

Abstract
During the past decade, pharmaceutical engineering of unimolecular agents has revealed the therapeutic potential of glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism. From this work, one of the most intriguing findings is that engagement of GIPR enhances the weight loss profile of glucagon-like peptide 1 (GLP-1)-based therapeutics. Consequently, this pharmacological approach, in combination with novel Gipr mouse models, has provided evidence indicating that activation of GIPR in certain areas of the brain that regulate energy balance is required for the synergistic weight loss of dual GIPR and GLP-1 receptor (GLP-1R) agonism. This has led to significant interest in understanding how GIPR activity in the brain functions to reduce caloric intake, induce negative energy balance, and drive weight loss. Herein, we review key findings in this field and provide a novel perspective explaining how GIP may act in the brain to affect energy balance both alone and in concert with GLP-1R agonism.
The Challenge
Type 2 diabetes (T2D) is the most prevalent form of diabetes worldwide. Traditionally classed as adult onset, the rising incidence of T2D in ever younger proportions of the population is being driven by another pandemic: obesity. For patients suffering from metabolic disease, weight loss can significantly improve glycemic control and, in some cases, leads to the reversal of T2D (1). Thus, therapies that promote weight loss, in addition to improving blood glucose levels, are important for the treatment of diabetes. Although strategies aimed at promoting healthier lifestyles are important interventions for curtailing the rise in obesity and related comorbidities, it is evident that such public health policy efforts alone are not enough, as rates of obesity and T2D continue to rise. As current trends predict that 20% of the world’s population will be obese in 2025 (2), pharmaceutical approaches to combat obesity and diabetes are a necessary next step to address this burgeoning health crisis. Mechanistically, it is increasingly evident that obesity is driven by aberrations in signaling pathways of the central nervous system (CNS), with most single nucleotide polymorphisms associated with high BMI localizing to genes encoding proteins that exert their functions in the brain (3). Central signaling pathways therefore represent promising targets for antiobesity therapeutics.
Incretin-Based Therapies in Metabolic Disease
Glucagon-like peptide 1 (GLP-1) is an incretin hormone that regulates postprandial glycemia by augmenting glucose-dependent insulin secretion, delaying gastric emptying, and suppressing appetite (4). The broad effects ascribed to the GLP-1 axis are the foundation of its metabolic utility and led to the development of GLP-1–based medicines. The GLP-1 receptor agonist (GLP-1RA) drug class is now firmly established as a pharmacologic therapy not only for the treatment of T2D but also for obesity (5) and may have further benefit for patients with T2D experiencing cardiorenal dysfunction (4,6). However, despite the compelling attributes of GLP-1 therapeutics, many patients still do not reach their glycemic and weight loss goals (7). This combined with the sheer scale of the obesity and T2D pandemics drives the need to identify agents capable of complementing and enhancing GLP-1R agonism (8).
Next Generation of T2D Therapeutics
It is anticipated that the next generation of T2D medications will be designed to engage complementary mechanisms through novel pharmaceutical engineering of unimolecular agents (9). One such strategy is centered on generating single peptide agonists created to harness GLP-1R activity while also targeting the glucose-dependent insulinotropic polypeptide receptor (GIPR). The aim of this new approach is to further augment metabolic benefits while minimizing adverse events (10). Importantly, this approach has resulted in enhanced glycemic control and unprecedented weight loss in T2D patients, highlighting the GIPR signaling axis as a promising and effective cotarget (11). Consequently, these dual agonists have spurred renewed interest in understanding how engagement of the GIPR contributes to enhanced metabolic control, with particular focus on the apparently beneficial effects on weight loss (10).
The Comeback of GIP as a Therapeutic Target
GIP is a gut hormone produced in enteroendocrine cells lining the upper small intestine and is secreted in response to the consumption of dietary nutrients (12). Similar to GLP-1, GIP plays a key role in the regulation of glucose homeostasis following meals, with evidence today pointing toward GIP being the primary incretin in man (13). In addition, GIP is implicated in postprandial lipid metabolism (12), a phenomenon that has contributed to the hypothesis that GIP could promote obesity (14). Consequently, this has led to the suggestion that blocking GIP action may be a viable therapeutic approach for promoting weight loss (15). To date, however, pharmacological approaches to augmenting GIPR activity have demonstrated neither weight gain nor metabolic dysregulation (16–18). In fact, over the last 10 years, advancements in therapeutic peptide design have led to the discovery that rather than driving weight gain, GIPR agonism induces negative energy balance (11,19,20).
Herein, we discuss recent insights into the previously underappreciated role of the central GIPR signaling axis in regulating energy balance. Further, a novel rationale is presented explaining how GIPR agonism in the CNS may enhance the weight loss profile of GLP-1R agonism.
GIP Does Not Promote a Positive Energy Balance
The historical framing of GIP as an obesogenic factor centers on its activity in the adipocyte. The coupling of lipid intake with its appropriate storage within white adipose tissue (WAT) is essential to maintaining metabolic health (21). One of the primary signals to induce GIP secretion is the ingestion of dietary lipid (22). The GIPR is expressed by adipocytes (23), where GIP acts to enhance both insulin-dependent and -independent triglyceride synthesis (24). In preclinical models and in healthy humans and humans with diabetes, GIP promotes postprandial triglyceride clearance in WAT (25–30). WAT storage of dietary triglyceride is essential to glycemic control, and since GIP has been shown to help regulate this process, it would be expected that inhibiting GIP action could impair lipid storage and result in ectopic lipid accumulation in other metabolic organs. Consistent with this, blockade of GIPR impairs postprandial lipid clearance in rodents and man (28,29). Further, both white adipocyte– and brown adipocyte–specific Gipr-null animals display impaired lipid tolerance (26,27). Thus, these findings support the notion that GIP is a gut-derived factor that couples the ingestion of dietary triglyceride with its proper storage within WAT (10).
Despite early studies recognizing the therapeutic potential of GIP to promote storage of dietary triglyceride in WAT (31), data collected from genetic knockout studies (Gip or Gipr) suggested that in the absence of a functional GIP axis, mice are protected from diet-induced obesity (14,32). This inspired the hypothesis that GIP is an obesogenic factor and the suggestion that inhibiting GIP activity is a viable approach for treating obesity and T2D (15). Undeniably the inhibition of GIP signaling protects mice from diet-induced obesity (33). It is unlikely, however, that the protection from weight gain through either genetic or pharmacological blockade of the GIP axis is due to the ability of GIP to augment the lipid-buffering capacity of WAT. This is because the only mechanisms underlying body weight gain include increased caloric intake and/or reduced caloric expenditure (34), with the amount of lipid stored within WAT reflecting the difference between these two components of energy homeostasis (34). Therefore, for GIP to drive weight gain, it would have to impact either one or both sides of the energy balance equation. To date, however, there is little evidence indicating that GIP promotes positive energy balance (16,18,35–37). To the contrary, acute infusion of supraphysiological levels of GIP has no effect on body weight, food intake, or caloric expenditure in adult humans (13). Further, chronic treatment with short- and long-acting GIPR agonists does not drive weight gain, promote hyperphagia, or decrease metabolic rate in either lean or obese mice (16,19,20,36). Lastly, it should be noted that genetic or pharmacological blockade of the GLP-1R also offers protection against diet-induced obesity in mice (38,39), demonstrating that both incretin hormones are subject to the same agonist/antagonist paradox.
GIP Induces a Negative Energy Balance
Although there are no reports showing that GIPR agonism increases adiposity, over the last 10 years, it has become apparent that GIP can induce a negative energy balance (20,40). Overexpression of GIP in mice reduces body weight due to both reduced caloric intake and increased energy expenditure (40). Further, administration of long-acting GIPR agonists in some instances suppresses appetite and reduces body weight in obese mice (19,20). Similarly, cotreatment with GIPR agonist and GLP-1RA reduces food intake and body weight beyond that achieved by either agent given alone in obese mice (16,36), and importantly, dual GIPR and GLP-1R agonism has been demonstrated to deliver superior weight loss in T2D patients when compared with a selective GLP-1RA (16,36). The relative contribution of GIPR signaling to the success of GIPR/GLP-1R dual agonists in altering energy balance is still uncertain, as, in preclinical models, both GIPR agonists and antagonists enhance appetite suppression and weight loss mediated by GLP-1RA treatment in mice (16,33,36). These novel pharmacological approaches have, however, clearly presented GIPR as an important candidate drug target for regulating energy homeostasis. In light of these findings, there is now significant interest in understanding how GIP functions to suppress appetite and, thereby, promote negative energy balance (10). The ability of GIP-based therapeutics to reduce food intake suggests that the effects of modulating GIPR activity on body weight may be due to central GIPR signaling that recruits neural networks regulating energy balance and feeding behavior. This apparent involvement of the CNS has reinvigorated interest in GIP as a previously overlooked player in the gut-brain axis.
The GIPR Is Present in Regions of the Brain Known to Regulate Energy Balance
A prerequisite for GIP and GIP-based agonists to directly engage central pathways regulating energy homeostasis is the localization of GIPR within brain regions governing energy balance. Gipr expression has been measured in homogenates of brain tissue with quantitative PCR and visualized in whole brain slices with use of in situ hybridization, and the presence of functional GIPR has been demonstrated through radio ligand binding (41–43). To achieve a high-resolution neuroanatomical atlas of Gipr expression at the single-cell level, we recently developed a transgenic mouse line expressing Cre recombinase under the control of the Gipr promotor (Gipr-Cre) (44). In Gipr-Cre mice crossed with an EYFP Cre reporter strain, EYFP is expressed in cells transcribing Gipr, enabling the detection of Gipr-expressing (Gipr+) cells via immunostaining for EYFP, circumventing limitations of current immunohistochemical reagents. In these studies, Gipr was found to be widely expressed throughout the brain, and Gipr+ cells were mapped to several brain regions that are key to energy balance, including the paraventricular, dorsomedial, and arcuate nuclei of the hypothalamus and the area postrema (AP) and nucleus tractus solitarius of the hindbrain (44). These nuclei are implicated in the control of meal patterning, food seeking, and nausea and are essential to the regulation of energy homeostasis (reviewed in 45,46). The expression of Gipr in these regions supports a role for the GIPR signaling axis in the central control of energy balance. Moving forward, the relative contributions of different Gipr+ subpopulations (defined by their neuroanatomical location) to GIPR agonist–induced anorexia will be an important area of investigation in coming years.
Having identified putative sites of action for GIP-based pharmacology in the CNS, there is an urgent need to elucidate the mechanism(s) underpinning the metabolic benefit of GIPR agonism. Here, we present three hypotheses (see Fig. 1) that may explain how central GIPR activity affects energy balance: 1) GIP engages neurons that control appetite, 2) GIP functions as an antiemetic agent, and 3) GIP regulates access of peripheral factors to brain regions that control feeding.
Figure 1.
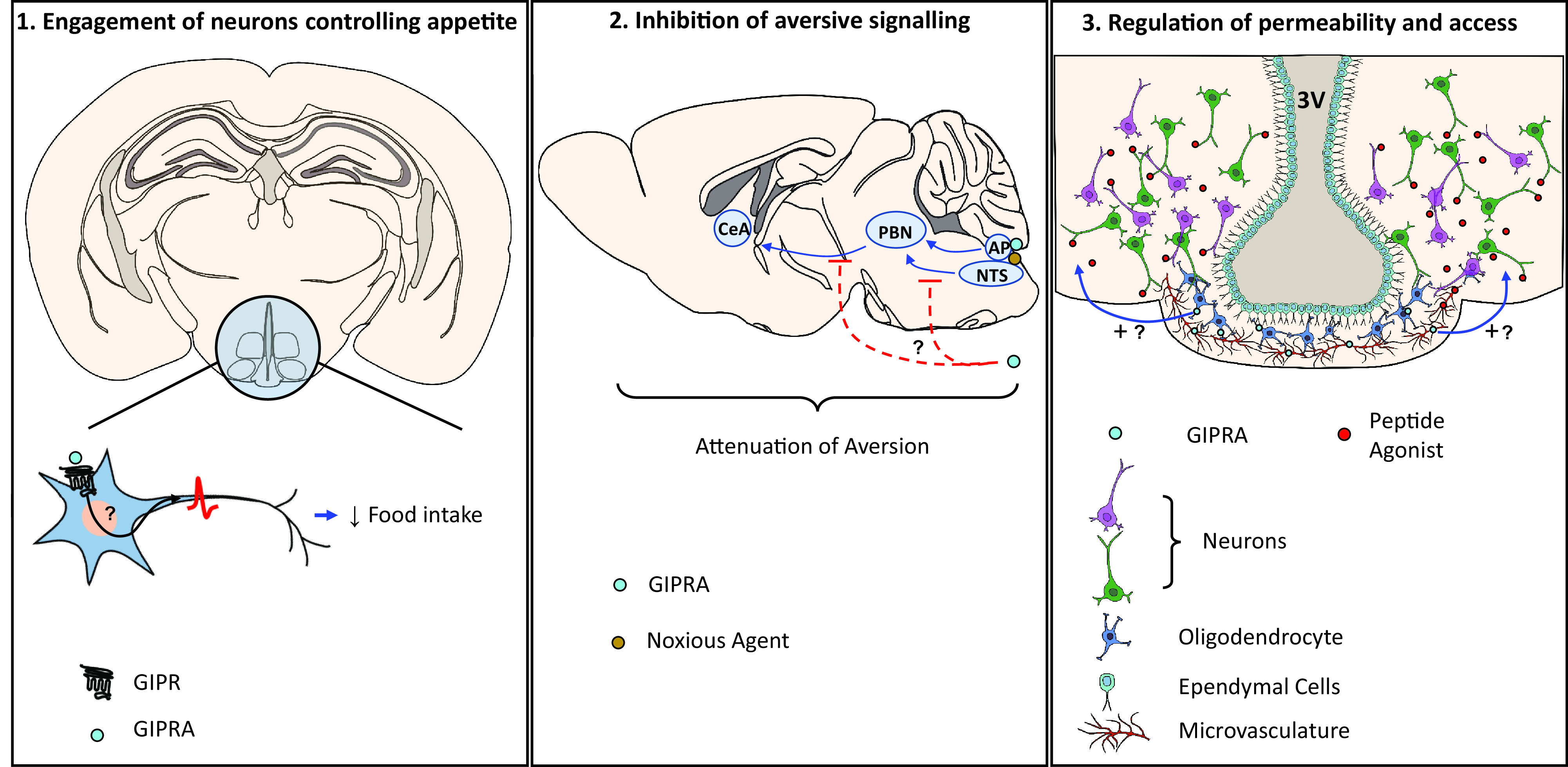
Proposed central actions of GIP and GIPR agonists. 1) Gipr is expressed by neurons in brain regions with established roles in controlling food intake, namely, the hypothalamus and the AP. Chemogenetic activation of hypothalamic Gipr+ neurons suppresses food intake in mice, confirming their potential to regulate feeding behavior. GIPR agonism may therefore recruit neurocircuitry that controls appetite and body weight regulatory pathways by directly activating Gipr+ neurons in these key regions. 2) GIPR agonists alleviate emesis and aversive behaviors induced by nauseating agents. The activation of Gipr+ circuitry may therefore integrate and/or inhibit central relays promoting nausea. 3) Gipr is expressed by oligodendrocytes and cell types constituting the neurovascular unit. As these cell types are known to regulate the permeability and perfusion of the brain parenchyma, activation of GIPR on oligodendrocytes and cells of the microvasculature may increase the access of peripheral signals—such as peptide agonists and endogenous gut hormones—to feeding centers that are normally protected by the diffusional barriers of the brain. CeA, central nucleus of the amygdala; GIPRA, GIPR agonist; NTS, nucleus tractus solitarius; PBN, parabrachial nucleus. Blue arrows indicate promotion of activation, and red lines indicate inhibition.
1) GIP Engages Neurons That Control Appetite
Peripherally administered GIPR agonists have been shown to induce neuronal activation (c-Fos) in the arcuate and ventromedial nuclei of the hypothalamus, confirming that the brain is a target organ for GIP-based pharmacology (19). While these data cannot confirm whether GIP-activated neuronal populations express Gipr, the c-Fos–activated regions in part overlap with Gipr+ nuclei (44). Given their location in the median eminence and AP, as well as brain regions directly apposing circumventricular organs (CVOs), Gipr+ cells are well placed anatomically to act as homeostatic signaling hubs within the brain, sensing circulating factors to indicate the nutritional status of an organism and relaying this information to central nuclei that regulate energy balance. Indeed, we recently highlighted the potential of Gipr+ cells to impact feeding behavior by showing that the chemogenetic activation of Gipr+ neurons in the hypothalamus reduces dark-phase food intake in both fed and fasted animals (44). Further, the importance of central Gipr+ cells in engaging appetite and body weight regulatory pathways has been demonstrated with a CNS Gipr knockout model. These studies showed that central Gipr expression is necessary for long-acting GIPR agonists to reduce body weight and food intake, the latter of which is likely due to enhanced satiation as measured by reduced meal size (19). While these reports showed that central GIPR signaling is required for weight loss, future studies are needed to elucidate the specific areas of the brain and/or neuronal populations targeted by GIPR agonists to suppress appetite and drive weight loss.
While chemogenetic and central Gipr knockout data indicate that hypothalamic Gipr+ cells integrate with networks controlling appetite, these circuits are, as yet, largely undefined. Although there is coexpression of the Gipr and Glp1r in the arcuate and dorsomedial nuclei of the hypothalamus in mice and humans, a large proportion of Gipr+ cells do not express Glp1r (44), suggesting that GIP may engage neurocircuitry that is separate from that triggered by GLP-1 for inducing changes in feeding behavior. Consistent with this, ICV administration of GIP or GLP-1 induces neuronal activity (c-Fos) in distinct as well as overlapping neurons in the hypothalamus (47). Further exemplifying that GIP and GLP-1 engage distinct neurocircuitry, in the hindbrain, Zhang et al. (48) recently demonstrated that Glp1r+ neurons of the AP are predominantly glutamatergic, whereas those expressing Gipr are principally GABAergic. Critically, however, it is clear, that both Gipr+ and Glp1r+ circuitry are necessary for the superior weight loss of GIPR/GLP-1R dual agonists, as CNS-specific knockout of Gipr expression ablates the ability of dual agonists to reduce body weight beyond what is achieved by GLP-1R agonism alone (19). Thus, although these studies demonstrated the necessity of central GIPR engagement in synergistic weight loss, further characterization of Gipr+ versus Glp1r+ neurons, as well as their separate but complementary neurocircuits, will be paramount to understanding of how GIP-based therapeutics drive pronounced weight loss.
In the hypothalamus, preliminary insight into which neuroregulatory circuits are engaged by GIP analogs is provided by single-cell RNA sequencing analyses. Transcriptomic characterization of hypothalamic Gipr+ neurons indicates that the majority are somatostatinergic and that >40% also express Avp and Cartpt—genes encoding neuromodulators with established anorectic function (44,49,50). Calcium imaging in isolated Gipr+ neurons confirmed functional expression of cell surface receptors encoded by Cckbr and Ghsr, suggesting shared circuitry among GIP, CCK, and ghrelin. GIPR activity is also likely to signal through leptin-sensing pathways, modulating hypothalamic leptin sensitivity (51). Further, initial transcriptomic profiling of Gipr+ neurons of the AP indicates that Gipr is expressed by neurons distinct from those responsive to satiety and aversive agents such as GLP-1, GDF15, and amylin (19). Collectively, these studies begin to inform a model for Gipr+ neuronal circuitry and signaling mechanisms. In the coming years, more work will be necessary to validate this model and to further clarify how Gipr+ cells contribute to the integration of peripheral signals into neural signaling cascades affecting feeding behavior.
2) GIP Functions as an Antiemetic Agent
GLP-1RAs reduce food intake by targeting neuronal circuits that suppress appetite or induce nausea and vomiting (52). Consequently, a barrier to achieving the full therapeutic potential of GLP-1–based medicines is the occurrence of dose-limiting nausea and emesis (53). Interestingly, GIPR agonists have been shown to reduce the emetic response of nauseating agents such as cisplatin and morphine in ferrets (54). These findings subsequently prompted the hypothesis that GIPR agonism enhances the weight loss profile of GLP-1–based therapeutics by negating their nauseating activity, thereby enabling higher dosing, while simultaneously potentiating appetite suppression and thereby expanding the therapeutic index and maximizing patient compliance (10). Indeed, it has recently been shown that GIPR agonism blocks the emetic effect of a long-acting GLP-1RA in the house musk shrew (55). Although speculation, whether GIP plays a physiological role in regulating nausea, and whether GIPR antagonism reduces food intake as a result of loss of GIP-dependent antiaversive action to factors like GLP-1 and GDF15, requires further investigation. The CNS control of nausea occurs via the detection of potentially toxic stimuli by AP or vagus/nucleus tractus solitarius neurons that subsequently communicate this information to the parabrachial nucleus (45), a known relay center for aversive stimuli that functions to regulate the defense against the consumption of toxic substances (56). Thus, future studies should investigate whether GIP reduces caloric intake and protects from nausea and emesis by targeting the same and/or distinct neuronal circuits. The finding that GIPR agonism blocks GLP-1–mediated nausea while simultaneously enhancing appetite suppression could have clinical implications for GLP-1 pharmacotherapy. Further investigation should also explore the potential value of GIPR agonism used in combination with other weight loss agents similarly hindered by nausea-dependent tolerability issues in nonhuman primates and man. Additional research is needed to validate this intriguing hypothesis and to establish whether the antiemetic effects of GIPR agonism represent a purely pharmacological phenomenon or whether GIP acts physiologically to engage aversive pathways.
3) GIP Regulates Access of Peripheral Factors to Brain Regions That Control Feeding
A major challenge of agents designed to suppress appetite is gaining access to their cognate receptors located in areas of the brain implicated in regulating energy homeostasis. GLP-1RAs have been reported to access the CNS via CVOs, with limited permeability to brain regions that are fully protected by the blood-brain barrier (57,58). Therefore, enhancing the permeability of the blood-brain barrier and diffusion barriers of the brain offers potential as a strategy to maximize weight loss that is induced by GLP-1R agonism (57,58). Notably, the passage of circulating factors from CVOs into the surrounding parenchyma and neighboring ventricular spaces is policed by an assembly of glia and vascular cells (59,60). Following single-cell RNA sequencing analysis of purified Gipr+ cells from the hypothalamus, we found that Gipr is expressed in oligodendrocytes—a cell type that is proposed to regulate access of peripheral signals to neuronal populations controlling appetite within the mediobasal hypothalamus (61)—as well as in cell types that make up the neurovascular unit (44). Further, two recent transcriptomic surveys of cells from the hindbrain confirmed that Gipr is expressed in oligodendrocytes in the dorsal vagal complex and that these cells are highly responsive to nutritional state (62,63). It is therefore tempting to speculate that nonneuronal Gipr+ cells may affect central perfusion and permeability and that, potentially, GIPR agonism may enhance the weight loss profile of GLP-1RAs by facilitating greater access to known target areas within the hypothalamus and brainstem and possibly allowing access to neuronal populations deeper within the brain that regulate energy balance. However, at present this proposed mechanism is based solely on gene expression data and further work is needed to provide concrete physiological data in support of this hypothesis.
Future Perspectives
A decade on from the first study proposing that GIP/GLP-1 combinatorial therapies may reduce body weight more effectively than GLP-1RAs alone, we have yet to fully elucidate the mechanisms of action of this promising therapeutic mechanism. Given the success of GIPR/GLP-1R dual agonists in clinical trials, future diabetes therapies leveraging elements of the GIPR signaling axis are sure to follow. To understand the superior efficacy of this candidate drug class, and therefore to continue building on its success, the mechanism by which GIPR signaling influences energy balance must be fully deciphered. Importantly, collective efforts in recent years implicate central GIPR signaling pathways in mediating the effect of dual agonism on weight loss. Although our understanding of the central mechanisms that are engaged by the GIPR axis is still in its nascence, information garnered from preclinical models using a combination of pharmacological and genetic approaches is beginning to provide a working model. As such, we propose that therapeutic GIP analogs modulate the CNS through multiple pathways to induce a negative energy balance, directly engaging neurons that integrate with circuitry governing appetite and nausea and affecting the access and permeability of areas of the brain to peripheral factors through glial and vascular cells. Future efforts will further address and build on these mechanistic themes. It is without doubt that the importance of GIPR signaling in the CNS will remain an active and fruitful area of investigation.
Article Information
Funding. Research in the laboratory of F.M.G. and F.R. is supported by the Medical Research Council (MRC_MC_UU_12012/3) and the Wellcome Trust (106262/Z/14/Z and 106263/Z/14/Z).
Duality of Interest. A.E.A. is supported by Eli Lilly and Company. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. R.J.S. and A.E.A. researched data and wrote the manuscript. K.W.S., F.M.G., and F.R. contributed to reviewing and editing the manuscript. A.E.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this work were presented at the 81st Scientific Sessions of the American Diabetes Association, virtual meeting, 25–29 June 2021.
Footnotes
R.J.S. and A.E.A. contributed equally.
References
- 1. Wilding JP. The importance of weight management in type 2 diabetes mellitus. Int J Clin Pract 2014;68:682–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Global Obesity Observatory . 2020. Obesity: missing the 2025 global targets. Accessed 15 March 2021. Available from https://www.worldobesity.org/resources/resource-library/world-obesity-day-missing-the-targets-report
- 3. Locke AE, Kahali B, Berndt SI, et al.; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium . Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 2018;27:740–756 [DOI] [PubMed] [Google Scholar]
- 5. Wilding JPH, Batterham RL, Calanna S, et al.; STEP 1 Study Group . Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med 2021;384:989. [DOI] [PubMed] [Google Scholar]
- 6. Müller TD, Finan B, Bloom SR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab 2019;30:72–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sikirica MV, Martin AA, Wood R, Leith A, Piercy J, Higgins V. Reasons for discontinuation of GLP1 receptor agonists: data from a real-world cross-sectional survey of physicians and their patients with type 2 diabetes. Diabetes Metab Syndr Obes 2017;10:403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gimeno RE, Briere DA, Seeley RJ. Leveraging the gut to treat metabolic disease. Cell Metab 2020;31:679–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sloop KW, Briere DA, Emmerson PJ, Willard FS. Beyond glucagon-like peptide-1: is G-protein coupled receptor polypharmacology the path forward to treating metabolic diseases? ACS Pharmacol Transl Sci 2018;1:3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Samms RJ, Coghlan MP, Sloop KW. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab 2020;31:410–421 [DOI] [PubMed] [Google Scholar]
- 11. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018;392:2180–2193 [DOI] [PubMed] [Google Scholar]
- 12. Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009;52:289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gasbjerg LS, Helsted MM, Hartmann B, et al. Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes 2019;68:906–917 [DOI] [PubMed] [Google Scholar]
- 14. Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002;8:738–742 [DOI] [PubMed] [Google Scholar]
- 15. Gasbjerg LS, Gabe MBN, Hartmann B, et al. Glucose-dependent insulinotropic polypeptide (GIP) receptor antagonists as anti-diabetic agents. Peptides 2018;100:173–181 [DOI] [PubMed] [Google Scholar]
- 16. Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med 2013;5:209ra151. [DOI] [PubMed] [Google Scholar]
- 17. Gault VA, Porter DW, Irwin N, Flatt PR. Comparison of sub-chronic metabolic effects of stable forms of naturally occurring GIP(1-30) and GIP(1-42) in high-fat fed mice. J Endocrinol 2011;208:265–271 [DOI] [PubMed] [Google Scholar]
- 18. Nørregaard PK, Deryabina MA, Tofteng Shelton P, et al. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab 2018;20:60–68 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Q, Delessa CT, Augustin R, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab 2021;33:833–844.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mroz PA, Finan B, Gelfanov V, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab 2019;20:51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frayn KN, Karpe F. Regulation of human subcutaneous adipose tissue blood flow. Int J Obes 2014;38:1019–1026 [DOI] [PubMed] [Google Scholar]
- 22. Lardinois CK, Starich GH, Mazzaferri EL. The postprandial response of gastric inhibitory polypeptide to various dietary fats in man. J Am Coll Nutr 1988;7:241–247 [DOI] [PubMed] [Google Scholar]
- 23. Rudovich N, Kaiser S, Engeli S, et al. GIP receptor mRNA expression in different fat tissue depots in postmenopausal non-diabetic women. Regul Pept 2007;142:138–145 [DOI] [PubMed] [Google Scholar]
- 24. Beck B, Max JP. Gastric inhibitory polypeptide enhancement of the insulin effect on fatty acid incorporation into adipose tissue in the rat. Regul Pept 1983;7:3–8 [DOI] [PubMed] [Google Scholar]
- 25. Wasada T, McCorkle K, Harris V, Kawai K, Howard B, Unger RH. Effect of gastric inhibitory polypeptide on plasma levels of chylomicron triglycerides in dogs. J Clin Invest 1981;68:1106–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beaudry JL, Kaur KD, Varin EM, et al. Physiological roles of the GIP receptor in murine brown adipose tissue. Mol Metab 2019;28:14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Killion EA, Chen M, Falsey JR, et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat Commun 2020;11:4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ebert R, Nauck M, Creutzfeldt W. Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm Metab Res 1991;23:517–521 [DOI] [PubMed] [Google Scholar]
- 29. Asmar M, Asmar A, Simonsen L, et al. The gluco- and liporegulatory and vasodilatory effects of glucose-dependent insulinotropic polypeptide (GIP) are abolished by an antagonist of the human GIP receptor. Diabetes 2017;66:2363–2371 [DOI] [PubMed] [Google Scholar]
- 30. Asmar M, Arngrim N, Simonsen L, et al. The blunted effect of glucose-dependent insulinotropic polypeptide in subcutaneous abdominal adipose tissue in obese subjects is partly reversed by weight loss. Nutr Diabetes 2016;6:e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yip RG, Wolfe MM. GIP biology and fat metabolism. Life Sci 2000;66:91–103 [DOI] [PubMed] [Google Scholar]
- 32. Kanemaru Y, Harada N, Shimazu-Kuwahara S, et al. Absence of GIP secretion alleviates age-related obesity and insulin resistance. J Endocrinol 2020;245:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Killion EA, Wang J, Yie J, et al. Anti-obesity effects of GIPR agonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med 2018;10:eaat3392. [DOI] [PubMed] [Google Scholar]
- 34. Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR. Energy balance and its components: implications for body weight regulation. Am J Clin Nutr 2012;95:989–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Varol C, Zvibel I, Spektor L, et al. Long-acting glucose-dependent insulinotropic polypeptide ameliorates obesity-induced adipose tissue inflammation. J Immunol 2014;193:4002–4009 [DOI] [PubMed] [Google Scholar]
- 36. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab 2018;18:3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamont BJ, Drucker DJ. Differential antidiabetic efficacy of incretin agonists versus DPP-4 inhibition in high fat fed mice. Diabetes 2008;57:190–198 [DOI] [PubMed] [Google Scholar]
- 38. Hansotia T, Maida A, Flock G, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 2007;117:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Svendsen B, Capozzi ME, Nui J, et al. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol Metab 2020;32:44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim S-J, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS One 2012;7:e40156–e40156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaplan AM, Vigna SR. Gastric inhibitory polypeptide (GIP) binding sites in rat brain. Peptides 1994;15:297–302 [DOI] [PubMed] [Google Scholar]
- 42. Paratore S, Ciotti MT, Basille M, et al. Gastric inhibitory polypeptide and its receptor are expressed in the central nervous system and support neuronal survival. Cent Nerv Syst Agents Med Chem 2011;11:210–222 [DOI] [PubMed] [Google Scholar]
- 43. Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993;133:2861–2870 [DOI] [PubMed] [Google Scholar]
- 44. Adriaenssens AE, Biggs EK, Darwish T, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab 2019;30:987–996.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 2012;16: 296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blouet C, Schwartz GJ. Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res 2010;209:1–12 [DOI] [PubMed] [Google Scholar]
- 47. NamKoong C, Kim MS, Jang BT, Lee YH, Cho YM, Choi HJ. Central administration of GLP-1 and GIP decreases feeding in mice. Biochem Biophys Res Commun 2017;490:247–252 [DOI] [PubMed] [Google Scholar]
- 48. Zhang C, Kaye JA, Cai Z, Wang Y, Prescott SL, Liberles SD. Area postrema cell types that mediate nausea-associated behaviors. Neuron 2021;109:461–472.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pei H, Sutton AK, Burnett KH, Fuller PM, Olson DP. AVP neurons in the paraventricular nucleus of the hypothalamus regulate feeding. Mol Metab 2014;3:209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farzi A, Lau J, Ip CK, et al. Arcuate nucleus and lateral hypothalamic CART neurons in the mouse brain exert opposing effects on energy expenditure. eLife 2018;7:e36494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaneko K, Fu Y, Lin HY, et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J Clin Invest 2019;129:3786–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology 2012;62:1916–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bettge K, Kahle M, Abd El Aziz MS, Meier JJ, Nauck MA. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: a systematic analysis of published clinical trials. Diabetes Obes Metab 2017;19:336–347 [DOI] [PubMed] [Google Scholar]
- 54. Asami T, Nishizawa N, Niida A, et al. Takeda Pharmaceutical Company Limited. GIP receptor activating peptide, WO/2018/181864 A1 [Google Scholar]
- 55. Geisler C, Borner T, Gaisinky J, et al. GIP receptor agonism attenuates GLP-1 receptor agonist induced nausea in rodents (Abstract). Obesity (Silver Spring) 2020;28(S2):40–18731774254 [Google Scholar]
- 56. Carter ME, Han S, Palmiter RD. Parabrachial calcitonin gene-related peptide neurons mediate conditioned taste aversion. J Neurosci 2015;35:4582–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest 2014;124:4473–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gabery S, Salinas CG, Paulsen SJ, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2020;5:133429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neuwelt EA, Bauer B, Fahlke C, et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci 2011;12:169–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mullier A, Bouret SG, Prevot V, Dehouck B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol 2010;518:943–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kohnke S, Lam B, Buller, et al. Nutritional signals rapidly activate oligodendrocyte differentiation in the adult hypothalamic median eminence. 1 September 2019 [preprint]. bioRxiv:751198 [Google Scholar]
- 62. Dowsett GKC, Lam BYH, Tadross J, et al. A survey of the mouse hindbrain in the fed and fasted state using single-nucleus RNA sequencing. 12 March 2021 [preprint]. bioRxiv:2021.03.11.434948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ludwig MQ, Cheng W, Gordian D, et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat Metab 2021;3:530–545 [DOI] [PMC free article] [PubMed] [Google Scholar]