than 15 years with numerous negative studies of both small molecule inhibitors and monoclonal antibodies. Clearly EFGR is expressed by many breast cancers and there is a well-defined minor subset of patients who respond well to EGFR targeted drugs.17 To target EGFR in breast cancer will likely require better antibodies or biomarkers or perhaps BsAb’s or cellular therapies. Perhaps BsAb’s targeted at EGFR may be a part of the solution.
Breast cancers occasionally express other targets of interest. As summarized in Table 1, MUC1, Notch, CEACAM, MAGE, Trop-2 and antibody-based therapies are being explored and developed. Also, PD-L1 is detected on some breast cancers (across all breast cancer receptor types) along with other potentially immune engaging targets such as GPNMB, CSF1R, Ptch, Smo, FZD, GSPG4, Jagged, androgen receptor, beta-catenin.18 While many BsAb constructs use CD3 or CD16 as the immune co-targeting, other receptors on immune effector cells are being explored (Tables 1–3). Abstract and patent searches reveal BsAb’s reported or proposed for co-targeting of PD-1, PD-L1, CD19, CD123, CD47/SIRPα, TGFβ, CD27, LAG3, CTLA4, ICOS, and CD20.
Table 3:
HER2 BsAb’s in clinical development. The details of Table 3 are derived from http://clinicaltrials.gov/.
| Agent | Listings | N | Phase | Target |
|---|---|---|---|---|
| ZW25 (Zymeworks) |
NCT04276493
NCT02892123 |
50 234 |
Ib/2 | 2 distinct sites on HER2 |
| ZW49 (Zymeworks) | NCT03821233 | 174 | 1 | 2 sites on HER2 (plus ADC delivery) |
| KN026 (NCT04165993, NCT04521179, NCT03847168, NCT04040699, NCT03619681) (Jiangsu Aphamab) | (5 studies) | 187 | 1 & 2 | Biparatropic HER2 |
| MCLA-128/zenocutuzumab (Merus) |
NCT03321981
NCT02912949 |
101 250 |
1 1/2 |
HER2 and HER3 |
| ISB-1302 (Ichnos Sciences) | NCT03983395 | 158 | 1/2 | HER2 and CD3 |
| HER2Bi ATC (BiAb armed activated Tcells (Transtarget) |
NCT03272334
NCT03661424 |
33 16 |
1 1 |
HER2 and CD3 HER2/CD3 in leptomeningeal dz |
| NCT01022138 | 32 | 2 | HER2/CD3 (ER/PR+/HER2 Neg) | |
| MBS301 (Beijing Mabworks) | NCT03842085 | 34 | 1 | 2 sites on HER2, afucosylated |
| M802 (Wuhan YZY) | NCT04501770 | 32 | 1 | HER2 and CD3 |
| BCD-147 (Biocad) | NCT03912441 | 15 | 1 | 2 sites on HER2 |
| DF1001 (Dragonfly) | NCT04143711 | 220 | 1/2 | HER2 and undisclosed NK engager |
| BTRC4017A (Merck) | NCT03448042 | 449 | 1/2 | HER2 and CD3 |
| IBI315 (Innovent) | NCT04162327 | 191 | 1a/1b | HER2 and PD-L1 |
3. Approaches for Breast Cancer:
Advances in recombinant DNA technology allow the production of a variety of BsAbs with unique designs (Figure 2). Commonly, a bivalent IgG bispecific antibody is engineered using knob in hole technology domain19 and contains fragment antigen binding (fab) regions capable of recognizing multiple antigens and a fragment crystallized region (Fc) capable of mediating effector functions. Creative approaches to BsAb construction include the use of a silent Fc region or formats with no Fc region at all. Some examples of bispecific constructs include the dual affinity re-targeting (DART), the diabody, the bispecific T cell engager (BiTE) and the bispecific killer cell engager (BiKE). As mentioned above blinatumomab is the only approved agent for cancer and it is a BiTE design. As shown in the diagram, BiTE’s are engineered with an immunoglobulin derived single chain variable fragment (scFV) containing variable heavy (VH) and variable light (VL) chain regions directed against a tumor associated antigen and linked to the scFv of an antibody targeting a T-cell. The concept is that this BsAb would bring T lymphocytes into close proximity to cancer cells and redirect cytotoxic T-cell activity against the tumor cells in the tumor microenvironment. The key function of a BiTE’s is to redirect the non-MHC restricted cytotoxicity of T-cells mediated by perforin/granzyme B that is independent of T-cell receptor specificity. Based upon a favorable safety profile, the blinatumomab BiTE agent is FDA approved and several other emerging BiTE’s appear safe in their early development. Modifications in binding affinities, size, inclusion of Fc-receptors and/or linkers may help to achieve the desired pharmacodynamics and anti-tumor effects.
BsAbs are being explored as vehicles for toxic payload delivery. Breast cancer researchers have made efforts to develop radioimmuno- BsAb platforms as well as antibody-drug conjugate (ADC) based BsAbs. There is also the principle of engineering BsAbs that bind to two different receptors or epitopes on the same cancer cell. One example is ZW25 which is a biparatopic BsAb targeting two different epitopes on the extracellular domains of HER2 (NCT03929666). The concept of targeting multiple receptors or epitopes not only improves targeting specificity, but also minimizes toxicity to healthy tissues and occasionally leads to new biology. Targeting two different epitopes on same death receptor-5 (DR5) using a BsAb led to a newly undiscovered mechanism of DR5 clustering.20 Finally, BsAb are being used to arm ex vivo expanded T-cells so that they can be targeted to cancer sites in patients with widely metastatic disease.21,22
4. Agents in human clinical trials:
As mentioned above, there are several bispecific agents at various stages of development in breast cancer. The first agent which was aggressively developed in breast cancer was Ertumaxomab, which targeted a unique HER2 epitope (different from trastuzumab) on tumor cells. Ertumaxomab was CD3 co-targeting and acted via Fc fragment resulting in macrophage activation and antibody dependent cellular cytotoxicity (ADCC). It was thus effectively a trifunctional antibody believed to temporarily attract immune effector cells to tumor cells.23,24 Ertumaxomab was manufactured by a quadroma cell line fusion of rat (anti-CD3) and mouse hybridoma cell line (anti-HER2). The heavy chain contains murine IgG2a and rat IgG2b subclasses and there is binding selectivity to Fcγ type I/III. The rationale for activity of this BsAb agent is that both T cells and accessory cells might complex together with a HER2+ cancer cell. The T cells would be activated by the CD3 binding and would release cytokines and perforin. At the same time the T cells would be prevented from anergy by the release of costimulatory cytokines and cell surface receptors on accessory monocytes/macrophages. Finally, phagocytosis of tumor cells by macrophages and dendritic cells should result in uptake, processing and presentation of any additional antigens beyond HER2. Preclinical studies with ertumaxomab showed that it could kill HER2+ cell lines in vitro. In a translational study, ertumaxomab eliminated HER2+ tumor cells from leukapheresis products of patients with breast cancer.25 Two phase I studies were then performed.24,26
In the first phase I clinical trial in patients with metastatic breast cancer, 15 patients were treated with escalating doses of ertumaxomab BsAb. Ultimately 100 μg/kg was deemed the maximum tolerated dose (MTD), because at greater dosages, the patients developing cytokine release syndrome including hypotension, respiratory distress syndrome, systemic inflammatory response syndrome, acute renal failure and heart failure. One third of patients experienced clinical benefit and there was one complete response reported. Immune correlative studies showed that IL-6, IL-2, TNF-α and IFN-γ responses were elevated in the serum. Unfortunately human anti-mouse/anti-rat antibodies developed in a third of the patients.24 In a separate phase I trial, 14 patients with HER2 expressing solid tumors (e.g. breast, gastric, rectal cancer) were given ertumaxomab with a build-up schedule prior to testing target doses.26 One partial response to ertumaxomab was observed out of 11 patients.26 A phase II trial of ertumaxomab weekly at 100 mcg was initiated in 2007 and terminated in 2011 after enrolling 19 patients and no results were made public. The development of ertumaxomab was discontinued by Trion pharma/Fresenius in 2011.27
Another agent, MM-111 (Merrimack pharmaceuticals), is a different approach to a BsAb. MM-111 specifically targets the HER2/HER3 heterodimer and blocks the binding of heregulin and HER3, thus inhibiting HER3 downstream signaling pathways.28 In preclinical murine studies, MM-111 with trastuzumab potently blocked tumor growth. A phase I study in combination with trastuzumab was completed in 2014 and presented at ASCO 2014 where it was reported that an maximum tolerated dose (MTD) was not reached in a study of 86 patients, although some anemia, neutropenia, mucosal inflammation and stomatitis were reported (NCT01304784). MM-111 was also studied in combination with paclitaxel (NCT01774851) and with trastuzumab (NCT01097460) and as monotherapy (NCT00911898). In the monotherapy study, 20 patients were treated and three had severe adverse events (SAEs). In the Herceptin® trial, only two of 16 patients experienced SAE’s. No efficacy results have been made public and the agent was recently was out-licensed with no disclosed future development plans.
EpCAM is a cell surface glycoprotein detected in over 90% of breast cancers that is associated with poor prognosis and chemotherapy resistance in triple negative breast cancer (TNBC).29 EpCAM is an attractive target for BsAb approaches in breast cancer. Catumaxomab, the EpCAM-CD3 BsAb that was EMA/FDA approved (2009) and later withdrawn. It has recently been re-examined in updated phase III trials including 156 patients with malignant ascites (NCT04222114). In addition to catumaxomab, there is a recent competing EpCAM BiTE construct known as MuS110/MT110/solitomab. It decreased tumor sizes in 4T1 and MCF-7 tumor bearing mice models.30 The MT110 BsAb also achieved lysis in EpCAM+ cells from malignant pleural effusions.31 In a human phase I study in 65 patients with solid tumors, MT110 treatments resulted in >95% of patients with treatment related adverse events (AE) which were grade 3 or greater.32 There was one grade 5 treatment related AE and no confirmed responses. No further development is reported publicly given the toxicities experienced with this MT110/solitomab agent.
Interestingly, there are two TGFβ targeting agents in clinical trials. The first is the EGFR x TGFβ BsAb known as BCA101 (Bicara Therapeutics). In the ongoing study BCA101 is given alone and with pembrolizumab (NCT04429542). The study is for TNBC and a few other cancer types known to express EGFR; the TNBC cohort requires central confirmation of EGFR expression. It is enrolling 292 patients for combined dose escalation and expansion (see Table 2). The preclinical data for this BsAb has not yet published.
Table 2:
Ongoing clinical trials of bispecific antibodies in Breast cancer not for HER2. The details of Table 2 are derived from http://Clinicaltrials.gov/.
| Study | NCT number | N | Phase | Status | Targets |
|---|---|---|---|---|---|
| Activated CIK armed with anti-CD3-MUC1 Bispecific Ab in breast cancer | NCT03524261 | 90 | 2 | Recruiting | CD3 and MUC1 |
| Pilot Study for Optimization of Immuno-PET Pretargeted With Anti-CEA Bispecific Antibody X Anti-HSG TF2 and the Peptide IMP-288 Radiolabeled With Gallium-68 -Pharmacokinetic and Imaging for Patients With a Recurrence of HER2 Negative Breast Carcinoma Expressing CEA | NCT01730612 | 23 | 1 | Not recruiting | CEA |
| A Multicenter, Open-label, Dose-escalating, Phase I Trial With GEM3PSCA, a PSCA Targeted BsAb Engaging T-cells, in Patients With Progressive Disease After Standard Systemic Therapy in Cancers With Positive PSCA Marker | NCT03927573 | 24 | 1 | Recruiting | PSCA |
| First-in-human, Open-label, Dose-escalation Trial With Expansion Cohorts to Evaluate Safety of GEN1044 in Subjects With Malignant Solid Tumors | NCT04424641 | 378 | 1/2 | Recruiting | CD3 and 5T4 |
| A Phase 1 Study of the PD-L1xCD27 BsAb CDX-527 in Patients With Advanced Malignancies | NCT04440943 | 96 | 1 | Recruiting | PDL1 and CD27 |
| Phase 2a Study of ZW25 in Combination With Palbociclib Plus Fulvestrant | NCT04224272 | 86 | 2a | Recruiting | 2 distinct sites on HER2 |
| A Phase 1 Multiple-Dose Study to Evaluate the Safety and Tolerability of XmAb®22841 Monotherapy and in Combination With Pembrolizumab in Subjects With Selected Advanced Solid Tumors (DUET-4) | NCT03849469 | 242 | 1 | Recruiting | CTLA4 and LAG3 |
| First-in-Human, Phase 1/1b, Open-label, Multicenter Study of Bifunctional EGFR/TGFβ Fusion Protein BCA101 Alone and in Combination With Pembrolizumab in Patients With EGFR-Driven Advanced Solid Tumors | NCT04429542 | 292 | 1/1b | Recruiting | EGFR and TGFβ |
| A Phase 1 Multiple-Dose Study to Evaluate the Safety and Tolerability of XmAb®23104 in Subjects With Selected Advanced Solid Tumors | NCT03752398 | 164 | 1 | Recruiting | PD1 and ICOS |
| M7824 in Treating Patients With Stage II-III HER2 Positive Breast Cancer | NCT03620201 | 20 | 1 | Recruiting | TGFb and PD-L1 |
A competing TGB-β construct is bintrafusp alpha. The bintrafusp alpha construct is an anti-PD-L1 fused to TGF-βPII. The TGF-βRII moiety binds three different TGF-β isoforms (TGF-β1, TGF- β 2 and TGF- β 3).33 It is conceptualized as a TGF- β trap which suppresses the TGF- β signaling and likely blocks epithelial to mesenchymal transition (EMT) in breast cancer stem cells. This purported anti-EMT function is supported by reduction in vimentin expression (a mesenchymal marker) in murine studies of bintrafusp alpha. Additionally, bintrafusp alpha was shown to decrease Treg infiltration into breast cancers and to increase CD4+ T cell proliferation. The phase I study of 19 patients was completed at the National Cancer Institute in Bethesda, MD with one patient experiencing a complete response and two patients who had durable partial responses.34 Clinicaltrials.gov lists 28 studies with this agent as of 3/2/2021 with notable trials in TNBC (NCT04489940), ER+ breast cancer (NCT03524170) and HER2+ (NCT04489940 and NCT03620201).
HER2 targeting BsAbs are recently reporting phase I and phase II results. The most notable results are zenocutuzumab which reported a phase II result in ER+, HER2low metastatic breast cancer.22 In a study of 48 patients, the disease control rate was 45% comprising two partial responses and 19 stable disease results. The adverse events were asthenia, diarrhea, and nausea and no drop in left ventricular ejection fraction was seen. Additional HER2 targeting antibodies in development are summarized in Table 2.
5. Combinatorial therapies with bispecific antibodies
Since some, but not all BsAbs, have run into problems with cytokine storm or other toxicities, novel delivery and combination approaches are noteworthy. One approach has been to use BsAbs to arm T cells (Figure 3). The first report of using BsAb to arm T cells for breast cancer was published in 200335 followed by a similar report from a different group in 2005.36 In the first trial, 23 patients with metastatic breast cancer were treated with autologous T cells which were armed with a CD3-HER2 bispecific antibody prior to infusion.37 A cell dose of 160 x 109 armed activated T cells was deemed safe and feasible. The cells were shown to persist in the blood for weeks and were shown to traffic to the tumor sites. The overall survival was 36.2 months and immune correlates suggested that favorable Th1 responses led to tumor recognition. In the second published trial of the same CD3xHER2 construct, there were eight patients with advanced breast cancer who were treated with serial infusions of BsAb-armed T cells targeting HER2.38 The BsAb-armed T cell treatments were followed with autologous stem cell transplant and T cell boosting.38 The study observed a time to progression of 11.2 months and an overall survival of 32.0 months in advanced disease setting. The third study is a phase II study and it was recently completed (NCT01022138) and enrolled 32 patients (24 ER+, HER2- and 8 TNBC).
Figure 3:
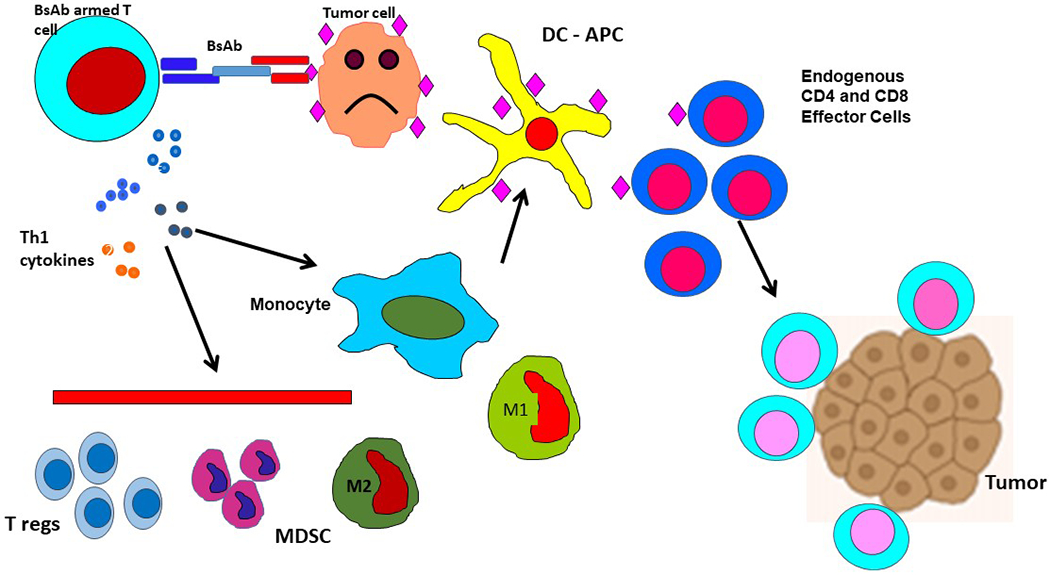
The proposed mechanism of BsAb armed T cell therapy. In this figure, the BsAb armed T cells are a population of T cells collected by apheresis and expanded ex vivo. Those T cells are then armed with an anti-CD3 BsAb with secondary specificity for a tumor antigen. Upon re-infusion into the body, the BsAb armed T cells preferentially collocate at the site of tumor antigen presence where cancer cells exist in the body. Th1 cytokine release ensues and cooperates in tumor cytolysis. The secondary process of additional tumor antigen release and presentation occurs and results in specific activation of endogenous, non-BsAb-armed CD4 and CD8 T cells and an expansion of tumor specific immune response as shown at the bottom right.
Another BsAb related T cell combinatorial study36 differed from the first three. The study involved use of autologous T cells similar to above, but they were administered 48hours before BsAb infusions. The study challenged the existing paradigm by delivering both a HER2 x CD3 BsAb and also an EpCAM x CD3 targeting BsAb to the same patients following delivery of autologous T cells. The outcome for dual BsAb + autologous T cells in heavily pretreated metastatic breast cancer is similar to the three armed T cell trials above which incubated the BsAb with the T cells prior to delivery. Acceptable safety profiles were observed and an OS of 27.7 months was reported. As per table 2, there are currently two ongoing clinical trials of BsAb armed T cells in breast cancer.
6. Preclinical Bispecific Antibody Highlights
The BsAb field is rapidly evolving and is increasingly diverse in terms of targets, constructs and delivery methods. It is impossible to capture all preclinical projects for review, but some of the most promising are discussed below.
A CD16 x EpCAM BsAb targeting NK cells was successful in killing several breast cancer cell lines in chromium release assays.39 The targeting has been formulated into what is being called a tetra-specific killer engager (TetraKE) with specificity for CD16, EpCAM and CD133 with an IL-15 crosslinker. The TetraKE lysed breast cell lines, BT-474 and SK-BR-3.
Another interesting approach features TIGIT (T cell immunoreceptor with Ig and ITIM domains). TIGIT is a type I transmembrane protein with expression on several T cell types including activated T cells, regulatory T cells memory T cells and NK and NKT cells.40,41 TIGIT’s ligand is polio virus receptor (PVR or CD155) which would normally act as a costimulatory marker for DNAM-1. In the presence of TIGIT, the TIGIT receptor outcompetes DNAM-1 and thus the overall immune response when TIGIT is present is a suppressive signaling cascade. Using in vitro assays, it was shown that TIGIT inhibition using an EFGR co-targeting BiTE antibody enhanced cytotoxic killing effects in several breast cancer models.41 A TIGIT x PD-L1 antibody has been published.42
The EGFR receptor has been thoroughly tested by standard univalent monoclonal antibodies in TNBC with several renowned failures as previously reviewed.43 More recently, there have been variety of EGFR BsAb’s nearing or at the phase I stage of development and some are relevant to breast cancer. For example there is an EGFR x HER3 agent, MEHD7945A, (NCT01577173, NCT01652482) which sensitized TNBC cells to PI3K inhibitors.44 Also of interest is a bispecific diabody-Fc fusion protein joining EGFR and HER3 targeting. This agent successfully inhibited the expansion and the survival of TNBC stem cells in an orthotopic MDA-MD-468 mouse model. Furthermore this approach increased the sensitivity of TNBC cells to PI3K inhibitors.44,45 Similarly EGFR x Notch BsAb’s also synergize with PI3Ki’s pre-clinically and are moving forward in development pipelines.46 Preclinical work demonstrated enhanced cytotoxic activity of the CD3 x EGFR BiTE against EGFR expressing TNBC cells compared to EGFR targeting alone.41
Another interesting preclinical BiTE which engages immune cells includes a mesothelin targeting moiety and CD16 (FcγRIII). Since CD16 is an activating receptor on NK cells, the Fc region of an antibody found to a tumor antigen should elicit ADCC as was shown in an orthotopic xenograft NSG mouse model of TNBC.47
Novel epitopes on known breast cancer cell surface receptors are emerging as new targets for BsAb’s. The most notable example is a BsAb that targets the novel HER2 antigen known as p95HER2. The p95 epitope is expressed by about 40% of HER2+ tumors. The p95HER2 is synthesized by alternative initiation of translation of the full HER2 transcript. Since antibodies which recognize this alternate epitope appear to have increased specificity for breast cancer cells over normal HER2 expressing cells of the body, it is an appealing target. The Arribas group from Spain first synthesized a BsAb to p95HER2 and CD3.48 Interestingly the engineering permits bivalent binding to two p95HER2 epitopes and one CD3 epitope at a time. In vivo assays showed that MCF7 cells expressing p95HER2 xenografts and also human PDX models showed tumor regression when treated with this BsAb.48
Finally, a significant portion of TNBC patients express apoptotic inducible DR5. Tigatuzumab, a DR5 agonist antibody which triggers apoptosis in human TNBC cells without cross-linking has been in tested unsuccessfully in phase-II clinical trials as a single agent or in combination of nab paclitaxel49. We have identified an unexpected activation of PD-L1 immune evasion mechanism by Tigatuzumab and other DR5 agonists, a potential contributor to their clinical failure.20 Importantly, a preclinical BsAb targeting DR5 and PD-L150 not only overcomes immune evasion mechanism in TNBC tumors but also effectively activates the highest degree of apoptotic cytotoxicity. An anti-PD-L1 antibody atezolizumab was recently FDA approved for metastatic TNBC patients expressing PD-L151, and since significant TNBC patients also express elevated DR5 levels52, it will be interesting to test the survival outcome by PD-L1-DR5 antibody BsAb in TNBC clinical trials.
It is worth pointing out that while some preclinical successes in BsAb work have been able to move into the clinic, a much larger proportion have not translated well to human clinical relevance. Speculation exists as to the faithfulness of mouse models especially in relation to breast cancer. Common criticisms of mouse models relate to the divergent immune milieu in the mouse model as well as the inherent difference in murine breast tumors which form early in the development of the mouse, but breast cancers naturally form later in humans (median age of human breast cancer is 64 years). The human breast microenvironment has been heavily studied to better understand the highly immunosuppressive nature of the stromal cells and frequency of tolerogenic responses seen in many human breast cancers, as those are other barriers to successful translation of bench research. Attempts at 3D in vitro modeling has also failed to recapitulate the immune milieu of the breast adequately to develop reliable pre-clinical predicators of future drug success.
EXPERT OPINION:
Breast cancer was one of the first anatomic locations to be described as “immune infiltrated” and one of the first solid tumor sites to benefit from monoclonal antibody therapy (trastuzumab). The breast paradoxically turns out to be one of the least responsive sites to other immunotherapies beyond trastuzumab. Even the more immunologically favorable breast cancer subtype, TNBC, is subdivided into several groups with varying degrees of sensitivity to immune approaches. Despite TIL density for neo-adjuvant therapy and PD-L1 for metastatic TNBC having recently emerged as biomarkers for breast cancer, there are still no approved checkpoint inhibitors for ER positive or HER2 positive disease and limited roles for checkpoint inhibitors in TNBC. This clinical situation exemplifies how the manipulations of the tumor infiltrating lymphocytes in the tumor microenvironment and the retargeting of T or NK cells with BsAbs remains challenging.
Zitvogel et al analyzed why immunotherapies and especially BsAbs have not achieved strong immune stimulation in breast cancer.53 They concluded that the immunosuppressed tumor microenvironment after chemotherapy may be due to loss of MHC class I molecules from the metastatic clones and impaired or delayed recovery of T-cell receptor repertoire clones directed at breast cancer antigen after chemotherapy. The activation status and responses of effector T cells coming into the TME can be modulated by Tregs, myeloid derived suppressor cells, suppressor macrophages, and regulatory polymorphonuclear leukocytes. The use of BsAb’s that target T cells and breast cancer may circumvent the barriers in the microenvironment by co-engaging cytotoxic effector T cells that kill in a non-MHC restricted fashion and release Th1 cytokines. This mode of targeted killing may convert immunologically cold tumors into hot tumors by switching “back on” exhausted or quiescent effector cells in the microenvironment or recruiting new endogenous effector cells. BsAb’s hold promise for binding novel antigens or activating receptors on effector cells while employing emerging immune engagers beyond CD3 and CD16.
The number of combinations and variations on bi- and tri- specificity and beyond are unlimited. In addition, the principles of BsAb delivery have been adapted to nanotechnologies, antibody-drug conjugates, radio-conjugates, and optimized Fc approaches. In clinical trials, there are over 30 trials of BsAb’s relevant to breast cancer (Tables 2–3). The development of BsAb armed T cells targeting HER2 positive and negative breast cancer are also progressing through clinical evaluations. Although such trials require greater resources to collect, expand, arm T cells, and deliver the T cells to the patients, the regulatory challenges are manageable. Based on early phase data, it is difficult to predict which agents/platforms will receive regulatory approvals. If the volume of enrollment is an indicator of what might be approved, the bintrafusp alpha, ZW25, KN-026 and GEN1044 BsAb may be leading in trial accrual, though accrual metrics are poor predictors of eventual clinical/commercial success.
As mentioned above there has been success for the blinatumumab BsAb in ALL and there are successful second and third generation HER2 targeting antibodies (ado-trastuzumab, pertuzumab, margetuximab), but there are no approved BsAb therapies in breast cancer. The major challenge facing BsAb’s for all disease types is the risk of cytokine release syndrome and/or autoimmune toxicities associated with infusions of BsAb targeting CD3 and those that target co-stimulation.21,54 Two major areas of concern are the “on-target/on-tumor” toxicity and the “on-target/off-tumor” toxicity. Tumor antigen engagement of the TCR that drives cytokine release (“on-target/on-tumor”) can usually be managed by steroids and by drug dose/distribution modifications. Cytokine release syndrome is often mediated by transient increases in TNFα, IL-6, IFNγ, and monocyte chemoattractant protein-1 whereas “on-target/off-tumor” toxicity of CD3 based BsAb’s to normal tissue is often harder to address and is impacted by target distribution, level of expression on normal tissue and cellular localization of the target. Modeling in animals for BsAb driven toxicities have not been predictive of toxicities in patients. The intensity of the CRS may be proportional to the level of expression of the target antigen on normal tissues. Histologic changes of lymphocytic infiltrates, acute inflammatory changes and single-cell necrosis seen after infusions of CD3 platform effector-based BsAbs is being documented in ongoing trials. The clinical trial results will, in turn, inform the type of effector cells being targeted in vivo and the dosing schedule (single or multiple).
Another challenge to BsAb development is that in vitro efficacy does not translate into in vivo efficacy. It is critical to ascertain which receptor-ligand axis in each disease model is applicable. As mentioned above, several agents in clinical development were halted for lack of efficacy or dose limiting toxicities despite encouraging preclinical data. Along those lines, any insufficient clinical efficacy underscores the importance of not only uncovering BsAb driven novel biology but also selecting the correct patient population or clinical endpoints when designing trials of a BsAb. Thus reliable biomarkers might better guide future clinical development. As mentioned above, the relative target molecule expression on cell surface is interesting for some targeted antibody therapies. Biomarkers for downstream signaling and markers of immune activation are other popular biomarkers being evaluated currently.
To improve immune-targeted BsAb success, the affinity, antigen density on the target tumor, isotype, the pharmacokinetics of the BsAb, and dose are all critically important to T cell responses. Those same factors are relevant toxicities, cytokine secretion/release, and cytotoxicity. Changing the valency may alter the ability of the anti-CD3-based BsAb’s to induce specific T cell functions. In a series of scFv anti-HER2 constructs, increasing binding affinity led to increasing cytotoxicity.55 For example, double valency directed at anti-CD3 would more tightly bind the BsAb construct to T cells that were armed with BsAb ex vivo in adoptive transfer approaches. The same BsAb may lead to excessive TCR activation and CRS if given as an IV infusion. If the adverse side effects can be controlled with engineering of the BsAb constructs or the use of desatinib (a tyrosine kinase inhibitor)56, the BsAb redirected immune effector cells together with antigen presenting cells can present TAA peptides/antigens derived necrotic and apoptotic tumor cells to the endogenous immune system leading in situ vaccination against a wide array of TAAs.37,38
Combining checkpoint inhibitors with BsAb infusions or the T cells armed with BsAb may provide additional anti-breast cancer effects without increasing side effects since the arming dose of BsAb, the number effector cell infusions, and the timing of checkpoint inhibitor infusions can all be dose and schedule controlled. In the near future, the application of BsAbs for breast cancer is poised to further clinical development and possibly may result in new clinical drug approvals. Bintrafusp alpha, ZW25, Zenocutuzumab and others are showing evidence of clinical activity without the toxicities associated prior generations of BsAb’s. Although, the anti-HER2 targeting clinical space is saturated with anti-HER2 drugs, one or more of these well designed HER2-targeted BsAbs may find a niche in clinical space. HER2 targeting may not only be applicable to tumors expressing high amount of HER2 but show activity against very low expressing or nil expressers of HER2. On the other hand, the TNBC space is ripe with potential antigens to target and has a dearth of active agents approved. Finally, the ER positive space in breast cancer remains attractive for new agents since it makes up 70% of breast cancers and lacks good therapies once patients fail to respond to endocrine agents. Unfortunately, immunotherapies in ER positive disease have historically fared poorly, thus rendering ER+ disease as the toughest subtype of breast cancer to target with BsAbs. The lower mutational burden and slower growth rates of many ER+ tumors are oft-cited reasons for the lack of checkpoint inhibitor therapy efficacy in this setting.
In conclusion, there is a unique history for the development of BsAb’s in breast cancer with areas of unmet need still existing in the breast cancer field (such as chemotherapy de-escalation, treatment of ER+ disease, and treatment of residual disease after neo-adjuvant therapy). There is a recent surge of new BsAb’s in clinical and preclinical development and mounting evidence that some BsAb and platforms have clinical activity and adequate safety profiles. Lead agents focus on HER2 related targets that make up only 20% of the population with breast cancer whereas there is a paucity of BsAbs for HER2 negative disease (hormone receptor positive or TNBC). Combinatorial approaches with autologous T cells, chemotherapy agents and other immunotherapy agents are encouraging. Novel designs with BsAb based ADC’s, nanotechnologies and novel biomarkers are in development.
Article Highlights:
Breast Cancer is targetable by antibody-based therapy as demonstrated by prior HER2 targeting agents.
Many novel targets for antibodies and bispecific antibodies are recently emerging in the breast cancer field including optimized HER2 targets, immune-relevant targets, and glycoproteins such as Trop-2.
Bispecific antibodies for breast cancer have been explored both preclinically and in early phase human clinical trials.
Bispecific antibodies in breast cancer carry the same risks of cytokine storm as observed in other clinical settings.
Reasons why bispecific antibodies might not translate from bench to bedside might include challenges in developing immune response in low mutation burden setting, tolerizing microenvironment, or limits on the faithfulness of preclinical models.
Funding
This study was in part by funded from DHHS R01 CA 140314 and in part by R01 CA182526 and P30CA044579 at the University of Virginia. LG Lum is supported in part by funding from DHHS R01 CA 092344, R01 CA 140314, R01 CA 182526.
Acronym Glossary:
- ADC
Antibody Drug Conjugate
- ADCC
Antibody Dependent Cellular Cytotoxicity
- ALL
Acute Lymphoblastic Leukemia
- BiKE
Bispecific Killer cell engager
- BiTE
Bispecific T cell engager
- BsAb
Bispecific antibody
- CRS
Cytokine Release Syndrome
- CTLA4
Cytotoxic T-lymphocyte associated Protein 4
- DART
Dual Affinity Re-targeting
- DR5
Death Receptor 5
- Fc
Fragment crystallizable region
- HER2
Human Epidermal Growth Factor Receptor 2
- EGFR
Epidermal Growth Factor Receptor
- EMA
European Medicines Agency
- EMT
Epithelial to Mesenchymal Transition
- EpCAM
Epithelial Cell Adhesion Molecule
- ER+
Estrogen Receptor positive
- FDA
Food and Drug Administration
- GSPG4
General secretion pathway protein G
- ICOS
Inducible T-cell COStimulator
- IFN-γ
Interferon Gamma
- LAG3
Lymphocyte Activating gene 3
- MCF-7
Michigan Cancer Foundation 7 cell line
- MRD
Minimal Residual Disease
- MUC1
Mucin 1
- MTD
Maximum Tolerate Dose
- NK
Natural Killer
- NSG
NOD scid gamma mouse
- PD-L1
Programmed Death Ligand 1
- PDX
Patient Derived Xenograft
- PIK3
Phosphoinositide-3 Kinase
- SAE
Serious Adverse Event
- scFV
single chain variable fragment
- SIRPα
Signal Regulatory Protein alpha
- TNBC
Triple Negative Breast Cancer
- TNF-α
Tumor Necrosis Factor alpha
- TGF-β
Transforming Growth Factor-beta
- TCR
T cell receptor
- TAA
Tumor associated antigen
- TetraKE
Tetra-specific killer engager
- Th1
T-helper cell type 1
- TIGIT
T cell Immunoreceptor with Ig and ITIM domains
Footnotes
Declaration of interests
LG Lum is co-founder of Transtarget Inc. P Dillon is a site investigator for breast cancer trials sponsored by Pfizer, Novartis, Merck, Seattle Genetics, Radius, Syndax, Tesaro, Tolero. P Dillon has institutional clinical trial participation with Merck, Seattle Genetics, Tolero, Pfizer, Abb-vie, Radius. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Aalberse RC, van der Gaag R, van Leeuwen J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. Journal of immunology (Baltimore, Md : 1950). 1983;130(2):722–726. [PubMed] [Google Scholar]
- 2.Nisonoff A, Rivers MM. Recombination of a mixture of univalent antibody fragments of different specificity. Archives of biochemistry and biophysics. 1961;93:460–462. [DOI] [PubMed] [Google Scholar]
- 3.Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314(6012):628–631. [DOI] [PubMed] [Google Scholar]
- 4.Pullarkat V, Deo Y, Link J, et al. A phase I study of a HER2/neu bispecific antibody with granulocyte-colony-stimulating factor in patients with metastatic breast cancer that overexpresses HER2/neu. Cancer immunology, immunotherapy : CII. 1999;48(1):9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Repp R, van Ojik HH, Valerius T, et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcgammaRI x anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. British journal of cancer. 2003;89(12):2234–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burges A, Wimberger P, Kümper C, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(13):3899–3905. [DOI] [PubMed] [Google Scholar]
- 7.Löffler A, Kufer P, Lutterbüse R, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95(6):2098–2103. [PubMed] [Google Scholar]
- 8.Li L, Wang Y. Recent updates for antibody therapy for acute lymphoblastic leukemia. Experimental hematology & oncology. 2020;9(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makhoul I, Atiq M, Alwbari A, Kieber-Emmons T. Breast Cancer Immunotherapy: An Update. Breast cancer : basic and clinical research. 2018;12:1178223418774802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dillon PM, Petroni GR, Smolkin ME, et al. A pilot study of the immunogenicity of a 9-peptide breast cancer vaccine plus poly-ICLC in early stage breast cancer. Journal for immunotherapy of cancer. 2017;5(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MJ, Choi JR, Tae N, et al. Novel Antibodies Targeting MUC1-C Showed Anti-Metastasis and Growth-Inhibitory Effects on Human Breast Cancer Cells. International journal of molecular sciences. 2020;21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young AR, Duarte JDG, Coulson R, et al. Immunoprofiling of Breast Cancer Antigens Using Antibodies Derived from Local Lymph Nodes. Cancers. 2019;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchiò C, Annaratone L, Marques A, Casorzo L, Berrino E, Sapino A. Evolving concepts in HER2 evaluation in breast cancer: Heterogeneity, HER2-low carcinomas and beyond. Seminars in cancer biology. 2020. [DOI] [PubMed] [Google Scholar]
- 14.Cardillo TM, Rossi DL, Zalath MB, et al. Predictive biomarkers for sacituzumab govitecan efficacy in Trop-2-expressing triple-negative breast cancer. Oncotarget. 2020;11(43):3849–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossi EA, Rossi DL, Cardillo TM, Chang CH, Goldenberg DM. Redirected T-cell killing of solid cancers targeted with an anti-CD3/Trop-2-bispecific antibody is enhanced in combination with interferon-α. Molecular cancer therapeutics. 2014;13(10):2341–2351. [DOI] [PubMed] [Google Scholar]
- 16.Zaman S, Jadid H, Denson AC, Gray JE. Targeting Trop-2 in solid tumors: future prospects. OncoTargets and therapy. 2019;12:1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer metastasis reviews. 2016;35(4):575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakhjavani M, Hardingham JE, Palethorpe HM, Price TJ, Townsend AR. Druggable Molecular Targets for the Treatment of Triple Negative Breast Cancer. Journal of breast cancer. 2019;22(3):341–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–621. [DOI] [PubMed] [Google Scholar]
- 20.Mondal T, Shivange GN, Tihagam RG, et al. Unexpected PD-L1 immune evasion mechanism in TNBC, ovarian, and other solid tumors by DR5 agonist antibodies. EMBO molecular medicine. 2021;13(3):e12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lum LG, Thakur A. Targeting T cells with bispecific antibodies for cancer therapy. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2011;25(6):365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pistilli B, Wildiers H, Hamilton EP, et al. Clinical activity of MCLA-128 (zenocutuzumab) in combination with endocrine therapy (ET) in ER+/HER2-low, non-amplified metastatic breast cancer (MBC) patients (pts) with ET-resistant disease who had progressed on a CDK4/6 inhibitor (CDK4/6i). Journal of Clinical Oncology. 2020;38(15_suppl):1037–1037. [Google Scholar]
- 23.Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. Journal of hematology & oncology. 2015;8:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiewe P, Thiel E. Ertumaxomab: a trifunctional antibody for breast cancer treatment. Expert opinion on investigational drugs. 2008;17(10):1553–1558. [DOI] [PubMed] [Google Scholar]
- 25.Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98(8):2526–2534. [DOI] [PubMed] [Google Scholar]
- 26.Haense N, Atmaca A, Pauligk C, et al. A phase I trial of the trifunctional anti Her2 × anti CD3 antibody ertumaxomab in patients with advanced solid tumors. BMC cancer. 2016;16:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.2021; NCT00522457.
- 28.McDonagh CF, Huhalov A, Harms BD, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Molecular cancer therapeutics. 2012;11(3):582–593. [DOI] [PubMed] [Google Scholar]
- 29.Keller L, Werner S, Pantel K. Biology and clinical relevance of EpCAM. Cell stress. 2019;3(6):165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong R, Zhou Y, Tian X, Wang L, Wu X. Selective inhibition of IDO1, D-1-methyl-tryptophan (D-1MT), effectively increased EpCAM/CD3-bispecific BiTE antibody MT110 efficacy against IDO1(hi)breast cancer via enhancing immune cells activity. International immunopharmacology. 2018;54:118–124. [DOI] [PubMed] [Google Scholar]
- 31.Witthauer J, Schlereth B, Brischwein K, et al. Lysis of cancer cells by autologous T cells in breast cancer pleural effusates treated with anti-EpCAM BiTE antibody MT110. Breast cancer research and treatment. 2009;117(3):471–481. [DOI] [PubMed] [Google Scholar]
- 32.Kebenko M, Goebeler ME, Wolf M, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8):e1450710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lind H, Gameiro SR, Jochems C, et al. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: status of preclinical and clinical advances. Journal for immunotherapy of cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strauss J, Heery CR, Schlom J, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(6):1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lum LG, Rathore R, Cummings F, et al. Phase I/II study of treatment of stage IV breast cancer with OKT3 x trastuzumab-armed activated T cells. Clinical breast cancer. 2003;4(3):212–217. [DOI] [PubMed] [Google Scholar]
- 36.Stemmler HJ, Salat C, Lindhofer H, et al. Combined treatment of metastatic breast cancer (MBC) by high-dose chemotherapy (HDCT) and bispecific antibodies: a pilot study. Anticancer research. 2005;25(4):3047–3054. [PubMed] [Google Scholar]
- 37.Lum LG, Thakur A, Al-Kadhimi Z, et al. Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(10):2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakur A, Rathore R, Kondadasula SV, Uberti JP, Ratanatharathorn V, Lum LG. Immune T cells can transfer and boost anti-breast cancer immunity. Oncoimmunology. 2018;7(12):e1500672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallera DA, Zhang B, Gleason MK, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer biotherapy & radiopharmaceuticals. 2013;28(4):274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer cell. 2014;26(6):923–937. [DOI] [PubMed] [Google Scholar]
- 41.Stamm H, Oliveira-Ferrer L, Grossjohann EM, et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology. 2019;8(12):e1674605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma L, Gai J, Qiao P, et al. A novel bispecific nanobody with PD-L1/TIGIT dual immune checkpoint blockade. Biochemical and biophysical research communications. 2020;531(2):144–151. [DOI] [PubMed] [Google Scholar]
- 43.Gupta GK, Collier AL, Lee D, et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers. 2020;12(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao JJ, Castel P, Radosevic-Robin N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Science signaling. 2014;7(318):ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rau A, Lieb WS, Seifert O, et al. Inhibition of Tumor Cell Growth and Cancer Stem Cell Expansion by a Bispecific Antibody Targeting EGFR and HER3. Molecular cancer therapeutics. 2020;19(7):1474–1485. [DOI] [PubMed] [Google Scholar]
- 46.Fu W, Lei C, Yu Y, et al. EGFR/Notch Antagonists Enhance the Response to Inhibitors of the PI3K-Akt Pathway by Decreasing Tumor-Initiating Cell Frequency. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019;25(9):2835–2847. [DOI] [PubMed] [Google Scholar]
- 47.Del Bano J, Florès-Florès R, Josselin E, et al. A Bispecific Antibody-Based Approach for Targeting Mesothelin in Triple Negative Breast Cancer. Frontiers in immunology. 2019;10:1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rius Ruiz I, Vicario R, Morancho B, et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Science translational medicine. 2018;10(461). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forero-Torres A, Varley KE, Abramson VG, et al. TBCRC 019: A Phase II Trial of Nanoparticle Albumin-Bound Paclitaxel with or without the Anti-Death Receptor 5 Monoclonal Antibody Tigatuzumab in Patients with Triple-Negative Breast Cancer. Clin Cancer Res. 2015;21(12):2722–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shivange G, Urbanek K, Przanowski P, et al. A Single-Agent Dual-Specificity Targeting of FOLR1 and DR5 as an Effective Strategy for Ovarian Cancer. Cancer Cell. 2018;34(2):331–345 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mavratzas A, Seitz J, Smetanay K, Schneeweiss A, Jager D, Fremd C. Atezolizumab for use in PD-L1-positive unresectable, locally advanced or metastatic triple-negative breast cancer. Future Oncol. 2020;16(3):4439–4453. [DOI] [PubMed] [Google Scholar]
- 52.Forero-Torres A, Shah J, Wood T, et al. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer Biother Radiopharm. 2010;25(1):13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Messaoudene M, Mourikis TP, Michels J, et al. T-cell bispecific antibodies in node-positive breast cancer: novel therapeutic avenue for MHC class I loss variants. Annals of oncology : official journal of the European Society for Medical Oncology. 2019;30(6):934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamperschroer C, Shenton J, Lebrec H, Leighton JK, Moore PA, Thomas O. Summary of a workshop on preclinical and translational safety assessment of CD3 bispecifics. Journal of immunotoxicology. 2020;17(1):67–85. [DOI] [PubMed] [Google Scholar]
- 55.McCall AM, Shahied L, Amoroso AR, et al. Increasing the affinity for tumor antigen enhances bispecific antibody cytotoxicity. Journal of immunology (Baltimore, Md : 1950). 2001;166(10):6112–6117. [DOI] [PubMed] [Google Scholar]
- 56.Mestermann K, Giavridis T, Weber J, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Science translational medicine. 2019;11(499). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang CH, Wang Y, Li R, et al. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer research. 2017;77(19):5384–5394. [DOI] [PubMed] [Google Scholar]
- 58.Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. Journal of the American Chemical Society. 2018;140(48):16413–16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang J, Li B, Howard CB, et al. Multifunctional lipid-coated calcium phosphate nanoplatforms for complete inhibition of large triple negative breast cancer via targeted combined therapy. Biomaterials. 2019;216:119232. [DOI] [PubMed] [Google Scholar]
- 60.Kamada H, Taki S, Nagano K, et al. Generation and characterization of a bispecific diabody targeting both EPH receptor A10 and CD3. Biochemical and biophysical research communications. 2015;456(4):908–912. [DOI] [PubMed] [Google Scholar]
- 61.Fisher TS, Hooper AT, Lucas J, et al. A CD3-bispecific molecule targeting P-cadherin demonstrates T cell-mediated regression of established solid tumors in mice. Cancer immunology, immunotherapy : CII. 2018;67(2):247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng YA, Wu TH, Wang YM, et al. Humanized bispecific antibody (mPEG × HER2) rapidly confers PEGylated nanoparticles tumor specificity for multimodality imaging in breast cancer. Journal of nanobiotechnology. 2020;18(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Vries Schultink AHM, Doornbos RP, Bakker ABH, et al. Translational PK-PD modeling analysis of MCLA-128, a HER2/HER3 bispecific monoclonal antibody, to predict clinical efficacious exposure and dose. Investigational new drugs. 2018;36(6):1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.G B, D D, R G, NH S, ML J, MM D. A phase I/II first-in-human study of a novel anti-MAGE-A4 TCR/anti-CD3 bispecific (IMC-C103C) as monotherapy and in combination with atezolizumab in HLA-A*02:01-positive patients with MAGE-A4-positive advanced solid tumors (IMC-C103C-101). ASCO; 2020; 5/25/20, 2020; Chicago, IL. [Google Scholar]
- 65.Li Y, Zhou C, Li J, et al. Single domain based bispecific antibody, Muc1-Bi-1, and its humanized form, Muc1-Bi-2, induce potent cancer cell killing in muc1 positive tumor cells. PloS one. 2018;13(1):e0191024. [DOI] [PMC free article] [PubMed] [Google Scholar]