

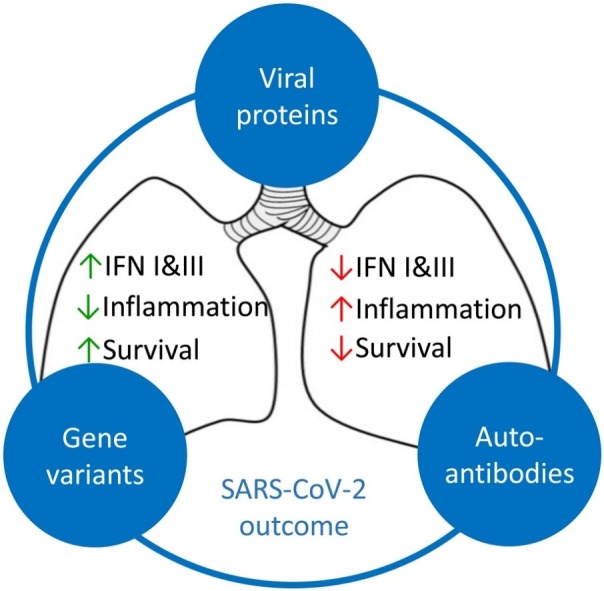
Graphical abstract
Abbreviations: ACE-2, angiotensin-converting enzyme 2; ARDS, acute respiratory distress syndrome; CoV, coronavirus; COVID-19, coronavirus disease 2019; CRS, cytokine release syndrome; IFN, interferon; IL, interleukin; IRF, interferon regulatory factor; ISG, interferon-stimulated gene; JAK, janus kinase; KPNA2, karyopherin α2; LY6E, lymphocyte antigen 6 complex, locus E; MAVS, mitochondrial antiviral-signaling protein; MDA5, melanoma-differentiated gene 5; MERS, Middle East respiratory syndrome; NF-κB, nuclear factorκB; N/L, neutrophil to lymphocyte; NSP, non-structural protein; OAS, oligoadenylate synthetase; ORF, open reading frame; pDC, plasmacytoid dendritic cell; PRR, pattern-recognition receptor; RIG, retinoic acid-inducible gene 1; RLR, RIG-I like receptor; SARS, severe acute respiratory syndrome; S, spike; STAT, signal transducer and activator of transcription; TBK1, TANK binding kinase 1; TLR, toll-like receptor; TMPRSS2, transmembrane serine protease 2; TNF-α, tumor necrosis factor alpha
Keywords: Interferons, Antiviral innate immunity, SARS-CoV-2, COVID-19
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a recently emerged pathogen that has caused coronavirus disease 2019 (COVID-19), the worst pandemic of our times leading to tremendous loss of human life and unprecedented measures of social distancing. COVID-19 symptom manifestations range from asymptomatic disease to severe and lethal outcomes. Lack of previous exposure and immunity to SARS-CoV-2, and high infectivity of the virus have contributed to its broad spread across the globe. In the absence of specific adaptive immunity, innate immune mechanisms are crucial for efficient antiviral defenses and control of the infection. Accumulating evidence now suggests that the remarkable heterogeneity in COVID-19 disease manifestations is due to variable degrees of impairment of innate immune mechanisms. In this review, we summarize recent findings describing both viral and host intrinsic factors that have been linked to defective innate immune responses and account for severe COVID-19. We also discuss emerging therapeutic opportunities for targeting innate immunity for the treatment of COVID-19.
1. Introduction
Coronavirus disease 2019 (COVID-19) is the worst pandemic of our times that has resulted in tremendous loss of human life and has triggered unprecedented restrictions of free movement and social distancing. It is caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1], for which humanity has no prior immunity. Although limited cross-reactivity due to the seasonal circulating coronaviruses has been reported [2], effective B and T cell responses against SARS-Cov-2 can be mounted only following vaccination or active infection. Therefore, the course of COVID-19 disease, following an individual’s first encounter with SARS-Cov-2, largely depends on the host’s innate immune response and its ability to restrict infection early on, preventing the development of exuberant inflammation. Remarkably, there is huge heterogeneity in the symptomatology of COVID-19 patients, ranging from asymptomatic or ambulatory disease to very severe or even lethal outcomes. This raised from the beginning of the pandemic the idea that defects in an individual’s innate immune response are the main culprits for the development of severe or lethal disease, and more broadly for the vast clinical variability observed. Indeed, several reports in the literature have documented impairments in the innate antiviral immune mechanisms that have been linked to severe COVID-19. We herein review all evidence linking impaired innate antiviral defenses to disease severity in COVID-19, and discuss its implications for disease clinical phenotypes and novel approaches to therapy.
2. Innate antiviral mechanisms against SARS-CoV-2
Sensing of viruses by innate immune mechanisms serves as the first line of antiviral defense of an organism and is absolutely crucial for an effective antiviral response [3]. SARS-CoV-2, the etiological factor of COVID-19, is a betacoronavirus, and like other related CoVs, is an enveloped positive-sense single-stranded RNA (+ssRNA) virus, that can infect mammals and birds, causing mostly respiratory tract infections, or gastroenteritis in certain cases [4]. Its tropism is primarily for the ciliated cells of the nasal and bronchial epithelium and the type 2 alveolar pneumocytes [5]. Soon after its characterization, angiotensin-converting enzyme 2 (ACE-2) was described to be SARS-CoV-2 receptor for viral entry into the cells, similar to SARS-CoV [6]. Through its spike (S) protein, SARS-CoV-2 binds to ACE-2 receptor of the host, to enter the cells with the help of the host cell transmembrane serine protease 2 (TMPRSS2), that promotes viral uptake and fusion at the cellular or endosomal membrane, followed by the viral RNA release in the cytoplasm [7].
In addition to ACE-2, several other host receptors have been proposed to alternatively mediate SARS-CoV-2 entry into the cells. Neuropilin-1 was identified by two groups to facilitate SARS-CoV-2 entry, especially in cells with low ACE-2 expression, such as the olfactory epithelium [8,9]. As SARS-CoV-2 spike protein contains a cleavage site for the protease furin that is absent from SARS-CoV, neuropilin-1 was found to bind the furin-cleaved S1 fragment in epithelial cell lines in vitro or in the mouse olfactory epithelium in vivo [8,9]. Similarly, the lectins CD209 L/L-SIGN, CD209/DC-SIGN and CD169/SIGLEC1, expressed in trans [[10], [11], [12]], and the phosphatidylserine receptors KIM-1 [13] and AXL [14], were reported to either act as alternative receptors or to facilitate SARS-CoV-2 entry into the cells in the presence of ACE-2, acting more as attachment receptors to enhance the interaction with ACE2, rather than being alternative primary receptors for viral entry [10]. Nevertheless, some of these receptors were described to be responsible for SARS-CoV-2 entry in other target organs, beyond the lung epithelium, such as the vascular endothelial cells [11], and the renal epithelial cells [13]. Heparan sulfate, a highly negatively charged linear polysaccharide, which is part of the cell’s glycocalyx, was found to interact with SARS-CoV-2 spike protein through the receptor-binding domain (RBD) on a docking site adjacent to the ACE-2-binding domain, suggesting the formation of a ternary complex between heparane sulfate, ACE-2, and S [15]. Finally, although CD147/basigin was proposed to be an alternative receptor for SARS-CoV-2 entry [16], leading to a clinical trial with an anti-CD147 humanized antibody, Meplazumab [17], the finding was later disputed by another group [18].
Within the cell, sensing of RNA viruses, such as SARS-CoV-2, is mediated through the engagement of viral structures by pattern-recognition receptors (PRRs) that include Toll-like receptors (TLRs), and retinoic acid-inducible gene 1 (RIG)-I like receptors (RLRs). Although the exact sensors that recognize SARS-CoV-2 have not been described, most likely the endosomal TLR3 and TLR7/8, as well as the cytocolic RIG-I and melanoma-differentiated gene 5 (MDA5) receptors, all recognizing different structures of foreign RNA [19], possibly play a major role. In fact, inhibitors for TLR3 and TLR7 reduced the production of IFNs and pro-inflammatory cytokines in in vitro cultures of human lung cell lines infected with SARS-CoV-2, suggesting a role of these receptors in viral recognition [20]. Further, plasmacytoid dendritic cells (pDCs) isolated from individuals with TLR3 and TLR7 genetic variants predicted to be loss-of-function, exhibited impaired type I IFN production in response to SARS-CoV-2 in vitro [21,22]. With regards to the cytocolic sensors, MDA5, in particular, together with LGP2, but not RIG-I, were shown to play a role in IFN induction in response to SARS-CoV-2 infection in lung epithelial Calu-3 cells, through the activation of IRF3, IRF5 and RelA (p65) [23]. To the contrary, another study suggested that SARS-CoV-2 RNA activates RIG-I-mitochondrial antiviral-signaling protein (MAVS) pathway, and not MDA5, to induce type I IFN production in HEK293 T [24]. The different cellular systems used by the researches might account for this discrepancy.
Engagement of PRRs initiates downstream signaling cascades that lead to the activation of transcription factors, most notably interferon regulatory factors (IRFs) and nuclear factor κB (NF-κB). These trigger the secretion of cytokines, such as the proinflammatory cytokines tumor necrosis factor alpha (TNF-α), and interleukin-1 (IL-1), IL-6 and IL-18, as well as a variety of chemokines that orchestrate the recruitment of leucocyte subsets. Interferons (IFNs) of the type I and III families, however, are considered to be the most important cytokines produced for the antiviral defense.
2.1. Interferons
Two major cytokine families, type I and III IFNs, constitute the host’s initial natural defense to viral infections. These cytokines are produced in response to viral infection, and induce antiviral programs both in an autocrine and paracrine manner, while at the same time they potentiate the adaptive immune response. Type I IFNs constitute a family of cytokines originally described over 60 years ago and comprise 13 IFN-α subtypes, IFN-β and IFN-ω, -κ, and –ε. They signal through the IFNαR1/IFNαR2 heterodimeric complex to up-regulate hundreds of genes collectively known as interferon-stimulated genes (ISGs). These exhibit multiple antiviral functions including inhibition of viral replication, degradation of viral nucleic acids and induction of viral resistance to neighboring cells, and account for the potent antiviral activity of type I IFNs [25].
Type III IFNs, also termed lambda IFNs (IFN-λ) or interleukins-28 and -29 (IL-28/29), share homology, expression patterns and antiviral functions with type I IFNs [26]. They consist of four members in humans (IFN-λ1/IL-29, IFN-λ2/IL-28A, IFN-λ3/IL-28B and IFN-λ4) and two in mice (IFN-λ2/IL-28A, IFN-λ3/IL-28B) [[27], [28], [29]] and are abundantly expressed at mucosal interfaces such as the respiratory and gastrointestinal tracts following microbial infection [26,30]. Interestingly, although IFN-λ signal through a unique heterodimeric receptor complex consisting of IFN-λRα (IL28Rα), which confers ligand specificity, and the common IL-10Rβ chain, they induce downstream signaling that appears remarkably similar to that of type I IFNs, and involves the phosphorylation of Janus kinase (JAK)-family kinases, and the activation of signal transducer and activator of transcription (STAT) and IRF transcription factors, driving the expression of ISGs and the induction of antiviral responses [26,31]. Despite this similarity, one major difference of these two IFN families is their receptor availability. While the IFNAR is expressed ubiquitously in all cell types, expression of the IFNLR is largely limited to epithelial cells and few immune cell subsets, suggesting an important role at mucosal surfaces. Indeed, we have described a unique non-redundant role of type III IFNs in the respiratory track, and a subtle mechanism by which type I and type III IFN cooperate to fine tune the antiviral response towards optimal protection and minimal tissue damage [32]. Following respiratory viral infection, IFN-λ are induced first, before and in excess levels to these of type I IFNs, and act locally at the epithelial barrier to limit initial viral spread without activating inflammation. In contrast, type I IFNs come into play later on, once infection escapes IFN-λ control, to provide further antiviral resistance but at the same time induce pro-inflammatory responses essential for dealing with infection but also mediating tissue damage [32]. This indicates that IFN-λ are at the front line of antiviral defense in the respiratory tract, preceding that of type I IFNs and providing early infection control.
2.2. Antiviral functions of interferons against SARS-CoV-2
Early evidence already demonstrated that SARS-CoV-2 is sensitive to type I [[33], [34], [35]] and III [36] IFN treatment in vitro, and perhaps to a greater degree than SARS-CoV [34]. Although the exact ISG products that inhibit CoVs are not yet well characterized in detail, Pfaender et al. showed that the ISG lymphocyte antigen 6 complex, locus E (LY6E) potently restricted infection by multiple CoVs, including SARS-CoV, SARS-CoV-2 and Middle East respiratory syndrome (MERS)-CoV, by interfering with S protein-mediated membrane fusion and thus inhibiting CoV entry into the cells [37].
Given the fact that ACE-2 is the receptor for SARS-CoV-2 entry into the cells [6], it was surprising that ACE-2 was shown to be an ISG in vitro in human primary upper airway basal epithelial cells [38]. This would suggest that the antiviral response, via IFN production, would favor viral entry. However, a truncated isoform of ACE2, which was named as deltaACE2 (dACE2), was later identified and shown to be the one induced by type I IFNs [39]. This dACE2 isoform was proven to be non-functional in binding the SARS-CoV-2 spike protein, rendering it unlikely to contribute to viral entry [39]. The importance of these findings in the course of COVID-19 infection remains to be elucidated.
3. Impaired innate antiviral mechanisms in COVID-19
Clinical manifestation of the course of SARS-CoV-2 infection is highly variable, ranging from asymptomatic disease to lethal outcomes. This variability intrigued scientists who are since seeking to address the factors that may underlie the individual’s response to SARS-CoV-2 infection. The primary risk factor for worse outcome is age with a log-linear increase of death by age [40]. Further, many other factors or co-morbidities have been described, such as male gender, obesity, diabetes, cancer and others [41,42]. Nevertheless, these risk factors alone could not provide sufficient explanation for the variability of phenotypes within age groups or specific categories. This led to the hypothesis that impairments in the host defense could account for this variability. Impaired immune responses to SARS-CoV-2 may be due to a number of factors, but direct inhibition by viral products and inborn genetic errors are the most likely (Fig. 1 ).
Fig. 1.

Mechanisms of impairment of the IFN response. SARS-CoV-2, after entering the cells, is recognized by PRRs, like TLR3, TLR7, RIG-I and MDA5, to initiate signaling pathways that induce that production of type I and III IFNs. IFNs bind to their receptors in the same or nearby cells to activate pathways that lead to the induction of ISGs and the initiation of the antiviral response. SARS-CoV-2 viral products have been shown to inhibit various steps of these processes, depicted in red, suppressing thus the IFN response. Moreover, genetic mutations in key molecules of these pathways, depicted in blue, have been linked to impaired immune responses in severe COVID-19 patients. Finally, auto-antibodies against type I IFN family members, depicted in green, constitute another mechanism of impaired IFN response detected in severe cases of COVID-19. Numbers in brackets [#] correspond to literature references of this review.
3.1. Suppression of the interferon response by SARS-CoV-2
Given the fact that IFNs are a major obstacle to viral infection, several viruses have evolved strategies to evade them, including CoVs [43]. Previous studies have demonstrated the suppressive ability of SARS-CoV in IFN production both in vitro and in vivo [[44], [45], [46]]. These evasion mechanisms employed by CoVs antagonize steps from PRR sensing to IFN signal transduction. In the case of SARS-CoV-2, four different viral non-structural proteins (NSPs) were identified to inhibit protein translation after infection. More specifically, NSP16 was shown to disrupt mRNA splicing, NSP1 to bind to the entry channel of the 18S ribosomal RNA, while NSP8 and NSP9 to bind to signal recognition particle and disrupt protein trafficking to the cell membrane. All these disruptions in protein production were eventually shown to suppress the IFN response during infection [47,48]. In support of that, an in-depth protein interaction analysis of SARS-CoV-2 proteins with human proteins, identified a multitude of protein–protein interactions that may affect several biological processes, including mRNA translation [5]. A general shutdown of translation, including synthesis of antiviral proteins, was also reported to be induced by NSP14 [49], or a preferential blocking of the host proteins alone [50].
Xia et al. were able to show a direct inhibition of the IFN pathway by SARS-CoV-2 viral proteins. More specifically, NSP6 was found to bind TANK binding kinase 1 (TBK1) to suppress IRF3 phosphorylation, NSP13 could bind and block TBK1 phosphorylation, and open reading frame 6 (ORF6) could bind importin Karyopherin α2 (KPNA2) to inhibit IRF3 nuclear translocation [51,52]. M protein was also shown to interact with RIG-I, MAVS, and TBK1, and to prevent the formation of the RIG-I, MAVS, TRAF3 and TBK1 multiprotein complex, and subsequently impeding the phosphorylation, nuclear translocation, and activation of IRF3, thus inhibiting type I and III IFN production [53]. In a similar mechanism, ORF6 was found to block STAT1 and STAT2 nuclear translocation and inhibit the IFN response [52,54]. Additionally, two sets of viral proteins were found to antagonize type I IFN signaling through blocking of STAT1/STAT2 activation; NSP1, NSP6, NSP13, ORF3a, ORF7b, and M inhibited STAT1 phosphorylation, while NSP6, NSP13, ORF7a, and ORF7b inhibited STAT2 phosphorylation [52]. Finally, nucleocapsid (N) protein was also reported to antagonize IFN signaling, by inhibiting the phosphorylation of STAT1 and STAT2 [55].
With regard to the NF-κB pathway, ORF9b, which is an alternative ORF within the N gene, was shown to target NEMO and interrupt its K63-linked polyubiquitination upon viral stimulation, thereby inhibiting the canonical IκB kinase alpha (IKKα)/β/γ-NF-κB signaling and subsequent IFN production [24]. ORF9b was additionally shown to inhibit type I IFN production by interacting with TOM70 localized on mitochondria [[56], [57], [58]]. Finally, ORF9b was shown to overall impair antiviral immunity, by targeting multiple molecules of the innate antiviral response, including RIG‐I, MDA‐5, MAVS, TRIF, STING and TBK1, as well as impeding the phosphorylation and nuclear translocation of IRF3, eventually inhibiting type I and III IFN production [59].
Further, SARS-CoV-2 papain-like protease was shown to antagonize ISG15-dependent activation by MDA5 [60,61]. Another interesting viral product is ORF3b, which has the ability to inhibit type I IFN production originating from a variety of CoVs, including SARS-CoV-2 [62]. Of note, a longer ORF3b variant with increased ability to suppress IFN induction was isolated from two patients with severe disease [62]. Along the same line, a SARS-CoV-2 variant containing a deletion in the NSP1-coding region (Δ500-532) correlated with higher RT-PCR cycle thresholds and lower serum IFN-β in the samples detected [63]. Although the physiological relevance of these observations in patients remains to be established, it still demonstrates the propensity of SARS-CoV-2 to inhibit IFN production and IFN-mediated antiviral responses.
3.2. Host impairments in interferon response
Despite its suppressive mechanisms in the IFN response, SARS-CoV-2 is still able to induce production of IFNs in vitro in human airway epithelial cells [33,64]. When compared to SARS-CoV, SARS-CoV-2 exhibited higher proliferative ability in ex vivo human lung tissue explants, but induced less IFNs and pro-inflammatory cytokines and chemokines [65].
In COVID-19 patients, a longitudinal analysis of the immune profiling detected increased levels of IFN-α and IFN-λ during the first week of symptoms that were sustained also later on in patients with severe disease, while they declined in patients with moderate disease [66]. We and others, however, have reported impaired and delayed production of type I and III IFNs in the serum or plasma of severe COVID-19 patients [[67], [68], [69]]. We further found that, in those COVID-19 patients where IFN-λ was detected, higher IFN-λ concentrations correlated with lower viral load in bronchial aspirates and faster viral clearance, while a higher IFN-λ to type I IFN ratio correlated with improved outcome in critically ill patients [68]. Our findings are in agreement with the important antiviral role IFN-λ play at mucosal surfaces, without inducing pro-inflammatory responses, as opposed to type I IFNs [32,70]. In some reports, impaired IFN production was accompanied by a defective induction of ISGs in severe versus mild or moderate cases [33], or by an early transient expression [67]. Also, whole-blood single-cell analysis showed that induction of ISGs was only present in mild and moderate cases, while it was completely absent from severe ones [71]. In our analysis, however, in peripheral blood ISGs were also strongly induced in severe cases of COVID-19 independently of IFN production [68].
According to other researchers both the type and the localization of IFNs may play a role in the severity of the disease. Sposito et al. found that IFNs, and especially type III IFNs, are over-represented in the lower airways of patients with severe COVID-19, while high levels of type I and III IFNs where detected in the upper airways of patients with high viral burden but reduced disease risk or severity [72]. This could be explained by the fact that although type III IFNs are generally characterized as less inflammatory when compared to type I IFNs [32,70], their presence in the lung during the repair phase of the respiratory tract has been linked to an impairment in epithelial proliferation [73,74].
In the light of impairments in the host immune response, large-scale GWAS studies have been performed, in order to identify genetic contributors to the SARS-CoV-2 response. Analysis of a large number of COVID-19 patients with severe disease, identified a 3p21.31 gene cluster as a genetic susceptibility locus in COVID-19 patients with respiratory failure [75]. Interestingly, the majority of genes in this cluster included immune genes, namely CCR9, CXCR6, XCR1, and FYCO1, that are involved in T-cell and dendritic-cell function, pinpointing the prominent role the host’s adaptive immune response play in the severity of COVID-19. Another study, the GenOMICC study, identified several interesting genetic loci linked to severe COVID-19, including 12q24.13 (rs10735079) that harbors the genes encoding for antiviral 2’,5’-oligoadenylate synthetase (OAS) enzymes, OAS1, OAS2, and OAS3, which are ISGs activating the latent form of ribonuclease L (RNaseL), 21q22.1 (rs2236757) within the interferon receptor gene IFNAR2, and 19p13.2 (rs74956615) near the gene that encodes tyrosine kinase 2 (TYK2), which is required for IFN signaling [76]. These findings support the hypothesis that dysfunctions in the IFN pathway are crucial for the severe course of COVID-19. Indeed, this is further supported by a recent study that identified several gene variants predicted to be loss-of-function variants, which are enriched in severe COVID-19 patients versus asymptomatic or mild disease patients. These included important contributors of the IFN pathway, namely TLR3, UNC93B1, TICAM1, TBK1, IRF3, IRF7, IFNAR1 and IFNAR2 [22]. In in vitro studies, pDCs from patients with a dysfunctional TLR7 did not produce type I IFNs [21], while fibroblasts from patients with deficient TLR3, IRF7 or IFNAR1 were susceptible to SARS-CoV-2 infection [22]. Especially for TLR7, mutations were reported to be associated with severe COVID-19 in male patients aged under 60 years old, while these genetic variants were not detected in asymptomatically or mildly infected male individuals of any age [21]. This was linked to impaired type I IFN production by blood pDCs in response to SARS-CoV-2 in vitro [21]. Overall these findings suggest that genetics in crucial pathways of the innate immune response may determine the clinical course of COVID-19 infection.
Further to genetic mutations, of note, Bastard et al. identified that at least 10 % of patients with severe COVID-19 pneumonia had high titers of pre-existing neutralizing autoantibodies against type I IFN-α2 and IFN-ω in plasma. These autoantibodies were not detected in asymptomatic or mild patients nor in healthy individuals, and could neutralize 10 ng/mL of type I IFNs [77]. Moreover, in a subsequent study the same group showed that at least 15 % of critical cases globally, 20 % of cases in patients > 80 years old, and 20 % of fatal COVID-19 across ages had levels of autoantibodies that could neutralize lower more physiologically relevant type I IFN concentrations [78]. This proportion is unprecedented among infectious diseases and indicates the magnitude of the problem.
Interestingly, the use of a mouse model for SARS-CoV-2 infection, showed that type I IFNs minimally contributed in the control of SARS-CoV-2 replication in vivo, while they were significant drivers of pathological responses [79]. This is in accordance to a previously described mouse model for SARS-CoV where delayed type I IFN signaling was observed after viral infection, and this correlated with an increased monocytic inflammatory response that caused lung immunopathology and reduced survival [80].
3.3. Altered cytokine patterns in COVID-19
Severe cases of COVID-19 appear to be linked to an exacerbated inflammatory response. This is actually directly linked to a defective type I IFN immunity, as this may allow the uncontrolled replication of the virus and its spread to the lungs, leading to an excessive leukocyte recruitment, and subsequent overt inflammation [81]. Indeed, a variety of cytokines of innate origin have been reported to have altered profiles of production in COVID-19. Pro-inflammatory cytokines, such as IL-1b, IL-6 and TNF-α are detected in increased and sustained levels in the serum or plasma of COVID-19 patients in several studies [66,68,82]. Although these cytokines are important for activating different branches of the immune response that are essential for confronting infection, their increased presence in the lung leads to a ‘cytokine storm’ or ‘cytokine release syndrome (CRS)’, which is linked to the development of acute respiratory distress syndrome (ARDS), a type of respiratory failure, which consists the main cause of mortality of COVID-19 patients with severe illness [83].
These altered cytokine patterns are directly linked to the imbalanced immune response driven by SARS-CoV-2, characterized by heightened neutrophil and low lymphocyte blood counts, leading to an increased neutrophil to lymphocyte (N/L) ratio. Indeed, a high N/L ratio can serve as a clinical marker for predicting complications related to ARDS in COVID-19 [84]. These circulating neutrophils include precursors, as well as dysfunctional mature neutrophils, as a hallmark of emergency myelopoiesis [85,86]. In addition to neutrophils, COVID-19 patient peripheral blood was characterized by disappearance of the non-classical CD14lowCD16high monocytic subset, and the accumulation of HLA-DRlow classical monocytes and HLA-DRhighCD11chigh inflammatory monocytes [85,86].
Findings observed systemically in the blood correlate well with the milieu in the lung of COVID-19 patients which is characterized by high levels of inflammatory cytokines and chemokines, and increased neutrophil and monocyte recruitment, as documented by single-cell analysis of the bronchoalveolar fluid of patients with COVID-19 [87].
3.4. Other innate soluble mediators
In addition to cytokines, other innate mediators have been described to be dysregulated in COVID-19. The state of high inflammation observed in severe cases of COVID-19 is accompanied by an excessive elevation of acute phase reactants such as C-reactive protein (CRP) and ferritin [88,89]. Proteome and metabolome analysis of sera of COVID-19 patients identified 93 differentially expressed proteins in severe COVID-19 patients, 50 of which belonged to three major pathways, namely activation of the complement system, macrophage function, and platelet degranulation [90]. Increased complement activation, in particular, has been detected in nasopharyngeal swabs of COVID-19 patients in a proportional manner to viral load [91], while in another study it was characterized as a unique immunological feature of COVID-19 patients with severe illness, as it was not observed in patients suffering from other respiratory diseases, such as influenza infection or non-COVID-19 respiratory failure [92]. These findings place the complement as a potential therapeutic target for COVID-19 [93].
Further, 204 metabolites correlated with COVID-19 disease severity that included increases in steroid hormones, or metabolites associated with the kynurenine pathway [90]. Finally, in a lipidome analysis, specific lipid mediators with immunomodulatory function were identified to be expressed in moderate and severe cases of COVID-19. In particular, moderate disease associated with lipid mediators that require cyclooxygenase activity as well as certain eicosapentaenoic acid products of ALOX12 such as resolvin E3, while severe disease was linked to lipid mediators that require activity of ALOX5 and cytochrome p450 enzymes [94]. As lipid mediators are dependent on the timing of the sampling and analysis, and might be affected by factors such as age, BMI or medical conditions that consist risk factors for COVID-19 disease outcome, it is important to interpret these data with caution.
4. Therapeutic restoration of impaired innate responses
Understanding the pathophysiological mechanisms underlying the immune response to SARS-CoV-2, is crucial for the identification of prognostic markers and the discovery of therapeutic schemes. As several research findings link severity of COVID-19 to impaired innate immune responses, restoration of these pathways may prove an effective therapeutic approach for these patients. Targeting specific pathways may be a more favorable approach than the use of general anti-inflammatory drugs that have been introduced in current clinical practice, such as dexamethasone that non-selectively inhibits the immune response [95].
As IFNs constitute major antiviral proteins, and their impaired production has been linked with severe cases of COVID-19, as discussed in this review, restoring its levels might be an efficient therapeutic approach. Various IFN family members have been tested in clinical protocols. In a retrospective multicenter cohort study of 446 COVID-19 hospitalized patients, early administration of IFN-α2b was associated with reduced in-hospital mortality in comparison with no administration of IFN-α2b, whereas late administration of IFN-α2b was associated with increased mortality, pinpointing also the importance of the timing of administration and the different roles IFN may have during the progress of COVID-19 [96]. In a different study, inhaled IFN-α2b treatment administered in moderate COVID-19 patients correlated with accelerated viral clearance in the upper airways, and in a reduction in serum IL-6 and CRP [97].
IFN-β1b, when examined in combination with lopinavir–ritonavir and ribavirin treatment, led to a shorter median time from start of study treatment to negative nasopharyngeal swab compared to the control group [98]. When tested alone, administered by inhalation, it improved recovery over the placebo group [99]. A more recent study, however, argues against the beneficial effect of IFN-β, as administration of IFN-β1a in combination with remdesivir did not have any effect in non-hospitalized COVID-19 patients when compared to the placebo group, while it even worsened the outcome of patients who required high-flow oxygen at baseline and received corticosteroids, suggesting a caution at its use [100].
Type III IFNs or IFN-λ constitute promising alternatives of type I IFNs for COVID-19 treatment as they lack many of the pro-inflammatory activities type I IFNs exhibit [101]. Thus, a pegylated version of IFN-λ (peginterferon lambda) was administered in outpatients diagnosed with COVID-19, and although it was found in one study to accelerate viral decline, increasing the proportion of patients with viral clearance by day 7 [102], in another study it displayed no efficacy [103]. It is possible that that this was due to the higher viral load patients exhibited in the former study [102].
These data suggest that an early administration of IFNs, in the timing of infection when IFNs are naturally produced, might be beneficial in COVID-19 patients. These data are of particular importance, as currently there is only one antiviral agent, remdesivir, approved for the treatment of COVID-19 [104].
As COVID-19 is characterized by an excessive inflammatory response leading, in severe cases, to ARDS and potentially to death, pharmacological interventions that prevent or restrain the overt inflammation, such as anti-inflammatory drugs, were hypothesized to be therapeutic. Indeed, in clinical practice, physicians have used immune-modulatory treatments such as IL-1 and IL-6 antagonists in COVID-19 patients, therapies commonly prescribed to individuals with autoimmune rheumatic diseases. For example, tocilizumab is an anti-IL6 monoclonal antibody that blocks IL-6 binding to its receptors. When used in severe COVID-19 patients, it ameliorated several clinical parameters, such as lymphocyte counts and C-reactive protein values, as well as CT opacities, while it reduced oxygen uptake [105]. In other studies, however, there was not a clear clinical benefit [106,107].
IL-1β is a master cytokine of local and systemic inflammation, the blocking of which could inhibit the activation of the innate immune response. There are currently three agents available for targeting the IL-1 pathway either by blocking IL-1 binding to its receptor (anakinra) or by binding directly to IL-1 (rilonacept and canakinumab). Anakinra blocks the binding of both IL-1α and IL-1β, and is the one most broadly used. When anakinra was used in patients with severe COVID-19, it showed a significant increase in survival without severe adverse effects [108,109]. Interestingly, anakinra showed a significant survival benefit when given without dexamethasone than when co-administrated [109]. In parallel, anakinra was used in a study that stratified patients on the basis of high plasma suPAR levels, thus at high risk for respiratory failure, and led to decreased 28-day mortality, and shortened hospital stay [110]. On the other hand, a randomized clinical trial that addressed the therapeutic potential of anakinra in mild-to-moderate COVID-19 patients, was stopped before completion, as no significant difference was found between the groups [108]. Therefore, anakinra could be considered a safe therapeutic option for reducing mortality risk in COVID-19 patients presenting with hyperinflammation, but not in less severe cases. The therapeutic potential of the other two IL-1 inhibitors, rilonacept and canakinumab, in COVID-19 remains to be evaluated.
Baricitinib is a JAK inhibitor that blocks the activation of the JAK-STAT pathway, and eventually cytokine production, that was predicted to be therapeutic in COVID-19 via artificial intelligence [111]. A clinical trial assessing the effect of baricitinib in COVID-19 patients showed a profound reduction in mortality, that was superior when compared to other immune-modulatory treatments [112]. Importantly, the survival benefits of baricitinib were independent of the concomitant use of steroids, namely dexamethasone.
In summary, mild-to-moderate cases of COVID-19 would perhaps profit more from an early administration of IFNs, while in severe cases, administration of an anti-inflammatory agent would be more beneficial. Therefore, the identification of patients that would most benefit from each treatment, as well as the timing of administration are key parameters in selecting the proper therapeutic regime in COVID-19.
5. Conclusions
Innate immune mechanisms are crucial for the antiviral defense to SARS-CoV-2, due to the lack of pre-existing adaptive immunity to this newly emerging virus. Variable phenotypes of COVID-19 disease could be explained by differential innate immune responses to SARS-CoV-2. Indeed, a variety of defects in innate immune mechanisms, including genetic mutations, gene variants, or secretion of autoantibodies, has been linked in recent studies to severity of COVID-19. These findings provide valuable information for the identification of key mechanisms in the defense against SARS-CoV-2, as well as possible therapeutic interventions. Such an example are IFNs, a group of key innate antiviral proteins, the defective response of which is associated with severe COVID-19. Treatments based on the administration of IFNs may thus prove to be of value but will need fine-tuning. As outbreaks of virus infections with the potential to cause global pandemics constitute an increasing threat, in the absence of a specific vaccine or a pathogen-specific agent, broad-spectrum antivirals, such as IFNs, would be valuable in limiting viral spread, consisting thus ideal candidates as general broad-spectrum antivirals [113].
Declaration of Competing Interest
The authors report no declarations of interest.
Acknowledgments
EA is supported by research grants from the European Commission (IMMUNAID, No 779295), the Hellenic Foundation for Research and Innovation (INTERFLU, No 1574) and Janssen Pharmaceuticals. IEG is supported by a research grant from the Hellenic Foundation for Research and Innovation (RELIEVE, No 506).
Data availability
No data was used for the research described in the article.
References
- 1.Wu F., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipsitch M., et al. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat. Rev. Immunol. 2020;20(11):709–713. doi: 10.1038/s41577-020-00460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O., Akira S. Innate immunity to virus infection. Immunol. Rev. 2009;227(1):75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon D.E., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann M., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. doi: 10.1016/j.cell.2020.02.052. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.V’Kovski P., et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19(3):155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantuti-Castelvetri L., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daly J.L., et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lempp F.A., et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature. 2021;598(7880):342–347. doi: 10.1038/s41586-021-03925-1. [DOI] [PubMed] [Google Scholar]
- 11.Amraei R., et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent. Sci. 2021;7(7):1156–1165. doi: 10.1021/acscentsci.0c01537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thepaut M., et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLoS Pathog. 2021;17(5):e1009576. doi: 10.1371/journal.ppat.1009576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichimura T., et al. KIM-1/TIM-1 is a receptor for SARS-CoV-2 in lung and kidney. medRxiv. 2020 [Google Scholar]
- 14.Wang S., et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021;31(2):126–140. doi: 10.1038/s41422-020-00460-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clausen T.M., et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057. doi: 10.1016/j.cell.2020.09.033. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020;5(1):283. doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bian H., et al. Safety and efficacy of meplazumab in healthy volunteers and COVID-19 patients: a randomized phase 1 and an exploratory phase 2 trial. Signal Transduct. Target. Ther. 2021;6(1):194. doi: 10.1038/s41392-021-00603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shilts J., et al. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci. Rep. 2021;11(1):413. doi: 10.1038/s41598-020-80464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen S., Thomsen A.R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012;86(6):2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bortolotti D., et al. TLR3 and TLR7 RNA sensor activation during SARS-CoV-2 infection. Microorganisms. 2021;9(9) doi: 10.3390/microorganisms9091820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asano T., et al. X-linked recessive TLR7 deficiency in ∼1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021;6(62) doi: 10.1126/sciimmunol.abl4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q., et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370(6515) doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin X., et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34(2):108628. doi: 10.1016/j.celrep.2020.108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J., et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021;34(7):108761. doi: 10.1016/j.celrep.2021.108761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNab F., et al. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15(2):87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazear H.M., Schoggins J.W., Diamond M.S. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50(4):907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotenko S.V., et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003;4(1):69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 28.Prokunina-Olsson L., et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013;45(2):164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheppard P., et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003;4(1):63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 30.Andreakos E., Zanoni I., Galani I.E. Lambda interferons come to light: dual function cytokines mediating antiviral immunity and damage control. Curr. Opin. Immunol. 2019;56:67–75. doi: 10.1016/j.coi.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galani I.E., Koltsida O., Andreakos E. Type III interferons (IFNs): emerging master regulators of immunity. Adv. Exp. Med. Biol. 2015;850:1–15. doi: 10.1007/978-3-319-15774-0_1. [DOI] [PubMed] [Google Scholar]
- 32.Galani I.E., et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46(5):875–890. doi: 10.1016/j.immuni.2017.04.025. e6. [DOI] [PubMed] [Google Scholar]
- 33.Blanco-Melo D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–1045. doi: 10.1016/j.cell.2020.04.026. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lokugamage K.G., et al. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020;94(23) doi: 10.1128/JVI.01410-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mantlo E., et al. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res. 2020;179:104811. doi: 10.1016/j.antiviral.2020.104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanifer M.L., et al. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 2020;32(1):107863. doi: 10.1016/j.celrep.2020.107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfaender S., et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020;5(11):1330–1339. doi: 10.1038/s41564-020-0769-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegler C.G.K., et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016–1035. doi: 10.1016/j.cell.2020.04.035. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onabajo O.O., et al. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020;52(12):1283–1293. doi: 10.1038/s41588-020-00731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Driscoll M., et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature. 2021;590(7844):140–145. doi: 10.1038/s41586-020-2918-0. [DOI] [PubMed] [Google Scholar]
- 41.Piroth L., et al. Comparison of the characteristics, morbidity, and mortality of COVID-19 and seasonal influenza: a nationwide, population-based retrospective cohort study. Lancet Respir. Med. 2021;9(3):251–259. doi: 10.1016/S2213-2600(20)30527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson E.J., et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584(7821):430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22(2):176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minakshi R., et al. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One. 2009;4(12):e8342. doi: 10.1371/journal.pone.0008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siu K.L., et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009;284(24):16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wathelet M.G., et al. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81(21):11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banerjee A.K., et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183(5):1325–1339. doi: 10.1016/j.cell.2020.10.004. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schubert K., et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27(10):959–966. doi: 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- 49.Hsu J.C., et al. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. U. S. A. 2021;118(24) doi: 10.1073/pnas.2101161118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finkel Y., et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature. 2021;594(7862):240–245. doi: 10.1038/s41586-021-03610-3. [DOI] [PubMed] [Google Scholar]
- 51.Lei X., et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020;11(1):3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia H., et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33(1):108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng Y., et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020;5(1):299. doi: 10.1038/s41392-020-00438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miorin L., et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. U. S. A. 2020;117(45):28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mu J., et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020;6(1):65. doi: 10.1038/s41421-020-00208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang H.W., et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020;17(9):998–1000. doi: 10.1038/s41423-020-0514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gordon D.E., et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science. 2020;370(6521) doi: 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stukalov A., et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021;594(7862):246–252. doi: 10.1038/s41586-021-03493-4. [DOI] [PubMed] [Google Scholar]
- 59.Han L., et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J. Med. Virol. 2021;93(9):5376–5389. doi: 10.1002/jmv.27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu G., et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021;6(4):467–478. doi: 10.1038/s41564-021-00884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin D., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konno Y., et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32(12):108185. doi: 10.1016/j.celrep.2020.108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin J.W., et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe. 2021;29(3):489–502. doi: 10.1016/j.chom.2021.01.015. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banerjee A., et al. Experimental and natural evidence of SARS-CoV-2-infection-induced activation of type I interferon responses. iScience. 2021;24(5):102477. doi: 10.1016/j.isci.2021.102477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chu H., et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020;71(6):1400–1409. doi: 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucas C., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584(7821):463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arunachalam P.S., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369(6508):1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galani I.E., et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021;22(1):32–40. doi: 10.1038/s41590-020-00840-x. [DOI] [PubMed] [Google Scholar]
- 69.Hadjadj J., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davidson S., et al. IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol. Med. 2016;8(9):1099–1112. doi: 10.15252/emmm.201606413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Combes A.J., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591(7848):124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sposito B., et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell. 2021;184(19):4953–4968. doi: 10.1016/j.cell.2021.08.016. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Broggi A., et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369(6504):706–712. doi: 10.1126/science.abc3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Major J., et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712–717. doi: 10.1126/science.abc2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ellinghaus D., et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 2020;383(16):1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pairo-Castineira E., et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 77.Bastard P., et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515) doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bastard P., et al. Autoantibodies neutralizing type I IFNs are present in ∼4% of uninfected individuals over 70 years old and account for ∼20% of COVID-19 deaths. Sci. Immunol. 2021;6(62) doi: 10.1126/sciimmunol.abl4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Israelow B., et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 2020;217(12) doi: 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Channappanavar R., et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Q., et al. Life-threatening COVID-19: defective interferons unleash excessive inflammation. Med (N Y) 2020;1(1):14–20. doi: 10.1016/j.medj.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang C., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de la Rica R., Borges M., Gonzalez-Freire M. COVID-19: in the eye of the cytokine storm. Front. Immunol. 2020;11:558898. doi: 10.3389/fimmu.2020.558898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao D., et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: a retrospective cohort study. Lancet Haematol. 2020;7(9):e671–e678. doi: 10.1016/S2352-3026(20)30217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schulte-Schrepping J., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182(6):1419–1440. doi: 10.1016/j.cell.2020.08.001. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Silvin A., et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182(6):1401–1418. doi: 10.1016/j.cell.2020.08.002. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liao M., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26(6):842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 88.Guan W.J., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou F., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen B., et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182(1):59–72. doi: 10.1016/j.cell.2020.05.032. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramlall V., et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020;26(10):1609–1615. doi: 10.1038/s41591-020-1021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma L., et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. bioRxiv. 2021 doi: 10.1126/sciimmunol.abh2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Risitano A.M., et al. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020;20(6):343–344. doi: 10.1038/s41577-020-0320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwarz B., et al. Cutting edge: severe SARS-CoV-2 infection in humans is defined by a shift in the serum lipidome, resulting in dysregulation of eicosanoid immune mediators. J. Immunol. 2021;206(2):329–334. doi: 10.4049/jimmunol.2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Horby P., et al. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 2021;384(8):693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang N., et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe. 2020;28(3):455–464. doi: 10.1016/j.chom.2020.07.005. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Q., et al. Interferon-alpha2b treatment for COVID-19 is associated with improvements in lung abnormalities. Viruses. 2020;13(1) doi: 10.3390/v13010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hung I.F., et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020;395(10238):1695–1704. doi: 10.1016/S0140-6736(20)31042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Monk P.D., et al. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021;9(2):196–206. doi: 10.1016/S2213-2600(20)30511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalil A.C., et al. Efficacy of interferon beta-1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID-19: a double-bind, randomised, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2021 doi: 10.1016/S2213-2600(21)00384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andreakos E., Tsiodras S. COVID-19: lambda interferon against viral load and hyperinflammation. EMBO Mol. Med. 2020;12(6):e12465. doi: 10.15252/emmm.202012465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Feld J.J., et al. Peginterferon lambda for the treatment of outpatients with COVID-19: a phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021;9(5):498–510. doi: 10.1016/S2213-2600(20)30566-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jagannathan P., et al. Peginterferon Lambda-1a for treatment of outpatients with uncomplicated COVID-19: a randomized placebo-controlled trial. Nat. Commun. 2021;12(1):1967. doi: 10.1038/s41467-021-22177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beigel J.H., et al. Remdesivir for the treatment of Covid-19 - final report. N. Engl. J. Med. 2020;383(19):1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xu X., et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. U. S. A. 2020;117(20):10970–10975. doi: 10.1073/pnas.2005615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Campochiaro C., et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: a single-centre retrospective cohort study. Eur. J. Intern. Med. 2020;76:43–49. doi: 10.1016/j.ejim.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salvarani C., et al. Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern. Med. 2021;181(1):24–31. doi: 10.1001/jamainternmed.2020.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): a randomised controlled trial. Lancet Respir. Med. 2021;9(3):295–304. doi: 10.1016/S2213-2600(20)30556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cavalli G., et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325–e331. doi: 10.1016/S2665-9913(20)30127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kyriazopoulou E., et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat. Med. 2021 doi: 10.1038/s41591-021-01499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stebbing J., et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol. Med. 2020;12(8):e12697. doi: 10.15252/emmm.202012697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marconi V.C., et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021 doi: 10.1016/S2213-2600(21)00331-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang B.X., Fish E.N. Global virus outbreaks: interferons as 1st responders. Semin. Immunol. 2019;43:101300. doi: 10.1016/j.smim.2019.101300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.