

Abstract
It is increasingly appreciated that cancer cell heterogeneity and plasticity constitute major barriers to effective clinical treatments and long-term therapeutic efficacy. Research in the past two decades suggest that virtually all treatment-naive human cancers harbor subsets of cancer cells that possess many of the cardinal features of normal stem cells. Such stem-like cancer cells, operationally defined as cancer stem cells (CSCs), are frequently quiescent and dynamically change and evolve during tumor progression and therapeutic interventions. Intrinsic tumor cell heterogeneity is reflected in a different aspect in that tumors also harbor a population of slow-cycling cells (SCCs) that are not in the proliferative cell cycle and thus are intrinsically refractory to anti-mitotic drugs. In this Perspective, we focus our discussions on SCCs in cancer and on various methodologies that can be employed to enrich and purify SCCs, compare the similarities and differences between SCCs, CSCs and cancer cells undergoing EMT, and present evidence for the involvement of SCCs in surviving anti-neoplastic treatments, mediating tumor relapse, maintaining tumor dormancy and mediating metastatic dissemination. Our discussions make it clear that an in-depth understanding of the biological properties of SCCs in cancer will be instrumental to developing new therapeutic strategies to prevent tumor relapse and distant metastasis.
Keywords: Slow-cycling cells, Quiescence, Cancer stem cells, Therapy resistance, Metastasis
1. INTRODUCTION: The cell cycle and slow-cycling cells (SCCs)
A fundamental hallmark of cancer is uncontrolled cell proliferation and loss of sensitivity to anti-proliferative signals. Once transformed (by genetic mutations together with epigenetic alterations), the founder tumor cell, or the cell-of-origin of tumor [1], establishes a clonal growth that gradually develops into a clinically incipient tumor, which then undergoes spatiotemporal evolution accompanying tumor progression and therapeutic treatments [2,3]. Therefore, deregulation in cell cycle control represents a critical driver of tumor formation and progression.
The cell cycle in normal mammalian cells consists of 4 well-defined phases, G1, S, G2 and M [4–12] (Figure 1). Cells in the G1, S and G2 phases are often said to be in the interphase whereas cells undergoing mitotic divisions in the M-phase (Figure 1A). The G1 cell-cycle phase could be further divided into the early and late G1 phases, demarcated by the Restriction (R) point [5,8,9] (Figure 1A–B). It is generally thought that once cells have passed the R point, they are irreversibly committed to entering the full cell cycle and ending with cell division. Regardless, cells in any of these 4 cell-cycle phases (G1, S, G2 and M) are often termed cycling cells or cells in the (cell) cycle. In contrast, some cells in the bulk population may reside in a non-cycling, resting and quiescent phase termed G0 (Figure 1A–C), and cells could enter G0 right after mitosis (Figure 1C) or, stochastically, from early G1 (Figure 1A–B). In general, the G1 phase is the longest whereas M phase, which consists of prophase, metaphase, anaphase, telophase and cytokinesis, the shortest (Figure 1C). Embryonic stem cells (ESCs) are well known to have very short G1 phase and undergo rapid symmetrical cell division generating more ESCs. On the other hand, a somatic stem cell (SC) may undergo asymmetric cell division (ACD) to generate a larger differentiated daughter cell while retaining a smaller SC (Figure 1A).
Fig. 1.
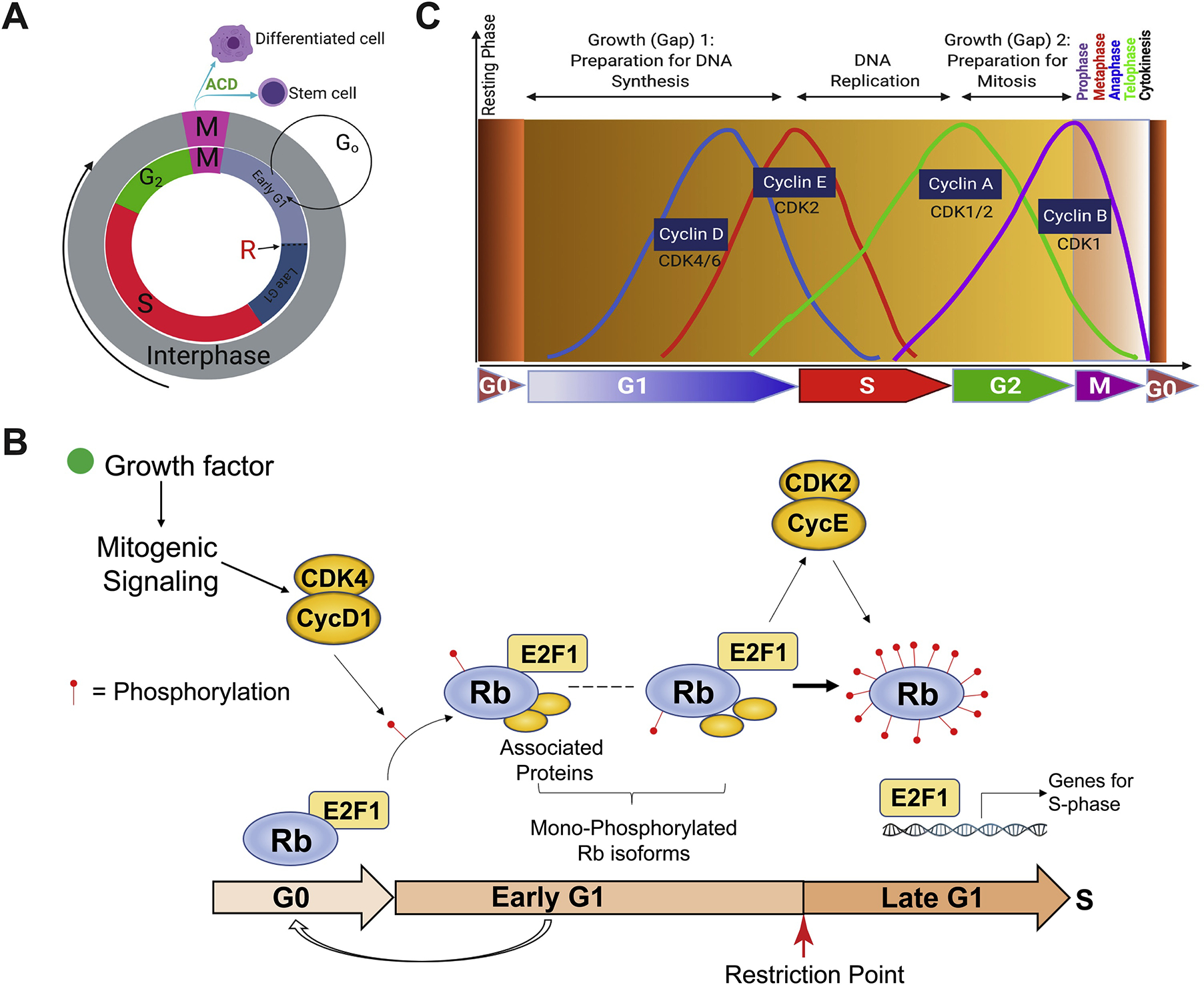
Cell cycle and cell-cycle phases in normal cells. (A) Schematic depicting the 4 cell-cycle phases (G1, S, G2 and M). Depicted also is the mitotic division that has generated two different daughter cells as a result of asymmetric cell division (ACD). Cells upon mitosis may enter the early G1 or G0 phase. R indicates the Restriction point that separates the early vs. late G1. (B) The level of Rb phosphorylation dictates the G0, early G1, and late G1 phase (based on [5]). Cells in the G0 have un-phosphorylated Rb. In response to growth factor-initiated mitogenic signaling, the cyclin D/CDK4 complex may mono-phosphorylate the Rb at one of the 14 sites and push the cells into the early G1 phase. The cyclin E/CDK2 then hyper-phosphorylate the Rb in all 14 sites thus pushing the cells over the R point and irreversibly committing cells to the rest of the cell cycle (see Text). (C) A more detailed presentation of the 4 cell-cycle phases driven by cyclin/CDK activities as well as the quiescent G0 phase. M-phase represents the shortest cell-cycle phase in which the dividing cell goes through prophase, metaphase, anaphase and telophase, and ends with cytokinesis.
Cell-cycle progression along the phases is regulated by cyclin/cyclin-dependent kinases (CDK) in response to the availability of mitogens, growth factors and nutrients in the extracellular environment (Figure 1B). The current thinking is that mitogenic signaling from growth factors would increase cyclin D1 synthesis and activate CDK4/6, which in turn hypo-phosphorylates the Rb tumor suppressor at one of the 14 sites and pushes the cells out of G0 and into early G1 [5,11,12] (Figure 1B). Therefore, cells at the G0 have un-phosphorylated Rb whereas cells at early G1 have one of the 14 mono-phosphorylated Rb isoforms [5,11,12] (Figure 1B). Cyclin E/CDK2 then hyper-phosphorylates Rb at all 14 sites and subsequently release E2F1, pushing cells over the R point to enter late G1 and leading to ‘irreversible’ cell-cycle commitment [4,5,9,11,12] (Figure 1B). Consequently, the CDK2 activity has been proposed to demarcate quiescent (i.e., G0 and early G1) vs. proliferative state of normal mammalian cells [4–12]. The S-phase is where cells replicate their DNA; consequently, nucleotide analog-based DNA incorporation has been commonly utilized to assay the proliferative proportion of cells in the S-phase (see below). Upon DNA replication, cells would finish cell cycle by entering the G2 phase, driven by cyclin A/CDK1/2, and, eventually, the M (mitosis) phase, driven by cyclin B/CDK1 activity (Figure 1C). Upon completion of chromosome duplication and segregation in M-phase, the mitotic (parent) cell will undergo cytokinesis (Figure 1C) generating two daughter cells that are either identical (not shown) or have different fates (Figure 1A). The well-delineated cell-cycle progression in normal cells frequently becomes deregulated in transformed and cancer cells due to, e.g., genetic mutations or deletions of key cell-cycle regulators such as Rb. It can be easily conceived that Rb mutation/deletion would abrogate the R point control leading to constitutive cell-cycle entry and increased cancer cell proliferation (Figure 1B).
G0 is a somewhat ‘cryptic’ phase as cells are not in the cell cycle (i.e., resting) but remain metabolically active and have un-phosphorylated Rb ‘signature’ (Figure 1B). There are two main types (states) of G0: irreversible (or permanent) and reversible (Figure 2). The prototypical example of permanently G0-arrested cells are the terminally differentiated cells in post-mitotic adult human tissues and organs, e.g., the brain, heart, and skeletal muscle, in which the parenchymal cells like neurons are irreversibly arrested in the quiescent G0 phase (Figure 2). Another type of permanently G0-arrested cells are senescent cells, which can be physiological due to telomere shortening or prematurely induced by, e.g., oncogene activation, DNA damage, and oxidative and culture stresses (Figure 2). In contrast to these irreversibly G0-arrested, differentiated or senescent cells, many cells may be transiently arrested in the G0 (or early G1) phase. For instance, some adult tissues such as the skin, small intestine and blood have fast turnover and constantly renew themselves; however, even in these fast-renewing tissues, there are primitive stem and early progenitor cells that are in a dormant and reversible G0 state [13] (Figure 2). These quiescent stem/progenitor cells can enter the cell cycle in response to tissue damage and loss of differentiated functional cells, and are thus slow-cycling cells or SCCs (Figure 2). Apparently, the slow-cycling stem/progenitor cells are to be distinguished from terminally differentiated cells (such as neurons) and senescent cells, both of which are permanently arrested in G0 (Figure 2). Finally, some special cell types such as hepatocytes and endothelial cells are well known to be largely quiescent (Figure 2).
Fig. 2.
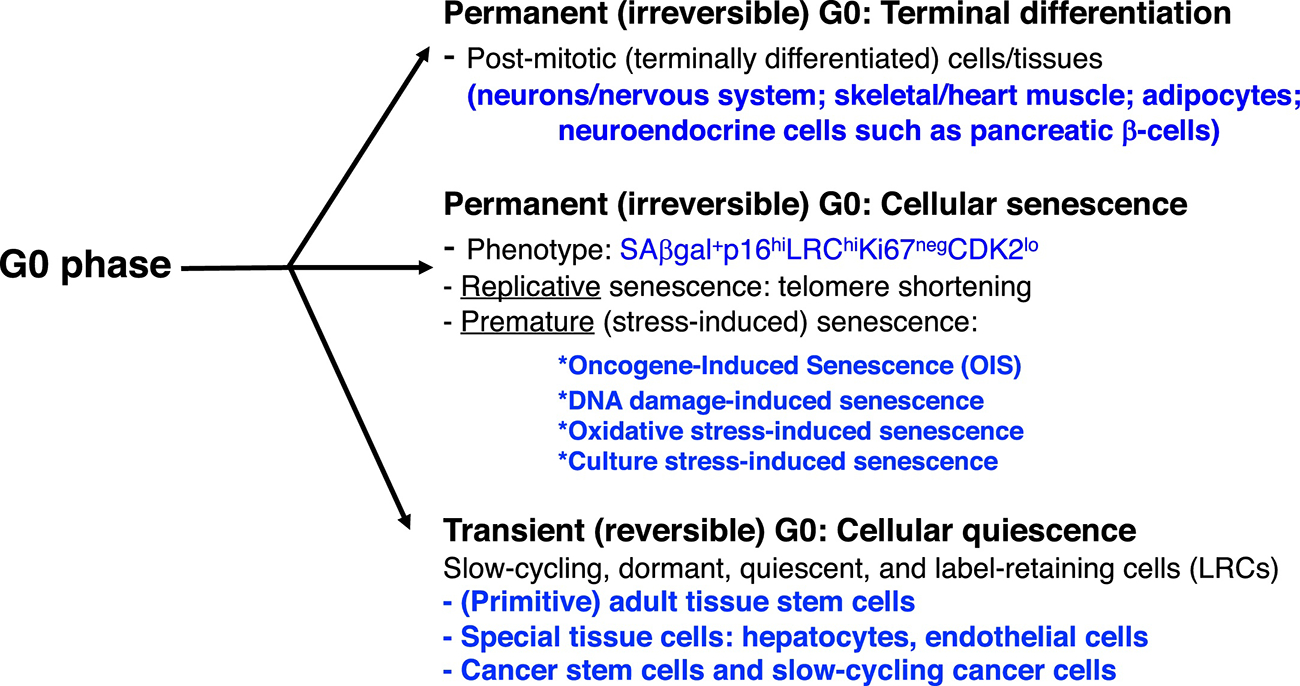
G0 phase. Shown are two different G0 phases, reversible and irreversible. See Text for details.
Interestingly, it has been observed for decades that human tumors, which resemble a disorganized organ with an overall fast turnover, also harbor subpopulations of SCCs [13–19] despite uncontrolled cell proliferation at the population level. These SCCs in cancer (Figure 2) are defined as nonproliferating quiescent/dormant cancer cells that are TRANSIENTLY arrested at the G0 or early G1 cell-cycle phase but can re-enter the proliferative cell cycle, stochastically or in response to mitogenic stimuli. In this review, we adhere to this definition and, frequently and interchangeably, use “slow-cycling,” “dormant,” “quiescent” and “label-retaining” to describe the SCCs in cancer.
2. Experimental approaches used to identify, isolate and study SCCs
2.1). . Nucleotide-based pulse and chase.
Before we discuss SCCs in cancer, we first describe several commonly used experimental methods to identify and purify SCCs (Figure 3). As mentioned earlier, cells incorporate nucleotides for DNA replication during the S-phase. Thus, nucleotide analogs such as BrdU (5-bromo-2’-deoxyuridine) and EdU (5-ethynyl-2’-deoxyuridine), or tritiated thymidine [3HT], can be used to pulse normal and cancer cells, in vitro or in vivo, for a short period of time (e.g., 20 min – 2 h depending on the overall growth kinetics of the culture) followed by cell fixation and staining using antibodies against BrdU or EdU. Cells that took up [3HT] can be identified by radiography. The fraction of BrdU+, EdU+ or [3HT]+ cells represents the % S-phase cells (Figure 3A; left). The nucleotide incorporation assays can be adapted to identify SCCs by continuously pulsing the cultures or tissues over a long period of time such that virtually all (or the majority of) cells become labeled; then such long-term pulsed cultures will be followed by a chase (i.e., washing off of the nucleotides) for different intervals of time (Figure 3A; right). Rapid-cycling cells (RCCs) would dilute out the label upon a short time of chase whereas SCCs will continue to retain the label even upon a long-term (LT) chase (Figure 3A; right). As a result, SCCs are also frequently called label-retaining cells or LRCs (Figure 3A; right). In principle, the LRCs identified upon chases for different time intervals should represent distinct SCC populations with different degrees of quiescence; in other words, short-term (ST) LRCs will identify relatively quiescent progenitor cells whereas LT-LRCs should identify the small population of deeply dormant (stem) cells arrested in G0 or early G1 (Figure 1; Figure 2). The nucleotide analog-based pulse/chase strategy was among the oldest method used to identify SCCs in studies of spermatogenesis in rat testis [20]. BrdU-based pulse/chase strategy was later adapted by Cotsarelis et al to study the slow-cycling epidermal SCs located in the hair follicles and the epidermis below sebaceous glands [21], and by Potten et al to study slow-cycling intestinal stem cells [22].
Fig. 3.
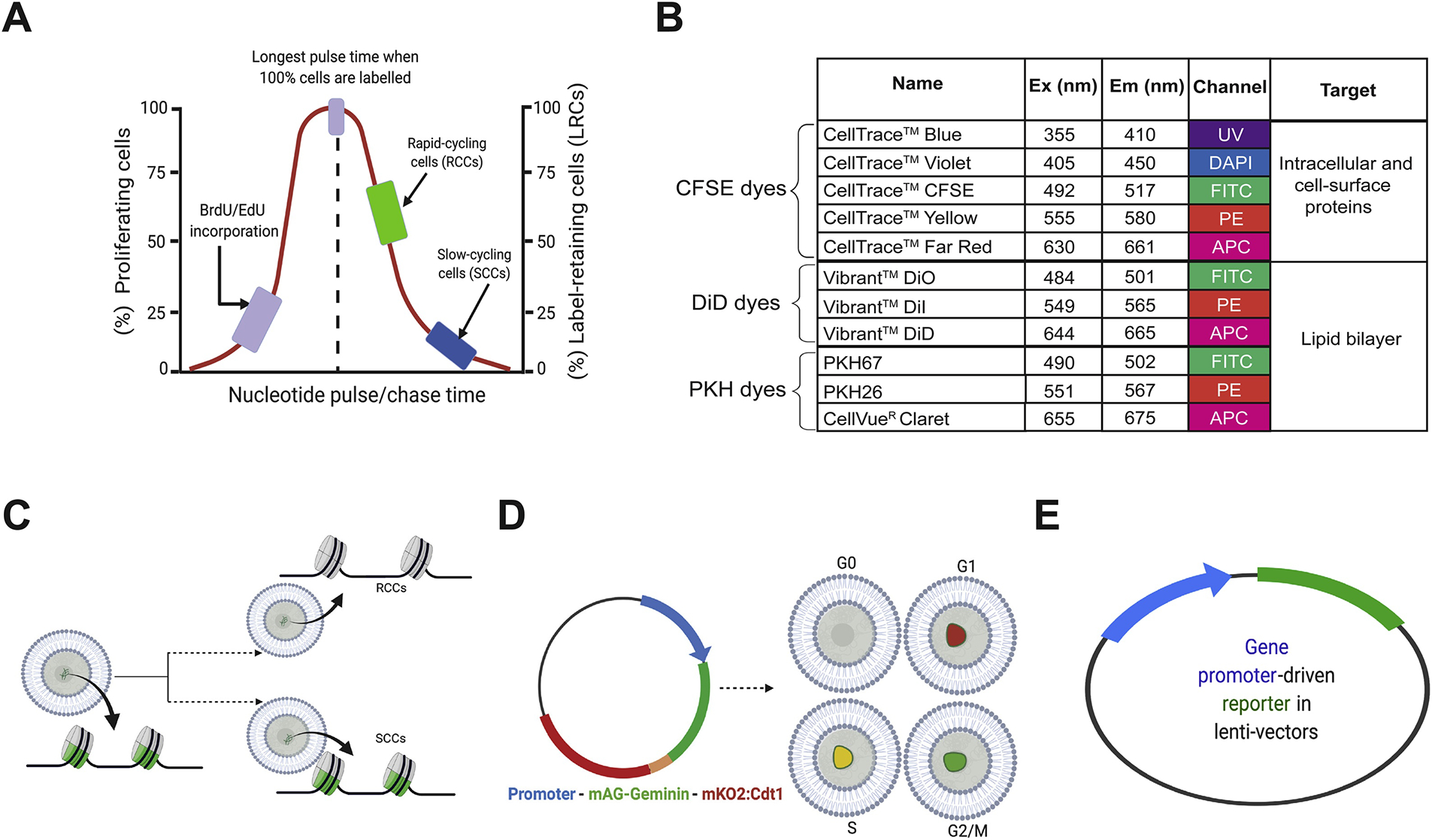
Experimental strategies to identify and purify SCCs. (A) Nucleotide analog-based pulse-chase strategy to identify cells in the S-phase (left) or the slow-cycling, long-term label-retaining cells (LRCs; right). This method does not allow purification of live cells for functional studies. (B) Dye retention-based methods to enrich and purify live SCCs (in cancer). (C) Identification and purification of SCCs based on H2B-GFP dilution. (D) FUCCI system used to separate cells in different cell-cycle phases including SCCs in G0. (E) Fluorescent reporters used to identify/purify SCCs using SCC-specific gene promoters.
Since 2003, our group has been employing prostate cancer (PCa) as a model to meticulously dissect cancer cell heterogeneity and plasticity [1,13,23–60], and our studies have led to the identification of several best defined PCa stem cell (PCSC) populations, e.g., the CD44+/hi [24–26,30,40,42,45] and PSA−/lo [33,40,42] PCSC populations. Importantly, most cells in the PCSC populations are slow-cycling (see below). In fact, our studies employing nucleotide analog-based pulse-chase strategy have provided direct evidence for SCCs in PCa spheres and prostate tumors in vivo (Figure 4; Figure 5). For instance, the LAPC4 spheres that had been chased for 1–5 days (d) had ~25% of LRCs, which began to decrease with a 10d chase, and ~1% LT-LRCs were observed after a 33d chase (Figure 4A). In contrast, the LNCaP cell spheres showed a prominent chase-dependent decrease in the number of LRCs such that by 25d and 46d after chase, only ~8% and 0.5% of the cells, respectively, retained BrdU (Figure 4B). Spheres derived from human glioma (U373), breast cancer (MCF7) and melanoma (WM562) cells also contained LRCs that can be identified upon different periods of BrdU chase (Figure 4A). We performed in vivo LRC experiments in 3 human PCa xenograft tumors, i.e., Du145, LAPC4, and LAPC9, in which we i.p injected 100 μl BrdU solution (20 mM stock; 4x/48 h) into NOD/SCID mice bearing these xenograft tumors. Tumors were then chased in mice for 20–68d until harvest. As shown in Figure 4C, we identified ~0.01 – 0.1% LRCs in all three tumors. A similar in vivo BrdU-based pulse-chase experiment in the autochthonous TRAMP tumors also identified 0.1% of LT-LRCs in the dorsal prostate (DP) and 0.1–1% of LT-LRCs in the ventral prostate (VP) of the TRAMP mice 3 months after chase (Figure 5). These studies demonstrate the presence of SCCs in both human PCa and a genetic mouse model of prostate tumor.
Fig. 4.
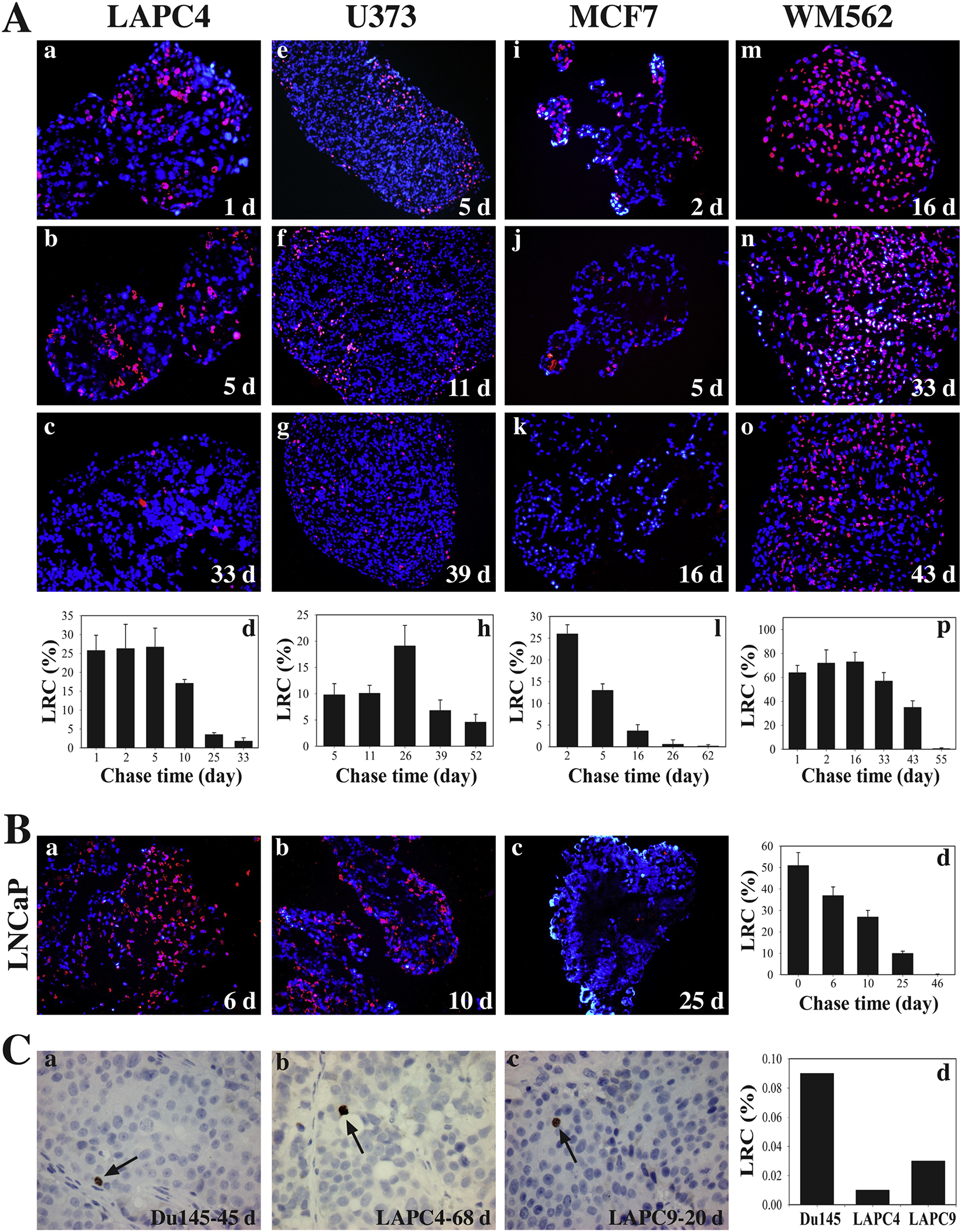
Human cancer cell spheres and prostate cancer xenografts harbor LT-LRCs (SCCs). (A-B) Human cancer cell spheres harbor LT-LRCs (i.e., SCCs) with distinct levels of quiescence. Human PCa (LAPC4 and LNCaP), glioblastoma (U373), breast cancer (MCF7) and melanoma (YVM562) cell-derived spheres were pulsed with 10 μM BrdU for 43 h, after which BrdU was washing off and sphere cultures chased for the days (d) indicated. The endpoint spheres were harvested and embedded in paraffin, and sections used in BrdU staining. At least 6 sections were analyzed and a total of 1,500 – 3,000 cells counted under an epifluorescence microscope for each cell type. The BrdU+ (i.e., label-retaining) cells were presented as bar graphs (mean ± S.D). (C) Human PCa xenografts have raie SCCs (LT-LRCs) in vivo. To identify LRCs in xenograft prostate tumors, 100 pi of BrdU solution (20 mM stock) was i.p injected (4x over 43 h) into the NOD/SCID mice bearing Du145, LAPC4, or LAPC9 xenograft tumors. Tumors were then “chased” for 45, 68 or 20 days (d), respectively, until harvest. Tumors were embedded in paraffin and serial sections of 5 pm were cut and used in immunohistochemical staining of BrdU. At least 6 sections were analyzed and counted for each tumor type and the BrdU ‘ cells scored under a microscope.
Fig. 5.
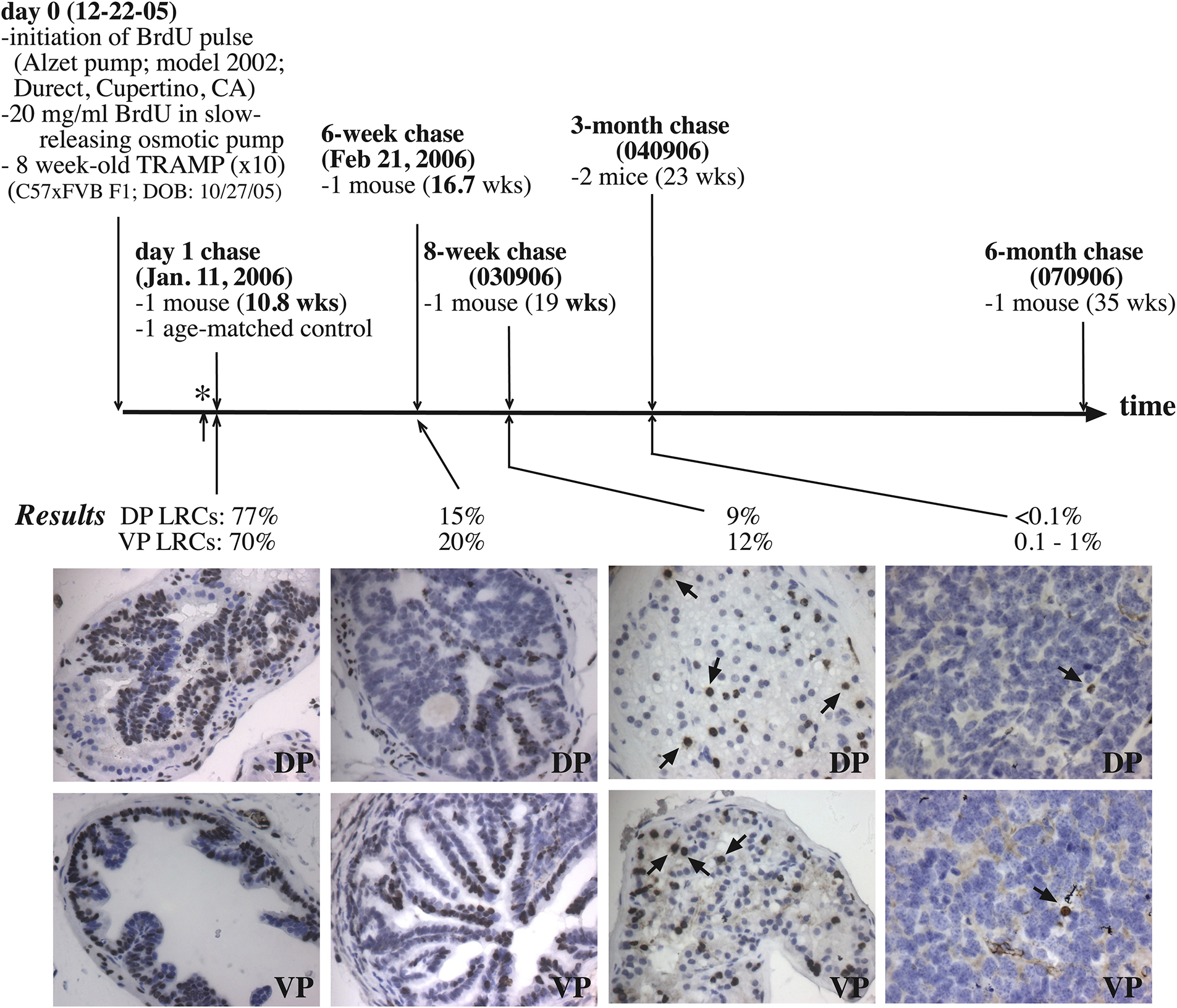
Autochthonous TRAMP tumors harbor LT-LRCs. Eight-week old TRAMP tumors in C57/FVB mice were continuously pulsed with 20 mg/ml BrdU using the slow-releasing Alzet pumps for 19 days (i.e., from Dec. 22 of 2005 to Jan. 10 of 2006) and then the pumps were removed (i.e., termination of pulse; indicated by *). Prostates chased for 1 day, 6 or 8 weeks (wks), or 3 or 6 months were harvested and embedded in paraffin. Serial sections of 5 μm were cut and used in BrdU IHC staining. At least 5 sections were analyzed and BrdU+ cells counted. Shown are the average BrdU+ cells in the dorsal prostate (DP) and ventral prostate (VP), respectively.
The obvious shortfall of the nucleotide analog-based pulse-chase strategy is its inability to allow purification of LIVE LRCs for transcriptomic profiling and functional studies. This is because to visualize the incorporated nucleotide analogs such as BrdU, cells/tissues need to be fixed and permeabilized for antibody labelling and along the way, the LRCs are killed.
2.2). Dye-based labeling and chase: Lipid and protein labeling
Live slow-cycling cells, on the other hand, can be enriched and purified via fluorescent dyes that preferentially bind membrane or intracellular proteins (e.g., CellTrace or CFSE) or membrane lipids (e.g., PKH and DiD) (Figure 3B). Among the first fluorescent dyes used to characterize cells with unique cycling parameters, PKH-based membrane labelling dye was first developed by Paul Horan in 1989 [61] and subsequently used by many to label and track cells with differing proliferative capacities [62,63] or with different proliferative kinetics [64–66] and to label extracellular vesicles [67,68]. Among the lipid-labeling dyes, PKH26 is the most commonly used. In general, PKH dyes are composed of a fluorescent carbocyanine polar head group and a hydrophobic tail that can be stably incorporated in the membrane lipid bilayer through non-covalent interactions. Once incorporated, PKH dyes are distributed throughout the membrane periphery by lateral distribution. PKH dyes, when used at non-toxic concentrations, provide rapid homogeneous staining in a salt-free environment but do not affect cell viability nor alter cell biological functions. When incubated for a long time, PKH dyes can label intracellular compartments through membrane lipid exchange. As the PKH dyes generally do not leak into the neighboring cells, dye dilution only takes place during cell division during which daughter cells inherit the membrane label from the mother cells. Thus, each cell division, in theory, would be accompanied by loss of half of the fluorescence, and dye dilution kinetics can be graphically presented as a function of cell proliferation. Slow-cycling stem-like cells rarely undergo cell division and can thus be isolated as PKH26-retaining LRCs, similar to BrdU-retaining cells but with native fluorescence permitting non-destructive cell identification and isolation via flow cytometry.
Cells can also be protein-labeled using CellTrace dyes such as CFSE (carboxy fluorescein succinyl diester) dyes (Figure 3B). Unlike PKH26, CellTrace dyes are initially colorless but, when diffused inside the cells, become metabolized by intracellular esterase to form a fluorescent product that interacts with membrane peptide via an amide bond. Like PKH dyes, the CSFE dye is compatible for both microscopic and flow cytometric analyses, at least for up to 1 week or so. Once incorporated into the cellular (membrane) proteins, CFSE can produce trackable fluorescent signal in the daughter cells for several rounds of cell divisions. Currently, CellTrace dyes may comprise five different fluorophores: blue, violet, green, yellow, and red (Figure 3B), which, in principle, can be combined with other stains for multiplexing.
The fluorescent dye-based labeling strategies have some limitations. For example, the two classes of cell-labeling dyes (Figure 3B) may identify and preferentially enrich different subpopulations of LRCs. Also, multiplexing using the two classes of fluorescent dyes, together with other fluorescence-based staining protocol(s), may entail significant optimization. In addition, using membrane-labeled cells for downstream immunofluorescence analysis along with other intracellular markers could be problematic as requisite permeabilization may cause dye leakage. Since none of the membrane labels are soluble in water-based solvents, PKH dyes are supplied in ethanol and CellTrace dyes are dissolved in DMSO. These solvents (i.e., ethanol, DMSO) could potentially be toxic to the cells.
2.3). Labeling and chase via H2B-GFP turnover
All of the aforementioned dyes may confound cell viability by virtue of either the diluent, dye loading stress (e.g., osmotic stress) or potentially the label itself. A dye alternative free of cell viability and behavioral impacts would thus be highly desirable. Histone 2B (H2B) is part of the histone octamer that has a long turnover with a half-life of 4–6 weeks. Taking advantage of the slow-cycling nature of H2B, Wahl and colleagues were the first to make a H2B-GFP fusion to study chromosome dynamics in mammalian cells [69]. Subsequently, the H2B-GFP based cell labeling and chase (Figure 3C) have been adapted to study label-retaining slow-cycling cells in multiple tissues and organs in vitro and in vivo as well as SCCs in cancer. For example, Puig et al [70] recently created a Doxycycline (DOX) inducible H2B-eGFP lentiviral reporter that can be integrated within the genome through lentiviral-mediated infection. Following transduction, cells can be induced by DOX to mark those that have incorporated the GFP-tagged H2B. During subsequent chase when the cells are grown in DOX-free medium, H2B-GFP will be partitioned between mother and daughter cells and thus reduced by 50% with every cell division. SCCs do not undergo rapid proliferation and will thus retain more fluorescence and can be isolated/sorted using FACS. In the follow-up experiments, authors identified TET2 dioxygenase, which is important for DNA demethylation at the cytosine residue, as a regulator of SCCs that mediate therapy resistance and tumor maintenance in melanoma, GBM and colorectal cancer models [70].
Several considerations need to be taken into account when using lentiviral-based H2B-GFP reporter system for enriching and isolating SCCs. First of all, the expression of H2B-GFP is obviously affected by the doses and time of DOX treatment; therefore, pilot studies are required for each cell model. Secondly, only the average multiplicity of infection (MOI) at the population level can be estimated and absolute lentiviral particle integration per cell is unattainable. Thirdly, H2B-GFP labelling may not be uniform across the cell population as the chromosomal environment and ploidy may affect lentiviral integration and H2B-GFP expression. Finally, precisely because SCCs proliferatively quiescent, they may not initially incorporate the H2B-GFP thus leading to the inability to study deeply dormant SCCs.
One significant advantage of the H2B-GFP system is its utility in in vivo models. We have recently adapted a DOX-repressible variant of the H2B-GFP labeling system to identify and study slow-cycling luminal epithelial cells in the mouse prostate (Figure 6; [54]). This system involves generating a double transgenic (or bigenic) mouse model by crossing the ARR2Pb-TetR.VP16 line with the TRE-mCMV/H2B-GFP reporter line (Figure 6C). The resultant bigenic mouse prostate will be fully labeled by H2B-GFP due to the binding of TetR.VP16 to the TRE (Figure 6A–C). As the tetracycline-repressible VP16 transactivator (TetR.VP16) is under the control of ARR2Pb promoter, most GFP-labeled cells in the unchased prostate, as expected, are luminal epithelial cells although a subset of the basal cells (2–8%) also become labeled (Figure 6D) [54]. Upon addition of DOX in the chow or drinking water, DOX will bind to and repress the TetR.VP16 thus turning off H2B-GFP transgene expression and initiating the chase (Figure 6C; right). Fast proliferating cells will be rapidly chased out of GFP whereas SCCs will retain GFP. We generally start chasing the male mice at 6–8 weeks and the initial luminal cell-labeling efficacy is around 80% for the VP and dorsolateral prostate (DLP) whereas the labeling efficiency for the anterior prostate (AP) seems much lower around 40–50% [54] (Figure 6D). As expected, upon chase, the mouse prostate showed chase time-dependent decreases in overall fluorescence intensity and the abundance of GFP+ cells [54] (Figure 6A). GFP+ luminal cells in the VP dropped from 80% to <20% upon a 9-week chase, to 2–6% in all prostate lobes upon a 12-week chase, and to undetectable by 14-week chase [54] (Figure 6E). Utilizing the 12-week chased GFP+ cells as the LT-LRCs for a large battery of functional studies, we have shown that these slow-cycling luminal epithelial cells possess the stem/progenitor cell gene expression profile and biological properties [54]. Significantly, these SCCs are intrinsically resistant to castration [54], likely due to their dormant nature and to the fact that many of these LT-SCCs are negative for AR [54] (Figure 6E)
Fig. 6.
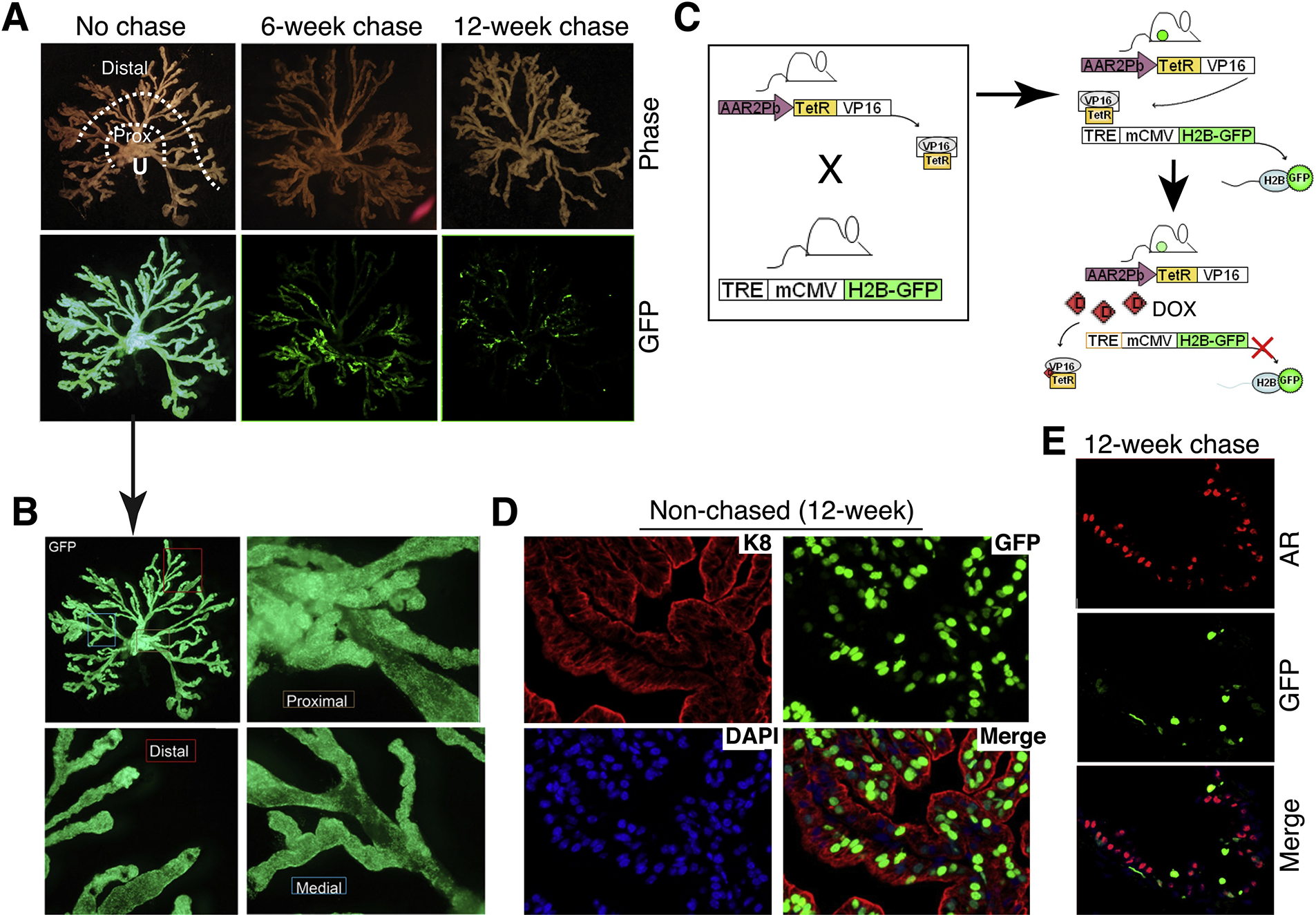
Adapting the inducible H2B-GFP labeling/chasing system to identify and study slow-cycling luminal progenitor cells in the mouse prostate. (A) Chase time-dependent decrease in GFP fluorescence intensity and in GFP+ cells. Shown on top are micro-dissected mouse prostate alveolar-ductal tree structure in relation to the urethra (U). Distal (secretory) alveoli and proximal (Prox) ducts close to the urethra are indicated. Shown below are the corresponding GFP images in non-chased (left; 20-week), 6-week chased (middle) and 12-week chased prostates. Note that the no-chase and 6-week chase images were adapted from [54] with permission. (B) The non-chased prostate in (A; the arrow) is shown in higher magnifications, to illustrate the homogeneous GFP labeling in the proximal, distal and medial regions of the mouse prostate. (C) Schematic of the model (see Text). (D) Analysis of a 12-week non-chased anterior prostate shows that the GFP+ cells are largely K8+ luminal epithelial cells. (E) Analysis of a 12-week chased ventral prostate reveals significantly reduced numbers of GFP+ cells, which are mostly AR−.
2.4). Cell-cycle phase reporters
An alternative method for isolating SCCs in the G1 cell-cycle phase is the utilization of the Fluorescent Ubiquitination-based Cell-Cycle Indicator or FUCCI (Figure 3D), developed initially by Sakaue-Sawano and colleagues in 2008 [71] and subsequently adapted and further developed by many groups for studying cell-cycle dynamics in vitro and in vivo [72–76]. The FUCCI system utilizes the expression of two cell-cycle restricted proteins: the Cdt1 protein, which is tagged with the monomeric Kusabira Orange (mKO2-hCdt1) and marks the cells with red fluorescence when they accumulate during the G1 phase; and the Geminin protein, which is tagged by monomeric Azami Green (mAG-hGem) and marks the cells green when they accumulate during the late S/G2/M phase (Figure 3D). Both Geminin and Cdt1 are regulated by ubiquitination in a cell-cycle dependent manner. Thus, the Anaphase Promoting Factor (APC), along with its regulator Cdh1, forms the APC/CCdh1 complex that leads to the ubiquitin-mediated degradation of Geminin, thereby inhibiting GFP expression during the G1 phase and the G1 cells will be identified as red (Figure 3D). In contrast, the S phase kinase associated protein 2 (Skp2) complexed with SCF is responsible for degrading Cdt1-mKO2 and cells in the G2/M phase will be labeled by the green fluorescence (Figure 3D). Cells in transition between G1 and G2/M, i.e., the S-phase cells, will be labeled as yellow (Figure 3D). Thus, in the FUCCI system, the APC/CCdh1 complex regulates SCFSkp2, and the latter also regulates APC/CCdh1 through a feedback mechanism.
The two-part FUCCI system is potentially advantageous over the above-mentioned pulse-chase strategy that traditionally only allows the identification and isolation of one cell population, i.e., SCCs, whereas FUCCI system can facilitate the tracking and purification in real time of both proliferating (S/G2/M) and nonproliferating (G1) cells (Figure 3D). Applying the FUCCI system in melanoma models, Puig et al [70] showed that the SCCs tend to localize at the center of the melanoma whereas the fast-cycling cells are distributed more towards the periphery of the tumor mass. On the other hand, FUCCI system does have its own limitations. Because the FUCCI expression vectors utilize lentiviral delivery, like H2B-GFP, the reporter expression will depend on the MOI of the vectors and the genomic locus and context of the vector insertion. Additionally, the two cell-cycle indicators in the FUCCI system, i.e., Cdt1 and Geminin, are normally expressed in a gradient, rather than a bimodal, fashion. Therefore, their ‘activation’ may not be completely regulated in a cell-cycle dependent manner, and chances of false positives could be high. Importantly, FUCCI does not permit discrimination of cells in G1 vs G0 eliminating the potential of FUCCI alone to identify authentic SCCs.
2.5). Gene promoter-driven reporters
Endogenous and exogenously introduced gene promoters have long been utilized to study and/or trace the development of a (stem) cell population in vitro and in vivo [1], and the basic principle has been adapted to study cancer cell populations including CSCs and SCCs (Figure 3E). As discussed earlier, cell-cycle progression is driven forward by cyclin/CDKs (Figure 1C). For example, CDK2 activity demarcates cells in quiescence vs. in proliferative cycle and CDK2 activation drives cells into the S-phase, after which cells would generally finish the cell cycle (Figure 1B–C). Consequently, CDK2 activity-based reporters could potentially separate cells in the cycle vs. cells in deep G1 or G0. Indeed, CDK2 promoter-based reporters have been used to fractionate bulk cells with CDK2lo cells representing G0 cells [4,10], including senescent cells permanently arrested in G0 phase (Figure 2). On the other hand, cell-cycle progression is also negatively regulated by the two families of CDK inhibitors (CDKIs), i.e., the INK4 family (p15, p16, p18 and p14ARF), which acts at the beginning of cell-cycle entry into the G1 phase, and the Cip/Kip family (p21, p27, and p57), which impedes the progression throughout the cell cycle. Therefore, the promoters of p21 and p27 have been employed to drive reporters such as GFP to enrich and purify quiescent cells. The best example is p16INK4A, which is significantly upregulated in G0 and deep G1 cells. As a result, reporters such as GFP and β-galactosidase (β-gal) have been knocked into the endogenous p16INK4A gene locus to identify and purify p16+ (i.e., β-gal+) senescent cells arrested in G0 [77–79].
Gene promoter-driven reporters have also been adapted in different flavors to identify and purify SCCs or CSCs. For example, if a new gene has been uncovered to be a critical regulator of SCCs, that gene promoter could potentially be used to drive a reporter in a lentivector to infect the bulk cells and identify/enrich the slow-cycling cell subpopulation (Figure 3E). Such is the case with the KDM5B (JARID1B) promoter, which is used to identify and study the transient population of SCCs in melanoma [80,81]. Our lab has adapted the concept of gene promoter-driven reporters to enrich and isolate live PCSCs. Specifically, we employed a lentiviral reporter system in which the gene promoter of PSA (Prostate Specific Antigen), a differentiation marker of prostate and PCa cells, was used to drive GFP or dsRed expression. By employing this reporter system and performing a series of biological studies, we have demonstrated that the phenotypically undifferentiated, i.e., PSA−/lo, PCa cell population harbors long-term self-renewing PCSCs that are largely dormant and intrinsically refractory to castration and chemotherapeutic drugs [33, 40, 42]. In contrast, the differentiated PSA+/hi cell population has more limited tumor-propagating capabilities and is sensitive to antiandrogens, anti-mitotic drugs as well as radiation and prooxidants.
3. SCCs, CSCs and EMT-like cancer cells: Similarities and distinctions
Most normal primitive SCs localized in their natural niches, e.g., long-term hematopoietic SCs (HSC) in the bone marrow, are quiescent, and they only enter the proliferative cell cycle when needed. This suggests that in normal SCs, stemness and quiescence may go hand-in-hand. CSCs, or stem-like cancer cells that possess some or many normal SC properties (e.g., self-renewal, differentiation, proliferative potential), have been reported in virtually all human cancers [1,13,23–60, 82–100]. Likewise, mitotically quiescent SCCs have now been reported in most cancer types including, among others, melanoma [80,81,101], lung cancer [102,103], glioblastoma [104–108], breast cancer [109–112], multiple myeloma [113], leukemia [114], and colorectal [115,116], pancreatic [117], skin [118,119] and gastric [120] cancers. PCa cells that have disseminated into the bone marrow are also known to be dormant [121–128], although very little is known about SCCs in primary prostate tumors and castration-resistant PCa (CRPC) disseminated into the other sites, especially visceral organs. Finally, cancer cells manifest inherently high cellular plasticity such that a subpopulation of cancer cells in the tumor may be constantly undergoing epithelial-to-mesenchymal transition or EMT [129–131]. Notably, EMT represents a process during which some cancer cells are still going through the process (sometimes called EMT’ing cells) whereas others have completed EMT and become fully ‘transformed’ into a mesenchymal lineage (i.e., full-EMT cells).
The relationship between SCCs, CSCs and EMT-like cells can be technically intricate, oftentimes semantic, and potentially confusing (Table 1). First and foremost, they are defined differently (Table 1). CSCs are most often phenotypically defined by hypothesis-driven unique marker expression profile (e.g., CD44hi, BMI-1hi, PSA−/lo) and functionally defined by their enhanced tumor-initiating and, importantly, long-term tumor-propagating properties [1,13,26,34,55]. In contrast, SCCs are always defined by their fundamental nature of being quiescent whereas EMT-like cells by their prototypical fibroblast morphology coupled with loss of epithelial phenotypes, markers and regulators (Table 1). While EMT-like cells generally assume a mesenchymal cell morphology, both SCCs and CSCs could be either mesenchymal or epithelial (Table 1). Indeed, in both prostate and breast cancers, the ALDHhi CSCs are epithelial and proliferative whereas the CD44hi CSCs are mesenchymal, invasive and quiescent [40,132,133]. This points highlights another difference between the three cell types: whereas both SCCs and EMT-like cells are dormant, CSCs can be either dormant or highly proliferative (Table 1). Consequently, in tumor xenotransplantation experiments under limiting-dilution conditions, SCCs and EMT-like cancer cells may ‘paradoxically’ display low tumor-initiating (tumor-regenerating) activities, in contrast to most CSCs [1] (Table 1).
Table 1.
SCCs, CSCs and EMT-like cells: Common traits and distinct features*
| Properties | SCCs | CSCs | EMT-like cells |
|---|---|---|---|
| Definition | Quiescence & dormancy | - Generally by markers - Great tumor-initiating potential - Long-term tumor-propagating capability |
- Cells may be undergoing (EMT’ing cells) or may have finished EMT. - Lost epithelial markers |
| Cell phenotype(s) | Can be epithelial or mesenchymal. | Can be epithelial or mesenchymal | Mesenchymal |
| Proliferation/Quiesc ence | Non-proliferative, quiescent | Possess enormous proliferative potential BUT can be proliferating or quiescent. | Non-proliferative, quiescent |
| Tumor-initiating (-regenerating) capability | May be low because SCCs are quiescent. | May be high for epithelial-type (ALDHhi) CSCs but low for mesenchymal-type (CD44hi) CSCs. | May be low because EMT-like cells are mostly non-proliferative. |
| Therapy resistance | Inherently resistant to multiple therapies | - Many CSCs are intrinsically resistant to various therapies. - Proliferative (e.g., ALDHhi) CSCs are sensitive to anti-mitotic therapies. |
- Full EMT cells are chemoresistant. - EMT’ing cells may be sensitive to inhibitors that target the RhoGTPases. |
| Invasiveness | Some SCCs are invasive. | Some CSCs are invasive. | Very invasive |
| Metastasis | - Have been implicated in mediating distant dissemination & metastasis. - SCCs may stay in the dormant state for months, years or even decades. |
- The mesenchymal type of CSCs may be highly invasive and metastatic. - The epithelial type (e.g., ALDHhi) of CSCs may not be metastatic. |
Cells undergoing EMT are important in the intravasation, extravasation and initial colonization phases of metastasis. But for the clinically overt macrometastases to establish, cells must undergo MET. |
| Inter-relationship | - May only partially overlap with CSCs, both phenotypically and functionally. - May be EMT-like or epithelial-like. - Pre-exist in tumors but can be selected for by therapies. - Representing a cellular state (i.e., random entry into G0/early G1 quiescent state) |
- Not all CSCs are slow-cycling. - May be EMT-like or epithelial-like. - Pre-exist in tumors but can be selected for by therapies. - Representing a cellular state (i.e., SC-like epigenetic state and having the ability to differentiate into a clonal progeny) |
- May partially overlap with SCCs and mesenchymal-type CSCs. - Pre-exist in tumors but can be selected for by therapies. - Representing a cellular state (i.e., migratory & quiescent in G0/early G1) |
| Regulation by TME | Yes | Yes | Yes |
| Major intrinsic signaling mechanism(s) | - Regulators of cellular quiescence - High negative cell-cycle regulators such as p16 and p21 - Low positive cell-cycle regulators such as CDK2 - Others: p38, TET2, KDM5B, NR2F1, etc |
- Regulated by stemness factors and developmental pathways - Examples include NOTCH, HH, FGF and WNT - May possess typical SC epigenetic landmarks such as EZH2/PRC |
- Driven by ‘master’ EMT regulators such as ZEB, SNAIL, SLUG and TGF-β - May also be induced by developmental signals such as FGF and WNT |
Commonly shared properties in 2 or 3 cell types are marked in blue while distinct features unique to each population are indicated in black.
To some degree and in certain circumstances, all three cancer cell types could well be describing the same population of cells in different cellular (epigenetic) states (Table 1). For instance, some (but NOT all) SCCs may possess phenotypic and functional properties of CSCs and, vice versa, some (but NOT all) CSCs may be dormant and slow-cycling. Thus, SCCs in many tumors have been shown to possess CSC-related properties, e.g., niche dependence, enhanced tumor-propagating ability, therapy resistance, and promotion of tumor relapse and metastasis [13,16–19,116,117]. Vice versa, many functionally validated CSCs are quiescent [e.g., 13,24,33,92]. The best examples are the CD44+/hi [24,26] and PSA−/lo [33,40] PCSC populations that we have extensively studied (Figure 7). For example, using the BrdU-based pulse/chase strategy, we found that the LAPC4 spheres harbor chase time-dependent LRCs (Figure 4A; Figure 7A). Upon 10, 25, and 33 days of chase, there were approximately 17%, 4% and 1% LRCs, respectively, in the LAPC4 spheres (Figure 4A; Figure 7A). Strikingly, most CD44+/hi PCSCs in the spheres colocalize with the LRCs chased for 10 and 25 days, and the CD44+/hi LRCs actually decreased by 33 days after chase (Figure 7B; [24]), suggesting that the CD44+/hi PCSCs in the LAPC4 PCa model have an intermediate level of quiescence. Similarly, most CD44+/hi cells in Du145 holoclones, which we have shown to harbor long-term self-renewing PCSCs [27], are nonproliferating, i.e., BrdU-negative, upon a short-term (4 h) BrdU pulse (Figure 7C; [26]), again suggesting that the CD44+/hi PCSCs in the Du145 model are relatively quiescent. Analogously, the PSA−/lo PCa cell population harbors long-term tumor-propagating cells and, of clinical significance, PSA−/lo (but not PSA+) PCa cells preferentially survive ADT and can initiate robust tumor regeneration in androgen-ablated hosts [33,40]. Importantly, time-lapse video-microscopy analysis of single cells has revealed that most PSA+ PCa cells undergo rapid symmetrical cell divisions generating an all-PSA+ cell clone in relatively a short time [33,40] (Figure 7D, top). In contrast, 5–8% of the PSA−/lo PCa cells have hardcore PCSC properties in that they undergo ACD generating both a PSA+ and a PSA−/lo daughter cell in their first division [33,40] (Figure 7D, bottom). Strikingly, in such PSA−/lo PCSC-derived ACD clones, the differentiated PSA+ daughter cell would embark on rapid subsequent cell divisions such that most cells in the endpoint clones are PSA+ with only one PSA−/lo PCSC [33,40] (Figure 7D). Consequently, at the population level, the PSA−/lo PCa cells have significantly longer cell-cycle transit times (Figure 7E) and less population doublings (Figure 7F) than PSA+ PCa cells [33,40], suggesting that the PSA−/lo PCSC population is largely quiescent. Notably, the PSA−/lo PCSCs are not enriched in EMT genes [33,40]. These discussions on the CD44+/hi and PSA−/lo PCSC populations vividly illustrate the intricate relationship between marker-defined CSCs, cellular quiescence (proliferation status) and the mesenchymal state (Table 1).
Fig. 7.
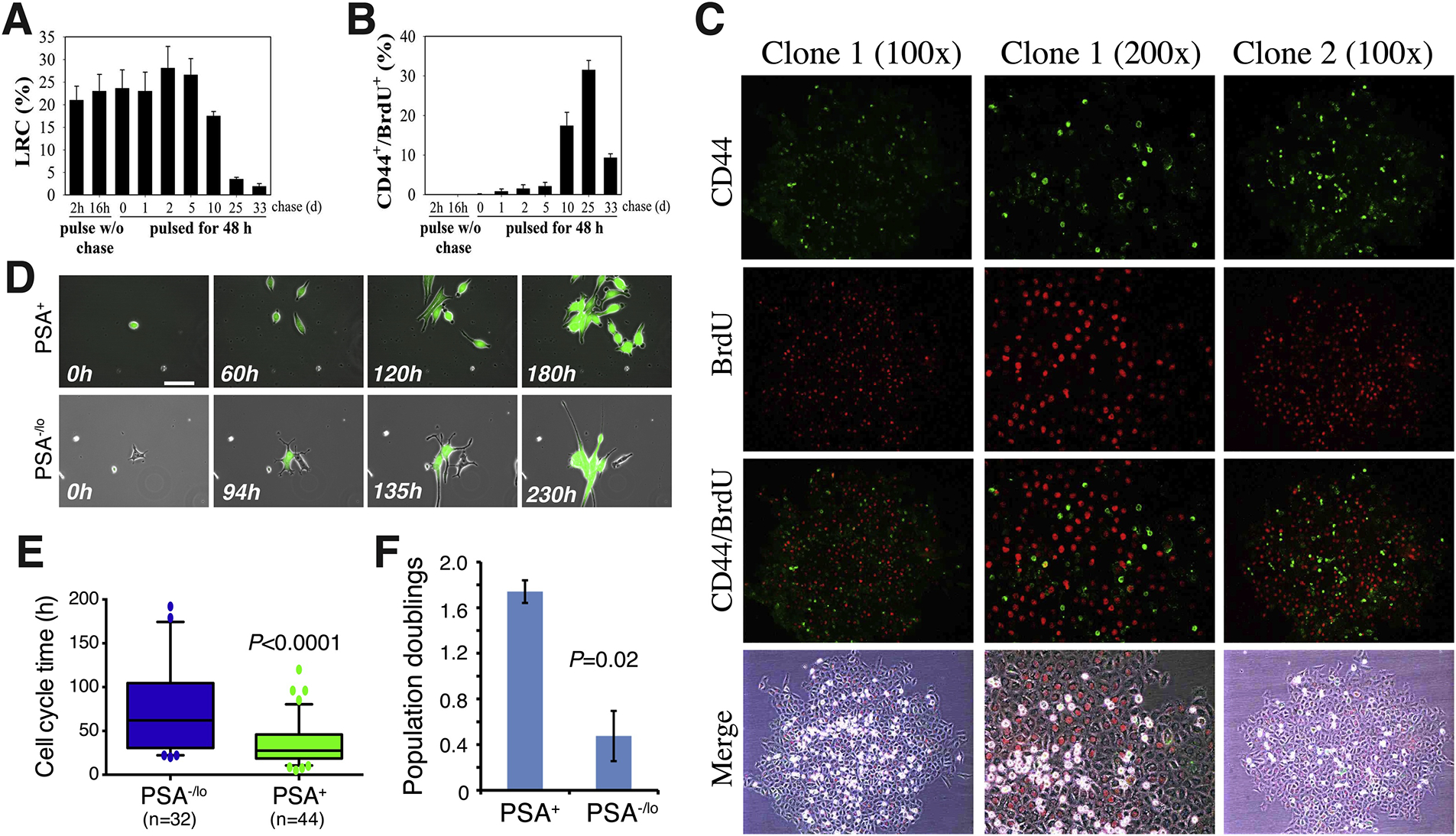
Human PCSCs are generally slow-cycling. (A-B) The CD44+ PCSCs have intermediate slow-cycling properties. The LAPC4 spheres were either pulsed with BrdU for 2 or 16 h without chase or pulsed for 48 h and then chased for up to 33 days. The BrdU-retaining cells, i.e., LRCs, were quantified (A). In B, the CD44 and BrdU double-positive cells were quantified from serial sections obtained from at least 12 spheres. The bars are the mean ± SEM obtained from three independent experiments. Data were from [24] with permission. (C) CD44+/hi PCa cells are relatively quiescent. Shown are two Du 145 cell holoclones pulsed with BrdU for 4 h followed by BrdU (red) and CD44 (green) staining. As can be seen, most CD44+/hi Du145 cells in the holoclones are BrdU-negative. Taken from [26] with permission. (D) The PSA−/lo PCSCs are quiescent compared to differentiated PSA+ PCa cells. Shown are time-lapse images of one PSA+ (green) LNCaP cell recorded for 180 h and one PSA−/lo LNCaP cell recorded for 230 h (taken from [33] with permission). (E-F) PSA−/lo LNCaP cells have much longer cell-cycle transit time (E) and lower population doublings (F) compared to the corresponding PSA+ LNCaP cells. Adapted from [40] with permission.
On the other hand, although SCCs, CSCs and EMT-like cells may each represent (overlapping) cellular states in a population continuum, the underlying mechanisms defining their cellular states may be different (Table 1). For example, not all CSC populations are dormant, as illustrated by the proliferative ALDHhi CSCs in many solid tumors including prostate and breast cancers [1,13,40,132,133] (Table 1). The co-existence of quiescent and proliferative CSC subpopulations in many tumors may be analogous to the existence of both quiescent AND cycling normal SC populations in some tissues such as the small intestine [13]. In principle, CSCs in treatment-naïve primary tumors, much like stem cells in normal tissues and organs, should be hierarchical, meaning that CSCs are on top of the cancer cell hierarchy having the ability to differentiate into more mature daughter cancer cell progeny (Table 1). In contrast, SCCs in unperturbed primary tumors may represent a random and stochastic, rather than hierarchical, cell population, meaning that SCCs at any moment may stochastically enter the proliferative cycle and, vice versa, cycling cells at any moment may enter the quiescent state (Table 1). And it is currently unclear whether SCCs and EMT-like cells in various cancers can undergo ACD generating phenotypically ‘differentiated’ progeny, which is a cardinal property of normal tissue SCs.
In spite of many nuanced differences between SCCs, CSCs and EMT-like cells, they all play critical roles in regulating tumor cell subpopulation dynamics and contributing to the generation of tumor cell heterogeneity along tumor evolution and accompanying therapeutic treatments. All three cell populations pre-exist in treatment-naïve tumors (or cultures) and can become greatly enriched (i.e., selected for) by therapeutic interventions (Table 1). Importantly, the SCCs, CSCs and EMT-like cancer cells have all been evinced to be intrinsically therapy-resistant and thus implicated in tumor relapse, and to be highly invasive and implicated in dissemination and metastasis (Table 1; see below for SCCs). SCCs, CSC and EMT-like cells are epigenetically regulated cancer cell populations and their cellular states are all dictated and significantly influenced by the neighboring cellular milieu and tumor microenvironment or TME [e.g., 13, 16,18,19,55,85,86–94,98–100,132,133] (Table 1). Consequently, the manifestation of the biological properties of SCCs, CSCs and EMT-like cells could be cell-autonomous (or cell-intrinsic) and/or be regulated by cell-extrinsic factors including non-CSCs and non-SCCs as well as various (non-cancer) cells and soluble/diffusible molecules in the TME (Table 1). Thus, many of the reported cell-intrinsic molecular regulators of SCCs, exemplified by LRIG1 [59], NR2F1 [129], TET2 [70], KDM5B [80,81], SOX9 [134] and TGFβ [135], as well as the proinflammatory/immune-suppressive TME [e.g., 98,136–138], are all well-known regulators of CSCs and EMT inducers (Table 1; also see below)..
4. SCCs in therapy resistance, tumor recurrence, and distant metastasis
The involvement of SCCs in mediating tumor dormancy, therapy resistance, tumor relapse and cancer cell dissemination and metastasis has been extensively discussed in a series of excellent reviews [14–19]; therefore, here we only briefly expound on this topic. The concept that SCCs represent a transient, epigenetically regulated cell population [102] is best illustrated by the KDM5Bhi melanoma cells, which are slow-cycling, possess some CSC properties, and, importantly, are shown to be required for long-term tumor maintenance [80]. In theory, if KDM5B is simply an enforcer of melanoma cell quiescence, knocking down KDM5B, a histone demethylase and transcriptional epigenetic regulator, should lead to increased tumor cell proliferation and tumor growth. Surprisingly, however, KDM5B knockdown attenuated tumorigenicity in a passage-dependent manner [80]. Further biological studies indicate that KDM5B+ SCCs are not maintained in a hierarchial fashion but instead seem to function as a cellular reservoir for maintaining the inexhaustible replicative potential of the bulk tumor [80]. These results highlight the overlapping but not identical nature of SCCs and CSCs (Table 1).
In the context of SCCs mediating therapy resistance, Hata et al generated several therapy-resistant slow-cycling clones of PC9 non-small cell lung cancer cells by exposing them to increasing drug concentrations [103]. Surprisingly, while some clones rapidly (i.e., 2–3 weeks) developed resistance, other clones took much longer time, i.e., 40 weeks, to develop therapy resistance. Next-generation sequencing together with DNA barcoding analysis comparing early-resistant (GR2) to late-resistant (GR3) clones revealed that GR2 clones were derived from pre-existing parental population wheras the GR3 clones acquired therapy resistance by modulating signalling, including BCL-2 and MAPK, pathways [103]. As a result, GR3 clones were sensitive to BCL2 inhibitors whereas GR2 clones were insensitive [103]. Walens et al also identified two therapy-resistant cell populations in triple negative breast cancer (TNBC): pre-existent and the therapy-induced populations [6]. Interestingly, the pre-existent population does not readily switch the cell-cycle pattern from slow-cycling to rapid-cycling following drug withdrawal. Rather, this population might function as a therapy-resistant reservoir and influence the neighboring microenvironment through the activation of IL-6-mediated paracrine signaling and STAT3 phosphorylation [6]. Such studies [6,103] make it clear that therapy-resistant SCCs preexist in treatment-naïve tumors (Table 1), and therapeutic treatments may further induce a de novo subset of therapy-refractory SCCs through cellular reprogramming. Pre-existent SCCs may mechanistically function in two different ways to mediate therapy resistance and tumor relapse: as the intrinsically resistant ‘seeds’ for the population survival and via microenvironment-mediated paracrine signaling to help induce the de novo therapy-resistant SCC population.
SCCs, presumably due to their non-proliferative and mesenchymal features, are highly invasive and likely represent the metastatic seeds [e.g., 101,106,111,112,116] (Table 1). On the flip side, there is also substantial evidence that disseminated tumor cells (DTC), which frequently manifest CSC phenotypes and properties and are thus called metastatic stem cells [136–138], in the secondary organs are mostly quiescent, and the best example perhaps is the metastatic PCa cells disseminated to the bone marrow. Early work from Taichman and co-workers showed that, strikingly, the bone-homing metastatic PCSCs usurp the normal HSC niche for their initial residence in which they live a mitotically quiescent life [121]. Subsequent work from the group implicated the TYRO3/AXL/MER (TAM) receptor kinase family and their ligand Gas6, TGF-β2 and TBK1 in mediating and maintaining the quiescence of the disseminated PCa cells [123–127]. Their recent work suggests that, intriguingly, the dormant DTCs in the bone marrow in PCa patients might be re-awoken by signaling from the sympathetic nervous system [128].
An important question has been on how disseminating and disseminated SCCs escape immune surveillance and sustain long-term survival. Some normal tissue stem cells are ‘immune-privileged’ and not susceptible to immune-mediated recognition and rejection [55,94]. For example, T cell therapy is highly effective against Lgr5+ stem cells in the gut, ovaries, and mammary gland where the epithelial layer is continuously replenished by the underlying SCs [94]. However, SCs residing at the hair follicles and muscle tissues rarely undergo proliferation and thus remain unaffected by T cell therapy [94]. CSCs in many tumors are also intrinsically immunodeficient [55,94] and CSCs in some other tumors are protected from immune surveillance by the TME factors such as tenascin-C [87] and TME cells such as mesenchymal stem cells [88,89] and cancer-associated fibroblasts or CAFs [98–100,136]. Similar mechanisms might be in operation to keep SCCs from immune attack [92,109,136,138]. For example, many surviving breast cancer patients, upon curative treatments, live with the so-called minimal residual disease (MRD) for decades. In such patients, disseminated breast cancer cells remain in the dormant state via downregulating the expression of cell surface glycoprotein ULBP1, which acts as a ligand and an activator of natural killer (NK) cells, thus allowing the SCCs to escape from innate immune system [109]. SCCs may also utilize T cell exhaustion as a potential anti-immune defense mechanism for survival [93]. For instance, in small cell lung carcinoma, the LRCs utilize the TGF-β pathway to express CD80, which is a cell surface ligand for the key immune checkpoint factor CTLA4. CD80 and CTLA4 interactions exhaust the cytotoxic activity of T cells allowing the survival of slow-cycling cancer cells [93].
5. Concluding remarks
It has been increasingly appreciated that human tumors are highly heterogeneous and plastic harboring many cancer cell subsets including SCCs, which undergo dynamic evolution driven by epigenetic mechanisms during tumor progression. SCCs may share many biological properties ascribed to CSCs (and EMT-like cells) but also have features distinct from CSCs (Table 1). Like CSCs, SCCs may evade immune surveillance and preferentially survive anti-cancer treatments, thus mediating therapy failure, tumor relapse and metastasis. Unlike CSCs, SCCs are defined by their fundamental trait, i.e., quiescence, and SCCs may not be hierarchical but instead represent a transient cell population stochastically entering and exiting the G0/early G1 phase [e.g., 80] (Table 1). Regardless, it has become clear that definitive cancer cure would entail the elimination of both CSCs and SCCs, the overlapping evil ‘twin sisters’ that underlie the MRD and metastatic recurrence. To eradicate CSCs, we’ll have to understand the core regulators of stemness and design strategies to specifically target these malignant, long-term tumor-maintaining and metastasis-propagating cells (Table 1). To make SCCs clinically irrelevant, we could perhaps consider, a priori, strategies to keep them in a permanently quiescent (i.e., irreversible G0) state or to selectively mobilize them out of the quiescent state followed by anti-mitotic bombardment. One way or another, more work is needed to uncover the core regulators of quiescence in slow-cycling cancer cells.
Acknowledgements
Work in the authors’ lab was supported, in part, by grants from the U.S National Institutes of Health (NIH) National Cancer Institute (NCI) R01CA237027, R01CA240290, R21CA237939, and R21CA218635, and Department of Defense (W81XWH-16–1-0575) (all to DGT), and by the Roswell Park Comprehensive Cancer Center (RPCCC) and NCI center grant P30CA016056. We also acknowledge the assistance of the M.D Anderson Science Park Flow Cytometry and Cell Imaging Core supported by CPRIT funding RP170628. We acknowledge the support of RPCCC Shared Resources (SR) including BSGSR, ETM, FICSR, GSR, LASR, and PNSR. We thank Mr. M. Macaluso for some early studies in bigenic H2B-GFP animals and other Tang lab members for contributions to the SCC project and for helpful discussions.
Funding Source
All sources of funding should also be acknowledged and you should declare any involvement of study sponsors in the study design; collection, analysis and interpretation of d ta; the writing of the manuscript; the decision to submit the manuscript for publication. If the study sponsors had no such involvement, this should be stated.
Footnotes
Conflict of interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Rycaj K and Tang DG, Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations, Cancer Res 75(19) (2015) 4003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McGranahan N and Swanton C, Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future, Cell. 168(4) (2017) 613–28. [DOI] [PubMed] [Google Scholar]
- [3].Walens A, Lin J, Damrauer JS, McKinney B, Lupo R, Newcomb R, Fox DB, Mabe NW, Gresham J, Sheng Z, Sibley AB, De Buysscher T, Kelkar H, Mieczkowski PA, Owzar K and Alvarez JV, Adaptation and selection shape clonal evolution of tumors during residual disease and recurrence, Nat Commun 11(1) (2020) 5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL and Meyer T, The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit, Cell. 155(2) (2013) 369–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P and Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation, eLife 3 (2014) e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Overton KW, Spencer SL, Noderer WL, Meyer T and Wang CL, Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states, Proc Natl Acad Sci USA. 111(41) (2014) E4386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miller I, Min M, Yang C, Tian C, Gookin S, Carter D and Spencer SL, Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence, Cell Rep 24(5) (2018) 1105–12 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Moser J, Miller I, Carter D and Spencer SL, Control of the restriction point by Rb and p21. Proc. Natl. Acad Sci. USA 115(35) (2018) E8219–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schwarz C, Johnson A, Kõivomägi M, Zatulovskiy E, Kravitz CJ, Doncic A, Skotheim JM, A precise Cdk activity threshold determines passage through the restriction point. Mol Cell 18;69(2) (2018) 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Min M and Spencer SL, Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways, PLoS Biol 17(3) (2019) e3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pennycook BR and Barr AR, restriction point regulation at the crossroads between quiescence and cell proliferation. FEBS Lett 2020. Jun 21. Doi: 10.1002 (Online ahead of print). [DOI] [PubMed] [Google Scholar]
- [12].Novak B and Tyson JJ, Mechanisms of signalling-memory governing progression through the eukaryotic cell cycle, Curr Opin Cell Biol 69 (2021) 7–16. [DOI] [PubMed] [Google Scholar]
- [13].Tang DG, Understanding cancer stem cell heterogeneity and plasticity, Cell research. 22(3) (2012) 457–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aguirre-Ghiso JA, Models, mechanisms and clinical evidence for cancer dormancy, Nat Rev Cancer. 7(11) (2007) 834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moore N, Houghton J and Lyle S, Slow-cycling therapy-resistant cancer cells, Stem Cells Dev. 21(10) (2012) 1822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sosa MS, Bragado P and Aguirre-Ghiso JA, Mechanisms of disseminated cancer cell dormancy: an awakening field, Nat Rev Cancer. 14(9) (2014) 611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ahn A, Chatterjee A and Eccles MR, The Slow Cycling Phenotype: A Growing Problem for Treatment Resistance in Melanoma, Mol Cancer Ther 16(6) (2017) 1002–09. [DOI] [PubMed] [Google Scholar]
- [18].Phan TG and Croucher PI, The dormant cancer cell life cycle, Nat Rev Cancer. 20(7) (2020) 398–411. [DOI] [PubMed] [Google Scholar]
- [19].Shen S, Vagner S and Robert C, Persistent Cancer Cells: The Deadly Survivors, Cell. 183(4) (2020) 860–74. [DOI] [PubMed] [Google Scholar]
- [20].Clermont Y and Leblond CP, Renewal of spermatogonia in the rat, Am J Anat 93(3) (1953) 475–501. [DOI] [PubMed] [Google Scholar]
- [21].Cotsarelis G, Sun TT and Lavker RM, Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis, Cell. 61(7) (1990) 1329–37. [DOI] [PubMed] [Google Scholar]
- [22].Potten CS, Kellett M, Roberts SA, Rew DA and Wilson GD, Measurement of in vivo proliferation in human colorectal mucosa using bromodeoxyuridine, Gut. 33(1) (1992) 71–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K and Tang DG, Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic, Cancer Res 65(14) (2005) 6207–19. [DOI] [PubMed] [Google Scholar]
- [24].Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L and Tang DG, Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells, Oncogene. 25(12) (2006) 1696–708. [DOI] [PubMed] [Google Scholar]
- [25].Patrawala L, Calhoun-Davis T, Schneider-Broussard R and Tang DG, Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells, Cancer Res 67(14) (2007) 6796–805. [DOI] [PubMed] [Google Scholar]
- [26].Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R and Jeter C, Prostate cancer stem/progenitor cells: identification, characterization, and implications, Mol Carcinog 46(1) (2007) 1–14. [DOI] [PubMed] [Google Scholar]
- [27].Li H, Chen X, Calhoun-Davis T, Claypool K and Tang DG, PC3 human prostate carcinoma cell holoclones contain self-renewing tumor-initiating cells, Cancer Res 68(6) (2008) 1820–5. [DOI] [PubMed] [Google Scholar]
- [28].Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, Calhoun-Davis T, Zaehres H, Daley GQ and Tang DG, Functional evidence that the self-renewal gene NANOG regulates human tumor development, Stem Cells. 27(5) (2009) 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ and Tang DG, NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation, Oncogene 30(36) (2011) 3833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D and Tang DG, The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44, Nat Med 17(2) (2011) 211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu C and Tang DG, MicroRNA regulation of cancer stem cells, Cancer Res 71(18) (2011) 5950–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu C, Kelnar K, Vlassov AV, Brown D, Wang J and Tang DG, Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7, Cancer Res 72(13) (2012) 3393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, Li W, Suraneni MV and Tang DG, The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration, Cell Stem Cell 10(5) (2012) 556–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen X, Rycaj K, Liu X, and Tang DG, New insights into prostate cancer stem cells. Cell Cycle 12(4) (2013) 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Badeaux MA, Jeter CR, Gong S, Liu B, Suraneni MV, Rundhaug J, Fischer SM, Yang T, Kusewitt D and Tang DG, In vivo functional studies of tumor-specific retrogene NanogP8 in transgenic animals, Cell Cycle. 12(15) (2013) 2395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jin M, Zhang T, Liu C, Badeaux MA, Liu B, Liu R, Jeter C, Chen X, Vlassov AV and Tang DG, miRNA-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiating cells, Cancer Res 74(15) (2014) 4183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang T, Rycaj K, Liu ZM and Tang DG, Cancer stem cells: constantly evolving and functionally heterogeneous therapeutic targets, Cancer Res. 74(11) (2014) 2922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rycaj K and Tang DG, Cancer stem cells and radioresistance, Int J Radiat Biol. 90(8) (2014) 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jeter CR, Yang T, Wang J, Chao HP and Tang DG, NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions, Stem Cells. 33(8) (2015) 2381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu X, Chen X, Rycaj K, Chao HP, Deng Q, Jeter C, Liu C, Honorio S, Li H, Davis T, Suraneni M, Laffin B, Qin J, Li Q, Yang T, Whitney P, Shen J, Huang J and Tang DG, Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells, Oncotarget 6(27) (2015) 23959–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li Q, Rycaj K, Chen X and Tang DG, Cancer stem cells and cell size: A causal link? Semin Cancer Biol 35 (2015) 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K, Zhang D, Liu B, Jeter C, Calhoun-Davis T, Lin K, Lu Y, Chao HP, Shen J and Tang DG, Defining a Population of Stem-like Human Prostate Cancer Cells That Can Generate and Propagate Castration-Resistant Prostate Cancer, Clin Cancer Res 22(17) (2016) 4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jeter CR, Liu B, Lu Y, Chao HP, Zhang D, Liu X, Chen X, Li Q, Rycaj K, Calhoun-Davis T, Yan L, Hu Q, Wang J, Shen J, Liu S and Tang DG, NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis, Cell Discov 2((2016) 16041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu R, Liu C, Zhang D, Liu B, Chen X, Rycaj K, Jeter C, Calhoun-Davis T, Li Y, Yang T, Wang J and Tang DG, miR-199a-3p targets stemness-related and mitogenic signaling pathways to suppress the expansion and tumorigenic capabilities of prostate cancer stem cells, Oncotarget 7(35) (2016) 56628–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rycaj K, Cho EJ, Liu X, Chao HP, Liu B, Li Q, Devkota AK, Zhang D, Chen X, Moore J, Dalby KN and Tang DG, Longitudinal tracking of subpopulation dynamics and molecular changes during LNCaP cell castration and identification of inhibitors that could target the PSA-/lo castration-resistant cells, Oncotarget 7(12) (2016) 14220–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, Chen X, Liu X, Chao HP, Whitney P, Calhoun-Davis T, Takata Y, Shen J, Iyer VR and Tang DG, Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer, Nat Commun 7((2016) 10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang D, Lin K, Lu Y, Rycaj K, Zhong Y, Chao HP, Calhoun-Davis T, Shen J and Tang DG, Developing a Novel Two-Dimensional Culture System to Enrich Human Prostate Luminal Progenitors that Can Function as a Cell of Origin for Prostate Cancer, Stem Cells Transl Med 6(3) (2017) 748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu C, Liu R, Zhang D, Deng Q, Liu B, Chao HP, Rycaj K, Takata Y, Lin K, Lu Y, Zhong Y, Krolewski J, Shen J and Tang DG, MicroRNA-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes, Nat Commun 8((2017) 14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rycaj K, Li H, Zhou J, Chen X and Tang DG, Cellular determinants and microenvironmental regulation of prostate cancer metastasis, Semin Cancer Biol 44((2017) 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rycaj K and Tang DG, Molecular determinants of prostate cancer metastasis, Oncotarget 8(50) (2017) 88211–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, Simic V, Zheng YL, Sripadhan P, Chen C, Lu J, Hou TW, Choudhury S, Kallakury B, Tang DG, Darling T, Thangapazham R, Timofeeva O, Dritschilo A, Randell SH, Albanese C, Agarwal S and Schlegel R, Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens, Nat Protoc 12(2) (2017) 439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu B, Gong S, Li Q, Chen X, Moore J, Suraneni MV, Badeaux MD, Jeter CR, Shen J, Mehmood R, Fan Q and Tang DG, Transgenic overexpression of NanogP8 in the mouse prostate is insufficient to initiate tumorigenesis but weakly promotes tumor development in the Hi-Myc mouse model, Oncotarget 8(32) (2017) 52746–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Guo Y, Zhang K, Cheng C, Ji Z, Wang X, Wang M, Chu M, Tang DG, Zhu HH and Gao WQ, Numb(-/low) Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling, Clin Cancer Res 23(21) (2017) 6744–56. [DOI] [PubMed] [Google Scholar]
- [54].Zhang D, Jeter C, Gong S, Tracz A, Lu Y, Shen J and Tang DG, Histone 2B-GFP Label-Retaining Prostate Luminal Cells Possess Progenitor Cell Properties and Are Intrinsically Resistant to Castration, Stem Cell Reports. 10(1) (2018) 228–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang D, Tang DG and Rycaj K, Cancer stem cells: Regulation programs, immunological properties and immunotherapy, Semin Cancer Biol 52(Pt 2) (2018) 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Skvortsov S, Skvortsova II, Tang DG and Dubrovska A, Prostate Cancer Stem Cells: Current Understanding, Stem Cells. 36(10) (2018) 1457–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, Liu B, Tang GW, Zhang D, Tracz A, Jeter C, Rycaj K, Calhoun-Davis T, Huang J, Rubin MA, Beltran H, Shen J, Chatta G, Puzanov I, Mohler JL, Wang J, Zhao R, Kirk J, Chen X and Tang DG, Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses, Nat Commun 9(1) (2018) 3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang D, Zhao S, Li X, Kirk JS and Tang DG, Prostate Luminal Progenitor Cells in Development and Cancer, Trends Cancer. 4(11) (2018) 769–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li Q, Liu B, Chao HP, Ji Y, Lu Y, Mehmood R, Jeter C, Chen T, Moore JR, Li W, Liu C, Rycaj K, Tracz A, Kirk J, Calhoun-Davis T, Xiong J, Deng Q, Huang J, Foster BA, Gokhale A, Chen X and Tang DG, LRIG1 is a pleiotropic androgen receptor-regulated feedback tumor suppressor in prostate cancer, Nat Commun 10(1) (2019) 5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu Y, Tracz A, Kirk J, Buonamici S, Zhu P, Wang J, Liu S and Tang DG, Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer, Nat Commun 11(1) (2020) 2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Teare GF, Horan PK, Slezak SE, Smith C and Hay JB, Long-term tracking of lymphocytes in vivo: the migration of PKH-labeled lymphocytes, Cell Immunol 134(1) (1991) 157–70. [DOI] [PubMed] [Google Scholar]
- [62].Wallace PK, Tario JD Jr., Fisher JL, Wallace SS, Ernstoff MS and Muirhead KA, Tracking antigen-driven responses by flow cytometry: monitoring proliferation by dye dilution, Cytometry A. 73(11) (2008) 1019–34. [DOI] [PubMed] [Google Scholar]
- [63].Tario JD Jr., Humphrey K, Bantly AD, Muirhead KA, Moore JS and Wallace PK, Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes, J Vis Exp 70) (2012) e4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Askenasy N and Farkas DL, Optical imaging of PKH-labeled hematopoietic cells in recipient bone marrow in vivo, Stem Cells. 20(6) (2002) 501–13. [DOI] [PubMed] [Google Scholar]
- [65].Ford JW, Welling TH 3rd, Stanley JC and Messina LM, PKH26 and 125I-PKH95: characterization and efficacy as labels for in vitro and in vivo endothelial cell localization and tracking, J Surg Res 62(1) (1996) 23–8. [DOI] [PubMed] [Google Scholar]
- [66].Pasto A, Marchesi M, Diamantini A, Frasson C, Curtarello M, Lago C, Pilotto G, Parenti AR, Esposito G, Agostini M, Nitti D and Amadori A, PKH26 staining defines distinct subsets of normal human colon epithelial cells at different maturation stages, PLoS One. 7(8) (2012) e43379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Puzar Dominkus P, Stenovec M, Sitar S, Lasic E, Zorec R, Plemenitas A, Zagar E, Kreft M and Lenassi M, PKH26 labeling of extracellular vesicles: Characterization and cellular internalization of contaminating PKH26 nanoparticles, Biochim Biophys Acta Biomembr 1860(6) (2018) 1350–61. [DOI] [PubMed] [Google Scholar]
- [68].Dehghani M, Gulvin SM, Flax J and Gaborski TR, Systematic Evaluation of PKH Labelling on Extracellular Vesicle Size by Nanoparticle Tracking Analysis, Sci Rep 10(1) (2020) 9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kanda T, Sullivan KF and Wahl GM, Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells, Curr Biol 8(7) (1998) 377–85. [DOI] [PubMed] [Google Scholar]
- [70].Puig I, Tenbaum SP, Chicote I, Arques O, Martinez-Quintanilla J, Cuesta-Borras E, Ramirez L, Gonzalo P, Soto A, Aguilar S, Eguizabal C, Caratu G, Prat A, Argiles G, Landolfi S, Casanovas O, Serra V, Villanueva A, Arroyo AG, Terracciano L, Nuciforo P, Seoane J, Recio JA, Vivancos A, Dienstmann R, Tabernero J and Palmer HG, TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence, J Clin Invest 128(9) (2018) 3887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H and Miyawaki A, Visualizing spatiotemporal dynamics of multicellular cell-cycle progression, Cell. 132(3) (2008) 487–98. [DOI] [PubMed] [Google Scholar]
- [72].Nakayama M, Kaida A, Deguchi S, Sakaguchi K and Miura M, Radiosensitivity of early and late M-phase HeLa cells isolated by a combination of fluorescent ubiquitination-based cell cycle indicator (Fucci) and mitotic shake-off, Radiat Res 176(3) (2011) 407–11. [DOI] [PubMed] [Google Scholar]
- [73].Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, Keighren MA, Hohenstein P, Miyawaki A and Jackson IJ, Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice, Cell Cycle. 13(17) (2014) 2681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bajar BT, Lam AJ, Badiee RK, Oh YH, Chu J, Zhou XX, Kim N, Kim BB, Chung M, Yablonovitch AL, Cruz BF, Kulalert K, Tao JJ, Meyer T, Su XD and Lin MZ, Fluorescent indicators for simultaneous reporting of all four cell cycle phases, Nat Methods. 13(12) (2016) 993–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Koh SB, Mascalchi P, Rodriguez E, Lin Y, Jodrell DI, Richards FM and Lyons SK, A quantitative FastFUCCI assay defines cell cycle dynamics at a single-cell level, J Cell Sci 130(2) (2017) 512–20. [DOI] [PubMed] [Google Scholar]
- [76].Sakaue-Sawano A, Yo M, Komatsu N, Hiratsuka T, Kogure T, Hoshida T, Goshima N, Matsuda M, Miyoshi H and Miyawaki A, Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle, Mol Cell. 68(3) (2017) 626–40 e5. [DOI] [PubMed] [Google Scholar]
- [77].Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH and Sharpless NE, Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model, Cell. 152(1–2) (2013) 340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sorrentino JA, Krishnamurthy J, Tilley S, Alb JG Jr., Burd CE and Sharpless NE, p16INK4a reporter mice reveal age-promoting effects of environmental toxicants, J Clin Invest 124(1) (2014) 169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV and Sharpless NE, Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence, Proc Natl Acad Sci U S A. 116(7) (2019) 2603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T and Herlyn M, A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth, Cell. 141(4) (2010) 583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T and Herlyn M, Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells, Cancer Cell. 23(6) (2013) 811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ and Clarke MF, Prospective identification of tumorigenic breast cancer cells, Proc Natl Acad Sci U S A. 100(7) (2003) 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C and De Maria R, Identification and expansion of human colon-cancer-initiating cells, Nature. 445(7123) (2007) 111–5. [DOI] [PubMed] [Google Scholar]
- [84].Hermann PC, Bhaskar S, Cioffi M and Heeschen C, Cancer stem cells in solid tumors, Semin Cancer Biol 20(2) (2010) 77–84. [DOI] [PubMed] [Google Scholar]
- [85].Korkaya H, Liu S and Wicha MS, Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots, Clin Cancer Res 17(19) (2011) 6125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Castano Z, Fillmore CM, Kim CF and McAllister SS, The bed and the bugs: interactions between the tumor microenvironment and cancer stem cells, Semin Cancer Biol 22(5–6) (2012) 462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, Piras IS, Restuccia U, Calcinotto A, Freschi M, Bachi A, Galli R and Bellone M, Tenascin-C Protects Cancer Stem-like Cells from Immune Surveillance by Arresting T-cell Activation, Cancer Res 75(10) (2015) 2095–108. [DOI] [PubMed] [Google Scholar]
- [88].McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR and Buckanovich RJ, Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production, J Clin Invest 121(8) (2011) 3206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S and Chang C, Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling, Oncogene. 33(21) (2014) 2768–78. [DOI] [PubMed] [Google Scholar]
- [90].Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D and Holland EC, Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells, Cell Stem Cell. 6(2) (2010) 141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A and Gilbertson RJ, A perivascular niche for brain tumor stem cells, Cancer Cell. 11(1) (2007) 69–82. [DOI] [PubMed] [Google Scholar]
- [92].Govaere O, Wouters J, Petz M, Vandewynckel YP, Van den Eynde K, Van den Broeck A, Verhulst S, Dollé L, Gremeaux L, Ceulemans A, Nevens F, van Grunsven LA, Topal B, Vankelecom H, Giannelli G, Van Vlierberghe H, Mikulits W, Komuta M and Roskams T, Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche, J Hepatol 64(3) (2016) 609–17. [DOI] [PubMed] [Google Scholar]
- [93].Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD and Fuchs E, Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells, Cell. 177(5) (2019) 1172–86 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A, Merad M and Brown BD, Quiescent Tissue Stem Cells Evade Immune Surveillance, Immunity. 48(2) (2018) 271–85 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yang ZF, Ngai P, Ho DW, Yu WC, Ng MN, Lau CK, Li ML, Tam KH, Lam CT, Poon RT and Fan ST, Identification of local and circulating cancer stem cells in human liver cancer, Hepatology. 47(3) (2008) 919–28. [DOI] [PubMed] [Google Scholar]
- [96].Tang B, Raviv A, Esposito D, Flanders KC, Daniel C, Nghiem BT, Garfield S, Lim L, Mannan P, Robles AI, Smith WI Jr., Zimmerberg J, Ravin R and Wakefield LM, A flexible reporter system for direct observation and isolation of cancer stem cells, Stem Cell Reports. 4(1) (2015) 155–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Srinivasan T, Walters J, Bu P, Than EB, Tung KL, Chen KY, Panarelli N, Milsom J, Augenlicht L, Lipkin SM and Shen X, NOTCH Signaling Regulates Asymmetric Cell Fate of Fast- and Slow-Cycling Colon Cancer-Initiating Cells, Cancer Res 76(11) (2016) 3411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jorgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Puré E, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT and Werb Z, A framework for advancing our understanding of cancer-associated fibroblasts, Nature Reviews Cancer. 20(3) (2020) 174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, Yu SL, Yuan SS, Chen YJ, Lin CY, Pan SH, Chou HY, Chen YJ, Chang GC, Chu WC, Lee YM, Lee JY, Lee PJ, Li KC, Chen HW and Yang PC, Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling, Nat Commun 5((2014) 3472. [DOI] [PubMed] [Google Scholar]
- [100].Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, Wulf-Goldenberg A and Birchmeier W, Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors, Cancer Res 77(8) (2017) 2134–47. [DOI] [PubMed] [Google Scholar]
- [101].Perego M, Maurer M, Wang JX, Shaffer S, Muller AC, Parapatics K, Li L, Hristova D, Shin S, Keeney F, Liu S, Xu X, Raj A, Jensen JK, Bennett KL, Wagner SN, Somasundaram R and Herlyn M, A slow-cycling subpopulation of melanoma cells with highly invasive properties, Oncogene. 37(3) (2018) 302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M and Settleman J, A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations, Cell. 141(1) (2010) 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, Lockerman E, Kalsy A, Lee D, Keating CL, Ruddy DA, Damon LJ, Crystal AS, Costa C, Piotrowska Z, Bardelli A, Iafrate AJ, Sadreyev RI, Stegmeier F, Getz G, Sequist LV, Faber AC and Engelman JA, Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition, Nat Med 22(3) (2016) 262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG and Parada LF, A restricted cell population propagates glioblastoma growth after chemotherapy, Nature. 488(7412) (2012) 522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Campos B, Gal Z, Baader A, Schneider T, Sliwinski C, Gassel K, Bageritz J, Grabe N, von Deimling A, Beckhove P, Mogler C, Goidts V, Unterberg A, Eckstein V and Herold-Mende C, Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma, J Pathol 234(1) (2014) 23–33. [DOI] [PubMed] [Google Scholar]
- [106].Hoang-Minh LB, Siebzehnrubl FA, Yang C, Suzuki-Hatano S, Dajac K, Loche T, Andrews N, Schmoll Massari M, Patel J, Amin K, Vuong A, Jimenez-Pascual A, Kubilis P, Garrett TJ, Moneypenny C, Pacak CA, Huang J, Sayour EJ, Mitchell DA, Sarkisian MR, Reynolds BA and Deleyrolle LP, Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma, EMBO J 37(23) (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sabelstrom H, Quigley DA, Fenster T, Foster DJ, Fuchshuber CAM, Saxena S, Yuan E, Li N, Paterno F, Phillips JJ, James CD, Norling B, Berger MS and Persson AI, High density is a property of slow-cycling and treatment-resistant human glioblastoma cells, Exp Cell Res 378(1) (2019) 76–86. [DOI] [PubMed] [Google Scholar]
- [108].Rusu P, Shao C, Neuerburg A, Acikgoz AA, Wu Y, Zou P, Phapale P, Shankar TS, Doring K, Dettling S, Korkel-Qu H, Bekki G, Costa B, Guo T, Friesen O, Schlotter M, Heikenwalder M, Tschaharganeh DF, Bukau B, Kramer G, Angel P, Herold-Mende C, Radlwimmer B and Liu HK, GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell. 25(2) (2019) 241–57 e8. [DOI] [PubMed] [Google Scholar]
- [109].Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E and Massague J, Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT, Cell. 165(1) (2016) 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Quayle LA, Ottewell PD and Holen I, Chemotherapy resistance and stemness in mitotically quiescent human breast cancer cells identified by fluorescent dye retention, Clin Exp Metastasis. 35(8) (2018) 831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hosseini H, Obradovic MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, Nanduri LK, Werno C, Ehrl C, Maneck M, Patwary N, Haunschild G, Guzvic M, Reimelt C, Grauvogl M, Eichner N, Weber F, Hartkopf AD, Taran FA, Brucker SY, Fehm T, Rack B, Buchholz S, Spang R, Meister G, Aguirre-Ghiso JA and Klein CA, Early dissemination seeds metastasis in breast cancer, Nature. 540(7634) (2016) 552–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, Avivar-Valderas A, Nagi C, Girnius N, Davis RJ, Farias EF, Condeelis J, Klein CA and Aguirre-Ghiso JA, Mechanism of early dissemination and metastasis in Her2(+) mammary cancer, Nature. 540(7634) (2016) 588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, Chai RC, De Veirman K, Owen KL, Opperman KS, Vandyke K, Clark JR, Seckinger A, Kovacic N, Nguyen A, Mohanty ST, Pettitt JA, Xiao Y, Corr AP, Seeliger C, Novotny M, Lasken RS, Nguyen TV, Oyajobi BO, Aftab D, Swarbrick A, Parker B, Hewett DR, Hose D, Vanderkerken K, Zannettino ACW, Amit I, Phan TG and Croucher PI, A niche-dependent myeloid transcriptome signature defines dormant myeloma cells, Blood. 134(1) (2019) 30–43. [DOI] [PubMed] [Google Scholar]
- [114].Lee S, Micalizzi D, Truesdell SS, Bukhari SIA, Boukhali M, Lombardi-Story J, Kato Y, Choo MK, Dey-Guha I, Ji F, Nicholson BT, Myers DT, Lee D, Mazzola MA, Raheja R, Langenbucher A, Haradhvala NJ, Lawrence MS, Gandhi R, Tiedje C, Diaz-Munoz MD, Sweetser DA, Sadreyev R, Sykes D, Haas W, Haber DA, Maheswaran S and Vasudevan S, A post-transcriptional program of chemoresistance by AU-rich elements and TTP in quiescent leukemic cells, Genome Biol 21(1) (2020) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wu FH, Mu L, Li XL, Hu YB, Liu H, Han LT and Gong JP, Characterization and functional analysis of a slow-cycling subpopulation in colorectal cancer enriched by cell cycle inducer combined chemotherapy, Oncotarget 8(45) (2017) 78466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Francescangeli F, Contavalli P, De Angelis ML, Careccia S, Signore M, Haas TL, Salaris F, Baiocchi M, Boe A, Giuliani A, Tcheremenskaia O, Pagliuca A, Guardiola O, Minchiotti G, Colace L, Ciardi A, D’Andrea V, La Torre F, Medema J, De Maria R and Zeuner A, A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer, J Exp Clin Cancer Res 39(1) (2020) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dembinski JL and Krauss S, Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma, Clin Exp Metastasis. 26(7) (2009) 611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Sachs N, Secades P, van Hulst L, Kreft M, Song JY and Sonnenberg A, Loss of integrin alpha3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells, Proc Natl Acad Sci U S A. 109(52) (2012) 21468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Sanchez-Danes A, Larsimont JC, Liagre M, Munoz-Couselo E, Lapouge G, Brisebarre A, Dubois C, Suppa M, Sukumaran V, Del Marmol V, Tabernero J and Blanpain C, A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy, Nature. 562(7727) (2018) 434–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ishimoto T, Oshima H, Oshima M, Kai K, Torii R, Masuko T, Baba H, Saya H and Nagano O, CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis, Cancer Sci 101(3) (2010) 673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ and Taichman RS, Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow, J Clin Invest 121(4) (2011) 1298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Chery L, Lam HM, Coleman I, Lakely B, Coleman R, Larson S, Aguirre-Ghiso JA, Xia J, Gulati R, Nelson PS, Montgomery B, Lange P, Snyder LA, Vessella RL and Morrissey C, Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways, Oncotarget 5(20) (2014) 9939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, Nobre AR, Aguirre-Ghiso JA, Jung Y and Taichman RS, Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow, Sci Rep 6((2016) 36520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, Yumoto K, Berry JE, Shiozawa Y and Pienta KJ, GAS6 receptor status is associated with dormancy and bone metastatic tumor formation, PLoS One. 8(4) (2013) e61873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, Krebsbach PH, Pienta KJ, Shiozawa Y and Taichman RS, TBK1 regulates prostate cancer dormancy through mTOR inhibition, Neoplasia. 15(9) (2013) 1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jung Y, Decker AM, Wang J, Lee E, Kana LA, Yumoto K, Cackowski FC, Rhee J, Carmeliet P, Buttitta L, Morgan TM and Taichman RS, Endogenous GAS6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow, Oncotarget 7(18) (2016) 25698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Cackowski FC, Eber MR, Rhee J, Decker AM, Yumoto K, Berry JE, Lee E, Shiozawa Y, Jung Y, Aguirre-Ghiso JA and Taichman RS, Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy, J Cell Biochem 118(4) (2017) 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Decker AM, Jung Y, Cackowski FC, Yumoto K, Wang J and Taichman RS, Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow, Mol Cancer Res 15(12) (2017) 1644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019. Dec;19(12):716–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Navas T, Kinders RJ, Lawrence SM, Ferry-Galow KV, Borgel S, Hollingshead MG, Srivastava AK, Alcoser SY, Makhlouf HR, Chuaqui R, Wilsker DF, Konaté MM, Miller SB, Voth AR, Chen L, Vilimas T, Subramanian J, Rubinstein L, Kummar S, Chen AP, Bottaro DP, Doroshow JH, Parchment RE. Clinical Evolution of Epithelial-Mesenchymal Transition in Human Carcinomas. Cancer Res 2020. Jan 15;80(2):304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori G, Dedhar S, Derynck R, Ford HL, Fuxe J, García de Herreros A, Goodall GJ, Hadjantonakis AK, Huang RJY, Kalcheim C, Kalluri R, Kang Y, Khew-Goodall Y, Levine H, Liu J, Longmore GD, Mani SA, Massagué J, Mayor R, McClay D, Mostov KE, Newgreen DF, Nieto MA, Puisieux A, Runyan R, Savagner P, Stanger B, Stemmler MP, Takahashi Y, Takeichi M, Theveneau E, Thiery JP, Thompson EW, Weinberg RA, Williams ED, Xing J, Zhou BP, Sheng G; EMT International Association (TEMTIA). Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020. Jun;21(6):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, Ginestier C, Charafe-Jauffret E, Clouthier SG, Birnbaum D, Wong ST, Zhan M, Chang JC, Wicha MS. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2013. Dec 27;2(1):78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Brooks MD, Burness ML, Wicha MS. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell. 2015. Sep 3;17(3):260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, Morrissey C, Chung CY, Farias EF, Bernstein E and Aguirre-Ghiso JA, NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes, Nat Commun 6(2015) 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Nakamura T, Shinriki S, Jono H, Guo J, Ueda M, Hayashi M, Yamashita S, Zijlstra A, Nakayama H, Hiraki A, Shinohara M and Ando Y, Intrinsic TGF-beta2-triggered SDF-1-CXCR4 signaling axis is crucial for drug resistance and a slow-cycling state in bone marrow-disseminated tumor cells, Oncotarget 6(2) (2015) 1008–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Cho J, Lee HJ, Hwang SJ, Min HY, Kang HN, Park AY, Hyun SY, Sim JY, Lee HJ, Jang HJ, Suh YA, Hong S, Shin YK, Kim HR and Lee HY, The Interplay between Slow-Cycling, Chemoresistant Cancer Cells and Fibroblasts Creates a Proinflammatory Niche for Tumor Progression, Cancer Res 80(11) (2020) 2257–72. [DOI] [PubMed] [Google Scholar]
- [137].Roato I and Ferracini R, Cancer Stem Cells, Bone and Tumor Microenvironment: Key Players in Bone Metastases, Cancers (Basel). 10(2) (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Oskarsson T, Batlle E and Massague J, Metastatic stem cells: sources, niches, and vital pathways, Cell Stem Cell. 14(3) (2014) 306–21. [DOI] [PMC free article] [PubMed] [Google Scholar]