

Abstract
Background
Squamous cell carcinoma of the anal canal (SCCA) is an uncommon malignancy with limited therapeutic options. Nivolumab and pembrolizumab show promising results in patients with SCCA. Human papillomavirus (HPV)-negative tumors are frequently TP53-mutated (TP53-MT) and often resistant to therapy.
Methods
We present a large molecularly-profiled cohort of SCCA, exploring the underlying biology of SCCA, differences between TP53-wild type (TP53-WT) and TP53-MT tumors, and differences between local and metastatic tumors. SCCA specimens (n=311) underwent multiplatform testing with immunohistochemistry (IHC), in situ hybridization (ISH) and next-generation sequencing (NGS). Tumor mutational burden (TMB) was calculated using only somatic nonsynonymous missense mutations. Chi-square testing was used for comparative analyses.
Results
The most frequently mutated genes included PIK3CA (28.1%), KMT2D (19.5%), FBXW7 (12%), TP53 (12%) and PTEN (10.8%). The expression of PD-1 was seen in 68.8% and PD-L1 in 40.5% of tumors. High TMB was present in 6.7% of specimens. HER2 IHC was positive in 0.9%, amplification by chromogenic in situ hybridization (CISH) was seen 1.3%, and mutations in ERBB2 were present in 1.8% of tumors. The latter mutation has not been previously described in SCCA. When compared with TP53-WT tumors, TP53-MT tumors had higher rates of CDKN2A, EWSR1, JAK1, FGFR1 and BRAF mutations. PD-1 and PD-L1 expression were similar, and high TMB did not correlate with PD-1 (P=0.50) or PD-L1 (P=0.52) expression.
Conclusions
Molecular profiling differences between TP53-MT and TP53-WT SCCA indicate different carcinogenic pathways which may influence response to therapy. Low frequency mutations in several druggable genes may provide therapeutic opportunities for patients with SCCA.
Keywords: Squamous cell carcinoma of the anal canal (SCCA), anal carcinoma, targeted therapy, genomic profiling
Introduction
Anal cancers are relatively rare, with an estimated 8,590 new cases in the US in 2020, although the incidence is rising (1). The most common histological subtype of anal cancer is squamous cell carcinoma of the anal canal (SCCA) (2). Although SCCA occurs more frequently in women than men, its incidence is rising in men who have sex with men and persons infected with HIV (3). Despite this, treatment for locoregional SCCA has not changed in many years and consists of the Nigro protocol, first described in 1974, of definitive concurrent chemoradiation with 5-fluorouracil (5-FU) and mitomycin-C, with surgery reserved for the salvage setting (4). A phase III EORTC study confirmed the use of combined modality therapy with chemoradiation (5-FU plus mitomycin-C), demonstrating improved local control and colostomy-free survival compared to radiation alone (5). Until recently, metastatic SCCA was treated with the same decades-old chemotherapy regimen of 5-FU plus cisplatin (6). This combination was recently challenged by carboplatin plus weekly paclitaxel in the phase II InterACCT trial (n=91), results at a median follow-up of 28.6 months revealed that carboplatin plus paclitaxel had comparable response rates to 5-FU plus cisplatin; the overall response rate (ORR) was 59% (95% CI, 42.1% to 74.4%) for carboplatin plus paclitaxel versus 57% (95% CI, 39.4% to 73.7%) for cisplatin plus 5-FU, with reduced serious events (36% compared to 62% P=0.016) and improved progression-free survival [PFS, 8.1 months (95% CI, 6.6 to 8.8 months) versus 5.7 months (95% CI, 3.3 to 9.0 months)]. Median overall survival (OS) was 20 months (95% CI, 12.7 months to not reached) versus 12.3 months (95% CI, 9.2 to 17.7 months, HR 2.00, 95% CI, 1.15 to 3.47; P=0.014) (7). These data indicate carboplatin plus paclitaxel should be considered as a new standard of care. The persistence of these standards of care is in part due to the rarity of SCCA and the difficulty of completing large, randomized clinical trials. Moreover, patients with local disease have variable responses to chemoradiation. New insights into the molecular biology of SCCA reveal the underlying mechanisms of carcinogenesis and are changing the treatment paradigm.
Human papillomavirus (HPV) is involved in 85–88% of SCCA (8). SCCA that is not related to HPV infection represents a small minority of cases. These cases are notoriously refractory to chemoradiation and frequently harbor TP53 mutations (9). Deeper sequencing techniques on 392 HPV-PCR negative cervival cancer specimens, detected HPV in 43.1% of specimens (n=169) suggesting HPV as the oncogenic driver even if previously testing negative (10). Interestingly, many precursor lesions (anal intraepithelial neoplasia II/III) are also positive for HPV (especially HPV16), suggesting that HPV infection is an early event in potential oncogenesis; however, few of these precursor lesions will actually become malignant (11). It remains unclear why some precursor lesions progress while others do not, but immunosuppression (especially from HIV infection) plays a role via the evasion of HPV-specific CD4+ and CD8+ T cells (12,13). While HPV infection is often transient in immunocompetent individuals, HIV-positive patients often have a persistent HPV infection (9). The HPV viral genes E6 and E7 contribute to oncogenic transformation. The E6 oncoprotein encoded by the HPV genome directly targets and inactivates the p53 protein, which usually functions as a tumor suppressor (14). Consequently, p53 is unable to induce apoptosis. The E7 oncoprotein effectively deregulates the cell-cycle; E7 forms complexes with the phosphorylated retinoblastoma protein (pRb), which is responsible for inhibiting transcriptional activity in the G1 phase of the cell cycle. Once complexed with E7, pRb is degraded, leading to progression of the cell through the S phase (15). Multiple other signaling pathways have been implicated in SCCA carcinogenesis, including epidermal growth factor receptor (EGFR), PI3K/AKT/mTOR, VEGF, hedgehog and Bcl-2 (15). Given the few treatment options for metastatic SCCA, knowledge of the activity of these pathways in individual tumors is vital to effective therapeutic selection.
A previous study of 199 recurrent or metastatic SCCA tumors was notable for frequent mutations in PIK3CA (33%) and TP53 (15%). The tumors also had high immunohistochemistry (IHC) expression of EGFR (89%), TOP2A (85%), TOPO1 (67%) and low ERCC1 (49%), potentially conferring sensitivity to anti-EGFR antibodies, anthracyclines, irinotecan and platinum-based chemotherapies, respectively (16). Few tumors in this study were tested for PD-1 or PD-L1, and PD-L1 expression is known to correlate with a worse prognosis in SCCA (17). Another study preformed comprehensive genomic profiling on 574 SCCA tumors to analyze the prevalence as well as mutational profiling details of tumor suppressor gene CYLD (18). CYLD mutations were seen in 13% of patients, and correlated with lower tumor mutational burden (TMB) and less alterations in PIK3CA. Additional comprehensive molecular profiling of SCCA are necessary to establish clinically useful biomarkers.
We sought to expand on this work through several avenues. First, we analyzed a large known cohort of molecularly-profiled SCCA. Second, by understanding the differences between profiles of primary and metastatic SCCA, pathways affecting metastasis can be identified. Third, there is recent data evaluating the molecular characteristics of the difference between TP53-WT versus TP53-MT tumors and our cohort adds to this growing field (19,20). Thus, continued knowledge expansion of molecular pathways that confer this resistance to standard therapy would help guide the selection of alternative treatments. Finally, the exploding field of immunotherapy holds particular promise in metastatic SCCA. Because SCCA tends to flourish in an immunosuppressed environment, there is significant hope that checkpoint inhibition can be utilized to harness the immune system in attacking SCCA.
In this study, we aimed to establish the true prevalence of PD-1 and PD-L1 expression in a larger repository of SCCA tumors. In addition, we sought to analyze the mutational burden in SCCA tumors because a higher mutational burden may lead to greater diversity in neoantigens and increase the likelihood of an effective immune response (21). Mutational burden has proven to be a useful biomarker for efficacy of immunotherapy in non-small cell lung cancer, urothelial cancer, melanoma and mismatch repair (MMR)-deficient tumors (22-25). We present the following article in accordance with the MDAR reporting checklist (available at https://dx.doi.org/10.21037/jgo-20-610).
Methods
A multi-institutional database was searched to identify patients diagnosed with SCCA from January 1, 2015, through March 31, 2019. All patient data were deidentified, negating the need for patient informed consent. Each archived formalin-fixed, paraffin-embedded tumor sample or newly obtained biopsy sample was assessed by the Caris Life Sciences (Phoenix, AZ, USA) multiplatform profiling service. Tumor samples were accompanied by limited patient demographic and clinical information, and this is shown in Table 1.
Table 1. Patient demographics.
| Parameter | Number |
|---|---|
| Total cases (n) | 311 |
| Age (years) | |
| Median age | 61 |
| Interquartile range | 31–89 |
| Gender, n (%) | |
| Female | 225 (72.3) |
| Male | 86 (27.7) |
| Location of primary tumor, n (%) | |
| Anus | 117 (56.9) |
| Anal canal | 92 (29.6) |
| Anorectum | 16 (5.1) |
| Overlapping lesion of rectum, anus, anal canal | 10 (3.2) |
| Rectum | 7 (2.3) |
| Anorectal junction | 6 (1.9) |
| Skin of anus | 1 (0.3) |
| Perianal skin | 1 (0.3) |
| Cloacagenic zone | 1 (0.3) |
| Specimen site, n (%) | |
| Local | 210 (67.5) |
| Distant metastasis | 101 (32.5) |
Molecular analysis
All 311 SCCA specimens underwent multiplatform testing with the test selection based on the primary oncologist’s recommendations. Testing included protein expression through IHC assay (n=302), gene amplification through chromogenic in situ hybridization (CISH, n=90), copy number alteration (CNA, n=215), fragment analysis (FA, n=15), fusion analysis via RNA sequencing (n=50), transcriptome sequencing (n=10) and next-generation sequencing [NGS, hot spot NGS (n=64) and Illumina NextSeq, 592 gene, NGS (n=226)]. TMB (n=209) was calculated using only somatic nonsynonymous missense mutations, and TMB-high was defined as ≥17 mutations (26). Details of the samples’ testing are shown in Figure 1.
Figure 1.

Specimen testing demographics. SCCA, squamous cell carcinoma of the anal canal; IHC, immunohistochemistry; NGS, next-generation sequencing; CISH, chromogenic in situ hybridization; CNA, copy number alteration; FA, fragment analysis.
IHC was performed on full formalin-fixed paraffin-embedded (FFPE) sections of glass slides, using automated staining techniques, per the manufacturer’s instructions, and were optimized and validated per CLIA/CAO and ISO requirements. Staining was scored for intensity (0, no staining; 1+, weak staining; 2+, moderate staining; 3+, strong staining) and staining percentage (0–100%). Results were categorized as positive or negative by defined thresholds specific to each marker based on published clinical literature that associates biomarker status with patient responses to therapeutic agents. A board-certified pathologist evaluated all IHC results independently. The primary antibody used against PD-L1 was SP142 (Spring Biosciences, Pleasanton, CA, USA). The staining was regarded as positive if its intensity on the membrane of the tumor cells was ≥2+ and the percentage of positively stained cells was >5%. The primary antibody used for PD-1 was MRQ-22 (Ventana) and staining was scored as positive if the number of PD-1+ TIL was >1 cell per high-power field. PD-1 TIL density was evaluated using a hotspot approach. The whole tumor sample was reviewed at a low power (4× objective), and the areas of highest density of TIL in direct contact with malignant cells of the tumor at 400× visual field (40× objective × 10× ocular) were enumerated.
Statistical analysis
The frequency of the protein expression, mutation and amplification data was compared between tumor samples that were TP53-WT vs. TP53-MT and primary vs. metastatic via either Chi-square tests or Fisher’s exact tests when appropriate. SAS software version 9.4 (SAS Ins., Cary, NC, USA) was used for the analysis. P values <0.05 were considered statistically siginificant.
Ethical statement
Informed consent was not required for this study; these analyses were conducted using de-identified patient data obtained from Caris Life Sciences (Phoenix, AZ, USA) database. It was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Results
Baseline patient characteristics
We performed an analysis of 311 SCCA cases. The median patient age was 61 years (interquartile range, 31–89 years). The majority of patients (72.3%) were female, and specimens were examined from both primary and metastatic sites (67.5% and 32.5%, respectively), with the most common metastatic site being the liver (51.5%, n=52). Local specimen lesions include the anus, anal canal, anorectum, overlapping lesion of rectum/anus/anal canal, rectum, anorectal junction, skin of anus, perianal skin, cloacogenic zone, vagina/labia, urethra, skin of thigh, gluteal skin and inguinal lymph node. Distant metastatic specimen sites include liver, small bowel, omental, ovary, brain, paraspinal, lung and pleural lesions as well as neck, cervical, supraclavicular, periaortic, paratracheal, subcarinal, scalene and retroperitoneal lymph nodes. Table 1 provides the details of the patient demographic characteristics.
Recurrent mutation and biomarker incidence
Recurrent mutations were seen in 82 genes (Table 2). In summary, the most frequently mutated genes included PIK3CA (28.1%), KMT2D (19.5%), FBXW7 (12%), TP53 (12%) and PTEN (10.8%). Of the 220 tumors subjected to Illumina NextSeq (592 gene) testing, the most common mutations were PIK3CA (28.1%), KMT2D (19.5%), KMT2C (16.2%), TP53 (12.0%), PTEN (10.8%), FBXW7 (10.3%), ARID1A (4.8%), ATRX (4.1%), APC (3.8%) and NOTCH1 (3.4%). IHC analysis showed frequent expression of EGFR (97.7%) and PTEN (68.1%). MMR deficiency frequency tested by IHC of MLH1, PMS2, MSH2 and MSH6 was 1.1%. Expression of PD-1 was seen in 68.8% (44/64) of tumors, and PD-L1 was seen in 40.5% (119/294). HER2 IHC was positive in 0.9% (1/106) of samples, and amplification by CISH was seen 1.3% (1/78) of samples. Mutations in ERBB2 were present in 1.8% (4/219) of samples.
Table 2. Mutation and biomarker incidence.
| Name | Incidence (%) | Incidence ratio | Technique |
|---|---|---|---|
| MSH2 | 100 | 92/92 | IHC |
| MSH6 | 100 | 90/90 | IHC |
| MLH1 | 98.9 | 91/92 | IHC |
| PMS2 | 98.9 | 87/88 | IHC |
| EGFR | 97.7 | 43/44 | IHC |
| PD-1 | 68.8 | 44/64 | IHC |
| PTEN | 68.1 | 64/94 | IHC |
| PD-L1 | 40.5 | 119/294 | IHC |
| PIK3CA | 28.1 | 62/221 | NGS |
| PIK3CA | 24.2 | 15/62 | NGS hot spot |
| KMT2D | 19.5 | 36/185 | NGS |
| KMT2C | 16.2 | 19/177 | NGS |
| FBXW7 | 12.9 | 8/62 | NGS hot spot |
| TP53 | 12. | 25/208 | NGS |
| PTEN | 10.8 | 22/204 | NGS |
| FBXW7 | 10.3 | 20/195 | NGS |
| TMB | 6.7 | 14/209 | NGS |
| PIK3CA | 5.4 | 11/205 | CNA |
| TP53 | 5 | 3/60 | NGS hot spot |
| ARID1A | 4.8 | 4/84 | NGS |
| ATRX | 4.1 | 3/73 | NGS |
| APC | 3.8 | 8/212 | NGS |
| NOTCH1 | 3.4 | 7/204 | NGS |
| RB1 | 3.3 | 2/61 | NGS hot spot |
| AKT1 | 3.2 | 2/62 | NGS hot spot |
| KRAS | 3.2 | 2/62 | NGS hot spot |
| TRK A/B/C | 3.1 | 2/64 | IHC |
| BCL6 | 2.9 | 6/205 | CNA |
| NFIB | 2.9 | 6/205 | CNA |
| PBRM1 | 2.9 | 6/206 | NGS |
| EP300 | 2.9 | 5/174 | NGS |
| CDKN2A | 2.8 | 6/216 | NGS |
| KRAS | 2.7 | 6/221 | NGS |
| FGF19 | 2.5 | 5/204 | CNA |
| TFRC | 2.4 | 5/205 | CNA |
| FGF4 | 2.4 | 5/208 | CNA |
| FGF3 | 2.4 | 5/209 | CNA |
| KDM5C | 2.4 | 3/126 | NGS |
| FGFR3 | 2.3 | 5/214 | NGS |
| AKT1 | 2.3 | 5/218 | NGS |
| BAP1 | 2.3 | 5/219 | NGS |
| CYLD | 2.1 | 4/191 | NGS |
| RB1 | 2 | 4/197 | NGS |
| PIK3R1 | 2 | 4/204 | NGS |
| KLHL6 | 2 | 4/205 | CNA |
| CCND1 | 1.9 | 4/210 | CNA |
| BRCA2 | 1.9 | 4/215 | NGS |
| MUTYH | 1.8 | 4/217 | NGS |
| ERBB2 | 1.8 | 4/219 | NGS |
| ZNF703 | 1.5 | 3/203 | CNA |
| CALR | 1.5 | 3/205 | CNA |
| ETV5 | 1.5 | 3/205 | CNA |
| LPP | 1.5 | 3/205 | CNA |
| LYL1 | 1.5 | 3/205 | CNA |
| PSIP1 | 1.5 | 3/205 | CNA |
| SMAD2 | 1.5 | 3/205 | CNA |
| EGFR | 1.4 | 3/208 | CNA |
| FH | 1.4 | 3/215 | NGS |
| CTNNB1 | 1.4 | 3/216 | NGS |
| HRAS | 1.4 | 3/221 | NGS |
| MSI | 1.3 | 2/156 | NGS |
| CREBBP | 1.1 | 2/188 | NGS |
| NSD1 | 1 | 2/198 | NGS |
| ARID2 | 1 | 2/200 | NGS |
| MYB | 1 | 2/202 | CNA |
| ADGRA2 | 1 | 2/203 | CNA |
| FANCG | 1 | 2/204 | CNA |
| PRRX1 | 1 | 2/204 | CNA |
| KEAP1 | 1 | 2/205 | CNA |
| KRAS | 1 | 2/205 | CNA |
| MALT1 | 1 | 2/205 | CNA |
| NSD3 | 1 | 2/205 | CNA |
| SMAD4 | 1 | 2/205 | CNA |
| SS18 | 1 | 2/205 | CNA |
| FGFR1 | 1 | 2/209 | CNA |
| MSH6 | 1 | 2/210 | NGS |
| ERCC2 | 0.9 | 2/213 | NGS |
| ATM | 0.9 | 2/214 | NGS |
| STK11 | 0.9 | 2/216 | NGS |
| BRAF | 0.9 | 2/217 | NGS |
| ERBB3 | 0.9 | 2/218 | NGS |
| BRCA1 | 0.9 | 2/219 | NGS |
IHC, immunohistochemistry; NGS, next-generation sequencing; CNA, copy number alteration.
TMB and microsatellite instability (MSI)
Mean TMB was 8.6 mutations per megabase. High TMB, as tested by NGS, was present in 6.7% of tumors (n=209). MSI-high tumors by FA and NGS were present in 6.7% (1/15), and 1.3% (2/156) of specimens, respectively.
Mutational differences between TP53-WT and TP53-MT
A comparison of TP53-MT to TP53-WT samples is summarized in Table 3. The incidence rates of statistically non-significant mutations and biomarkers are summarized in Table S1. CDKN2A mutations, known to encode both p16 (INK4a) and p14 (ARF) genes, were exclusively seen in TP53-MT samples (6/25). Additional mutations seen only in TP53-MT samples were BRAF (2/25, NGS), EWSR1 (1/24, CNA), JAK1 (1/25, NGS) and FGFR1 (2/24, CNA). When comparing biomarkers for immunotherapy, they were slightly less prevalent in TP53-MT compared to TP53-WT samples, although this difference was not statistically significant: IHC for PD-1, 50% (2/4) vs. 70% (42/60), P=0.583; IHC for PD-L1, 32.1% (9/28) vs. 41.4% (110/266), P=0.345; NGS for MSI, 0% (0/25) vs. 1.53% (2/131); and NGS for FA, 0% (0/0) vs. 6.67% (1/15), P=1.000. All HER2-positive samples were from TP53-WT. When testing for APC, TP53-MT samples had a significantly higher incidence of APC than TP53-WT samples by NGS (20%, 5/25 compared to 1.6%, 3/187, P<0.001) and by NGS hotspot (33.33%, 1/3 compared to 0%, 0/59, P=0.048). NOTCH1 incidence was also significantly higher in TP53-MT (12%, 3/25) than TP53-WT (2.23%, 4/179) with P=0.04 for NGS. The incidence of CALR by CNA was significantly higher in TP53-MT (8.33%, 2/24) compared to TP53-WT (0.55%, 1/181) tumors, P=0.037. FGFR1 incidence was significantly higher in TP53-MT (8.33%, 2/24) than in TP53-WT tumors (0%, 0/185), P=0.013, and ZNF703 incidence was higher in TP53-MT (8.33%, 2/24) compared to TP53-WT samples (0.59%, 1/170), P=0.041. PIK3CA incidence by NGS was significantly higher in TP53-WT (30.6%, 60/196) compared to TP53-MT samples (8%, 2/25), P=0.018.
Table 3. Incidence rates of statistically significant mutations and biomarkers in TP53-MT (n=28) tumors versus TP53-WT (n=238).
| Alteration | Test | TP53-MT (n=28) incidence (%) | TP53-MT incidence ratio | TP53-WT (n=238) incidence (%) | TP53-WT incidence ratio | P value |
|---|---|---|---|---|---|---|
| Tumor suppressor genes | ||||||
| CDKN2A | NGS | 24 | 6/25 | 0 | 0/191 | 0.0000 |
| Immune | ||||||
| APC | NGS | 20 | 5/25 | 1.6 | 3/187 | 0.0007 |
| APC | NGS hot spot | 33.33 | 1/3 | 0 | 0/59 | 0.0484 |
| RAS/RAF/MEK | ||||||
| BRAF | NGS | 8 | 2/25 | 0 | 0/192 | 0.0128 |
| PI3K related kinases | ||||||
| PIK3CA | NGS | 8 | 2/25 | 30.6 | 60/196 | 0.0178 |
| Other | ||||||
| NOTCH1 | NGS | 12 | 3/25 | 2.23 | 4/179 | 0.0410 |
| CAL-R | CNA | 8.33 | 2/24 | 0.55 | 1/181 | 0.0367 |
| FGFR1 | CNA | 8.33 | 2/24 | 0 | 0/185 | 0.0127 |
| ZNF703 | CNA | 8.33 | 2/24 | 0.55 | 1/170 | 0.0409 |
TP53-MT, TP53-mutated; TP53-WT, TP53-wild type; NGS, next-generation sequencing; CNA, copy number alteration.
Mutational differences between local and metastatic lesions
The analysis of both local and metastatic lesions is summarized in Table 4, with Figure 2 illustrating the statistically significant differences in incidence rates between local and metastatic tumor samples. The incidence rates of statistically non-significant mutations and biomarkers are summarized in Table S2. Local tumors showed higher incidence of PD-1 80.9% (38/47 IHC) compared to 35.3% (6/17), P<0.001 in metastatic lesions. Local tumors also showed a higher incidence of PTEN 13.8% (19/138 NGS) compared to 4.5% (3/66 NGS) in metastatic lesions, P=0.047. Metastatic lesions had significantly greater incidence of FGFR3 mutations, 6.8% (5/73 NGS) compared to 0% (0/141) in local tumors, P=0.004. All HER2-positive samples were from metastatic lesions.
Table 4. Incidence rates of statistically significant mutations and biomarkers in local versus metastatic locations.
| Alteration | Test | Metastatic incidence (%) | Metastatic incidence ratio | Local incidence (%) | Local incidence ratio | P value |
|---|---|---|---|---|---|---|
| Immune | ||||||
| PD-1 | IHC | 35.29 | 6/17 | 80.85 | 38/47 | 0.0005 |
| SMAD pathway | ||||||
| SMAD2 | CNA | 4.35 | 3/69 | 0 | 0/137 | 0.0365 |
| PI3K related kinases | ||||||
| PTEN | NGS | 4.55 | 3/66 | 13.77 | 19/138 | 0.0470 |
| Other | ||||||
| FGFR3 | NGS | 6.85 | 5/73 | 0 | 0/141 | 0.0042 |
IHC, immunohistochemistry; CNA, copy number alteration; NGS, next-generation sequencing.
Figure 2.
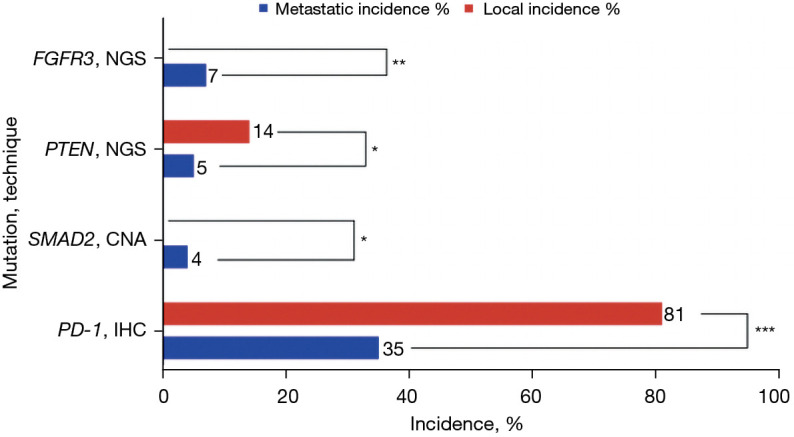
Incidence of mutations in PI3K related kinases in local and metastatic cases of SCCA. *, P<0.05; **, P<0.01; ***, P<0.001. SCCA, squamous cell carcinoma of the anal canal; NGS, next-generation sequencing; CNA, copy number alteration; IHC, immunohistochemistry.
Discussion
Although SCCA is a rare malignancy, its incidence and morbidity are increasing in the United States. Here we report an extensive comprehensive genomic profiling with NGS and gene expression profiling for patients with SCCA. Several tumor suppressor genes and oncogenes were newly identified in our population of SCCA.
Within this study, we were unable to identify HPV related cases by p16 overexpression given the lack of such testing’s availability at the time of our cohort’s analysis, other HPV driven malignancies such as head and neck cancer, cervical cancer and other gynecologic cancers utilize p16 overexpression as a confirmation for HPV infection as well as a prognostic marker related to PFS and OS [8–11]. The prognostic value of HPV-DNA and p16 expression in SCCA has also been explored, with results revealing that patients testing positively for both HPV-DNA and p16 overexpression have longer OS [18–19]. Newer unlike other gastrointestinal malignancies such as colon or pancreatic adenocarcinomas, SCCA rarely harbors KRAS mutations, with an incidence of 3% or less, which is confirmed in this comprehensive analysis (27,28). Similar to smaller studies, PIK3CA was the most common mutation identified. PIK3CA mutations are seen in approximately one-third of all specimens, frequently occurring in TP53-WT tumors (see Figure 3), suggesting that HPV may drive the accumulation of activating mutations in PIK3CA, which have been linked to higher risks of relapse (16,27-30). PIK3CA mutations have been identified in other HPV driven malignancies, including SCC of the head and neck (31). In addition, MLL2/KMT2D and MLL3/KMT2C are important in histone modification and are associated with oncogenesis as well as TP53 expression in preclinical studies. Prior SCCA studies identified mutations in these genes (29,32,33). Our analysis revealed high rates of mutations in both epigenetic regulators MLL2/KMT2D (19.5%) and MLL3/KMT2C (16.2%), the majority of both mutations in TP53-WT specimens.
Figure 3.

Incidence of mutations in TP53-MT (n=28) tumors and TP53-WT (n=238). *, P<0.05. CNA, copy number alteration; NGS, next-generation sequencing; TP53-MT, TP53-mutated; TP53-WT, TP53-wild type.
The tumor suppressor gene TP53, responsible for cell cycle regulation and apoptosis, is known to be overexpressed in SCCA. Many studies have seen a correlation between TP53 mutations and worse locoregional control and reduced disease-free survival in patients with SCCA (34-37). One study (n=119) found 4% of SCCA expressed exon 5 TP53 mutations (30). Another recent study (n=106) confirmed the correlation with HPV-negativity, p53 staining, and TP53 mutation in SCCA are associated with inferior OS as well as reduced recurrence-free survival (19). Our study discovered TP53 mutations in 12% of NGS samples (25/208) and 5% of NGS hot spot samples (3/60).
Rare mutations noted in this study include HRAS, found in 1.4% (3/221 NGS) of samples, all of which were in local TP53-WT specimens. HRAS was first noted in a smaller SCCA whole-exome sequencing cohort, along with ARID1A mutations, and were felt to be driver mutations of SCCA (38). ARID1A was prevalent in 4.8% (4/84 NGS) of our study specimens, all of which were TP53-WT, mostly localized tumors.
Potentially targetable mutations
Outside of immunotherapy, there are currently no clinically established biological markers to guide therapy for patients with SCCA. Table 5 reviews potentially targetable mutations identified in this analysis.
Table 5. Potentially targetable mutations outside of immunotherapy.
| Potentially targetable mutation/test | Treatment | Incidence |
|---|---|---|
| DDR pathways | Synthetic lethality with agents combined with a PARPi | |
| BRCA 1/NGS | 2/219 (0.9%) | |
| BRCA 2/NGS | 4/215 (1.9%) | |
| ATM/NGS | 2/214 (0.9%) | |
| ATRX/NGS | 3/73 (4.1%) | |
| PTEN/IHC | 64/94 (68.1%) | |
| PIK3CA | ||
| PIK3CA/NGS | 62/221 (28.1%) | |
| PIK3CA/NGS hot spot | 15/62 (24.2%) | |
| PIK3CA/CNA | 11/205 (5.4%) | |
| BRAF | BRAF/MEK inhibitors | |
| BRAF NGS | 2/217 (0.9%) | |
| HER2 | Targeted HER2 agents | |
| ERBB2 NGS | 4/219 (1.8%) | |
| HER2/IHC | 1/106 (0.9%) | |
| HER2/CISH | 1/78 (1.3%) | |
| EGFR | EGFR inhibitors | |
| EGFR/IHC | 43/44 (97.7%) | |
| EGFR/CNA | 3/208 (1.4%) | |
| FGFR | FGFR inhibitors | |
| FGFR3/NGS | 5/214 (2.3%) | |
| FGFR2/NGS | 1/215 (0.05%) |
DDR, DNA damage response; NGS, next-generation sequencing; IHC, immunohistochemistry; CNA, copy number alteration; EGFR, epidermal growth factor receptor; FGFR, Fibroblast growth factor receptor.
PI3KCA
As noted above, PIK3CA was the most common mutation in this collection of SCCA samples, with an incidence of 28.1% (62/221) by NGS, and 24.2% (15/62) by NGS hot spot. PI3K and AKT are located downstream of the EGFR receptor. Following the binding of EGFR to its receptor and subsequent activation of PI3K and AKT, the cell signals to survive and proliferate. Mutations in PIK3CA, a portion of the PI3K kinase, result in constitutive PI3K activity, subsequent activation of AKT, and downstream oncogenic activity (29,30). In one PDX mouse model of SCCA, there was no did response to treatment with a PI3K inhibitor, suggesting that the high PIK3CA mutation rate and abnormally high PI3K activity plays an important role in anal carcinogenesis (29,38). The high prevalence of PIK3CA mutations seen in this analysis invites further analysis of PI3K and mTOR inhibitors as potential therapeutic options.
EGFR
Overexpression of EGFR is believed to contribute to tumor development and unregulated cell proliferation and is seen in many epithelial cancers. Our IHC results showed that 97.7% of SCCA samples express EGFR (43/44), although the rate of EGFR mutations, by CNA, is low at 1.4% (3/208). No EGFR mutations were found in 219 NGS or 62 NGS hot spot samples. Similar combinations of high rates of EGFR expression with low mutational rates have been seen in smaller SCCA studies (39-42). Cetuximab is an IgG1 chimeric monoclonal antibody against EGFR that is utilized in other gastrointestinal malignancies when no downstream K-RAS/N-RAS/BRAF mutations are present. The use of cetuximab in patients with SCCA has been studied without success. The phase II ECOG 3,205 and AMC0 45 trials both evaluated the addition of cetuximab to standard frontline 5-FU plus cisplatin concurrent with radiation (43,44). Unfortunately, substantial patient toxicity was seen with added cetuximab; 26–32% of patients experienced grade 4 toxicities (compared to a historic rate of 20% in the RTOG 98-11 trial) (45). Similar studies utilizing cetuximab to treat SCCA also revealed significant toxicities (46-48). However, it is possible that cetuximab may be useful in the PIK3CA wild-type subgroup.
HER2
In the same tyrosine kinase receptor family as EGFR is HER2/ERBB2, ERBB3 and ERBB4. HER2 expression has been well described and documented in many malignancies. It is prevalent in breast cancer and gastric cancer and seen less commonly in salivary, vaginal, bladder, endometrial, cervical and colorectal cancers (49-56). HER2 has emerged as a successful therapeutic target transforming outcomes for patients with breast and gastric cancers. The MY PATHWAY basket trial studied the efficacy of pertuzumab and trastuzumab in HER2-positive tumors, and responses were seen in 9 different tumor types, giving further indication that HER2 is an actionable mutation across many malignancies (57). Our molecular analysis is the first to identify HER2 expression and ERBB2 mutations in SCCA. HER2 IHC was positive in 0.9%, amplification by CISH was seen 1.3%, and mutations in ERBB2 were present in 1.8% of tumors from our population. These findings could lead to novel therapeutic options for this patient population, and the adoption of widespread tumor molecular profiling would allow patients to participate in clinical trials of molecularly-matched therapies.
BRAF
BRAF is located downstream of EGFR. The BRAF protein is a serine/threonine protein kinase responsible for regulating the MAPK/ERK signaling pathway during cell proliferation, differentiation, growth and apoptosis (58). Mutations in the BRAF gene result in downstream activation of the MAPK pathway via phosphorylation of MEK and ERK. The most common type of BRAF mutation is the V600E missense mutation.
In this population of SCCA, the BRAF mutation rate was relatively low, with an overall incidence of 0.9% (n=2) by NGS. This is consistent with previous reports of SCCA mutational analyses, which found most SCCA to be BRAF wild-type on whole-exome sequencing (28,29).
FGFR
Fibroblast growth factor receptors (FGFRs) are a subset of tyrosine kinases unregulated in cancer cells leading to increased proliferation, angiogenesis and cell survival. FGFR inhibitors are approved in biliary and urothelial cancers with FGFR alterations and appear to have activity in other malignancies with FGFR alterations (59-61). Fusions for FGFR2 and FGFR3 were not identified in the 12 samples checked. FGFR2 mutations were identified in 0.05% of samples (n=215) by NGS samples. FGFR3 mutations had an incidence of 2.3% (n=214) by NGS. The incidence of FGFR mutations and fusions in SCCA is low, but warrants further investigation of efficacy with FGFR inhibitors as an innovative therapeutic strategy to overcome drug resistance in the disease.
Immunotherapy
PD-1 blockade with nivolumab or pembrolizumab has shown promising results in SCCA patients (13,62), and we highlight data on the prevalence of TMB, PD-1 and PD-L1 expression in a larger repository of SCCA tumors. PD-L1 expression was found to be high in SCCA, which is likely related to the immune response to HPV oncoproteins (62). A recently published study preformed PD-L1 staining on 62 SCCA tumors and found expression (CPS ≥1) in 32% of cases (19). Amongst those who were HPV-negative with PD-L1 expression, there was a trend toward shorter OS (9.8 vs. 40.6 months; P=0.064). The first trial to establish the therapeutic benefits of immunotherapy in SCCA was the phase II NCI9673 study. This trial investigated the efficacy of nivolumab (3 mg/kg every 2 weeks) in patients with unresectable/metastatic SCCA, who had progressed on prior chemotherapy. The results revealed an objective response rate of 24% and a disease control rate of 72%, including 2 complete and 7 partial responses (13). Thirty patients underwent pretreatment cell-free DNA analysis which revealed mutations in TP53 (n=8, 27%) and PIK3CA (n=7, 23%) (13). Median PFS was 4.1 months, and median OS was 11.5 months. Responders had a higher percentage of CD3+ and CD8+ T cells and higher expression of granzyme-B, PD-1 and PD-L1 on tumor cells (13). Another study was the multicohort phase 1B KEYNOTE-028 trial for PD-L1 positive (≥1%) tumors, in which patients were treated with pembrolizumab at a dose of 10 mg/kg IV once every 2 weeks. Study investigators treated 24 patients with advanced SCCA and observed an ORR of 17% (95% CI, 5.0–37%) and a disease control rate of 58%. The median PFS was 3.0 months (95% CI, 1.7–7.3 months), with 6- and 12-month PFS rates of 31.6% and 19.7%, respectively (62). Results from these trials led to preferential selection of immunotherapy in the second-line for patients with advanced SCCA (63).
Cancers with high TMB, including melanoma and colorectal cancers, respond well to immune checkpoint inhibition, indicating that mutational load correlates with response to checkpoint inhibitors (62). Data suggest only a moderate mutation rate in SCCA, which is accordant with other HPV-associated cancers (13,62). Hence SCCA response to immune checkpoint inhibition is more likely related to an inflammatory tumor microenvironment than to TMB (13). Further evaluation of the efficacy of immune checkpoint inhibitors is ongoing in several clinical trials, as outlined in Table 6.
Table 6. Review of ongoing clinical trials utilizing immunotherapy or targetable mutations.
| Clinical trial | Size (n) | Molecular target | Title | Status |
|---|---|---|---|---|
| NCT03233711 | 200 | PD-1; PD-L1 | A Randomized Phase III Study of Nivolumab After Combined Modality Therapy (CMT) in High Risk Anal Cancer | Recruiting April 17, 2020 |
| NCT02314169 | 137 | PD-1; CTLA-4 | A Multi-Institutional Phase II Study of Nivolumab or Nivolumab in Combination with Ipilimumab in Refractory Metastatic SCCA | Recruiting April 10, 2020 |
| NCT03074513 | 160 | PD-L1 | A Phase II, Single-Arm Open-Label Study of the Combination of Atezolizumab and Bevacizumab in Rare Solid Tumors | Recruiting June 13, 2019 |
| NCT02919969 | 32 | PD-L1; VEGF | A Multicenter Phase 2 Clinical Trial of Pembrolizumab in Refractory Metastatic Anal Center | Recruiting July 25, 2019 |
| NCT04287868 | 29 | HPV vaccine; PDL1/TGF-b IL-12 | Phase I/II Trial of Combination Immunotherapy in Subjects with Advanced HPV Associated Malignancies | Recuriting June 11, 2020 |
SCCA, squamous cell carcinoma of the anal canal; HPV, human papillomavirus.
Limitations
Limitations of this study include the lack of broad HPV-DNA and p16 testing to correlate with HPV infection in the setting of SCCA and lack of clinical treatment and outcomes data to correlate with mutational analysis. The first is due to the lack of such testing’s availability as well as lack of relevance in SCCA at the time of our population’s diagnosis. Other limitations include lack of each patient clinical or pathologic staging, ability to obtain germline DNA testing and lack of details regarding specimen histopathologic findings.
Conclusions
In conclusion, we report on the largest comprehensive molecular profiling study of patient SCCA, which provides insights into the pathogenesis of SCCA and identifies potential therapeutic targets.
Acknowledgments
We would like to thank Marion L. Hartley, PhD, for her valuable edits and suggestions during the composition of the manuscript.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). Informed consent was not required. These analyses were conducted using de-identified patient data obtained from Caris Life Sciences database.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at https://dx.doi.org/10.21037/jgo-20-610
Peer Review File: Available at https://dx.doi.org/10.21037/jgo-20-610
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/jgo-20-610). DA, JX, ZG are employed by Caris Life Sciences. The other authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7-30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2.Williams GR, Talbot IC. Anal carcinoma--a histological review. Histopathology 1994;25:507-16. 10.1111/j.1365-2559.1994.tb01370.x [DOI] [PubMed] [Google Scholar]
- 3.Silverberg MJ, Lau B, Justice AC, et al. Risk of anal cancer in HIV-infected and HIV-uninfected individuals in North America. Clin Infect Dis 2012;54:1026-34. 10.1093/cid/cir1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nigro ND, Vaitkevicius VK, Buroker T, et al. Combined therapy for cancer of the anal canal. Dis Colon Rectum 1981;24:73-5. 10.1007/BF02604287 [DOI] [PubMed] [Google Scholar]
- 5.Bartelink H, Roelofsen F, Eschwege F, et al. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J Clin Oncol 1997;15:2040-9. 10.1200/JCO.1997.15.5.2040 [DOI] [PubMed] [Google Scholar]
- 6.Faivre C, Rougier P, Ducreux M, et al. 5-fluorouracile and cisplatinum combination chemotherapy for metastatic squamous-cell anal cancer. Bull Cancer 1999;86:861-5. [PubMed] [Google Scholar]
- 7.Rao S, Sclafani F, Eng C, et al. International Rare Cancers Initiative Multicenter Randomized Phase II Trial of Cisplatin and Fluorouracil Versus Carboplatin and Paclitaxel in Advanced Anal Cancer: InterAAct. J Clin Oncol 2020;38:2510-8. 10.1200/JCO.19.03266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vuyst H, Clifford GM, Nascimento MC, et al. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int J Cancer 2009;124:1626-36. 10.1002/ijc.24116 [DOI] [PubMed] [Google Scholar]
- 9.Meulendijks D, Tomasoa NB, Dewit L, et al. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br J Cancer 2015;112:1358-66. 10.1038/bjc.2015.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arroyo Mühr LS, Lagheden C, Lei J, et al. Deep sequencing detects human papillomavirus (HPV) in cervical cancers negative for HPV by PCR. Br J Cancer 2020;123:1790-5. 10.1038/s41416-020-01111-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoots BE, Palefsky JM, Pimenta JM, et al. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer 2009;124:2375-83. 10.1002/ijc.24215 [DOI] [PubMed] [Google Scholar]
- 12.Gervaz P, Hirschel B, Morel P. Molecular biology of squamous cell carcinoma of the anus. Br J Surg 2006;93:531-8. 10.1002/bjs.5376 [DOI] [PubMed] [Google Scholar]
- 13.Morris VK, Salem ME, Nimeiri H, et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol 2017;18:446-53. 10.1016/S1470-2045(17)30104-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990;63:1129-36. 10.1016/0092-8674(90)90409-8 [DOI] [PubMed] [Google Scholar]
- 15.Bernardi MP, Ngan SY, Michael M, et al. Molecular biology of anal squamous cell carcinoma: implications for future research and clinical intervention. Lancet Oncol 2015;16:e611-21. 10.1016/S1470-2045(15)00292-2 [DOI] [PubMed] [Google Scholar]
- 16.Smaglo BG, Tesfaye A, Halfdanarson TR, et al. Comprehensive multiplatform biomarker analysis of 199 anal squamous cell carcinomas. Oncotarget 2015;6:43594-604. 10.18632/oncotarget.6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gujja S, Williamson SK, Batra A, et al. Programmed cell death-ligand 1 (PD-L1) expression and outcome in patients with squamous cell cancer of the anal canal (SCCAC). J Clin Oncol 2015;33 suppl:abstr 3523.
- 18.Williams EA, Montesion M, Sharaf R, et al. CYLD-mutant cylindroma-like basaloid carcinoma of the anus: a genetically and morphologically distinct class of HPV-related anal carcinoma. Mod Pathol 2020;33:2614-25. 10.1038/s41379-020-0584-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X, Jamshed S, Zou J, et al. Molecular and immunophenotypic characterization of anal squamous cell carcinoma reveals distinct clinicopathologic groups associated with HPV and TP53 mutation status. Mod Pathol 2021;34:1017-30. 10.1038/s41379-020-00729-y [DOI] [PubMed] [Google Scholar]
- 20.Chung JH, Sanford E, Johnson A, et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann Oncol 2016;27:1336-41. 10.1093/annonc/mdw152 [DOI] [PubMed] [Google Scholar]
- 21.Qiu P, Pang L, Arreaza G, et al. Data Interoperability of Whole Exome Sequencing (WES) Based Mutational Burden Estimates from Different Laboratories. Int J Mol Sci 2016;17:651. 10.3390/ijms17050651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509-20. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909-20. 10.1016/S0140-6736(16)00561-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stadler ZK, Battaglin F, Middha S, et al. Reliable Detection of Mismatch Repair Deficiency in Colorectal Cancers Using Mutational Load in Next-Generation Sequencing Panels. J Clin Oncol 2016;34:2141-7. 10.1200/JCO.2015.65.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cacheux W, Rouleau E, Briaux A, et al. Mutational analysis of anal cancers demonstrates frequent PIK3CA mutations associated with poor outcome after salvage abdominoperineal resection. Br J Cancer 2016;114:1387-94. 10.1038/bjc.2016.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin V, Zanellato E, Franzetti-Pellanda A, et al. EGFR, KRAS, BRAF, and PIK3CA characterization in squamous cell anal cancer. Histol Histopathol 2014;29:513-21. [DOI] [PubMed] [Google Scholar]
- 29.Morris V, Rao X, Pickering C, et al. Comprehensive Genomic Profiling of Metastatic Squamous Cell Carcinoma of the Anal Canal. Mol Cancer Res 2017;15:1542-50. 10.1158/1541-7786.MCR-17-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel H, Polanco-Echeverry G, Segditsas S, et al. Activation of AKT and nuclear accumulation of wild type TP53 and MDM2 in anal squamous cell carcinoma. Int J Cancer 2007;121:2668-73. 10.1002/ijc.23028 [DOI] [PubMed] [Google Scholar]
- 31.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 2013;3:761-9. 10.1158/2159-8290.CD-13-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu D, Gao X, Morgan MA, et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 2013;33:4745-54. 10.1128/MCB.01181-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider R, Bannister AJ, Myers FA, et al. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 2004;6:73-7. 10.1038/ncb1076 [DOI] [PubMed] [Google Scholar]
- 34.Allal AS, Waelchli L, Bründler MA. Prognostic value of apoptosis-regulating protein expression in anal squamous cell carcinoma. Clin Cancer Res 2003;9:6489-96. [PubMed] [Google Scholar]
- 35.Bonin SR, Pajak TF, Russell AH, et al. Overexpression of p53 protein and outcome of patients treated with chemoradiation for carcinoma of the anal canal: a report of randomized trial RTOG 87-04. Radiation Therapy Oncology Group. Cancer 1999;85:1226-33. [DOI] [PubMed] [Google Scholar]
- 36.Ogunbiyi OA, Scholefield JH, Smith JH, et al. Immunohistochemical analysis of p53 expression in anal squamous neoplasia. J Clin Pathol 1993;46:507-12. 10.1136/jcp.46.6.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong CS, Tsao MS, Sharma V, et al. Prognostic role of p53 protein expression in epidermoid carcinoma of the anal canal. Int J Radiat Oncol Biol Phys 1999;45:309-14. 10.1016/S0360-3016(99)00188-1 [DOI] [PubMed] [Google Scholar]
- 38.Shin S, Park HC, Kim MS, et al. Whole-exome sequencing identified mutational profiles of squamous cell carcinomas of anus. Hum Pathol 2018;80:1-10. 10.1016/j.humpath.2018.03.008 [DOI] [PubMed] [Google Scholar]
- 39.Alvarez G, Perry A, Tan BR, et al. Expression of epidermal growth factor receptor in squamous cell carcinomas of the anal canal is independent of gene amplification. Mod Pathol 2006;19:942-9. 10.1038/modpathol.3800608 [DOI] [PubMed] [Google Scholar]
- 40.Hui YZ, Noffsinger AE, Miller MA, et al. Strong Epidermal Growth Factor Receptor Expression But Not HER2/neu Expression Correlates with Cell Proliferation in Anal Canal Carcinomas. Int J Surg Pathol 1999;7:193-203. 10.1177/106689699900700402 [DOI] [Google Scholar]
- 41.Lê LH, Chetty R, Moore MJ. Epidermal growth factor receptor expression in anal canal carcinoma. Am J Clin Pathol 2005;124:20-3. 10.1309/X4UADHVN317V2XMW [DOI] [PubMed] [Google Scholar]
- 42.Paliga A, Onerheim R, Gologan A, et al. EGFR and K-ras gene mutation status in squamous cell anal carcinoma: a role for concurrent radiation and EGFR inhibitors? Br J Cancer 2012;107:1864-8. 10.1038/bjc.2012.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garg MK, Zhao F, Sparano JA, et al. Cetuximab Plus Chemoradiotherapy in Immunocompetent Patients With Anal Carcinoma: A Phase II Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group Trial (E3205). J Clin Oncol 2017;35:718-26. 10.1200/JCO.2016.69.1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sparano JA, Lee JY, Palefsky J, et al. Cetuximab Plus Chemoradiotherapy for HIV-Associated Anal Carcinoma: A Phase II AIDS Malignancy Consortium Trial. J Clin Oncol 2017;35:727-33. 10.1200/JCO.2016.69.1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ajani JA, Winter KA, Gunderson LL, et al. Fluorouracil, mitomycin, and radiotherapy vs fluorouracil, cisplatin, and radiotherapy for carcinoma of the anal canal: a randomized controlled trial. JAMA 2008;299:1914-21. 10.1001/jama.299.16.1914 [DOI] [PubMed] [Google Scholar]
- 46.Deutsch E, Lemanski C, Pignon JP, et al. Unexpected toxicity of cetuximab combined with conventional chemoradiotherapy in patients with locally advanced anal cancer: results of the UNICANCER ACCORD 16 phase II trial. Ann Oncol 2013;24:2834-8. 10.1093/annonc/mdt368 [DOI] [PubMed] [Google Scholar]
- 47.Levy A, Azria D, Pignon JP, et al. Low response rate after cetuximab combined with conventional chemoradiotherapy in patients with locally advanced anal cancer: long-term results of the UNICANCER ACCORD 16 phase II trial. Radiother Oncol 2015;114:415-6. 10.1016/j.radonc.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 48.Olivatto LO, Vieira FM, Pereira BV, et al. Phase 1 study of cetuximab in combination with 5-fluorouracil, cisplatin, and radiotherapy in patients with locally advanced anal canal carcinoma. Cancer 2013;119:2973-80. 10.1002/cncr.28045 [DOI] [PubMed] [Google Scholar]
- 49.Khasraw M, Bell R. Primary systemic therapy in HER2-amplified breast cancer: a clinical review. Expert Rev Anticancer Ther 2012;12:1005-13. 10.1586/era.12.62 [DOI] [PubMed] [Google Scholar]
- 50.Hechtman JF, Polydorides AD. HER2/neu gene amplification and protein overexpression in gastric and gastroesophageal junction adenocarcinoma: a review of histopathology, diagnostic testing, and clinical implications. Arch Pathol Lab Med 2012;136:691-7. 10.5858/arpa.2011-0168-RS [DOI] [PubMed] [Google Scholar]
- 51.Yan M, Parker BA, Schwab R, et al. HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev 2014;40:770-80. 10.1016/j.ctrv.2014.02.008 [DOI] [PubMed] [Google Scholar]
- 52.English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther 2013;17:85-99. 10.1007/s40291-013-0024-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diver EJ, Foster R, Rueda BR, et al. The Therapeutic Challenge of Targeting HER2 in Endometrial Cancer. Oncologist 2015;20:1058-68. 10.1634/theoncologist.2015-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valtorta E, Martino C, Sartore-Bianchi A, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol 2015;28:1481-91. 10.1038/modpathol.2015.98 [DOI] [PubMed] [Google Scholar]
- 55.Conradi LC, Styczen H, Sprenger T, et al. Frequency of HER-2 positivity in rectal cancer and prognosis. Am J Surg Pathol 2013;37:522-31. 10.1097/PAS.0b013e318272ff4d [DOI] [PubMed] [Google Scholar]
- 56.Salem ME, Weinberg BA, Xiu J, et al. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017;8:86356-68. 10.18632/oncotarget.21169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol 2018;36:536-42. 10.1200/JCO.2017.75.3780 [DOI] [PubMed] [Google Scholar]
- 58.Hussain MR, Baig M, Mohamoud HS, et al. BRAF gene: From human cancers to developmental syndromes. Saudi J Biol Sci 2015;22:359-73. 10.1016/j.sjbs.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med 2019;381:338-48. 10.1056/NEJMoa1817323 [DOI] [PubMed] [Google Scholar]
- 60.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671-84. 10.1016/S1470-2045(20)30109-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chae YK, Hong F, Vaklavas C, et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J Clin Oncol 2020;38:2407-17. 10.1200/JCO.19.02630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ott PA, Piha-Paul SA, Munster P, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with recurrent carcinoma of the anal canal. Ann Oncol 2017;28:1036-41. 10.1093/annonc/mdx029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.NCCN. NCCN Guidelines for Anal Cancer 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/anal.pdf