

Keywords: Alzheimer’s disease, hypertension, stroke, microcirculation, neurovascular coupling
Abstract
Brain function critically depends on a close matching between metabolic demands, appropriate delivery of oxygen and nutrients, and removal of cellular waste. This matching requires continuous regulation of cerebral blood flow (CBF), which can be categorized into four broad topics: 1) autoregulation, which describes the response of the cerebrovasculature to changes in perfusion pressure; 2) vascular reactivity to vasoactive stimuli [including carbon dioxide (CO2)]; 3) neurovascular coupling (NVC), i.e., the CBF response to local changes in neural activity (often standardized cognitive stimuli in humans); and 4) endothelium-dependent responses. This review focuses primarily on autoregulation and its clinical implications. To place autoregulation in a more precise context, and to better understand integrated approaches in the cerebral circulation, we also briefly address reactivity to CO2 and NVC. In addition to our focus on effects of perfusion pressure (or blood pressure), we describe the impact of select stimuli on regulation of CBF (i.e., arterial blood gases, cerebral metabolism, neural mechanisms, and specific vascular cells), the interrelationships between these stimuli, and implications for regulation of CBF at the level of large arteries and the microcirculation. We review clinical implications of autoregulation in aging, hypertension, stroke, mild cognitive impairment, anesthesia, and dementias. Finally, we discuss autoregulation in the context of common daily physiological challenges, including changes in posture (e.g., orthostatic hypotension, syncope) and physical activity.
CLINICAL HIGHLIGHTS
Lassen's classic autoregulation curve is often misinterpreted to mean that cerebral blood flow (CBF) is always kept constant. However, this is only true for relatively gradual changes in blood pressure (BP), for example, during the normal nocturnal fall in BP or reductions in BP following treatment of hypertension. For progressively faster changes in BP (minutes, seconds), CBF becomes incrementally more unstable and may show large fluctuations. Such rapid changes in BP may occur, for example, during surgery, critical illness, everyday postural changes, and some forms of exercise.
It is commonly thought that chronic hypertension and aging, which often occur together, cause impairment in autoregulation, such that brain blood vessels adapt and may even become dependent on higher BP to maintain perfusion. This misconception carries the risk of undertreatment of hypertension, especially in older patients. Increasing evidence suggests autoregulation is maintained in hypertension and aging, and treatment to lower BP is not associated with reductions in CBF.
Autoregulation, CO2 reactivity, and neurovascular coupling (NVC) are sometimes confused or used interchangeably. For example, a study that finds impairment in NVC may be reported as impaired autoregulation. However, impaired NVC or CO2 reactivity do not automatically translate into impairment in how CBF responds to changes in BP (i.e., autoregulation).
1. INTRODUCTION: REGULATION OF CEREBRAL BLOOD FLOW IN HUMANS
1.1. Historical Perspective
Some of the earliest knowledge regarding cerebrovascular anatomy, and specifically the circle of Willis, dates back to 1664 when Thomas Willis published his thesis “Cerebri Anatome” (1). In 1783, Alexander Monro (2), who also named the “foramen of Monro” in the brain’s ventricles, deduced the brain’s high demand for blood flow. He did so by taking the weight of the arm and the diameter of the subclavian artery, its source of blood supply, and comparing this with the weight of the brain and its blood supply, the combined diameters of the carotid and vertebral arteries (2). Monro, however, is perhaps best remembered for the Monro-Kellie doctrine (3), which states that because the brain is enclosed in the skull, there must be an equilibrium between its volumetric components (brain parenchyma, interstitial and cerebrospinal fluid, and arterial and venous blood volume). In the early 1900s, this doctrine led to the view that there was no regulation of cerebral blood flow (CBF). Rather, CBF, and changes in CBF, were restrained by this equilibrium. In other words, large increases in CBF were thought to be impossible because it would cause disequilibrium in intracranial pressure (ICP) (3), a view supported by the influential physiologists Hill (4) and Bayliss et al. (5). Their idea was that there was no active control of CBF, rather CBF passively followed changes in blood pressure (BP). If the brain demanded an increase in CBF, this was achieved by vasoconstriction in the rest of the body, increasing BP and thereby CBF (4).
In 1868, the physiologist F. C. Donders (6) may have been the first to hint at active regulation of CBF in response to cognitive activation (i.e., neuronal stimulation), a concept now referred to as neurovascular coupling (NVC) or functional hyperemia (see also sects. 1.3 and 3.1 for further information on NVC). Donders (6) wrote “[because]… hemorrhage, or reduced cardiac function, are associated with loss of consciousness [we conclude] that a regular supply of blood to the brain is a prerequisite for cognitive processes, indicating a central role for brain metabolism [in cognitive activation]…which consumes oxygen and produces carbon dioxide.” In the late 19th century, the physiologist Angelo Mosso (7) published the first work related to regulation of CBF in humans. Mosso recorded pulsations of the brain in a patient with skull defects following a neurosurgical procedure, in which Mosso reported immediate increases in pulsations whenever the subject was spoken to, or when the subject began to think actively (e.g., arithmetic). Mosso (8) also introduced the “human circulation balance,” which represents a measurement technique using a balanced table that tipped downwards if the weight on either end were increased. Using this balance, he found that the moment emotional or intellectual activity began in a subject, the balance would tip down at the head end, reflecting a redistribution of blood toward the brain.
An experiment by Roy and Sherrington in 1890 (9), wherein they injected a solution of homogenized brain intravenously in a dog, revealed an increase in brain volume (used as an index of increased CBF) without any increase in BP. This experiment formed the first evidence for the metabolic hypothesis of NVC (3). The interpretation was that this brain extract, from a dog that had died from hemorrhagic shock 4 h earlier, contained chemical substances that induced cerebral vasodilation. They hypothesized these substances had been located in the perivascular fluid. Of note, however, this experiment could not be replicated by Bayliss et al. (5).
These early experiments (by Donders, Mosso, Roy, and Sherrington) were largely ignored in their time, or seen as incorrect, because Hill (4) found contradictory results and was convinced there was no relationship between brain function and its circulation and therefore no active regulation of CBF (10).
While the study by Roy and Sherrington from 1890 (9) is often cited for the experiment (based on just one animal) that suggested the presence of NVC, their paper contains many other experiments and represents one of the more comprehensive publications on early integrative physiology of the cerebral circulation. In a series of studies using dogs, cats, and rabbits, they investigated effects of sensorimotor stimulation, vagal-sympathetic stimulation, brain stem stimulation, hypoxia, hemorrhagic shock, acid-base changes, and several drugs (9). They may also have been the first to indirectly describe autoregulation, as will be discussed in sect. 2.1. It is also interesting to read their description and recognition of brain lymphatics and perivascular spaces, topics for which there is now renewed interest (11, 12).
In 1895, Bayliss et al. (5) reported contrasting findings compared with the work from Roy and Sherrington (9). Bayliss and colleagues (5) examined the brain circulation by performing simultaneous recordings of arterial and venous pressure, intracranial pressure (ICP), and cerebral venous pressure with a design comparable to Roy and Sherrington. They concluded “there is no evidence supporting the existence of cerebral local vasomotor mechanisms”, and that “the cerebral circulation passively follows the changes in the general arterial and venous pressures” (5). Bayliss is well known for the “Bayliss effect,” which refers to the response of vascular muscle to changes in pressure (see sect. 3.4) based on experiments in the peripheral circulation but not the cerebral circulation. Indeed, combining experiments, Bayliss et al. (5) concluded that “there is, up to the present, no satisfactory evidence of the presence of a vasomotor supply to the brain; so that when an increased blood-supply is required in that organ it is provided by constriction of vessels throughout the rest of the body, which is, in this respect, so to speak the slave of the brain.” The Bayliss effect was in his opinion not present in the brain but only in the systemic circulation, most notably the splanchnic circulation.
Contrary to the belief of Hill and Bayliss, the concept that the cerebral circulation is responsive to neuronal activation and changes in BP, as earlier suggested by Donders and Mosso and (indirectly) Roy and Sherrington, was confirmed in subsequent decades (13–15) [see review by Rosenblum (16)]. This research may have been sparked by work from Fulton (17), who obtained evidence of NVC in the occipital cortex upon visual stimulation in a patient with a cortical defect, very similar to the work of Mosso (3, 7, 10).
Although the observations by Mosso (7) in humans are now 140 yr old, thorough insight into the integrative nature of the regulation of CBF in humans has proven difficult for a variety of reasons. For many years, the presence of the skull made the brain something of a “black box” as it prohibited direct observation of blood vessels, measurements of blood flow, or blood flow-mediated changes in volume or pulsatility, approaches that were feasible in the systemic circulation.
A milestone in this field was the nitrous oxide method developed by Seymour Kety in 1948 (18, 19). This method applies the Fick principle (conservation of mass) and measures changes in cerebral arterial and venous concentrations of nitrous oxide following its inhalation. The Kety-Schmidt method yields an estimate of CBF averaged over 10–15 min. An adaptation of this method was introduced by Lassen and Ingvar in 1961 (20), who used intra-arterial injection of radioactive Krypton-85 or Xenon-133. Using a camera, the intracerebral concentration of the tracer could be recorded to derive both global CBF and estimates of regional CBF. Other adaptations of the Kety-Schmidt method have been adopted since, for example, in position emission tomography (PET) and single-photon emission computed tomography (SPECT).
A second important development was the introduction of advanced techniques from the 1970s onwards that could be applied safely in humans, starting with computed tomography (CT), followed by PET and magnetic resonance imaging (MRI) (21, 22). These techniques have significantly contributed to improved understanding of the anatomy, physiology, and pathophysiology of the cerebrovascular system and currently play a key role in diagnosis and research focused on diseases that affect the cerebral circulation. Despite continuous technological advances that provided more detailed insight, these techniques are still limited by a poor temporal resolution, that would be needed to record rapid changes in CBF in response to physiological or pathophysiological stimuli (21). Although the introduction of transcranial Doppler (TCD) ultrasound in the 1980s (23, 24) helped resolve this limitation because of its very high temporal resolution, the method has its own limitations in that it measures blood flow velocity, without knowledge of vessel diameter (or cross-sectional area). Therefore, any changes in vessel diameter during interventions and measurements affect estimates of CBF using TCD.
1.2. Relevance of CBF for Brain Function and Consequences of Hypoperfusion
The vertebrate brain is a unique organ. In the human, the brain represents only 2–3% of total body mass while requiring ∼15% of cardiac output and consuming ∼20% of the available O2 under normal conditions. The high metabolic rate of the brain, combined with limited energy stores, highlights the importance of CBF for nutrient and O2 delivery but also for removal of cellular, metabolic, or toxic by-products. The importance of controlling CBF is highlighted by the acute consequences that occur following substantial reductions in CBF, a change that can rapidly lead to unconsciousness, life-threatening complications and brain damage if sustained (25, 26). In a clinical context, acute reductions in global CBF can occur with cardiac arrest (26) or with a sudden profound reduction in BP, which can lead to global reductions in CBF, termed syncope (for a recent review and overview of guidelines on syncope, see Ref. 27 and sect. 6.1.2). Focal reductions in CBF occur in stroke, where there is an acute vessel occlusion leading to hypoperfusion and ischemia. Examples are the classic clinical stroke of the middle cerebral artery or downstream microinfarcts in the case of small vessel disease or migrating microthrombi. Experiments in nonhuman primates and other preclinical models have shown that a reduction in CBF to below ∼50% of normal induces an immediate loss of neuronal function (25, 26). Normal global CBF in awake humans is ∼50 mL/100 g/min, and loss of function is observed below ∼22 mL/100 g/min (25). This loss of function is responsible for the clinical symptoms of hypoperfusion, which include slowing of mental processing. These symptoms are followed by loss of consciousness, often within 10 s after the onset of hypoperfusion or ischemia (26). With rapid recovery of CBF, function can be restored without permanent cellular injury. In contrast, persistent lowering in CBF causes irreversible damage. There are no exact thresholds beyond which ischemic injury occurs, but estimates are that a sustained fall to 20–30% of baseline CBF causes ischemia within minutes (26, 28, 29). This estimation is obviously an oversimplification, because effects of ischemia vary between different populations of brain cells, between the core and penumbra in the case of stroke, and because apart from these immediate effects of hypoperfusion on cell function, a cascade of responses are triggered that lead to further damage and delayed ischemia, hours to days later (26). A detailed description of such changes is beyond the scope for this review, we refer to the extensive review by Lipton (26).
Other than acute effects of ischemia, an increasing literature describes the clinical relevance of chronic, but more modest, reductions in CBF that potentially contribute to loss of brain health in neurodegenerative diseases such as Alzheimer’s disease, vascular dementia, or mixed dementia (see sect. 5). Here, hypertension is suggested to play a central role through its effects on CBF. Hypertension is a key risk factor for overall disease burden and health loss worldwide (30, 31). Hypertension is associated with chronic mild cerebral hypoperfusion, which may explain why hypertension is a risk factor for Alzheimer’s disease. Recent research (see sect. 5.2) suggests that BP-lowering treatment in hypertension may restore CBF to normal, which may explain why antihypertensive treatment can reduce the risk of Alzheimer’s disease. This clinical example illustrates the complexity and implications of regulation of cerebral perfusion. Both hypertension itself and medications used to treat hypertension can affect CBF, most likely via effects on vascular function, vascular structure, or vascular mechanics (32), as well as effects on the autonomic nervous system. In addition, hypertension increases the risk of orthostatic hypotension, which poses an additional challenge to autoregulation (see sects. 6.1.1 and 6.1.2). This example, focusing on hypertension, highlights the importance of understanding the integrative nature of CBF regulation and how alterations in BP may contribute to the development and progression of a series of clinical conditions that share a final common pathway involving CBF (FIGURE 1).
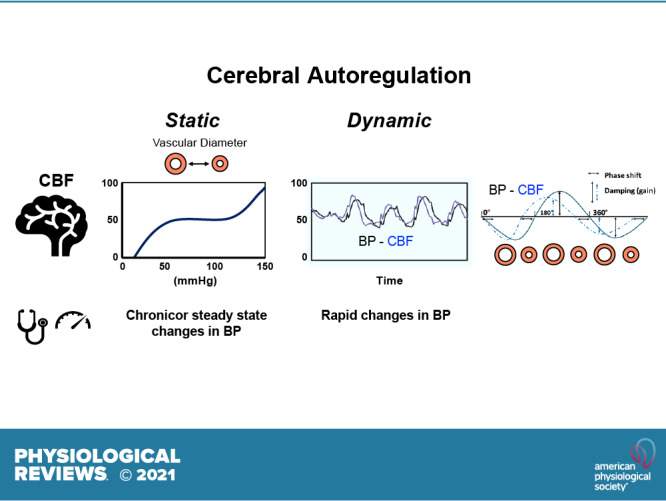
FIGURE 1.
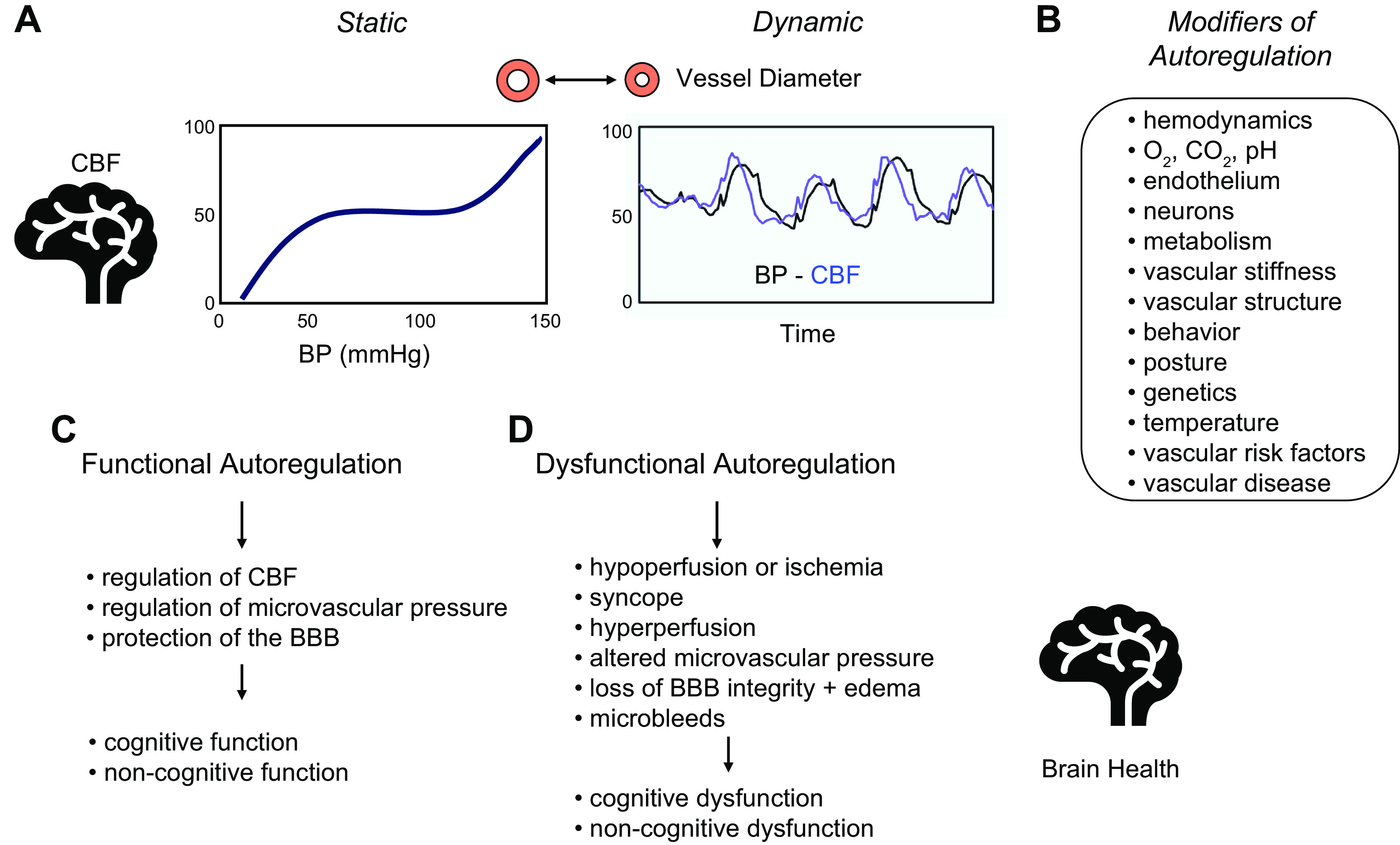
Overview of cerebral autoregulation. General overview of autoregulation and key summary points for this review. A: 2 graphs illustrating the concepts of static and dynamic autoregulation. B: summary of modifiers or factors that have a direct influence on cerebral blood flow (CBF) or on the relationship between blood pressure (BP) and CBF (i.e., autoregulation). Examples include hemodynamics (including the rate of rise or temporal profile of changes in BP) and behavior (sedentary versus exercise). C: summary of the impact of normally functioning autoregulation. D: summary of consequences of impairment in autoregulation in relation to brain health. See text for further details. BBB, blood brain barrier.
1.3. Integrative Approach to Regulation of CBF
Control of CBF involves a spectrum of overlapping regulatory mechanisms that collectively facilitate optimal O2 and nutrient delivery to individual brain cells (21, 33). Mechanisms that influence CBF include arterial blood gases [partial pressure of arterial CO2 () and arterial ()] tissue levels of pco2 and Po2 (34, 35), central hemodynamics (BP, including effects of hydrostatic pressure gradients), cerebral metabolism, and neural mechanisms including extrinsic autonomic and sensory nerves, and intrinsic neurons in close association with the vasculature within the brain parenchyma (36). In addition to their individual impact, there are strong interactions between these regulators. Some of these mechanisms are unique to the brain, explaining the generalized observation that the peripheral vasculature represents a poor model or predictor for the cerebral vasculature in health or disease (37).
An important characteristic of CBF regulation is mechanistic redundancy, i.e., overlapping mechanisms contribute to maintaining CBF under highly challenging conditions. Studies exploring the regulation of CBF are importantly impacted by this, because the overlap in pathways makes it difficult to explore the relative importance of individual pathways or identify key contributors. From a teleological perspective, this redundancy makes the regulation of CBF a robust system where multiple strategies are present to ensure precise control and thus protect against potential brain damage.
The regulation of CBF can be affected by certain processes in the rest of the body, because they can influence the sensitivity of the various systems that affect CBF. For example, and are determined primarily by pulmonary gas exchange, local lung perfusion, and body posture, yet they have a major influence on cerebrovascular resistance (34, 35) (FIGURE 1).
Using a simplified approach, one can divide mechanisms that regulate CBF into four distinct components or adaptive responses: autoregulation, chemoregulation, neuronal regulation, and endothelium-dependent regulation. The first mechanism, termed autoregulation, relates to the response of the cerebral circulation to changes in cerebral perfusion pressure (21, 33, 38–42). Cerebral perfusion pressure is determined by BP, ICP, and venous pressure. Under physiological conditions, cerebral perfusion pressure is mainly determined by arterial BP and body posture (supine or prone vs. seated or standing in humans), because venous BP and ICP are relatively low and normally exhibit only small changes. Some key exceptions are traumatic brain injury or intracranial hemorrhage (ICH), where ICP can be substantially elevated and affects perfusion pressure. Cerebral autoregulation (henceforward referred to simply as “autoregulation”) will be the focus of this review and will be discussed in detail in sects. 1, 5, and 6.
The second mechanism is chemoregulation, another class of vascular reactivity. This includes vascular responses to changes in CO2 (and subsequently pH), Po2, or O2 content. Changes in these variables (e.g., hypocapnia and hypercapnia) elicit strong CBF responses (34, 35, 43). Of these mechanisms, reactivity to CO2 has been the most widely studied in controlled laboratory-based setting in humans and has also been termed cerebral vasomotor reactivity to CO2 (35).
The third mechanism consists of the influence of nerves or neurons on CBF. This includes NVC, the local CBF response to nearby intrinsic neuronal activation within the brain parenchyma (44, 45), but also includes effects of autonomic and sensory nerves (extrinsic innervation) on the extraparenchymal segments of the cerebral vasculature (36).
A fourth mechanism relates to the effects of vascular endothelial cells on vascular tone and therefore CBF. In response to hemodynamic stimuli (e.g., shear stress), ions, neurotransmitters, metabolic stimuli, and therapeutic agents, among other factors (32, 46–49). This ability of endothelial cells to cause vasorelaxation importantly contributes to NVC. Studies using selective injury of endothelium or cell-specific genetic manipulation have shown that this cell plays an essential role in propagated (or ascending) vasodilation during NVC, eventually reaching arterioles and arteries on the pial surface (49, 50). Other studies that examined genetic alterations of signaling molecules in endothelial cells but did not focus on propagated vasodilation provide further direct evidence that endothelial cells play a major role in the vascular component of NVC in pial and parenchymal arterioles (51, 52). While still supporting a key role for endothelium, one study (52) provided some evidence against a role for endothelial cells in propagated vasodilation (from capillaries to small parenchymal arterioles) in that vasodilation in response to whisker stimulation in mice occurred in precapillary arterioles before changes in diameter of capillaries. Endothelial dysfunction, a feature of many forms of large and small vessel disease, can therefore have multiple consequences, not only on CBF, but also on other endpoints related to brain health.
Unlike myogenic responses (discussed below), in which a single cell type mediates most (or all) of the response to changes in transmural pressure, endothelium-dependent responses cannot occur without endothelial cells exerting effects on vascular muscle or other cellular targets via molecular or electrical signaling. Furthermore, endothelium-dependent effects on platelets, immune cells, neurons, and glia, each involve target cells, highlighting that this signaling is not based on a single cell response (45).
As a final short note to illustrate the complexity of endothelial function in the brain, endothelial cells normally also inhibit processing of β-amyloid and phosphorylation of tau: both major contributors to Alzheimer’s disease (48, 53).
Because autoregulation, i.e., the focus of this review, does not operate in isolation, we have attempted to present an integrative approach, briefly discussing other interacting mechanisms. Studies of autoregulation need to consider overall integrated responses. For example, many studies of autoregulation only consider effects of BP on CBF. However, other parameters affecting CBF when measurements are made cause “physiological noise” in the BP-CBF relationship (54). For this reason, studies of autoregulation need to control variables such as the amount of exercise and caffeine intake before testing, room temperature, cognitive activation, or mental stress during the experiment and measure or end-tidal CO2 during experiments to ascertain if this parameter remained stable or to take changes in CO2 into consideration when interpreting results (55).
Another reason for this integrative approach is that mechanisms can be confused in the literature. For example, some studies use the term autoregulation when describing vasomotor reactivity to CO2. Others use “pressure reactivity” to describe autoregulation, which can be confused with CO2 reactivity.
1.4. Other Roles for the Cerebral Circulation outside the Scope of This Review
The focus of this review is on the role of the cerebral circulation in delivering blood flow to the brain, with the main purpose of delivering O2 and removing CO2. Other functions of the cerebral circulation receive little attention in this review. For example, the blood-brain barrier (BBB) plays an essential role in controlling or limiting the movement of ions, molecules, amino acids, nutrients, cells, and so forth, into and out of the brain parenchyma (56–58). Damage or dysfunction of the BBB can be caused by aging, abnormal hemodynamics (i.e., acute hypertension), and various diseases (e.g., stroke, chronic hypertension, dementias, traumatic brain injury, multiple sclerosis, neuroinflammation, and so forth) (57, 58). Such damage or dysfunction can in turn have multiple consequences, such as impaired transport of essential substances into the brain, reduced removal of waste products or toxins from the brain, and the entrance of molecules or cells (e.g., toxins, infectious agents, drugs, proteins, and immune cells) that are neurotoxic and/or cause cellular damage. This topic is outside the scope of this review, so we refer to other recent reviews for further reading (57, 58).
Another proposed function of the cerebral circulation that has reemerged recently and has some overlap with BBB function relates to clearance of waste and toxins from the brain (45, 59). Three mechanisms, or pathways, have been proposed to play key roles. It should be noted that there is still controversy regarding the physiological and pathophysiological role of these pathways (60, 61).
One mechanism is the transvascular pathway, where toxins are actively transported across the BBB, from the brain and into the blood. An example is the transportation of amyloid-β from the perivascular space into the blood vessel by low-density receptor-related protein (LRP1). Amyloid-β and other waste/toxins reach the perivascular space by diffusion from the brain parenchyma (45, 59). The second mechanism is the perivascular pathway. Here, toxins and waste that have diffused into the perivascular spaces that surround penetrating arterioles and are transported alongside these same arterioles toward arteries on the surface of the brain, followed by drainage into cerebral lymphatics (located in the dura mater and meninges) and eventually into cervical lymph nodes (45, 59). Pulsations in blood flow, influenced by the dynamics of vascular distention, or spontaneous vasomotion, may have a role as a pump that drives this perivascular flow of brain interstitial fluid (ISF) (11, 62, 63). The third proposed mechanism is paravascular transportation. This concept has recently been termed the “glymphatic system” (11). Here, cerebral spinal fluid (CSF) is hypothesized to travel into the brain parenchyma, alongside penetrating arteries and arterioles (therefore also in the perivascular space, but in opposite direction from the perivascular pathway), where it would clear the ISF and then exit alongside venous perivascular spaces. This paravascular pathway is thought to rely on aquaporin-4 channels in astrocytic end-feet in the perivascular spaces, to transport the CSF into the ISF (45, 59). Changes in hemodynamics, structure, or mechanics of the cerebral circulation may affect these three pathways in several ways. For example, changes in the pulsatile dynamics, which occurs in hypertension with increased vascular stiffness, may affect perivascular transportation. A feature of small vessel disease are changes in the microvascular perivascular spaces, associated with hypertension and expansion of perivascular spaces (Virchow-Robin spaces), which may also contribute to impaired ISF dynamics (12). Using an animal model, others have suggested recently that spontaneous slow oscillations (vasomotion) in arteriolar diameter may drive clearance or drainage of solutes from the brain parenchyma (64). This clearance was enhanced when vasomotion was increased by visual activation of the occipital cortex and was reduced in the context of vessel wall dysfunction (e.g., cerebral amyloid angiopathy).
2. AUTOREGULATION: THE STATIC AND DYNAMIC RESPONSES OF CBF TO CHANGES IN BP
2.1. Historical Perspective
Lassen’s publication in 1959 (38) in this journal can be marked as the “birth” of autoregulation research in humans, although Roy and Sherrington (9) provided a description of what we now call autoregulation in 1890. Roy and Sherrington were unable to directly measure CBF, but estimated changes in CBF from changes in brain volume or increases in cerebral venous pressure. They noted that changes in CBF could be smaller than changes in BP or show an earlier return toward baseline (9). Others such as Fog (13) provided early data that confirmed and extended the concept of autoregulation. Using a cranial window approach in anesthetized cats, Fog (13) described rapid changes in the diameter of pial arteries and arterioles in response to experimental manipulation in arterial BP.
Lassen’s paper (38) introduced a more integrative approach of linking central (systemic) hemodynamics and regulation of CBF. Lassen presented a now very well-known figure that plotted CBF against BP for 11 groups of patients with varying levels of BP. That figure revealed a plateau in CBF across a relatively large range of BP (mean arterial BP between ∼50 and 150 mmHg) and was the first description of effective maintenance of CBF over a considerable range of BP, a concept that Lassen termed autoregulation, to suggest the dominance of local mechanisms. Lassen’s conclusions regarding autoregulation, in combination with many subsequent studies in the following decades, evolved into a widely accepted concept in vascular biology and medicine regarding regulation of CBF in humans and in preclinical models. His work continues to be cited frequently in the literature.
2.2. Critical Appraisal of Lassen’s Autoregulation Curve
Lassen’s original curve was an extrapolation, based on a cross-sectional comparison of 376 individuals from 7 publications. It featured a straight line drawn across average CBF data from 9 of the 11 groups, which varied around a mean CBF of 55 mL/100g/min, for mean arterial BP between 50 and 175 mmHg. The graph also featured a downward line that connected two data points well below and to the left of the horizontal line, from two studies with mean CBF of 30 mL/100 g/min and mean BP values of 40 mmHg. Lassen’s interpretation of these latter two data points was that they marked the “lower limit of autoregulation.” There were no data at higher BP values, and therefore no “upper limit of autoregulation”; that concept was added later. Questions have been raised about the validity of some of the data points used to fit Lassen’s curve (65). For example, one of the seven publications could not be traced and verified, and one of the data points had been plotted in error. A corrected plot would no longer feature a straight line but a line with a slight upward slope (65). Furthermore, it is important to realize that the original curve contained no within-subject data on changes in BP with repeated measurements of CBF. The curve consisted entirely of group averaged CBF values for a set of patients with a specific BP level, linked to specific medical conditions. Lassen’s curve nonetheless was commonly interpreted as reflecting what would happen to CBF if, in a group of people or within a single individual, BP was reduced or increased across the wide range depicted in the figure.
More direct evidence to support this interpretation of the autoregulatory curve came after this original publication, consisting mainly of findings from preclinical models. A series of generally well-controlled experiments in nonhuman primates and common laboratory species confirmed the substantial autoregulatory capacity of the cerebral circulation, where despite large changes in BP, CBF is held relatively stable (66–80). FIGURE 2 provides examples of such experiments (67, 72, 73, 75, 77, 79, 81–83). All these studies reveal a range of BP in which CBF was relatively stable. In some studies, there is a portion of the curve where a slope is not apparent. In others, there is a small positive slope: an increase in CBF as BP is increased. Upper and lower limits of autoregulation are suggested or can be seen in some of these data (FIGURE 2).
FIGURE 2.
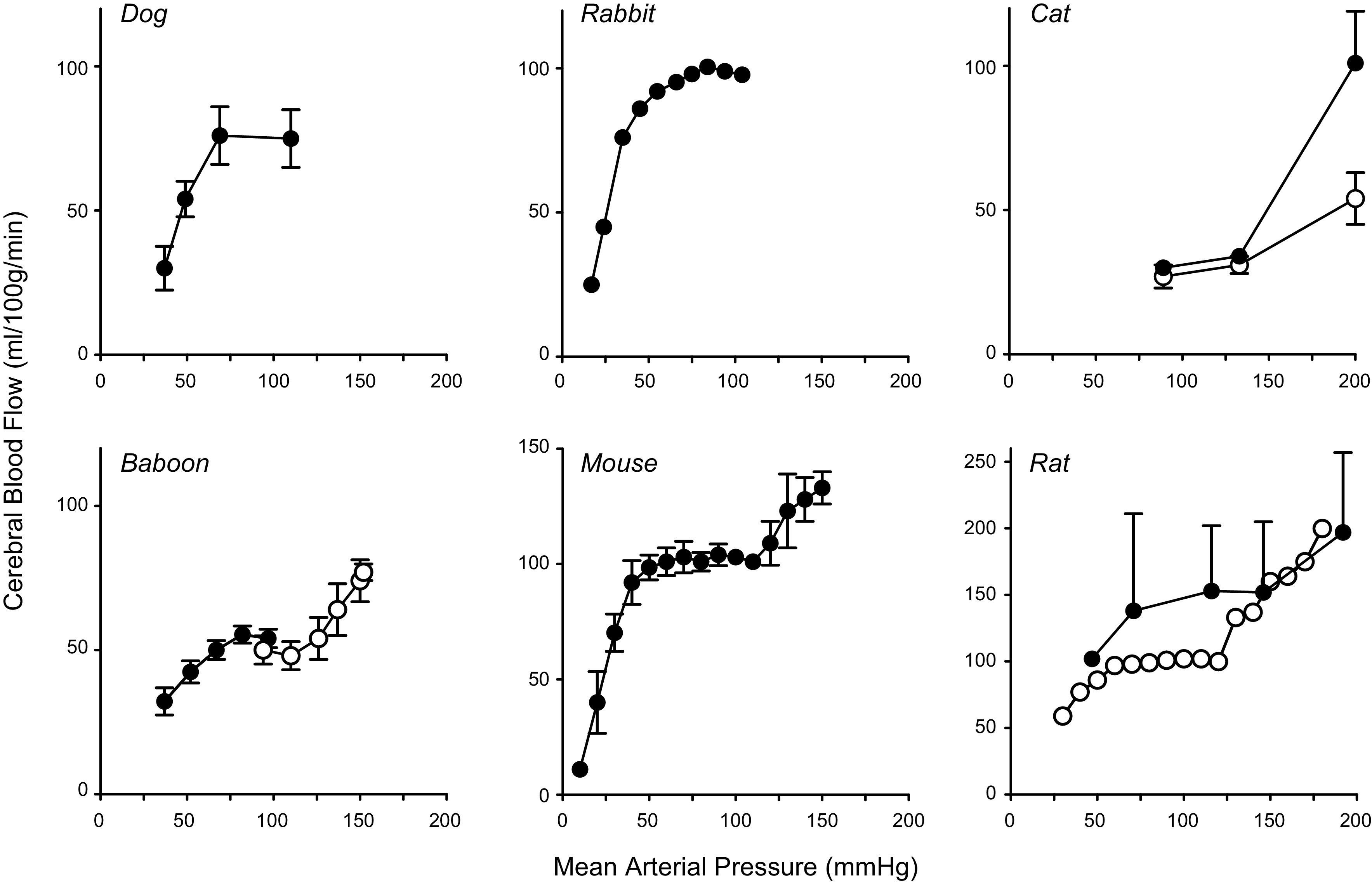
Static autoregulation: summary of animal models. Data for a range of experimental models, replotted from the original publications, illustrate the relationship between cerebral blood flow (CBF) and mean arterial pressure (MAP). Many studies either examined the low end or the high end of the autoregulatory curve, but a few quantified both ends of the curve within a single publication. For all species, there was a substantial range of MAP where CBF was relatively stable (72,73, 75, 77, 79, 81–84). For clarity, we have not plotted all studies of this type, but for reference cite some additional examples (66, 78, 80). For the panels with data from dogs, cats, baboons, and rats (filled circles), CBF is expressed in ml/min/100 g. For panels with data from rabbits, mice, and rats (open circles), CBF is expressed as percent change with the control value set at 100.
In conclusion, if one combines animal and human experimental data (discussed below), the overall concept of Lassen’s autoregulatory curve can be maintained, albeit with some important caveats and modifications. The existence of an autoregulatory range of BP, where autoregulation is highly effective and where CBF is relatively stable, is undisputed. We suggest it should no longer be called a “plateau phase,” because this implies a horizontal line with zero slope. In reality, the relationship between BP and CBF within this autoregulatory range can vary between an upward or even a downward slope (85–87). A better description than “plateau” therefore might be the term “gradient.” We suggest a more precise description for this “gradient phase of autoregulation” would be that there is a range of BP where autoregulation minimizes variations in CBF when BP is altered. How wide this range may be cannot be precisely defined from the available studies, as a large number only looked at one end of the autoregulatory curve or a limited range of change in BP. Studies that would be needed to obtain such data may be impossible to perform in humans, for both physiological and ethical reasons. Even if such data did exist, it is unlikely that a group-averaged range would apply to all individuals. What we do know, however, is that in most studies relatively slow reductions or increases in mean arterial BP between approximately −20 and +20 mmHg are associated with a stable CBF in humans (for details and references, see sect. 5.2.2 on hypertension and static autoregulation).
2.3. The Importance of Time: Autoregulation Is Strongly Affected by the Rate at Which BP Changes
One of the reasons why it is difficult to study a wide range of BP changes in humans is the efficacy of the baroreflex. The baroreflex normally helps to maintain BP within a narrow range during normal daily activities and under pathophysiological conditions (88). As such, this neural reflex will limit or at least dampen large fluctuations in BP (88). Commonly, this only allows for the assessment of relatively small changes in BP that can be maintained for a longer period. An example is lowering or increasing BP over weeks by starting or stopping antihypertensive medication. Larger, but acute, changes in BP are difficult to achieve and can only be maintained for shorter periods, usually only during an experimental setting. Examples are administration of intravenous drugs to acutely lower or increase BP (86, 89). Although pharmacological manipulations are a common experimental strategy to alter BP, it is important to realize that depending on the agent used and whether it activates endothelial cell receptors or crosses the BBB to reach vascular muscle, pharmacological agents sometimes have direct effects on the cerebral vasculature, independent of changes in BP.
Numan et al. (87) recently reanalyzed 40 studies in healthy humans that examined the relation between BP and CBF above and below resting mean BP. Largely independent of the technique used to assess CBF (i.e., TCD, MRI, PET, or Xenon-133 technique), these studies confirmed that the cerebrovasculature has autoregulatory capacity but not that CBF was completely stable over an autoregulatory range. In fact, they reported a relatively small range of mean BP that is associated with a stable CBF within subjects. Moreover, the efficacy to regulate CBF seemed to differ based on the direction of change in BP, with a more effective capacity to stabilize CBF during acute (transient) periods of hypertension, compared with hypotension. However, the studies summarized in that review used a variety of techniques to increase and decrease BP, leading to potential confounders, such as effects of medications, autonomic activation, and differences in . Many studies applied BP changes over relatively short periods of time (i.e., minutes), as in the study by Liu et al. (86). This affects the shape of the classic autoregulation curve, because these faster changes in BP lead to a smaller range of BP wherein CBF is kept relatively stable (the “gradient phase” noted above) and to a steeper gradient within that range. The importance of time in autoregulatory mechanisms is captured in the terms static and dynamic autoregulation (90). Understanding these two concepts, and the difference between them, is important for a proper understanding of autoregulation.
2.4. Static and Dynamic Autoregulation
2.4.1. Static autoregulation.
The classic relationship between mean arterial BP and CBF, derived from Lassen’s publication (38), should now be referred to as “static” autoregulation (40, 41, 90–94). This does not relate to physiological characteristics per se but rather refers to experimental characteristics that only measure the steady-state relationship between CBF and BP. The term static refers to the concept that BP and CBF are measured under conditions where individual variables (BP, CBF) have reached a steady state (40, 95). BP may have been experimentally increased or decreased, but it has reached a new stable, steady-state level (40–42). BP and CBF are measured over longer time intervals (10 min or more), and the resulting values represent the average BP and CBF during that period. This approach was a consequence of the fact that historically, measurements of CBF in humans required at least 10 min to perform. Due to this averaging, both BP and CBF will not display their short-term variability. With the use of this definition of static autoregulation, especially when applied to between-subject changes in BP and CBF, CBF is held relatively stable between various levels of BP under normal physiological conditions. This interpretation is a close approximation of Lassen’s data presentation and conclusions (see also FIGURE 2).
2.4.2. Dynamic autoregulation.
The concept of dynamic autoregulation only emerged once techniques with high temporal resolution became available to measure faster changes in CBF (40–42). Now, instead of values that took 10 min or more to obtain, these techniques allowed for measurements of changes in BP and CBF (or CBF velocity) occurring over seconds (96–98). It became clear that during more rapid changes in BP, there was much greater variability in CBF than was hitherto assumed based on Lassen’s concept of static autoregulation (FIGURES 3 and 4). The ability of autoregulation to respond to rapid changes in BP across seconds or minutes (e.g., during postural changes, coughing, or physical activity) is referred to as dynamic autoregulation (40, 41, 90–94) (FIGURES 3, 4, and 5).
FIGURE 3.
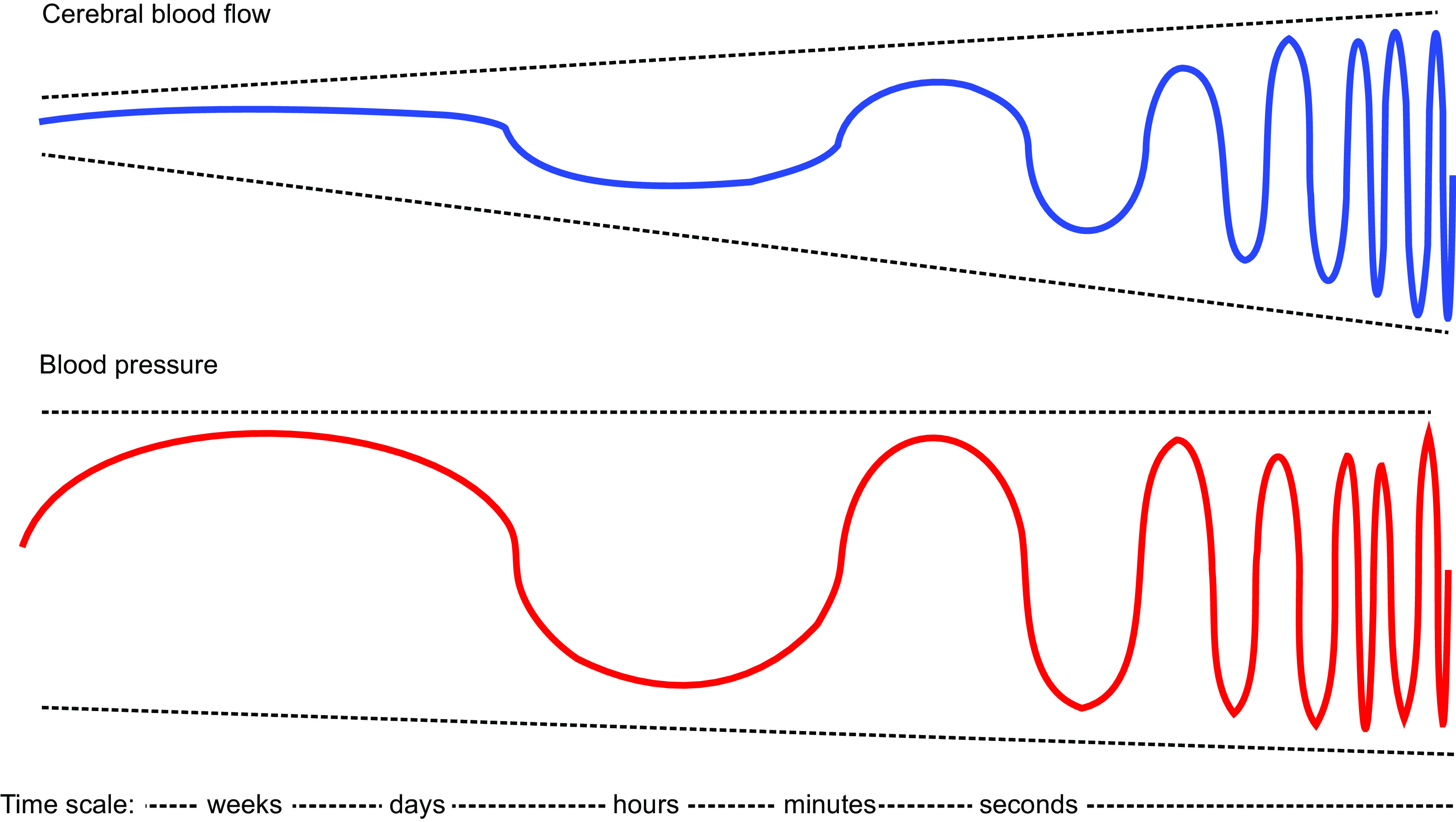
Effect of the time scale of blood pressure (BP) changes on autoregulation of cerebral blood flow (CBF). Schematic representation explaining how the time period in which changes in BP occur affects autoregulation of CBF. Slower changes (e.g., weeks, such as with chronic hypertension or treatment of hypertension) have minimal effects on CBF, whereas with more rapid changes the effects on CBF can increase such that there is essentially no effective autoregulation with very fast BP changes occurring within seconds. This presentation represents a hypothetical model of the transition from static autoregulation (slow changes in BP) to dynamic autoregulation (faster changes in BP).
FIGURE 4.

Oscillations in blood pressure (BP) and cerebral blood flow (CBF) induced by repeated sit to stand maneuvers. An example of repeated sit-to-stand maneuvers to induce strong oscillations in BP and CBF from a patient in a study on autoregulation in Alzheimer’s disease (99). Starting with a baseline recording while seated, the patient was asked to stand up for 10 s, then to sit down for 10 s, followed by standing up for 10 s, and so on [using methods proposed previously (100)]. With a cycle duration of 20 s, large oscillations in BP and CBF (blood velocity in the left and right middle cerebral artery) are induced at 0.05 Hz, a frequency where dynamic autoregulation is active (see text for details). Changes in systolic BP, which had a seated baseline value of ≈150 mmHg, oscillated between ≈110 and ≈190 mmHg and CBF velocity, which had a seated baseline value of ≈50 cm/s, oscillated between 40 and 70 cm/s. SBP, systolic blood pressure recorded using Finapres; CBFV, blood velocity in the left (L) and right (R) middle cerebral arteries, measured using transcranial Doppler.
FIGURE 5.

With the use of a zoomed-in section of the data presented in FIGURE 4, this graph illustrates the relationship between changes in blood pressure (BP; black line) and cerebral blood flow (CBF) (blue line) induced by the repeated sit-to-stand maneuvers at 0.05 Hz, see details in the legend for FIGURE 4. Bottom: the timing of these sit-stand maneuvers is schematically illustrated in the graph. Note that the BP and CBF graphs are not fully synchronous; this is an effect of autoregulation. The leftward shift of CBF [here, represented by the left middle cerebral artery blood velocity (CBFVL) signal recorded with transcranial Doppler] compared with BP [systolic BP (SBP) measured using Finapres] is referred to as phase shift. Phase shift is one of the parameters that follows from transfer function analysis. Top: inserted graph is a schematic explanation of phase shift. If we consider the repeated changes in BP and CBF as an oscillatory signal, resembling a sinusoid with a period of 360°, the leftward shift of CBF can be quantified in degrees (or converted to radians), in this example ∼40–50°, which in this frequency (0.05 Hz, the very-low-frequency range) indicates normal dynamic autoregulation. The parameter gain is also illustrated, representing damping, where effective dynamic autoregulation will result in relatively smaller changes in CBF compared with BP. Because BP and CBF have different physical units, gain is not necessarily below 1.
TCD to measure CBF velocity (typically using the middle cerebral artery), with simultaneous beat-to-beat recordings of BP, has become a popular technique to measure dynamic autoregulation (24). Aaslid and coworkers (24, 101, 102) examined the temporal relationships between BP and CBF in response to deflation of BP cuffs placed around the thigh to induce rapid, transient hypotension . The resulting drop in BP was accompanied by a proportional decrease in middle cerebral artery blood flow velocity, which was assumed to provide an accurate index for changes in CBF. In healthy individuals, CBF recovered more quickly than BP. These observations highlighted in humans that autoregulatory mechanisms were unable to maintain CBF perfectly stable during rapid changes in BP. Instead, dynamic autoregulation results in a relative, time-dependent buffering of changes in CBF.
This idea of a relative CBF buffering capacity for rapid changes in BP was later expanded by Birch et al. (103). They identified that the capacity to buffer changes in CBF was strongly dependent on the speed of BP changes (103). The slower the change in BP, the smaller the impact on CBF, to a point where CBF becomes almost unaffected. However, for more rapid changes in BP, the buffering capacity is progressively reduced and changes in CBF become larger, until a point where changes in CBF become as large as the change in BP. At that point, CBF passively follows BP. This concept is illustrated in FIGURE 3, where oscillations (a change occurring at a specific frequency) are transmitted or blocked depending on the frequency, and is comparable to a high-pass filter that transmits high frequencies but blocks low frequencies (104, 105).
This relationship between CBF and BP is analogous to the application in acoustics, where a high-pass filter transmits high frequency signals from an audio source to a tweeter. This led Giller (106) to apply transfer function analysis (TFA) to dynamic autoregulation, a mathematical concept based on this example of acoustic signals. TFA quantifies how different frequencies and amplitudes in BP are transmitted (transferred) to CBF. Specifically, Giller (106) applied the concept of coherence [i.e., a measure of the strength of the relationship between oscillations (frequencies) in BP and CBF]. As a simplified explanation, coherence can be compared with r in a linear regression. A coherence of 0 means no relationship between oscillations in BP and CBF. A coherence of 1 indicates that the oscillation in CBF is fully explained by the oscillation in BP. A coherence of 1 also means that CBF is passively following BP and there is no dynamic autoregulation. Giller therefore proposed coherence as a measure of autoregulation efficacy: indeed, coherence increased for faster oscillations (higher frequencies). Further applications of TFA to dynamic autoregulation involved the use of TFA gain and phase by Panerai et al. (95, 107, 108), Blaber et al. (109), and Zhang et al. (110). In summary, these groups reported that TFA characteristics between oscillations (fluctuations) in BP and CBF resemble a high-pass filter, wherein higher frequency fluctuations are more linearly transferred (i.e., higher coherence) to the cerebral circulation than lower frequency fluctuations (41). The parameter gain, as in acoustics, quantifies damping (i.e., smaller oscillations in CBF compared with BP oscillations). Phase, or more appropriate, phase shift, indicates that the oscillations in BP and CBF are no longer synchronized. In other words, because in every oscillation, CBF returns toward baseline before BP does, the CBF oscillations show a phase shift compared with the BP oscillations (see FIGURES 4 and 5). TFA, with estimates of coherence, phase, and gain, has become the most widespread method to quantify dynamic autoregulation (41, 55, 111, 112).
2.5. How Is Dynamic Autoregulation Assessed?
TFA has become popular because it allows assessment of dynamic autoregulation from a 5-min (or longer) recording of BP and CBF, without the need for an experimental intervention to change BP (55, 95, 110). This approach is feasible because BP normally exhibits spontaneous changes (oscillations) that occur at specific frequencies. These BP changes influence CBF, but how they are transmitted is determined by dynamic autoregulation (113). The benefits of this method are being able to perform measurements while subjects rest quietly, either supine or sitting. This makes the technique more suitable for patients who cannot tolerate or perform interventions to experimentally manipulate BP (55). These benefits are in part offset by a number of disadvantages (114). The assessment of dynamic autoregulation can be hindered by a low signal-to-noise ratio. This can occur because BP oscillations can sometimes be too small (115) in relation to the level of noise that is always present when performing noninvasive recordings (54, 116, 117). The signal-to-noise ratio is also reduced because CBF is affected not only by BP, but by various other parameters during these measurements, such as changes in or cognitive activation (54). This has led to the development of methods to enhance BP oscillations (114, 118). Several methods are now used, including repeated squat-stand (118, 119) or repeated sit-stand maneuvers (99, 100), paced breathing (104, 120), repeated occlusion/deflation of thigh cuffs (121), or oscillatory lower body negative pressure (122). Each procedure has in common that the frequency of the maneuver determines the frequency of the BP oscillation.
An example of how this method can be used to study dynamic autoregulation is illustrated in FIGURE 4. This figure shows an example of repeated sit-stand maneuvers that cause large oscillations in BP and CBF (middle cerebral artery blood velocity) at a frequency of 0.05 Hz. Visual inspection might indicate that BP changes are passively transmitted to CBF, implying absent or poor autoregulation. Upon closer inspection, with each oscillation in BP, CBF recovers before BP. This leads to a phase shift between the BP and CBF signals (FIGURE 5). FIGURE 5 also illustrates the concept of gain (or the ability for damping) by comparing the relative magnitude of CBF oscillations to those in BP. With normal dynamic autoregulation, a faster return toward baseline of CBF compared with BP will result in damping of CBF oscillations (41).
The frequency of 0.05 Hz is one where dynamic autoregulation is very effective. Autoregulation can be even more effective with slower changes in BP (lower frequencies). This phenomenon can, at least theoretically, be extended to changes in BP that become progressively slower, occurring over hours, days, and weeks and resulting in progressively smaller changes in CBF. These very low frequency fluctuations in BP represent the transition between dynamic (faster changes) and static (very slow changes) autoregulation, represented in FIGURE 3. Empirical evidence supporting this notion was recently published, in experiments wherein progressively slower oscillations in BP were induced with oscillatory lower body negative pressure (123) but is also supported by long-duration observations of spontaneous changes in BP (124).
2.6. Static versus Dynamic Autoregulation
The concept that dynamic and static autoregulation are two ends of a spectrum has several consequences. First, a measurement at one end of the spectrum may not correlate with measurements at the other end. More precisely, dynamic autoregulation may not reflect static autoregulation (89), even though this was originally suggested when dynamic autoregulation was introduced (90). Methodological differences may explain the opposite findings of de Jong et al. (89), who found no association between static and dynamic autoregulation when tested in the same subjects, versus Tiecks et al. (90) who found a strong linear relationship between static and dynamic autoregulation. The main methodological difference is that Tiecks et al. performed experiments during surgery, where isoflurane anesthesia was used to impair autoregulation (see sect. 5.7). This drug-induced impairment of autoregulation may have driven the correlation between static and dynamic autoregulation (89). Further research is needed to evaluate how assessment of dynamic autoregulation relates to static autoregulation. For example, can a finding of normal dynamic autoregulation be used to predict a normal static autoregulation response (e.g., to BP-lowering treatment)? It is conceivable that in an individual, impairment in autoregulation manifests as a slowing of the adaptive response, starting with impairment to counteract faster changes in BP (i.e., impaired dynamic autoregulation), while the adaptation to slow changes in BP is still intact (i.e., normal static autoregulation) (125). Nonetheless, in the study by de Jong et al. (89), the lack of association between dynamic and static autoregulation was observed in subjects with normal static and normal dynamic autoregulation. Ideally, studies performing repeated within-subject comparisons of static and dynamic autoregulation could shed more light on this controversy.
A second consequence of the “two ends of a spectrum” concept is that the outcome of studies in humans that try to measure static autoregulation are affected by the timing and magnitude of BP changes. A clear example of this is the study by Tan (126) that plotted a static autoregulation curve using induced BP (and CBF) oscillations at a frequency of 0.03 Hz. This frequency is much more in the dynamic range than in the static range, which explains why that study found a very narrow gradient phase (“autoregulation plateau”) of only 10 mmHg, and a steep slope for this gradient (126).
2.7. Revisiting Historical Experiments
When we revisit early studies with the current knowledge of dynamic autoregulation, we can explain initial observations of Roy and Sherrington (9) and Bayliss et al. (5). Based on the heart beat traces in the figures in the Bayliss and Hill publication, we can tell that the perfusion pressure changes in the various experiments were of short duration (i.e., seconds). We know now that for such rapid changes in perfusion pressure, isolated cerebral blood vessels and CBF passively, but transiently, follow such BP changes, without effective regulation of CBF. FIGURE 6 compares one of the figures of Bayliss et al. (5) with a recent human experiment wherein rapid changes in BP were provoked by repeated transitions between squatting and standing (118). In both experiments, the sudden increases and decreases in BP occur within 15 s, and these transient changes in BP appear to be passively followed by CBF, as interpreted by Bayliss et al. (5). Closer inspection however indicates a more rapid return of the CBF trace in both experiments. In retrospect, a more precise interpretation of Bayliss’s experiments should have been that this control was unable to maintain a stable CBF under the experimental conditions that were used (rather than that there was no active vasomotor control in the cerebral circulation). Another important message from the experiments of the Bayliss et al. is the value of presenting individual findings, something that is often omitted in the current literature. In this case, presenting individual data made it possible to reinterpret findings, even >100 yr after the original publication.
FIGURE 6.

Comparison of hemodynamic traces from one of Bayliss’s 1895 animal experiments with those from a human experiment performed in 2009. A: original figure from Bayliss et al. (5). The transient increase in carotid arterial blood pressure (BP) appears to be passively followed by the trace labeled cerebral venous pressure (CVP), which was used in this experiment as a proxy for cerebral blood fluid (CBF). Systemic venous pressure (SVP) was recorded simultaneously and remained stable (to indicate that the cerebrovascular changes were not secondary to systemic changes). B: tracings from a human experiment from Claassen et al. (118). The transient increase in BP (evoked by a squat-stand maneuver) also appears to be passively followed by middle cerebral artery blood velocity (CBFV; see also the legends for FIGURES 4 and 5), a proxy for CBF. Closer inspection and analysis, as described in FIGURES 4 and 5, are required to appreciate effects of autoregulation.
2.8. Mechanisms That Underlie Autoregulation
Some key phenotypic features of autoregulation have been known for many decades. For example, using a cranial window to measure vascular responses in anesthetized cats, Fog (13) described rapid changes in the diameter of pial arteries and arterioles (within 1–2 min) in response to experimental manipulation of arterial BP. These changes included vasoconstriction, with vascular diameter decreasing below baseline values, in response to a rise in BP and vasodilation following a reduction in BP (13). This work provided direct early examples of the rapidity of the cerebrovascular response following increases or decreases in arterial BP. In addition to insight regarding the temporal nature of the response, these studies revealed that BP-induced changes in the diameter of smaller arterioles were greater on a percentage basis than in larger arteries. Although it was not yet known that arteries and arterioles in this segment of the cerebral circulation are resistance vessels (45, 72, 127, 128), Fog (13) speculated that reductions in vessel diameter during increases in BP would prevent a rise in capillary pressure and thus protect against brain edema. This concept remains widely accepted today (FIGURE 1) (129, 130).
With respect to autoregulation-related terminology, myogenic tone is defined as the state of partial vasoconstriction that occurs in an isolated blood vessel when maintained at a constant pressure (129). Arteries and arterioles in the pial and parenchymal circulation respond to pressurization with development of myogenic tone (129). As an example, isolated brain parenchymal arterioles from both humans and mice develop substantial myogenic tone when pressurized to physiological levels (127, 131–133). Myogenic reactivity reflects changes in vascular tone in response to changes in pressure (129). The term myogenic response seems to be used as a more general or global term, representing alterations in vascular tone that result from changes in intravascular or transmural pressure in vitro or in vivo. Myogenic responses of isolated cerebral blood vessels are more commonly studied during increases in pressure (rather than reductions in pressure). Many in vitro studies have confirmed the basic concept described in vivo, that changes in vessel diameter occur relatively rapidly in response to changes in intravascular BP (127, 132–138).
Arteries or arterioles from the pial or parenchymal circulation exhibit myogenic reactivity following changes in transmural pressure (129, 139). In response to reductions in arterial BP, these vessels respond by reducing myogenic tone and increasing lumen diameter, thus reducing resistance through each vessel. Conversely, an increase in arterial BP activates mechanisms that increase myogenic tone and reduce vascular diameter, thus increasing vascular resistance. This suggests that rather than regulating vessel diameter per se, the physiological variable that is being regulated is transmural wall tension (129). With increases in BP, a reduction in vessel diameter helps to normalize wall tension. This concept is supported by the fact that similar phenotypic changes are seen in isolated vessels studied under pressurized conditions in vitro, but where blood flow is absent. Such behavior of isolated arteries or arterioles in vitro during increases or decreases in transmural pressure (127, 132–138) confirm the changes in vascular diameter that have been described by many laboratories performing in vivo experiments in which arterial BP was altered (for examples, see Refs. 13, 71, 72, 76, 83, 134). It is important to stress that, as the name implies, the myogenic response is intrinsic to vascular muscle. For example, this response remains intact if endothelial cells are removed or selectively injured (129, 134–138).
An important step in the development of the myogenic response relates to depolarization of vascular muscle, which has profound effects on vascular tone (FIGURE 7, A and B). Work by Nelson and others (140, 141) demonstrated the high sensitivity of vascular tone to changes in membrane potential of vascular muscle. Data based on direct recordings during alterations in intravascular pressure demonstrated changes in membrane potential of only a few millivolts. Under physiological conditions (i.e., baseline −40 mV), such changes cause significant alterations in intracellular Ca2+ and subsequently vascular diameter (FIGURE 7A) (140–142).
FIGURE 7.

Mechanisms that contribute to myogenic responses and the impact of these mechanisms on regulation of vascular tone, the distribution of intravascular pressure in brain, and autoregulation of cerebral blood flow (CBF). Several mechanisms have been implicated in regulation of myogenic responses. Vascular diameter is highly sensitive to changes in the cellular membrane potential (A). Membrane potential is determined by the integrated effects of mechanisms that produce depolarization or hyperpolarization of the cell membrane (A). Two key regulators in the context of pressure-induced changes in vascular tone are voltage-dependent calcium channels (e.g., CaV2.1) and large conductance potassium channels (BKCa) (A). Several molecular mechanotransducers have been proposed to function as sensors for changes in pressure and initiators of depolarization of vascular muscle, resulting in increased intracellular Ca2+ (B). Increases in intracellular Ca2+ can also occur due to Ca2+ release from the sarcoplasmic reticulum (SR) (local Ca2+ signals) resulting in activation of contractile proteins [myosin light chain kinase (MLCK)], phosphorylation of myosin (MLCP), contraction of vascular muscle, and a reduction in diameter of resistance vessels (B). Local release of Ca2+ sparks from the SR can activate BKCa, producing local hyperpolarization and feedback that limits the degree of vasoconstriction. In addition to activating CaV2.1, mechanotransduction activates guanine nucleotide exchange factors that activate RhoA (RhoGEF) and a RhoA target, Rho kinase (ROCK). ROCK exerts inhibitory effects on MLC phosphatase. As discussed in the main text, there are several candidate sensors (or mechanotransducers) for pressure changes (C) as well as modulators of autoregulation (D). Integrated changes in myogenic responses and thus vascular tone influence the distribution of intravascular pressure that normally occurs along the vascular tree in brain (E) as well as the efficacy of autoregulation of CBF (F). See text for further details. GPCR, G protein-coupled receptor; TRP, transient receptor potential.
FIGURE 8.

Effects of antihypertensive treatment on cerebral blood flow (CBF) in hypertensive patients. Summary of studies that measured effects of antihypertensive treatment on blood pressure (BP) and CBF, using different techniques to measure CBF: transcranial Doppler (TCD; A), MRI arterial spin labeling (B), and Xenon-133 CT (C). A: data from a study investigating antihypertensive treatment in patients with mild or moderate hypertension, after 2–3 wk and after 3 mo of treatment (390). B: summary from 3 studies. Two of these studies (label 1: Ref. 391; label 3: Ref. 392) investigated effects of standard versus intensive BP-lowering treatment. One study (label 2: Ref. 393) investigated effects of stopping antihypertensive treatment (causing BP to increase). C: the study that compared different β-blocking agents to lower BP (394). See text for details on all these studies. CBFV, middle cerebral artery blood velocity.
Increases in intraluminal pressure within an isolated artery or arteriole results in mechanotransduction, depolarization of vascular muscle, and Ca2+ mobilization (FIGURE 7B). The increase in intracellular Ca2+ following a rise in intravascular pressure is due primarily to influx of Ca2+ across the plasmalemma (mainly via voltage-dependent Ca2+ channel, CaV1.2) or release of Ca2+-stores from the sarcoplasmic reticulum (local Ca2+-signals) via IP3 or ryanodine receptors (129, 140), resulting in activation of contractile proteins [phosphorylation of myosin light chain (MLC)] and a reduction in vascular diameter (FIGURE 7B). When voltage-dependent Ca2+ channels are blocked, or when extracellular Ca2+ is removed, the vasoconstrictor response to pressure (development of myogenic tone) is abolished, thus confirming an essential role of Ca2+ (141). Conversely, hyperpolarization of the cell membrane, due to stimuli that include Ca2+ sparks, closes CaV1.2 channels, reducing Ca2+ entry and intracellular Ca2+ concentrations, resulting in vasodilation (FIGURE 7, A and B) (129, 140, 141). Together, these changes are believed to be major contributors to the maintenance or relative stability of CBF during increases or decreases in perfusion pressure in vivo (129, 140) (FIGURE 7). In addition to changes in subcellular concentrations of Ca2+, myogenic tone is regulated by mechanisms that influence Ca2+ sensitivity, including protein kinase C and Rho kinase (ROCK) (FIGURE 7B) (129, 139). For example, in brain parenchymal arterioles, pharmacological inhibition of ROCK, or the ROCK2 subtype, eliminates the vast majority of myogenic tone (127, 143).
In relation to the control of myogenic responses and autoregulation of CBF, a major unanswered question continues to be: “what is the sensor responsible for mechanotransduction underlying the myogenic response and subsequent depolarization of vascular muscle?” Although this aspect of myogenic responses remains relatively poorly defined (particularly in vivo), multiple candidates have been proposed to be involved. These candidates include integrins (e.g., α5β1), mechanosensitive G protein-coupled receptors (GPCRs) [e.g., angiotensin AT1, sphingosine-1-phosphate (S1P), purinergic (P2Y)], G-protein subunits (G12/G13) linked to activation of ROCK, and stretch-activated ion channels such as transient receptor potential (TRP) (e.g., TRPC6, TRPM4) and epithelial Na+ channels (βENaC) (FIGURE 7C) (51, 129, 139, 144–150). Another potential mediator, the PIEZO1 stretch-activated ion channel in vascular muscle, has been investigated but does not appear to play a role in regulating myogenic tone in isolated cerebral arteries (151).
In addition to multiple pressure-sensitive candidates, defining myogenic mechanisms is further complicated by evidence that the role of putative mechanotranducers may vary depending on the vascular segment or zone in question. For example, pharmacological inhibition of the AT1 receptor has been reported to reduce myogenic tone in cerebral arteries but not parenchymal arterioles (144), despite the fact that parenchymal arterioles develop greater levels of myogenic tone (127, 129, 131, 143, 152, 153).
Although myogenic responses are intrinsic to vascular muscle, there are additional determinants or modulators of this response including genomics, endothelial-, metabolic-, neural-, and immune-related signaling, as well as other ion channels and signaling networks (FIGURE 7D) (129, 140, 150). For example, large conductance Ca2+-activated K+ channels (BKCa) in the cell membrane are activated by local release of Ca2+ sparks from the sarcoplasmic reticulum, resulting in local membrane hyperpolarization and attenuation of myogenic tone (FIGURE 7B) (140, 142). The cystic fibrosis transmembrane regulator (a membrane protein and chloride channel) exerts inhibitory effects on myogenic tone by attenuating S1P-dependent signaling (154). Collectively, such mechanisms may limit the development of excessive myogenic tone in vivo.
While vascular muscle and the myogenic response appear to play a predominant role in relation to autoregulation of CBF during changes in BP in vivo, it is not the only mechanism or cell type involved, making its precise contribution to overall autoregulation difficult to quantify. Thus, when new mechanotransducers, cell types, or signaling events are implicated, based on studies of myogenic responses in isolated vessels, one cannot assume that the relative impact of a given mechanism studied in vitro is the same in vivo. For example, during increases in arterial BP (the upper half of the autoregulatory curve), several lines of evidence suggest sympathetic and trigeminal nerves influence the vascular response (see sect. 3.4). Mechanisms that control vessel diameter and vascular resistance during reductions in arterial BP are likely not mediated through inhibition of these mechanisms that are activated when BP is increased. With decreases in perfusion pressure, mechanisms involving tissue hypoxia, adenosine, calcitonin gene-related peptide (CGRP), ATP-sensitive K+ channels, and endothelial nitric oxide synthase (eNOS) have all been implicated to play a role (47, 76, 150, 155–159). In this context, genetic deficiency in NOS3 (eNOS) shifts the lower end of the autoregulatory curve to the right, consequently leading to augmented reductions in CBF in response to decreases in BP (155). A loss of function mutation in the NOS3 gene is associated with impaired autoregulation in response to carotid artery compression in humans (150). In contrast, other genetic variants enhance NO (eNOS-derived) signaling and are associated with long-term beneficial effects, including reducing the risk for stroke in humans (160). We speculate that such protective effects may relate, in part, to positive effects on autoregulation of CBF.
Based on a model in which vessels within a brain slice are perfused and pressurized in vitro, a role for astrocytes in modulating myogenic tone has been suggested (FIGURE 7D) (161). The potential impact of these cells in this context is difficult to define because the model elevated intravascular pressure by increasing vascular perfusion within the slice. In other words, arteriolar responses were not studied at constant pressure (with constant flow) making interpretation less clear. The slice model is also complicated by the fact it was studied under hypothermic conditions, which are known to affect myogenic tone and CBF (162).
Recent studies continue to provide insight into novel potential sensors or signaling events that may contribute to myogenic responses in isolated vessels. Unfortunately, the majority have not tested the relative contribution of each candidate mechanism in vivo. For example, the AT1 receptor is reported to be mechanosensitive, with activation of the receptor increasing myogenic tone in isolated cerebral arteries (144, 163). In other studies, however, inhibition of this receptor does not significantly alter baseline diameter of pial arterioles or resting CBF or impair autoregulation of CBF during increases in arterial pressure (79, 164–167). It seems logical to assume that in vivo myogenic responses contribute to resting tone of cerebral arterioles, baseline CBF, and changes in CBF with increased BP. Thus the contribution of AT1 receptors in relation to autoregulation of CBF remains unclear (FIGURE 7B).
There are exceptions to the approach of only studying myogenic mechanisms in isolated vessels in vitro. For example, a recent study examined effects of smooth muscle-specific deficiency in G12/G13 protein subunits or the Rho guanine nucleotide exchange factor ARHGEF12, both in vitro and in vivo (145). Overall, the study suggested a dual mechanism for development of myogenic tone, one that involves Ca2+-dependent and G12/G13 G protein subunits, along with ROCK-dependent signaling in vascular muscle (FIGURE 7B) (145). Additionally, links between ROCK and TRPM4 had been suggested previously (143). The investigators also observed that genetic deficiency in G12/G13 subunits in vascular muscle increased baseline CBF (145), a phenotype that is consistent with reduced myogenic tone, although autoregulatory responses per se were not examined in vivo.
2.9. Where in the Vascular Bed Does Autoregulation Take Place?
Basic laws of hemodynamics dictate that blood flow is determined by pressure gradients and vascular resistance. As a consequence, autoregulation operates by changing vascular resistance in response to alterations in BP, to maintain blood flow (168, 169). For blood vessels with a fixed length and blood with a fixed viscosity, vascular resistance is determined by vessel diameter (170). Changes in the radius “r” of a blood vessel (i.e., due to vasodilation, vasoconstriction, or vascular remodeling) affect its resistance by r4. This means that even small changes in vessel diameter can lead to large changes in vascular resistance, and therefore in blood flow (33, 168, 169, 171, 172). For example, an increase in vessel diameter of only 20% (at a constant pressure), which is within the physiological ability of arteries for acute changes in diameter, will increase blood flow by 207%.
The degree of myogenic tone that develops under normal conditions varies along the vascular tree. Based on work using isolated blood vessels, there is less myogenic tone under baseline conditions in larger cerebral arteries than in smaller pial and parenchymal arterioles (127, 129, 131, 143, 152, 153). These findings are consistent with the observation that effects of changes in arterial pressure on vessel diameter are size dependent in pial arteries and arterioles in vivo (13, 76, 83). This suggests that, during autoregulation (i.e., changes in vascular resistance during changes in perfusion pressure in vivo), differences may exist in the relative importance between segments. Direct measurements of CBF and intravascular pressure under baseline conditions indicate that vascular resistance is distributed over different vascular segments (or zones), resulting in a reduction in pressure along the vascular tree. For example, intravascular pressure in pial arterioles on the surface of the brain is approximately half of systemic arterial BP (i.e., central aortic BP) (FIGURE 7E) (45, 46, 71, 72, 127). As part of this arrangement, pulsatile effects of the beating heart on intravascular pressure (pulse pressure) are attenuated as blood flows down the vascular tree and into the parenchymal circulation and pial venules (67, 71, 72, 128, 173). Furthermore, the hemodynamic profile within the parenchyma may be influenced by precapillary sphincters or other structures at the transition from small arterioles to capillaries (174, 175). Venules and larger veins contribute modestly to overall vascular resistance under normal conditions (67, 71, 72, 173).
Increases or decreases in BP evoke changes in resistance of large arteries, arterioles on the pial surface, and within the brain parenchyma (13, 72, 73, 76, 83, 84, 128, 176). From these experiments, we have learned that small vessel resistance, which was calculated based on measurements of intravascular pressure in pial arterioles and pial venules, accounts for approximately half of total cerebrovascular resistance (FIGURE 7E) (84, 128).
To our knowledge, the relative contribution of arterioles, capillaries, and venules within the parenchyma to overall vascular resistance has never been directly determined in vivo. Because most of the decline in intravascular pressure in the pial circulation occurs at the level of arteries and arterioles (FIGURE 7E) (45, 127, 128, 177), we assume arterioles and capillaries within the parenchyma are the major contributors to the small vessel resistance component of total cerebrovascular resistance (84, 128). Indeed, during moderate increases in arterial BP (∼40 mmHg), small vessel resistance increased by ∼50% and CBF was maintained at baseline levels (84).
Although myogenic tone and reactivity have been studied in isolated parenchymal arterioles in vitro, little is known regarding the responses of these same arterioles to changes in BP in vivo. In one study that used multiphoton imaging, a reduction in cerebral perfusion pressure was produced by increasing ICP by 10–15 mmHg, resulting in vasodilation of penetrating arterioles by ∼12% (178). Although this experiment demonstrates an autoregulatory response in vivo, details regarding its influence on CBF and effects of changes in perfusion pressure on the various subtypes of parenchymal arterioles (179, 180) are lacking.
As described elsewhere (sect. 5), the clinical consequences of failed autoregulation and the mechanisms involved differ depending on which portion of the autoregulatory curve is affected (FIGURE 7F). Failure at the left side of the curve results in hypoperfusion, potential ischemia, and even death (FIGURE 1). If autoregulatory mechanisms are overwhelmed and fail at the right end of the curve, hyperperfusion, increases in pressure in arterioles, capillaries, and venules, disruption of the BBB, edema, increased ICP, and death can result (FIGURE 1) (129, 130, 150, 181). With moderate increases in arterial pressure (∼40 mmHg), elevations in vascular resistance are sufficient to maintain CBF at normotensive levels and prevent increases in microvascular pressure in pial venules (84). However, when the ability to autoregulate is exceeded during acute severe hypertension, CBF increases substantially along with elevated pressure in pial venules (FIGURE 7, E and F) (84, 173, 182). These microvascular hemodynamic changes are important because acute increases in venular pressure are key determinants of disruption of the BBB, much of which occurs at the levels of venules (173, 182, 183). Although the BBB is present in cerebral arterioles, capillaries, and venules (184), proteins that comprise tight and adherent junctions between endothelial cells exhibit heterogenous features, including looser junctional strands in venules (184). These looser venular junctional strands may explain why the BBB in venules appears to be predisposed to disruption during acute episodes of hypertension that exceeds the autoregulatory capacity of the circulation (FIGURE 7, E and F). The clinical aspects of hypertensive emergencies are discussed in sect. 5.2.
Anatomical differences in the cerebrovascular bed or other factors, either in physiological or pathological conditions, can result in regional differences in intravascular pressure. For example, intravascular pressure is predicted to be higher in lenticulostriate arteries than in the intraparenchymal arteries in the parietal lobe (185). In addition, regional differences in vascular anatomy and patterns of the vascular tree can affect the control of microvascular pressure and vascular resistance. This could cause regional differences in susceptibility to ischemic events, such as lacunar infarction, which is predominantly seen in brain regions supplied by the lenticulostriate arteries (185, 186). Under pathophysiological conditions, such as small vessel disease, distinct changes in structure and function of small arterioles in the parenchyma may increase local intravascular pressure in upstream arterioles (175). This increase in arteriolar pressure may increase local wall stress and contribute to the development of arteriolar rupture and the pathogenesis of microbleeds (175).
In some, but not all studies, changes in diameter of capillaries have also been described during vasoactive stimuli (49, 174, 187–190). When changes in capillary diameter are detected in vivo, to what extent these changes are active or passive, that is, due to alterations in the contractile state of local pericytes versus changes in transmural capillary pressure, remains largely unstudied and a possibility that is not generally considered (191).
Under some conditions, changes in diameter or cross-sectional area of venules may also occur (192). For example, increases in diameter of small venules occur during hypercapnia (192). A caveat when measuring venous or venular diameter is that veins often do not maintain a circular shape at low pressures (or when not pressurized), exhibiting an oval or collapsed shape (193). A similar limitation is a potential issue when measuring diameter of any vessel using in vitro brain slice preparations, where no pressurization is typically present.
Overall, multiple lines of evidence indicate that vascular muscles in cerebral arteries and arterioles are sensitive to changes in BP and that smooth muscle is the primary effector cell driving acute changes in vascular resistance in response to increases or decreases in BP (FIGURE 7, A, B, E, and F). Based on the assumption that the primary mechanism contributing to autoregulation of CBF is myogenic responses of vascular muscle, then cerebral arteries and arterioles (pial and parenchymal) play a significant role in mediating substantial changes in myogenic tone. Capillaries, despite their large surface area, make a smaller contribution to total vascular resistance (FIGURE 7) and are unlikely to have a major contribution to autoregulation as capillaries do not actively alter their diameter in response to changes in transmural pressure. Whether pericytes can respond to physiological changes in capillary pressure by actively adjusting local capillary diameter is an area of ongoing investigation.
In relation to potential neural control of autoregulation, a few studies have examined whether specific regions within the brain may influence autoregulation of CBF globally. For example, electrolytic lesions of the fastigial nucleus in the cerebellum had little or no effect on resting CBF or autoregulation of CBF during reductions in arterial BP (produced by hemorrhagic hypotension) in conscious rats (194). In contrast, electrolytic lesions of the nucleus tractus solitarii (NTS) in the medulla impaired autoregulation in multiple brain regions during phenylephrine-induced increases in arterial BP in anesthetized rats (195). This suggests that neurons that originate or pass through the NTS may play an important role. What mechanism(s) could account for such an effect remains unclear as the neural pathways from the NTS do not project to most of the brain regions that were affected (195).
The hypothesis that central pathways might be essential for intact autoregulation contrasts with the concept that local myogenic responses play a major role in this adaptive vascular response. That isolated arteries and arterioles studied in vitro exhibit myogenic responses that phenocopy key elements of autoregulation argues that the presence of specific brain nuclei or dependent signaling pathways are not essential. Having said that, modulation of autoregulation by neural pathways (e.g., sympathetic nerves) has been described and will be further discussed (sect. 3.4). Thus neural modulation of autoregulation via specific central pathways remains a possibility, albeit one that remains poorly defined at this time.
In conclusion, because of the physiological and evolutionary relevance of maintaining CBF during changes in perfusion pressure that follow from the various postural challenges in daily living, it is not surprising that autoregulation is normally so effective and that multiple mechanisms and cell types modulate the response (FIGURE 7). From an evolutionary perspective, the choice for relatively simple (but robust) myogenic responses as the main mechanism for autoregulation is logical, as they appear well positioned to withstand the effects of aging and some diseases.
2.10. Summary
Various methods currently exist to 1) assess CBF (or CBF velocity), 2) measure and manipulate BP, and 3) quantify the efficacy of autoregulatory mechanisms through several analytical approaches, each with their respective limitations and caveats that must be considered. Consequently, research examining autoregulation can be challenging, which may contribute to results that appear to be conflicting at times. Nonetheless, collectively these approaches have provided strong evidence that autoregulatory mechanisms maintain CBF relatively stable across a range of BP values in humans.
In the remainder of this review, we focus on improving our understanding of dynamic autoregulation of CBF in humans under resting conditions, in the presence of relevant disease states, as well as during daily life challenges. We first address other important mechanisms that affect CBF (e.g., arterial blood gases, cerebral metabolism, neurogenic stimuli) but also the interrelationships between these stimuli. We discuss how and where these mechanisms or stimuli regulate CBF (i.e., at the level of large arteries and/or the microcirculation). With that background, we then focus on autoregulation, discussing clinical implications that may arise as a consequence of impaired autoregulation. We address clinical conditions where CBF seems to play an important role: hypertension, vascular dementia, Alzheimer’s disease, mixed dementia, ischemic cerebrovascular disease, and cerebral hemorrhage (FIGURE 1). Finally, we discuss the consequences of daily life challenges for CBF, such as postural challenges (e.g., orthostatic hypotension, syncope) and the impact of physical activity (or inactivity).
3. OTHER MECHANISMS THAT REGULATE CEREBRAL BLOOD FLOW
Factors that we discuss below collectively influence CBF regionally or globally, serving to match CBF to demands for O2 and removal of CO2, or other mechanisms including the impact of blood gases or neural mechanisms (21). Many textbooks and reviews state that autoregulation operates through “myogenic, metabolic, and neurogenic” mechanisms, implying that all these mechanisms are involved. It may therefore appear confusing if we list metabolic and neurogenic mechanisms as “additional factors.” For autoregulation, in the definition of the adaptation of CBF to changes in BP, it remains uncertain whether all these mechanisms contribute in an orchestrated manner or whether they represent redundant or fail-safe mechanisms (134, 196–199). For example, if BP falls, and a major mechanism influencing autoregulation (myogenic responses) should fail to maintain CBF, cerebral metabolism may be affected. Such a change could prompt metabolic mechanisms to promote vasodilation and restore CBF. Similarly, in response to a large rise in BP, increases in myogenic tone and activation of sympathetic nerves promote protective vasoconstrictor responses, thus attenuating increases in CBF and hemodynamic consequences that include disruption of the BBB and edema. With such a combination of mechanisms, all working synergistically and showing redundancy, it is difficult to discern which could be viewed as a primary mechanism in autoregulation or simply an independent mechanism that provides redundancy or synergy due to overlapping functional effects. For example, the role of the autonomic nervous system in autoregulation can be investigated through inhibition or activation (e.g., pharmacological, denervation, or genetic approaches), followed by studying the response of CBF to changes in BP under such contrasting conditions (200–205). However, such experiments can be riddled with confounding factors. For example, sympathetic blockade causes hypotension and affects respiratory control, causing changes in (9). Similar “side effects” may be observed in experiments that aim to inhibit endothelial function (134, 206, 207). This means that, even though some experiments have suggested that myogenic responses are independent of endothelial cells or the autonomic nervous system in vitro, experimental designs that can definitely exclude a role for endothelial cells or nerves are difficult to achieve in vivo. We have therefore chosen to discuss these factors or determinants of CBF that may operate independently from autoregulation (even leading to confounding, or “physiological noise,” under some conditions) (54, 55, 117).
3.1. Cerebral Metabolism and NVC
The regulation of CBF is tightly coupled to cerebral metabolism (208–211). The observation that both variables are closely linked has been acknowledged for more than a century (6, 9), although the precise mechanisms underlying this coupling are not fully understood. The term metabolism suggests a role for reductions in tissue molecular O2 and increases in CO2 (which result from metabolic activity) in this coupling (6). The effects on CBF of O2, CO2, and associated changes in pH will be discussed in sect. 3.2. In addition to O2 and CO2, changes in metabolism may also affect CBF through local production of other vasoactive substances, including adenosine or potassium ion (45).
The metabolic “coupling” pathway has been described as a “feedback system,” where changes in O2, CO2, pH, and other vasoactive substances [i.e., adenosine, nitric oxide (NO), prostanoids, and potassium ion] drive local vasodilation and increase CBF (45). For at least some brain regions, these factors alone are unlikely to account for the local increase in CBF that occurs in response to increases in neural activity (45). Indeed, the increase in CBF following neural activation can exceed metabolic demand (212). Moreover, increases in CBF following neuronal activation also occur under conditions of an abundance in glucose and O2 (213). Metabolic processes alone may be too slow to explain the fast increase in CBF following changes in neural activity. Therefore, a “feedforward” theory has also been proposed, wherein coupling largely takes place between neurons, glia (responsible for consuming O2 and the demand for nutrients), and the microvasculature (responsible for supplying blood, nutrients, and O2), within elements of what is commonly referred to as a neurovascular unit (45). Excitatory and inhibitory neurons form synapses on both astrocytes and GABAergic interneurons (36). With neural activation and NVC, endothelium-dependent propagation of vascular signals occurs, which leads to remote vasodilation of upstream arterioles and arteries, including within the pial circulation (49, 50, 214, 215). These actions participate in mechanisms that maintain a close coupling between neuronal activation and local CBF. For example, activation of the occipital cortex by visual stimulation leads to a rapid increase in posterior cerebral artery blood flow, which supplies the occipital lobe (216, 217). This coupling can be observed through various techniques and allows for detailed insight into the ability to closely match regional demand with the supply of blood and O2 (217). Recent work indicates that there may be regional differences in the timing and the precise nature of vascular responses to neural activation, but it should be noted that it remains uncertain how experimental conditions (e.g., anesthesia) may affect these results (187).
It is now thought that the coupling of CBF to brain activity derives from a combination of the “feedback system” and “feedforward” mechanisms (45). Our overview of all factors that influence CBF includes factors that play a role in the CBF response to changes in metabolic or neural activity (NVC): arterial blood gasses, pH, endothelium, vascular muscle, and (autonomic) neural innervation.
Research into defining precise mechanisms that underlie NVC is an active area that is still unfolding (45, 49, 51, 174) and has implicated a role for neurons, endothelial cells, astrocytes, pericytes, and more recently, precapillary sphincters (45, 49, 51, 174). These topics are beyond the scope for this review. Instead, we refer to two excellent reviews covering these topics (45, 187).
In summary, cerebral metabolism and NVC regulate local CBF in response to neural activation. The constant and rapid changes in brain activity explain the relevance of this system, including a hypothetical condition where BP is fully stable and autoregulation has no active role in regulating CBF. Therefore, we consider metabolic control of CBF and NVC as mechanisms of their own, independent from autoregulation, despite some obvious redundancies. Components of NVC will be discussed in sects. 3.2.2, 3.2.3, and 3.2.4.
3.2. Regulation by Arterial Blood Gases and pH
3.2.1. Partial pressure of carbon dioxide.
Brain perfusion is highly sensitive to changes in or tissue levels of CO2 (34, 35, 43, 218–221). An increase in (hypercapnia) produces vasodilation, reductions in cerebrovascular resistance, and increases in CBF, whereas a drop in (hypocapnia) increases cerebrovascular resistance and reduces CBF (34, 43, 219–222). As summarized recently (35, 223), studies adopting TCD for assessment of the middle and posterior cerebral arteries or duplex ultrasound of internal carotid and vertebral arteries (extracranial conduit arteries) all demonstrated an ∼3–6% increase in CBF per mmHg rise in and a 1–3% decrease in CBF per mmHg reduction in (43, 102, 222, 224). Differences in methodological design, assessment of CBF, analysis of the responses, magnitude of manipulation of , and correction for , and especially differences in BP, as changes can affect BP (43, 225, 226), may contribute to varying results between studies. Nonetheless, the primary observation has consistently been that CBF shows a high sensitivity to changes in . This behavior of the cerebral circulation is clearly distinct from most of the systemic circulation (227–229). This basic observation in humans is consistent with many studies using preclinical models and multiple species, with only a small representation presented here (34, 74, 230–236).
Sensitivity to changes in can be detected throughout the cerebrovascular tree, from large arteries, pial arteries and arterioles, and parenchymal arterioles (67, 230, 235, 237–242). Based on changes relative to the original baseline diameter, the greatest response to hypercapnia occurs in the smaller arterioles (230, 231, 239, 241, 243). This may be related, in part, to a relatively larger content of vascular muscle and to a higher degree of resting tone under baseline conditions. In a study using SPECT, the relative reductions and increases in CBF following hypocapnia and hypercapnia showed no relevant regional differences across various brain regions including gray and white matter (244). Note however that regional differences in baseline CBF (e.g., much lower CBF in white matter compared with gray matter) cause regional differences in absolute CBF responses to hypocapnia and hypercapnia. More recently, MRI techniques have been developed to measure global and regional responses in CBF to changes in (for review see Ref. 245).
Acidosis is known to be responsible for many of the biological effects of increased CO2 (34). Classic work by Kontos et al. (243) demonstrated that reduced extracellular pH, not increases in CO2 per se, are responsible for vasodilation to local changes in pco2. This suggests that vascular responses activated by increases in relate to the diffusion of CO2 across the BBB, subsequently leading to reductions in pH in the extracellular space and cerebrospinal fluid. Ultimately, a reduction in pH represents the primary stimulus that causes vasodilation of vascular muscle. While intermediate pathways involved in the response to hypercapnia have been studied for years, and include NO and prostanoids (34, 230, 234, 246, 247), molecular sensors that detect changes in extracellular pH have been more difficult to define. Recent studies suggest activation of a proton-gated cation channel [acid-sensing ion channel-1A (ASIC1A)], particularly in neurons, by extracellular acidosis plays a critical role in CO2-induced vasodilation in brain (248). With very high levels of hypercapnia, additional mechanisms may be recruited and contribute to increases in CBF (248, 249).
3.2.2. Partial pressure of oxygen.
The cerebrovasculature is sensitive to variations in (or tissue Po2). When drops below ∼50 mmHg (equivalent to an arterial O2 saturation of ∼80%), this leads to cerebral vasodilation. This response also depends on the prevailing . The simultaneous presence of hypercapnia (i.e., another vasodilator) increases the sensitivity to hypoxia, strengthening the vasodilator response, whereas the presence of hypocapnia (i.e., a vasoconstrictor) attenuates the CBF response to hypoxia (101, 236, 250–257). The practical consequence of this interdependency of cerebrovascular responses to changes in and is that examining the impact of hypoxia in humans leads to a ventilatory response (i.e., hyperventilation), with resulting hypocapnia and vasoconstriction. In addition, regional differences in sensitivity to may be present. As a result, studies on the topic of cerebrovascular reactivity to hypoxia have produced results with significant variations (252). Although the overall impact of hypoxia is clear, these variations and dependency on alterations in highlight the importance for simultaneous assessment of levels of and in experimental conditions to correctly interpret outcomes.
A discussion of the impact of on cerebral perfusion should not omit the role of arterial oxygen content (). The magnitude of changes in CBF during hypoxia are such that O2 delivery (the product of CBF and ) is maintained at or near normal levels despite reductions in , and appear linked to , rather than per se (258). Situations that produce a reduction in (even in the presence of preserved ), such as carbon monoxide exposure, acute or chronic anemia, or hemodilution procedures, lead to an increased CBF that maintains delivery at or near normal levels (258).
Several processes are suggested to play a role in the increase in CBF during hypoxia. The first relates to local decreases in Po2 within neurons, glia, or vascular cells. Second, lack of local oxygen (Po2 or O2 delivery) may contribute to anaerobic metabolism, leading to extracellular acidosis, another vasodilator stimulus. Third, direct vascular mechanisms may be in place, where local hypoxia leads to the production and release of vasodilator substances. In this respect, adenosine is a popular and frequently studied vasodilator, partly because of various studies reporting increases in adenosine in response to hypoxemia (259). Indeed, several studies have observed increases in the CBF response to hypoxia when adenosine is enhanced and reduced CBF responses to hypoxia when adenosine is antagonized (e.g., by theophylline, caffeine, or more specific antagonists), although not all studies show similar results (259, 260). These partly conflicting data suggest that, although adenosine may contribute to cerebral vascular responses to hypoxia, redundancy in vasodilator pathways may be present and vasodilation may also be mediated through alternative mechanisms.
In summary, arterial blood gases can markedly affect CBF but are not considered to be a mechanistic component of the autoregulatory response. These variables are however of clear relevance for studies of autoregulation, as changes in arterial blood gases affect autoregulatory control of CBF. This interaction is important in preclinical studies, as well as in clinical conditions where blood gases are altered, such as cardiopulmonary diseases, artificial ventilation (intensive care), anesthesia, exposure to high altitude, or exercise. Even everyday changes in posture, from lying to standing for example, cause changes in ventilation and lung perfusion, affecting blood gases and thereby CBF (and hence valid evaluation of autoregulation).
3.3. Role of Endothelium, Vascular Muscle, and Pericytes
All arteries, arterioles, capillaries, venules, and veins are lined by endothelial cells (45–47). Arteries contain multiple layers of vascular muscle, with the smallest arterioles containing a single layer of smooth muscle (261, 262). During functional hyperemia or NVC (see also 3.1), arterioles in the parenchyma dilate and this response is propagated in a retrograde direction so that the integrated response includes vasodilation of arterioles and arteries on the brain surface (49, 215, 217). Whether the diameter of capillaries increases or not during NVC (or other increases in local CBF) has been considered, but is currently controversial (49, 187–191). For example, recent studies indicate that red blood cells can rapidly deform under conditions of reduced O2 (i.e., increased O2 consumption during activation), thereby increasing the flow of red blood cells through the capillary, without the necessity for capillary dilation (187). Below, the role of endothelium and vascular muscle are discussed in relation to regulation of regional CBF.
Regulation of vascular tone by endothelial cells occurs via several mechanisms, including the release of signaling molecules that affect adjacent vascular muscle (47, 129, 263) but also the spread of electrical signals from endothelial cells to smooth muscle via myoendothelial gap junctions (263, 264). A family of endothelium-derived molecules have been identified that directly affect the contractile state of vascular muscle. The major molecular messenger in this context is NO. In addition, potassium ion, hydrogen peroxide, ATP, and other, yet unidentified, endothelium-derived hyperpolarizing factors may contribute to affecting vascular smooth muscle tone under some circumstances (46). These vasoactive substances are released by the endothelium in response to mechanical shear stress, activation of endothelial cell receptors, and other mechanisms (46, 263). Collectively, these factors play an important role in the endothelium-dependent control of vascular tone, along with elements of vascular structure, vascular mechanics, antithrombotic activity, as well as function of neurons, synapses, and glia (32, 45–50, 129, 138, 263, 265, 266). Despite these advances, our understanding of this system is still relatively limited, particularly in relation to integration of responses between and within different cell types and segments (or zones) of the vasculature. Studies that specifically focus on cerebral arteries, arterioles, capillaries, and venules, preferably in an in vivo setting (187), are required to truly understand the diverse impact of endothelium-dependent mechanisms. The importance of better understanding these mechanisms is emphasized by overwhelming evidence from experimental and clinical studies highlighting the central role of endothelial dysfunction in mediating and accelerating the process of atherosclerosis (267, 268).
Emerging evidence suggests endothelial dysfunction is also a major player in the pathogenesis of small vessel disease in brain (58, 265, 269). As one example, endothelial-dependent vasodilation plays a key role in NVC (50–52). To what extent endothelial cells modulate autoregulation in vivo is less studied (155), but most evidence to date suggests that myogenic responses in vitro are largely independent from endothelial function (129, 134–138, 270–272).
Morphological and molecular characteristics of pericytes have been reviewed in detail (273, 274). Although anatomical relationships of pericytes in the cerebral circulation are reasonably well defined (with the exception of transition zones such as precapillary arterioles and postcapillary venules), uncertainties regarding their molecular characteristics and key aspects of their potential functional importance remain unclear (187, 274, 275). In contrast to vascular muscle, where closely associated cells are circularly arranged on a regular basis and oriented perpendicular to blood flow with essentially a zero-degree pitch (261), pericytes often extend processes down the long axis of capillaries, occasionally encircling some or all of its circumference (273, 275). Activation of contractile pericytes at the capillary level could potentially reduce local diameter at specific sites and thus affect individual capillary blood flow. However, the concept that changes in capillary diameter due to activation or relaxation of pericytes occur in response to physiological stimuli is supported by some studies but not by all (49, 187–190, 274). In addition, much of the work on pericyte control of capillary diameter has used brain slices in vitro, a model that some experts conclude is not adequate to mimic in vivo conditions (187). Furthermore, changes in capillary diameter are often quite modest in magnitude, raising questions as to whether commonly used microscopic techniques (multiphoton microscopy for example) can reliably detect physiological changes in capillary diameter (187). With respect to autoregulation, we are not aware of direct evidence supporting an active role for pericytes in regulation of CBF during changes in BP. However, pericytes might affect capillary blood flow under conditions of ischemia, where some studies suggest pericytes are sensitive to ischemia and respond with prolonged contraction, causing local entrapment of red blood cells (81, 276, 277). This concept is controversial, as others found that precapillary arteriolar smooth muscles, rather than capillary pericytes, contract under ischemic conditions (189).
3.4. Sympathetic, Parasympathetic, and Sensory (Trigeminal) Innervation
Large arteries and arterioles in the pial circulation are richly innervated by adrenergic (neurotransmitters: epinephrine, norepinephrine, dopamine, and neuropeptide Y), cholinergic (neurotransmitters: acetylcholine and vasoactive intestinal polypeptide), and sensory (neurotransmitters: calcitonin gene-related peptide, substance P, and pituitary adenylate cyclase-activity peptide) fibers within the sympathetic, parasympathetic, and trigeminal nerve systems, respectively (36, 278, 279). This innervation does not follow blood vessels as they dive into the brain parenchyma (36, 279). The presence of adventitial sympathetic, parasympathetic, and sensory fibers, innervating a major segment of the cerebrovasculature, makes them distinct from most peripheral organs (280). In addition to this extrinsic innervation of the pial circulation, intrinsic innervation of parenchymal vessels is also present in brain (36). Extrinsic innervation of extraparenchymal arteries and arterioles originates mainly from the superior cervical ganglia (sympathetic innervation), otic, and sphenopalatine ganglia (parasympathetic innervation), and the trigeminal ganglion (sensory nerves). Upon entry into the brain parenchyma, arterioles and other microvessels are under influence of innervation from brain stem and other nuclei, including the nucleus basalis of Meynert (36, 281, 282). In addition to this innervation, sympathetic nerves potentially influence CBF through effects of circulating neurotransmitters (283), although an intact BBB may substantially limit such effects.
3.4.1. Sympathetic nervous system.
When exploring effects of the sympathetic nervous system in the regulation of CBF, activation of these nerves is sometimes associated with a decrease in CBF. Studies that have examined patients that, as part of their treatment, underwent superior cervical ganglionectomy demonstrate that this procedure leads to an increase in CBF (284–286). Studies adopting local or systemic pharmacological ganglionic blockade have produced mixed results, although the majority report an increase in CBF (42). Methodological factors related to the pharmacological procedure used to block the ganglia (including potentially only partial inhibition but also effects on BP and ) may contribute to variations in findings. Nonetheless, it is generally accepted that the sympathetic nervous system prevents increases in CBF under resting conditions (77, 278, 279, 287). However, mechanisms other than the sympathetic nervous system (e.g., endothelial, chemical, or metabolic) are likely to be more important for the regulation of resting CBF (283).
While the contribution of the sympathetic nervous system to resting CBF may be modest, a key role for the sympathetic nervous system is its influence during rapid or acute increases in arterial BP. For example, sympathetic blockade doubled CBF during a Valsalva maneuver (204) and prevented the decrease in CBF associated with head-up tilt (288), although this finding was not replicated (289). In addition, nonselective α-adrenergic receptor blockade led to a stronger increase in CBF when BP was rapidly increased using intravenous phenylephrine (290). Inhibiting sympathetic innervation through ganglionic blockade also led to increased transfer function gain and reduced transfer function phase, implying reduced dynamic autoregulation (205). A complicating aspect for this and related studies is that ganglionic blockade reduced BP and BP variability, thus affecting TFA. This was addressed by increasing BP using phenylephrine and by increasing BP variability using oscillatory lower body negative pressure (205). Such an example illustrates that it is nearly impossible to study effects of sympathetic activation or blockade in isolation, without affecting other confounding variables. Several other studies, adopting different paradigms or pharmaceutical strategies, but with the same caveats, suggest the sympathetic nervous system contributes to autoregulation of CBF during rapid changes in BP [reviewed by ter Laan et al. (283)] For example, during acute hypertension, the vasoconstrictor effects of the sympathetic nervous system buffers surges in downstream microvascular pressure, thus contributing to preservation of CBF and potentially the BBB (77, 278). It is important to realize that both the direct innervation of cerebral arteries, but also stimulation of adrenergic receptors by circulating neurotransmitters, provided they cross the BBB, may contribute to dampening increases in CBF associated with acute hypertension. Previous publications provide a more extensive discussion on whether or not autoregulation is influenced by the sympathetic nervous system (291, 292).
3.4.2. Parasympathetic nervous system.
Cholinergic nerve terminals are richly distributed throughout intracranial vessels, proximal to the Virchow-Robin spaces (36). Despite the anatomical presence of this innervation, a clear view on the role of the parasympathetic nervous system in the regulation of CBF in humans is still lacking. In animal models, cholinergic control of CBF seems species specific, with a role observed for the parasympathetic nervous system in dogs and rats (293, 294), among others. Although the traditional view considered cholinergic-mediated vasodilation to be of limited importance, some recent studies suggest the opposite. For example, systemic cholinergic blockade with glycopyrrolate led to increased transfer function coherence between BP and CBF, which could indicate a reduction in dynamic autoregulation (295). In addition, Seifert et al. (296) found that glycopyrrolate abolished the increase in middle cerebral artery blood velocity during cycling and static handgrip exercise, with no change in cerebral metabolism. Although these data support a role for cholinergic effects on the regulation of CBF, the question remains whether a role for the parasympathetic nervous system per se is indicated.
The role of the extrinsic parasympathetic nervous system must not be confused with the brain’s intrinsic cholinergic innervation that derives from the basal forebrain (nucleus basalis of Meynert) (36, 297, 298). In rat models and in human experiments, stimulation of the basal forebrain increases CBF through direct cholinergic and indirect interneuron (that release NO) vascular innervation (297). Of note, Alzheimer’s disease and Lewy body dementia are characterized by early and prominent degeneration of these intrinsic cholinergic neurons originating from the nucleus basalis of Meynert (281, 282). It has been hypothesized that the reduction in CBF that occurs in these dementias may be related, in part, to this loss of cholinergic innervation and thereby its vasodilator influence and that part of the clinical benefit of cholinesterase inhibitors may be explained by increased cholinergic-mediated vasodilation (281, 282). However, these relationships await further study. The current view is that in humans, cholinergic-mediated vasodilation is recognized for peripheral blood vessels but not generally considered physiologically important within the brain (36).
In summary, cholinergic projections influence parenchymal neurons and the vasculature by releasing their neurotransmitters [i.e., acetylcholine; a potent vasodilator (294, 297)]. In addition, cholinergic projections innervate interneurons that release NO locally (230, 281, 282, 297). This intrinsic cholinergic system can be pharmacologically influenced by cholinergic or anticholinergic drugs. Nonetheless, at this point, it seems unlikely that this system plays a major role in autoregulation.
3.4.3. Trigeminal sensory nerves.
In addition to sympathetic and parasympathetic innervation, cerebral arteries and pial arterioles are innervated by the trigeminovascular system (36, 299, 300). Roy and Sherrington (9) provided evidence that stimulation of the trigeminal nerve increased CBF without a change in BP. Trigeminal nerve fibers release calcitonin gene-related peptide (CGRP), substance P, and pituitary adenylate cyclase-activity peptide (36, 299, 300). Released by stimulation of meningeal afferents, CGRP is an extremely potent peptide that mediates most of the effects of the trigeminal nerve on vascular tone. It has therefore become a new therapeutic target for treatment of migraine (299).
Using several approaches, including unilateral trigeminal ganglionectomy in a preclinical model, insight into vascular effects of the trigeminal vascular system have emerged. For example, increases in pial arteriolar diameter and CBF in response to severe acute hypertension (levels of BP that exceed the upper limit of autoregulation) are mediated in part by these sensory fibers (301, 302). One implication of this work is that increases in CBF during severe acute hypertension are not only caused by autoregulatory breakthrough, with pressure-driven passive vasodilation (301, 302). In contrast, trigeminal ganglionectomy had no effect on regional CBF under baseline conditions (301, 302), suggesting the influence of this neural system on resting CBF is minimal.
4. OTHER FACTORS THAT INFLUENCE CEREBRAL BLOOD FLOW
4.1. Patterns of Blood Flow
In the peripheral circulation, a significant literature focuses on patterns of blood flow in large arteries. While resistance arteries and microvessels demonstrate a relatively smooth, continuous flow of blood, large conduit arteries exhibit fluctuations in blood flow across the cardiac cycle. These fluctuations relate to the functional and structural characteristics of large conduit arteries, blood volume, pressure gradients (within the artery and across the arterial wall), and blood characteristics (e.g., viscosity).
The pattern of blood flow represents an important hemodynamic stimulus in relation to adaptation of conduit arteries in the systemic circulation (303). Blood flow in peripheral arteries is characterized by a large antegrade component during systole (i.e., blood flows toward the periphery), followed by a short retrograde component during diastole (i.e., blood flows back to the heart). Acute increases in antegrade blood flow are linked to immediate vasodilation, upregulation of antiatherogenic gene expression and improvement of vascular function (304–306). In contrast, periods of increased retrograde blood flow led to stimulus-dependent impairment, upregulation of proatherogenic genes and impairment of vascular health (304, 307–309). Sustained exposure to antegrade or retrograde blood flow can ultimately lead to changes in vascular structure or stiffness (303). These findings highlight the impact of changes in blood flow patterns on vascular health in the peripheral circulation. To date, few studies have focused on blood flow waveform patterns in the cerebral circulation, and to what extent they impact cerebrovascular health.
The common and internal carotid arteries have been a popular site for ultrasound-based assessment of arterial characteristics, largely because they are relatively easy to access and exhibit a high susceptibility for development of atherosclerotic plaques (310). In addition, for clinical diagnostic purposes (e.g., to detect stenosis), ultrasound-based assessment of the common or internal carotid artery wall thickness demonstrates independent prognostic value for future cardiovascular or cerebrovascular disease (311). These structural characteristics may also affect blood flow profiles in the internal carotid arteries. Indeed, assessment of blood flow profiles is frequently used to evaluate the impact of a stenosis on downstream blood flow toward the brain. Analysis of intra- or extracranial artery blood flow and shear stress waveforms contribute to our understanding the development of atherosclerosis in these arterial segments, as low and turbulent levels of shear stress are associated with increased atherosclerotic plaque development and complexity in the internal carotid artery (312).
Structural (e.g., wall thickening, stenosis) or functional impairment (e.g., waveform) in large extracranial arteries supplying the brain may also affect regulation of CBF. Indirect evidence for this idea came from a meta-analysis that reported that carotid atherosclerosis is associated with white matter lesions and silent brain infarctions (313). Moreover, in a 7-yr prospective study in 6,025 dementia-free subjects, carotid artery plaque predicted development of vascular or mixed dementias (314). Whether development of atherosclerosis and plaques with resulting changes in blood flow patterns was a direct cause of progression of cognitive impairment remains uncertain. In this context, recent work on dolichoectasia, an age-associated vascular disease characterized by increased vessel diameter, increased tortuosity, and altered blood flow, is of interest. Older people with dolichoectasia carried a higher risk of progression to Alzheimer’s dementia (315).
More direct evidence for a link between cranial artery waveforms and cerebrovascular health was provided in a study reporting a correlation between diastolic (but not systolic) carotid artery wall shear stress and cerebral infarcts (316). Others confirmed that low carotid artery wall shear stress was associated with white-matter lesions and cognitive impairment in older humans (317). Moreover, it was recently reported that carotid artery wall shear stress is independently related to progression of cognitive decline and occurrence of white matter lesions across a 5.4-yr follow-up in 689 older humans (318).
The majority of work on blood flow and shear stress patterns in arteries supplying CBF comes from studies of the common and internal carotid arteries. Current techniques that lack high spatial and temporal resolution to measure cerebral artery diameter make it challenging to examine shear stress patterns in intracranial arteries. CBF pulsatility, estimated from waveform characteristics of the middle cerebral artery using TCD, was investigated across a large group of healthy subjects ranging from 22 to 80 yr (319). Advancing age was associated with a linear decline in middle cerebral artery CBF velocity but also an increase in pulsatility of the waveform in this artery that started after midlife. A reduction in wall shear stress in cerebral arteries with aging was associated with increases in vessel diameter and increased BP (320). The potential clinical relevance of these findings is highlighted by the observation that higher CBF pulsatility correlated with a greater white matter lesion volume in older adults (321) and the finding that subjects with Alzheimer’s disease exhibit increased levels of middle cerebral artery pulsatility (322). As a caveat, a “chicken or the egg” dilemma on causality applies here. Do altered flow patterns cause vascular injury and then cognitive deficits, or is it the other way around where increased pulsatility is caused by increased vascular resistance due to vascular pathology?
Some work in this area has also been performed on the basilar artery. A cross-sectional study reported that lower diastolic (but not systolic or mean) wall shear stress in the basilar artery was found in patients with mild cognitive impairment and Alzheimer’s disease, while levels of wall shear stress correlated with the level of cognitive decline (323). Here, it is interesting to note that work on dolichoectasia (with potential effects on CBF) hinted at a relationship between a larger basilar artery diameter and markers of cerebrovascular disease (324).
Taken together, these studies suggest a link between conduit artery waveforms and cerebrovascular health. The exact underlying mechanisms are difficult to summarize at this stage. Changes in waveforms in arteries may result from a combination of proximal, upstream alterations (e.g., systolic or diastolic and pulse pressure), local factors (e.g., endothelial functional, mechanical or structural characteristics), and downstream (distal) variables in the cerebrovascular tree (e.g., increased resistance). Whether information on wall shear stress from different intracranial arteries provide distinct prognostic information for cerebrovascular health is currently unknown and should be a topic for future research. For the purposes of this review, an interesting question is whether blood flow patterns play a role in autoregulation. Left ventricular assist devices (LVAD) for patients with heart failure offer an opportunity to investigate the effects of different flow patterns. Loss of pulsatile flow in patients with continuous-flow devices had no effect on dynamic autoregulation in a study of 9 patients with continuous flow LVAD, 5 with pulsatile flow LVAD, and 10 controls (325). More work will be required to better understand if and how patterns of blood flow, rather than just the quantity of blood flow per se, affect the effectiveness of autoregulation and brain health.
4.2. Brain-Heart Hydrostatic Gradient
The brain-heart hydrostatic gradient describes the potential influence of gravity on CBF. The influence of the brain-heart hydrostatic gradient has been debated since the earliest literature on the cerebral circulation (326–329). For an overview and detailed explanation of arguments for and against the relevance of a brain-heart hydrostatic gradient, we refer to a Point-Counterpoint discussion (327) and exchange in Anesthesia Patient Safety Foundation Newsletters in 2007–09. It is not the presence of the gradient itself that is debated, but whether or not the gradient affects CBF. The hydrostatic gradient between heart and brain refers to the pressure gradient in an imaginary fluid column between the level of the heart and the brain for a person in the upright position. There are currently two scientific opinions on how this gradient affects CBF. The first claims that the pressure gradient on the arterial side is compensated by the gradient on the venous side, so that the net effect on CBF is null (329, 330). The second opinion claims this assumption is not valid in the cerebral circulation, and therefore perfusion pressure is reduced in the upright body posture by the hydrostatic gradient (i.e., BP-hydrostatic pressure) (302, 326, 327).
Scientists that propose that there are no hydrostatic effects on CBF refer to the cerebral circulation as a closed-loop system, also referred to as a siphon model (329, 330). The siphon model is the concept of a length of rigid tubing containing fluid, where the pressure at either end is determined by the vertical position of these ends but not by the position of the rest of the tubing. The closed-loop system is similar but contains a pump that circulates fluid through a length of tubing. If this system is brought from a horizontal plane to a vertical plane, the pump will not require extra energy against the hydrostatic pressure gradient, because the returning flow now returns with a higher pressure.
Opponents of this view indicate that this comparison does not hold for the cerebral circulation, for reasons that include nonrigid tubing (collapsible veins), pressure differences across the system (intrathoracic pressure, ICP), and the cerebral circulation itself with its wide range of vessel diameters (326, 327).
This ongoing debate extends from humans to giraffes to sauropods (with their extraordinary hydrostatic gradient) (330–332) to species of snakes [the heart-brain distance is longer in snakes with a mostly horizontal habitat (i.e., aquatic) vs. snakes in a vertical habitat (i.e., arboreal)] (333).
It is very complex to perform measurements to prove or disprove either theory. For example, one could study the effect of a change in posture from supine to upright on CBF. If the only parameter to change were the hydrostatic gradient, a reduction in CBF following the reduction in perfusion pressure (BP minus hydrostatic gradient) from supine to upright would suggest the hydrostatic gradient affects CBF. Indeed, CBF decreases in human subjects between supine and standing (upright) position (331, 332, 334–337). Unfortunately, this change in body position affects multiple factors that also influence CBF, such as BP, central blood volume, , and ICP (334–337). This array of changes makes it difficult to isolate the single effect of a hydrostatic reduction in perfusion pressure on CBF. In addition, dynamic autoregulation will modify the effect of a change in perfusion pressure on CBF (see sect. 2.4 on dynamic autoregulation).
Parabolic flight experiments offer the unique opportunity to modify effects of gravity without changing posture (338, 339). In a parabola following normal gravity (1 G), a period of ≈22 s of 0 G is preceded and followed by ≈30 s of hypergravity (1.8 G). These changes in gravity occur quickly (in ∼1 s). For a hydrostatic gradient in a subject seated upright in this plane, this pressure gradient suddenly nearly doubles for 30 s (from 1 to 1.8 G) and then equally suddenly drops to zero for 20 s (from 1.8 G to 0 G), only to rapidly return to double for another 30 s (from 0 to 1.8 G), after which it returns to normal (338–342). For autoregulation, these conditions create a series of step-wise, very fast changes in perfusion pressure (under the hypothesis that the gradient affects perfusion pressure) of ≈30 mmHg. Under the opposing hypothesis (siphon), these changes in gravity should have no effect on perfusion pressure. Unfortunately (yet unsurprisingly), the sudden changes in gravity affect multiple physiological processes (338, 339, 341–343). For example, hypergravity increases BP whereas zero gravity reduces BP, but also ICP, and (344).
Nonetheless, in 16 healthy volunteers, with 15 parabolas for each subject, the averaged traces of BP and CBF velocity revealed stepwise changes in CBF velocity during the transitions from hypergravity to zero-gravity and back, while BP remained stable during these changes (343). Plots for BP corrected for changes in hydrostatic gradient identified stepwise changes in perfusion pressure that could explain (from the viewpoint of autoregulation) the stepwise changes and subsequent adaptation in CBF velocity (343). This is circumstantial evidence that is not meant to end this debate. However, from a clinical perspective, incorrectly dismissing effects of a hydrostatic gradient on CBF carries more risk than incorrectly dismissing the siphon hypothesis. For example, in a patient with hypotension (mean arterial BP of 50 mmHg), if the siphon hypothesis is correct, the position of the head compared with the rest of the body (e.g., upright, supine or head-down tilt for example) has no effect on perfusion pressure, and therefore no effect on CBF. However, if the siphon hypothesis is false, the upright position may reduce perfusion pressure to ≈30 mmHg, whereas a head-down tilt could increase it to ≈70 mmHg. This simplified example ignores effects of body position on BP and ICP, but hopefully serves to explain our point. For further reading on such clinical implications, see Pohl and Cullen (345) and the call-out text for clinicians.
5. REGULATION OF CEREBRAL BLOOD FLOW AND CLINICAL IMPLICATIONS
Despite redundancy in mechanisms that regulate CBF, diverse clinical conditions have been linked to acute or chronic changes in brain perfusion. In this section, we describe the clinical implications of autoregulation in diseases commonly associated with cerebrovascular pathology [e.g., ischemic stroke, hypertension, and dementias (vascular dementia, Alzheimer’s disease, and mixed dementia)] and dysregulation of CBF. Since older age is one of the most common risk factors in these clinical conditions, we start by discussing the impact of aging on regulation of CBF.
5.1. Cerebral Perfusion Across the Age Span
Although the impact of aging on CBF has been frequently studied, our understanding of the biology of vascular aging is relatively modest compared with its clinical impact (265). Independent of the techniques and designs used to measure CBF, blood flow to gray and white matter (or global CBF) decreases by ∼0.5% per year from early adulthood (346–348). The potential relevance of a lower CBF may not only relate to its regulation but may also be associated with age-related changes in cognitive function (349) and specific domains of cognitive performance (350). Recently, a prospective cohort study in 4,759 participants from the general Dutch population with a 6.9-yr follow-up found that cerebral hypoperfusion at baseline was related to accelerated cognitive decline and increased risk for dementia, independent of known risk factors for dementia (351). Separate findings from the Alzheimer’s Disease Neuroimaging Initiative reinforced these conclusions, reporting that a higher index of cerebrovascular resistance (ergo lower CBF) predicted cognitive decline (independent of reduced metabolism) and brain atrophy (independent of β-amyloid) in older adults across a 2-yr period (352).
Mechanisms that account for age-related decreases in CBF are not fully understood but are likely multifactorial. One potential explanation relates to changes in the cerebral metabolic rate (see sect. 3.1), which has been demonstrated to decline by ∼0.5% per annum (348). Older age may impair neuronal or glial mitochondrial metabolism. Indeed, metabolic rates for neuronal tricarboxylic acid and glutamate-glutamine cycles are lower in older adults (353). However, other studies found progressive reductions in CBF that were independent of changes in cerebral O2 consumption (318, 350, 352, 354). These findings suggest that age-related changes in metabolism cannot fully explain the progressive decline in CBF that occurs as individuals age.
Somewhat related to cerebral metabolic rate is the presence of cerebral atrophy (and therefore lower cerebral metabolism). Even in healthy individuals, advanced age is associated with cerebral atrophy. Interestingly, age-related atrophy shows regional variability and seems more prominent in frontal and temporal regions (355, 356). Regional differences in cerebral hypoperfusion may also be present with older age (357, 358). These findings raise the hypothesis that regions of cerebral atrophy and hypoperfusion with older age may match. To understand this potential link, the spatial pattern of age-related changes in cerebral atrophy and CBF were explored (358, 359). While these studies confirmed the presence of regional variation in age-related changes in CBF and brain atrophy, regional effects of age on CBF differed from that of gray-matter atrophy and were not related to metabolism. Therefore, dissociation may be present between age-related hypoperfusion and cerebral atrophy in older humans. Atrophy may also affect the reliability of imaging through partial volume effects. This may affect imaging-based measurements of CBF and lead to either underestimation or overestimation of CBF (348).
5.1.1. Aging and autoregulation, CO2 reactivity, and baroreflex function.
Changes in hemodynamics may also contribute to the age-related decline in CBF. Aging is associated with a gradual increase in arterial BP, which may be related to several mechanisms including endothelial dysfunction, aortic stiffening, and an increase in systemic vascular resistance (360). In addition, assuming ICP does not change significantly (361), an increase in arterial BP leads to an increase in cerebral perfusion pressure with aging. To compensate for such changes and prevent hyperperfusion, an increase in cerebrovascular resistance is required.
Another factor that may contribute to age-related reductions in CBF is impairment of autoregulation or related mechanisms. Oudegeest-Sander et al. (362) examined the dynamic responses of CBF velocity (using TCD) and cortical oxygenation (using near infrared spectroscopy) to changes in BP in response to sit-to-stand maneuvers and in cognitively healthy individuals: young, elderly, and very elderly. While this study confirmed the presence of an age-related decrease in middle cerebral artery CBF velocity, the relative changes in CBF in response to these paradigms were similar across age groups, suggesting preserved dynamic autoregulation as well as preserved CO2 reactivity with older age. Others have also reported preserved dynamic autoregulation in healthy older populations (100, 363–370).
Previous studies have linked older age and neurodegeneration with impaired baroreflex sensitivity (88). Interestingly, such work suggested that baroreflex and dynamic autoregulation may interact in the regulation of CBF to counteract acute changes in arterial BP. One study found an inverse correlation between baroreflex sensitivity and autoregulation (371), suggesting counterregulatory functionalities between these mechanisms. However, a recent study examined both dynamic autoregulation and baroreflex sensitivity in a cohort of 136 healthy individuals (370). This study confirmed a reduction in CBF and impaired baroreflex sensitivity in the older population. However, autoregulation was unimpaired with aging, and there was no relationship between baroreflex function and autoregulation. Similar observations were found in a large data set of healthy participants across a wide age range, showing preserved autoregulation and no relationship between autoregulation and baroreflex function (372). Together, these data do not support a link between baroreflex sensitivity and autoregulation in the older population.
Studies have also examined the impact of aging on cerebrovascular reactivity to changes in . While some studies found that increases in CBF during hypercapnia are impaired with aging (46, 373), others did not (265, 362). These conflicting findings may result from differences in the methods used to measure CBF (TCD, MRI), the range of tested, the extent of aging, and the protocol used to induce hypo- and/or hypercapnia (223). Although some studies do not provide clear insight into the impact of older age on vascular reactivity, these results are largely in line with studies that examined dynamic autoregulation, in that older age does not demonstrate a clear age-related decline in the ability to regulate CBF in response to stimuli affecting BP, arterial blood gases, or pH.
It is important to note that studies of CBF in aging have a potential confounder due to age-associated neurodegenerative disease (e.g., Alzheimer’s disease, vascular dementia, and mixed dementia). Because the prevalence of neurodegenerative diseases increases sharply with advancing age (374), it is likely that some studies have unknowingly included older people with lower CBF caused by neurodegenerative disease. This can occur because many of these diseases remain asymptomatic or at least not clinically recognized, in their early stages.
5.2. Autoregulation in Hypertension
There are two widespread, yet incorrect, concepts about autoregulation in hypertension. The first is that chronic hypertension (which is often combined with aging) leads to impaired autoregulation (static and dynamic), through hypertension-associated vascular dysfunction (see Ref. 375 or Ref. 376 for examples from review articles where this concept is presented as a fact). The second, in partial contrast with the first, is that static autoregulation remains intact in hypertension; however, its lower and upper levels are shifted toward higher pressure levels (377–379). In other words, the autoregulation curve has shifted rightwards (376, 380). With this concept, the brain can tolerate higher BP but becomes more sensitive to low BP (380). The translation of these two concepts to clinical practice is explained in the following example. A 75-yr-old hypertensive patient has a habitual systolic BP of 160 mmHg. According to the first concept, further increases or decreases in BP, albeit fast or slow, would affect CBF because static and dynamic autoregulation is impaired. According to the second concept, an increase in systolic BP to 180 mmHg will not affect CBF, because the upper limit of the static autoregulation curve has shifted upward. However, a reduction in systolic BP to 120 mmHg may fall below the lower limit of autoregulation, which has also shifted to a higher pressure, leading to a reduction in CBF. With both concepts, older hypertensive patients are considered to be vulnerable to cerebral hypoperfusion when BP is reduced (380–382). Common clinical examples of BP reduction are those associated with antihypertensive treatment (mostly slow, gradual changes) and with postural changes in BP (fast changes, see also sect. 6.1 on postural changes).
5.2.1. Dynamic autoregulation.
One of the first studies of dynamic autoregulation in hypertension tested the following hypothesis: “… that age and hypertension would impair dynamic autoregulation, resulting in relative cerebral hypoperfusion during acute hypotensive stress” (369). Ten young (24 yr of age), 10 normotensive (mean BP of 125/68 mmHg), and 10 hypertensive (mean BP of 153/90 mmHg) older adults (72 yr of age) underwent assessment of dynamic autoregulation during an active stand from sitting. This maneuver causes a transient reduction in BP, which usually reaches a maximum around 15 s after standing and recovers within 30–40 s (100, 369, 383). Mean BP fell by 25%, leading to a reduction in CBF of 15% in old adults and 19% in young adults. Autoregulation during spontaneous fluctuations in BP was also assessed using TFA, during 5 min of quiet sitting or quiet standing. An impairment in dynamic autoregulation would manifest as a lower phase and a higher gain (see section 2.4). However, phase was normal and gain was lower in hypertensive older adults. In contrast with the hypothesis, the authors concluded that there were no differences in dynamic autoregulation between young, older, and older hypertensive adults (369).
These results were confirmed by a study in 21 hypertensive patients (49 yr of age) and 21 normotensive controls (384). Here, transient hypotension was evoked by standing up from a squatting position, which leads to a stronger drop in BP compared with sit-to-stand. The average 35–40% transient decrease in BP led to a reduction in CBF of 21% in hypertensive adults and 28% in controls (384). In the hypertensive group, there were no differences between those with well controlled BP (136/78 mmHg) and uncontrolled BP (154/95 mmHg) (384).
In a study of acute BP lowering in hypertensive crisis (385), 28 patients (55 yr of age) with a mean systolic BP of 200 mmHg were randomized to rapid BP reduction with sublingual captopril or nifedipine. CBF velocity was not significantly reduced following treatment with captopril but was modestly reduced by nifedipine (385).
These three studies with relatively small sample sizes were followed by a larger study in 35 normotensive (mean BP, 124/76 mmHg) and 45 hypertensive older adults (mean BP, 152/89 mmHg, 68 yr of age) (386). In this study, the range of systolic BP was wide in the hypertensive adults, up to 206 mmHg. The authors investigated a wider spectrum of dynamic autoregulation by looking at effects of decreases and increases in BP that were sustained for up to 3 min, as well as more rapid decreases and increases in BP. For all procedures, the changes in BP (up or down) were between 15 and 20 mmHg. The authors found no differences in dynamic autoregulation between hypertensives and controls (386).
A second large study recruited 60 older adults (72 yr of age); 22 normotensives (BP <140/90 mmHg, no medication), 20 controlled hypertension (BP <140/90 mmHg on medication), and 18 uncontrolled hypertension (systolic BP >160 mmHg with or without medication) (387). Dynamic autoregulation was examined using TFA of spontaneous changes in BP at rest and by studying transient BP changes during a single sit to stand protocol. This study was comparable in design to the smaller study by Lipsitz et al. (369). Following standing, the transient decrease in BP ranged between 20 and 25 mmHg. A transient reduction in CBF was observed, 15% in normotensives and 10% in controlled and uncontrolled hypertensives (387). TFA found similar values for phase in all groups, whereas, as in the earlier study by Lipsitz et al. (369), the gain was lower in hypertensives.
5.2.1.1. summary of dynamic autoregulation studies in hypertension.
In five studies with 240 participants and a range of ages from young, middle aged, to older, dynamic autoregulation was intact in hypertension during nonpharmacological changes in BP. Limitations of these studies are that no patients over 80 yr old were included and that participants were nonfrail and had limited comorbidity.
5.2.2. Static autoregulation.
5.2.2.1. studies using tcd.
For clinical translation, the studies described above related mainly to faster BP changes evoked by daily life challenges such as postural changes. An unexplored area was how slower, more gradual reductions in BP using antihypertensive treatment would affect CBF. There are two arguments why this is relevant. First, antihypertensive medication could have direct effects on cerebrovascular function that may impair autoregulation. Second, BP lowering using antihypertensive treatment could reduce BP to the lower limit of autoregulation, such that any further reduction in BP (e.g., during a postural challenge) would lead to hypoperfusion. Ideally, therefore, studies exploring these relationships would combine measures of static and dynamic autoregulation.
In one study, normal static autoregulation was observed in 42 young (34 yr of age) hypertensive patients (388). BP was reduced with atenolol from a mean arterial BP value between 100 and 105 mmHg to between 80 and 85 mmHg. CBF velocity was measured after 30 and 60 days and was not reduced (388). In another study, older hypertensive patients were studied (389) in three groups: normotensive (n = 19), controlled hypertension (n = 18), and uncontrolled hypertension (n = 14), with a mean age of 70.4, 72.4, and 72.4 yr and ≈50% female. At baseline, mean systolic BP in the three groups was 124, 135, and 160 mmHg, respectively. Only the uncontrolled hypertensive group was treated, to a systolic BP target <140 mmHg. After 6 mo of antihypertensive therapy, the group with uncontrolled hypertension had, on average, a 17-mmHg decline in systolic BP, while CBF velocity increased significantly. At baseline, dynamic autoregulation was similar between groups, and this did not change following BP lowering in the uncontrolled hypertensive group (389). Thus preserved static autoregulation can be deduced from the observation that there was no decline in CBF following BP reduction. The increase in CBF following BP reduction is not explained by autoregulation but may point toward a negative effect of chronic hypertension on CBF. Chronic hypertension could impair vascular compliance and through this mechanism reduce CBF. Indeed, antihypertensive treatment led to increased carotid artery distensibility, a measure of vascular compliance (389).
Static combined with dynamic autoregulation were addressed in a study in 21 newly diagnosed (but untreated) hypertensive patients, 49 yr of age (range from 27 to 66), and 9 controls (390). Of the 21 hypertensive patients, 12 had mild (BP of 143/88 mmHg), and 9 had moderate (BP of 163/101 mmHg) hypertension, diagnosed with 24-h ambulatory BP monitoring. Within 2 wk, BP was reduced using antihypertensive medications (losartan-hydrochlorothiazide) to 126/77 mmHg in mild and to 134/84 mmHg in moderately hypertensive patients, (i.e., an average reduction of systolic BP of 20–30 mmHg), thus lowering BP to or near the lower limit of autoregulation. At that point, CBF was measured during an orthostatic stress test using head-up tilt. This was repeated after longer term treatment (3–4 mo). Acute (1–2 wk) reductions in BP brought about by antihypertensive treatment did not reduce CBF. Furthermore, CBF remained stable during the orthostatic stress test. After 3–4 mo of treatment, BP remained at its reduced level, and CBF remained stable (390). Thus, combining assessments of static and dynamic autoregulation, this study found no evidence for impairment in autoregulation. There was also no evidence of an upward shift of the lower limit of autoregulation (FIGURE 8). Similar to the findings of Lipsitz et al. (390) and Serrador et al. (387), the patients with hypertension (but not those with only mild hypertension) had lower gain values than controls at baseline. An interesting observation is that gain increased to similar values as in the control group following treatment. Lower gain could be interpreted as better autoregulation; however, it may also be explained by differences in vascular compliance, supported by the observation in this study, but also several other studies of autoregulation (99, 387, 389, 390, 395), that increases in cerebrovascular resistance are associated with lower gain and that normalization of cerebrovascular resistance increases gain (387, 390).
5.2.2.2. summary of studies on static (combined with dynamic) autoregulation in hypertension.
In n = 123 subjects, including young, middle-aged, and old individuals, static and dynamic autoregulation was investigated before and after pharmacological treatment to lower BP. Treatment had no effect on dynamic autoregulation, and follow-up CBF remained stable, suggesting normal static autoregulation. Beneficial effects of BP lowering on CBF (i.e., increases in CBF) were observed, possibly due to improvement in arterial compliance.
5.2.2.3. studies using xenon-133.
In all studies described above, CBF was evaluated using TCD to measure changes in cerebral blood velocity in middle cerebral arteries. That artery supplies ∼70% of the blood flow to the cerebral cortex. Investigators in this area use TCD because it has the high temporal resolution that is needed to measure dynamic autoregulation in humans. For static autoregulation, additional techniques are available that can provide measurements of global or regional CBF, some of which have been around for more than half a century. Perhaps the first studies to dispel the notion of impaired static autoregulation in hypertension were published in the late 1970s and 1980s (394, 396) but appear to have received little attention. Conen et al. (396) investigated the effects of short-term (hours) and longer term (4 wk) BP lowering on CBF, where CBF was measured using the Xenon-133 method: the same technique used by Lassen (38) and Harper et al. (70) in their work on static autoregulation. Patients (n = 10, 37 to 70 yr of age) with a mean arterial BP of ≈ 130 mmHg, requiring emergency BP lowering (for hypertensive encephalopathy, ICH, fundal hemorrhage, or diabetic retinopathy) were studied. BP was reduced on average from 220 mmHg to 150 mmHg systolic in ∼1 h using intravenous nifedipine or clonidine, whereas CBF (≈60 mL/100 g/min) remained stable overall (396). In that same study, 21 patients (10 female) with mild to moderate hypertension (50–89 yr of age), were treated for 4wk with nitrendipine, verapamil, or chlorthalidone. Systolic BP was reduced from ≈170 mmHg to 145 mmHg, while CBF remained stable (≈50 mL/100 g/min) (396).
In 1979, Griffith et al. (394) investigated the effects of BP lowering using different antihypertensive agents (all β-blockers) in a large number of hypertensive patients (27 per group, >100 in total). Mean BP was lowered from values between 135–140 mmHg to between 110–115 mmHg. CBF was measured using Xenon-133. In all patients, and for all β-blockers, despite this substantial BP lowering, CBF remained stable (394) (see also FIGURE 8).
A small study in 1987 using Xenon-133 in eight older hypertensive patients found no reduction in CBF following BP lowering with prazosin (397). In addition, in 23 older hypertensives (66–91 yr of age), systolic BP was lowered using amlodipine from 177 to 156 mmHg (24-h ambulatory BP) for daytime measurements, and from 157 to 140 mmHg for overnight measurements, in 8 wk. There was no reduction in global or regional CBF described using SPECT (Tc-HM-PAO) (398). However, in most instances (as in this study) SPECT only allows qualitative estimates of CBF patterns, not quantitative CBF, unless tracer kinetics are applied (398). Lastly, in 15 patients (9 men, 60–79 yr of age), with hypertension and carotid artery stenosis, CBF (measured using Xenon-133) before and 2 h after BP lowering (mean arterial BP from 110 to 102 mmHg) remained stable (399).
5.2.2.4. studies using mri.
In 37 older (74 yr of age) hypertensive patients (systolic BP >150 mmHg), CBF was measured using MRI (arterial spin labeling) before and after antihypertensive treatment in two different regimens: standard and intensive (392). After 12 wk, systolic BP was reduced by an average of 15 mmHg in the standard treatment group and 27 mmHg in the intensive treatment group. BP lowering did not result in lower CBF (FIGURE 8). In fact, in the intensive BP-lowering group, CBF actually increased by ∼10% (392).
The following is an illustration of how persistent the concept of impaired autoregulation in hypertension is in the literature. Despite accumulating evidence indicating preserved autoregulation in older hypertensive patients, a study was initiated in 2013 with the hypothesis that older hypertensive patients have impaired autoregulation, and their antihypertensive treatment would therefore cause cerebral hypoperfusion (400). It was also hypothesized that stopping their medication would increase BP and CBF, leading to improved cognitive function (400). This was a large important study in 102 older, hypertensive adults (81 yr of age). Participants were randomized to continuation of antihypertensive treatment (n = 47) or to stopping all antihypertensive medication (n = 55). BP and CBF (arterial spin labeling) were evaluated at baseline and after 4 mo. It was noteworthy that already at baseline, the findings were in contrast with the original hypothesis (393). Hypertension (i.e., higher systolic BP) was associated with lower, not higher, CBF. At follow-up, discontinuation of antihypertensive medication led to an expected increase in BP of ∼10 mmHg systolic. Again, in contrast with the original hypothesis, this did not lead to an increase in CBF (FIGURE 8) (393). An important contribution of this study is that it included subgroups of people with small vessel disease, diabetes, and mild cognitive impairment. In these subgroups, there was no evidence of impaired autoregulation (393).
Whether autoregulation is impaired in older hypertensive patients with cerebrovascular disease (small vessel disease) was further explored in the PRESERVE trial (391). Patients with severe small vessel disease were included, i.e., a combination of clinically defined lacunar infarcts and white matter disease (a Fazekas score of 2 or 3 out of 3). Sixty-two patients (69 yr of age) were included, with relevant comorbidity such as diabetes, cognitive impairment, depression, and smoking. With treatment, mean BP was reduced from 150/83 to 141/79 mmHg in the standard group and from 154/88 to 126/75 mmHg in the intensive treatment group over a period of 3 mo. In both groups, CBF (measured using MRI arterial spin labeling) was unaffected by BP lowering (391).
5.2.2.5. hypertensive emergency.
In hypertensive emergencies, also described in the literature as hypertensive crisis, malignant hypertension, or hypertensive encephalopathy, BP levels are extremely high and exceed the upper limit of autoregulation, (e.g., systolic BP of >180 mmHg) (130). In this situation, an excessive increase in CBF and ICP may occur, which explains the signs and symptoms associated with hypertensive emergency: altered mental state, encephalopathy, infarction or bleeding, optic disk swelling, and retinal bleeds. The presence of signs or symptoms of acute end-organ damage in a patient with very high BP levels (for example, diastolic BP >130 mmHg, systolic >200 mmHg) should alert the clinician that the upper limit of autoregulation may have been exceeded, and therefore, autoregulation is impaired (130). Impaired autoregulation (static and dynamic) in patients with hypertensive emergency was demonstrated in a small study using TCD (401), which reported a reduction in CBF following BP-lowering treatment. It is possible however that the magnitude of reduction in CBF was overestimated due to changes in minimum alveolar concentration (MCA) diameter, affecting blood-velocity measurements (401). CBF was unaffected in a small study using Xenon-133, where systolic BP was reduced from 220 to 150 mmHg in 1 h (396). The clinical relevance is that in a patient with a hypertensive emergency, a very fast and very large reduction in BP must be avoided to prevent cerebral hypoperfusion. Current guidelines recommend a 20–25% reduction in BP (e.g., from 220 to 165 mmHg) in the first hour, followed by a target BP of 160/(100–110) mmHg in hours 2–6, using intra-arterial BP measurements and intravenous antihypertensive agents (130). The vascular biology in hypertensive emergency is discussed in sect. 2.8.
A hypertensive crisis is not only determined by how high BP levels are but also by how fast they have risen (130). This follows logically from autoregulation: with a slow or gradual increase in BP, autoregulation adapts by a rightward shift of the upper limit of autoregulation, the result being that autoregulation can function effectively, even at very high BP levels (e.g., systolic BP of 200 mmHg). In contrast, in an individual with a habitual systolic BP of 120 mmHg, a sudden sustained increase in BP to 180 mmHg can exceed the upper limit of autoregulation and trigger a hypertensive crisis. In clinical practice, hypertensive crisis tends to be overdiagnosed because there are many (older) hypertensive patients with chronically elevated BP and because the clinical symptoms in such individuals (altered mental state, confusion, encephalopathy) fully overlap with those characteristics of delirium and dementia, which have a high prevalence in older patients in acute care settings.
5.2.2.6. summary of all studies on autoregulation in hypertension.
Between 1979 and 2018, 747 hypertensive patients have been investigated. In 240 patients, dynamic autoregulation was studied (without evaluation of treatment). All studies found that autoregulation was similar to normotensive controls. In 507 older patients, autoregulation was evaluated before and after BP-lowering treatment. In 384 patients, this was an evaluation of static autoregulation. In 160 patients, CBF was measured using Xenon-133 CT, in 23 with SPECT, and in 201 patients with MRI (arterial spin labeling). In 123 patients, both static and dynamic autoregulation were evaluated before and after treatment, using TCD to measure changes in CBF velocity. Individuals that were included were not only otherwise healthy and nonfrail but included a substantial number of people over 70 and even 80 yr of age with comorbidities (vascular disease, including cerebral small vessel disease, diabetes, depression) and cognitive impairment. All these studies came to the conclusion that autoregulation is not impaired in hypertension. FIGURE 8 provides a summary of studies that investigated static autoregulation in hypertension.
5.2.3. Hypertension and adverse outcomes.
Evidence suggesting normal autoregulation during hypertension can also be obtained from studies that did not measure CBF. Such studies investigated adverse outcomes associated with cerebral hypoperfusion, the feared outcome of BP lowering if autoregulation is impaired. Examples of adverse outcomes related to cerebral hypoperfusion include falls, dizziness, cerebrovascular lesions, mild cognitive impairment, or dementias. Interestingly, in a prospective cohort of almost 600 older hypertensive patients (70–97 yr of age), the use of antihypertensive medication to lower BP was associated with a substantially lower odds ratio (0.6) for the risk of falls (402). This result strongly argues against the concept of impaired autoregulation with aging and hypertension and also against the concept of an upward shift of the lower limit of autoregulation (402). The increased risk of falls in patients not using antihypertensive medication may be explained by the observation that untreated hypertension (in older people) increases orthostatic hypotension. Orthostatic hypotension is a strong risk factor for falls (403, 404). The findings by Lipsitz at al. (138) have subsequently been confirmed by others, as summarized elsewhere (404). These studies highlight that hesitance to treat hypertension in older people, based on the assumption of impaired autoregulation and fear of induced cerebral hypoperfusion, is unsupported and may in fact have the opposite effect on the risk of falls (402, 404). Untreated hypertension can increase the risk for orthostatic hypotension, with large transient reductions in BP, which may not be prevented by autoregulation and cause cerebral hypoperfusion and falls.
There have also been prospective, randomized trials that have evaluated the outcome of BP lowering on cognitive decline. The SPRINT trial was a study of intensive versus standard antihypertensive therapy. It included patients >75 yr old, with a subgroup of moderately frail participants. The SPRINT-MIND substudy (n = 9361) was specifically designed to evaluate cognitive function (405). If autoregulation were impaired in hypertension, the expected outcome would have been that intensive BP lowering, through cerebral hypoperfusion, would promote cognitive decline. The contrary was found. The intensive treatment group had less risk of developing mild cognitive impairment or dementia than the standard treatment group (405). Equally, there was no increase in risk of falls or fractures in the SPRINT study (405, 406). The prevalence of syncope was not increased (see sect. 6.1.2: note that syncope is a disorder of BP regulation, much more than a disorder of autoregulation). In the subgroup of patients >80 yr old, intensive treatment was associated with increased risk of a decline in kidney function (406). The clinical consequences of this reduction in kidney function (which was defined as a 30% or more reduction in estimated clearance) remain uncertain. A recent study indicates that these reductions in estimated creatinine clearance do no really represent clinically relevant reductions in renal function (407). In addition, this older subgroup demonstrated reductions in cardiovascular events and mortality and reductions in cognitive decline with intensive BP lowering, without increased risk of reduced mobility (gait speed) or falls (406). Of note, the benefit was influenced by baseline cognitive function: in those with poor cognitive function at baseline (defined as a Montreal Cognitive Assessment score below 18 or 20 depending on level of education), there was no or limited benefit of intensive treatment on all endpoints, including cardiovascular events. This seems to emphasize the role of early start of antihypertensive treatment in the prevention of cognitive decline.
The INFINITY trial also investigated effects of intensive versus standard antihypertensive therapy in older patients (199 randomized patients, 54% women, 80.4 yr of age) (408). At study entry, subjects had a systolic BP >170 mmHg, or between 150 and 170 mmHg on antihypertensive medication. All subjects had evidence of white matter disease on MRI. They were then treated from a mean systolic BP (based on 24-h ambulatory monitoring) of 149 mmHg to 128 mmHg (intensive) or 144 mmHg (standard), with a 3-yr follow-up (408). Outcome measures were gait speed, cerebrovascular lesions, volume of white matter lesions, falls, and syncope. Impairment of autoregulation would be predicted to manifest as a slowing of gait, an increase in white matter lesion volume, and/or an increase in falls. However, these were not found. In contrast, the time-dependent increase in white matter lesion volume was smallest in the intensively treated group (408). Finally, a meta-analysis of 31,090 individual participant data from trials performed between 1980 and 2019 investigated the risk of development of dementia associated with use of antihypertensive medication (409). Participants were 55 yr and older (≈50% were >75 yr, but only ≈5% were >85) and were followed for at least 5 yr. In ∼15,000 participants with a baseline systolic BP <140 mmHg, antihypertensive use did not increase dementia risk. In the ∼15,000 participants with baseline systolic BP >140 mmHg, use of antihypertensive medication even reduced dementia risk (RR 0.88) (409). There were no differences between classes of antihypertensive medication.
5.2.3.1. conclusions.
Despite the evidence presented above, statements claiming that autoregulation is impaired in hypertension, and that older hypertensive patients require higher BP because of a rightward shift of the autoregulation curve, are still found frequently in the literature. Very often, they are presented as facts, sometimes without references. Equally, there is an abundance of cross-sectional studies dealing with BP levels and treatment of hypertension, sometimes with, but often without, measurements of CBF, that nevertheless draw inferences regarding the relationship between BP and CBF. The bias in such studies almost always leads to observations that people with low BP have worse outcomes in mortality or cognition than people with high BP. Such anticipated outcomes may be explained because BP levels often decline in cancer, heart failure, or dementia. Nevertheless, and almost without exception, such studies end with a conclusion that the negative outcome can be explained by a low CBF, caused by low BP and impaired autoregulation.
However, based on prospective studies that measured CBF as an outcome in >700 older hypertensive patients, there is no convincing evidence that autoregulation is impaired in hypertension, nor is there evidence that older hypertensive patients require higher levels of BP to preserve CBF. This conclusion is further supported by the outcomes of three large prospective studies, and one recent large meta-analysis, of antihypertensive treatment (combining >25,000 patients >75 yr of age) that found no evidence for adverse effects that could be attributed to cerebral hypoperfusion, including falls, cerebrovascular lesions, or cognitive decline and risk of dementia.
5.3. Cerebral Perfusion and Stroke: Immediate and Long-Term Changes
The key role of BP control in the management of acute ischemic and hemorrhagic stroke makes assessment of autoregulation a top research priority to inform clinical decision-making, with the aim of preventing secondary damage due to extremes of hypo- or hyperperfusion (FIGURE 1) (410, 411). The need for a better understanding of autoregulation following stroke has led to a substantial literature in this area as reflected by the number of reviews dedicated to the topic (32, 41, 139, 379, 410, 412–421). The main aim of this section is to assess progress with the understanding of autoregulation pathophysiology in human stroke and to identify priorities for future research.
Ischemia impairs CBF regulatory mechanisms, including autoregulation (44, 416, 418, 422), and hence, it is not surprising that most studies of autoregulation have shown that autoregulatory mechanisms are often impaired following both ischemic stroke and ICH (379, 410, 412, 414, 415, 417, 420, 423). In addition, autoregulation has been shown to be impaired in most studies following aneurysmal subarachnoid hemorrhage (SAH) (424–440). The finding that autoregulation tends to be impaired in the main types of stroke (ischemic or hemorrhagic) has come from controlled and observational studies where patients have been compared with healthy controls as distinct groups. However, this cannot be generalized to all stroke patients as some studies, mainly involving mild strokes, have shown that many stroke patients maintain intact autoregulation (410, 412, 423, 441–447). Moreover, even in the case of patients with moderate to severe strokes, autoregulation is not impaired at all time points. Indeed, one of the main items of interest is the temporal evolution of autoregulatory efficacy poststroke to inform treatment decisions in a timely fashion (423). In addition to the need to understand the time course of impairment of autoregulation following stroke, individual heterogeneity due to phenotype, stroke etiology, severity, subtype, location, comorbidities, and early intervention, as well as the different protocols and techniques used for assessment of autoregulation, represents a highly complex, multifaceted problem that has not been addressed in a comprehensive manner. The extent of impairment of autoregulation, and its poststroke temporal evolution, also likely depend on the volumes of the core and penumbra regions (sect. 1.2) and patterns of reperfusion (26, 415, 448, 449). The role of these multiple factors is well illustrated by one of the first studies of autoregulation in ischemic stroke that proposed a quantitative measure of static autoregulation, as the ratio of changes in CBF [measured with hydrogen clearance and cerebral (A-V) ] and cerebral perfusion pressure (invasive measurements of BP and ICP) (450). Meyer et al. (450) found that dysregulation was associated with severity and location. In other words, there was worse autoregulation with brainstem or subcortical lesions as compared with cortical strokes. Moreover, they reported an inverse correlation with time of measurement (day 1 to 44) after ictus, suggesting less dysregulation with longer duration since ictus, without an influence of age or hypertension.
5.3.1. Methodological aspects of autoregulation assessment in stroke.
Before reviewing the different factors that can influence autoregulation in ischemic or hemorrhagic stroke, it is important to call attention to the diversity of protocols and measures of both static and dynamic autoregulation that have been used in these studies and their potential to confound findings. Assessment of autoregulation based on static methods requires induction of changes in mean BP that have been performed by means of tilting (450) or the use of vasoactive drugs such as nicardipine or labetalol (442, 443). In addition to changes in physiological processes that can result from these manipulations of BP, such as changes in with tilting, there are also concerns about estimating the slope of Lassen’s static autoregulation curve using only two points, which can potentially lead to substantial errors (40).
The majority of the literature is dominated by studies using the dynamic autoregulation approach. Although originally proposed in combination with sudden reductions in BP induced by the rapid release of compressed thigh cuffs (90, 101), dynamic autoregulation assessment has been increasingly based on spontaneous fluctuations in BP, whose convenience is particularly relevant in the acute stroke setting (113), where patients may be unable to perform or tolerate postural changes or other interventions to manipulate their BP. Concern about the sufficiency of BP variability in spontaneous cardiovascular fluctuations (114) has led many investigators to revert to the thigh cuff approach (429, 451–454) or to use alternative ways to provoke rapid changes in BP, such as rhythmic handgrip (455), tilting (453), paced breathing (434, 456, 457), Valsalva maneuver (458), rapid changes in head position (459), brief carotid artery occlusion (424, 433, 435, 438, 460), or elbow flexion (461). The concomitant changes that these maneuvers can induce in covariates of autoregulation, such as , heart rate, cardiac output, and activity of the sympathetic nervous system, need to be kept in mind when comparing studies using different protocols (113). A few studies used spontaneous fluctuation recordings but attempted to improve reliability of autoregulation parameters by using only larger pressor or depressor transients in BP identified in the data (452, 453, 462, 463).
A relatively large number of indexes have been adopted for estimation of autoregulation efficiency (91). By far, the main approach has been TFA, in conjunction with spontaneous fluctuations in BP, with the use of the amplitude (gain) and phase frequency responses as metrics of dynamic autoregulation (95, 110, 434, 464–467). At each frequency, gain reflects the ratio of output (i.e., CBF or CBF velocity) to input (i.e., BP) amplitudes, while the phase expresses the time delay between output and input. With failing autoregulation, one would expect the gain to increase (that is reduced buffering of BP fluctuations) and the phase to decrease (that is the tendency of output to follow the input). A recent systematic review and meta-analysis has shown that phase is a more reliable parameter than gain to detect changes in autoregulation in ischemic stroke (414), something that had been highlighted in previous reviews (55, 91). When comparing studies that used phase or gain, it is important to consider the different settings that are required in TFA, which may confound comparability. To address this problem, a White Paper proposed standardization of TFA settings aimed at improving comparability of future studies (55). When BP changes were induced by means of thigh cuffs, efficiency of autoregulation was expressed with the rate of regulation (101) and autoregulation index (ARI) (90, 429, 451–454) indexes. The rate of regulation was adapted to express the rate of recovery of the CBF velocity response to a step change in BP (calculated via TFA) (464, 468, 469) and also to studies of autoregulation in stroke based on MRI estimates of CBF (470). Following the demonstration that the ARI can be calculated by means of TFA, using spontaneous fluctuations in BP (108), several studies have used this approach in both ischemic and hemorrhagic stroke (441, 446, 454, 459, 471–473). This alternative has a number of advantages as it abbreviates several choices that need to be made when using phase or gain directly (55).
When using spontaneous fluctuations in BP, one alternative to TFA is the mean velocity index, usually referred to as Mx (or Mxa) that has been adopted in autoregulation studies of both ischemic and hemorrhagic stroke (including SAH) (421, 426, 428, 437, 440, 444, 445, 449, 467). A variant of the Mx, the Sx index, using the systolic value of CBF velocity, instead of the mean value, has been adopted in SAH studies (426, 427, 437, 460). The Mx/Mxa is correlated with the ARI (474), but hitherto it has not been scrutinized regarding its sensitivity to noise, measurement errors, and choice of parameter settings. In situations when measurements of ICP are available, the PRx index, expressed by the correlation coefficient between mean arterial BP and ICP, has also been used, mainly in studies of SAH (430, 475–477). In a few studies, indirect measures of CBF were obtained with methods reflecting the utilization of O2, leading to correlation indexes, similar to the Mx or Sx, labeled TOx (427, 440, 460, 476) or ORx (430, 432), from measurements with near-infrared spectroscopy (NIRS) or an invasive tissue O2 tension probe, respectively.
Finally, modeling of autoregulation in stroke has been performed with the multimodal pressure-flow (MMPF) method (458, 478, 479). This approach generates estimates of phase differences between fluctuations in BP and CBF velocity (either spontaneous or from Valsalva maneuvers), but these should not be confounded with phase differences derived by TFA as there are substantial differences in the data processing involved. In fact, the authors have shown that MMPF seems to provide greater sensitivity than TFA in detecting deterioration of autoregulation in stroke and other conditions (95, 110, 458, 464, 465, 467, 480, 481).
5.3.2. Temporal course of changes in autoregulation following stroke.
Most studies of autoregulation in ischemic stroke have performed assessments in the acute (<48 h) and subacute phases (<14 days after ictus) (410, 412), with a much smaller number of studies performing measurements several weeks or months after stroke onset (450, 452, 453, 455, 458, 459, 473, 478, 479, 482, 483). Given the considerable heterogeneity of patient conditions and autoregulation assessment methodology, inferences about longitudinal changes in autoregulatory metrics can only be made from a relatively small number of studies that performed two or more intrapatient measurements. In addition to the time period encompassing measurements, it is also relevant to take into consideration whether autoregulation parameters were found to be altered in comparison with controls, or not. In studies that did not have a control group, or indications that autoregulation was impaired at any time following stroke, autoregulatory metrics were found to deteriorate within the first 5 days following stroke (444, 449). In contrast, others detected improvement over time, mainly in the acute and subacute stages (445, 484), although Kwan et al. (455) found increased values of TFA phase and decreased gain 3 mo after stroke onset. When control groups were included, measures of autoregulation indicated that impairment was present from the acute to the subacute stages, up to 28 days after stroke onset (452, 471, 485, 486). Noteworthy, two studies reported normalization of the ARI after 3 mo, following significantly reduced values of this index at approximately 5 days (459) and after 2 wk (473), respectively. On the other hand, Guo et al. (483) obtained reduced values of TFA phase, suggesting impaired dynamic autoregulation, at <48 h after onset that were not altered 6 mo later. Although serial assessments were not performed, Novak et al. (458) and Hu et al. (479, also found that the phase, derived with the MMPF method, was lower in comparison with controls, an average of 18 and 6 mo after stroke onset, respectively, but other investigators reported normal values of autoregulation parameters 1 mo up to 63 mo after ictus (453, 482). In summary, most studies assessing the evolution of autoregulation parameters in ischemic stroke, have shown a deterioration of autoregulation metrics in the first 2 wk after ictus, with return to normal values between 1 and 3 mo later.
Although the corresponding number of studies in ICH is much smaller, there seems to be a similar pattern. Based on TFA phase and the ARI, respectively, Oeinck et al. (465) and Minhas et al. (472) have not observed any changes in the TFA phase and ARI, respectively, in the first 5 to 14 days after ictus. Reinhard et al. (467) reported Mx index values not different from controls, on day 1, but by day 5, higher values of the Mx index (indicating worse dynamic autoregulation) were a significant predictor of outcome. A longer follow up (464) indicated that TFA phase was lower than controls from days 1–2, reaching a minimum by days 10–12, but then recovering to comparable values as controls at 30 days after ictus. Therefore, these data on TFA phase and ARI support the earlier temporal changes in autoregulation after stroke, with a decline in the first 2 wk after ictus and a return to normal values thereafter.
In SAH, several studies reported impairment of autoregulation from days 1 to 4 after admission (424–426, 428–430, 432, 435, 436, 439, 440, 466), in some cases with temporary improvement, followed by further deterioration around days 7–14 (427, 429, 430, 434, 435). The occurrence of vasospasm, often followed by delayed cerebral ischemia (DCI), shows an interaction with autoregulation impairment.
The temporal evolution of effectiveness of autoregulation, as suggested by different metrics, needs to be taken into account to inform the clinical management of patients with either ischemic or hemorrhagic stroke but cannot be considered in isolation from a number of other factors, such as the severity of stroke.
5.3.3. Influence of stroke severity and subtype on estimates of autoregulation.
When examining the influence of stroke severity, controlled studies in ischemic and hemorrhagic stroke indicate that indexes of autoregulation show normal values in mild strokes but have a greater tendency to present abnormal values in moderate and severe strokes (410, 412, 415, 416, 421). The volumes of the core infarct and penumbral regions (26) likely influence the relationship between autoregulation and stroke severity, but distinguishing the core and penumbral regions has been elusive (415). In many studies, the association of autoregulation with severity is incidental without formal statistical testing. However, a reasonable number of studies have used a nominal or ordinal scale to assess the relationship between indexes of autoregulation and corresponding measures of severity. The National Institutes of Health Stroke Scale (NIHSS) is the main tool for quantifying severity in stroke, with values 1–5 indicating a mild stroke and very severe strokes corresponding to values >25 (441, 444, 446, 462, 486). Other measures of severity that have been used are the Glasgow Coma Score (GCS) (443, 464, 465, 467), the Barthel Index (462), degree of stenosis (456, 468, 487, 488), or other scales (450). With the use of MRI or CT imaging, classification of severity has also been based on the volume of infarct or hematoma (442, 443, 448, 449, 467, 489, 490), the presence of brain atrophy (478), hemorrhagic transformation in ischemic stroke (489), or ventricular hemorrhage in ICH (467, 472). In SAH, the most common measures of severity are the GCS, the Hunt and Hess score, the World Federation of Neurological Surgeons Scale (WFNS), or the Fisher scale (491). In summary, independent of the measures used to express stroke severity, or the parameter adopted for assessment of autoregulation, these studies found no association between autoregulatory metrics and stroke severity for mild strokes (442–444, 446, 462, 486, 487). However, in moderate and severe strokes, there is a positive association between stroke severity and worse autoregulation, independently of stroke etiology (441, 446, 448–450, 456, 464, 465, 467, 468, 478, 487–489, 492). The influence of stroke severity on autoregulation in SAH is more complex, due to the development of vasospasm and DCI, entities that by themselves are dependent on severity. Direct association of the Hunt and Hess, WFNS, Fisher, or GCS with autoregulation was reported in only a few cases (435). On the other hand, multiple studies suggested an association of indexes of autoregulation with the occurrence of vasospasm (424, 429, 434, 436, 437, 477) or DCI (426, 428, 430, 433, 435, 436, 493). Given that autoregulation was found to be depressed after SAH, often before the development of vasospasm and DCI, several authors have proposed predictive models, showing that alterations in autoregulation indexes were independent predictors of vasospasm and/or DCI (424, 428, 435, 436, 460, 466). The interdependence of stroke subtypes, severity, phenotype, and comorbidity makes it difficult to unravel the separate influences of these factors on autoregulation. Studies specifically designed to assess the influence of stroke subtypes on autoregulation have adopted either the Oxford Community Stroke Project (OCSP) or the TOAST criteria for classifying stroke subtype (494) but have not controlled for co-factors and usually involved a relatively small number of participants (441, 451, 454, 462, 469, 486, 494). Most of these studies have not found a significant, multiclass association of autoregulation status with either the OCSP or the TOAST classifications, with the exception of Ma et al. (486) who reported lower values of TFA phase in strokes due to large artery atherosclerosis when compared with small vessel occlusions. A larger number of studies however investigated interhemispherical comparisons of autoregulation parameters for different subtypes of ischemic stroke. In general, small vessel disease and lacunar strokes result in bilateral impairment of autoregulation, as might be expected given their more diffuse distribution (191, 458, 463, 469, 483, 495). For large vessel occlusions though, a more complex pattern emerges, given the expectation that unilateral lesions should lead to impairment of autoregulation in the affected hemisphere, when compared with the unaffected hemisphere. This was shown to be the case in some studies (463, 469, 484, 490), but in other studies autoregulation was depressed in both hemispheres, despite unilateral occlusion (486, 495). In the majority of studies though, the population included a mixture of stroke subtypes, with small vessel disease or lacunar infarcts representing a variable proportion (usually <50%) of the sample. What is startling is that in all these studies, both hemispheres had depressed autoregulation, independently of the metric adopted for autoregulation (441, 442, 451, 454, 455, 459, 462, 470, 471, 473, 478, 479, 496). In ischemic strokes of undetermined etiology, Tutaj et al. (457) found impaired autoregulation only in the unaffected hemisphere. In ICH, most studies did not find a difference in autoregulation parameters between sides, and an association with hematoma location was not reported (448, 464, 465, 467, 472). Of note, in some of these studies, the autoregulation metric was not different from controls. For SAH, we could not find any reports of an association between the extent of impairment of autoregulation and the location of the ruptured aneurysm. One possible explanation for this lack of specificity, is that the diffusion of blood in the subarachnoid space, and its effects of triggering endothelial dysfunction, reactive oxygen species formation, and hypersensitivity of vascular muscle to contraction (139, 419, 420, 497), make the corresponding loss of autoregulation independent of the site of bleeding.
5.3.3.1. interim summary.
Autoregulation parameters after ischemic or hemorrhagic stroke tend to show similar values in both hemispheres, independently of location or subtype, with only a few studies reporting unilateral impairment. The difficulty of explaining the extent of autoregulation impairment based on stroke subtype is undoubtedly related to the intervening effects of stroke severity. A few studies highlight this interdependence (446, 488, 495), but much more work is needed to improve our understanding of how autoregulation is influenced by the confluence of stroke subtype and severity.
5.3.4. The association of autoregulation with stroke outcome.
The potential association of autoregulation efficiency with the outcome of stroke is of considerable interest, not only for its more immediate importance to guide patient management, but also to shed light on the causal mechanisms involved in the pathophysiology underlying alterations of autoregulation. The relevance of this association is reflected by the recent increase in the number of studies describing the relationship between autoregulation and outcomes. In these studies, outcome has been nearly unanimously expressed with the modified Rankin Scale (mRS) (498) at 90 days after stroke onset, dichotomized as good (0–2) or poor (3–6). In ischemic stroke, poor outcomes were associated with parameters indicating impairment of autoregulation; reduced TFA phase (456, 486, 490, 496), increasing Mx index (445, 449), increased TFA gain (499), or lower ARI (446). The short-term prognostic value of depressed dynamic autoregulation, lower TFA phase 6 h after stroke onset, was shown by its association with hemorrhagic transformation and cerebral edema (489). Similar associations of TFA phase and the Mx index with outcome were observed in ICH (464, 465, 467). In patients with SAH, outcome was often assessed with the mRS at discharge, as well as at 3, or sometimes 6, mo after discharge, with the majority of studies reporting a strong association with autoregulation (426, 429, 430, 432, 434, 435, 475, 476). A measure of functional independence and the efficiency of rehabilitation were also associated to severity of SAH and the shape of the autoregulation curve (425).
5.3.4.1. interim summary.
Taken together, the accumulated evidence that autoregulation is often impaired in ischemic stroke, ICH, and SAH, with significant associations to severity and outcome, suggests that further consideration should be given to incorporate autoregulatory assessment as part of routine protocols to manage patients with stroke. To progress in that direction though, there are important gaps in our knowledge about the pathophysiology of autoregulation in humans that need to be addressed.
The association of different indexes of autoregulation with severity and outcome of ischemic and hemorrhagic stroke would suggest that one possible strategy to improve stroke treatment is to take autoregulation efficacy into account in treatment protocols. This approach has been gaining momentum in the critical care of patients with severe brain injury and also in premature newborns through the concept of optimal cerebral perfusion pressure or optimal BP (493, 500). Given the long monitoring times required by this approach, indexes of autoregulation have been estimated with NIRS or from the BP-ICP relationship, with the pressure-reactivity index (501), rather than with indexes derived from CBF velocity (TCD). By observing changes in pressure-reactivity index or a number of different indexes obtained from NIRS (500), a U-shaped curve can be derived as a function of BP or cerebral perfusion pressure, with its minimum indicating the optimal value to maximize autoregulation efficacy (502). A recent NIRS-based study has demonstrated the feasibility of using the optimal-BP approach in ischemic stroke, in patients undergoing mechanical thrombectomy (503). A similar approach in SAH has shown a strong association between outcome and the time patients spent outside the boundaries of the optimal BP interval (475, 476). The possibility of individualizing optimal BP management, as compared with the adoption of fixed BP targets, and the better outcomes of the former (475, 476, 503), warrant further research, including large scale clinical trials. On the other hand, the fairly long observation times required with the optimal BP approach, which was 28 ± 18 h (503), 68 ± 51 h (476), or an average of 95 h per patient (475), might be a critical limitation for extending this approach to all stroke patients. Moreover, unless mean BP is manipulated, its spontaneous variation might not necessarily cover a sufficient range to show a U-shaped curve and hence the identification of the optimal BP target. Alternatively, a simpler approach to restoring autoregulation to normality was proposed in the context of ICH (472), by using hyperventilation to improve dynamic autoregulation by means of hypocapnia (101, 223). The possibility of adopting a similar strategy in ischemic stroke, could be limited by the observation that spontaneous hypocapnia already seems to be prevalent in this population (504). Furthermore, the effects of hypocapnia on CBF are transient, as CBF starts to return to normal after 4-6 h of hyperventilation (505).
Although restoration of autoregulation efficacy seems an intuitive target in the treatment of ischemic and hemorrhagic stroke, its judicious application to patients with different phenotypes, stroke subtypes, location, and severity, will undoubtedly require more work to fill in the gaps in knowledge identified in the sections above. Considerably more data are needed to characterize the temporal course of autoregulation stratified by stroke subtypes, location, and severity, for example. Likewise, the recovery in autoregulation, observed between 1 and 3 mo after ischemic stroke onset (459, 473), needs to be better understood, in particular how individual time courses relate to outcome, which hitherto remains unexplored.
Although targeting improvement of autoregulation as a potential coadjutant in stroke therapy is an avenue that needs further exploration, the overall conclusion so far is that advances in clinical or surgical management of patients with ischemic or hemorrhagic stroke are unlikely to be achieved without due consideration for the key role of autoregulation in poststroke pathophysiology and management.
5.3.5. Autoregulation and blood pressure control in stroke.
Although the role of autoregulation has not been directly investigated in large randomized controlled trials (RCT) of BP management in ischemic or hemorrhagic stroke, some indirect evidence about autoregulation efficacy in stroke can be inferred from the results of several RCTs, and important lessons can be learned for improving the design of future trials. Following ischemic stroke, a significant number of patients present with hypertension (411, 506), which has been the main drive to test different approaches to lowering BP in the acute and subacute stages (507–511). Theories about autoregulation in stroke play an important role in the rationale behind such trials. For example, the increase in BP can be hypothesized as serving a purpose of maintaining CBF (specifically in the penumbra) and lowering BP may therefore be disadvantageous. On the other hand, if autoregulation is impaired in the penumbra, the increase in BP following stroke may cause hyperperfusion injury in the infarcted area, and BP-lowering interventions may prove beneficial. However, excessive BP lowering may cause hypoperfusion injury if autoregulation is impaired. This reasoning also extends to ICH, where antihypertensive therapy has been the focus of many trials in ICH in the attempt to reduce the risk of hematoma expansion and intraventricular hemorrhage due to elevated BP (512–514). Many other hypotheses on what would constitute optimal BP management in stroke can be formulated based on assumptions of autoregulation in the affected infarct area, affected hemisphere, or unaffected hemisphere. The setting of optimal BP targets, following ischemic stroke, is also complicated by differences in the extent of the collateral circulation in individual patients, which will play a key role in reperfusion of the penumbra (411, 515). In summary, in both types of stroke, the underlying assumption for many trials has been that autoregulation would be impaired and CBF would then need to be maintained within safe limits by control of BP.
Overall, different strategies for lowering BP, involving different BP target levels, timing of intervention, or types of vasodepressor drugs, have shown significant reductions in systolic and diastolic BP, in comparison with placebo or other controls, but could not demonstrate a difference in favorable clinical outcomes, usually based on mRS <3 (507–514, 516). On the other hand, most trials have not detected any adverse effects of antihypertensive therapy either. Noteworthy, the Controlling Hypertension and Hypotension Immediately PostStroke (CHHIPS) trial, comparing the use of labetalol or lisinopril to placebo, reported a reduction in mortality at 3 mo from 20.3% (placebo) to 9.7% in the treatment group (509). A pooled analysis of two previous large RCTs showed that achieving early and stable systolic BP was associated with favorable outcomes in patients with ICH (513).
The lack of impact of BP-lowering therapy on the outcome from stroke, as reported by most trials, would suggest that, contrary to the underlying assumptions, autoregulation was intact in the populations studied and the reductions in BP did not influence cerebral perfusion. This interpretation, however, needs reconsideration, taking into account three key aspects of the involvement of autoregulation in ischemic and hemorrhagic stroke: 1) severity of stroke, 2) extent of BP reduction achieved, and 3) timing of intervention and assessment. The association of stroke severity with impairment of autoregulation would suggest that most large RCT samples would involve a mixture of patients with autoregulation ranging from intact to severely affected and that the accruing benefits of BP lowering could then be diluted by the prevalence of less severe strokes. As an example, in the pooled analysis of the INTERACT-2 and ATACH-2 trials, the patients showing improved outcomes were mainly those with mild-to-moderate ICH (513). Although most trials demonstrated a significant reduction in BP in the treated group compared with controls, the amplitude of the difference is likely to have an association with outcome. In the RIGHT-2 trial, early application of transdermal glyceryl trinitrate led to a mean reduction in BP of only 5.8 mmHg in comparison with patients receiving a sham patch (508), while the trial in ICH, where improvements in outcome were observed, had a sustained mean systolic BP reduction of 29 mmHg (513). In line with this latter observation, the CHHIPS study, where a difference in mortality was detected, had a systolic BP reduction of 31 mmHg (at 2 wk) compared with placebo (509). Although a “dose-response” relationship remains to be investigated, it is more likely that differences in outcome would be observed with larger BP reductions, as long as the mean BP remains above the lower limit of autoregulation. Following this, positive effects of larger BP reductions may have been obscured in these trials by patients in whom BP was lowered too much, i.e., below the lower limit of autoregulation. Knowledge of a patient’s prestroke habitual BP is essential to define optimal BP targets poststroke, but this information is often not known or not taken into account. The most critical aspect of the trials under discussion is the timing of interventions, especially in relation to our current knowledge of the temporal evolution of autoregulation poststroke. Autoregulation tends to be impaired at ∼2 wk after stroke onset, followed by recovery by 3 mo. Given this time course, it could be expected that maximum benefit would be obtained by achieving BP targets in the most critical period, that is between 1 and 3 wk after onset. This was the case with the CHHIPS trial (509), but in most RCTs intervention took place <36 h after onset (507, 508, 511, 512), and it was not demonstrated that reduced BP levels were sustained for more than a few days following treatment.
This mismatch between BP control and the time-dependent deterioration of autoregulation poststroke is likely to have contributed to the absence of significant impact of BP-lowering therapy on stroke outcomes (517). An ongoing RCT (CAARBS) will perform multiple observations of BP, following administration of Ca2+ channel blockers or angiotensin-converting enzyme inhibitors, which might allow a better assessment of outcome taking into account the timing of BP reduction in comparison with the expected evolution of autoregulation efficacy poststroke (510).
In summary, the main lesson learned from scrutiny of RCTs aiming to improve outcome by means of BP control is that inclusion of autoregulation assessment provides the “missing link” to allow objective interpretation of results that could lead to better informed, individualized treatment of patients with ischemic or hemorrhagic stroke.
5.4. Autoregulation in Cognitive Impairment and Dementias
5.4.1. Vascular dementia.
Vascular dementia represents a heterogeneous group of brain disorders in which cognitive impairment is attributable to cerebrovascular abnormalities and is responsible for ∼20% of all cases of dementia (518). When mixed dementia is taken into consideration (the combination of neurodegeneration and vascular disease), the contribution of vascular changes to all dementias is much more substantial (374, 519–523). Vascular dementia can present in several ways clinically (376, 518, 524–526). For brevity, we will divide them broadly into two forms: 1) a typical, easily recognized presentation; and 2) a common, but atypical and less distinct presentation. In the typical presentation and characteristic form, the patient will have a history of clinical stroke followed by the onset of cognitive impairment, or a history of repeated clinical strokes followed by a stepwise deterioration in cognitive function. On examination, these patients have signs of stroke (e.g., hemiparesis) and have characteristic neuropsychological deficits, specifically in executive function and processing speed. Their memory deficits benefit from cues, and typically they function well in a structured environment. In contrast, the common but more atypical presentation of vascular dementia is that of a gradual cognitive decline (more similar to Alzheimer’s disease) combined with the identification on MRI of significant vascular brain injury (e.g., diffuse white matter lesions) (374, 376, 518, 523, 526). Often, this vascular brain injury has been clinically silent in the sense that there is no history of transient ischemia attacks or stroke. Clinically, the vascular lesions may not manifest as hemiparesis but as a more subtle disorder in balance, reduced motor coordination, vascular Parkinsonism, or loss of urinary control (518). A complicating factor is that there is considerable overlap in cognitive profiles with which patients with vascular dementia or Alzheimer’s disease may present. For example, vascular dementia patients can have predominant memory dysfunction, which is a characteristic of Alzheimer’s disease, whereas Alzheimer’s disease patients can have prominent executive dysfunction, which is characteristic of vascular dementia (527).
In line with coronary and peripheral arterial disease, impaired endothelial function contributes to cerebrovascular disease with its unique features (32, 46, 48, 50, 263, 265, 521, 523, 528). Not surprisingly, a large number of studies, independent of the technique used to examine CBF, have demonstrated that patients with vascular dementia present with lower CBF compared with cognitively healthy peers (529, 530). Two review papers have provided overviews of TCD studies in vascular dementia (compared with Alzheimer’s disease and controls) and found that resting CBF is reduced (both in vascular dementia and Alzheimer’s disease) and that pulsatility index (a marker of vascular compliance) is increased in both dementias but more so in vascular dementia. CO2 reactivity is impaired in both dementias, but again, more so in vascular dementia (531, 532). In vascular dementia, resting CBF seems especially depressed in areas of white matter lesions. Interestingly, cerebral hypoperfusion has also been described in normal appearing white matter (533), suggesting that local blood flow reductions precede and, accordingly, may contribute to damage of white matter and its connectome (http://www.humanconnectomeproject.org). To support this hypothesis, cerebral hypoperfusion and impaired cerebrovascular reactivity to hypercapnia are associated with a larger volume of white matter lesions (534, 535), suggesting that reductions in CBF are present before the onset of dementias, supporting an important role for cerebral hypoperfusion in the pathogenesis of vascular (and potentially other) dementias.
To date, there have been no studies on autoregulation in vascular dementia to our knowledge, only studies on autoregulation in stroke patients and in patients with small vessel disease described in the sections on autoregulation in stroke and hypertension.
5.4.2. Alzheimer’s disease.
There is a vast body of literature, mainly preclinical, that describes impairment of cerebrovascular function in Alzheimer’s disease (44, 58, 323, 374, 536–545). Changes in both cerebrovascular function and structure appear to be involved in this form of dementia (58). This literature started with the rare genetic forms of Alzheimer’s disease and covers animal models mimicking genetic forms in humans, as well as clinical studies addressing genetic causes of human Alzheimer’s disease. More recently, evidence of cerebrovascular involvement has also been repeatedly demonstrated in the much more common late onset, sporadic form of Alzheimer’s disease (58, 539, 546). Because cerebrovascular disease is commonly present in late onset Alzheimer’s disease, it can be difficult to distinguish whether cerebrovascular dysfunction is a consequence of Alzheimer’s pathology or vascular disease independent of Alzheimer’s pathology (547, 548). Advances in in vivo imaging now allow better phenotyping of patients, which helps in differentiating vascular effects caused by Alzheimer’s disease from vascular effects that result simply from vascular comorbidities associated with aging (549). Also, ongoing research on rare human genetic forms of Alzheimer’s disease may help to shed light on this issue, because cerebrovascular dysfunction can be studied in the early stages of Alzheimer’s disease in a population with, because of the young age of onset, limited vascular comorbidities.
Before we discuss what is currently known about autoregulation in Alzheimer’s disease, we will first briefly summarize the literature on global changes in CBF, CO2 reactivity, and NVC in Alzheimer’s disease. These aspects of CBF regulation have been studied much more extensively than autoregulation. However, these concepts are sometimes confused, with the result that some studies of NVC or CO2 reactivity have been reported to reflect changes in autoregulation, which is incorrect.
5.4.2.1. cerebral blood flow.
CBF is essential to support changes in activity of neurons and other brain cells. Disruption of CBF regulation, baseline, temporal, or regional homeostasis, may represent a major factor in the development and progression of Alzheimer’s disease and other dementias. Previous work from animal models found that mild-to-moderate cerebral hypoperfusion impairs neuronal protein synthesis, while cerebral ischemia ultimately leads to an increase in local glutamate concentrations and contributes to the accumulation of neurotoxic molecules (e.g., amyloid-β) (545). Reported increases in amyloid-β following ischemia appear to be short-lived however, and it remains debated whether this mechanism can explain the sustained widespread accumulation of amyloid-β in Alzheimer’s disease (550). Nonetheless, these vascular-derived insults might contribute to neuronal degeneration in Alzheimer’s disease. A reduction in CBF in Alzheimer’s disease compared with healthy controls has been observed in multiple studies using different imaging modalities: Xenon-133, SPECT, PET, TCD, extracranial ultrasound, and arterial spin labeling MRI (551, 552).
Importantly, reductions in CBF have repeatedly been shown in the preclinical stages of Alzheimer’s disease (541, 548). The reduction in CBF shows a regional pattern and has been proposed as a diagnostic biomarker, comparable to FDG-PET, which is a labeled-glucose imaging modality that can identify regional hypometabolism (551, 553). Such data raise the question if lower levels of CBF in Alzheimer’s disease may reflect synaptic failure (359, 554), resulting in reduced metabolism and reduced demand for CBF. This synaptic failure then continues throughout the course of the disease and is associated with the cognitive decline in the later stages (555). In this scenario, reduced CBF is not a causal factor in neurodegeneration but a consequence of loss of synaptic function. Support for this notion comes from recent studies that reported that cerebral global hypoperfusion in the general population is related to accelerated cognitive decline (351, 352) and increased risk for dementia (351). Specifically related to Alzheimer’s disease, cross-sectional studies found that lower CBF is present in those with worse cognition (556) and in patients that are rapidly declining (557). In a prospective study, cerebral hypoperfusion, measured using MRI (arterial spin labeling), predicted progression from mild cognitive impairment to Alzheimer’s disease (558). Moreover, reduced CBF was also associated with a faster 2-yr cognitive decline in a cohort of 88 patients with dementia due to Alzheimer’s disease (559). After adjustment for subject demographics (e.g., age, sex, and education) and brain structure (e.g., gray matter volume, atrophy, white matter lesions), whole brain and parietal hypoperfusion is associated with accelerated cognitive decline. In another study, these authors confirmed that reduced regional CBF was associated with a decline in global cognitive function but also executive function (560). These studies may reflect a close matching of CBF to synaptic function, making CBF an early biomarker of disease and a predictor of disease progression.
Alternatively, reduced CBF may be caused by cerebrovascular dysfunction and contribute to loss of brain function and disease progression through a mismatch of CBF to neuronal demand. Reduced levels of CBF, measured by MRI-arterial spin labeling, represented the earliest sign of late-onset Alzheimer’s disease, present even before traditional biomarkers (e.g., cerebrospinal fluid β-amyloid, cerebral hypometabolism, or brain atrophy) (541). These findings and others (529, 548) leave open the possibility that cerebral hypoperfusion has a direct causal role in the development of Alzheimer’s disease.
5.4.2.2. co2 reactivity and nvc.
Impaired vascular reactivity to CO2 may already be present at an early, preclinical stage of Alzheimer’s disease. Indeed, individuals carrying the APOE4 gene (561), and those with early stage probable Alzheimer’s disease (562), exhibit impaired cerebrovascular responses to hypercapnia measured with TCD, compared with cognitively normal controls. These findings were confirmed in a study in younger, cognitively healthy APOE4 carriers that showed reduced responses to hypercapnia using blood O2 level-dependent (BOLD)-functional (f)MRI (563). In addition to changes in cerebrovascular reactivity, other studies suggest impaired NVC in Alzheimer’s disease, which may also already be present in the preclinical stage (44, 45, 548). With the use of BOLD-fMRI, diminished local CBF responses in brain regions engaged during cognitively demanding tasks in individuals with genetic predisposition for Alzheimer’s disease or in early stage Alzheimer’s disease have been observed (564). Moreover, fMRI studies indicate disrupted resting-state neural connectivity in brain areas susceptible to atrophy in Alzheimer’s disease (e.g., hippocampus, medial prefrontal, and cingulate cortex) (564).
5.4.2.3. autoregulation.
Evidence pointing toward progressive impairment in cerebrovascular function and structure in Alzheimer’s disease led to the hypothesis that autoregulation is impaired in Alzheimer’s disease (565). Clinically, the most relevant consequence of impairment in autoregulation in patients with Alzheimer’s disease would be their increased vulnerability to cerebral hypoperfusion following reductions in BP. This increased vulnerability could be a double-edged sword. With impaired autoregulation for a given reduction in BP, the reduction in CBF would be greater in patients with Alzheimer’s disease. Added to this, effects of Alzheimer’s pathology and hypoperfusion on neurons, glia, and other cell types could be greater than in healthy brain (58, 518, 548, 566). From a clinical perspective, the most commonly encountered reductions in BP in patients with Alzheimer’s disease are those that occur in the context of hypertension, antihypertensive treatment, daily postural changes including orthostatic hypotension, and during perioperative care.
5.4.2.3.1. Studies of autoregulation in Alzheimer’s disease.
The oldest study of autoregulation in dementia known to us was from 1971 using the methods proposed by Simard et al. (567). Global and regional CBF was measured using the intra-arterial Xenon-133. Baseline CBF was studied in 24 dementia patients (39–74 yr of age); 13 with early onset dementia (<65 yr), of whom 5 had biopsy-confirmed Alzheimer pathology; and 9 patients with onset >65 yr, including four Korsakoff and three vascular dementia patients. In 15 patients (6 young, 9 old), autoregulation was measured before and after pharmacologically induced BP changes. It is uncertain if any of these 15 patients had Alzheimer’s disease. The average reduction in mean arterial BP was 28%, with an associated mean reduction in CBF of 4%. In two cases (young onset dementia), large reductions in BP were obtained (69 and 44%). These changes were associated with reductions in CBF of 25 and 19%, respectively. Increases in BP (in 2 cases) had no effect on CBF. No control group was included, although the authors could draw from their numerous previous experiments using this technique for comparison. Despite the obvious limitations related to the heterogeneity of the sample (in both age and diagnosis), the authors found no evidence for significant impairment of autoregulation in any of the 15 patients with dementia, regardless of age or underlying diagnosis (567).
The first study of autoregulation in patients with a certain clinical diagnosis of Alzheimer’s disease was published in 2009 (568). Nine patients with Alzheimer’s disease (3 with mild cognitive impairment, CDR 0.5, 6 with mild dementia, CDR 1, mean age 68 yr) and eight age- and sex-matched controls were studied. The diagnosis was confirmed by neuropsychological evaluation, MRI, and CSF biomarkers. Dynamic autoregulation was evaluated using both spontaneous changes in BP and changes induced by repeated squatting and standing maneuvers (118). These maneuvers induce large (20–30%) changes in BP and mimic the effects of daily life postural changes on BP and CBF (118). CBF velocity, and its changes, were measured using TCD. Alzheimer’s disease patients had lower CBF and higher cerebrovascular resistance, while there were no significant differences in total brain volume between patients and controls. Despite these cerebrovascular differences, dynamic autoregulation was equally effective in counteracting BP changes in Alzheimer’s patients and controls (118).
Preserved autoregulation was confirmed in a study in 20 Alzheimer’s disease patients (no controls), 74.6 yr of age, 16 with CDR 0.5, 4 with CDR 1, and 17 with a positive amyloid-PET (569). This study evaluated static autoregulation, where CBF was measured using O15-PET before and after BP lowering induced by intravenous nicardipine. CBF was measured after a reduction in BP that was maintained for 7–10 min. While mean BP was reduced on average from 109 to 92 mmHg, global CBF was unaffected (44 vs. 43 mL/100 g/min). Moreover, there were no regional reductions in CBF, including areas with more white matter lesions, in watershed regions, and in regions with the highest β-amyloid accumulation (569).
A study of dynamic autoregulation used TCD and evaluated 10 Alzheimer’s disease patients (73 yr of age) and 17 controls during simple, every day postural challenges: standing up from sitting (100, 570). Standing up from sitting causes transient (<30 s) reductions in BP, between 15 and 20 mmHg in mean arterial BP. When performed as repeated maneuvers, similar to repeated squat-stand maneuvers, they induce strong oscillations in BP and CBF, with the benefit that sit-stand maneuvers are easier to perform in older patients than squat-stand maneuvers (100). CBF was lower and cerebrovascular resistance was higher in Alzheimer’s disease patients compared with controls. Despite these baseline differences, the CBF response to the orthostatic challenges was similar in patients and controls (570, 571). Of note, cortical oxygenation [measured using near-infrared spectroscopy (NIRS)] decreased more in Alzheimer’s disease patients, despite similar changes in CBF, which may point toward reduced O2 supply or increased O2 extraction in the microcirculation (571). However, the NIRS technique requires further validation.
Other authors confirmed normal dynamic autoregulation in 17 patients with Alzheimer’s dementia, 19 with mild cognitive impairment and 20 controls (395). However, the first potentially discrepant observation was made in a study in 12 patients (of whom 10 were also included in Refs. 570, 571). Even though a normal CBF response to a BP reduction following standing was confirmed, the transient increase in BP (20% on average) that followed the return to sitting after standing was associated with a greater increase in CBF in Alzheimer’s disease patients versus controls (24 vs. 13%). Such a difference was also observed during repeated sit- stand maneuvers, which led to repeated 20% increases and decreases in BP from baseline. In Alzheimer patients, these BP changes led to slightly larger changes in CBF than in controls (27% vs. 22%). However, assessment of dynamic autoregulation using TFA again revealed no differences between patients and controls (562).
Thus, after combinination of these smaller studies, 58 patients with Alzheimer’s disease (38 dementia, 20 mild cognitive impairment) were studied with TCD, indicating normal dynamic autoregulation, except for 1 study with partially conflicting results (562). In addition, 20 patients with mild cognitive impairment were studied using PET and had normal static autoregulation.
Recently, the largest studies on autoregulation in Alzheimer’s disease to date were published (99, 572) wherein both dynamic and static autoregulation were investigated. In 53 patients with Alzheimer’s dementia (73 yr of age), 37 patients with mild cognitive impairment (suspected early stage Alzheimer’s disease, 69 yr of age) and 47 controls (69 yr of age), dynamic autoregulation, measured with TCD, was evaluated during spontaneous changes in BP and during larger induced changes in BP using repeated sit-to-stand maneuvers (99). These induced changes led to increases and decreases in mean BP of on average 25%. This large study confirmed that dynamic autoregulation was similar in Alzheimer’s disease patients (mild cognitive impairment and dementia) and controls. This conclusion was despite clear evidence for other cerebrovascular changes in Alzheimer’s disease (reduced CBF, higher cerebrovascular resistance, and reduced CO2 reactivity).
Next, from the 53 patients with Alzheimer’s dementia, 44 patients also underwent CBF measurements using MRI-arterial spin labeling with a repeated measurement after six months of BP lowering with nilvadipine versus placebo (572). In 22 patients on nilvadipine, systolic BP was reduced by an average of 11 mmHg, while global and regional CBF were not reduced (572). There was a nonsignificant trend toward a 10% increase in global CBF following the BP reduction, which is of interest because two other studies of antihypertensive treatment in patients of similar age, but without Alzheimer’s disease, found (in larger sample sizes) a significant increase in CBF of 10% following BP-lowering treatment (see sect. 5.2.2).
5.4.2.3.2. Autoregulation: indirect evidence of normal autoregulation in Alzheimer’s disease.
There have been several large prospective studies that have measured outcomes that offer indirect evidence suggesting autoregulation is normal, similar to what was discussed in sect. 4.2 in relation to hypertension and autoregulation. These are outcomes that capture the expected consequences of impaired autoregulation, such as cognitive decline, cerebrovascular events, and falls or syncope. Regarding falls and particularly regarding syncope, we repeat the caveat that these often represent a failure of BP regulation (88) and not necessarily a failure of autoregulation (sect. 6.1).
Recent meta-analyses of antihypertensive treatment show that BP lowering is associated with a lower risk to develop dementia’s, including Alzheimer’s disease (409). In addition, the SPRINT-MIND study found a lower risk of mild cognitive impairment and possibly dementia when intensive BP-lowering treatment (to a systolic BP of 120 mmHg or below) was compared with standard treatment (405). Given the relatively short follow-up times in these studies, and the knowledge that Alzheimer’s disease is a slowly progressive disease with an asymptomatic, clinically unrecognized phase that can last 10–20 yr, we can assume that many of the participants with an outcome of mild cognitive impairment or dementia due to Alzheimer’s disease already had asymptomatic Alzheimer’s disease at the time of inclusion. If Alzheimer’s disease were associated with impaired autoregulation already in an early stage of the disease, as suggested by animal models (573), impairment in autoregulation would be predicted to be present at study inclusion in these participants. If so, exposure to BP-lowering treatment would have increased the risk for cerebral hypoperfusion and therefore increased the risk to develop progressive cognitive decline and reach the end-point of mild cognitive impairment or dementia. In reality however, the reverse was found, with a reduced risk (hazard ratio 0.84) to develop dementia (405). Moreover, in the subgroup of older (>80 yr old) patients with low cognitive scores on a validated screening test (Montreal Cognitive Assessment), indicating a subgroup that will include many patients with mild cognitive impairment or even unrecognized dementia, there was no increased risk of cognitive deterioration following intensive BP reduction (406).
5.4.2.3.3 Contrast with animal models.
These findings of preserved autoregulation in patients with late onset Alzheimer’s disease are in contrast with observations from an animal model (573). Indications that autoregulation was impaired in Alzheimer’s disease came from other studies performed in preclinical models [reviewed elsewhere (565)]. For example, impaired static autoregulation was observed at a young age in transgenic mice that overexpress amyloid precursor protein, even before the occurrence of parenchymal amyloid-β deposition (573). Impaired autoregulation in these animals was attributed to vascular dysfunction due to effects of β-amyloid, impaired contraction of vascular muscle (with increased arterial BP), and altered vascular architecture (564, 565, 573).
There are several potential explanations for the discrepancy between the animal model results and human observations. The animal model for Alzheimer’s disease is not a model for late onset Alzheimer’s disease, but rather for the rare young onset genetic form. Even there, this represents only a partial model, as they do not involve tau pathology. In addition, animal models sometimes do not develop significant neurodegeneration or cognitive decline. These and other limitations of preclinical models for Alzheimer’s disease have been discussed elsewhere (574) and likely contribute to the discrepancies described to date between studies performed in animals and humans. This also highlights the difficulty of translating observations from animals to clinical populations in humans.
5.4.2.3.4. Summary and clinical interpretation.
There is consistent evidence for cerebrovascular changes in Alzheimer’s disease, which can be summarized as an increase in vascular resistance, an early reduction in global (and regional) CBF, and impaired NVC and CO2 reactivity. These changes appear to be somewhat selective, because there is now substantial evidence that autoregulation is preserved in patients with Alzheimer’s disease. The available studies were performed under conditions wherein BP was increased or decreased by ∼20–25% or less. This was done through BP-lowering medication or by postural challenges to decrease or increase BP. Thus we can translate these studies to clinical situations such as treatment of mild to moderate hypertension, or to orthostatic hypotension, but not to more extreme conditions such as malignant hypertension (hypertensive crisis), or septic or hypovolemic shock. The clinical implication is that patients with Alzheimer’s disease have no increased vulnerability to BP-lowering treatment and therefore do not require higher levels of BP to maintain normal CBF. For example, when a hypertension guideline advises a BP target of <140 mmHg systolic and <90 mmHg diastolic for a hypertensive patient, there is no evidence that if this patient also has Alzheimer’s disease, these targets should be raised to higher levels of BP to preserve CBF. There may of course be other arguments not to follow such guidelines, for example, time to benefit, patient preference, severe frailty, and so forth. However, available data indicated that fear of causing cerebral hypoperfusion should no longer be an argument. Of note, but not reviewed here (as it is beyond the current scope), there is also no evidence that Alzheimer’s disease patients have increased risk of orthostatic hypotension related to antihypertensive treatment (402, 570, 575, 576).
The impairment in NVC or CO2 reactivity that is associated with Alzheimer’s disease is not directly related to autoregulation. Higher levels of BP may not translate into better NVC, nor will lower levels (within the ranges described) further impair NVC. In contrast, longstanding untreated hypertension can promote cerebrovascular disease and thereby further impairment in NVC or CO2 reactivity. Some studies even suggest that it is not BP per se, but factors associated with hypertension, that cause impairment in NVC. For example, elevated levels of tissue or circulating angiotensin II (165, 528) may directly affect endothelial function, which could then contribute to impairment in NVC, independent of changes in BP. The clinical relevance of this discussion is that hypertension and Alzheimer’s disease often coexist. Around 40% of Alzheimer’s disease patients also have hypertension (577). Vice versa, the prevalence of hypertension increases to 60% with aging. Because aging is the strongest risk factor for Alzheimer’s disease (578), many older hypertensive patients may develop Alzheimer’s disease.
5.5. Autoregulation in Neurocritical Care
We have explained the strong capacity of the cerebrovasculature and dynamic autoregulation to protect against significant increases in CBF (hyperperfusion) during acute increases in BP or hypertensive emergencies. Clinical conditions that can cause such increases in BP include drug abuse (i.e., cocaine, amphetamines), anxiety or panic disorders, stroke, traumatic brain injury, aortic dissection, and hypertensive disorders of pregnancy (130). End-organ damage to brain (and the retina) can result if autoregulatory capacity is exceeded, resulting in marked increases in CBF, endothelial dysfunction, increased microvascular pressure, loss of blood-brain barrier integrity, and edema (130).
Neurointensive care is a field where protection of the brain against effects of either reduced or increased perfusion pressure is critical (130, 500, 579–581). In the majority of this review, we have discussed autoregulation and diseases related to autoregulation, generally in the context of conditions where ICP is normal and relatively stable. In neurocritical care, this situation can be very different. For example, in ICH or in traumatic brain injury, brain damage can result from elevated ICP. Under these conditions, monitoring and treatment are therefore not only focused on maintaining adequate CBF to supply O2 and glucose but also on adapting BP to changes in ICP. The importance of ICP monitoring and the availability of long-term measurements of ICP in the neurointensive care unit has led to monitoring of autoregulation based on the relationship between BP and ICP, as well as the relationship between BP and CBF (500, 579–581). This field of clinical research is unique in that it has provided long-term (hours) monitoring, where changes in autoregulation over time can be linked to clinical prognosis (500, 581–587). In this setting, additional factors come into play in relation to autoregulation (e.g., brain swelling and edema, CSF circulation, vascular or neuroinflammation, or changes in the meninges or choroid plexus: key sites of immune cell trafficking), making BP and ICP regulation, versus CBF regulation, topics that would require a review of its own. Fortunately, several excellent recent reviews on this topic are available (493, 500, 581).
5.6. Direct Control of the Brain on Blood Pressure?
Recently, the idea has resurfaced that the brain may directly control BP to preserve its function. This was the idea formulated by Hill (4) and Bayliss et al. (5) around 1890, who were convinced that there was no active autoregulation to maintain CBF, and therefore (paraphrased), the rest of the body functioned as a slave to the brain, increasing BP if the brain needed more CBF. Indeed, in more recent times, common examples such as the increase in BP following stroke has been interpreted as direct control of the brain on BP in response to the failing CBF. In a similar vein, the higher BP that is associated with aging has been considered to be caused by the brain in response to the waning CBF with aging (588). This idea was recently reframed (120 yr after Hill) as the “selfish brain” hypothesis (note that the “selfish brain” term has also been used for very different hypotheses for mood disorders, addiction, etc.) (588).
A recent study provided evidence for a direct effect of the brain on BP, following a change in ICP and cerebral hemodynamics (178). This study identified astrocytes that lie adjacent to brain autonomic centers as potential modifiers of BP to maintain brain function. There are however several methodological caveats to this study. First, the results are presented as if it is a reduction in CBF that triggered the astrocytes’ response (178). However, the reduction in CBF was brought about by an increase in ICP, raising the question of whether the astrocytic response was due to increased ICP or to reduced CBF (178). Raising ICP to lower CBF was perhaps the only reasonable design, because other options to reduce CBF (i.e., lowering BP) would have triggered baroreflex responses (88). Activation of the classic baroreflex response would have confounded the results, as the authors wished to test if astrocytes functioned as a “brain baroreflex.” The increase in ICP (and subsequent reduction in CBF) led to an increase in BP with a delay of ∼30 s, persisting for ∼30 min: the time scale of these changes clearly differs from the faster changes associated with the classic baroreflex. Interpretation of this work is further complicated by the fact that the somewhat moderate increased in ICP (10–15 mmHg) was associated with a 40% reduction in CBF, suggesting autoregulation in this model was rather ineffective. In contrast, multiple other studies have reported that CBF is maintained quite effectively during increases in ICP of this magnitude or more. These issues aside, this study of astrocytes is of potential interest, perhaps applicable to the neurointensive care unit setting, where elevated ICP is frequently observed (see sect. 5.5). Second, with regard to the discussion above, most other instances where CBF is reduced (e.g., following BP lowering, antihypertensive treatment, postural changes, hypovolemic, or septic shock), ICP decreases in parallel with CBF (344, 589). Therefore, it remains uncertain if a reduction in CBF, without an increase in ICP, would trigger a “brain-initiated” increase in BP, mediated through the autonomic nervous system.
5.7. Anesthesia and Autoregulation
From a clinical perspective, the topic of anesthesia and autoregulation involves not only the use of anesthetic drugs but also control of respiration and respiratory gases, BP, combined with the potential hemodynamic effects of surgery. The relevance of taking autoregulation into account to prevent cerebral ischemia during surgery has recently been reviewed (590) and is also explained in our call-out text box for clinicians. A discussion of complex cardiovascular or cardiothoracic surgery, with extracorporeal circulation and anterograde selective cerebral perfusion (591), is beyond the scope of this review. This also applies to the use of anesthesia in traumatic brain injury, where a recent review identified important knowledge gaps (592). For the effects of changes in concentration of respiratory gases (O2 and CO2) on CBF, we refer to sects. 1.3 and 3.2 of this review. Here we briefly discuss the effects of anesthetic agents on CBF and autoregulation and refer to a recent review for further reading (593).
Anesthetic agents affect brain metabolism (593, 594). For most agents, the suppression of consciousness reduces metabolism and hence demand for O2 consumption. If, as under normal conditions, the tight coupling between metabolism and CBF is maintained (sect. 1.2), this reduction in the cerebral metabolic rate of O2 (CMRO2) will equally reduce CBF. However, some anesthetic agents cause an uncoupling of metabolism and blood flow (593, 594). In some cases, there may also be direct effects of anesthetics on the cerebral vasculature, changing vascular tone and thus cerebrovascular resistance. The net outcome of such changes on CBF during anesthesia depends on whether metabolism and blood flow remain coupled, and on interaction with changes in respiratory gases. Although anesthetics affect CBF, most agents do not impair autoregulation, with the exception of volatile anesthetics (the fluranes) (593). The effects of commonly used anesthetic agents on CBF, metabolism (CMRO2), and autoregulation are briefly summarized below.
Isoflurane, desflurane, and sevoflurane (volatile agents) have a direct vasodilatory effect on cerebral blood vessels, and this effect is independent of the dose or depth of anesthesia (595). The effects on autoregulation however are dose dependent, with normal autoregulation at lower dose for isoflurane or desflurane (1 MCA), but impaired autoregulation above 1.5 MCA. For sevoflurane, autoregulation is already impaired at a dose above 1 MCA (593, 596).
Effects of fluranes or anesthetics are modified by CO2, where hypercapnia lowers the threshold for impairment in autoregulation, and hypocapnia increases it. These anesthetics all lower brain metabolism (CMRO2) and because they also induce vasodilation cause uncoupling between metabolism and CBF (i.e., CBF is higher than metabolism would demand) (593). Nitrous oxide, when used as a single agent (which is no longer in practice), increases CBF while CMRO2 remains stable; however, when used in combination with other anesthetics, results are inconclusive. There is no information on its effects on autoregulation (593, 596). Propofol reduces brain metabolism, which in turn leads to a reduction in CBF. Direct in vitro vascular application of propofol causes vasodilation. Autoregulation remains intact unless doses of propofol exceed 200 µgram/kg/min (593, 596). Ketamine increases CBF through direct vasodilation, but there is no evidence for effects on autoregulation (593, 596). Etomidate reduces CMRO2 and thereby CBF. This agent has a potential direct vasoconstrictor effect, but there are no known effects on autoregulation. Unique among anesthetics, etomidate does not lower BP (593). Dexmedetomidine reduces CBF, potentially by activation of postsynaptic α2-adrenergic receptors that induce vasoconstriction, and in addition reduce CMRO2 (525). Benzodiazepines (e.g., midazolam), nonbenzodiazepine agents that target GABA (e.g., zolpidem), and opioids (e.g., fentanyl) reduce CRMO2 and thereby CBF but do not affect autoregulation (593, 596).
Because of their effects on cerebral metabolism and on cerebrovascular tone, anesthetic agents can also affect NVC. From a research perspective, this has implications for the interpretation of studies on NVC in preclinical models, which are most commonly performed under anesthesia (594). Anesthesia may also affect NVC in humans (597, 598). New approaches that allow studies in awake animals have been proposed to address this potential limitation (594).
6. IMPACT OF DAILY LIFE CHALLENGES ON CEREBRAL BLOOD FLOW
6.1. Postural Challenges
6.1.1. Orthostatic hypotension.
Whenever we stand up from a lying or sitting position, the combination of our upright posture and Earth’s gravity causes large shifts in circulatory volume (300–500 mL) from the central body and head to the lower abdomen and legs (599). These fluid shifts lead to reductions in venous return to the heart, reducing cardiac output and BP. Actively standing up also causes reflex arterial vasodilation (to support muscle perfusion) that contributes to the reduction in BP. Maximum decreases in BP after standing are reached within 15 s and can easily be as large as 15–25% (599). The baroreflex usually restores BP within 30–40 s (88). Failure to normalize BP after 60 s is termed orthostatic hypotension, defined as a sustained reduction in systolic BP of 20 mmHg or more.
Even without orthostatic hypotension, normal BP responses pose daily challenges for autoregulation. Their speed of onset limits the potential for autoregulation to buffer these BP changes. This explains the common subtle symptoms we all may experience upon standing up. In orthostatic hypotension however, sustained reductions in systolic BP can be as large as 60 mmHg and exceed the lower limits of autoregulation, causing cerebral hypoperfusion, dizziness, falls, or syncope.
The longer we lie or sit, the stronger is this transient decrease in BP. Other postural challenges may also cause large reductions in BP. For example, getting up after squatting leads to strong reductions in BP (118). Combining standing with a Valsalva maneuver (e.g., singing, blowing a horn) may also elicit a strong fall in BP. Bending forward may cause abdominal vascular compression, more so in pregnant women or in obesity, and provoke hypotension (599).
These challenges occur on a daily basis and are an important reminder that BP is not a stable entity per se. Steady-state BP, measured sitting after 5 min of rest, should indeed be more or less constant in a healthy person if repeated with an interval of days or weeks. However, when measured over shorter time intervals throughout a day, BP constantly varies with postural changes. The strong impact of postural changes on both BP and CBF can easily be learned from studies using repeated sit-stand or squat-stand maneuvers (118, 570) (FIGURE 4).
6.1.2. Syncope.
Syncope (or fainting) is defined as a brief loss of consciousness caused by a transient reduction in CBF (600–602). This definition clearly sets syncope apart from the two other most common causes of transient loss of consciousness: seizures (epilepsy) and psychogenic syncope. The definition of syncope may have contributed to a common misconception that syncope is caused by a disorder in autoregulation. Syncope is however primarily caused by a failure to maintain stable and adequate BP (600–602). Theoretically, for the same reduction in BP, a person with impaired autoregulation could experience (pre)syncope earlier than someone with normal autoregulation. However, the influence of BP regulation in syncope outweighs that of autoregulation (603).
Syncope is caused by a failure to maintain BP at a level that is required to maintain sufficient CBF (604, 605). Autoregulation operates within two boundaries: the magnitude (static) and the speed (dynamic) of BP changes. The magnitude refers to the limits of static autoregulation. When BP falls below the lower limit of autoregulation (e.g., below a systolic BP of 70–80 mmHg), CBF will fall progressively with further reductions in BP. The speed refers to limitations in response time of dynamic autoregulation. Slow changes in BP are much more efficiently buffered than fast changes, and very fast changes (e.g., those occurring within 1–5 s) cannot be buffered at all.
BP is determined by cardiac output and total peripheral resistance (170, 604, 606–608). Cardiac output is in turn determined by stroke volume and heart rate, whereas peripheral vascular resistance is determined by vascular structure, vascular mechanics, and vascular tone of the systemic circulation (170, 604, 606–608). The baroreflex system plays an important role in short-term BP regulation, as it controls heart rate and peripheral vascular resistance through the parasympathetic (mainly vagus nerve) and sympathetic arms of the autonomic nervous system (88).
Even if baroreflex function is normal, syncope can occur when certain challenges exceed baroreflex capacity (27, 88, 609, 610). Examples are severe hypovolemia (bleeding, excessive fluid loss due to diarrhea, vomiting, exercise, or hot environments) or large volume shifts (when standing up after prolonged squatting, e.g., during gardening). This leads to a fast and large reduction in BP, that due to its speed of onset cannot be buffered by autoregulation and leads to hypoperfusion. Many cases of syncope occur during temporary baroreflex dysfunction (88). Called neurally mediated reflex, or vasovagal syncope (88), such disorders can cause the baroreflex to initiate bradycardia and peripheral vasodilation, leading to prolonged reductions in BP below the lower limit of autoregulation, and loss of consciousness. These circumstances include pain, fear, strong emotion, sustained orthostatic stress (standing still for long periods), among others. Baroreflex failure associated with disease is seen in autonomic failure (e.g., diabetes, Parkinson’s disease, primary autonomic failure, or spinal cord lesions (88). Finally, cardiac disorders (structural or rhythm) can cause syncope through their effects on BP.
6.2. Physical Activity
The spectrum of physical activity encompasses the areas of low-, moderate-, and vigorous-intensity physical activity but also the lower end of the spectrum including sedentary behavior (e.g., sitting and lying). The interest in understanding the impact of exercise-like physical activity on health (611), including the regulation of CBF (612) and its impact on brain health, dates back to the middle of the previous century. During the most recent decade, studies have started to acknowledge the importance of examining the impact of lifestyle or behavior, driven by the observation that increasing sedentary behavior, independent of exercise performance, is related to all-cause mortality and morbidity (613, 614). More recent work also suggested the potential of sedentary behavior to affect cardiovascular and cerebrovascular health and thus brain health (615–617).
6.2.1. Sedentary behavior.
In studies designed to better understand the immediate impact of prolonged sedentary behavior in healthy individuals, Carter et al. (618) examined the impact of 4- and 6-h periods of prolonged, uninterrupted sitting on CBF. With the use of TCD, prolonged, uninterrupted sitting led to a small (1.4–3.4 cm/s, equivalent to 2–5%) but significant decline in middle cerebral artery CBF velocity in healthy volunteers. Because there was a small (0.9 mmHg) reduction in end-tidal CO2, which may be the result of changes in pulmonary ventilation during prolonged sitting, it remains uncertain if a change in could have contributed to the reduction in CBF (618). In this range, even small changes in (e.g., 1 mmHg) can cause significant changes in CBF (3–8%) (223, 619). The effect of sitting on CBF has also been explored in groups at increased risk for cerebrovascular disease. A study in 12 older adults (70 yr of age) found a small reduction in CBF (620); however, or end-tidal CO2 was not measured in that study. In 22 older adults (78 yr of age, 9 women), there was no reduction in CBF after 3 h of sitting (621). CO2 was measured in that study and remained stable. Of note, there was an increase in BP with sitting. In 25 middle-aged workers with prehypertension (622), sitting, but not sitting alternated with periods of standing, was associated with a decline in CBF over the day (without measurements of or end-tidal CO2). Finally, a recent cross-sectional study found in 52 healthy older adults that greater sedentary time was significantly associated with lower CBF in lateral and medial frontal regions (623).
Some of these studies have also investigated if sedentary behavior may affect autoregulation or CO2 reactivity. While cerebrovascular responses to hypercapnia were preserved, changes in autoregulation during sit-to-stand maneuvers were found in one study (618). Prolonged uninterrupted sitting for 4 h resulted in a decrease in phase in the very-low-frequency range (618). The subsequent study that adopted a 6 h uninterrupted sitting protocol found an increase in normalized gain in the very-low-frequency range (618). The differential impairment in autoregulatory parameters between both studies may relate to a time-dependent, progressive impact of sitting on autoregulation. It is hypothesized that impairment in autoregulation may first affect phase (i.e., the latency of response), before affecting gain (i.e., the efficiency of the response) (624). However, a series of studies from our laboratory in an older population with or without increased cardiometabolic risk found no impact of prolonged, uninterrupted sitting on autoregulation, despite a significant decrease in CBF (621, 625). Therefore, it is unlikely that a change in autoregulation is responsible for the reductions in CBF during prolonged sitting that were found in some studies. One should also consider the potential impact of sitting on alternative interacting pathways or mechanisms, including baroreflex sensitivity, cerebral metabolism, endothelial function, or NVC.
Population (626)- and laboratory-based studies (627) aimed at better understanding the impact of prolonged sitting on health outcomes highlight the relative importance of breaking up sedentary behavior, independent of the duration and intensity of this physical activity break. For this purpose, studies have interrupted prolonged sitting and found that breaking up sitting prevents the decline in CBF in healthy volunteers (618). Interestingly, this is not a universal finding across all populations, since a recent study found that effects of 3 h of sitting on CBF were not prevented by physical activity breaks in subjects with prediabetes (625) or older individuals (621). Possibly, in subjects with a priori increased risk for cerebrovascular disease, low-intensity physical activity breaks are insufficient. Some support for this hypothesis is provided by a recent study that found that 30 min of moderate-intensity walking exercise prevented the decline in CBF associated with prolonged sitting (620). However, this study did not measure and control for changes in .
Taken together, recent studies suggest that prolonged sitting is associated with a stimulus-dependent decrease in CBF but with no consistent effect on dynamic autoregulation. The exact mechanisms underlying these changes in CBF are unclear but appear to result from the prolonged period of physical inactivity, since regularly interrupting sitting with short bouts of low-intensity physical activity or moderate-intensity walking exercise prevents these effects. However, changes in due to changes in ventilation and lung perfusion are expected with prolonged sitting and can be altered by the bouts of physical activity, and these changes alone may explain the effects on CBF. was not always measured and accounted for in these studies. Further work is required to better understand the impact of sedentary behavior at different levels of the cerebrovasculature, including the blood-brain barrier, or more chronic changes such as vascular remodeling or changes in vascular mechanics. Whether the effects of sitting are distinct in populations with impaired cerebrovascular health and hypoperfusion and translate to detrimental long-term effects is unclear at this time and requires further examination.
6.2.2. Exercise: role of intensity.
Exercise can be regarded as an ultimate, integrative stimulus for the brain to regulate CBF, especially since exercise is known to evoke large changes in key stimuli that affect CBF-O2 consumption, BP, cardiac output, blood gases (especially ), activity of sympathetic nerves, and brain activity. All these variables change with exercise, often with different magnitudes and patterns, constantly challenging the sufficient delivery of CBF to meet metabolic demand (but also to prevent hyperperfusion) (347, 628). Changes in CBF in response to incremental levels of exercise are typically described as biphasic. There is a progressive increase in CBF in response to whole body exercise, up to an exercise intensity of ∼60% of maximal O2 consumption, followed by a plateau in CBF and a decrease toward resting levels when reaching maximal intensity exercise (see review in Ref. 347). Our understanding of regulatory mechanisms that contribute to the biphasic CBF response to incremental levels of exercise is limited by the complexity and redundancy in processes that regulate blood flow to the brain. Below, we have summarized what are thought to be the most important factors contributing to the biphasic CBF response.
Early hypotheses assumed there was a key role for exercise-induced changes in blood gases, especially , contributing to changes in CBF with exercise. This idea was supported by the sensitivity of the cerebrovasculature to changes in but also the observation that incremental levels of exercise first caused an increase in , followed by a (hyperventilation-induced) decline in toward (near) maximal exercise intensity. Indeed, CBF was increased with increasing levels at baseline (629) or when mitigating the drop in during near maximal exercise (630). During submaximal exercise, however, increases in CBF seem less dependent on changes in , although this stimulus may still contribute to the regional distribution of CBF (631). In addition to , may also play a role in regulating CBF during exercise, especially when exercise is performed during acute or chronic hypoxia. For example, exercise during acute hypoxia elevates the CBF response, possibly as a compensatory response to a lower and maintenance of O2 delivery. In contrast, lower CBF responses to exercise are observed during chronic hypoxia. At a minimum, these studies highlight the importance of measuring both and when attempting to understand the regulation of CBF during exercise.
During exercise, marked increases in BP (30% or more), cardiac output (300–600%), and neural activity may support appropriate regional distribution of blood flow of the peripheral circulation and the brain. These factors all show a clear temporal linearity with increases in activity level, suggesting that BP, cardiac output, or neural activity alone are unlikely to dictate the biphasic CBF response during exercise. Moreover, any increases in BP should be buffered by autoregulation. Regarding cardiac output, studies have examined whether altered cardiac output during exercise, either within individuals [e.g., manipulation (632)] or between subjects [e.g., heart transplantation (633)], is followed by comparable changes in CBF. Despite increases in cardiac output with exercise, CBF remains largely unaffected, suggesting a relatively modest role for cardiac output in contributing to CBF changes with exercise. In addition, studies aimed at better understanding neurogenic mechanisms (e.g., by blocking activity or effects of parasympathetic nerves) provide somewhat conflicting results that may relate to methodological issues pertaining to these experiments (e.g., effectiveness of blockade). There is currently limited evidence supporting a key role for neurogenic mechanisms in mediating CBF changes during exercise (628).
A final factor for regulation of CBF relates to cerebral metabolism. While various techniques, exercise protocols and methodological designs have been used, the general consensus is that exercise leads to a gradual, dose-dependent increase in cerebral O2 uptake, reflective of elevated cerebral metabolism (628). Accordingly, increases in cerebral metabolism may contribute to the increased CBF at low- to moderate-intensity exercise. However, the lower CBF at near maximal exercise was not reflected by a decrease in cerebral metabolism (634). Possibly, a compensatory increase in O2 extraction and/or cerebral delivery of O2 may contribute to a further increase in cerebral metabolism with higher-intensity exercise. Alternatively, studies have suggested changes in substrate utilization may occur during exercise. More specifically, cerebral metabolism normally (under resting conditions) is thought to mainly depend on O2 and glucose. However, during exercise, there is an increased uptake of lactate, accompanied by a reduced uptake of O2 and glucose, which may suggest that the brain uses lactate as fuel, thus being less dependent on the consumption of glucose and O2 (635).
Since older age is an important risk factor for cerebrovascular pathology, several studies have explored the impact of aging on CBF during exercise. Studies examining these responses have previously been reviewed in detail (347) and suggest the presence of a similar biphasic CBF response to incremental levels of exercise as observed in healthy young individuals. However, CBF is markedly lower in older individuals compared with healthy young subjects, both at rest and during exercise (636–638). Despite these differences in absolute CBF between both groups, the magnitude of change in cerebral metabolism, including the dependency on the various metabolic substrates at different levels of exercise, are comparable between younger and older subjects. Similarly, comparable responses are found between younger and older individuals in central hemodynamics (e.g., BP, cardiac output) and neural activity. One potential factor, partly contributing to the age-related differences, may involve a lower in older individuals (638). Other factors, such as brain atrophy or altered vasodilator mechanisms, may also contribute to these age-related differences in CBF during exercise.
Taken together, the biphasic pattern of CBF during exercise seems to relate to multiple physiological mechanisms that have a complex integration, synergism, and redundancy to prevent impairment of CBF during exercise. Based on current evidence, the balance between cerebral metabolism and seem to contribute most to the biphasic pattern of CBF during incremental levels of exercise, which can be present in healthy young as well as in older populations. Nonetheless, a lower CBF in older individuals may be present at rest and during exercise, which in part may relate to a lower . Better insight into these responses of acute exercise may help to understand, but also to guide and optimize, the effectiveness of exercise training strategies to improve cerebrovascular and brain health.
6.2.3. Exercise training.
Several studies have examined the potential clinical impact of exercise training and/or physical activity in primary and secondary prevention of cerebrovascular diseases and have largely provided convincing evidence for a role of regular exercise in the clinical management of these diseases. Since this is beyond the scope of this review, we refer to excellent reviews in the literature that have covered this topic (639, 640). Here we discuss the impact of regular exercise training on CBF and autoregulation but also whether these effects depend on the population examined.
In contrast to a relatively large number of studies examining the acute effects of exercise on CBF, few have thoroughly examined the impact of regular exercise training on regional or global CBF. A recent study in healthy older men examined the impact of 8 wk of exercise training and examined global and regional CBF using MRI (641). Interestingly, this study found improved regional CBF in the frontal lobe, particularly the subcallosal and anterior cingulate gyrus, whereas a significant decrease was found in the temporal fusiform gyrus. These regional differences may explain the absence of an effect on global CBF in this study (641). Others also found regular exercise training to improve local CBF (642), including elevated bilateral blood flow to the hippocampus (643). When exercise training is causally linked to increases in local CBF, one may expect that cessation of exercise training would decrease CBF. Indeed, a study of 10-day cessation of exercise training in older master athletes found reductions in CBF in the hippocampus and gray matter (644). These results suggest that exercise training increases local CBF, specifically in regions that are linked to cognitive function.
In line with studies performed in healthy individuals, exercise training also seems to improve local CBF in clinical populations. For example, exercise training for 6 mo in stroke survivors improved CBF to grey matter and perfusion in the parietal lobe (645). However, increased local CBF is not a universal finding. For example, Alfini and coworkers (646) found increased CBF in the left insula in subjects with mild cognitive impairment, while 12 wk of endurance exercise training decreased CBF in the left insula and improved cognitive function. In subjects with cognitive impairment, changes in local CBF should be interpreted with caution, especially when examining brain regions that may be related to impaired cognitive function. Previous work has linked decreased regional CBF to the pathophysiology of cognitive impairment (351, 352). Future studies are warranted, preferably adopting a longitudinal design and concomitant assessment of cognitive function, to better understand the impact of exercise training on regional CBF and the impact of these regional changes in CBF for clinical populations.
Related to the central topic of this review, one may question whether regular exercise training impacts autoregulation. Aengevaeren et al. (363) was among the first to examine this question and compared autoregulation between older master athletes and age-matched sedentary controls. In this cross-sectional comparison, no differences in autoregulation were found between groups (363). Subsequent cross-sectional studies also found no significant differences in autoregulation between highly trained versus sedentary individuals (647–649). It is important to highlight that some recent studies suggest that changes in autoregulation may be present after high-intensity exercise (650). Such changes consisted of very small reductions in TFA phase at 0.1 Hz. Another study found no effects of high-intensity training on autoregulation (651). While work on exercise and autoregulation, largely coming from cross-sectional comparisons, suggests that regular exercise training has little impact on autoregulation in healthy individuals, one may question whether this concluvsion can be extrapolated to clinical populations. In one study, patients with chronic obstructive pulmonary disease exhibited preserved autoregulation compared with healthy controls, and 8 wk of exercise training did not alter autoregulation in either group (652).
Taken together, exercise training seems to have no major impact on autoregulation. However, regular exercise training is a potent stimulus for changes in global and regional CBF, in both healthy individuals as well as in those with cerebrovascular disease. Specifically, regular exercise training is associated with increased regional CBF, such as in the hippocampus and gray matter (i.e., regions that match with cognitive abilities like memory and attention). Within this respect, previous studies have linked cellular biological factors, e.g., brain-derived neurotrophic factor and myokines, to contribute to (hippocampal) neuroplasticity (653). Although aerobic exercise is known to have beneficial effects on the cerebral vasculature, brain health, and cognitive function, mechanisms that underlie these changes are still relatively poorly understood (654, 655). Several mechanisms are likely to be involved. Part of the benefit of exercise may be hemodynamic, including effects of shear stress on phosphorylation and activity of eNOS, reductions in oxidative stress, angiogenesis, and increased CBF (654, 655). Another mechanism may relate to effects of neurotrophic factors including brain-derived neurotrophic factor (BDNF). Exercise increases expression of BDNF, known to have positive effects on neuronal plasticity, neurogenesis, synaptogenesis, hippocampal and cognitive function (654, 655). Other data suggest that BDNF signaling may be dependent on activity of eNOS and production of NO by endothelial cells (654, 655). Considering the importance of the topic for brain health, further research is warranted to better define pathways explaining and facilitating optimization of exercise training to improve brain health.
7. SUMMARY AND CONCLUSION
The brain is a richly perfused organ with a dense vascular supply, characterized by relatively high blood flow and low vascular resistance. Its high metabolic demand, roughly one-fifth of the total body metabolism, in an organ that represents <2% of total body weight, depends on autoregulation to maintain adequate blood flow. A second important characteristic is that the brain is enclosed in the skull, which requires a much tighter control of tissue pressure (ICP) compared with other organs that have more room to expand. Autoregulation also plays an important role in the homeostasis of ICP, because an increase in cerebral blood volume will increase ICP and vice versa.
Autoregulation is the mechanism that safeguards blood flow to the brain in the context of changes in BP, the most important determinant of CBF. In this review, we have discussed physiological and pathophysiological examples where changes in BP occur. These include everyday changes in body position including effects of gravity on hemodynamics, physical activity, hypertension, and antihypertensive treatment. Autoregulation operates by regulating cerebrovascular resistance to counteract changes in BP. In relation to acute adjustments, these changes are thought to mainly occur due to actions of pressure-sensitive vascular muscle that alter vascular resistance through vasodilation or vasoconstriction (FIGURE 7).
The time frame over which changes in BP occur determines the effectiveness and stabilization of CBF by autoregulation. The effect of relatively slow changes in BP on CBF is best described by the model of static autoregulation, where it has been shown that CBF is held stable (within a margin of about ±10%) across a wide range of BP levels (≈50–150 mmHg in MAP). The effects of faster changes in BP on CBF are captured in the model of dynamic autoregulation, where progressively faster changes in BP lead to larger changes in CBF. A consequence of dynamic autoregulation is that even with optimally functioning autoregulatory mechanisms, there can still be variability in CBF caused by changes in BP.
In contrast to what is often stated, autoregulation is a robust mechanism that is not generally affected by aging or hypertension. Evolving research suggests it also remains relatively intact in Alzheimer’s disease. However, autoregulation does not function in isolation. We have described that CBF is affected and controlled by multiple factors, such as blood gases, neurovascular innervation, endothelial function, ICP, metabolism, and NVC. Conditions such as hypertension and Alzheimer’s disease can therefore affect CBF, even if they do not impair autoregulation. As an example, endothelial dysfunction associated with hypertension may lead to a chronic reduction in CBF, while autoregulation (which is independent from endothelial function) is still intact, and therefore, when BP is lowered by antihypertensive treatment, there is no risk of hypoperfusion, despite the lower baseline CBF. Thus a reduction in CBF or impairment in NVC, as has been observed in hypertension and in Alzheimer’s disease, should not be interpreted as evidence of failed autoregulation and should not justify attempts to raise BP levels to restore CBF or NVC. Vice versa, a finding of normal autoregulation in a clinical population (e.g., stroke, hypertension, Alzheimer’s disease) should not be interpreted as a guarantee that global or regional CBF, or NVC, will be normal. Instead, we proposed an integrative approach to study the regulation of CBF, hoping to stimulate research and clinical efforts to disentangle autoregulation from the multiple other mechanisms that influence CBF.
GRANTS
During the preparation of this article, F.M.F. was supported by National Institute of Neurological Disorders and Stroke Grants NS-096465 and NS-108409 and J.A.C. was supported by Alzheimer Nederland Grant WE.03-2015-15040.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.A.C. conceived and designed the review; J.A.C. and F.M.F. prepared figures; J.A.C., D.H.T., R.B.P., and F.M.F. drafted manuscript; J.A.C., D.H.T., R.B.P., and F.M.F. edited and revised manuscript; J.A.C., D.H.T., R.B.P., and F.M.F. approved final version of manuscript.
REFERENCES
- 1.Willis T. Cerebri Anatome: Cui Accessit Nervorum Descriptio Et Usus. London: James Flesher, Joseph Martyn and James Allestry, 1664. [Google Scholar]
- 2.Monro A. Observations on the Structure and Functions of the Nervous System. Edinburgh, Scotland: William Creech, 1783. [Google Scholar]
- 3.Friedland RP, Iadecola C. Roy and Sherrington (1890): a centennial reexamination of “On the regulation of the blood-supply of the brain”. Neurology 41: 10–14, 1991. doi: 10.1212/WNL.41.1.10. [DOI] [PubMed] [Google Scholar]
- 4.Hill L. The Physiology and Pathology of the Cerebral Circulation. An Experimental Research. London: J&A Churchill, 1896. [Google Scholar]
- 5.Bayliss WM, Hill L, Gulland GL. On intra-cranial pressure and the cerebral circulation: Part I. Physiological; Part II. Histological. J Physiol 18: 334–362, 1895. doi: 10.1113/jphysiol.1895.sp000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donders FC. Over de snelheid van psychische processen. Onderzoekingen gedaan in het Physiologisch Laboratorium der Utrechtsche Hoogeschool. Tweede Reeks 92-120: 1868–1869, 1868. [Google Scholar]
- 7.Mosso A. Ueber den Kreislauf des Blutes im Menschlichen Gehirn: Untersuchungen. Leipzig: Von Veit, 1881. [Google Scholar]
- 8.Sandrone S, Bacigaluppi M, Galloni MR, Cappa SF, Moro A, Catani M, Filippi M, Monti MM, Perani D, Martino G. Weighing brain activity with the balance: Angelo Mosso’s original manuscripts come to light. Brain 137: 621–633, 2014. doi: 10.1093/brain/awt091. [DOI] [PubMed] [Google Scholar]
- 9.Roy CS, Sherrington CS. On the regulation of the blood-supply of the brain. J Physiol 11: 85–158, 1890. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raichle ME. Behind the scenes of functional brain imaging: a historical and physiological perspective. Proc Natl Acad Sci U S A 95: 765–772, 1998. doi: 10.1073/pnas.95.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol 17: 1016–1024, 2018. doi: 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ, Tannenbaum A, Ballerini L, Rungta RL, Boido D, Sweeney M, Montagne A, Charpak S, Joutel A, Smith KJ, Black SE, colleagues from the Fondation Leducq Transatlantic Network of Excellence on the Role of the Perivascular Space in Cerebral Small Vessel Disease. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol 16: 137–153, 2020. doi: 10.1038/s41582-020-0312-z. [DOI] [PubMed] [Google Scholar]
- 13.Fog M. The relationship between the blood pressure and the tonic regulation of the pial arteries. J Neurol Psychiatry 1: 187–197, 1938. doi: 10.1136/jnnp.1.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forbes HS, Cobb SS. Vasomotor control of cerebral vessels. Brain 61: 221–232, 1938. doi: 10.1093/brain/61.2.221. [DOI] [Google Scholar]
- 15.Sokoloff L. The action of drugs on the cerebral circulation. Pharmacol Rev 11: 1–85, 1959. [PubMed] [Google Scholar]
- 16.Rosenblum WI. Cerebral microcirculation: a review emphasizing the interrelationship of local blood flow and neuronal function. Angiology 16: 485–507, 1965. doi: 10.1177/000331976501600809. [DOI] [PubMed] [Google Scholar]
- 17.Fulton JF. Vasomotor and reflex sequelae of unilateral cervical and lumbar ramisectomy in a case of Raynaud's disease, with observations on tonus. Ann Surg 88: 827–841, 1928. doi: 10.1097/00000658-192811000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kety SS. Quantitative determination of cerebral blood flow in man. Methods Med Res 1: 204–217, 1948. [PubMed] [Google Scholar]
- 19.Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest 27: 476–483, 1948. doi: 10.1172/JCI101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lassen NA, Ingvar DH. The blood flow of the cerebral cortex determined by radioactive krypton. Experientia 17: 42–43, 1961. doi: 10.1007/BF02157946. [DOI] [PubMed] [Google Scholar]
- 21.Payne S. Cerebral Blood Flow and Metabolism: A Quantitative Approach. Hackensack, NJ: World Scientific, 2018, p. 1–440. [Google Scholar]
- 22.Wintermark M, Sesay M, Barbier E, Borbély K, Dillon WP, Eastwood JD, Glenn TC, Grandin CB, Pedraza S, Soustiel JF, Nariai T, Zaharchuk G, Caillé JM, Dousset V, Yonas H. Comparative overview of brain perfusion imaging techniques. J Neuroradiol 32: 294–314, 2005. doi: 10.1016/s0150-9861(05)83159-1. [DOI] [PubMed] [Google Scholar]
- 23.Aaslid R, Huber P, Nornes H. Evaluation of cerebrovascular spasm with transcranial Doppler ultrasound. J Neurosurg 60: 37–41, 1984. doi: 10.3171/jns.1984.60.1.0037. [DOI] [PubMed] [Google Scholar]
- 24.Aaslid R, Markwalder TM, Nornes H. Noninvasive transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg 57: 769–774, 1982. doi: 10.3171/jns.1982.57.6.0769. [DOI] [PubMed] [Google Scholar]
- 25.del Zoppo GJ, Sharp FR, Heiss WD, Albers GW. Heterogeneity in the penumbra. J Cereb Blood Flow Metab 31: 1836–1851, 2011. doi: 10.1038/jcbfm.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipton P. Ischemic cell death in brain neurons. Physiol Rev 79: 1431–1568, 1999. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 27.Goldberger ZD, Petek BJ, Brignole M, Shen WK, Sheldon RS, Solbiati M, Deharo JC, Moya A, Hamdan MH. ACC/AHA/HRS versus ESC guidelines for the diagnosis and management of syncope: JACC guideline comparison. J Am Coll Cardiol 74: 2410–2423, 2019. doi: 10.1016/j.jacc.2019.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia–the ischemic penumbra. Stroke 12: 723–725, 1981. doi: 10.1161/01.STR.12.6.723. doi:. [DOI] [PubMed] [Google Scholar]
- 29.Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology . J Neurosurg 77: 169–184, 1992. doi: 10.3171/jns.1992.77.2.0169. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension 75: 285–292, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol 16: 223–237, 2020. doi: 10.1038/s41581-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, De Silva TM, Chen J, Faraci FM. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ Res 120: 449–471, 2017. doi: 10.1161/CIRCRESAHA.116.308427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Payne S. Cerebral autoregulation: control of blood flow in the brain. In: SpringerBriefs in Bioengineering. New York: Springer, 2016, p. 1–125. [Google Scholar]
- 34.Brian JE Jr. Carbon dioxide and the cerebral circulation. Anesthesiology 88: 1365–1386, 1998. doi: 10.1097/00000542-199805000-00029. [DOI] [PubMed] [Google Scholar]
- 35.Hoiland RL, Fisher JA, Ainslie PN. Regulation of the cerebral circulation by arterial carbon dioxide. Compr Physiol 9: 1101–1154, 2019. doi: 10.1002/cphy.c180021. [DOI] [PubMed] [Google Scholar]
- 36.Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol (1985) 100: 1059–1064, 2006. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- 37.Edvinsson L, Krause DN. Cerebral Blood Flow and Metabolism. London: Lippincott Williams & Wilkins: 2002, p. 1–520. [Google Scholar]
- 38.Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev 39: 183–238, 1959. doi: 10.1152/physrev.1959.39.2.183. [DOI] [PubMed] [Google Scholar]
- 39.Lassen NA. Control of cerebral circulation in health and disease. Circ Res 34: 749–760, 1974. doi: 10.1161/01.RES.34.6.749. [DOI] [PubMed] [Google Scholar]
- 40.Panerai RB. Assessment of cerebral pressure autoregulation in humans–a review of measurement methods. Physiol Meas 19: 305–338, 1998. doi: 10.1088/0967-3334/19/3/001. [DOI] [PubMed] [Google Scholar]
- 41.van Beek AH, Claassen JA, Rikkert MG, Jansen RW. Cerebral autoregulation: an overview of current concepts and methodology with special focus on the elderly. J Cereb Blood Flow Metabol 28: 1071–1085, 2008. doi: 10.1038/jcbfm.2008.13. [DOI] [PubMed] [Google Scholar]
- 42.Willie CK, Tzeng YC, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol 592: 841–859, 2014. doi: 10.1113/jphysiol.2013.268953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Claassen JA, Zhang R, Fu Q, Witkowski S, Levine BD. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. J Appl Physiol 102: 870–877, 2007. doi: 10.1152/japplphysiol.00906.2006. [DOI] [PubMed] [Google Scholar]
- 44.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100: 328–335, 2006. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 45.Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96: 17–42, 2017. doi: 10.1016/j.neuron.2017.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol 300: H1566–H1582, 2011. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faraci FM, Heistad DD. Regulation of the cerebral circulation: Role of endothelium and potassium channels. Physiol Rev 78: 53–97, 1998. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 48.Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J 35: 888–894, 2014. doi: 10.1093/eurheartj/eht544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20: 717–726, 2017. doi: 10.1038/nn.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EA. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc 3: e000787, 2014. doi: 10.1161/jaha.114.000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow BW, Nunez V, Kaplan L, Granger AJ, Bistrong K, Zucker HL, Kumar P, Sabatini BL, Gu C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 579: 106–110, 2020. doi: 10.1038/s41586-020-2026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogan-Cann AD, Lu P, Anderson CM. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc Natl Acad Sci U S A 116: 10229–10231, 2019. doi: 10.1073/pnas.1902647116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Austin SA, Katusic ZS. Loss of endothelial nitric oxide synthase promotes p25 generation and tau phosphorylation in a murine model of Alzheimer's disease. Circ Res 119: 1128–1134, 2016. doi: 10.1161/CIRCRESAHA.116.309686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanders ML, Elting JW, Panerai RB, Aries M, Bor-Seng-Shu E, Caicedo A, Chacon M, Gommer ED, Van Huffel S, Jara JL, Kostoglou K, Mahdi A, Marmarelis VZ, Mitsis GD, Muller M, Nikolic D, Nogueira RC, Payne SJ, Puppo C, Shin DC, Simpson DM, Tarumi T, Yelicich B, Zhang R, Claassen J. Dynamic cerebral autoregulation reproducibility is affected by physiological variability. Front Physiol 10: 865, 2019. doi: 10.3389/fphys.2019.00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Claassen JA, Meel-van den Abeelen AS, Simpson DM, Panerai RB, on behalf of the International Cerebral Autoregulation Research Network (CARNet). Transfer function analysis of dynamic cerebral autoregulation: a white paper from the International Autoregulation Research Network (CARNet). J Cereb Blood Flow Metab 36: 665–680, 2016. doi: 10.1177/0271678X15626425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41–53, 2006. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 57.Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: Important unanswered questions. J Exp Med 217: e20190062, 2020. doi: 10.1084/jem.20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction–The disregarded partner of Alzheimer's disease. Alzheimers Dement 15: 158–167, 2019. doi: 10.1016/j.jalz.2018.07.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Menard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11: 457–470, 2015. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin BJ, Smith AJ, Verkman AS. Spatial model of convective solute transport in brain extracellular space does not support a “glymphatic” mechanism. J Gen Physiol 148: 489–501, 2016. doi: 10.1085/jgp.201611684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith AJ, Yao X, Dix JA, Jin BJ, Verkman AS. Test of the “glymphatic” hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. eLife 6: e27679, 2017. doi: 10.7554/eLife.27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bakker EN, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AW, Weller RO, Carare RO. Lymphatic clearance of the brain: perivascular, paravascular and significance for neurodegenerative diseases. Cell Mol Neurobiol 36: 181–194, 2016. doi: 10.1007/s10571-015-0273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol Appl Neurobiol 29: 106–117, 2003. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 64.van Veluw SJ, Hou SS, Calvo-Rodriguez M, Arbel-Ornath M, Snyder AC, Frosch MP, Greenberg SM, Bacskai BJ. Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron 105: 549–561, 2020. doi: 10.1016/j.neuron.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heistad DD, Kontos HA. Cerebral circulation. In: Handbook of Physiology, edited by Shepherd JT. Bethesda, Maryland: American Physiological Society, 1983, p. 137–182. [Google Scholar]
- 66.Chemtob S, Beharry K, Rex J, Varma DR, Aranda JV. Prostanoids determine the range of cerebral blood flow autoregulation of newborn piglets. Stroke 21: 777–784, 1990. doi: 10.1161/01.STR.21.5.777. [DOI] [PubMed] [Google Scholar]
- 67.Faraci FM, Heistad DD, Mayhan WG. Role of large arteries in regulation of blood flow to brain stem in cats. J Physiol 387: 115–123, 1987. doi: 10.1113/jphysiol.1987.sp016566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fitch W, Ferguson GG, Sengupta D, Garibi J, Harper AM. Autoregulation of cerebral blood flow during controlled hypotension in baboons. J Neurol Neurosurg Psychiatry 39: 1014–1022, 1976. doi: 10.1136/jnnp.39.10.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fitch W, Jones JV, MacKenzie ET, Harper AM. Autoregulation of cerebral blood flow in chronically hypertensive baboons. Br J Surg 63: 663, 1976. [PubMed] [Google Scholar]
- 70.Harper AM, Lassen NA, MacKenzie ET, Rowan JO, Sengupta D, Strandgaard S. Proceedings: the upper limit of ‘autoregulation’ of cerebral blood flow in the baboon. J Physiol 234: 61P–62P, 1973. [PubMed] [Google Scholar]
- 71.Harper SL, Bohlen HG. Microvascular adaptation in the cerebral cortex of adult spontaneously hypertensive rats. Hypertension 6: 408–419, 1984. doi: 10.1161/01.HYP.6.3.408. doi:. [DOI] [PubMed] [Google Scholar]
- 72.Harper SL, Bohlen HG, Rubin MJ. Arterial and microvascular contributions to cerebral cortical autoregulation in rats. Am J Physiol Heart Circ Physiol 246: H17–H24, 1984. doi: 10.1152/ajpheart.1984.246.1.H17. [DOI] [PubMed] [Google Scholar]
- 73.Heistad DD, Marcus ML, Abboud FM. Role of large arteries in regulation of cerebral blood flow in dogs. J Clin Invest 62: 761–768, 1978. doi: 10.1172/JCI109187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heistad DD, Marcus ML, Piegors DJ, Armstrong ML. Regulation of cerebral blood flow in atherosclerotic monkeys. Am J Physiol Heart Circ Physiol 239: H539–H544, 1980. doi: 10.1152/ajpheart.1980.239.4.H539. [DOI] [PubMed] [Google Scholar]
- 75.Hoffman WE, Edelman G, Kochs E, Werner C, Segil L, Albrecht RF. Cerebral autoregulation in awake versus isoflurane-anesthetized rats. Anesth Analg 73: 753–757, 1991. doi: 10.1213/00000539-199112000-00013. [DOI] [PubMed] [Google Scholar]
- 76.Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL Jr.. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol Heart Circ Physiol 234: H371–H383, 1978. doi: 10.1152/ajpheart.1978.234.4.H371. [DOI] [PubMed] [Google Scholar]
- 77.Marcus ML, Heistad DD. Effects of sympathetic nerves on cerebral blood flow in awake dogs. Am J Physiol Heart Circ Physiol 236: H549–H553, 1979. doi: 10.1152/ajpheart.1979.236.4.H549. [DOI] [PubMed] [Google Scholar]
- 78.Sadoshima S, Heistad DD. Regional cerebral blood flow during hypotension in normotensive and stroke-prone spontaneously hypertensive rats: effect of sympathetic denervation. Stroke 14: 575–579, 1983. doi: 10.1161/01.STR.14.4.575. [DOI] [PubMed] [Google Scholar]
- 79.Strandgaard S, MacKenzie ET, Sengupta D, Rowan JO, Lassen NA, Harper AM. Upper limit of autoregulation of cerebral blood flow in the baboon. Circ Res 34: 435–440, 1974. doi: 10.1161/01.RES.34.4.435. [DOI] [PubMed] [Google Scholar]
- 80.Tureen JH, Dworkin RJ, Kennedy SL, Sachdeva M, Sande MA. Loss of cerebrovascular autoregulation in experimental meningitis in rabbits. J Clin Invest 85: 577–581, 1990. doi: 10.1172/JCI114475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dalkara T, Irikura K, Huang Z, Panahian N, Moskowitz MA. Cerebrovascular responses under controlled and monitored physiological conditions in the anesthetized mouse. J Cereb Blood Flow Metab 15: 631–638, 1995. doi: 10.1038/jcbfm.1995.78. [DOI] [PubMed] [Google Scholar]
- 82.Jones JV, Fitch W, MacKenzie ET, Strandgaard S, Harper AM. Lower limit of cerebral blood flow autoregulation in experimental renovascular hypertension in the baboon. Circ Res 39: 555–557, 1976. doi: 10.1161/01.RES.39.4.555. [DOI] [PubMed] [Google Scholar]
- 83.Tuor UI, Farrar JK. Pial vessel caliber and cerebral blood flow during hemorrhage and hypercapnia in the rabbit. Am J Physiol Heart Circ Physiol 247: H40–H51, 1984. doi: 10.1152/ajpheart.1984.247.1.H40. [DOI] [PubMed] [Google Scholar]
- 84.Faraci FM, Mayhan WG, Heistad DD. Segmental vascular responses to acute hypertension in cerebrum and brain stem. Am J Physiol Heart Circ Physiol 252: H738–H742, 1987. doi: 10.1152/ajpheart.1987.252.4.H738. [DOI] [PubMed] [Google Scholar]
- 85.Claassen JA. The plateau phase is a slippery slope: Raising blood pressure may lower brain perfusion. J Physiol 594: 2783, 2016. doi: 10.1113/JP272121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu J, Tseng BY, Khan MA, Tarumi T, Hill C, Mirshams N, Hodics TM, Hynan LS, Zhang R. Individual variability of cerebral autoregulation, posterior cerebral circulation and white matter hyperintensity. J Physiol 594: 3141–3155, 2016. doi: 10.1113/JP271068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Numan T, Bain AR, Hoiland RL, Smirl JD, Lewis NC, Ainslie PN. Static autoregulation in humans: A review and reanalysis. Med Eng Phys 36: 1487–1495, 2014. doi: 10.1016/j.medengphy.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 88.Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Baroreflex dysfunction. N Engl J Med 382: 163–178, 2020. doi: 10.1056/NEJMra1509723. [DOI] [PubMed] [Google Scholar]
- 89.de Jong DL, Tarumi T, Liu J, Zhang R, Claassen J. Lack of linear correlation between dynamic and steady-state cerebral autoregulation. J Physiol 595: 5623–5636, 2017. doi: 10.1113/JP274304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tiecks FP, Lam AM, Aaslid R, Newell DW. Comparison of static and dynamic cerebral autoregulation measurements. Stroke 26: 1014–1019, 1995. doi: 10.1161/01.STR.26.6.1014. [DOI] [PubMed] [Google Scholar]
- 91.Panerai RB. Cerebral autoregulation: from models to clinical applications. Cardiovasc Eng 8: 42–59, 2008. doi: 10.1007/s10558-007-9044-6. [DOI] [PubMed] [Google Scholar]
- 92.Panerai RB, Carey BJ, Potter JF. Short-term variability of cerebral blood flow velocity responses to arterial blood pressure transients. Ultrasound Med Biol 29: 31–38, 2003. doi: 10.1016/S0301-5629(02)00698-1. [DOI] [PubMed] [Google Scholar]
- 93.Panerai RB, Dawson SL, Eames PJ, Potter JF. Cerebral blood flow velocity response to induced and spontaneous sudden changes in arterial blood pressure. Am J Physiol Heart Circ Physiol 280: H2162–H2174, 2001. doi: 10.1152/ajpheart.2001.280.5.H2162. [DOI] [PubMed] [Google Scholar]
- 94.Panerai RB, Eames PJ, Potter JF. Variability of time-domain indices of dynamic cerebral autoregulation. Physiol Meas 24: 367–381, 2003. doi: 10.1088/0967-3334/24/2/312. [DOI] [PubMed] [Google Scholar]
- 95.Panerai RB, Rennie JM, Kelsall AW, Evans DH. Frequency-domain analysis of cerebral autoregulation from spontaneous fluctuations in arterial blood pressure. Med Biol Eng Comput 36: 315–322, 1998. doi: 10.1007/BF02522477. [DOI] [PubMed] [Google Scholar]
- 96.Greenfield JC Jr, Rembert JC, Tindall GT. Transient changes in cerebral vascular resistance during the Valsalva maneuver in man. Stroke 15: 76–79, 1984. doi: 10.1161/01.STR.15.1.76. [DOI] [PubMed] [Google Scholar]
- 97.Nornes H, Knutzen HB, Wikeby P. Cerebral arterial blood flow and aneurysm surgery. J Neurosurg 47: 819–827, 1977. doi: 10.3171/jns.1977.47.6.0819. [DOI] [PubMed] [Google Scholar]
- 98.Nornes H, Wikeby P. Cerebral arterial blood flow and aneurysm surgery. Part 1: Local arterial flow dynamics. J Neurosurg 47: 810–818, 1977. doi: 10.3171/jns.1977.47.6.0810. [DOI] [PubMed] [Google Scholar]
- 99.de Heus RA, de Jong DL, Sanders ML, van Spijker GJ, Oudegeest-Sander MH, Hopman MT, Lawlor BA, Olde Rikkert MG, Claassen J. Dynamic regulation of cerebral blood flow in patients with Alzheimer disease. Hypertension 72: 139–150, 2018. doi: 10.1161/HYPERTENSIONAHA.118.10900. [DOI] [PubMed] [Google Scholar]
- 100.van Beek AH, Rikkert O, Pasman JW, Hopman MT, Claassen JA. Dynamic cerebral autoregulation in the old using a repeated sit-stand maneuver. Ultrasound Med Biol 36: 192–201, 2010. doi: 10.1016/j.ultrasmedbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 101.Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke 20: 45–52, 1989. [PMC doi: 10.1161/01.STR.20.1.45. [DOI] [PubMed] [Google Scholar]
- 102.Markwalder TM, Grolimund P, Seiler RW, Roth F, Aaslid R. Dependency of blood flow velocity in the middle cerebral artery on end-tidal carbon dioxide partial pressure–a transcranial ultrasound Doppler study. J Cereb Blood Flow Metab 4: 368–372, 1984. doi: 10.1038/jcbfm.1984.54. [DOI] [PubMed] [Google Scholar]
- 103.Birch AA, Dirnhuber MJ, Hartley-Davies R, Iannotti F, Neil-Dwyer G. Assessment of autoregulation by means of periodic changes in blood pressure. Stroke 26: 834–837, 1995. doi: 10.1161/01.STR.26.5.834. [DOI] [PubMed] [Google Scholar]
- 104.Diehl RR, Linden D, LüCke D, Berlit P. Phase relationship between cerebral blood flow velocity and blood pressure. A clinical test of autoregulation. Stroke 26: 1801–1804, 1995. doi: 10.1161/01.STR.26.10.1801. [DOI] [PubMed] [Google Scholar]
- 105.Diehl RR, Linden D, Lucke D, Berlit P. Spontaneous blood pressure oscillations and cerebral autoregulation. Clin Auton Res 8: 7–12, 1998. doi: 10.1007/BF02267598. [DOI] [PubMed] [Google Scholar]
- 106.Giller CA. The frequency-dependent behavior of cerebral autoregulation. Neurosurgery 27: 362–368, 1990. doi: 10.1227/00006123-199009000-00004. [DOI] [PubMed] [Google Scholar]
- 107.Panerai RB, Kelsall AW, Rennie JM, Evans DH. Analysis of cerebral blood flow autoregulation in neonates. IEEE Trans Biomed Eng 43: 779–788, 1996. doi: 10.1109/10.508541. [DOI] [PubMed] [Google Scholar]
- 108.Panerai RB, White RP, Markus HS, Evans DH. Grading of cerebral dynamic autoregulation from spontaneous fluctuations in arterial blood pressure. Stroke 29: 2341–2346, 1998. doi: 10.1161/01.STR.29.11.2341. [DOI] [PubMed] [Google Scholar]
- 109.Blaber AP, Bondar RL, Stein F, Dunphy PT, Moradshahi P, Kassam MS, Freeman R. Transfer function analysis of cerebral autoregulation dynamics in autonomic failure patients. Stroke 28: 1686–1692, 1997. doi: 10.1161/01.STR.28.9.1686. [DOI] [PubMed] [Google Scholar]
- 110.Zhang R, Zuckerman JH, Giller CA, Levine BD. Transfer function analysis of dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol 274: H233–H241, 1998. doi: 10.1152/ajpheart.1998.274.1.H233. [DOI] [PubMed] [Google Scholar]
- 111.Meel-van den Abeelen AS, Simpson DM, Wang LJ, Slump CH, Zhang R, Tarumi T, Rickards CA, Payne S, Mitsis GD, Kostoglou K, Marmarelis V, Shin D, Tzeng YC, Ainslie PN, Gommer E, Muller M, Dorado AC, Smielewski P, Yelicich B, Puppo C, Liu X, Czosnyka M, Wang CY, Novak V, Panerai RB, Claassen JA. Between-centre variability in transfer function analysis, a widely used method for linear quantification of the dynamic pressure-flow relation: The CARNet Study. Med Eng Phys 36: 620–627, 2014. doi: 10.1016/j.medengphy.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meel-van den Abeelen AS, Van Beek AH, Slump CH, Panerai RB, Claassen JA. Transfer function analysis for the assessment of cerebral autoregulation using spontaneous oscillations in blood pressure and cerebral blood flow. Med Engineering Physics 36: 563–575, 2014. doi: 10.1016/j.medengphy.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 113.Tzeng YC, Panerai RB. CrossTalk proposal: dynamic cerebral autoregulation should be quantified using spontaneous blood pressure fluctuations. J Physiol 596: 3–5, 2018. doi: 10.1113/JP273899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Simpson D, Claassen J. CrossTalk opposing view: dynamic cerebral autoregulation should be quantified using induced (rather than spontaneous) blood pressure fluctuations. J Physiol 596: 7–9, 2018. doi: 10.1113/JP273900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van den Brule JM, van Kaam CR, van der Hoeven JG, Claassen J, Hoedemaekers CW. Influence of induced blood pressure variability on the assessment of cerebral autoregulation in patients after cardiac arrest. Biomed Res Int 2018: 8153241, 2018. doi: 10.1155/2018/8153241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elting JW, Sanders ML, Panerai RB, Aries M, Bor-Seng-Shu E, Caicedo A, Chacon M, Gommer ED, Van Huffel S, Jara JL, Kostoglou K, Mahdi A, Marmarelis VZ, Mitsis GD, Muller M, Nikolic D, Nogueira RC, Payne SJ, Puppo C, Shin DS, Simpson DM, Tarumi T, Yeliich B, Zhang R, Claassen JA. Assessment of dynamic cerebral autoregulation in humans: is reproducibility dependent on blood pressure variability? PLoS One 15: e0227651, 2020. doi: 10.1371/journal.pone.0227651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sanders ML, Claassen J, Aries M, Bor-Seng-Shu E, Caicedo A, Chacon M, Gommer ED, Van Huffel S, Jara JL, Kostoglou K, Mahdi A, Marmarelis VZ, Mitsis GD, Muller M, Nikolic D, Nogueira RC, Payne SJ, Puppo C, Shin DC, Simpson DM, Tarumi T, Yelicich B, Zhang R, Panerai RB, Elting JW. Reproducibility of dynamic cerebral autoregulation parameters: a multi-centre, multi-method study. Physiol Meas 39: 125002, 2018. doi: 10.1088/1361-6579/aae9fd. [DOI] [PubMed] [Google Scholar]
- 118.Claassen JA, Levine BD, Zhang R. Dynamic cerebral autoregulation during repeated squat-stand maneuvers. J Appl Physiol 106: 153–160, 2009. doi: 10.1152/japplphysiol.90822.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smirl JD, Hoffman K, Tzeng YC, Hansen A, Ainslie PN. Methodological comparison of active- and passive-driven oscillations in blood pressure; implications for the assessment of cerebral pressure-flow relationships. J Appl Physiol 119: 487–501, 2015. doi: 10.1152/japplphysiol.00264.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diehl RR. Cerebral autoregulation studies in clinical practice. Eur J Ultrasound 16: 31–36, 2002. doi: 10.1016/s0929-8266(02)00048-4. [DOI] [PubMed] [Google Scholar]
- 121.Katsogridakis E, Simpson DM, Bush G, Fan L, Birch AA, Allen R, Potter JF, Panerai RB. Coherent averaging of pseudorandom binary stimuli: is the dynamic cerebral autoregulatory response symmetrical? Physiol Meas 38: 2164–2175, 2017. doi: 10.1088/1361-6579/aa9086. [DOI] [PubMed] [Google Scholar]
- 122.Hamner JW, Cohen MA, Mukai S, Lipsitz LA, Taylor JA. Spectral indices of human cerebral blood flow control: responses to augmented blood pressure oscillations. J Physiol 559: 965–973, 2004. doi: 10.1113/jphysiol.2004.066969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hamner JW, Ishibashi K, Tan CO. Revisiting human cerebral blood flow responses to augmented blood pressure oscillations. J Physiol 597: 1553–1564, 2019. doi: 10.1113/JP277321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang R, Zuckerman JH, Levine BD. Spontaneous fluctuations in cerebral blood flow: Insights from extended-duration recordings in humans. Am J Physiol Heart Circ Physiol 278: H1848–1855, 2000. doi: 10.1152/ajpheart.2000.278.6.H1848. [DOI] [PubMed] [Google Scholar]
- 125.Panerai RB, Robinson TG, Minhas JS. The upper frequency limit of dynamic cerebral autoregulation. J Physiol 597: 5821–5833, 2019. doi: 10.1113/JP278710. [DOI] [PubMed] [Google Scholar]
- 126.Tan CO. Defining the characteristic relationship between arterial pressure and cerebral flow. J Appl Physiol (1985) 113: 1194–1200, 2012. doi: 10.1152/japplphysiol.00783.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.De Silva TM, Faraci FM. Microvascular dysfunction and cognitive impairment. Cell Mol Neurobiol 36: 241–258, 2016. doi: 10.1007/s10571-015-0308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res 66: 8–17, 1990. doi: 10.1161/01.RES.66.1.8. [DOI] [PubMed] [Google Scholar]
- 129.Cipolla MJ. The cerebral circulation. Colloquium Series on Integrated Systems Physiology: from Molecule to Function (2nd ed.). London: Morgan & Claypool, 2016, p. 1–67. doi: 10.4199/C00141ED2V01Y201607ISP066. [DOI] [Google Scholar]
- 130.Peixoto AJ. Acute severe hypertension. N Engl J Med 381: 1843–1852, 2019. doi: 10.1056/NEJMcp1901117. [DOI] [PubMed] [Google Scholar]
- 131.Dabertrand F, Kroigaard C, Bonev AD, Cognat E, Dalsgaard T, Domenga-Denier V, Hill-Eubanks DC, Brayden JE, Joutel A, Nelson MT. Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci USA 112: E796–805, 2015. doi: 10.1073/pnas.1420765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Silva TM, Modrick ML, Dabertrand F, Faraci FM. Changes in cerebral arteries and parenchymal arterioles with aging: role of Rho kinase 2 and impact of genetic background. Hypertension 71: 921–927, 2018. doi: 10.1161/HYPERTENSIONAHA.118.10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Elhusseiny A, Hamel E. Muscarinic–but not nicotinic–acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cereb Blood Flow Metabol 20: 298–305, 2000. doi: 10.1097/00004647-200002000-00011. [DOI] [PubMed] [Google Scholar]
- 134.Faraci FM, Baumbach GL, Heistad DD. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl 7: S61–64, 1989. [PubMed] [Google Scholar]
- 135.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol 269: H348–H355, 1995. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 136.McCarron JG, Osol G, Halpern W. Myogenic responses are independent of the endothelium in rat pressurized posterior cerebral arteries. Blood Vessels 26: 315–319, 1989. doi: 10.1159/000158780. [DOI] [PubMed] [Google Scholar]
- 137.Wallis SJ, Firth J, Dunn WR. Pressure-induced myogenic responses in human isolated cerebral resistance arteries. Stroke 27: 2287–2290, 1996. doi: 10.1161/01.str.27.12.2287. [DOI] [PubMed] [Google Scholar]
- 138.Ward ME, Yan L, Kelly S, Angle MR. Flow modulation of pressure-sensitive tone in rat pial arterioles: Role of the endothelium. Anesthesiology 93: 1456–1464, 2000. doi: 10.1097/00000542-200012000-00018. [DOI] [PubMed] [Google Scholar]
- 139.Lidington D, Kroetsch JT, Bolz SS. Cerebral artery myogenic reactivity: the next frontier in developing effective interventions for subarachnoid hemorrhage. J Cereb Blood Flow Metabol 38: 17–37, 2018. doi: 10.1177/0271678X17742548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Harraz OF, Hill-Eubanks D, Nelson MT. PIP2: a critical regulator of vascular ion channels hiding in plain sight. Proc Natl Acad Sci U S A 117: 20378–20389, 2020. doi: 10.1073/pnas.2006737117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 143.Li Y, Brayden JE. Rho kinase activity governs arteriolar myogenic depolarization. J Cereb Blood Flow Metab 37: 140–152, 2017. doi: 10.1177/0271678X15621069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brayden JE, Li Y, Tavares MJ. Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 33: 293–299, 2013. doi: 10.1038/jcbfm.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chennupati R, Wirth A, Favre J, Li R, Bonnavion R, Jin YJ, Wietelmann A, Schweda F, Wettschureck N, Henrion D, Offermanns S. Myogenic vasoconstriction requires G12/G13 and LARG to maintain local and systemic vascular resistance. Elife 8: e49374, 2019. doi: 10.7554/eLife.49374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res 95: 922–929, 2004. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 147.Lim M, Choi SK, Cho YE, Yeon SI, Kim EC, Ahn DS, Lee YH. The role of sphingosine kinase 1/sphingosine-1-phosphate pathway in the myogenic tone of posterior cerebral arteries. PLoS One 7: e35177, 2012. doi: 10.1371/journal.pone.0035177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.VanLandingham LG, Gannon KP, Drummond HA. Pressure-induced constriction is inhibited in a mouse model of reduced βENaC. Am J Physiol Regul Integr Comp Physiol 297: R723–R728, 2009. doi: 10.1152/ajpregu.00212.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90: 248–250, 2002. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 150.Zeiler FA, Thelin EP, Donnelly J, Stevens AR, Smielewski P, Czosnyka M, Hutchinson PJ, Menon DK. Genetic drivers of cerebral blood flow dysfunction in TBI: a speculative synthesis. Nat Rev Neurol 15: 25–39, 2019. doi: 10.1038/s41582-018-0105-9. [DOI] [PubMed] [Google Scholar]
- 151.Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, Demolombe S, Patel A, Honore E. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep 13: 1161–1171, 2015. doi: 10.1016/j.celrep.2015.09.072. [DOI] [PubMed] [Google Scholar]
- 152.Coucha M, Abdelsaid M, Ward R, Abdul Y, Ergul A. Impact of metabolic diseases on cerebral circulation: Structural and functional consequences. Compr Physiol 8: 773–799, 2018. doi: 10.1002/cphy.c170019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.De Silva TM, Ketsawatsomkron P, Pelham C, Sigmund CD, Faraci FM. Genetic interference with peroxisome proliferator-activated receptor γ in smooth muscle enhances myogenic tone in the cerebrovasculature via a Rho kinase-dependent mechanism. Hypertension 65: 345–351, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lidington D, Fares JC, Uhl FE, Dinh DD, Kroetsch JT, Sauve M, Malik FA, Matthes F, Vanherle L, Adel A, Momen A, Zhang H, Aschar-Sobbi R, Foltz WD, Wan H, Sumiyoshi M, Macdonald RL, Husain M, Backx PH, Heximer SP, Meissner A, Bolz SS. CFTR therapeutics normalize cerebral perfusion deficits in mouse models of heart failure and subarachnoid hemorrhage. JACC Basic Transl Sci 4: 940–958, 2019. doi: 10.1016/j.jacbts.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab 16: 981–987, 1996. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 156.Kusano Y, Echeverry G, Miekisiak G, Kulik TB, Aronhime SN, Chen JF, Winn HR. Role of adenosine A2 receptors in regulation of cerebral blood flow during induced hypotension. J Cereb Blood Flow Metab 30: 808–815, 2010. doi: 10.1038/jcbfm.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rocha MP, Campos MO, Mattos JD, Mansur DE, Rocha HN, Secher NH, Nobrega AC, Fernandes IA. KATP channels modulate cerebral blood flow and oxygen delivery during isocapnic hypoxia in humans. J Physiol 598: 3343–3356, 2020. doi: 10.1113/JP279751. [DOI] [PubMed] [Google Scholar]
- 158.Shin HK, Hong KW. Importance of calcitonin gene-related peptide, adenosine and reactive oxygen species in cerebral autoregulation under normal and diseased conditions. Clin Exp Pharmacol Physiol 31: 1–7, 2004. doi: 10.1111/j.1440-1681.2004.03943.x. [DOI] [PubMed] [Google Scholar]
- 159.Toyoda K, Fujii K, Ibayashi S, Kitazono T, Nagao T, Fujishima M. Role of ATP-sensitive potassium channels in brain stem circulation during hypotension. Am J Physiol Heart Circ Physiol 273: H1342–H1346, 1997. doi: 10.1152/ajpheart.1997.273.3.H1342. [DOI] [PubMed] [Google Scholar]
- 160.Emdin CA, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A, Haas M, Aragam K, Ardissino D, Wilson JG, Schunkert H, McPherson R, Watkins H, Elosua R, Bown MJ, Samani NJ, Baber U, Erdmann J, Gormley P, Palotie A, Stitziel NO, Gupta N, Danesh J, Saleheen D, Gabriel S, Kathiresan S. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 137: 222–232, 2018. doi: 10.1161/CIRCULATIONAHA.117.028021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA, Filosa JA. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci 35: 8245–8257, 2015. doi: 10.1523/JNEUROSCI.4486-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kuluz JW, Prado R, Chang J, Ginsberg MD, Schleien CL, Busto R. Selective brain cooling increases cortical cerebral blood flow in rats. Am J Physiol Heart Circ Physiol 265: H824–H827, 1993. doi: 10.1152/ajpheart.1993.265.3.H824. [DOI] [PubMed] [Google Scholar]
- 163.Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J 27: 3092–3103, 2008. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Arrick DM, Sharpe GM, Sun H, Mayhan WG. Losartan improves impaired nitric oxide synthase-dependent dilatation of cerebral arterioles in type 1 diabetic rats. Brain Res 1209: 128–135, 2008. doi: 10.1016/j.brainres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through Nox-2-derived radicals. Arterioscler Thromb Vasc Biol 26: 826–832, 2006. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 166.Matulla B, Streit G, Pieh S, Findl O, Entlicher J, Graselli U, Eichler HG, Wolzt M, Schmetterer L. Effects of losartan on cerebral and ocular circulation in healthy subjects. Br J Clin Pharmacol 44: 369–375, 1997. doi: 10.1046/j.1365-2125.1997.t01-1-00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Warrington JP, Fan F, Duncan J, Cunningham MW, LaMarca BB, Dechend R, Wallukat G, Roman RJ, Drummond HA, Granger JP, Ryan MJ. The angiotensin II type I receptor contributes to impaired cerebral blood flow autoregulation caused by placental ischemia in pregnant rats. Biol Sex Differ 10: 58, 2019. doi: 10.1186/s13293-019-0275-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Early CB, Dewey RC, Pieper HP, Hunt WE. Dynamic pressure-flow relationships of brain blood flow in the monkey. J Neurosurg 41: 590–596, 1974. doi: 10.3171/jns.1974.41.5.0590. [DOI] [PubMed] [Google Scholar]
- 169.Kety SS, Hafkenschiel JH, Jeffers WA, Leopold IH, Shenkin HA. The blood flow, vascular resistance, and oxygen consumption of the brain in essential hypertension. J Clin Invest 27: 511–514, 1948. doi: 10.1172/JCI101998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Burton AC. Physiology and Biophysics of the Circulation. Chicago, IL: Year Book Medical Publishers, 1972, p. 1–226. [Google Scholar]
- 171.Panerai RB, Coughtrey H, Rennie JM, Evans DH. A model of the instantaneous pressure-velocity relationships of the neonatal cerebral circulation. Physiol Meas 14: 411–418, 1993. doi: 10.1088/0967-3334/14/4/002. [DOI] [PubMed] [Google Scholar]
- 172.Payne S, Morris H, Rowley A. A combined haemodynamic and biochemical model of cerebral autoregulation. Conf Proc IEEE Eng Med Biol Soc 2005: 2295–2298, 2005. doi: 10.1109/iembs.2005.1616923. [DOI] [PubMed] [Google Scholar]
- 173.Mayhan WG, Faraci FM, Heistad DD. Mechanisms of protection of the blood-brain barrier during acute hypertension in chronically hypertensive rats. Hypertension 9: 101–105, 1987. doi: 10.1161/01.HYP.9.6_Pt_2.III101. [DOI] [PubMed] [Google Scholar]
- 174.Grubb S, Cai C, Hald BO, Khennouf L, Murmu RP, Jensen AG, Fordsmann J, Zambach S, Lauritzen M. Precapillary sphincters maintain perfusion in the cerebral cortex. Nat Commun 11: 395, 2020. doi: 10.1038/s41467-020-14330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ratelade J, Klug NR, Lombardi D, Angelim M, Dabertrand F, Domenga-Denier V, Al-Shahi Salman R, Smith C, Gerbeau JF, Nelson MT, Joutel A. Reducing hypermuscularization of the transitional segment between arterioles and capillaries protects against spontaneous intracerebral hemorrhage. Circulation 141: 2078–2094, 2020. doi: 10.1161/CIRCULATIONAHA.119.040963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Baumbach GL, Heistad DD. Effects of sympathetic stimulation and changes in arterial pressure on segmental resistance of cerebral vessels in rabbits and cats. Circ Res 52: 527–533, 1983. doi: 10.1161/01.RES.52.5.527. [DOI] [PubMed] [Google Scholar]
- 177.Laurent S, Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circ Res 116: 1007–1021, 2015. doi: 10.1161/CIRCRESAHA.116.303596. [DOI] [PubMed] [Google Scholar]
- 178.Marina N, Christie IN, Korsak A, Doronin M, Brazhe A, Hosford PS, Wells JA, Sheikhbahaei S, Humoud I, Paton JF, Lythgoe MF, Semyanov A, Kasparov S, Gourine AV. Astrocytes monitor cerebral perfusion and control systemic circulation to maintain brain blood flow. Nature Commun 11: 131, 2020. doi: 10.1038/s41467-019-13956-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hariharan A, Weir N, Robertson C, He L, Betsholtz C, Longden TA. The ion channel and GPCR toolkit of brain capillary pericytes. Front Cell Neurosci 14: 601324, 2020. doi: 10.3389/fncel.2020.601324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rungta RL, Chaigneau E, Osmanski BF, Charpak S. Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron 99: 362–375, 2018. doi: 10.1016/j.neuron.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gonzalez-Marrero I, Hernandez-Abad LG, Castaneyra-Ruiz L, Carmona-Calero EM, Castaneyra-Perdomo A. Changes in the choroid plexuses and brain barriers associated with high blood pressure and ageing. Neurologia S0213-4853(18)30175-0, 2018. doi: 10.1016/j.nrl.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 182.Mayhan WG, Heistad DD. Role of veins and cerebral venous pressure in disruption of the blood-brain barrier. Circ Res 59: 216–220, 1986. doi: 10.1161/01.RES.59.2.216. [DOI] [PubMed] [Google Scholar]
- 183.Auer L. Origin and localization of Evans blue extravasations in acutely-induced hypertension in cats. Eur Neurol 17: 211–215, 1978. doi: 10.1159/000114947. [DOI] [PubMed] [Google Scholar]
- 184.Wilhelm I, Nyul-Toth A, Suciu M, Hermenean A, Krizbai IA. Heterogeneity of the blood-brain barrier. Tissue Barriers 4: e1143544, 2016. doi: 10.1080/21688370.2016.1143544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Spence JD. Blood pressure gradients in the brain: their importance to understanding pathogenesis of cerebral small vessel disease. Brain Sci 9: 21, 2019. doi: 10.3390/brainsci9020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Blanco PJ, Muller LO, Spence JD. Blood pressure gradients in cerebral arteries: A clue to pathogenesis of cerebral small vessel disease. Stroke Vasc Neurol 2: 108–117, 2017. doi: 10.1136/svn-2017-000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grutzendler J, Nedergaard M. Cellular control of brain capillary blood flow: in vivo imaging veritas. Trends Neurosci 42: 528–536, 2019. doi: 10.1016/j.tins.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508: 55–60, 2014. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87: 95–110, 2015. doi: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wei HS, Kang H, Rasheed ID, Zhou S, Lou N, Gershteyn A, McConnell ED, Wang Y, Richardson KE, Palmer AF, Xu C, Wan J, Nedergaard M. Erythrocytes are oxygen-sensing regulators of the cerebral microcirculation. Neuron 91: 851–862, 2016. doi: 10.1016/j.neuron.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Faraci FM. Watching small vessel disease grow. Circ Res 122: 810–812, 2018. doi: 10.1161/CIRCRESAHA.118.312762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mayhan WG, Faraci FM, Heistad DD. Effects of vasodilatation and acidosis on the blood-brain barrier. Microvasc Res 35: 179–192, 1988. doi: 10.1016/0026-2862(88)90061-1. [DOI] [PubMed] [Google Scholar]
- 193.Katz AI, Chen Y, Moreno AH. Flow through a collapsible tube. Experimental analysis and mathematical model. Biophys J 9: 1261–1279, 1969. doi: 10.1016/S0006-3495(69)86451-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Takahashi S, Crane AM, Jehle J, Cook M, Kennedy C, Sokoloff L. Role of the cerebellar fastigial nucleus in the physiological regulation of cerebral blood flow. J Cereb Blood Flow Metab 15: 128–142, 1995. doi: 10.1038/jcbfm.1995.15. [DOI] [PubMed] [Google Scholar]
- 195.Ishitsuka T, Iadecola C, Underwood MD, Reis DJ. Lesions of nucleus tractus solitarii globally impair cerebrovascular autoregulation. Am J Physiol Heart Circ Physiol 251: H269–H281, 1986. doi: 10.1152/ajpheart.1986.251.2.H269. [DOI] [PubMed] [Google Scholar]
- 196.Liu Y, Harder DR, Lombard JH. Interaction of myogenic mechanisms and hypoxic dilation in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol 283: H2276–H2281, 2002. doi: 10.1152/ajpheart.00635.2002. [DOI] [PubMed] [Google Scholar]
- 197.Panerai RB. System identification of human cerebral blood flow regulatory mechanisms. Cardiovasc Eng 4: 59–71, 2004. doi: 10.1023/B:CARE.0000025123.43747.e1. [DOI] [Google Scholar]
- 198.Panerai RB, Hanby MF, Robinson TG, Haunton VJ. Alternative representation of neural activation in multivariate models of neurovascular coupling in humans. J Neurophysiol 122: 833–843, 2019. doi: 10.1152/jn.00175.2019. [DOI] [PubMed] [Google Scholar]
- 199.Payne SJ. Identifying the myogenic and metabolic components of cerebral autoregulation. Med Eng Phys 58: 23–30, 2018. doi: 10.1016/j.medengphy.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 200.Ainslie PN, Brassard P. Why is the neural control of cerebral autoregulation so controversial? F1000Prime Rep 6: 14, 2014. doi: 10.12703/P6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bernardi L, Cencetti S, Cipriani M, Fattorini L, Bandinelli G, WdowczyckSzulc JL, Lagi A. Assessment of autonomic control of cerebral blood flow: comparison with peripheral circulation. Cerebrovasc Dis 7: 303–304, 1997. [Google Scholar]
- 202.Goadsby PJ. Autonomic nervous system control of the cerebral circulation. In: Handbook of Clinical Neurology Autonomic Nervous System, edited by Buuijs RM, Swaab DF.. Amsterdam, The Netherlands: Elsevier, 2013, p. 193–201. [DOI] [PubMed] [Google Scholar]
- 203.Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA. Sympathetic control of the cerebral vasculature in humans. Stroke 41: 102–109, 2010. doi: 10.1161/STROKEAHA.109.557132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhang R, Crandall CG, Levine BD. Cerebral hemodynamics during the Valsalva maneuver: Insights from ganglionic blockade. Stroke 35: 843–847, 2004. doi: 10.1161/01.STR.0000120309.84666.AE. [DOI] [PubMed] [Google Scholar]
- 205.Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation 106: 1814–1820, 2002. doi: 10.1161/01.CIR.0000031798.07790.FE. [DOI] [PubMed] [Google Scholar]
- 206.Ngai AC, Winn HR. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circ Res 77: 832–840, 1995. doi: 10.1161/01.res.77.4.832. [DOI] [PubMed] [Google Scholar]
- 207.White RP, Vallance P, Markus HS. Effect of inhibition of nitric oxide synthase on dynamic cerebral autoregulation in humans. Clin Sci (Lond) 99: 555–560, 2000. doi: 10.1042/CS20000064,10.1042/cs0990555. doi:. [DOI] [PubMed] [Google Scholar]
- 208.Buxton RB, Uludağ K, Dubowitz DJ, Liu TT. Modeling the hemodynamic response to brain activation. Neuroimage 23, Suppl 1: S220–233, 2004. doi: 10.1016/j.neuroimage.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 209.Gotoh J, Kuang TY, Nakao Y, Cohen DM, Melzer P, Itoh Y, Pak H, Pettigrew K, Sokoloff L. Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am J Physiol Heart Circ Physiol 280: H821–H829, 2001. doi: 10.1152/ajpheart.2001.280.2.H821. [DOI] [PubMed] [Google Scholar]
- 210.Raichle ME. Functional brain imaging and human brain function. J Neurosci 23: 3959–3962, 2003. doi: 10.1523/JNEUROSCI.23-10-03959.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sokoloff L. Energetic of functional activation in neural tissues. Neurochem Res 24: 321–329, 1999. doi: 10.1023/A:1022534709672. [DOI] [PubMed] [Google Scholar]
- 212.Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci 29: 449–476, 2006. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- 213.Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci 25: 621–625, 2002. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- 214.Fujii K, Heistad DD, Faraci FM. Flow-mediated dilatation of the basilar artery in vivo. Circ Res 69: 697–705, 1991. doi: 10.1161/01.res.69.3.697. [DOI] [PubMed] [Google Scholar]
- 215.Iadecola C, Yang G, Ebner TJ, Chen G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J Neurophysiol 78: 651–659, 1997. doi: 10.1152/jn.1997.78.2.651. [DOI] [PubMed] [Google Scholar]
- 216.Aaslid R. Visually evoked dynamic blood flow response of the human cerebral circulation. Stroke 18: 771–775, 1987. doi: 10.1161/01.STR.18.4.771. [DOI] [PubMed] [Google Scholar]
- 217.O'Herron P, Chhatbar PY, Levy M, Shen Z, Schramm AE, Lu Z, Kara P. Neural correlates of single-vessel haemodynamic responses in vivo. Nature 534: 378–382, 2016. doi: 10.1038/nature17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ainslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol 296: R1473–R1495, 2009. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- 219.Greenberg JH, Alavi A, Reivich M, Kuhl D, Uzzell B. Local cerebral blood volume response to carbon dioxide in man. Circ Res 43: 324–331, 1978. doi: 10.1161/01.res.43.2.324. [DOI] [PubMed] [Google Scholar]
- 220.Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest 27: 484–492, 1948. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Reivich M. Arterial Pco2 and cerebral hemodynamics. Am J Physiol 206: 25–35, 1964. doi: 10.1152/ajplegacy.1964.206.1.25. [DOI] [PubMed] [Google Scholar]
- 222.Ide K, Poulin MJ. A protocol to study the relationship between middle cerebral artery blood velocity and end-tidal Pco2 in the hypocapnic-hypercapnic range in humans. FASEB J 17: A872–A872, 2003. [DOI] [PubMed] [Google Scholar]
- 223.Minhas JS, Panerai RB, Robinson TG. Modelling the cerebral haemodynamic response in the physiological range of PaCO2. Physiol Meas 39: 065001, 2018. doi: 10.1088/1361-6579/aac76b. [DOI] [PubMed] [Google Scholar]
- 224.Dahl A, Russell D, Nyberg-Hansen R, Rootwelt K, Mowinckel P. Simultaneous assessment of vasoreactivity using transcranial Doppler ultrasound and cerebral blood flow in healthy subjects. J Cereb Blood Flow Metab 14: 974–981, 1994. doi: 10.1038/jcbfm.1994.130. [DOI] [PubMed] [Google Scholar]
- 225.Edwards MR, Lin DC, Hughson RL. Modeling the interaction between perfusion pressure and CO2 on cerebral blood flow. Front Modeling Control Breathing 499: 285–290, 2001. doi: 10.1007/978-1-4615-1375-9_45. [DOI] [PubMed] [Google Scholar]
- 226.Hetzel A, Braune S, Guschlbauer B, Dohms K. CO2 reactivity testing without blood pressure monitoring? Stroke 30: 398–401, 1999. doi: 10.1161/01.str.30.2.398. [DOI] [PubMed] [Google Scholar]
- 227.Ainslie PN, Ashmead JC, Ide K, Morgan BJ, Poulin MJ. Differential responses to CO2 and sympathetic stimulation in the cerebral and femoral circulations in humans. J Physiol 566: 613–624, 2005. doi: 10.1113/jphysiol.2005.087320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kontos HA, Richardson DW, Patterson JL Jr.. Effects of hypercapnia on human forearm blood vessels. Am J Physiol 212: 1070–1080, 1967. doi: 10.1152/ajplegacy.1967.212.5.1070. [DOI] [PubMed] [Google Scholar]
- 229.Raper AJ, Richardson DW, Kontos HA, Patterson JL Jr.. Circulatory responses to breath holding in man. J Appl Physiol 22: 201–206, 1967. doi: 10.1152/jappl.1967.22.2.201. [DOI] [PubMed] [Google Scholar]
- 230.Faraci FM, Brian JE Jr.. Nitric oxide and the cerebral circulation. Stroke 25: 692–703, 1994. doi: 10.1161/01.STR.25.3.692. [DOI] [PubMed] [Google Scholar]
- 231.Golding EM, Steenberg ML, Contant CF Jr, Krishnappa I, Robertson CS, Bryan RM Jr.. Cerebrovascular reactivity to CO2 and hypotension after mild cortical impact injury. Am J Physiol Heart Circ Physiol 277: H1457–H1466, 1999. doi: 10.1152/ajpheart.1999.277.4.H1457. [DOI] [PubMed] [Google Scholar]
- 232.Harris AP, Ohata H, Koehler RC. Role of nitric oxide in cerebrovascular reactivity to NMDA and hypercapnia during prenatal development in sheep. Int J Dev Neurosci 26: 47–55, 2008. doi: 10.1016/j.ijdevneu.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Helou SM, Hudak ML, Jones MD Jr.. Cerebral blood flow response to hypercapnia in immature fetal sheep. Am J Physiol Heart Circ Physiol 261: H1366–H1370, 1991. doi: 10.1152/ajpheart.1991.261.5.H1366. [DOI] [PubMed] [Google Scholar]
- 234.Iadecola C, Zhang F. Permissive and obligatory roles of NO in cerebrovascular responses to hypercapnia and acetylcholine. Am J Physiol Regul Integr Comp Physiol 271: R990–R1001, 1996. doi: 10.1152/ajpregu.1996.271.4.R990. [DOI] [PubMed] [Google Scholar]
- 235.Kidoguchi K, Tamaki M, Mizobe T, Koyama J, Kondoh T, Kohmura E, Sakurai T, Yokono K, Umetani K. In vivo X-ray angiography in the mouse brain using synchrotron radiation. Stroke 37: 1856–1861, 2006. doi: 10.1161/01.STR.0000226904.96059.a6. [DOI] [PubMed] [Google Scholar]
- 236.Massik J, Jones MD Jr, Miyabe M, Tang YL, Hudak ML, Koehler RC, Traystman RJ. Hypercapnia and response of cerebral blood flow to hypoxia in newborn lambs. J Appl Physiol (1985) 66: 1065–1070, 1989. doi: 10.1152/jappl.1989.66.3.1065. [DOI] [PubMed] [Google Scholar]
- 237.Coverdale NS, Badrov MB, Shoemaker JK. Impact of age on cerebrovascular dilation versus reactivity to hypercapnia. J Cereb Blood Flow Metab 37: 344–355, 2017. doi: 10.1177/0271678X15626156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hotta H, Masamoto K, Uchida S, Sekiguchi Y, Takuwa H, Kawaguchi H, Shigemoto K, Sudo R, Tanishita K, Ito H, Kanno I. Layer-specific dilation of penetrating arteries induced by stimulation of the nucleus basalis of Meynert in the mouse frontal cortex. J Cereb Blood Flow Metab 33: 1440–1447, 2013. doi: 10.1038/jcbfm.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Oizumi XS, Akisaki T, Kouta Y, Song XZ, Takata T, Kondoh T, Umetani K, Hirano M, Yamasaki K, Kohmura E, Yokono K, Sakurai T. Impaired response of perforating arteries to hypercapnia in chronic hyperglycemia. Kobe J Med Sci 52: 27–35, 2006. [PubMed] [Google Scholar]
- 240.Verbree J, Bronzwaer AS, Ghariq E, Versluis MJ, Daemen MJ, van Buchem MA, Dahan A, van Lieshout JJ, van Osch MJ. Assessment of middle cerebral artery diameter during hypocapnia and hypercapnia in humans using ultra-high-field MRI. J Appl Physiol (1985) 117: 1084–1089, 2014. doi: 10.1152/japplphysiol.00651.2014. [DOI] [PubMed] [Google Scholar]
- 241.Wei EP, Kontos HA, Patterson JL Jr.. Dependence of pial arteriolar response to hypercapnia on vessel size. Am J Physiol 238: 697–703, 1980. [DOI] [PubMed] [Google Scholar]
- 242.Willie CK, Macleod DB, Shaw AD, Smith KJ, Tzeng YC, Eves ND, Ikeda K, Graham J, Lewis NC, Day TA, Ainslie PN. Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol 590: 3261–3275, 2012. doi: 10.1113/jphysiol.2012.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kontos HA, Raper AJ, Patterson JL. Analysis of vasoactivity of local pH, Pco2, and bicarbonate on pial vessels. Stroke 8: 358–360, 1977. doi: 10.1161/01.str.8.3.358. [DOI] [PubMed] [Google Scholar]
- 244.Rostrup E, Knudsen GM, Law I, Holm S, Larsson HB, Paulson OB. The relationship between cerebral blood flow and volume in humans. Neuroimage 24: 1–11, 2005. doi: 10.1016/j.neuroimage.2004.09.043. [DOI] [PubMed] [Google Scholar]
- 245.Liu P, De Vis JB, Lu H. Cerebrovascular reactivity (CVR) MRI with CO2 challenge: a technical review. Neuroimage 187: 104–115, 2019. doi: 10.1016/j.neuroimage.2018.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hurn PD, Traystman RJ. Changes in arterial gas tension. In: Cerebral Blood Flow and Metabolism, edited by Edvinsson LK. New York: Lippincott Williams & Wilkens, 2001, p. 384–394. [Google Scholar]
- 247.Okamoto H, Hudetz AG, Roman RJ, Bosnjak ZJ, Kampine JP. Neuronal NOS-derived NO plays permissive role in cerebral blood flow response to hypercapnia. Am J Physiol Heart Circ Physiol 272: H559–H566, 1997. doi: 10.1152/ajpheart.1997.272.1.H559. [DOI] [PubMed] [Google Scholar]
- 248.Faraci FM, Taugher RJ, Lynch C, Fan R, Gupta S, Wemmie JA. Acid-sensing ion channels: Novel mediators of cerebral vascular responses. Circ Res 125: 907–920, 2019. doi: 10.1161/CIRCRESAHA.119.315024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wenzel J, Hansen CE, Bettoni C, Vogt MA, Lembrich B, Natsagdorj R, Huber G, Brands J, Schmidt K, Assmann JC, Stolting I, Saar K, Sedlacik J, Fiehler J, Ludewig P, Wegmann M, Feller N, Richter M, Muller-Fielitz H, Walther T, Konig GM, Kostenis E, Raasch W, Hubner N, Gass P, Offermanns S, de Wit C, Wagner CA, Schwaninger M. Impaired endothelium-mediated cerebrovascular reactivity promotes anxiety and respiration disorders in mice. Proc Natl Acad Sci U S A 117: 1753–1761, 2020. doi: 10.1073/pnas.1907467117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Aubineau P, Sercombe R, Seylaz J. Synergy of effects of PaO2 and PaCO2 on local cerebral blood flow: Evidence against extrinsic neural mediation of hypoxia-induced cerebral vasodilation. In: Cerebral Blood Flow: Effects of Nerves and Neurotransmitters, edited by Heistad DD, Marcus ML.. Amsterdam, The Natherlands: Elsevier North Holland, Inc., 1982, p. 463–471. [Google Scholar]
- 251.Fortune JB, Feustel PJ, deLuna C, Graca L, Hasselbarth J, Kupinski AM. Cerebral blood flow and blood volume in response to O2 and CO2 changes in normal humans. J Trauma 39: 463–471, 1995. doi: 10.1097/00005373-199509000-00012. [DOI] [PubMed] [Google Scholar]
- 252.Hoiland RL, Bain AR, Rieger MG, Bailey DM, Ainslie PN. Hypoxemia, oxygen content, and the regulation of cerebral blood flow. Am J Physiol Regul Integr Comp Physiol 310: R398–R413, 2016. doi: 10.1152/ajpregu.00270.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hudetz AG. Regulation of oxygen supply in the cerebral circulation. In: Oxygen Transport to Tissue XIX, edited by Harrison D. New York: Plenum Press, 1997, pp. 513–520. [DOI] [PubMed] [Google Scholar]
- 254.Ogoh S, Nakahara H, Ainslie PN, Miyamoto T. The effect of oxygen on dynamic cerebral autoregulation: critical role of hypocapnia. J Appl Physiol (1985) 108: 538–543, 2010. doi: 10.1152/japplphysiol.01235.2009. [DOI] [PubMed] [Google Scholar]
- 255.Poulin MJ, Liang PJ, Robbins PA. Dynamics of the cerebral blood flow response to step changes in end-tidal Pco2 and Po2 in humans. J Appl Physiol (1985) 81: 1084–1095, 1996. doi: 10.1152/jappl.1996.81.3.1084. [DOI] [PubMed] [Google Scholar]
- 256.Rosenberg AA, Narayanan V, Jones MD Jr.. Comparison of anterior cerebral artery blood flow velocity and cerebral blood flow during hypoxia. Pediatr Res 19: 67–70, 1985. doi: 10.1203/00006450-198501000-00018. [DOI] [PubMed] [Google Scholar]
- 257.Ursino M, Magosso E. Acute cardiovascular response to isocapnic hypoxia. I. A mathematical model. Am J Physiol Heart Circ Physiol 279: H149–H165, 2000. doi: 10.1152/ajpheart.2000.279.1.H149. [DOI] [PubMed] [Google Scholar]
- 258.Jones MD Jr, Traystman RJ, Simmons MA, Molteni RA. Effects of changes in arterial O2 content on cerebral blood flow in the lamb. Am J Physiol Heart Circ Physiol 240: H209–H215, 1981. doi: 10.1152/ajpheart.1981.240.2.H209. [DOI] [PubMed] [Google Scholar]
- 259.Phillis JW. Adenosine and adenine nucleotides as regulators of cerebral blood flow: roles of acidosis, cell swelling, and KATP channels. Crit Rev Neurobiol 16: 237–270, 2004. doi: 10.1615/CritRevNeurobiol.v16.i4.20. [DOI] [PubMed] [Google Scholar]
- 260.Kontos HA, Wei EP. Oxygen-dependent mechanisms in cerebral autoregulation. Ann Biomed Eng 13: 329–334, 1985. doi: 10.1007/BF02584251. [DOI] [PubMed] [Google Scholar]
- 261.Chan SL, Sweet JG, Bishop N, Cipolla MJ. Pial collateral reactivity during hypertension and aging: Understanding the function of collaterals for stroke therapy. Stroke 47: 1618–1625, 2016. doi: 10.1161/STROKEAHA.116.013392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Holm A, Heumann T, Augustin HG. Microvascular mural cell organotypic heterogeneity and functional plasticity. Trends Cell Biol 28: 302–316, 2018. doi: 10.1016/j.tcb.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 263.Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT. Vascular TRP channels: performing under pressure and going with the flow. Physiology (Bethesda) 29: 343–360, 2014. doi: 10.1152/physiol.00009.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Aydin F, Rosenblum WI, Povlishock JT. Myoendothelial junctions in human brain arterioles. Stroke 22: 1592–1597, 1991. doi: 10.1161/01.STR.22.12.1592. [DOI] [PubMed] [Google Scholar]
- 265.De Silva TM, Faraci FM. Contributions of aging to cerebral small vessel disease. Annu Rev Physiol 82: 275–295, 2020. doi: 10.1146/annurev-physiol-021119-034338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Sobey CG, Faraci FM. Effect of nitric oxide and potassium channel agonists and inhibitors on basilar artery diameter. Am J Physiol Heart Circ Physiol 272: H256–H262, 1997. doi: 10.1152/ajpheart.1997.272.1.H256. [DOI] [PubMed] [Google Scholar]
- 267.SchäChinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 101: 1899–1906, 2000. doi: 10.1161/01.CIR.101.16.1899. [DOI] [PubMed] [Google Scholar]
- 268.Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149–1160, 2003. doi: 10.1016/S0735-1097(03)00994-X. [DOI] [PubMed] [Google Scholar]
- 269.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol 12: 483–497, 2013. doi: 10.1016/S1474-4422(13)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Adib-Samii P, Rost N, Traylor M, Devan W, Biffi A, Lanfranconi S, et al. 17q25 Locus is associated with white matter hyperintensity volume in ischemic stroke, but not with lacunar stroke status. Stroke 44: 1609–1615, 2013. doi: 10.1161/STROKEAHA.113.679936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Jahshan S, Dayan L, Jacob G. Nitric oxide-sensitive guanylyl cyclase signaling affects CO2-dependent but not pressure-dependent regulation of cerebral blood flow. Am J Physiol Regul Integr Comp Physiol 312: R948–R955, 2017. doi: 10.1152/ajpregu.00241.2016. [DOI] [PubMed] [Google Scholar]
- 272.Zhang R, Wilson TE, Witkowski S, Cui J, Crandall GG, Levine BD. Inhibition of nitric oxide synthase does not alter dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol 286: H863–H869, 2004. doi: 10.1152/ajpheart.00373.2003. [DOI] [PubMed] [Google Scholar]
- 273.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215, 2011. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 274.Berthiaume AA, Hartmann DA, Majesky MW, Bhat NR, Shih AY. Pericyte structural remodeling in cerebrovascular health and homeostasis. Front Aging Neurosci 10: 210, 2018. doi: 10.3389/fnagi.2018.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Uemura MT, Maki T, Ihara M, Lee VM, Trojanowski JQ. Brain microvascular pericytes in vascular cognitive impairment and dementia. Front Aging Neurosci 12: 80, 2020. doi: 10.3389/fnagi.2020.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Dalkara T. Pericytes: A novel target to improve success of recanalization therapies. Stroke 50: 2985–2991, 2019. doi: 10.1161/STROKEAHA.118.023590. [DOI] [PubMed] [Google Scholar]
- 277.Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 15: 1031–1037, 2009. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 278.Faraci FM, Mayhan WG, Werber AH, Heistad DD. Cerebral circulation: effects of sympathetic nerves and protective mechanisms during hypertension. Circulation Res 61: 102–106, 1987. [PubMed] [Google Scholar]
- 279.Heistad DD. Protection of cerebral vessels by sympathetic nerves. Physiologist 23: 44–49, 1980. [PubMed] [Google Scholar]
- 280.Parati G, Mancia G, Rienzo MD, Castiglioni P. Point:Counterpoint: Cardiovascular variability is/is not an index of autonomic control of circulation. J Appl Physiol 101: 676–682, 2006. doi: 10.1152/japplphysiol.00446.2006. [DOI] [PubMed] [Google Scholar]
- 281.Claassen JA, Jansen RW. Cholinergically mediated augmentation of cerebral perfusion in Alzheimer's disease and related cognitive disorders: the cholinergic-vascular hypothesis. J Gerontol A Biol Sci Med Sci 61: 267–271, 2006. doi: 10.1093/gerona/61.3.267. [DOI] [PubMed] [Google Scholar]
- 282.Van Beek AH, Claassen JA. The cerebrovascular role of the cholinergic neural system in Alzheimer's disease. Behav Brain Res 221: 537–542, 2011. doi: 10.1016/j.bbr.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 283.ter Laan M, van Dijk JM, Elting JW, Staal MJ, Absalom AR. Sympathetic regulation of cerebral blood flow in humans: a review. Br J Anaesth 111: 361–367, 2013. doi: 10.1093/bja/aet122. [DOI] [PubMed] [Google Scholar]
- 284.Hafkenschiel JH, Crumpton CW, Shenkin HA, Moyer JH, Zintel HA, Wendel H, Jeffers WA. The effects of twenty degree head-up tilt upon the cerebral circulation of patients with arterial hypertension before and after sympathectomy. J Clin Invest 30: 793–798, 1951. doi: 10.1172/JCI102494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Jeng JS, Yip PK, Huang SJ, Kao MC. Changes in hemodynamics of the carotid and middle cerebral arteries before and after endoscopic sympathectomy in patients with palmar hyperhidrosis: preliminary results. J Neurosurg 90: 463–467, 1999. doi: 10.3171/jns.1999.90.3.0463. [DOI] [PubMed] [Google Scholar]
- 286.Suzuki J, Iwabuchi T, Hori S. Cervical sympathectomy for cerebral vasospasm after aneurysm rupture. Neurol Med Chir (Tokyo) 15: 41–50, 1975. doi: 10.2176/nmc.15pt1.41. [DOI] [PubMed] [Google Scholar]
- 287.Edvinson L. Neurogenic mechanisms in the cerebrovascular bed. Autonomic nerves, amine receptors and their effects on cerebral blood flow. Acta Physiol Scand Suppl 427: 1–35, 1975. [PubMed] [Google Scholar]
- 288.Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension 36: 383–388, 2000. doi: 10.1161/01.HYP.36.3.383. [DOI] [PubMed] [Google Scholar]
- 289.Zhang R, Levine BD. Autonomic ganglionic blockade does not prevent reduction in cerebral blood flow velocity during orthostasis in humans. Stroke 38: 1238–1244, 2007. doi: 10.1161/01.STR.0000260095.94175.d0. [DOI] [PubMed] [Google Scholar]
- 290.Kimmerly DS, Tutungi E, Wilson TD, Serrador JM, Gelb AW, Hughson RL, Shoemaker JK. Circulating norepinephrine and cerebrovascular control in conscious humans. Clin Physiol Funct Imaging 23: 314–319, 2003. doi: 10.1046/j.1475-0961.2003.00507.x. [DOI] [PubMed] [Google Scholar]
- 291.Levine BD, Zhang R. Comments on Point:Counterpoint: Sympathetic activity does/does not influence cerebral blood flow. J Appl Physiol 105: 1369–1373, 2008. doi: 10.1152/japplphysiol.zdg-8199.pcpcomm.2008. [DOI] [PubMed] [Google Scholar]
- 292.van Lieshout JJ, Secher NH, Strandgaard S, Sigurdsson ST. Point:Counterpoint: Sympathetic activity does/does not influence cerebral blood flow. J Appl Physiol 105: 1364–1368, 2008. doi: 10.1152/japplphysiol.90597.2008. [DOI] [PubMed] [Google Scholar]
- 293.D'Alecy LG, Rose CJ. Parasympathetic cholinergic control of cerebral blood flow in dogs. Circ Res 41: 324–331, 1977. doi: 10.1161/01.res.41.3.324. [DOI] [PubMed] [Google Scholar]
- 294.Scremin OU, Rovere AA, Raynald AC, Giardini A. Cholinergic control of blood flow in the cerebral cortex of the rat. Stroke 4: 233–239, 1973. doi: 10.1161/01.STR.4.2.232. [DOI] [PubMed] [Google Scholar]
- 295.Hamner JW, Tan CO, Tzeng YC, Taylor JA. Cholinergic control of the cerebral vasculature in humans. J Physiol 590: 6343–6352, 2012. doi: 10.1113/jphysiol.2012.245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Seifert T, Fisher JP, Young CN, Hartwich D, Ogoh S, Raven PB, Fadel PJ, Secher NH. Glycopyrrolate abolishes the exercise-induced increase in cerebral perfusion in humans. Exp Physiol 95: 1016–1025, 2010. doi: 10.1113/expphysiol.2010.054346. [DOI] [PubMed] [Google Scholar]
- 297.Lecrux C, Hamel E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos Trans R Soc Lond B Biol Sci 371: 20150350, 2016. doi: 10.1098/rstb.2015.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Vaucher E, Hamel E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: Electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J Neurosci 15: 7427–7441, 1995. doi: 10.1523/JNEUROSCI.15-11-07427.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ashina H, Schytz HW, Ashina M. CGRP in human models of migraine. Handb Exp Pharmacol 255: 109–120, 2019. doi: 10.1007/164_2018_128. [DOI] [PubMed] [Google Scholar]
- 300.Ashina M, Hansen JM, Do TP, Melo-Carrillo A, Burstein R, Moskowitz MA. Migraine and the trigeminovascular system–40 years and counting. Lancet Neurol 18: 795–804, 2019. doi: 10.1016/S1474-4422(19)30185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Moskowitz MA, Wei EP, Saito K, Kontos HA. Trigeminalectomy modifies pial arteriolar responses to hypertension or norepinephrine. Am J Physiol Heart Circ Physiol 255: H1–H6, 1988. doi: 10.1152/ajpheart.1988.255.1.H1. [DOI] [PubMed] [Google Scholar]
- 302.Sakas DE, Moskowitz MA, Wei EP, Kontos HA, Kano M, Ogilvy CS. Trigeminovascular fibers increase blood flow in cortical gray matter by axon reflex-like mechanisms during acute severe hypertension or seizures. Proc Natl Acad Sci U S A 86: 1401–1405, 1989. doi: 10.1073/pnas.86.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Green DJ, Hopman MT, Padilla J, Laughlin MH, Thijssen DH. Vascular adaptation to exercise in humans: Role of hemodynamic stimuli. Physiol Rev 97: 495–528, 2017. doi: 10.1152/physrev.00014.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Newcomer SC, Thijssen DH, Green DJ. Effects of exercise on endothelium and endothelium/smooth muscle cross talk: role of exercise-induced hemodynamics. J Appl Physiol (1985) 111: 311–320, 2011. doi: 10.1152/japplphysiol.00033.2011. [DOI] [PubMed] [Google Scholar]
- 305.Tinken TM, Thijssen DH, Hopkins N, Black MA, Dawson EA, Minson CT, Newcomer SC, Laughlin MH, Cable NT, Green DJ. Impact of shear rate modulation on vascular function in humans. Hypertension 54: 278–285, 2009. doi: 10.1161/HYPERTENSIONAHA.109.134361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Tinken TM, Thijssen DH, Hopkins N, Dawson EA, Cable NT, Green DJ. Shear stress mediates endothelial adaptations to exercise training in humans. Hypertension 55: 312–318, 2010. doi: 10.1161/HYPERTENSIONAHA.109.146282. [DOI] [PubMed] [Google Scholar]
- 307.Schreuder TH, Green DJ, Hopman MT, Thijssen DH. Impact of retrograde shear rate on brachial and superficial femoral artery flow-mediated dilation in older subjects. Atherosclerosis 241: 199–204, 2015. doi: 10.1016/j.atherosclerosis.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 308.Thijssen DH, Dawson EA, Tinken TM, Cable NT, Green DJ. Retrograde flow and shear rate acutely impair endothelial function in humans. Hypertension 53: 986–992, 2009. doi: 10.1161/HYPERTENSIONAHA.109.131508. [DOI] [PubMed] [Google Scholar]
- 309.Thijssen DH, Schreuder TH, Newcomer SW, Laughlin MH, Hopman MT, Green DJ. Impact of 2-weeks continuous increase in retrograde shear stress on brachial artery vasomotor function in young and older men. J Am Heart Assoc 4: e001968, 2015. doi: 10.1161/jaha.115.001968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.de Weerd M, Greving JP, Hedblad B, Lorenz MW, Mathiesen EB, O'Leary DH, Rosvall M, Sitzer M, Buskens E, Bots ML. Prevalence of asymptomatic carotid artery stenosis in the general population: An individual participant data meta-analysis. Stroke 41: 1294–1297, 2010. doi: 10.1161/STROKEAHA.110.581058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lorenz MW, Schaefer C, Steinmetz H, Sitzer M. Is carotid intima media thickness useful for individual prediction of cardiovascular risk? Ten-year results from the Carotid Atherosclerosis Progression Study (CAPS). Eur Heart J 31: 2041–2048, 2010. doi: 10.1093/eurheartj/ehq189. [DOI] [PubMed] [Google Scholar]
- 312.Seneviratne A, Hulsmans M, Holvoet P, Monaco C. Biomechanical factors and macrophages in plaque stability. Cardiovasc Res 99: 284–293, 2013. doi: 10.1093/cvr/cvt097. [DOI] [PubMed] [Google Scholar]
- 313.Moroni F, Ammirati E, Magnoni M, D'Ascenzo F, Anselmino M, Anzalone N, Rocca MA, Falini A, Filippi M, Camici PG. Carotid atherosclerosis, silent ischemic brain damage and brain atrophy: A systematic review and meta-analysis. Int J Cardiol 223: 681–687, 2016. doi: 10.1016/j.ijcard.2016.08.234. [DOI] [PubMed] [Google Scholar]
- 314.Carcaillon L, Plichart M, Zureik M, Rouaud O, Majed B, Ritchie K, Tzourio C, Dartigues JF, Empana JP. Carotid plaque as a predictor of dementia in older adults: The Three-City Study. Alzheimers Dement 11: 239–248, 2015. doi: 10.1016/j.jalz.2014.07.160. [DOI] [PubMed] [Google Scholar]
- 315.Gutierrez J, Guzman V, Khasiyev F, Manly J, Schupf N, Andrews H, Mayeux R, Brickman AM. Brain arterial dilatation and the risk of Alzheimer's disease. Alzheimers Dement 15: 666–674, 2019. doi: 10.1016/j.jalz.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mutsaerts HJ, Palm-Meinders IH, de Craen AJ, Reiber JH, Blauw GJ, van Buchem MA, van der Grond J, Box FM, PROSPER Study Group. Diastolic carotid artery wall shear stress is associated with cerebral infarcts and periventricular white matter lesions. Stroke 42: 3497–3501, 2011. doi: 10.1161/STROKEAHA.111.614453. [DOI] [PubMed] [Google Scholar]
- 317.Liu Z, Zhao Y, Wang X, Zhang H, Cui Y, Diao Y, Xiu J, Sun X, Jiang G. Low carotid artery wall shear stress is independently associated with brain white-matter hyperintensities and cognitive impairment in older patients. Atherosclerosis 247: 78–86, 2016. doi: 10.1016/j.atherosclerosis.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 318.Zhang H, Liu H, Dong Y, Wang J, Zhao Y, Cui Y, Chai Q, Liu Z. Low carotid wall shear stress independently accelerates the progression of cognitive impairment and white matter lesions in the elderly. Oncotarget 9: 11402–11413, 2018. doi: 10.18632/oncotarget.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Tarumi T, Ayaz KM, Liu J, Tseng BY, Parker R, Riley J, Tinajero C, Zhang R. Cerebral hemodynamics in normal aging: central artery stiffness, wave reflection, and pressure pulsatility. J Cereb Blood Flow Metabol 34: 971–978, 2014. doi: 10.1038/jcbfm.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Zhao X, Zhao M, Amin-Hanjani S, Du X, Ruland S, Charbel FT. Wall shear stress in major cerebral arteries as a function of age and gender–a study of 301 healthy volunteers. J Neuroimaging 25: 403–407, 2015. doi: 10.1111/jon.12133. [DOI] [PubMed] [Google Scholar]
- 321.Webb AJ, Simoni M, Mazzucco S, Kuker W, Schulz U, Rothwell PM. Increased cerebral arterial pulsatility in patients with leukoaraiosis: Arterial stiffness enhances transmission of aortic pulsatility. Stroke 43: 2631–2636, 2012. doi: 10.1161/STROKEAHA.112.655837. [DOI] [PubMed] [Google Scholar]
- 322.Roher AE, Garami Z, Tyas SL, Maarouf CL, Kokjohn TA, Belohlavek M, Vedders LJ, Connor D, Sabbagh MN, Beach TG, Emmerling MR. Transcranial Doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer's disease. Alzheimers Dement 7: 445–455, 2011. doi: 10.1016/j.jalz.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.van Es AC, van der Flier WM, Box FM, Middelkoop HA, Westendorp RG, van Buchem MA, van der Grond J. Carotid and basilar artery wall shear stress in Alzheimer's disease and mild cognitive impairment. Dement Geriatr Cogn Disord 28: 220–224, 2009. doi: 10.1159/000237740. [DOI] [PubMed] [Google Scholar]
- 324.Tanaka M, Sakaguchi M, Miwa K, Okazaki S, Furukado S, Yagita Y, Mochizuki H, Kitagawa K. Basilar artery diameter is an independent predictor of incident cardiovascular events. Arterioscler Thromb Vasc Biol 33: 2240–2244, 2013. doi: 10.1161/ATVBAHA.113.301467. [DOI] [PubMed] [Google Scholar]
- 325.Cornwell WK 3rd, Tarumi T, Aengevaeren VL, Ayers C, Divanji P, Fu Q, Palmer D, Drazner MH, Meyer DM, Bethea BT, Hastings JL, Fujimoto N, Shibata S, Zhang R, Markham DW, Levine BD. Effect of pulsatile and nonpulsatile flow on cerebral perfusion in patients with left ventricular assist devices. J Heart Lung Transplant 33: 1295–1303, 2014. doi: 10.1016/j.healun.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 326.Dawson EA, Secher NH, Dalsgaard MK, Ogoh S, Yoshiga CC, Gonzalez-Alonso J, Steensberg A, Raven PB. Standing up to the challenge of standing: a siphon does not support cerebral blood flow in humans. Am J Physiol Regul Integr Comp Physiol 287: R911–R914, 2004. doi: 10.1152/ajpregu.00196.2004. [DOI] [PubMed] [Google Scholar]
- 327.Gisolf J, Gisolf A, van Lieshout JJ, Karemaker JM. The siphon controversy: an integration of concepts and the brain as baffle. Am J Physiol Regul Integr Comp Physiol 289: R627–R629, 2005. doi: 10.1152/ajpregu.00709.2004. [DOI] [PubMed] [Google Scholar]
- 328.Hill L. The influence of the force of gravity on the circulation of the blood. J Physiol 18: 15–53, 1895. doi: 10.1113/jphysiol.1895.sp000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Munis JR, Lozada LJ. Giraffes, siphons, and starling resistors. Cerebral perfusion pressure revisited. J Neurosurg Anesthesiol 12: 290–296, 2000. doi: 10.1097/00008506-200007000-00029. [DOI] [PubMed] [Google Scholar]
- 330.Hicks JW, Badeer HS. Siphon mechanism in collapsible tubes: application to circulation of the giraffe head. Am J Physiol Regul Integr Comp Physiol 256: R567–R571, 1989. doi: 10.1152/ajpregu.1989.256.2.R567. [DOI] [PubMed] [Google Scholar]
- 331.Seymour RS. Cardiovascular physiology of dinosaurs. Physiology (Bethesda) 31: 430–441, 2016. doi: 10.1152/physiol.00016.2016. [DOI] [PubMed] [Google Scholar]
- 332.Seymour RS, Lillywhite HB. Why vascular siphons with sub-atmospheric pressures are physiologically impossible in sauropod dinosaurs. J Exp Biol 219: 2078–2079, 2016. doi: 10.1242/jeb.140988. [DOI] [PubMed] [Google Scholar]
- 333.Lillywhite HB. Snakes, blood-circulation and gravity. Sci Am 259: 92–98, 1988. doi: 10.1038/scientificamerican1288-92. 3070746 [DOI] [Google Scholar]
- 334.Gisolf J, van Lieshout JJ, van Heusden K, Pott F, Stok WJ, Karemaker JM. Human cerebral venous outflow pathway depends on posture and central venous pressure. J Physiol 560: 317–327, 2004. doi: 10.1113/jphysiol.2004.070409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Gisolf J, Wilders R, Immink RV, van Lieshout JJ, Karemaker JM. Tidal volume, cardiac output and functional residual capacity determine end-tidal CO2 transient during standing up in humans. J Physiol 554: 579–590, 2004. doi: 10.1113/jphysiol.2003.056895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Van Lieshout JJ, Wieling W, Karemaker JM, Secher NH. Syncope, cerebral perfusion, and oxygenation. J Appl Physiol (1985) 94: 833–848, 2003. doi: 10.1152/japplphysiol.00260.2002. [DOI] [PubMed] [Google Scholar]
- 337.Wieling W, Van Lieshout JJ, Ten Harkel AD. Dynamics of circulatory adjustments to head-up tilt and tilt-back in healthy and sympathetically denervated subjects. Clin Sci (Lond) 94: 347–352, 1998. doi: 10.1042/cs0940347. [DOI] [PubMed] [Google Scholar]
- 338.Foldager N, Andersen TA, Jessen FB, Ellegaard P, Stadeager C, Videbaek R, Norsk P. Central venous pressure in humans during microgravity. J Appl Physiol 81: 408–412, 1996. doi: 10.1152/jappl.1996.81.1.408. [DOI] [PubMed] [Google Scholar]
- 339.Latham RD, Fanton JW, Vernalis MN, Gaffney FA, Crisman RP. Central hemodynamics in a baboon model during microgravity induced by parabolic flight. Adv Space Res 14: 349–358, 1994. doi: 10.1016/0273-1177(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 340.Klein T, Wollseiffen P, Sanders M, Claassen J, Carnahan H, Abeln V, Vogt T, Struder HK, Schneider S. The influence of microgravity on cerebral blood flow and electrocortical activity. Exp Brain Res 237: 1057–1062, 2019. doi: 10.1007/s00221-019-05490-6. [DOI] [PubMed] [Google Scholar]
- 341.Ogoh S, Hirasawa A, Raven PB, Rebuffat T, Denise P, Lericollais R, Sugawara J, Normand H. Effect of an acute increase in central blood volume on cerebral hemodynamics. Am J Physiol Regul Integr Comp Physiol 309: R902–R911, 2015. doi: 10.1152/ajpregu.00137.2015. [DOI] [PubMed] [Google Scholar]
- 342.Waki H, Shimizu T, Katahira K, Nagayama T, Yamasaki M, Katsuda S. Effects of microgravity elicited by parabolic flight on abdominal aortic pressure and heart rate in rats. J Appl Physiol 93: 1893–1899, 2002. doi: 10.1152/japplphysiol.01064.2001. [DOI] [PubMed] [Google Scholar]
- 343.Klein T, Sanders M, Wollseiffen P, Carnahan H, Abeln V, Askew CD, Claassen JA, Schneider S. Transient cerebral blood flow responses during microgravity. Life Sci Space Res (Amst) 25: 66–71, 2020. doi: 10.1016/j.lssr.2020.03.003. [DOI] [PubMed] [Google Scholar]
- 344.Lawley JS, Petersen LG, Howden EJ, Sarma S, Cornwell WK, Zhang R, Whitworth LA, Williams MA, Levine BD. Effect of gravity and microgravity on intracranial pressure. J Physiol 595: 2115–2127, 2017. doi: 10.1113/JP273557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Pohl A, Cullen DJ. Cerebral ischemia during shoulder surgery in the upright position: A case series. J Clin Anesth 17: 463–469, 2005. doi: 10.1016/j.jclinane.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 346.Ainslie PN, Cotter JD, George KP, Lucas S, Murrell C, Shave R, Thomas KN, Williams MJ, Atkinson G. Elevation in cerebral blood flow velocity with aerobic fitness throughout healthy human ageing. J Physiol 586: 4005–4010, 2008. doi: 10.1113/jphysiol.2008.158279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Braz ID, Fluck D, Lip GY, Lundby C, Fisher JP. Impact of aerobic fitness on cerebral blood flow and cerebral vascular responsiveness to CO2 in young and older men. Scand J Med Sci Sports 27: 634–642, 2017. doi: 10.1111/sms.12674. [DOI] [PubMed] [Google Scholar]
- 348.Leenders KL, Perani D, Lammertsma AA, Heather JD, Buckingham P, Jones T, Healy MJ, Gibbs JM, Wise RJ, Hatazawa J, Herold S, Beaney RP, Brooks DJ, Spinks T, Rhodes C, Frackowiak RS. Cerebral blood flow, blood volume and oxygen utilization. Normal values and effect of age. Brain 113: 27–47, 1990. doi: 10.1093/brain/113.1.27. [DOI] [PubMed] [Google Scholar]
- 349.Xekardaki A, Rodriguez C, Montandon ML, Toma S, Tombeur E, Herrmann FR, Zekry D, Lovblad KO, Barkhof F, Giannakopoulos P, Haller S. Arterial spin labeling may contribute to the prediction of cognitive deterioration in healthy elderly individuals. Radiology 274: 490–499, 2015. doi: 10.1148/radiol.14140680. [DOI] [PubMed] [Google Scholar]
- 350.De VJ, Peng SL, Chen X, Li Y, Liu P, Sur S, Rodrigue KM, Park DC, Lu H. Arterial-spin-labeling (ASL) perfusion MRI predicts cognitive function in elderly individuals: a 4-year longitudinal study. J Magn Reson Imaging 48: 449–458, 2018. doi: 10.1002/jmri.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Wolters FJ, Zonneveld HI, Hofman A, van der Lugt A, Koudstaal PJ, Vernooij MW, Ikram MA, Heart-Brain Connection Collaborative Research Group. Cerebral perfusion and the risk of dementia. A population-based study. Circulation 136: 719–728, 2017. doi: 10.1161/CIRCULATIONAHA.117.027448. [DOI] [PubMed] [Google Scholar]
- 352.Yew B, Nation DA, Alzheimer's Disease Neuroimaging Initiative. Cerebrovascular resistance: effects on cognitive decline, cortical atrophy, and progression to dementia. Brain 140: 2001–2017, 1987. doi: 10.1093/brain/awx112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Boumezbeur F, Mason GF, de Graaf RA, Behar KL, Cline GW, Shulman GI, Rothman DL, Petersen KF. Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 30: 211–221, 2010. doi: 10.1038/jcbfm.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Amin-Hanjani S, Du X, Pandey DK, Thulborn KR, Charbel FT. Effect of age and vascular anatomy on blood flow in major cerebral vessels. J Cereb Blood Flow Metab 35: 312–318, 2015. doi: 10.1038/jcbfm.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Mander BA, Rao V, Lu B, Saletin JM, Lindquist JR, Ancoli-Israel S, Jagust W, Walker MP. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci 16: 357–364, 2013. doi: 10.1038/nn.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Oh H, Madison C, Villeneuve S, Markley C, Jagust WJ. Association of gray matter atrophy with age, β-amyloid, and cognition in aging. Cereb Cortex 24: 1609–1618, 2014. doi: 10.1093/cercor/bht017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Jefferson AL, Liu D, Gupta DK, Pechman KR, Watchmaker JM, Gordon EA, Rane S, Bell SP, Mendes LA, Davis LT, Gifford KA, Hohman TJ, Wang TJ, Donahue MJ. Lower cardiac index levels relate to lower cerebral blood flow in older adults. Neurology 89: 2327–2334, 2017. doi: 10.1212/WNL.0000000000004707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Lu H, Xu F, Rodrigue KM, Kennedy KM, Cheng Y, Flicker B, Hebrank AC, Uh J, Park DC. Alterations in cerebral metabolic rate and blood supply across the adult lifespan. Cereb Cortex 21: 1426–1434, 2011. doi: 10.1093/cercor/bhq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Chen JJ, Rosas HD, Salat DH. Age-associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 55: 468–478, 2011. doi: 10.1016/j.neuroimage.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Wahlin A, Nyberg L. At the heart of cognitive functioning in aging. Trends Cogn Sci 23: 717–720, 2019. doi: 10.1016/j.tics.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 361.Czosnyka M, Balestreri M, Steiner L, Smielewski P, Hutchinson PJ, Matta B, Pickard JD. Age, intracranial pressure, autoregulation, and outcome after brain trauma. J Neurosurg 102: 450–454, 2005. doi: 10.3171/jns.2005.102.3.0450. [DOI] [PubMed] [Google Scholar]
- 362.Oudegeest-Sander MH, van Beek AH, Abbink K, Olde Rikkert MG, Hopman MT, Claassen JA. Assessment of dynamic cerebral autoregulation and cerebrovascular CO2 reactivity in ageing by measurements of cerebral blood flow and cortical oxygenation. Exp Physiol 99: 586–598, 2014. doi: 10.1113/expphysiol.2013.076455. [DOI] [PubMed] [Google Scholar]
- 363.Aengevaeren VL, Claassen JA, Levine BD, Zhang R. Cardiac baroreflex function and dynamic cerebral autoregulation in elderly Masters athletes. J Appl Physiol (1985) 114: 195–202, 2013. doi: 10.1152/japplphysiol.00402.2012. [DOI] [PubMed] [Google Scholar]
- 364.Brodie FG, Panerai RB, Foster S, Evans DH, Robinson TG. Long-term changes in dynamic cerebral autoregulation: a 10 years follow up study. Clin Physiol Funct Imaging 29: 366–371, 2009. doi: 10.1111/j.1475-097X.2009.00880.x. [DOI] [PubMed] [Google Scholar]
- 365.Carey BJ, Eames PJ, Blake MJ, Panerai RB, Potter JF. Dynamic cerebral autoregulation is unaffected by aging. Stroke 31: 2895–2900, 2000. doi: 10.1161/01.STR.31.12.2895. [DOI] [PubMed] [Google Scholar]
- 366.Carey BJ, Panerai RB, Potter JF. Effect of aging on dynamic cerebral autoregulation during head-up tilt. Stroke 34: 1871–1875, 2003. doi: 10.1161/01.STR.0000081981.99908.F3. [DOI] [PubMed] [Google Scholar]
- 367.Dineen NE, Brodie FG, Panerai RB, Robinson TG. Effects of ageing on cerebral autoregulation assessed during respiratory manoeuvres. Age Ageing 40: 199–204, 2011. doi: 10.1093/ageing/afq170. [DOI] [PubMed] [Google Scholar]
- 368.Edlow BL, Kim MN, Durduran T, Zhou C, Putt ME, Yodh AG, Greenberg JH, Detre JA. The effects of healthy aging on cerebral hemodynamic responses to posture change. Physiol Meas 31: 477–495, 2010. doi: 10.1088/0967-3334/31/4/002. [DOI] [PubMed] [Google Scholar]
- 369.Lipsitz LA, Mukai S, Hamner J, Gagnon M, Babikian V. Dynamic regulation of middle cerebral artery blood flow velocity in aging and hypertension. Stroke 31: 1897–1903, 2000. doi: 10.1161/01.STR.31.8.1897. [DOI] [PubMed] [Google Scholar]
- 370.Xing CY, Tarumi T, Meijers RL, Turner M, Repshas J, Xiong L, Ding K, Vongpatanasin W, Yuan LJ, Zhang R. Arterial pressure, heart rate, and cerebral hemodynamics across the adult life span. Hypertension 69: 712–720, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Tzeng YC, Lucas SJ, Atkinson G, Willie CK, Ainslie PN. Fundamental relationships between arterial baroreflex sensitivity and dynamic cerebral autoregulation in humans. J Appl Physiol (1985) 108: 1162–1168, 2010. doi: 10.1152/japplphysiol.01390.2009. [DOI] [PubMed] [Google Scholar]
- 372.Maxwell JD. Novel Interventions to Improve Cerebral and Peripheral Vascular Function. Liverpool, UK: Liverpool John Moores University, 2020. [Google Scholar]
- 373.Kastrup A, Dichgans J, Niemeier M, Schabet M. Changes of cerebrovascular CO2 reactivity during normal aging. Stroke 29: 1311–1314, 1998. doi: 10.1161/01.STR.29.7.1311. [DOI] [PubMed] [Google Scholar]
- 374.Wiesmann M, Kiliaan AJ, Claassen JA. Vascular aspects of cognitive impairment and dementia. J Cereb Blood Flow Metab 33: 1696–1706, 2013. doi: 10.1038/jcbfm.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Shekhar S, Liu R, Travis OK, Roman RJ, Fan F. Cerebral autoregulation in hypertension and ischemic stroke: a mini review. J Pharm Sci Exp Pharmacol 2017: 21–27, 2017. [PMC free article] [PubMed] [Google Scholar]
- 376.Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312: H1–H20, 2017. doi: 10.1152/ajpheart.00581.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Strandgaard S. Autoregulation of cerebral blood flow in hypertensive patients. The modifying influence of prolonged antihypertensive treatment on the tolerance to acute, drug-induced hypotension. Circulation 53: 720–727, 1976. doi: 10.1161/01.cir.53.4.720. [DOI] [PubMed] [Google Scholar]
- 378.Strandgaard S, Paulson OB. Cerebral autoregulation. Stroke 15: 413–416, 1984. doi: 10.1161/01.str.15.3.413. [DOI] [PubMed] [Google Scholar]
- 379.Strandgaard S, Paulson OB. Regulation of cerebral blood flow in health and disease. J Cardiovasc Pharmacol 19: S89–93, 1992. doi: 10.1097/00005344-199219006-00014. [DOI] [PubMed] [Google Scholar]
- 380.Strandgaard S, Paulson OB. Cerebral blood flow in untreated and treated hypertension. Neth J Med 47: 180–184, 1995. doi: 10.1016/0300-2977(95)00065-u. [DOI] [PubMed] [Google Scholar]
- 381.Goodwin JS. Embracing complexity: a consideration of hypertension in the very old. J Gerontol A Biol Sci Med Sci 58: 653–658, 2003. doi: 10.1093/gerona/58.7.m653. [DOI] [PubMed] [Google Scholar]
- 382.Rigaud AS, Forette B. Hypertension in older adults. J Gerontol A Biol Sci Med Sci 56: M217–225, 2001. doi: 10.1093/gerona/56.4.m217. [DOI] [PubMed] [Google Scholar]
- 383.Sorond FA, Serrador JM, Jones RN, Shaffer ML, Lipsitz LA. The sit-to-stand technique for the measurement of dynamic cerebral autoregulation. Ultrasound Med Biol 35: 21–29, 2009. doi: 10.1016/j.ultrasmedbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Traon AP, Costes-Salon MC, Galinier M, Fourcade J, Larrue V. Dynamics of cerebral blood flow autoregulation in hypertensive patients. J Neurol Sci 195: 139–144, 2002. doi: 10.1016/s0022-510x(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 385.Gemici K, Baran I, Bakar M, Demircan C, Ozdemir B, Cordan J. Evaluation of the effect of the sublingually administered nifedipine and captopril via transcranial doppler ultrasonography during hypertensive crisis. Blood Press 12: 46–48, 2003. [PubMed] [Google Scholar]
- 386.Eames PJ, Blake MJ, Panerai RB, Potter JF. Cerebral autoregulation indices are unimpaired by hypertension in middle aged and older people. Am J Hypertens 16: 746–753, 2003. doi: 10.1016/s0895-7061(03)00947-6. [DOI] [PubMed] [Google Scholar]
- 387.Serrador JM, Sorond FA, Vyas M, Gagnon M, Iloputaife ID, Lipsitz LA. Cerebral pressure-flow relations in hypertensive elderly humans: Transfer gain in different frequency domains. J Appl Physiol (1985) 98: 151–159, 2005. doi: 10.1152/japplphysiol.00471.2004. [DOI] [PubMed] [Google Scholar]
- 388.Troisi E, Attanasio A, Matteis M, Bragoni M, Monaldo BC, Caltagirone C, Silvestrini M. Cerebral hemodynamics in young hypertensive subjects and effects of atenolol treatment. J Neurol Sci 159: 115–119, 1998. doi: 10.1016/S0022-510X(98)00147-6. [DOI] [PubMed] [Google Scholar]
- 389.Lipsitz LA, Gagnon M, Vyas M, Iloputaife I, Kiely DK, Sorond F, Serrador J, Cheng DM, Babikian V, Cupples LA. Antihypertensive therapy increases cerebral blood flow and carotid distensibility in hypertensive elderly subjects. Hypertension 45: 216–221, 2005. doi: 10.1161/01.HYP.0000153094.09615.11. [DOI] [PubMed] [Google Scholar]
- 390.Zhang R, Witkowski S, Fu Q, Claassen JA, Levine BD. Cerebral hemodynamics after short- and long-term reduction in blood pressure in mild and moderate hypertension. Hypertension 49: 1149–1155, 2007. doi: 10.1161/HYPERTENSIONAHA.106.084939. [DOI] [PubMed] [Google Scholar]
- 391.Croall ID, Tozer DJ, Moynihan B, Khan U, O'Brien JT, Morris RG, Cambridge VC, Barrick TR, Blamire AM, Ford GA, Markus HS, Team PS, for the PRESERVE Study Team. Effect of standard vs intensive blood pressure control on cerebral blood flow in small vessel disease: The PRESERVE randomized clinical trial. JAMA Neurol 75: 720–727, 2018. doi: 10.1001/jamaneurol.2017.5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Tryambake D, He J, Firbank MJ, O’Brien JT, Blamire AM, Ford GA. Intensive blood pressure lowering increases cerebral blood flow in older subjects with hypertension. Hypertension 61: 1309–1315, 2013. doi: 10.1161/HYPERTENSIONAHA.112.200972. [DOI] [PubMed] [Google Scholar]
- 393.Foster-Dingley JC, Moonen JE, de Craen AJ, de Ruijter W, van der Mast RC, van der Grond J. Blood pressure is not associated with cerebral blood flow in older persons. Hypertension 66: 954–960, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05799. [DOI] [PubMed] [Google Scholar]
- 394.Griffith DN, James IM, Newbury PA, Woollard ML. The effect of ß-adrenergic receptor blocking drugs on cerebral blood flow. Br J Clin Pharmacol 7: 491–494, 1979. doi: 10.1111/j.1365-2125.1979.tb00991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Gommer ED, Martens EG, Aalten P, Shijaku E, Verhey FR, Mess WH, Ramakers IH, Reulen JP. Dynamic cerebral autoregulation in subjects with Alzheimer’s disease, mild cognitive impairment, and controls: evidence for increased peripheral vascular resistance with possible predictive value. J Alzheimers Dis 30: 805–813, 2012. doi: 10.3233/JAD-2012-111628. [DOI] [PubMed] [Google Scholar]
- 396.Conen D, Ruttimann S, Noll G, Schneider K, Muller J. Short- and long-term cerebrovascular effects of nitrendipine in hypertensive patients. J Cardiovasc Pharmacol 12: S64–68, 1988. doi: 10.1097/00005344-198806124-00012. [DOI] [PubMed] [Google Scholar]
- 397.Ram CV, Meese R, Kaplan NM, Devous MD Sr, Bonte FJ, Forland SC, Cutler RE. Antihypertensive therapy in the elderly. Effects on blood pressure and cerebral blood flow. Am J Med 82: 53–57, 1987. doi: 10.1016/0002-9343(87)90145-8. [DOI] [PubMed] [Google Scholar]
- 398.Pandita-Gunawardena ND, Clarke SE. Amlodipine lowers blood pressure without affecting cerebral blood flow as measured by single photon emission computed tomography in elderly hypertensive subjects. Age Ageing 28: 451–457, 1999. doi: 10.1093/ageing/28.5.451. [DOI] [PubMed] [Google Scholar]
- 399.Patel RV, Ramadan NM, Levine SR, Welch KM, Fagan SC. Effects of ramipril and enalapril on cerebral blood flow in elderly patients with asymptomatic carotid artery occlusive disease. J Cardiovasc Pharmacol 28: 48–52, 1996. doi: 10.1097/00005344-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 400.Moonen JE, Foster-Dingley JC, de Ruijter W, van der Grond J, Bertens AS, van Buchem MA, Gussekloo J, Middelkoop HA, Wermer MJ, Westendorp RG, de Craen AJ, van der Mast RC. Effect of discontinuation of antihypertensive treatment in elderly people on cognitive functioning–the DANTE Study Leiden: a randomized clinical trial. JAMA Intern Med 175: 1622–1630, 2015. doi: 10.1001/jamainternmed.2015.4103. [DOI] [PubMed] [Google Scholar]
- 401.Immink RV, van den Born BJ, van Montfrans GA, Koopmans RP, Karemaker JM, van Lieshout JJ. Impaired cerebral autoregulation in patients with malignant hypertension. Circulation 110: 2241–2245, 2004. doi: 10.1161/01.CIR.0000144472.08647.40. [DOI] [PubMed] [Google Scholar]
- 402.Lipsitz LA, Habtemariam D, Gagnon M, Iloputaife I, Sorond F, Tchalla AE, Dantoine TF, Travison TG. Reexamining the effect of antihypertensive medications on falls in old age. Hypertension 66: 183–189, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Claassen J. Orthostatic hypotension in elderly patients. Ned Tijdschr Geneeskd 162: D1943, 2018. [PubMed] [Google Scholar]
- 404.Kahlaee HR, Latt MD, Schneider CR. Association between chronic or acute use of antihypertensive class of medications and falls in older adults. A systematic review and meta-analysis. Am J Hypertens 31: 467–479, 2018. doi: 10.1093/ajh/hpx189. [DOI] [PubMed] [Google Scholar]
- 405.Group S, Williamson JD, Pajewski NM, Auchus AP, Bryan RN, Chelune G, Cheung AK, Cleveland ML, et al. Effect of intensive vs standard blood pressure control on probable dementia: A randomized clinical trial. JAMA 321: 553–561, 2019. doi: 10.1001/jama.2018.21442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Pajewski NM, Berlowitz DR, Bress AP, Callahan KE, Cheung AK, Fine LJ, Gaussoin SA, Johnson KC, King J, Kitzman DW, Kostis JB, Lerner AJ, Lewis CE, Oparil S, Rahman M, Reboussin DM, Rocco MV, Snyder JK, Still C, Supiano MA, Wadley VG, Whelton PK, Wright JT Jr, Williamson JD. Intensive vs standard blood pressure control in adults 80 years or older: A secondary analysis of the systolic blood pressure intervention trial. J Am Geriatr Soc 68: 496–504, 2020. doi: 10.1111/jgs.16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Collard D, Brouwer TF, Olde Engberink RH, Zwinderman AH, Vogt L, van den Born BJ. BH. Initial estimated glomerular filtration rate decline and long-term renal function during intensive antihypertensive therapy: A post hoc analysis of the SPRINT and ACCORD-BP randomized controlled trials. Hypertension 75: 1205–1212, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.White WB, Wakefield DB, Moscufo N, Guttmann CR, Kaplan RF, Bohannon RW, Fellows D, Hall CB, Wolfson L. Effects of intensive versus standard ambulatory blood pressure control on cerebrovascular outcomes in older people (INFINITY). Circulation 140: 1626–1635, 2019. doi: 10.1161/CIRCULATIONAHA.119.041603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Ding J, Davis-Plourde KL, Sedaghat S, Tully PJ, Wang W, Phillips C, Pase MP, Himali JJ, Gwen WB, Griswold M, Gottesman R, Mosley TH, White L, Guethnason V, Debette S, Beiser AS, Seshadri S, Ikram MA, Meirelles O, Tzourio C, Launer LJ. Antihypertensive medications and risk for incident dementia and Alzheimer's disease: Ad meta-analysis of individual participant data from prospective cohort studies. Lancet Neurol 19: 61–70, 2020. doi: 10.1016/S1474-4422(19)30393-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Aries MJ, Elting JW, De Keyser J, Kremer BP, Vroomen PC. Cerebral autoregulation in stroke: a review of transcranial Doppler studies. Stroke 41: 2697–2704, 2010. doi: 10.1161/STROKEAHA.110.594168. [DOI] [PubMed] [Google Scholar]
- 411.Vitt JR, Trillanes M, Hemphill JC. Management of blood pressure during and after recanalization therapy for acute ischemic stroke. Front Neurol 10: 138, 2019. doi: 10.3389/fneur.2019.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Castro P, Azevedo E, Sorond FA. Cerebral autoregulation in stroke. Curr Atheroscler Rep 20: 37, 2018. doi: 10.1007/s11883-018-0739-5. [DOI] [PubMed] [Google Scholar]
- 413.Guo ZN, Jin H, Sun H, Zhao Y, Liu J, Ma H, Sun X, Yang Y. Antioxidant melatonin: Potential functions in improving cerebral autoregulation after subarachnoid hemorrhage. Front Physiol 9: 1146, 2018. doi: 10.3389/fphys.2018.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Intharakham K, Beishon L, Panerai RB, Haunton VJ, Robinson TG. Assessment of cerebral autoregulation in stroke: a systematic review and meta-analysis of studies at rest. J Cereb Blood Flow Metabol 39: 2105–2116, 2019. doi: 10.1177/0271678X19871013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Jordan JD, Powers WJ. Cerebral autoregulation and acute ischemic stroke. Am J Hypertens 25: 946–950, 2012. doi: 10.1038/ajh.2012.53. [DOI] [PubMed] [Google Scholar]
- 416.Kunz A, Iadecola C. Cerebral vascular dysregulation in the ischemic brain. Handb Clin Neurol 92: 283–305, 2009. doi: 10.1016/S0072-9752(08)01914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Minhas JS, Panerai RB, Ghaly G, Divall O, Robinson TG. Cerebral autoregulation in hemorrhagic stroke: A systematic review and meta-analysis of transcranial Doppler ultrasonographic studies. J Clin Ultrasound 47: 14–21, 2019. doi: 10.1002/jcu.22645. [DOI] [PubMed] [Google Scholar]
- 418.Palomares SM, Cipolla MJ. Myogenic tone as a therapeutic target for ischemic stroke. Curr Vasc Pharmacol 12: 788–800, 2014. doi: 10.2174/15701611113116660155. [DOI] [PubMed] [Google Scholar]
- 419.Sobey CG, Faraci FM. Subarachnoid haemorrhage: what happens to the cerebral arteries? Clin Exp Pharmacol Physiol 25: 867–876, 1998. doi: 10.1111/j.1440-1681.1998.tb02337.x. [DOI] [PubMed] [Google Scholar]
- 420.Xiao M, Li Q, Feng H, Zhang L, Chen Y. Neural vascular mechanism for the cerebral blood flow autoregulation after hemorrhagic stroke. Neural Plast 2017: 5819514, 2017. doi: 10.1155/2017/5819514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Xiong L, Liu X, Shang T, Smielewski P, Donnelly J, Guo ZN, Yang Y, Leung T, Czosnyka M, Zhang R, Liu J, Wong KS. Impaired cerebral autoregulation: Measurement and application to stroke. J Neurol Neurosurg Psychiatry 88: 520–531, 2017. doi: 10.1136/jnnp-2016-314385. [DOI] [PubMed] [Google Scholar]
- 422.Jackman K, Iadecola C. Neurovascular regulation in the ischemic brain. Antioxid Redox Signal 22: 149–160, 2015. doi: 10.1089/ars.2013.5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Markus HS. Transcranial Doppler ultrasound. Br Med Bull 56: 378–388, 2000. doi: 10.1258/0007142001903021. [DOI] [PubMed] [Google Scholar]
- 424.Al-Jehani H, Angle M, Marcoux J, Teitelbaum J. Early abnormal transient hyperemic response test can predict delayed ischemic neurologic deficit in subarachnoid hemorrhage. Crit Ultrasound J 10: 1, 2018. doi: 10.1186/s13089-017-0079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Brooks FA, Ughwanogho U, Henderson GV, Black-Schaffer R, Sorond FA, Tan CO. The link between cerebrovascular hemodynamics and rehabilitation outcomes after aneurysmal subarachnoid hemorrhage. Am J Phys Med Rehabil 97: 309–315, 2018. doi: 10.1097/PHM.0000000000000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Budohoski KP, Czosnyka M, Kirkpatrick PJ, Reinhard M, Varsos GV, Kasprowicz M, Zabek M, Pickard JD, Smielewski P. Bilateral failure of cerebral autoregulation is related to unfavorable outcome after subarachnoid hemorrhage. Neurocrit Care 22: 65–73, 2015. doi: 10.1007/s12028-014-0032-6. [DOI] [PubMed] [Google Scholar]
- 427.Budohoski KP, Czosnyka M, Smielewski P, Kasprowicz M, Helmy A, Bulters D, Pickard JD, Kirkpatrick PJ. Impairment of cerebral autoregulation predicts delayed cerebral ischemia after subarachnoid hemorrhage: A prospective observational study. Stroke 43: 3230–3237, 2012. doi: 10.1161/STROKEAHA.112.669788. [DOI] [PubMed] [Google Scholar]
- 428.Calviere L, Nasr N, Arnaud C, Czosnyka M, Viguier A, Tissot B, Sol JC, Larrue V. Prediction of delayed cerebral ischemia after subarchnoid hemorrhage using cerebral blood flow velocities and cerebral autoregulation assessment. Neurocrit Care 23: 253–258, 2015. doi: 10.1007/s12028-015-0125-x. [DOI] [PubMed] [Google Scholar]
- 429.Fontana J, Moratin J, Ehrlich G, Scharf J, Weiβ C, Schmieder K, Barth M. Dynamic autoregulatory response after aneurysmal subarachnoid hemorrhage and its relation to angiographic vasospasm and clinical outcome. Neurocrit Care 23: 355–363, 2015. doi: 10.1007/s12028-014-0104-7. [DOI] [PubMed] [Google Scholar]
- 430.Gaasch M, Schiefecker AJ, Kofler M, Beer R, Rass V, Pfausler B, Thome C, Schmutzhard E, Helbok R. Cerebral autoregulation in the prediction of delayed cerebral ischemia and clinical outcome in poor-grade aneurysmal subarachnoid hemorrhage patients. Crit Care Med 46: 774–780, 2018. doi: 10.1097/ccm.0000000000003016. [DOI] [PubMed] [Google Scholar]
- 431.Hattingen E, Blasel S, Dettmann E, Vatter H, Pilatus U, Seifert V, Zanella FE, Weidauer S. Perfusion-weighted MRI to evaluate cerebral autoregulation in aneurysmal subarachnoid haemorrhage. Neuroradiology 50: 929–938, 2008. doi: 10.1007/s00234-008-0424-4. [DOI] [PubMed] [Google Scholar]
- 432.Jaeger M, Soehle M, Schuhmann MU, Meixensberger J. Clinical significance of impaired cerebrovascular autoregulation after severe aneurysmal subarachnoid hemorrhage. Stroke 43: 2097–2101, 2012. doi: 10.1161/STROKEAHA.112.659888. [DOI] [PubMed] [Google Scholar]
- 433.Lam JM, Smielewski P, Czosnyka M, Pickard JD, Kirkpatrick PJ. Predicting delayed ischemic deficits after aneurysmal subarachnoid hemorrhage using a transient hyperemic response test of cerebral autoregulation. Neurosurgery 47: 819–825, 2000. doi: 10.1097/00006123-200010000-00004. [DOI] [PubMed] [Google Scholar]
- 434.Lang EW, Diehl RR, Mehdorn HM. Cerebral autoregulation testing after aneurysmal subarachnoid hemorrhage: the phase relationship between arterial blood pressure and cerebral blood flow velocity. Crit Care Med 29: 158–163, 2001. doi: 10.1097/00003246-200101000-00031. [DOI] [PubMed] [Google Scholar]
- 435.Ratsep T, Asser T. Cerebral hemodynamic impairment after aneurysmal subarachnoid hemorrhage as evaluated using transcranial Doppler ultrasonography: relationship to delayed cerebral ischemia and clinical outcome. J Neurosurg 95: 393–401, 2001. doi: 10.3171/jns.2001.95.3.0393. [DOI] [PubMed] [Google Scholar]
- 436.Santos GA, Petersen N, Zamani AA, Du R, LaRose S, Monk A, Sorond FA, Tan CO. Pathophysiologic differences in cerebral autoregulation after subarachnoid hemorrhage. Neurology 86: 1950–1956, 2016. doi: 10.1212/WNL.0000000000002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Soehle M, Czosnyka M, Pickard JD, Kirkpatrick PJ. Continuous assessment of cerebral autoregulation in subarachnoid hemorrhage. Anesth Analg 98: 1133–1139, 2004. doi: 10.1213/01.ane.0000111101.41190.99. [DOI] [PubMed] [Google Scholar]
- 438.Tseng MY, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke 36: 1627–1632, 2005. doi: 10.1161/01.STR.0000176743.67564.5d. [DOI] [PubMed] [Google Scholar]
- 439.Yundt KD, Grubb RL Jr, Diringer MN, Powers WJ. Autoregulatory vasodilation of parenchymal vessels is impaired during cerebral vasospasm. J Cereb Blood Flow Metab 18: 419–424, 1998. doi: 10.1097/00004647-199804000-00010. [DOI] [PubMed] [Google Scholar]
- 440.Zweifel C, Castellani G, Czosnyka M, Carrera E, Brady KM, Kirkpatrick PJ, Pickard JD, Smielewski P. Continuous assessment of cerebral autoregulation with near-infrared spectroscopy in adults after subarachnoid hemorrhage. Stroke 41: 1963–1968, 2010. doi: 10.1161/STROKEAHA.109.577320. [DOI] [PubMed] [Google Scholar]
- 441.Llwyd O, Salinet AS, Panerai RB, Lam MY, Saeed NP, Brodie F, Bor-Seng-Shu E, Robinson TG, Nogueira RC. Cerebral haemodynamics following acute ischaemic stroke: effects of stroke severity and stroke subtype. Cerebrovasc Dis Extra 8: 80–89, 2018. doi: 10.1159/000487514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Powers WJ, Videen TO, Diringer MN, Aiyagari V, Zazulia AR. Autoregulation after ischaemic stroke. J Hypertens 27: 2218–2222, 2009. doi: 10.1097/HJH.0b013e328330a9a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Powers WJ, Zazulia AR, Videen TO, Adams RE, Yundt KD, Aiyagari V, Grubb RL, Jr, Diringer MN. Autoregulation of cerebral blood flow surrounding acute (6 to 22 hours) intracerebral hemorrhage. Neurology 57: 18–24, 2001. doi: 10.1212/WNL.57.1.18. [DOI] [PubMed] [Google Scholar]
- 444.Reinhard M, Roth M, Guschlbauer B, Harloff A, Timmer J, Czosnyka M, Hetzel A. Dynamic cerebral autoregulation in acute ischemic stroke assessed from spontaneous blood pressure fluctuations. Stroke 36: 1684–1689, 2005. doi: 10.1161/01.STR.0000173183.36331.ee. [DOI] [PubMed] [Google Scholar]
- 445.Reinhard M, Wihler C, Roth M, Harloff A, Niesen WD, Timmer J, Weiller C, Hetzel A. Cerebral autoregulation dynamics in acute ischemic stroke after rtPA thrombolysis. Cerebrovasc Dis 26: 147–155, 2008. doi: 10.1159/000139662. [DOI] [PubMed] [Google Scholar]
- 446.Salinet AS, Silva NC, Caldas J, de Azevedo DS, de-Lima-Oliveira M, Nogueira RC, Conforto AB, Texeira MJ, Robinson TG, Panerai RB, Bor-Seng-Shu E. Impaired cerebral autoregulation and neurovascular coupling in middle cerebral artery stroke: influence of severity? J Cereb Blood Flow Metab 39: 2277–2285, 2019. doi: 10.1177/0271678X18794835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Truijen J, Rasmussen LS, Kim YS, Stam J, Stok WJ, Pott FC, van Lieshout JJ. Cerebral autoregulatory performance and the cerebrovascular response to head-of-bed positioning in acute ischaemic stroke. Eur J Neurol 25: 1365–e1117, 2018. doi: 10.1111/ene.13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Ma H, Guo ZN, Sun X, Liu J, Lv S, Zhao L, Guo W, Jin H, Yang Y. Hematoma volume is a predictive factor of disturbed autoregulation after spontaneous intracerebral hemorrhage. J Neurol Sci 382: 96–100, 2017. doi: 10.1016/j.jns.2017.09.035. [DOI] [PubMed] [Google Scholar]
- 449.Reinhard M, Rutsch S, Lambeck J, Wihler C, Czosnyka M, Weiller C, Hetzel A. Dynamic cerebral autoregulation associates with infarct size and outcome after ischemic stroke. Acta Neurol Scand 125: 156–162, 2012. doi: 10.1111/j.1600-0404.2011.01515.x. [DOI] [PubMed] [Google Scholar]
- 450.Meyer JS, Shimazu K, Fukuuchi Y, Ouchi T, Okamoto S, Koto A. Impaired neurogenic cerebrovascular control and dysautoregulation after stroke. Stroke 4: 169–186, 1973. doi: 10.1161/01.str.4.2.169. [DOI] [PubMed] [Google Scholar]
- 451.Dawson SL, Blake MJ, Panerai RB, Potter JF. Dynamic but not static cerebral autoregulation is impaired in acute ischaemic stroke. Cerebrovasc Dis 10: 126–132, 2000. doi: 10.1159/000016041. [DOI] [PubMed] [Google Scholar]
- 452.Eames PJ, Robinson TG, Panerai RB, Potter JF. The systemic haemodynamic and cerebral autoregulatory effects of bendrofluazide in the subacute post-stroke period. J Hypertens 22: 2017–2024, 2004. doi: 10.1097/00004872-200410000-00026. [DOI] [PubMed] [Google Scholar]
- 453.Gierthmuhlen J, Allardt A, Sawade M, Baron R, Wasner G. Dynamic cerebral autoregulation in stroke patients with a central sympathetic deficit. Acta Neurol Scand 123: 332–338, 2011. doi: 10.1111/j.1600-0404.2010.01424.x. [DOI] [PubMed] [Google Scholar]
- 454.Saeed NP, Panerai RB, Horsfield MA, Robinson TG. Does stroke subtype and measurement technique influence estimation of cerebral autoregulation in acute ischaemic stroke? Cerebrovasc Dis 35: 257–261, 2013. doi: 10.1159/000347075. [DOI] [PubMed] [Google Scholar]
- 455.Kwan J, Lunt M, Jenkinson D. Assessing dynamic cerebral autoregulation after stroke using a novel technique of combining transcranial Doppler ultrasonography and rhythmic handgrip. Blood Press Monit 9: 3–8, 2004. doi: 10.1097/00126097-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 456.Gong X, Liu J, Dong P, Zhang P, Li N, Zhao X, Wang Y. Assessment of dynamic cerebral autoregulation in patients with basilar artery stenosis. PLoS One 8: e77802, 2013. doi: 10.1371/journal.pone.0077802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Tutaj M, Miller M, Krakowska-Stasiak M, Piątek A, Hebda J, Lątka M, Strojny J, Szczudlik A, Słowik A. Dynamic cerebral autoregulation is compromissed in ischaemic stroke of undetermined aetiology only in the non-affected side. Neurol Neurochir Pol 48: 91–97, 2014. doi: 10.1016/j.pjnns.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 458.Novak V, Yang AC, Lepicovsky L, Goldberger AL, Lipsitz LA, Peng CK. Multimodal pressure-flow method to assess dynamics of cerebral autoregulation in stroke and hypertension. Biomed Eng Online 3: 39, 2004. doi: 10.1186/1475-925X-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Lam MY, Haunton VJ, Robinson TG, Panerai RB. Dynamic cerebral autoregulation measurement using rapid changes in head positioning: Experiences in acute ischemic stroke and healthy control populations. Am J Physiol Heart Circ Physiol 316: H673–H683, 2019. doi: 10.1152/ajpheart.00550.2018. [DOI] [PubMed] [Google Scholar]
- 460.Budohoski KP, Czosnyka M, Smielewski P, Varsos GV, Kasprowicz M, Brady KM, Pickard JD, Kirkpatrick PJ. Cerebral autoregulation after subarachnoid hemorrhage: comparison of three methods. J Cereb Blood Flow Metabol 33: 449–456, 2013. doi: 10.1038/jcbfm.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Salinet AS, Robinson TG, Panerai RB. Effects of cerebral ischemia on human neurovascular coupling, CO2 reactivity, and dynamic cerebral autoregulation. J Appl Physiol 118: 170–177, 2015. doi: 10.1152/japplphysiol.00620.2014. [DOI] [PubMed] [Google Scholar]
- 462.Eames PJ, Blake MJ, Dawson SL, Panerai RB, Potter JF. Dynamic cerebral autoregulation and beat to beat blood pressure control are impaired in acute ischaemic stroke. J Neurol Neurosurg Psychiatry 72: 467–472, 2002. doi: 10.1136/jnnp.72.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Immink RV, van Montfrans GA, Stam J, Karemaker JM, Diamant M, van Lieshout JJ. Dynamic cerebral autoregulation in acute lacunar and middle cerebral artery territory ischemic stroke. Stroke 36: 2595–2600, 2005. doi: 10.1161/01.STR.0000189624.06836.03. [DOI] [PubMed] [Google Scholar]
- 464.Ma H, Guo ZN, Liu J, Xing Y, Zhao R, Yang Y. Temporal course of dynamic cerebral autoregulation in patients with intracerebral hemorrhage. Stroke 47: 674–681, 2016. doi: 10.1161/STROKEAHA.115.011453. [DOI] [PubMed] [Google Scholar]
- 465.Oeinck M, Neunhoeffer F, Buttler KJ, Meckel S, Schmidt B, Czosnyka M, Weiller C, Reinhard M. Dynamic cerebral autoregulation in acute intracerebral hemorrhage. Stroke 44: 2722–2728, 2013. doi: 10.1161/STROKEAHA.113.001913. [DOI] [PubMed] [Google Scholar]
- 466.Otite F, Mink S, Tan CO, Puri A, Zamani AA, Mehregan A, Chou S, Orzell S, Purkayastha S, Du R, Sorond FA. Impaired cerebral autoregulation is associated with vasospasm and delayed cerebral ischemia in subarachnoid hemorrhage. Stroke 45: 677–682, 2014. doi: 10.1161/STROKEAHA.113.002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Reinhard M, Neunhoeffer F, Gerds TA, Niesen WD, Buttler KJ, Timmer J, Schmidt B, Czosnyka M, Weiller C, Hetzel A. Secondary decline of cerebral autoregulation is associated with worse outcome after intracerebral hemorrhage. Intensive Care Med 36: 264–271, 2010. doi: 10.1007/s00134-009-1698-7. [DOI] [PubMed] [Google Scholar]
- 468.Chen J, Liu J, Xu WH, Xu R, Hou B, Cui LY, Gao S. Impaired dynamic cerebral autoregulation and cerebrovascular reactivity in middle cerebral artery stenosis. PLoS One 9: e88232, 2014. doi: 10.1371/journal.pone.0088232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Guo ZN, Liu J, Xing Y, Yan S, Lv C, Jin H, Yang Y. Dynamic cerebral autoregulation is heterogeneous in different subtypes of acute ischemic stroke. PLoS One 9: e93213, 2014. doi: 10.1371/journal.pone.0093213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Panerai RB, Jara JL, Saeed NP, Horsfield MA, Robinson TG. Dynamic cerebral autoregulation following acute ischaemic stroke: comparison of transcranial Doppler and magnetic resonance imaging techniques. J Cereb Blood Flow Metabol 36: 2194–2202, 2016. doi: 10.1177/0271678X15615874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Atkins ER, Brodie FG, Rafelt SE, Panerai RB, Robinson TG. Dynamic cerebral autoregulation is compromised acutely following mild ischemic stroke but not transient ischaemic attack. Cerebrovasc Dis 29: 228–235, 2010. doi: 10.1159/000267845. [DOI] [PubMed] [Google Scholar]
- 472.Minhas JS, Panerai RB, Swienton D, Robinson TG. Feasibility of improving cerebral autoregulation in acute intracerebral hemorrhage (BREATHE-ICH) study: results from an experimental interventional study. Int J Stroke 15: 627–637, 2020. doi: 10.1177/1747493019873690. [DOI] [PubMed] [Google Scholar]
- 473.Salinet AS, Panerai RB, Robinson TG. The longitudinal evolution of cerebral blood flow regulation after acute ischaemic stroke. Cerebrovasc Dis Extra 4: 186–197, 2014. doi: 10.1159/000366017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Czosnyka M, Smielewski P, Lavinio A, Pickard JD, Panerai R. An assessment of dynamic autoregulation from spontaneous fluctuations of cerebral blood flow velocity: a comparison of two models, index of autoregulation and mean flow index. Anesth Analg 106: 234–239, 2008. doi: 10.1213/01.ane.0000295802.89962.13. [DOI] [PubMed] [Google Scholar]
- 475.Rasulo FA, Girardini A, Lavinio A, De Peri E, Stefini R, Cenzato M, Nodari I, Latronico N. Are optimal cerebral perfusion pressure and cerebrovascular autoregulation related to long-term outcome in patients with aneurysmal subarachnoid hemorrhage? J Neurosurg Anesthesiol 24: 3–8, 2012. doi: 10.1097/ANA.0b013e318224030a. [DOI] [PubMed] [Google Scholar]
- 476.Silverman A, Kodali S, Strander S, Gilmore EJ, Kimmel A, Wang A, Cord B, Falcone G, Hebert R, Matouk C, Sheth KN, Petersen NH. Deviation from personalized blood pressure targets is associated with worse outcome after subarachnoid hemorrhage. Stroke 50: 2729–2737, 2019. doi: 10.1161/STROKEAHA.119.026282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Tseng MY, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute treatment with statins on cerebral autoregulation in patients after aneurysmal subarachnoid hemorrhage. Neurosurg Focus 21: E10, 2006. doi: 10.3171/foc.2006.21.3.10. [DOI] [PubMed] [Google Scholar]
- 478.Aoi MC, Hu K, Lo MT, Selim M, Olufsen MS, Novak V. Impaired cerebral autoregulation is associated with brain atrophy and worse functional status in chronic ischemic stroke. PLoS One 7: e46794, 2012. doi: 10.1371/journal.pone.0046794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Hu K, Lo MT, Peng CK, Liu Y, Novak V. A nonlinear dynamic approach reveals a long-term stroke effect on cerebral blood flow regulation at multiple time scales. PLoS Comput Biol 8: e1002601, 2012. doi: 10.1371/journal.pcbi.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Hu K, Peng CK, Czosnyka M, Zhao P, Novak V. Nonlinear assessment of cerebral autoregulation from spontaneous blood pressure and cerebral blood flow fluctuations. Cardiovasc Eng 8: 60–71, 2008. doi: 10.1007/s10558-007-9045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Hu K, Peng CK, Huang NE, Wu Z, Lipsitz LA, Cavallerano J, Novak V. Altered phase interactions between spontaneous blood pressure and flow fluctuations in type 2 diabetes mellitus: Nonlinear assessment of cerebral autoregulation. Physica A 387: 2279–2292, 2008. doi: 10.1016/j.physa.2007.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Gommer ED, Staals J, van Oostenbrugge RJ, Lodder J, Mess WH, Reulen JP. Dynamic cerebral autoregulation and cerebrovascular reactivity: A comparative study in lacunar infarct patients. Physiol Meas 29: 1293–1303, 2008. doi: 10.1088/0967-3334/29/11/005. [DOI] [PubMed] [Google Scholar]
- 483.Guo ZN, Xing Y, Wang S, Ma H, Liu J, Yang Y. Characteristics of dynamic cerebral autoregulation in cerebral small vessel disease: diffuse and sustained. Sci Rep 5: 15269, 2015. doi: 10.1038/srep15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Petersen NH, Ortega-Gutierrez S, Reccius A, Masurkar A, Huang A, Marshall RS. Dynamic cerebral autoregulation is transiently impaired for one week after large-vessel acute ischemic stroke. Cerebrovasc Dis 39: 144–150, 2015. doi: 10.1159/000368595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Dawson SL, Panerai RB, Potter JF. Serial changes in static and dynamic cerebral autoregulation after acute ischaemic stroke. Cerebrovasc Dis 16: 69–75, 2003. doi: 10.1159/000070118. [DOI] [PubMed] [Google Scholar]
- 486.Ma H, Guo ZN, Yan JH, Liu X, Lv J, Zhang S, Sun P, Yang XY. Preliminary study of dynamic cerebral autoregulation in acute ischemic stroke: association with clinical factors. Front Neurol 9: 1006, 2018. doi: 10.3389/fneur.2018.01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Haubrich C, Kruska W, Diehl RR, Moller-Hartmann W, Klotzsch C. Dynamic autoregulation testing in patients with middle cerebral artery stenosis. Stroke 34: 1881–1885, 2003. doi: 10.1161/01.STR.0000080936.36601.34. [DOI] [PubMed] [Google Scholar]
- 488.Wang S, Guo ZN, Xing Y, Ma H, Jin H, Liu J, Yang Y. Dynamic cerebral autoregulation in asymptomatic patients with unilateral middle cerebral artery stenosis. Medicine 94: e2234, 2015. doi: 10.1097/MD.0000000000002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Castro P, Azevedo E, Serrador J, Rocha I, Sorond F. Hemorrhagic transformation and cerebral edema in acute ischemic stroke: link to cerebral autoregulation. J Neurol Sci 372: 256–261, 2017. doi: 10.1016/j.jns.2016.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Castro P, Serrador JM, Rocha I, Sorond F, Azevedo E. Efficacy of cerebral autoregulation in early ischemic stroke predicts smaller infarcts and better outcomes. Front Neurol 8: 113, 2017. doi: 10.3389/fneur.2017.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Rosen DS, Macdonald RL. Subarachnoid hemorrhage grading scales: a systematic review. Neurocrit Care 2: 110–118, 2005. doi: 10.1385/NCC:2:2:110. [DOI] [PubMed] [Google Scholar]
- 492.Dohmen C, Bosche B, Graf R, Reithmeier T, Ernestus RI, Brinker G, Sobesky J, Heiss WD. Identification and clinical impact of impaired cerebrovascular autoregulation in patients with malignant middle cerebral artery infarction. Stroke 38: 56–61, 2007. doi: 10.1161/01.STR.0000251642.18522.b6. [DOI] [PubMed] [Google Scholar]
- 493.Aries MJ, Czosnyka M, Budohoski KP, Steiner LA, Lavinio A, Kolias AG, Hutchinson PJ, Brady KM, Menon DK, Pickard JD, Smielewski P. Continuous determination of optimal cerebral perfusion pressure in traumatic brain injury. Crit Care Med 40: 2456–2463, 2012. doi: 10.1097/CCM.0b013e3182514eb6. [DOI] [PubMed] [Google Scholar]
- 494.Amarenco P, Bogousslavsky J, Caplan LR, Donnan GA, Hennerici MG. Classification of stroke subtypes. Cerebrovasc Dis 27: 493–501, 2009. doi: 10.1159/000210432. [DOI] [PubMed] [Google Scholar]
- 495.Xiong L, Tian G, Lin W, Wang W, Wang L, Leung T, Mok V, Liu J, Chen X, Wong KS. Is dynamic cerebral autoregulation bilaterally impaired after unilateral acute ischemic stroke? J Stroke Cerebrovasc Dis 26: 1081–1087, 2017. doi: 10.1016/j.jstrokecerebrovasdis.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 496.Chi NF, Hu HH, Wang CY, Chan L, Peng CK, Novak V, Hu CJ. Dynamic cerebral autoregulation is an independent functional outcome predictor of mild acute ischemic stroke. Stroke 49: 2605–2611, 2018. doi: 10.1161/STROKEAHA.118.022481. [DOI] [PubMed] [Google Scholar]
- 497.Deng W, Kandhi S, Zhang B, Huang A, Koller A, Sun D. Extravascular blood augments myogenic constriction of cerebral arterioles: implications for hemorrhage-induced vasospasm. J Am Heart Assoc 7: e008623, 2018. doi: 10.1161/jaha.118.008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: Implications for stroke clinical trials: a literature review and synthesis. Stroke 38: 1091–1096, 2007. doi: 10.1161/01.STR.0000258355.23810.c6. [DOI] [PubMed] [Google Scholar]
- 499.Castro P, Azevedo E, Rocha I, Sorond F, Serrador JM. Chronic kidney disease and poor outcomes in ischemic stroke: Is impaired cerebral autoregulation the missing link? BMC Neurol 18: 21, 2018. doi: 10.1186/s12883-018-1025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Czosnyka M, Hutchinson P, Smielewski P. Treatment targets based on autoregulation parameters in neurocritical care patients. Curr Opin Crit Care 26: 109–114, 2020. doi: 10.1097/MCC.0000000000000704. [DOI] [PubMed] [Google Scholar]
- 501.Czosnyka M, Smielewski P, Kirkpatrick P, Laing RJ, Menon D, Pickard JD. Continuous assessment of the cerebral vasomotor reactivity in head injury. Neurosurgery 41: 11–17, 1997. doi: 10.1097/00006123-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 502.Steiner LA, Czosnyka M, Piechnik SK, Smielewski P, Chatfield D, Menon DK, Pickard JD. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med 30: 733–738, 2002. doi: 10.1097/00003246-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 503.Petersen NH, Silverman A, Strander SM, Kodali S, Wang A, Sansing LH, Schindler JL, Falcone GJ, Gilmore EJ, Jasne AS, Cord B, Hebert RM, Johnson M, Matouk CC, Sheth KN. Fixed compared with autoregulation-oriented blood pressure thresholds after mechanical thrombectomy for ischemic stroke. Stroke 51: 914–921, 2020. doi: 10.1161/STROKEAHA.119.026596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Salinet AS, Minhas JS, Panerai RB, Bor-Seng-Shu E, Robinson TG. Do acute stroke patients develop hypocapnia? A systematic review and meta-analysis. J Neurol Sci 402: 30–39, 2019. doi: 10.1016/j.jns.2019.04.038. [DOI] [PubMed] [Google Scholar]
- 505.Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol 23: 394–403, 1970. doi: 10.1001/archneur.1970.00480290014002. [DOI] [PubMed] [Google Scholar]
- 506.Willmot M, Leonardi-Bee J, Bath PM. High blood pressure in acute stroke and subsequent outcome: a systematic review. Hypertension 43: 18–24, 2004. doi: 10.1161/01.HYP.0000105052.65787.35. [DOI] [PubMed] [Google Scholar]
- 507.Anderson CS, Huang Y, Lindley RI, Chen X, Arima H, Chen G, et al. Intensive blood pressure reduction with intravenous thrombolysis therapy for acute ischaemic stroke (ENCHANTED): an international, randomised, open-label, blinded-end-point, phase 3 trial. Lancet 393: 877–888, 2019. doi: 10.1016/S0140-6736(19)30038-8. [DOI] [PubMed] [Google Scholar]
- 508.Bath PM, Scutt P, Anderson CS, Ankolekar S, Appleton JP, Berge E, Cala L, et al. Prehospital transdermal glyceryl trinitrate in patients with ultra-acute presumed stroke (RIGHT-2): an ambulance-based, randomised, sham-controlled, blinded, phase 3 trial. Lancet 393: 1009–1020, 2019. doi: 10.1016/S0140-6736(19)30194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Potter JF, Robinson TG, Ford GA, Mistri A, James M, Chernova J, Jagger C. Controlling hypertension and hypotension immediately post-stroke (CHHIPS): A randomised, placebo-controlled, double-blind pilot trial. Lancet Neurol 8: 48–56, 2009. doi: 10.1016/S1474-4422(08)70263-1. [DOI] [PubMed] [Google Scholar]
- 510.Robinson TG, Davison WJ, Rothwell PM, Potter JF. Randomised controlled trial of a calcium channel or angiotensin converting enzyme inhibitor/angiotensin receptor blocker regime to reduce blood pressure variability following ischaemic stroke (CAARBS): a protocol for a feasibility study. BMJ Open 9: e025301, 2019. doi: 10.1136/bmjopen-2018-025301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Robinson TG, Potter JF, Ford GA, Bulpitt CJ, Chernova J, Jagger C, James MA, Knight J, Markus HS, Poulter NR, Cossacs I. Effects of anithypertensive treatment after acute stroke in the Continue or Stop Post-Stroke Antihypertensives Collaborative Study (COSSACS): a prospective, randomised, open, blinded end-point trial. Lancet Neurol 9: 767–775, 2010. doi: 10.1016/S1474-4422(10)70163-0. [DOI] [PubMed] [Google Scholar]
- 512.Chan E, Anderson CS, Wang X, Arima H, Saxena A, Moullaali TJ, Delcourt C, Wu G, Wang J, Chen G, Lavados PM, Stapf C, Robinson T, Chalmers J, The INTERACT Investigators. Early blood pressure lowering does not reduce growth of intraventricular hemorrhage following acute intracerebral hemorrhage: results of the INTERACT studies. Cerebrovasc Dis Extra 6: 71–75, 2016. doi: 10.1159/000448897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Moullaali TJ, Wang X, Martin RH, Shipes VB, Robinson TG, Chalmers J, Suarez JI, Qureshi AI, Palesch YY, Anderson CS. Blood pressure control and clinical outcomes in acute intracerebral haemorrhage: a preplanned pooled analysis of individual participant data. Lancet Neurol 18: 857–864, 2019. doi: 10.1016/S1474-4422(19)30196-6. [DOI] [PubMed] [Google Scholar]
- 514.Qureshi AI, Palesch YY, Barsan WG, Hanley DF, Hsu CY, Martin RL, Moy CS, Silbergleit R, Steiner T, Suarez JI, Toyoda K, Wang Y, Yamamoto H, Yoon BW, Investigators ATACH-2 Trial Investigators and the Neurological Emergency Treatment Trials Network . Intensive blood-pressure lowering in patients with acute cerebral hemorrhage. N Engl J Med 375: 1033–1043, 2016. doi: 10.1056/NEJMoa1603460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Palomares SM, Cipolla MJ. Vascular protection following cerebral ischemia and reperfusion. J Neurol Neurophysiol 2011: S1–S004, 2011. doi: 10.4172/2155-9562.S1-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Johnston KC, Durkalski-Mauldin VL. Considering prehospital stroke trials: Did TIGHT-2 get it right? Lancet 393: 963–965, 2019. doi: 10.1016/S0140-6736(19)30276-4. [DOI] [PubMed] [Google Scholar]
- 517.Starke RM, Peterson EC, Komotar RJ, Connolly ES. A randomized clinical trial of aggressive blood pressure control in patients with acute cerebral hemorrhage. Neurosurgery 79: N17–N18, 2016. doi: 10.1227/01.neu.0000508604.61565.ba. [DOI] [PubMed] [Google Scholar]
- 518.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S, American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42: 2672–2713, 2011. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 15: 934–943, 2016. doi: 10.1016/S1474-4422(16)30029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Cuadrado-Godia E, Dwivedi P, Sharma S, Ois Santiago A, Roquer Gonzalez J, Balcells M, Laird J, Turk M, Suri HS, Nicolaides A, Saba L, Khanna NN, Suri JS. Cerebral small vessel disease: A review focusing on pathophysiology, biomarkers, and machine learning strategies. J Stroke 20: 302–320, 2018. doi: 10.5853/jos.2017.02922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Poggesi A, Inzitari D, Pantoni L. Atrial fibrillation and cognition: epidemiological data and possible mechanisms. Stroke 46: 3316–3321, 2015. doi: 10.1161/STROKEAHA.115.008225. [DOI] [PubMed] [Google Scholar]
- 522.Qiu C, Fratiglioni L. A major role for cardiovascular burden in age-related cognitive decline. Nat Rev Cardiol 12: 267–277, 2015. doi: 10.1038/nrcardio.2014.223. [DOI] [PubMed] [Google Scholar]
- 523.Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, STandards for ReportIng Vascular changes on nEuroimaging (STRIVE v1), et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: 822–838, 2013. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Ballard C, Burns A, Corbett A, Livingston G, Rasmussen J. Helping you to assess cognition. A practical toolkit for clinicians. Alzheimer's Society March 2013: 1–40, 2013. [Google Scholar]
- 525.Makin SD, Turpin S, Dennis MS, Wardlaw JM. Cognitive impairment after lacunar stroke: Systematic review and meta-analysis of incidence, prevalence and comparison with other stroke subtypes. J Neurol Neurosurg Psychiatry 84: 893–900, 2013. doi: 10.1136/jnnp-2012-303645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Sachdev P, Kalaria R, O’Brien J, Skoog I, Alladi S, Black SE, Blacker D, Blazer DG, Chen C, Chui H, Ganguli M, Jellinger K, Jeste DV, Pasquier F, Paulsen J, Prins N, Rockwood K, Roman G, Scheltens P. Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis Assoc Disord 28: 206–218, 2014. doi: 10.1097/WAD.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Mathias JL, Burke J. Cognitive functioning in Alzheimer's and vascular dementia: a meta-analysis. Neuropsychology 23: 411–423, 2009. doi: 10.1037/a0015384. [DOI] [PubMed] [Google Scholar]
- 528.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27: 1908–1918, 2007. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 529.Iadecola C. The pathobiology of vascular dementia. Neuron 80: 844–866, 2013. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Vinciguerra L, Lanza G, Puglisi V, Pennisi M, Cantone M, Bramanti A, Pennisi G, Bella R. Transcranial Doppler ultrasound in vascular cognitive impairment–no dementia. PLoS One 14: e0216162, 2019. doi: 10.1371/journal.pone.0216162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Keage HA, Churches OF, Kohler M, Pomeroy D, Luppino R, Bartolo ML, Elliott S. Cerebrovascular function in aging and dementia: a systematic review of transcranial Doppler studies. Dement Geriatr Cogn Dis Extra 2: 258–270, 2012. doi: 10.1159/000339234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Sabayan B, Jansen S, Oleksik AM, van Osch MJ, van Buchem MA, van Vliet P, de Craen AJ, Westendorp RG. Cerebrovascular hemodynamics in Alzheimer's disease and vascular dementia: a meta-analysis of transcranial Doppler studies. Ageing Res Rev 11: 271–277, 2012. doi: 10.1016/j.arr.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 533.O'Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SC, Markus HS. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 59: 321–326, 2002. doi: 10.1212/wnl.59.3.321. [DOI] [PubMed] [Google Scholar]
- 534.Bakker SL, de Leeuw FE, de Groot JC, Hofman A, Koudstaal PJ, Breteler MM. Cerebral vasomotor reactivity and cerebral white matter lesions in the elderly. Neurology 52: 578–583, 1999. doi: 10.1212/wnl.52.3.578. [DOI] [PubMed] [Google Scholar]
- 535.Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Vrooman HA, Hofman A, Krestin GP, Breteler MM. Total cerebral blood flow and total brain perfusion in the general population: The Rotterdam Scan Study. J Cereb Blood Flow Metabol 28: 412–419, 2008. doi: 10.1038/sj.jcbfm.9600526. [DOI] [PubMed] [Google Scholar]
- 536.Bowler JV, Gorelick PB. Advances in vascular cognitive impairment. Stroke 40: e315-318–e318, 2009. doi: 10.1161/STROKEAHA.108.544650. [DOI] [PubMed] [Google Scholar]
- 537.Claassen JA. Alzheimer's disease: trade-off for increased survival with atherosclerosis? Ann Neurol 64: 475, 2008. doi: 10.1002/ana.21285. [DOI] [PubMed] [Google Scholar]
- 538.Claassen JA. Cognitive decline and dementia: are we getting to the vascular heart of the matter? Hypertension 65: 505–506, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04706. [DOI] [PubMed] [Google Scholar]
- 539.Claassen JA. New cardiovascular targets to prevent late onset Alzheimer disease. Eur J Pharmacol 763: 131–134, 2015. doi: 10.1016/j.ejphar.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 540.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 5: 347–360, 2004. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 541.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative. Alzheimer's disease neuroimaging I. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun 7: 11934, 2016. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab 31: 1354–1370, 2011. doi: 10.1038/jcbfm.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Pasha EP, Rutjes E, Tomoto T, Tarumi T, Stowe A, Claassen J, Munro Cullum C, Zhu DC, Zhang R. Carotid stiffness is associated with brain amyloid-β burden in amnestic mild cognitive impairment. J Alzheimers Dis 74: 925–935, 2020. doi: 10.3233/JAD-191073. [DOI] [PubMed] [Google Scholar]
- 544.Roher AE, Esh C, Rahman A, Kokjohn TA, Beach TG. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 35: 2623–2627, 2004. doi: 10.1161/01.STR.0000143317.70478.b3. [DOI] [PubMed] [Google Scholar]
- 545.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12: 723–738, 2011. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Iadecola C, Yaffe K, Biller J, Bratzke LC, Faraci FM, Gorelick PB, Gulati M, Kamel H, Knopman DS, Launer LJ, Saczynski JS, Seshadri S, Hazzouri ZA, American Heart Association Council on Hypertension; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Quality of Care and Outcomes Research; and Stroke Council. Impact of hypertension on cognitive function: a scientific statement from the American Heart Association. Hypertension 68: e67–e94, 2016. doi: 10.1161/hyp.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Gustavsson AM, van Westen D, Stomrud E, Engstrom G, Nagga K, Hansson O. Midlife atherosclerosis and development of Alzheimer or vascular dementia. Ann Neurol 87: 52–62, 2020. doi: 10.1002/ana.25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Iadecola C, Gottesman RF. Cerebrovascular alterations in Alzheimer disease. Circ Res 123: 406–408, 2018. doi: 10.1161/CIRCRESAHA.118.313400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R. , Contributors. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimer's Dement 14: 535–562, 2018. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, Frosch MP, Greenberg SM, Bacskai BJ. Cerebrovascular lesions induce transient ß-amyloid deposition. Brain 134: 3697–3707, 2011. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Alsop DC, Dai W, Grossman M, Detre JA. Arterial spin labeling blood flow MRI: Its role in the early characterization of Alzheimer's disease. JAD 20: 871–880, 2010. doi: 10.3233/JAD-2010-091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Wolk DA, Detre JA. Arterial spin labeling MRI: an emerging biomarker for Alzheimer's disease and other neurodegenerative conditions. Curr Opin Neurol 25: 421–428, 2012. doi: 10.1097/WCO.0b013e328354ff0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Xu G, Rowley HA, Wu G, Alsop DC, Shankaranarayanan A, Dowling M, Christian BT, Oakes TR, Johnson SC. Reliability and precision of pseudo-continuous arterial spin labeling perfusion MRI on 3.0 T and comparison with 15O-water PET in elderly subjects at risk for Alzheimer's disease. NMR Biomed 23: 286–293, 2010. doi: 10.1002/nbm.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Musiek ES, Chen Y, Korczykowski M, Saboury B, Martinez PM, Reddin JS, Alavi A, Kimberg DY, Wolk DA, Julin P, Newberg AB, Arnold SE, Detre JA. Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer's disease. Alzheimers Dement 8: 51–59, 2012. doi: 10.1016/j.jalz.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, Weiner MW, Jagust WJ, Alzheimer's Disease Neuroimaging Initiative. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 72: 578–586, 2012. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Binnewijzend MA, Benedictus MR, Kuijer JP, van der Flier WM, Teunissen CE, Prins ND, Wattjes MP, van Berckel BN, Scheltens P, Barkhof F. Cerebral perfusion in the predementia stages of Alzheimer's disease. Eur Radiol 26: 506–514, 2016. doi: 10.1007/s00330-015-3834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Nagahama Y, Nabatame H, Okina T, Yamauchi H, Narita M, Fujimoto N, Murakami M, Fukuyama H, Matsuda M. Cerebral correlates of the progression rate of the cognitive decline in probable Alzheimer's disease. Eur Neurol 50: 1–9, 2003. doi: 10.1159/000070851. [DOI] [PubMed] [Google Scholar]
- 558.Chao LL, Buckley ST, Kornak J, Schuff N, Madison C, Yaffe K, Miller BL, Kramer JH, Weiner MW. ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis Assoc Disord 24: 19–27, 2010. doi: 10.1097/WAD.0b013e3181b4f736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Benedictus MR, Leeuwis AE, Binnewijzend MA, Kuijer JP, Scheltens P, Barkhof F, van der Flier WM, Prins ND. Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer's disease. Eur Radiol 27: 1169–1175, 2017. doi: 10.1007/s00330-016-4450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Leeuwis AE, Benedictus MR, Kuijer JP, Binnewijzend MA, Hooghiemstra AM, Verfaillie SC, Koene T, Scheltens P, Barkhof F, Prins ND, van der Flier WM. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer's disease. Alzheimers Dement 13: 531–540, 2017. doi: 10.1016/j.jalz.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 561.Hajjar I, Sorond F, Lipsitz LA. Apolipoprotein E, carbon dioxide vasoreactivity, and cognition in older adults: effect of hypertension. J Am Geriatr Soc 63: 276–281, 2015. doi: 10.1111/jgs.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Abeelen A, Lagro J, Beek A, Claassen J. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res 11: 11–17, 2014. doi: 10.2174/1567205010666131119234845. [DOI] [PubMed] [Google Scholar]
- 563.Suri S, Mackay CE, Kelly ME, Germuska M, Tunbridge EM, Frisoni GB, Matthews PM, Ebmeier KP, Bulte DP, Filippini N. Reduced cerebrovascular reactivity in young adults carrying the APOE epsilon4 allele. Alzheimers Dement 11: 648–657, 2015. doi: 10.1016/j.jalz.2014.05.1755. [DOI] [PubMed] [Google Scholar]
- 564.Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: 419–434, 2017. doi: 10.1038/nrn.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Claassen JA, Zhang R. Cerebral autoregulation in Alzheimer's disease. J Cereb Blood Flow Metab 31: 1572–1577, 2011. doi: 10.1038/jcbfm.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci 17: 7655–7661, 1997. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Simard D, Olesen J, Paulson OB, Lassen NA, Skinhoj E. Regional cerebral blood flow and its regulation in dementia. Brain 94: 273–288, 1971. doi: 10.1093/brain/94.2.273. [DOI] [PubMed] [Google Scholar]
- 568.Claassen JA, Diaz-Arrastia R, Martin-Cook K, Levine BD, Zhang R. Altered cerebral hemodynamics in early Alzheimer disease: a pilot study using transcranial Doppler. J Alzheimers Dis 17: 621–629, 2009. doi: 10.3233/JAD-2009-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Zazulia AR, Videen TO, Morris JC, Powers WJ. Autoregulation of cerebral blood flow to changes in arterial pressure in mild Alzheimer's disease. J Cereb Blood Flow Metab 30: 1883–1889, 2010. doi: 10.1038/jcbfm.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.van Beek AH, Sijbesma JC, Jansen RW, Rikkert MG, Claassen JA. Cortical oxygen supply during postural hypotension is further decreased in Alzheimer's disease, but unrelated to cholinesterase-inhibitor use. J Alzheimers Dis 21: 519–526, 2010. doi: 10.3233/JAD-2010-100288. [DOI] [PubMed] [Google Scholar]
- 571.van Beek AH, Lagro J, Olde-Rikkert MG, Zhang R, Claassen JA. Oscillations in cerebral blood flow and cortical oxygenation in Alzheimer’s disease. Neurobiol Aging 33: e421-431, 2012. doi: 10.1016/j.neurobiolaging.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 572.de Jong DL, de Heus RA, Rijpma A, Donders R, Olde Rikkert MG, Gunther M, Lawlor BA, van Osch MJ, Claassen J. Effects of nilvadipine on cerebral blood flow in patients with Alzheimer disease. Hypertension 74: 413–420, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12892. [DOI] [PubMed] [Google Scholar]
- 573.Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283: H315–H323, 2002. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- 574.King A. The search for better animal models of Alzheimer's disease. Nature 559: S13–S15, 2018. doi: 10.1038/d41586-018-05722-9. [DOI] [PubMed] [Google Scholar]
- 575.de Heus RA, Donders R, Santoso AM, Olde RM, Lawlor BA, Claassen J, Nilvad S. G. Blood pressure lowering with nilvadipine in patients with mild-to-moderate Alzheimer disease does not increase the prevalence of orthostatic hypotension. J Am Heart Assoc 8: e011938, 2019. doi: 10.1161/jaha.119.011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.van Beek AH, Sijbesma JC, Olde Rikkert MG, Claassen JA. Galantamine does not cause aggravated orthostatic hypotension in people with Alzheimer's disease. J Am Geriatr Soc 58: 409–410, 2010. doi: 10.1111/j.1532-5415.2009.02712.x. [DOI] [PubMed] [Google Scholar]
- 577.de Heus RA, Olde Rikkert MG, Tully PJ, Lawlor BA, Claassen J, Group NS, NILVAD Study Group. Blood pressure variability and progression of clinical Alzheimer disease. Hypertension 74: 1172–1180, 2019. doi: 10.1161/HYPERTENSIONAHA.119.13664. [DOI] [PubMed] [Google Scholar]
- 578.Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, Ballard C, Banerjee S, Burns A, Cohen-Mansfield J, Cooper C, Fox N, Gitlin LN, Howard R, Kales HC, Larson EB, Ritchie K, Rockwood K, Sampson EL, Samus Q, Schneider LS, Selbaek G, Teri L, Mukadam N. Dementia prevention, intervention, and care. Lancet 390: 2673–2734, 2017. doi: 10.1016/S0140-6736(17)31363-6. [DOI] [PubMed] [Google Scholar]
- 579.Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: Facts, myths, and missing links. Neurocrit Care 10: 373–386, 2009. doi: 10.1007/s12028-008-9175-7. [DOI] [PubMed] [Google Scholar]
- 580.Liu X, Czosnyka M, Donnelly J, Budohoski KP, Varsos GV, Nasr N, Brady KM, Reinhard M, Hutchinson PJ, Smielewski P. Comparison of frequency and time domain methods of assessment of cerebral autoregulation in traumatic brain injury. J Cereb Blood Flow Metab 35: 248–256, 2015. doi: 10.1038/jcbfm.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Rivera-Lara L, Zorrilla-Vaca A, Geocadin R, Ziai W, Healy R, Thompson R, Smielewski P, Czosnyka M, Hogue CW. Predictors of outcome with cerebral autoregulation monitoring: A systematic review and meta-analysis. Crit Care Med 45: 695–704, 2017. doi: 10.1097/CCM.0000000000002251. [DOI] [PubMed] [Google Scholar]
- 582.Brady KM, Lee JK, Kibler KK, Smielewski P, Czosnyka M, Easley RB, Koehler RC, Shaffner DH. Continuous time-domain analysis of cerebrovascular autoregulation using near-infrared spectroscopy. Stroke 38: 2818–2825, 2007. doi: 10.1161/STROKEAHA.107.485706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Czosnyka M, Smielewski P, Kirkpatrick P, Menon DK, Pickard JD. Monitoring of cerebral autoregulation in head-injured patients. Stroke 27: 1829–1834, 1996. doi: 10.1161/01.STR.27.10.1829. [DOI] [PubMed] [Google Scholar]
- 584.Czosnyka M, Smielewski P, Piechnik S, Schmidt EA, Al-Rawi PG, Kirkpatrick PJ, Pickard JD. Hemodynamic characterization of intracranial pressure plateau waves in head-injury patients. J Neurosurg 91: 11–19, 1999. doi: 10.3171/jns.1999.91.1.0011. [DOI] [PubMed] [Google Scholar]
- 585.Czosnyka M, Smielewski P, Piechnik S, Schmidt EA, Seeley H, Al-Rawi P, Matta BF, Kirkpatrick PJ, Pickard JD. Continuous assessment of cerebral autoregulation–clinical verification of the method in head injured patients. Acta Neurochir Suppl 76: 483–484, 2000. doi: 10.1007/978-3-7091-6346-7_101. [DOI] [PubMed] [Google Scholar]
- 586.Kirkpatrick PJ, Czosnyka M, Pickard JD. Multimodal monitoring in neurointensive care. J Neurol Neurosurg Psychiatry 60: 131–139, 1996. doi: 10.1136/jnnp.60.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Lu CW, Czosnyka M, Shieh JS, Smielewska A, Pickard JD, Smielewski P. Complexity of intracranial pressure correlates with outcome after traumatic brain injury. Brain 135: 2399–2408, 2012. doi: 10.1093/brain/aws155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.McBryde FD, Malpas SC, Paton JF. Intracranial mechanisms for preserving brain blood flow in health and disease. Acta Physiol (Oxf) 219: 274–287, 2017. doi: 10.1111/apha.12706. [DOI] [PubMed] [Google Scholar]
- 589.Wilson MH. Monro-Kellie 2.0: the dynamic vascular and venous pathophysiological components of intracranial pressure. J Cereb Blood Flow Metabol 36: 1338–1350, 2016. doi: 10.1177/0271678X16648711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Zhou ZB, Meng L, Gelb AW, Lee R, Huang WQ. Cerebral ischemia during surgery: an overview. J Biomed Res 30: 83–87, 2016. doi: 10.7555/jbr.30.20150126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Spielvogel D, Kai M, Tang GH, Malekan R, Lansman SL. Selective cerebral perfusion: a review of the evidence. J Thorac Cardiovasc Surg 145: S59–62, 2013. doi: 10.1016/j.jtcvs.2012.11.073. [DOI] [PubMed] [Google Scholar]
- 592.Froese L, Dian J, Batson C, Gomez A, Unger B, Zeiler FA. Cerebrovascular response to propofol, fentanyl, and midazolam in moderate/severe traumatic brain injury: a scoping systematic review of the human and animal literature. Neurotrauma Rep 1: 100–112, 2020. doi: 10.1089/neur.2020.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Slupe AM, Kirsch JR. Effects of anesthesia on cerebral blood flow, metabolism, and neuroprotection. J Cereb Blood Flow Metab 38: 2192–2208, 2018. doi: 10.1177/0271678X18789273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Gao YR, Ma Y, Zhang Q, Winder AT, Liang Z, Antinori L, Drew PJ, Zhang N. Time to wake up: Studying neurovascular coupling and brain-wide circuit function in the un-anesthetized animal. Neuroimage 153: 382–398, 2017. doi: 10.1016/j.neuroimage.2016.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Matta BF, Heath KJ, Tipping K, Summors AC. Direct cerebral vasodilatory effects of sevoflurane and isoflurane. Anesthesiology 91: 677–680, 1999. doi: 10.1097/00000542-199909000-00019. [DOI] [PubMed] [Google Scholar]
- 596.Minhas JS, Rook W, Panerai RB, Hoiland RL, Ainslie PN, Thompson JP, Mistri AK, Robinson TG. Pathophysiological and clinical considerations in the perioperative care of patients with a previous ischaemic stroke: a multidisciplinary narrative review. Br J Anaesth 124: 183–196, 2020. doi: 10.1016/j.bja.2019.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Saxena N, Gili T, Diukova A, Huckle D, Hall JE, Wise RG. Mild propofol sedation reduces frontal lobe and thalamic cerebral blood flow: an arterial spin labeling study. Front Physiol 10: 1541, 2019. doi: 10.3389/fphys.2019.01541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Wu GR, Di Perri C, Charland-Verville V, Martial C, Carriere M, Vanhaudenhuyse A, Laureys S, Marinazzo D. Modulation of the spontaneous hemodynamic response function across levels of consciousness. Neuroimage 200: 450–459, 2019. doi: 10.1016/j.neuroimage.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 599.Gutkin M, Stewart JM. Orthostatic circulatory disorders: from nosology to nuts and bolts. Am J Hypertens 29: 1009–1019, 2016. doi: 10.1093/ajh/hpw023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.O' Brien H, Anne Kenny R. Syncope in the elderly. Eur Cardiol 9: 28–36, 2014. doi: 10.15420/ecr.2014.9.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Sutton R. Reflex syncope: diagnosis and treatment. J Arrhythm 33: 545–552, 2017. doi: 10.1016/j.joa.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Moya A, Sutton R, Ammirati F, Blanc JJ, Brignole M, Dahm JB, Deharo JC, Gajek J, Gjesdal K, Krahn A, Massin M, Pepi M, Pezawas T, Ruiz GR, Sarasin F, Ungar A, van Dijk JG, Walma EP, Wieling Task Force for the Diagnosis and Management of Syncope, European Society of Cardiology, European Heart Rhythm Association, Heart Failure Association, Heart Rhythm Society. W. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 30: 2631–2671, 2009. doi: 10.1093/eurheartj/ehp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Schondorf R, Stein R, Roberts R, Benoit J, Cupples W. Dynamic cerebral autoregulation is preserved in neurally mediated syncope. J Appl Physiol (1985) 91: 2493–2502, 2001. doi: 10.1152/jappl.2001.91.6.2493. [DOI] [PubMed] [Google Scholar]
- 604.Fu Q, Levine BD. Pathophysiology of neurally mediated syncope: role of cardiac output and total peripheral resistance. Auton Neurosci 184: 24–26, 2014. doi: 10.1016/j.autneu.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.van Wijnen VK, Ten Hove D, Gans RO, Nieuwland W, van Roon AM, Ter Maaten JC, Harms MP. Orthostatic blood pressure recovery patterns in suspected syncope in the emergency department. Emerg Med J 35: 226–230, 2018. doi: 10.1136/emermed-2017-207207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Iwasaki K, Zhang R, Perhonen MA, Zuckerman JH, Levine BD. Reduced baroreflex control of heart period after bed rest is normalized by acute plasma volume restoration. Am J Physiol Regul Integr Comp Physiol 287: R1256–R1262, 2004. doi: 10.1152/ajpregu.00613.2002. [DOI] [PubMed] [Google Scholar]
- 607.Levine BD, Giller CA, Lane LD, Buckey JC, Blomqvist CG. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation 90: 298–306, 1994. doi: 10.1161/01.cir.90.1.298. [DOI] [PubMed] [Google Scholar]
- 608.Zhang R, Zuckerman JH, Pawelczyk JA, Levine BD. Effects of head-down-tilt bed rest on cerebral hemodynamics during orthostatic stress. J Appl Physiol (1985) 83: 2139–2145, 1997. doi: 10.1152/jappl.1997.83.6.2139. [DOI] [PubMed] [Google Scholar]
- 609.Benditt DG, Fabian W, Iskos D, Lurie KG. Review article: Heart rate and blood pressure control in vasovagal syncope. J Interv Card Electrophysiol 2: 25–32, 1998. doi: 10.1023/a:1009756521965. [DOI] [PubMed] [Google Scholar]
- 610.Thijs RD, Wieling W, Kaufmann H, van Dijk G. Defining and classifying syncope. Clin Auton Res 14: i4–8, 2004. doi: 10.1007/s10286-004-1002-4. [DOI] [PubMed] [Google Scholar]
- 611.Morris JN, Heady JA, Raffle PA, Roberts CG, Parks JW. Coronary heart-disease and physical activity of work. Lancet 262: 1053–1057, 1953. doi: 10.1016/s0140-6736(53)90665-5. [DOI] [PubMed] [Google Scholar]
- 612.Lambertsen CJ, Owen SG, Wendel H, Stroud MW, Lurie AA, Lochner W, Clark GF. Respiratory and cerebral circulatory control during exercise at .21 and 2.0 atmospheres inspired Po2. J Appl Physiol 14: 966–982, 1959. doi: 10.1152/jappl.1959.14.6.966. [DOI] [PubMed] [Google Scholar]
- 613.Ekelund U, Steene-Johannessen J, Brown WJ, Fagerland MW, Owen N, Powell KE, Bauman A, Lee IM, Lancet Physical Activity Series 2 Executive Committee, Lancet Sedentary Behaviour Working Group. Does physical activity attenuate, or even eliminate, the detrimental association of sitting time with mortality? A harmonised meta-analysis of data from more than 1 million men and women. Lancet 388: 1302–1310, 2016. doi: 10.1016/S0140-6736(16)30370-1. [DOI] [PubMed] [Google Scholar]
- 614.van der Ploeg HP, Chey T, Korda RJ, Banks E, Bauman A. Sitting time and all-cause mortality risk in 222 497 Australian adults. Arch Intern Med 172: 494–500, 2012. doi: 10.1001/archinternmed.2011.2174. [DOI] [PubMed] [Google Scholar]
- 615.Carter S, Hartman Y, Holder S, Thijssen DH, Hopkins ND. Sedentary behavior and cardiovascular disease risk: mediating mechanisms. Exerc Sport Sci Rev 45: 80–86, 2017. doi: 10.1249/JES.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 616.Falck RS, Davis JC, Liu-Ambrose T. What is the association between sedentary behaviour and cognitive function? A systematic review. Br J Sports Med 51: 800–811, 2017. doi: 10.1136/bjsports-2015-095551. [DOI] [PubMed] [Google Scholar]
- 617.Wheeler MJ, Dempsey PC, Grace MS, Ellis KA, Gardiner PA, Green DJ, Dunstan DW. Sedentary behavior as a risk factor for cognitive decline? A focus on the influence of glycemic control in brain health. Alzheimers Dement (N Y) 3: 291–300, 2017. doi: 10.1016/j.trci.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Carter SE, Draijer R, Holder SM, Brown L, Thijssen DH, Hopkins ND. Regular walking breaks prevent the decline in cerebral blood flow associated with prolonged sitting. J Appl Physiol (1985) 125: 790–798, 2018. doi: 10.1152/japplphysiol.00310.2018. [DOI] [PubMed] [Google Scholar]
- 619.Claassen JA, Levine BD, Zhang R. Cerebral vasomotor reactivity before and after blood pressure reduction in hypertensive patients. Am J Hypertens 22: 384–391, 2009. doi: 10.1038/ajh.2009.2. [DOI] [PubMed] [Google Scholar]
- 620.Wheeler MJ, Dunstan DW, Smith B, Smith KJ, Scheer A, Lewis J, Naylor LH, Heinonen I, Ellis KA, Cerin E, Ainslie PN, Green DJ. Morning exercise mitigates the impact of prolonged sitting on cerebral blood flow in older adults. J Appl Physiol 126: 1049–1055, 2019. doi: 10.1152/japplphysiol.00001.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Maasakkers CM, Melis RJ, Kessels RP, Gardiner PA, Olde RM, Thijssen DH, Claassen J. The short-term effects of sedentary behaviour on cerebral hemodynamics and cognitive performance in older adults: a cross-over design on the potential impact of mental and/or physical activity. Alzheimers Res Ther 12: 76, 2020. doi: 10.1186/s13195-020-00644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Perdomo SJ, Gibbs BB, Kowalsky RJ, Taormina JM, Balzer JR. Effects of alternating standing and sitting compared with prolonged sitting on cerebrovascular hemodynamics. Sport Sci Health 15: 375–383, 2019. doi: 10.1007/s11332-019-00526-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Zlatar ZZ, Hays CC, Mestre Z, Campbell LM, Meloy MJ, Bangen KJ, Liu TT, Kerr J, Wierenga CE. Dose-dependent association of accelerometer-measured physical activity and sedentary time with brain perfusion in aging. Exp Gerontol 125: 110679, 2019. doi: 10.1016/j.exger.2019.110679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Wright AD, Smirl JD, Bryk K, van Donkelaar P. Systolic and diastolic regulation of the cerebral pressure-flow relationship differentially affected by acute sport-related concussion. Acta Neurochir Suppl 126: 303–308, 2018. doi: 10.1007/978-3-319-65798-1_59. [DOI] [PubMed] [Google Scholar]
- 625.Hartman YA, Tillmans LC, Benschop DL, Hermans AN, Nijssen KM, Eijsvogels TM, Willems P, Tack CJ, Hopman MT, Claassen J, Thijssen DH. Long-term and acute benefits of reduced sitting on vascular flow and function. Med Sci Sports Exerc 53: 341–350, 2021. doi: 10.1249/MSS.0000000000002462. [DOI] [PubMed] [Google Scholar]
- 626.Healy GN, Winkler EA, Brakenridge CL, Reeves MM, Eakin EG. Accelerometer-derived sedentary and physical activity time in overweight/obese adults with type 2 diabetes: cross-sectional associations with cardiometabolic biomarkers. PLoS One 10: e0119140, 2015. doi: 10.1371/journal.pone.0119140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Dunstan DW, Kingwell BA, Larsen R, Healy GN, Cerin E, Hamilton MT, Shaw JE, Bertovic DA, Zimmet PZ, Salmon J, Owen N. Breaking up prolonged sitting reduces postprandial glucose and insulin responses. Diabetes Care 35: 976–983, 2012. doi: 10.2337/dc11-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Smith KJ, Ainslie PN. Regulation of cerebral blood flow and metabolism during exercise. Exp Physiol 102: 1356–1371, 2017. doi: 10.1113/EP086249. [DOI] [PubMed] [Google Scholar]
- 629.Siebenmann C, Sorensen H, Jacobs RA, Haider T, Rasmussen P, Lundby C. Hypocapnia during hypoxic exercise and its impact on cerebral oxygenation, ventilation and maximal whole body O2 uptake. Respir Physiol Neurobiol 185: 461–467, 2013. doi: 10.1016/j.resp.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 630.Olin JT, Dimmen AC, Subudhi AW, Roach RC. Cerebral blood flow and oxygenation at maximal exercise: the effect of clamping carbon dioxide. Respir Physiol Neurobiol 175: 176–180, 2011. doi: 10.1016/j.resp.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Braz ID, Fisher JP. The impact of age on cerebral perfusion, oxygenation and metabolism during exercise in humans. J Physiol 594: 4471–4483, 2016. doi: 10.1113/JP271081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Ogoh S, Brothers RM, Barnes Q, Eubank WL, Hawkins MN, Purkayastha S, O-Yurvati A, Raven PB. The effect of changes in cardiac output on middle cerebral artery mean blood velocity at rest and during exercise. J Physiol 569: 697–704, 2005. doi: 10.1113/jphysiol.2005.095836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Smirl JD, Haykowsky MJ, Nelson MD, Altamirano-Diaz LA, Ainslie PN. Resting and exercise cerebral blood flow in long-term heart transplant recipients. J Heart Lung Transplant 31: 906–908, 2012. doi: 10.1016/j.healun.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 634.Smith KJ, MacLeod D, Willie CK, Lewis NC, Hoiland RL, Ikeda K, Tymko MM, Donnelly J, Day TA, MacLeod N, Lucas SJ, Ainslie PN. Influence of high altitude on cerebral blood flow and fuel utilization during exercise and recovery. J Physiol 592: 5507–5527, 2014. doi: 10.1113/jphysiol.2014.281212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Quistorff B, Secher NH, Van Lieshout JJ. Lactate fuels the human brain during exercise. FASEB J 22: 3443–3449, 2008. doi: 10.1096/fj.08-106104. [DOI] [PubMed] [Google Scholar]
- 636.Fisher JP, Hartwich D, Seifert T, Olesen ND, McNulty CL, Nielsen HB, van Lieshout JJ, Secher NH. Cerebral perfusion, oxygenation and metabolism during exercise in young and elderly individuals. J Physiol 591: 1859–1870, 2013. doi: 10.1113/jphysiol.2012.244905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Fisher JP, Ogoh S, Young CN, Raven PB, Fadel PJ. Regulation of middle cerebral artery blood velocity during dynamic exercise in humans: influence of aging. J Appl Physiol 105: 266–273, 2008. doi: 10.1152/japplphysiol.00118.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Fluck D, Beaudin AE, Steinback CD, Kumarpillai G, Shobha N, McCreary CR, Peca S, Smith EE, Poulin MJ. Effects of aging on the association between cerebrovascular responses to visual stimulation, hypercapnia and arterial stiffness. Front Physiol 5: 49, 2014. doi: 10.3389/fphys.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Duzel E, van Praag H, Sendtner M. Can physical exercise in old age improve memory and hippocampal function? Brain 139: 662–673, 2016. doi: 10.1093/brain/awv407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Erickson KI, Hillman C, Stillman CM, Ballard RM, Bloodgood B, Conroy DE, Macko R, Marquez DX, Petruzzello SJ, Powell KE, for Physical Activity Guidelines Advisory Council . Physical activity, cognition, and brain outcomes: a review of the 2018 Physical Activity Guidelines. Med Sci Sports Exerc 51: 1242–1251, 2019. doi: 10.1249/MSS.0000000000001936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Kleinloog JP, Mensink RP, Ivanov D, Adam JJ, Uludag K, Joris PJ. Aerobic exercise training improves cerebral blood flow and executive function: a randomized, controlled cross-over trial in sedentary older men. Front Aging Neurosci 11: 333, 2019. doi: 10.3389/fnagi.2019.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Lehmann N, Villringer A, Taubert M. Colocalized white matter plasticity and increased cerebral blood flow mediate the beneficial effect of cardiovascular exercise on long-term motor learning. J Neurosci 40: 2416–2429, 2020. doi: 10.1523/JNEUROSCI.2310-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Chapman SB, Aslan S, Spence JS, Keebler MW, DeFina LF, Didehbani N, Perez AM, Lu H., D'Esposito S. Distinct brain and behavioral benefits from cognitive vs. physical training: a randomized trial in aging adults. Front Hum Neurosci 10: 338, 2016. doi: 10.3389/fnhum.2016.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Alfini AJ, Weiss LR, Leitner BP, Smith TJ, Hagberg JM, Smith JC. Hippocampal and cerebral blood flow after exercise cessation in master athletes. Front Aging Neurosci 8: 184, 2016. doi: 10.3389/fnagi.2016.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Robertson AD, Marzolini S, Middleton LE, Basile VS, Oh PI, MacIntosh BJ. Exercise training increases parietal lobe cerebral blood flow in chronic stroke: An observational study. Front Aging Neurosci 9: 318, 2017. doi: 10.3389/fnagi.2017.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Alfini AJ, Weiss LR, Nielson KA, Verber MD, Smith JC. Resting cerebral blood flow after exercise training in mild cognitive impairment. J Alzheimers Dis 67: 671–684, 2019. doi: 10.3233/JAD-180728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Ichikawa D, Miyazawa T, Horiuchi M, Kitama T, Fisher JP, Ogoh S. Relationship between aerobic endurance training and dynamic cerebral blood flow regulation in humans. Scand J Med Sci Sports 23: e320-329–n/a, 2013. doi: 10.1111/sms.12082. [DOI] [PubMed] [Google Scholar]
- 648.Lind-Holst M, Cotter JD, Helge JW, Boushel R, Augustesen H, Van Lieshout JJ, Pott FC. Cerebral autoregulation dynamics in endurance-trained individuals. J Appl Physiol 110: 1327–1333, 2011. doi: 10.1152/japplphysiol.01497.2010. [DOI] [PubMed] [Google Scholar]
- 649.Perry BG, Cotter JD, Korad S, Lark S, Labrecque L, Brassard P, Paquette M, Le Blanc O, Lucas SJ. Implications of habitual endurance and resistance exercise for dynamic cerebral autoregulation. Exp Physiol 104: 1780–1789, 2019. doi: 10.1113/EP087675. [DOI] [PubMed] [Google Scholar]
- 650.Drapeau A, Labrecque L, Imhoff S, Paquette M, Le Blanc O, Malenfant S, Brassard P. Six weeks of high-intensity interval training to exhaustion attenuates dynamic cerebral autoregulation without influencing resting cerebral blood velocity in young fit men. Physiol Rep 7: e14185, 2019. doi: 10.14814/phy2.14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Tsukamoto H, Hashimoto T, Olesen ND, Petersen LG, Sorensen H, Nielsen HB, Secher NH, Ogoh S. Dynamic cerebral autoregulation is maintained during high-intensity interval exercise. Med Sci Sports Exerc 51: 372–378, 2019. doi: 10.1249/MSS.0000000000001792. [DOI] [PubMed] [Google Scholar]
- 652.Lewis N, Gelinas JC, Ainslie PN, Smirl JD, Agar G, Melzer B, Rolf JD, Eves ND. Cerebrovascular function in patients with chronic obstructive pulmonary disease: The impact of exercise training. Am J Physiol Heart Circ Physiol 316: H380–H391, 2019. doi: 10.1152/ajpheart.00348.2018. [DOI] [PubMed] [Google Scholar]
- 653.Kim TW, Park SS, Park JY, Park HS. Infusion of plasma from exercised mice ameliorates cognitive dysfunction by increasing hippocampal neuroplasticity and mitochondrial functions in 3xTg-AD mice. Int J Mol Sci 21: 3291, 2020. doi: 10.3390/ijms21093291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Bliss ES, Wong RH, Howe PR, Mills DE. Benefits of exercise training on cerebrovascular and cognitive function in ageing. J Cereb Blood Flow Metab 41: 447–470, 2021. doi: 10.1177/0271678X20957807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Stillman CM, Esteban-Cornejo I, Brown B, Bender CM, Erickson KI. Effects of exercise on brain and cognition across age groups and health states. Trends Neurosci 43: 533–543, 2020. doi: 10.1016/j.tins.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]