

Abstract
We have previously demonstrated a novel role of bone morphogenic protein 4 (BMP4) in inducing NOX1-dependent endothelial nitric oxide synthase (eNOS) uncoupling, endothelial dysfunction, and inflammatory activation in type 2 diabetes mellitus (T2DM). However, how BMP4 activates NOX1 and whether targeting the new mechanistic pathway revealed is effective in preserving endothelial function in T2DM remains unclear. In this study, we observed that BMP4 induced a marked, time-dependent increase in physiological binding between TLR2 and NOX1 in aortic endothelial cells as well as increased binding of TLR2 to NOXO1. In TLR2 knockout (Tlr2−/−) mice fed high-fat diet, body weight gain was significantly less compared with wild-type (WT) mice both in males and females. The high-fat diet–induced increases in fasting blood glucose levels, as well as in circulating insulin and leptin levels, were absent in Tlr2−/− mice. High-fat feeding induced increases in overall fat mass, and in fat mass of different pockets were abrogated in Tlr2−/− mice. Whereas energy intake was similar in high-fat–fed WT and Tlr2−/− mice, TLR2 deficiency resulted in higher energy expenditure attributable to improved physical activity, which was accompanied by restored skeletal muscle mitochondrial function. In addition, TLR2 deficiency recoupled eNOS, reduced total superoxide production, improved H4B and NO bioavailabilities in aortas, and restored endothelium-dependent vasorelaxation. Collectively, our data strongly indicate that TLR2 plays important roles in the development of metabolic features of T2DM and its related endothelial/vascular dysfunction. Therefore, targeting TLR2 may represent a novel therapeutic strategy for T2DM, obesity, and cardiovascular complications via specific inhibition of NOX1.
Introduction
Diabetes is increasingly prevalent worldwide, and has become a major public health challenge (1). According to the U.S. National Diabetes Statistics Report 2020, >34 million Americans have diabetes, and an additional 88 million people likely have prediabetes (2). Cardiovascular complications are the major cause of mortality in patients with diabetes (3). Of note, oxidative stress has been established to play an important role in the development and progression of diabetes and its cardiovascular complications (4–6). Accumulating evidence has demonstrated that endothelial dysfunction induced by oxidative stress is a critical and initiating factor in the pathogenesis of diabetic vascular diseases (7–11). However, the detailed molecular mechanisms remain to be fully understood.
We and others have shown that activation of NADPH oxidase (NOX) is responsible for increased reactive oxygen species (ROS) production in vascular diseases, such as hypertension (12–14), aortic aneurysms (15), atherosclerosis (16), and diabetic vascular complications (17–19), as well as in cardiac diseases, including ischemia-reperfusion injury of the heart (20), heart failure (21), and cardiac arrhythmias (22,23). Our previous studies have shown that angiotensin II (Ang II) activation of NOX in type 1 diabetes mellitus (T1DM) leads to initial superoxide production, which consequently oxidizes endothelial nitric oxide synthase (eNOS) cofactor tetrahydrobiopterin (H4B) to result in eNOS uncoupling as well as eNOS uncoupling–dependent endothelial dysfunction (9,17,18,24). Blocking Ang II signaling in vivo with an Ang II type 1 receptor antagonist or an ACE inhibitor attenuated NOX activity and eNOS uncoupling in streptozotocin (STZ)–treated T1DM mice (17). We have further demonstrated that specific activation of NOX isoform 1 (NOX1) precedes eNOS uncoupling in STZ-induced T1DM mice (18), which is dependent on Ang II signaling. In contrast, our data have revealed a differential mechanism by which bone morphogenic protein 4 (BMP4) mediates eNOS uncoupling and inflammatory protein expression in db/db T2DM mice via activation of NOX1 (19). Thus, the roles and mechanisms of NOX1 in mediating diabetic vascular complications in T2DM in relation to BMP4 signaling is important to fully understand.
Lee et al. (25) showed that Toll-like receptor 2 (TLR2) and NOX1 physically interact with each other based on evidence from a yeast two-hybrid system. TLRs play a pivotal role in the innate immune system by recognizing pathogen-associated molecular patterns derived from various microbes and endogenous damage-associated molecular patterns produced from damaged cells (26). TLRs signal through the recruitment of specific adaptor molecules, such as MyD88 and TIR domain–containing adapter-inducing interferon-β (TRIF), leading to activation of the transcription factor nuclear factor-κB and interferon regulatory factors, which dictate the outcome of innate immune responses (26). Levels of TLR2, a subclass of TLRs, are elevated in both patients with diabetes and experimental animal models of diabetes (27,28). The increased expression and activity of TLR2 promotes activation of inflammatory signaling, which in turn leads to diabetes and its complications (29). While expression of TLR2 is ubiquitous in immune cells (30), it is also expressed in cells of nonhematopoietic origin, such as vascular smooth muscle cells (31) and endothelial cells (32). However, the roles of TLR2 in initiating and facilitating vascular dysfunction in diabetes in vivo, especially via endothelial cell–originated mechanisms, have not been previously studied.
Importantly, we have previously demonstrated an intermediate role of NOX1 in vascular dysfunction in db/db T2DM mice, differentially in response to BMP4 rather than to Ang II as in T1DM (18,19). Combined with existing literature that supports physical binding between TLR2 and NOX1 by a yeast two-hybrid system (25), we hypothesized that TLR2 activation might contribute to vascular dysfunction in T2DM via activation of NOX1 specifically. Indeed, we found that BMP4 (but not Ang II) treatment induced a time-dependent increase in physiological binding between TLR2 and NOX1 in aortic endothelial cells as well as increased binding of TLR2 to NOXO1. To further elucidate a potential functional role of TLR2 in T2DM in vivo, we subjected TLR2 knockout (KO) (Tlr2−/−) mice to high-fat diet–induced T2DM/obesity. Of note, we found that TLR2 deficiency completely abrogated diabetic and obese phenotypes in response to high-fat feeding. In the meantime, TLR2 deficiency recoupled eNOS, reduced total superoxide production, restored H4B and nitric oxide (NO) bioavailabilities, and attenuated the impairment in endothelium-dependent vasorelaxation. Collectively, these data clearly demonstrate, for the first time, that TLR2 plays a remarkably important role in the development of T2DM/obesity and its associated endothelial/vascular dysfunction. Therapeutic approaches that target TLR2 may prove highly effective in treating T2DM and cardiovascular complications via inhibition of NOX1 activity.
Research Design and Methods
Cell Culture
Bovine aortic endothelial cells (BAECs) (Genlantis, San Diego, CA) were cultured in Media 199 (Thermo Fisher Scientific, Waltham, MA) containing 10% FBS to confluence, and starved in 5% media overnight before experiments.
Co-immunoprecipitation Assay
BAECs were treated with BMP4 for 0, 0.5, and 24 h and then lysed in immunoprecipitation lysis buffer (50 mmol/L Tris HCl, 150 mmol/L NaCl, 1% Nonidet P-40). Cell lysates (1 mg) were precleared with beads (Trueblot anti-goat IgG beads, #00-8844-25; Rockland, Limerick, PA) for 1 h and then centrifuged at 3,000g for 1 min. Supernatants were then incubated with 1 μg NOX1 antibody (#SC-5821; Santa Cruz Biotechnology, Dallas, TX), NOXO1 antibody (#600-401-899; Rockland, Limerick, PA), or p47phox antibody (#610354; BD Biosciences, San Jose, CA) for 24 h. Then, 25 μL beads (Trueblot goat beads, #88-1488-31; Rockland, Limerick, PA) were added for additional incubation for 24 h, and the beads were washed six times. Immunoprecipitates were eluted by adding 35 μL 1× Laemmli sample buffer, boiled for 10 min at 95°C, and centrifuged at 13,000 rpm for 5 min. The supernatant was subjected to a standard Western blotting analysis using antibodies for TLR2 (#251110, diluted 1:500; Abbiotec, Escondido, CA), NOX1 (diluted 1:500), NOXO1 (diluted 1:500), p47phox (diluted 1:250), and β-actin (#A5441, diluted 1:5,000; MilliporeSigma, St. Louis, MO).
Western Blotting
Western blot analyses were performed as previously described (17–19). In brief, freshly isolated mouse aortic samples were lysed in ice-cold Tris buffer (composition: 50 mmol/L Tris [pH 7.4], 2 mmol/L EDTA, 1 mmol/L EGTA, 50 mmol/L NaF, 10 mmol/L Na4P2O7, 1 mmol/L Na3VO4, 1 mmol/L dithiothreitol, and 1 mmol/L phenylmethylsulfonyl fluoride) containing 1% Triton X-100, 0.1% SDS, and 10 μL/mL protease inhibitor cocktail (#P2714; MilliporeSigma). Equal amounts of total protein (35 μg) were separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat milk and incubated overnight with the primary antibodies for TLR2, eNOS (#610297, diluted 1:1,000; BD Biosciences, San Jose, CA), or β-actin, and the membranes were washed and further incubated with horseradish peroxidase–conjugated secondary antibodies (Bio-Rad Laboratories, Hercules, CA) at room temperature for 1 h. The immune complexes were visualized by enhanced chemiluminescent methods, and the band intensity was quantified using Image J software (National Institutes of Health).
Animal Studies Using WT and TLR2 KO Mice
All animals and experimental procedures were approved by the institutional animal care and use committee at the University of California, Los Angeles. Breeders of mice deficient in TLR2 (Tlr2−/−, strain B6.129-Tlr2tm1kir/J) were purchased from The Jackson Laboratory (Bar Harbor, ME) in a C57BL/6 genetic background and bred in-house for experimental use. Studies were carried out using both Tlr2−/− male and female mice and age-matched wild-type (WT) C57BL/6 mice obtained from Charles River Laboratories (Wilmington, MA). Before experimentation, animals were kept in ventilated cages with free access to water and standard chow and cared for by Division of Laboratory Animal Medicine staff until 8 weeks old. Mice were randomly divided into two dietary groups and fed either regular chow (#5053, 13.2% fat; LabDiet, St. Louis, MO) or high-fat diet (#TD.88137, 42.0% fat; Harlan Laboratories, Madison, WI) for 6 weeks.
Genotyping of Tlr2−/− Mice
The genotyping of Tlr2−/− mice was carried out according to the instructions provided by The Jackson Laboratory, using the primers for common (5′-CTT CCT GAA TTT GTC CAG TAC A-3′), mutant (5′-GGG CCA GCT CAT TCC TCC CAC-3′), and WT (5′-ACG AGC AAG ATC AAC AGG AGA-3′) animals. Mouse genomic DNA was isolated using buffer containing 100 mmol/L Tris-HCl (pH 8.5), 5 mmol/L EDTA (pH 8.0), 200 mmol/L NaCl, and 0.2% SDS. PCR products were separated on 1.5% agarose gel, with a 334-base pair band indicating the presence of mutant allele. A 499-base pair product from PCR indicates WT allele (Supplementary Fig. 1).
Analyses of Fasting Blood Glucose Level and Plasma Insulin and Leptin Levels
All mice were fasted for 14 h before weekly collection of blood samples from the tail vein (250 μL). Fasting blood glucose levels were determined at baseline before initiation of regular chow or high-fat diet feeding and on a weekly basis thereafter using the OneTouch Ultra blood glucose meter (LifeScan, Milpitas, CA). Likewise, plasma insulin and leptin levels were analyzed using an Ultra-Sensitive Rat Insulin ELISA kit and a mouse leptin ELISA kit (Crystal Chem, Elk Grove Village, IL), respectively, at baseline and on a weekly basis (17–19).
Assessment of Body Weight and Body Composition
Body weight of individual animals was measured weekly until the end of the experimental protocol. Body composition (fat mass and lean mass) in live animals were analyzed by a rodent EchoMRI (Echo Medical Systems, Houston, TX), as described previously (33). The fat mass and lean mass gains were calculated by comparing with data at week 0. The subcutaneous, perivascular, epididymal, and retroperitoneal fat were isolated and weighed at the end of the experiment.
Metabolic Analysis
Food and water intake and physical activity were measured by the comprehensive laboratory animal monitoring system (Oxymax-CLAMS; Columbus Instruments Inc., Columbus, OH). Animals were housed individually in chambers with free access to food and water. Intake of food and water and physical activity were monitored for 3 days. Energy intake was calculated as 3.1 kcal/g food for normal diet and 4.5 kcal/g food for high-fat diet (34).
Mitochondrial Swelling Analysis
Isolation of mitochondria from mouse skeletal muscle was conducted by differential centrifugation as we previously described (20). Freshly isolated mitochondria were mixed in buffer containing 250 mmol/L sucrose and 10 mmol/L Tris (pH 7.4). The mixture was then incubated with 5 mmol/L succinate for 1 min at room temperature and then with 250 mmol/L of CaCl2. Absorbance at 540 nm was measured immediately for 20 min at 1-min intervals. Swelling was measured as a decrease in absorbance over time.
Electron Spin Resonance Detection of Mitochondrial Superoxide Production
Freshly isolated mitochondria were incubated with freshly prepared, superoxide-specific spin trap methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, 500 μmol/L; Alexis Corporation) solution containing diethyldithiocarbamic acid (DETC, 5 μmol/L; MilliporeSigma) and deferoxamine (25 μmol/L; MilliporeSigma). The mixture was then loaded into a glass capillary (Kimble, 71900-50; Thermo Fisher Scientific, Waltham, MA) for detection of superoxide signal in the presence or absence of 20 units/mL polyethylene glycol–conjugated superoxide dismutase (MilliporeSigma) using an electron spin resonance (ESR) spectrometer (eScan; Bruker, Billerica, MA) as we previously published (17–19,34–39). The ESR settings used were the following: center field 3479.00 G, sweep width 9.00 G, microwave frequency 9.79 GHz, microwave power 21.02 mW, modulation amplitude 2.47 G, 512 points of resolution, and receiver gain 1,000.
DHE Imaging of Aortic Superoxide Production
Freshly isolated mouse aortas were immediately embedded in optimal cutting temperature embedding compound (Thermo Fisher Scientific), frozen at −20°C, and then cut transversely into 5 μm sections. Freshly prepared frozen sections of aortic rings were incubated with dihydroethidium (DHE) (2 μmol/L; MilliporeSigma) for 1 h in the presence or absence of NOS inhibitor l-NG-nitro-l-arginine methyl ester (l-NAME) (100 μmol/L; MilliporeSigma) and then rinsed of excess reagent. The fluorescent images were captured using a Nikon A1R confocal microscope (Nikon, Tokyo, Japan).
ESR Determination of Aortic Superoxide Production and eNOS Uncoupling Activity
Aortic superoxide production was determined by ESR as we previously published (17–19,34–39). Briefly, freshly isolated aortas were cleaned of connecting tissues and fat in ice-cold modified Krebs/HEPES buffer (composition: 99 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.0 mmol/L KH2PO4, 1.9 mmol/L CaCl2, 25 mmol/L NaHCO3, 11.1 mmol/L glucose, and 20 mmol/L NaHEPES) and cut into 3-mm rings. The superoxide-specific spin trap CMH (1 mmol/L) solution was freshly prepared in nitrogen gas–bubbled Krebs/HEPES buffer containing 5 μmol/L DETC and 25 μmol/L deferoxamine. Aortic rings were mixed with spin trap solution, loaded into a glass capillary, and assayed for superoxide production using an ESR spectrophotometer by taking the difference in the presence or absence of superoxide dismutase (20 units/mL). To determine eNOS uncoupling activity, measurements were made with the addition of l-NAME (100 μmol/L). The ESR settings used were the following: center field 3479.00 G, sweep width 9.00 G, microwave frequency 9.79 GHz, microwave power 21.02 mW, modulation amplitude 2.47 G, 512 points of resolution, and receiver gain 1,000.
Determination of Aortic H4B Bioavailability Using High-Performance Liquid Chromatography
Aortic H4B bioavailability was determined using high-performance liquid chromatography (HPLC) as we previously published (18–20,35). In brief, freshly isolated aortas were lysed in H4B lysis buffer (0.1 mol/L phosphoric acid, 1 mmol/L EDTA, 10 mmol/L DL-dithiothreitol) and centrifuged at 12,000g for 3 min, and the lysates were subjected to differential oxidation in acidic (0.2 mol/L trichloroacetic acid with 2.5% I2 and 10% KI) and alkalytic (0.1 mol/L NaOH with 0.9% I2 and 1.5% KI) solutions. After centrifugation, 35 μL of the supernatant was injected into an HPLC system equipped with a fluorescence detector (Shimadzu America Inc., Carlsbad, CA). Excitation and emission wavelengths of 350 nm and 450 nm, respectively, were used to detect H4B levels. H4B concentration was calculated to an H4B standard curve (#T4425; MilliporeSigma) as previously described (18–20,35).
ESR Determination of Aortic NO Bioavailability
Aortic NO production was determined by ESR as we previously published (17–19,34–39). Freshly isolated aortic rings (2 mm) were incubated with freshly prepared NO-specific spin trap Fe2+(DETC)2 (0.5 mmol/L) in modified Krebs/HEPES buffer at 37°C for 60 min in the presence of calcium ionophore A23187 (10 μmol/L). After incubation, the aortic segments were snap frozen in liquid nitrogen and loaded into a finger Dewar for analysis with an ESR spectrophotometer. The instrument settings used were the following: center field 3440.00 G, sweep width 100.00 G, microwave frequency 9.79 GHz, microwave power 13.26 mW, modulation amplitude 9.82 G, 512 points of resolution, and receiver gain 356.
Assessment of Endothelium-Dependent and Endothelium-Independent Vasorelaxation
Endothelium-dependent and -independent vasorelaxation of aortic rings from high-fat diet–fed WT and Tlr2−/− mice was assessed using a Multi Wire Myograph System (Model 620M; Danish Myo Technology, Ann Arbor, MI). In brief, freshly prepared aortic rings (2 mm) were placed in a chamber containing physiological saline solution (PSS) bicarbonate buffer (composition: 130 mmol/L NaCl, 4.7 mmol/L KCl, 1.18 mmol/L KH2PO4, 1.17 mmol/L MgSO4, 14.9 mmol/L NaHCO3, 5 mmol/L d-glucose, 0.026 mmol/L EDTA, and 1.6 mmol/L CaCl2), aerated with a mixture of 95% O2/5% CO2, and maintained at 37°C for 30 min. After being kept under 12 mN tension for 5 min, the rings were then stimulated twice with 60 mmol/L potassium in PSS bicarbonate buffer (composition: 74.7 mmol/L NaCl, 60 mmol/L KCl, 1.18 mmol/L KH2PO4, 1.17 mmol/L MgSO4, 14.9 mmol/L NaHCO3, 5 mmol/L d-glucose, 0.026 mmol/L EDTA, and 1.6 mmol/L CaCl2) and then washed with PSS. After equilibration, the rings were precontracted with 1 μmol/L phenylephrine. Once the vessels reached a steady-state contraction, increasing concentrations of acetylcholine (1 nmol/L–1 μmol/L) were administered, and the response to each concentration was recorded. To evaluate endothelium-independent vasorelaxation, increasing doses of sodium nitroprusside (#567538, 1 nmol/L–1 μmol/L; Millipore Sigma) were applied to precontracted aortas with 1 μmol/L phenylephrine. Data acquisition was performed using Lab-Trax 4 hardware and Data-Trax 2 software (World Precision Instruments, Sarasota, FL).
Statistical Analysis
All data are presented as mean ± SEM and analyzed using GraphPad Prism 9.0 software. The statistical comparisons between two groups were analyzed by unpaired Student t test. The statistical significance among multiple groups was analyzed by one-way ANOVA with a Newman-Keuls post hoc test. In all of the analyses, P < 0.05 was considered statistically significant.
Data and Resource Availability
All data generated and/or analyzed for this study are available from the corresponding author upon reasonable request.
Results
BMP4 Treatment Induced TLR2 Interaction With NOX1 and NOXO1 in Aortic Endothelial Cells
In view of the potential physiological significance of TLR2 interaction with NOX1, we first examined whether BMP4, known to induce vascular dysfunction in T2DM via activation of NOX1 (19), is able to induce increased binding activity between TLR2 and NOX1. BAECs were treated with BMP4 (50 ng/mL) for 30 min and 24 h and then harvested for co-immunoprecipitation with antibodies for TLR2 and NOX1. As shown in Fig. 1A and B, BMP4 treatment induced a marked, time-dependent increase in physiological binding between TLR2 and NOX1, indicating a potential activating role of TLR2 on NOX1. Stimulation of BAECs with Ang II, however, had no effect on TLR2 and NOX1 binding activity (Fig. 1C and D). We have also examined binding activity between TLR2 and other regulatory partners of NOX1, NOXO1 and p47phox (18). We found that BMP4 treatment also time dependently increased binding of TLR2 to NOXO1 (Fig. 1E and F), but not to p47phox (Supplementary Fig. 2).
Figure 1.

BMP4 induces increased interaction between TLR2 and NOX1/NOXO1 in aortic endothelial cells. BAECs were treated with BMP4 (50 ng/mL) or Ang II (100 nmol/L) for 30 min and 24 h, and the cells were harvested for co-immunoprecipitation (co-IP). The co-IP of TLR2 by NOX1 or NOXO1 was determined by Western blotting and normalized to β-actin. A: Representative co-IP blots of TLR2 by anti-NOX1 in BMP4-stimulated endothelial cells. B: Grouped densitometric data of co-IP results in A (n = 5–6). C: Representative co-IP blots of TLR2 by anti-NOX1 in Ang II–stimulated endothelial cells. D: Grouped densitometric data of co-IP results in C (n = 5–6). E: Representative co-IP blots of TLR2 by anti-NOXO1 in BMP4-stimulated endothelial cells. F: Grouped densitometric data of co-IP results in E (n = 3). Data are shown as mean ± SEM. *P < 0.05; **P < 0.01. IB, immunoblot.
High-Fat–Fed Tlr2−/− Mice Had Less Body Weight Gain
The metabolic features of Tlr2−/− mice after high-fat feeding were phenotyped. As shown is Fig. 2A, the body weight of Tlr2−/− mice was significantly lighter than WT mice after 6 weeks of high-fat feeding. Of note, the body weight of Tlr2−/− mice was not different from WT mice at baseline (Supplementary Fig. 3). The body weight gain in male Tlr2−/− mice (8.0 ± 0.7 g) was only ∼50% of that of male WT mice (15.4 ± 0.6 g) (Fig. 2B and C). A similar trend in body weight gain (3.4 ± 0.4 vs. 7.3 ± 0.8 g for Tlr2−/− vs. WT mice) was observed in female mice (Fig. 2D and E).
Figure 2.

TLR2 KO attenuates high-fat diet feeding–induced increases in body weight and body weight gain. Eight-week-old WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. Body weight of individual animals was measured weekly until end of the experimental protocol. A: Representative pictures of male WT and Tlr2−/− mice fed with regular chow or high-fat diet for 6 weeks. B: Grouped data of weekly changes in body weight over the 6-week study period in WT and Tlr2−/− male mice fed high-fat diet (n = 7–8). C: Grouped data of weekly body weight gain in WT and Tlr2−/− male mice fed high-fat diet (n = 7–8). D: Grouped data of weekly changes in body weight over the 6-week study period in WT and Tlr2−/− female mice fed high-fat diet (n = 7). E: Grouped data of weekly body weight gain in WT and Tlr2−/− female mice fed high-fat diet (n = 7). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT + regular chow; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. WT + high-fat diet.
High-Fat–Fed Tlr2−/− Mice Had Lower Blood Glucose and Plasma Insulin Levels
To further investigate the role of TLR2 in glucose homeostasis, we measured levels of blood glucose and plasma insulin in high-fat–fed WT and Tlr2−/− mice weekly. High-fat feeding significantly increased fasting blood glucose and plasma insulin levels in a time-dependent manner in WT mice starting from week 2 (Fig. 3A and B), which were completely attenuated in Tlr2−/− mice. These results indicate that besides limiting body weight gain, TLR2 deficiency can alleviate high-fat feeding–induced increases in blood glucose and plasma insulin levels.
Figure 3.

TLR2 KO decreases blood glucose and plasma insulin levels in response to high-fat diet feeding. Eight-week-old male WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. All the mice were fasted for 14 h before blood collection. Blood samples were collected from the tail vein (250 μL) at baseline and on a weekly basis thereafter after high-fat feeding for determination of blood glucose and plasma insulin levels by blood glucose meter and ELISA for rat insulin, respectively. A: Grouped data of weekly changes in blood glucose levels over the 6-week study period in WT and Tlr2−/− mice fed high-fat diet (n = 7–9). B: Grouped data of weekly changes in plasma insulin levels over the 6-week study period in WT and Tlr2−/− mice fed high-fat diet (n = 5–6). Data are shown as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT + regular chow; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. WT + high-fat diet.
High-Fat–Fed Tlr2−/− Mice Had Lower Plasma Leptin Levels and Reduced Fat Mass
The adipocyte-derived hormone leptin is an important regulator of body weight and metabolism through activation of brain leptin receptors expressed in such regions as the hypothalamus (40). In the current study, we found that plasma leptin levels (Fig. 4A) and fat mass (Fig. 4B) were markedly increased in high-fat–fed WT mice, indicating leptin resistance. However, these responses were significantly abrogated in high-fat–fed Tlr2−/− mice (Fig. 4A and B). Of note, the lean mass was not changed in both WT and Tlr2−/− mice (Fig. 4C). After 6 weeks of high-fat feeding, we harvested the mice and measured organ weights and weights of different fat pockets. As shown in Fig. 4D, there was no significant difference in the weights of heart, spleen, lung, kidney, and pancreas between regular diet– and high-fat–fed WT mice and between high-fat–fed Tlr2−/− and WT mice. Although liver size increased slightly in high-fat–fed WT mice, and the increase was somewhat relieved in Tlr2−/− mice, there was no statistical difference. The small increase in liver size might be attributed to duration of high-fat-diet feeding for 6 weeks. However, the weights of fats isolated from subcutaneous, perivascular, epididymal, and retroperitoneal pockets were substantially less in Tlr2−/− mice compared with those of WT mice (Fig. 4E).
Figure 4.

TLR2 KO abrogates high-fat diet–induced increases in plasma leptin levels, total fat mass, and fat mass of different pockets. Eight-week-old male WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. Mice were fasted for 14 h before blood collection to measure plasma leptin levels at baseline and on a weekly basis thereafter after high-fat feeding using ELISA for mouse leptin. In parallel experiments, live mice were examined for weekly body composition (fat mass and lean mass) using EchoMRI. A: Grouped data of weekly changes in plasma leptin levels over the 6-week study period in WT and Tlr2−/− mice fed high-fat diet (n = 5–6). B: Grouped data of weekly changes in fat mass over the 6-week study period in WT and Tlr2−/− mice fed high-fat diet (n = 7–8). C: Grouped data of weekly changes in lean mass over the 6-week study period in WT and Tlr2−/− mice fed high-fat diet (n = 7–8). D: Grouped data of heart, liver, spleen, lung, kidney, and pancreas weights in WT and Tlr2−/− mice fed regular chow or high-fat diet for 6 weeks (n = 5–6). E: Grouped data of fat mass of subcutaneous (Sub), perivascular (Peri), epididymal (Epi), and retroperitoneal (Retro) pockets in WT and Tlr2−/− mice fed regular chow or high-fat diet for 6 weeks (n = 7–8). Data are shown as mean ± SEM. ***P < 0.001, ****P < 0.0001 vs. WT + regular chow; ###P < 0.001, ####P < 0.0001 vs. WT + high-fat diet.
Metabolic analyses with the Oxymax-CLAMS metabolic cages indicated that Tlr2−/− mice had similar food intake as WT mice fed with either regular chow or high-fat diet (Fig. 5A). Although the water intake decreased and calorie intake increased after high-fat feeding, no difference was found between WT and Tlr2−/− mice (Fig. 5B and C). At the same time, we observed a significant restoration of physical activity in Tlr2−/− mice compared with WT mice with high-fat feeding, who had reduced physical activity, an effect that was pronounced during dark phases (Fig. 5D–F). Since skeletal muscle mitochondrial dysfunction was shown to be responsible for impaired activity as a result of obesity in our previous study (34), we examined mitochondrial swelling activity and superoxide production in high-fat–fed WT and Tlr2−/− mice. Consistent with the restoration of physical activity in high-fat–fed Tlr2−/− mice, increased mitochondrial superoxide production (Fig. 5G) and swelling activity were both completely attenuated in Tlr2−/− mice (Fig. 5H).
Figure 5.

TLR2 KO preserves physical activity in response to high-fat diet feeding, which is attributable to restored skeletal muscle mitochondrial function. Eight-week-old male WT and Tlr2−/− mice were housed individually in the chambers of the Oxymax-CLAMS animal monitoring system with free access to food and water and randomly divided into two dietary groups to receive regular chow or high-fat diet for 3 days. In parallel experiments, male mice were fed regular chow or high-fat diet for 6 weeks to freshly isolate mitochondria for analyses of superoxide production and swelling activity. A: Grouped data of food intake in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). B: Grouped data of water intake in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). C: Grouped data of calorie intake in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). D: Grouped data of locomotor activity measured continuously over 36 h in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). Time: 0 h = 6:00 p.m. and represents starting of lights off; the dark cycles are highlighted in gray shades. E: Grouped data of total locomotor activity during the dark cycle in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). F: Grouped data of total locomotor activity during the light cycle in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 6–7). G: Grouped data of mitochondrial superoxide production measured by ESR in WT and Tlr2−/− mice fed regular chow or high-fat diet for 6 weeks (n = 3–4). H: Grouped data of mitochondrial swelling activity in WT and Tlr2−/− mice fed regular chow or high-fat diet for 6 weeks (n = 3–4). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. For D, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT + regular chow; ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. WT + high-fat diet. A540, absorbance at 540 nm.
High-Fat–Fed Tlr2−/− Mice Had Reduced ROS Production and Preserved eNOS Coupling Activity
We measured superoxide production using both DHE fluorescent imaging and ESR in the presence or absence of l-NAME, an inhibitor of NOS. If eNOS is functional and coupled, its inhibition by l-NAME will increase the measured superoxide as a result of inhibition of NO production from coupled eNOS to scavenge superoxide. However, if eNOS is dysfunctional, uncoupled and producing superoxide, inhibition with l-NAME will reduce measured superoxide. As seen in Fig. 6A–C, we found from DHE imaging analysis that eNOS was uncoupled in high-fat–fed WT mice, which was completely attenuated in high-fat–fed Tlr2−/− mice. At the same time, eNOS was also found, using advanced ESR technology as we previously published (17–19,34–39), to be uncoupled. As shown in Fig. 6D and E, inhibition with l-NAME in isolated intact aorta rings reduced measured superoxide in high-fat–fed WT mice, indicating uncoupling of eNOS, while this response was completely attenuated in high-fat–fed Tlr2−/− mice. It is important to note that eNOS protein upregulation, when uncoupled, is a deteriorating response. Upregulation of eNOS protein has been observed in various models of vascular diseases, including apolipoprotein E–null mice (41,42) and deoxycorticosterone acetate-salt hypertensive mice (43). Here, we found that eNOS protein expression was upregulated in high-fat–fed WT mice, which was abrogated in high-fat–fed Tlr2−/− mice (Fig. 6F and G).
Figure 6.
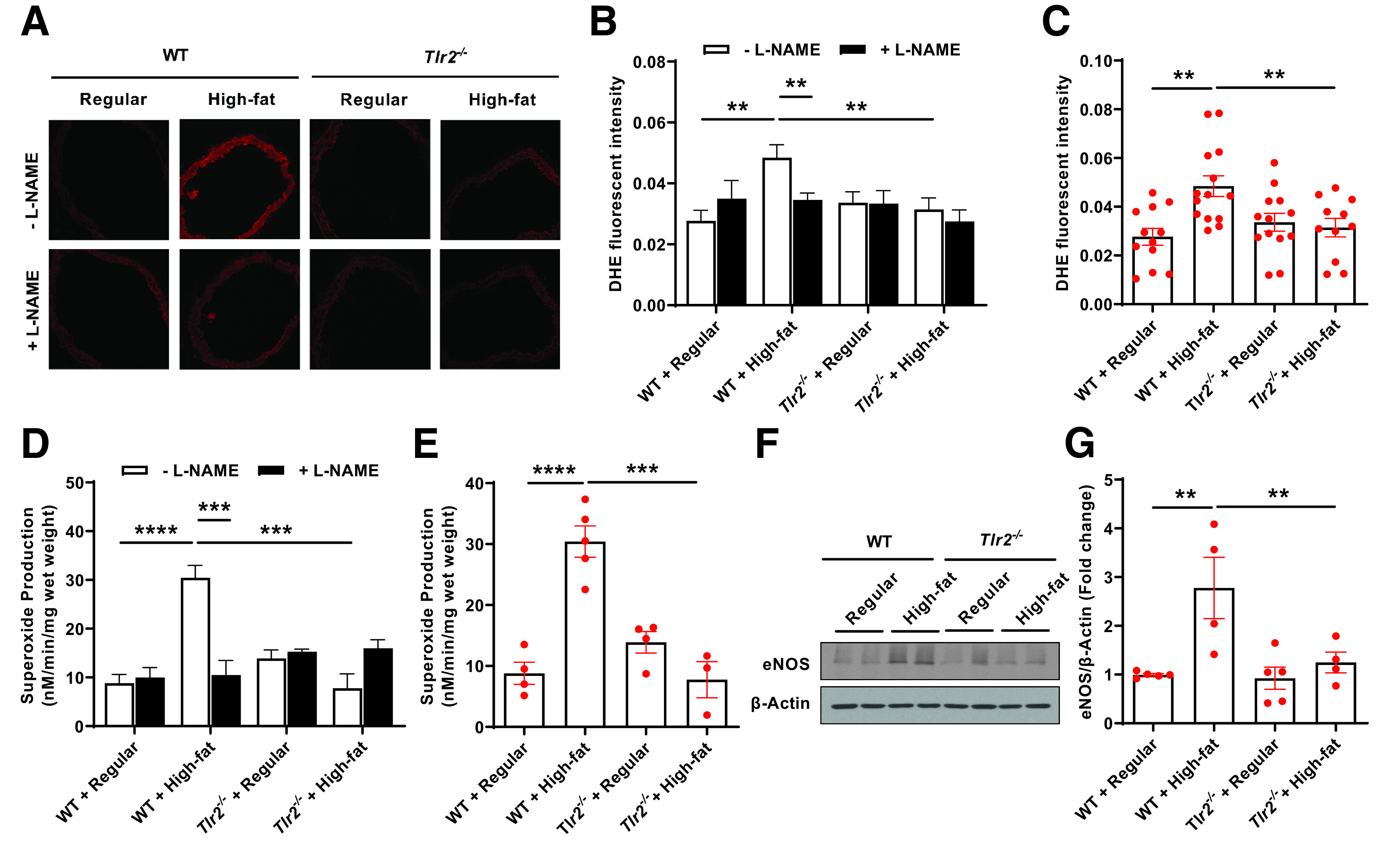
TLR2 KO attenuates high-fat diet–induced eNOS uncoupling activity and total superoxide production as determined by DHE imaging and ESR. Eight-week-old male WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. Freshly isolated aortas were subjected to DHE fluorescent imaging and ESR determination of eNOS uncoupling activity and total superoxide production, and Western blotting analysis of eNOS protein expression. A: Representative images of aortic DHE fluorescent staining in WT and Tlr2−/− mice fed regular chow or high-fat diet in the presence or absence of l-NAME. B: Grouped densitometric data of fluorescent intensities from A reflective of eNOS coupling/uncoupling status (n = 11–14). C: Grouped densitometric data of total superoxide production from A (without l-NAME) (n = 11–14). D: Grouped data of aortic superoxide production as assessed by ESR in the presence or absence of l-NAME reflective of eNOS coupling/uncoupling status (n = 3–5). E: Grouped data of aortic total superoxide production as assessed by ESR (n = 3–5). F: Representative Western blots of eNOS/β-actin in aortas of WT and Tlr2−/− mice fed regular chow or high-fat diet. G: Grouped densitometric data of eNOS expression from F (n = 4–5). Data are shown as mean ± SEM. **P < 0.01; ***P < 0.001; ****P < 0.0001.
High-Fat–Fed Tlr2−/− Mice Had Restored H4B and NO Bioavailability
eNOS uncoupling occurs consequent to a deficiency in its cofactor tetrahydrobiopterin (H4B). We also measured H4B bioavailability by HPLC as we previously published (18–20,35). The results indicate that high-fat feeding induced a deficiency in aortic H4B bioavailability, which was, however, absent in high-fat-fed Tlr2−/− mice (Fig. 7A). An important consequence of increased ROS production in vascular cells is oxidative inactivation of NO by superoxide anion, and that loss of NO triggers endothelial dysfunction to mediate pathogenesis of atherosclerosis, hypertension, and diabetic vascular disease (44). Therefore, we next measured aortic NO bioavailability in isolated aortas using ESR, as we previously published (17–19,34–39). As shown in Fig. 7B and C, we observed a significant reduction in aortic NO bioavailability in high-fat–fed WT mice, which was markedly alleviated in Tlr2−/− mice despite high-fat feeding.
Figure 7.
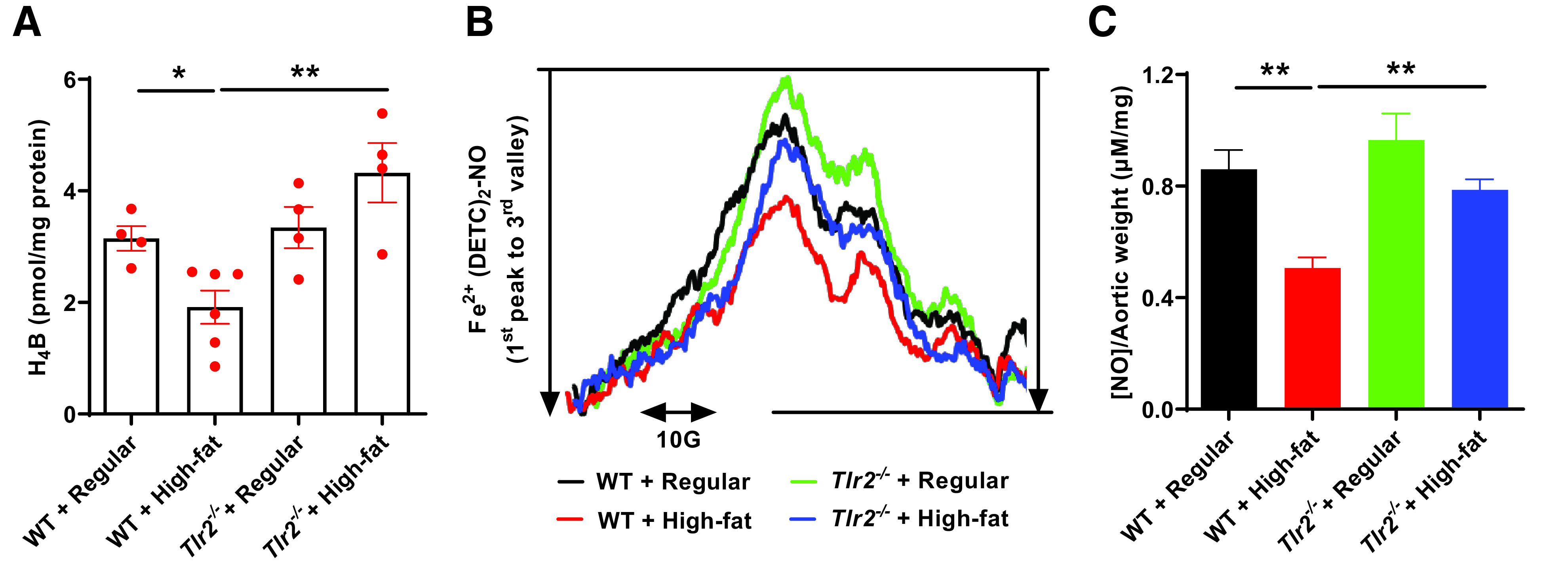
TLR2 KO restores H4B and NO bioavailabilities in response to high-fat feeding. Eight-week-old male WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. Mice were harvested for assessment of aortic H4B bioavailability and NO bioavailability using HPLC and ESR respectively. A: Grouped data of aortic H4B levels in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 4–6). B: Representative ESR spectra of NO in WT and Tlr2−/− mice fed regular chow or high-fat diet. C: Grouped data of aortic NO levels in WT and Tlr2−/− mice fed regular chow or high-fat diet (n = 5–6). Data are shown as mean ± SEM. *P < 0.05; **P < 0.01.
High-Fat–Fed Tlr2−/− Mice Had Preserved Endothelium-Dependent Vasorelaxation
Impairment in endothelium-dependent vasorelaxation is attributed to reduced NO bioavailability and is a hallmark of pathological processes of cardiovascular complications in diabetes (45). Since we observed preserved eNOS coupling activity and NO bioavailability in high-fat–fed Tlr2−/− mice, we further examined endothelium-dependent relaxation/vascular function after high-fat feeding in Tlr2−/− and WT mice. As shown in Fig. 8A, endothelium-dependent vasorelaxation induced by acetylcholine was markedly impaired in high-fat–fed WT mice, which was, however, substantially restored in high-fat–fed Tlr2−/− mice; the endothelium-independent vasorelaxation induced by sodium nitroprusside was not affected by either high-fat feeding or TLR2 deficiency (Fig. 8B). Taken together, these data implicate a novel, intermediate role of TLR2 in diabetic and obesity phenotypes and vascular dysfunction in high-fat–fed T2DM mice via activation of NOX1 to induce eNOS uncoupling, NO deficiency, and impaired endothelium-dependent vasorelaxation. This novel signaling cascade, as defined by our new observations, is illustrated in Fig. 9.
Figure 8.
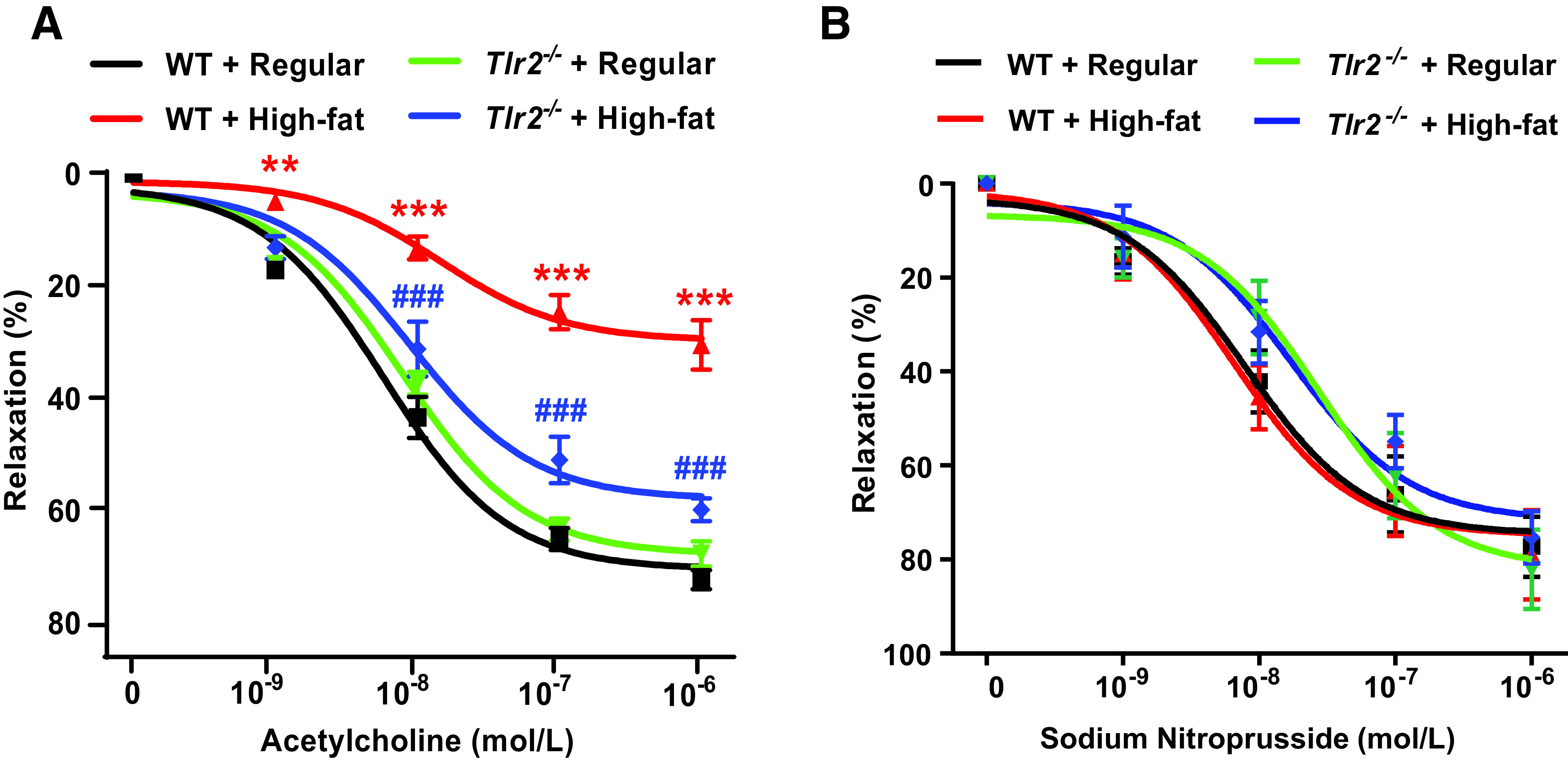
TLR2 KO reverses high-fat diet–induced impairment in endothelium-dependent vasorelaxation. Eight-week-old male WT and Tlr2−/− mice were randomly divided into two dietary groups and fed either regular chow or high-fat diet for 6 weeks. Freshly isolated aortic rings were mounted into an organ bath (Multi Wire Myograph System) under 12 mN tension for 5 min and then equilibrated by PSS bicarbonate buffer. The rings were precontracted with 1 μmol/L phenylephrine and then exposed to incremental concentrations of acetylcholine (1 nmol/L–1 μmol/L) or sodium nitroprusside (1 nmol/L–1 μmol/L) to evaluate endothelium-dependent or endothelium-independent vasorelaxation, respectively. A: Grouped data of endothelium-dependent vasorelaxation in response to acetylcholine in precontracted aortic rings (n = 10–11). Data are shown as mean ± SEM. **P < 0.01, ***P < 0.001 vs. WT + regular chow; ###P < 0.001 vs. WT + high-fat diet. B: Grouped data of endothelium-independent vasorelaxation in response to sodium nitroprusside in precontracted aortic rings (n = 5–7). Data are shown as mean ± SEM. Differences are nonsignificant among groups.
Figure 9.

A novel signaling cascade of TLR2/NOX1 in mediating BMP4-dependent endothelial/vascular dysfunction and diabetic/obesity phenotypes. Under normal conditions, NOX1 binds to p22phox on the cell membrane as the core catalytic unit, while TLR2 and NOXO1 are unbound in the cytosol. Under T2DM, increased levels of BMP4 promote TLR2 binding to NOXO1 and NOX1, resulting in activation of NOX1, oxidative stress, and inflammatory activation, which in turn lead to endothelial/vascular dysfunction. The TLR2/NOX1 axis also lies upstream of diabetic/obesity phenotypes, blocking of which can further eliminate endothelial/vascular dysfunction to prevent cardiovascular complications in diabetes. Therefore, targeting TLR2 may represent a novel therapeutic strategy for diabetes/obesity and its associated cardiovascular complications.
Discussion
The most significant finding of the current study is that TLR2 plays an important intermediate role in the development of T2DM/obesity and related vascular dysfunction via activation of NOX1: 1) BMP4 treatment induced a marked, time-dependent increase in physiological binding between TLR2 and NOX1 in aortic endothelial cells as well as increased binding of TLR2 to NOXO1; 2) TLR2 deficiency abrogated diabetic and obese phenotypes in mice fed high-fat diet; and 3) TLR2 deficiency recoupled eNOS, restored H4B and NO bioavailabilities, and preserved endothelium-dependent vasorelaxation in mice fed high-fat diet. We found that BMP4 treatment induced a physiological binding between TLR2 and NOX1 in aortic endothelial cells. Although Lee et al. (25) reported that TLR2 physically interacts with NOX1 based on a yeast two-hybrid analysis, our data for the first time confirm physiological significance of this interaction in mammalian aortic endothelial cells. At the same time, we found that BMP4 treatment also time dependently increased binding between TLR2 and NOXO1 but not with p47phox. We have previously shown that activation of NOX1 in endothelial cells requires binding of NOXO1 and p47phox (18,46), so our current results indicate that NOXO1, instead of p47phox, forms a complex with TLR2/NOX1 to participate in TLR2-dependent activation of NOX1 by BMP4. Of note, TLR2 expression was found to be increased in patients with diabetes (27,28), and in our current model of high-fat–induced T2DM mice (Supplementary Fig. 4), indicating a significant role of TLR2 in T2DM via regulation of NOX1. Importantly, we have previously shown that NOX1 is activated and responsible for endothelial dysfunction in T1DM and T2DM (17–19,34,38). Therefore, our current findings further revealed functional interaction between TLR2 and NOX1 in T2DM. We have previously shown that BMP4 specifically induces eNOS uncoupling, inflammatory protein expression, and endothelial dysfunction in a NOX1-dependent manner in db/db T2DM mice (19). However, the upstream regulators of NOX1 activation in diabetes remained previously unknown. Our current data for the first time demonstrate an upstream activating role of TLR2 on NOX1 in T2DM.
Rodent models of high-fat diet feeding are a common experimental model for studying T2DM/obesity, and its basic phenotypic characteristics are increases in body weight and fat mass (47). In the present study, we observed that after 6 weeks of high-fat feeding, body weight, body weight gain, and fat mass of Tlr2−/− mice were significantly less than that of WT mice. Our results are consistent with the findings by Kuo et al. (48), which indicate that TLR2 deficiency leads to a reduction in body weight and fat mass after a long period of high-fat feeding for 8 months. We further examined fat mass of different pockets and found that all were reduced in TLR2-deficient animals after high-fat feeding. Of note, the differences in experimental design, including high-fat diet composition and duration of feeding, might have an impact on experimental results, which likely explains the inconsistency in body weight gain with some previous data that indicated no difference in Tlr2−/− mice (49,50). Nonetheless, our findings are consistent with some classical reports that indicated reduced body weight gain in high-fat–fed Tlr2−/− mice (48).
Importantly, we used metabolic cages to examine metabolic changes in individually housed mice and found that energy intake was similarly increased by high-fat feeding in both WT and Tlr2−/− mice, suggesting that Tlr2−/− mice have normal energy absorption. Instead, the energy expenditure was increased in high-fat–fed Tlr2−/− mice because of restored physical activity, which was accompanied by restored skeletal muscle mitochondrial function featured by less mitochondrial superoxide production and less swelled mitochondria. These data indicate that Tlr2−/− mice have normal energy absorption but increased energy expenditure coming from increased physical activity, resulting in reduced fat deposition and content. Of note, the lean mass and weights of various organs after high-fat feeding were not significantly different between WT and Tlr2−/− mice, whereas there is a downward trend in liver size in Tlr2−/− mice.
In normal conditions, insulin stimulates leptin secretion from adipocytes, which in turn inhibit synthesis and secretion of insulin from pancreatic β-cells, thereby controlling the homeostasis of body weight and glucose metabolism (51). In the current study, we found that TLR2 deficiency can alleviate the increases in blood glucose and insulin levels induced by high-fat diet, which are consistent with previous observations (48). In addition, another interesting finding is that the increased circulating leptin levels by high-fat feeding in WT mice was also significantly alleviated in Tlr2−/− mice. Leptin, the product of ob/ob gene (52), is positively correlated with BMI (53) and known for its proinflammatory properties (54). Thus, based on our current results, we speculate that TLR2 deficiency leads to weaker inflammatory response in high-fat diet–fed T2DM mice. In view of our previous findings that NOX1 mediates inflammatory protein expression in db/db mice (19), the TLR2-NOX1 axis seems to play an important role in regulating inflammatory responses in T2DM via perhaps both ROS and leptin signaling.
In particular, our data revealed that TLR2 deficiency attenuated eNOS uncoupling and reduced aortic total superoxide production in T2DM mice fed high-fat diet. In addition, aortic H4B and NO bioavailabilities were completely restored in diabetic Tlr2−/− mice. More importantly, the impairment in endothelium-dependent vasorelaxation in high-fat–fed WT mice was also substantially reversed in high-fat–fed Tlr2−/− mice. We have previously shown that eNOS uncoupling is the primary source of superoxide production in the diabetic endothelium (17,38,44), whereas NOX enzymes remain active in subendothelial vascular smooth muscle cells (17). Uncoupling of eNOS represents a central mechanism by which NOX activation leads to activation of secondary enzymatic systems to produce excessive ROS to sustain oxidative stress in the pathogenesis of cardiovascular diseases (44). Endothelial dysfunction is hallmark of and a major contributor to vascular diseases in diabetes (45). Prior studies from our group have demonstrated that NOX1, but not other NOX isoforms, is selectively activated to mediate eNOS uncoupling and endothelial dysfunction in diabetes (17–19). We have previously found that BMP4 levels are elevated in db/db T2DM mice to activate NOX1 (19), whereas Ang II levels are differentially elevated in STZ-induced T1DM mice to activate NOX1 in comparison (17–19). These data indicate that mechanisms of NOX1 activation are different in T1DM and T2DM, while our present study identifies a novel upstream role of TLR2 in NOX1 activation in response to elevated BMP4 levels in T2DM. Considering the effects of TLR2 deficiency on eNOS uncoupling and endothelial dysfunction, we believe that the TLR2/NOX1 axis is critically important in mediating BMP4-dependent pathophysiological processes that lead to cardiovascular complications in T2DM.
In conclusion, our study reveals for the first time a previously undescribed role of TLR2 in activating NOX1-dependent eNOS uncoupling to result in impaired NO bioavailability and deficient endothelium-dependent vasorelaxation in T2DM, correction of which with TLR2 KO completely prevents these pathological responses. TLR2 deficiency also completely attenuated metabolic features of T2DM/obesity. In view of these novel findings, targeting TLR2 may prove to be an effective therapeutic strategy for T2DM and its cardiovascular complications.
Article Information
Funding. This study was supported by National Heart, Lung, and Blood Institute grants HL-077440, HL-088975, HL-142951, and HL-154754 to H.C.
Duality of Interest. No conflicts of interests relevant to this article were reported.
Author Contributions. Z.G. performed the experiments, analyzed data, and wrote and edited the manuscript. Y.Z., C.L., and J.Y.Y. performed the experiments, analyzed data, and wrote part of the manuscript. H.C. designed the studies and wrote, edited, revised, and finalized the manuscript. H.C. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Z.G., Y.Z., and C.L. contributed equally to this study.
This article contains supplementary material online at https://doi.org/10.2337/figshare.14754585.
References
- 1. King H, Aubert RE, Herman WH. Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 1998;21:1414–1431 [DOI] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention . National Diabetes Statistics Report, 2020. Atlanta, GA, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, 2020 [Google Scholar]
- 3. Paterson AD, Rutledge BN, Cleary PA, Lachin JM; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group . The effect of intensive diabetes treatment on resting heart rate in type 1 diabetes: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study. Diabetes Care 2007;30:2107–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J 2017;40:257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yaribeygi H, Butler AE, Barreto GE, Sahebkar A. Antioxidative potential of antidiabetic agents: a possible protective mechanism against vascular complications in diabetic patients. J Cell Physiol 2019;234:2436–2446 [DOI] [PubMed] [Google Scholar]
- 6. Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 2005;96:818–822 [DOI] [PubMed] [Google Scholar]
- 7. Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res 2005;68:26–36 [DOI] [PubMed] [Google Scholar]
- 8. Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 2001;88:E14–E22 [DOI] [PubMed] [Google Scholar]
- 9. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844 [DOI] [PubMed] [Google Scholar]
- 10. Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation 1993;88:2510–2516 [DOI] [PubMed] [Google Scholar]
- 11. Meigs JB, Hu FB, Rifai N, Manson JE. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004;291:1978–1986 [DOI] [PubMed] [Google Scholar]
- 12. Landmesser U, Cai H, Dikalov S, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002;40:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J, Youn JY, Kim AY, et al. NOX4-dependent hydrogen peroxide overproduction in human atrial fibrillation and HL-1 atrial cells: relationship to hypertension. Front Physiol 2012;3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu J, Saleh MA, Kirabo A, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest 2016;126:1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siu KL, Li Q, Zhang Y, et al. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol 2017;11:118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in pathogenesis of atherosclerosis. Circ Res 2017;120:713–735 [DOI] [PubMed] [Google Scholar]
- 17. Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 2007;56:118–126 [DOI] [PubMed] [Google Scholar]
- 18. Youn JY, Gao L, Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012;55: 2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Youn JY, Zhou J, Cai H. Bone morphogenic protein 4 mediates NOX1-dependent eNOS uncoupling, endothelial dysfunction and COX2 induction in type 2 diabetes mellitus. Mol Endocrinol 2015;29:1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siu KL, Lotz C, Ping P, Cai H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J Mol Cell Cardiol 2015;78:174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal 2013;18:1024–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youn JY, Zhang J, Zhang Y, et al. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol 2013;62:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Shimizu H, Siu KL, Mahajan A, Chen JN, Cai H. NADPH oxidase 4 induces cardiac arrhythmic phenotype in zebrafish. J Biol Chem 2014;289:23200–23208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 2003;24:471–478 [DOI] [PubMed] [Google Scholar]
- 25. Lee JH, Joo JH, Kim J, et al. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc Res 2013;99:483–493 [DOI] [PubMed] [Google Scholar]
- 26. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab 2008;93:578–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010;33:861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jialal I, Kaur H. The role of toll-like receptors in diabetes-induced inflammation: implications for vascular complications. Curr Diab Rep 2012;12:172–179 [DOI] [PubMed] [Google Scholar]
- 30. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol 2001;1:135–145 [DOI] [PubMed] [Google Scholar]
- 31. Lee GL, Chang YW, Wu JY, et al. TLR 2 induces vascular smooth muscle cell migration through cAMP response element-binding protein-mediated interleukin-6 production. Arterioscler Thromb Vasc Biol 2012;32:2751–2760 [DOI] [PubMed] [Google Scholar]
- 32. Wilhelmsen K, Mesa KR, Lucero J, Xu F, Hellman J. ERK5 protein promotes, whereas MEK1 protein differentially regulates, the Toll-like receptor 2 protein-dependent activation of human endothelial cells and monocytes. J Biol Chem 2012;287:26478–26494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhasin KK, van Nas A, Martin LJ, Davis RC, Devaskar SU, Lusis AJ. Maternal low-protein diet or hypercholesterolemia reduces circulating essential amino acids and leads to intrauterine growth restriction. Diabetes 2009;58:559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Youn JY, Siu KL, Lob HE, Itani H, Harrison DG, Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 2014;63:2344–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 2005;102:9056–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen A, Cai H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc Natl Acad Sci USA 2006;103:6530–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Youn JY, Wang T, Cai H. An ezrin/calpain/PI3K/AMPK/eNOSs1179 signaling cascade mediating VEGF-dependent endothelial nitric oxide production. Circ Res 2009;104:50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oak JH, Youn JY, Cai H. Aminoguanidine inhibits aortic hydrogen peroxide production, VSMC NOX activity and hypercontractility in diabetic mice. Cardiovasc Diabetol 2009;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H, Li Q, Zhang Y, et al. Novel treatment of hypertension by specifically targeting e2f for restoration of endothelial dihydrofolate reductase and eNOS function under oxidative stress. Hypertension 2019;73:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1995;1:1311–1314 [DOI] [PubMed] [Google Scholar]
- 41. Laursen JB, Somers M, Kurz S, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001;103:1282–1288 [DOI] [PubMed] [Google Scholar]
- 42. Siu KL, Miao XN, Cai H. Recoupling of eNOS with folic acid prevents abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E null mice. PLoS One 2014;9:e88899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Youn JY, Wang T, Blair J, et al. Endothelium-specific sepiapterin reductase deficiency in DOCA-salt hypertension. Am J Physiol Heart Circ Physiol 2012;302:H2243–H2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol 2020;17:170–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol 2000;130:963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siu KL, Gao L, Cai H. Differential roles of protein complexes NOX1-NOXO1 and NOX2-p47phox in mediating endothelial redox responses to oscillatory and unidirectional laminar shear stress. J Biol Chem 2016;291:8653–8662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 2007;15:798–808 [DOI] [PubMed] [Google Scholar]
- 48. Kuo LH, Tsai PJ, Jiang MJ, et al. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia 2011;54:168–179 [DOI] [PubMed] [Google Scholar]
- 49. Davis JE, Braucher DR, Walker-Daniels J, Spurlock ME. Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J Nutr Biochem 2011;22:136–141 [DOI] [PubMed] [Google Scholar]
- 50. Jang HJ, Kim HS, Hwang DH, Quon MJ, Kim JA. Toll-like receptor 2 mediates high-fat diet-induced impairment of vasodilator actions of insulin. Am J Physiol Endocrinol Metab 2013;304:E1077–E1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation 2008;117:3238–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425–432 [DOI] [PubMed] [Google Scholar]
- 53. Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995;1:1155–1161 [DOI] [PubMed] [Google Scholar]
- 54. Härle P, Straub RH. Leptin is a link between adipose tissue and inflammation. Ann N Y Acad Sci 2006;1069:454–462 [DOI] [PubMed] [Google Scholar]