

Abstract
Excess chronic contact between microbial motifs and intestinal immune cells is known to trigger a low-grade inflammation involved in many pathologies such as obesity and diabetes. The important skewing of intestinal adaptive immunity in the context of diet-induced obesity (DIO) is well described, but how dendritic cells (DCs) participate in these changes is still poorly documented. To address this question, we challenged transgenic mice with enhanced DC life span and immunogenicity (DChBcl-2 mice) with a high-fat diet. Those mice display resistance to DIO and metabolic alterations. The DIO-resistant phenotype is associated with healthier parameters of intestinal barrier function and lower intestinal inflammation. DChBcl-2 DIO-resistant mice demonstrate a particular increase in tolerogenic DC numbers and function, which is associated with strong intestinal IgA, T helper 17, and regulatory T-cell immune responses. Microbiota composition and function analyses reveal that the DChBcl-2 mice microbiota is characterized by lower immunogenicity and an enhanced butyrate production. Cohousing experiments and fecal microbial transplantations are sufficient to transfer the DIO resistance status to wild-type mice, demonstrating that maintenance of DCs’ tolerogenic ability sustains a microbiota able to drive DIO resistance. The tolerogenic function of DCs is revealed as a new potent target in metabolic disease management.
Introduction
The World Health Organization estimates that ∼34% of the population has developed or is at risk to develop a metabolic syndrome that predisposes to cardiovascular diseases and cancers (1). A major component that triggers metabolic syndrome is a chronic low-grade inflammation that originates, at least in part, from impaired intestinal barrier function (IBF) (2,3). Modifications in the composition of the intestinal microbiota induced by dietary changes have been shown to promote many pathologies, including metabolic disorders (2–5).
Although much progress has been made to better characterize the role of proinflammatory immune responses in the pathogenesis of metabolic dysfunctions, few studies have focused on the role of intestinal antigen-presenting cells (APCs) (6,7). Detection of enteric pathogens through pattern recognition receptors induces dendritic cell (DC) maturation, triggering proinflammatory adaptive immunity for pathogen clearance. Conversely, at steady states, mucosal DCs are known to promote tolerogenic immunity (8). Several environmental cues enable the maintenance of DC tolerogenic function. Particular microbiota-derived metabolites, such as butyrate, may influence DCs’ tolerogenic activity (9,10).
The constant dialog between the microbiota and the immune system is critical in regulating the composition of the intestinal microbiota throughout life. However, the contribution of DC–microbiota cross talk in orchestrating the progression of low-grade inflammation is unclear.
To decipher the impact of DCs, we used a mouse model developed in our laboratory for which the human antiapoptotic factor B-cell lymphoma 2 (Bcl-2) is expressed under the CD11c promoter to target DCs (DChBcl-2 mice). Targeting Bcl-2 in DCs is known to increase DC life span, resulting in a higher DC number in lymphoid organs, thus boosting the adaptive immune responses (11,12). Since it has been hypothesized that inappropriate intestinal T helper (Th) polarization could promote the deleterious proinflammatory immune environment associated with metabolic alterations (13), we assessed how the increased DC life span impacts DC–microbiota cross talk and host metabolism in the context of diet-induced obesity (DIO).
Research Design and Methods
Animal Experimentation
Transgenic male mice on the C57BL/6J background expressing the open reading frame of human BCL2 apoptosis regulator gene (hBcl2) under the murine CD11c (Itgax; Integrin alpha X) promoter (DChBcl-2) were obta-ined as previously described (12). Littermates at birth until weaning, heterozygous DChBcl-2 and wild-type (WT) controls were either cohoused or single housed depending on their genotype in individually ventilated cages. Mice were fed either a control chow diet (CCD) (E157451–347; ssniff Spezialdiätten GmbH, Soest, Germany) or a high-fat diet (HFD) (60% fat and 20% carbohydrates [kcal/kg]; E15742–347; ssniff Spezialdiätten GmbH) starting at 8 weeks old for 24 weeks. Mice had free access to food and water. Body composition was assessed by using 7.5-MHz time domain-nuclear magnetic resonance (LF90II MiniSpec; Bruker, Rheinstetten, Germany) at 0, 12, and 24 weeks of diet. All procedures involving mice were carried out according to the Guide for the Care and Use of Laboratory Animals published by the European Commission Directive 86/609/EEC. All animal studies were approved by the regional veterinary services of the Paris police headquarters (agreement number 75-751320) and by the Biological Services Unit of Sorbonne University.
Oral Glucose Tolerance Test
After 13 weeks of diet, overnight-fasted mice were treated with an oral gavage glucose load (2 g glucose/kg body wt). Blood glucose was measured at time 0, 15, 30, 60, and 90 min after the oral glucose load with a glucose meter (Accu-Chek; Roche, Basel, Switzerland) on blood samples collected from the tip of the tail vein. Plasma insulin concentration was determined using ELISA (Mercodia, Uppsala, Sweden). HOMA of insulin resistance (HOMA-IR) index was calculated according to the formula: fasting insulin (microunits per liter) × fasting glucose (nanomoles per liter)/22.5.
Insulin Tolerance Test
After 14 weeks of diet, mice were fasted for 6 h, and blood glucose levels were determined at time 0, 15, 30, 60, and 90 min after an intraperitoneal injection of regular human insulin (0.75 units/kg body wt; Humulin; Eli Lilly and Company, Indianapolis, IN).
Adipocyte Size Measurement and Stromal Vascular Fraction Isolation
The mean adipocyte diameters were determined from epididymal adipose tissue (AT) after 24 weeks of HFD. Epididymal AT was rapidly washed with physiologic saline and then incubated with collagenase (1 mg/mL; Sigma-Aldrich, Saint-Quentin-Fallavier, France) in PBS solution (pH 7.4) at 37°C and further processed for adipocyte size measurement or cytokine assay of the stromal vascular fraction (SVF). Mean diameter was defined as the mean value for the distribution of adipocyte diameters of 150 cells.
Intestinal Paracellular Permeability Test
Intestinal paracellular permeability measurement in vivo was based on the intestinal permeability to 4,000 Da fluorescent dextran-FITC (Sigma-Aldrich) as detailed in the Supplementary Material.
Fecal Calorimetry
After 20 weeks of diet, 1 week of feces was collected per cage of three to four mice each. During the same time, the food intake was monitored. The feces were dried overnight at 70°C and weighed. The total energy content of the feces was determined by bomb calorimetry (C200 bomb calorimeter; IKA, Staufen, Germany). and results were expressed as kilocalories per day.
Quantification of Fecal Lipocalin-2 and Fecal Albumin by ELISA
Frozen fecal pellets were reconstituted in PBS containing 0.1% Tween 20 (100 mg/mL) and vortexed for 20 min. Fecal homogenates were then centrifuged for 10 min at 10,000g and 4°C, and cleared supernatants were stored at −20°C and further processed through ELISA (Duoset Murine Lipocalin-2 ELISA kit from R&D Systems, Minneapolis, MN; and albumin from Bethyl Laboratories, Montgomery, AL).
Quantification of Fecal Secreted IgA by ELISA
Frozen fecal samples diluted fivefold (w/v) in protease inhibitor cocktail containing PMSF (5 mmol/L), EDTA (1 mmol/L), and pepstatin (1 µg/mL; Sigma-Aldrich) were homogenized, centrifuged for 10 min at 10,000g at 4°C to collect supernatants, and further processed through an ELISA (Supplementary Material).
Fecal Flagellin and Lipopolysaccharide Load Quantification
We quantified flagellin and lipopolysaccharide (LPS) using human embryonic kidney (HEK)-Blue-mouse (m)TLR5 and HEK-Blue-mTLR4 cells, respectively (Invivogen, San Diego, CA). We resuspended fecal material in PBS as mentioned above and applied the supernatants to the reporter cell lines following the manufacturer’s instructions.
Cytokine Secretion Assay
Single-cell suspensions were prepared from mesenteric lymph nodes (mLNs), small intestine lamina propria (SILP), and colon lamina propria (CLP) (Supplementary Material). Single-cell preparations were cultured in complete media (106 cells/mL) for 72 h in anti-CD3/anti-CD28–precoated (5 μg/mL; BD Biosciences, San Jose, CA) 96-flat-well plates (BD Biosciences). AT SVF single-cell preparation was cultured overnight in complete media (106 cells/mL). Cytokine secretion of supernatants was assessed using a Bio-Plex assay (Luminex MAGPIX Instrument; Bio-Rad Laboratories, Marnes-la-Coquette, France).
Immunolabeling for Flow Cytometry and Cell Sorting
Single-cell preparations were preincubated with Fc-blocking antibody (eBioscience/Thermo Fisher Scientific, Les Ulis, France) for 20 min at 4°C. Cells were then incubated for 30 min at 4°C with fixable viability dye eFluor 506 (eBioscience) and further stained for 30 min with antibodies to surface markers (Supplementary Material).
Analysis of Retinaldehyde Dehydrogenase Activity by Aldefluor Staining
Retinaldehyde dehydrogenase (RALDH) activity in individual cells was analyzed using the aldefluor staining kit (STEMCELL Technologies, Vancouver, British Columbia, Canada) following the manufacturer’s instructions.
16S rRNA Gene Sequencing and Analysis
Feces were collected at day 0 and 12 weeks after starting the diet, immediately frozen in liquid nitrogen, and then stored at −80°C. Fecal DNA was extracted as previously described (14) (Supplementary Material).
Fecal Microbiota Transplantation
Feces from donor mice were diluted (30–50 mg [1:10 w/v]) and homogenized in reduced sterile Ringer solution (VWR) containing l-cysteine (0.5 g/L; Sigma-Aldrich) as reducing agent. This solution was immediately administered to germ-free (GF) recipients by oral gavage once per week during the first 3 weeks of HFD.
Short-Chain Fatty Acid Analysis
Short-chain fatty acids (SCFAs) were measured by gas-liquid chromatography (Supplementary Material).
Microarray Analysis
After FACS, cells were counted and resuspended in TRIzol lysis reagent (Thermo Fisher Scientific), frozen in liquid nitrogen, and stored at −80°C. RNA extraction was performed using the RNeasy Micro Kit (Qiagen). Quality and quantity of RNA extraction were performed using the Bioanalyzer 2100 RNA 6000 Pico chip assay (Agilent Technologies). Total RNA (2.5 ng) was reverse-transcribed following the Ovation Pico WTA System V2 (NuGEN). cDNA hybridization was performed using the GeneChip Mouse Gene 2.0 ST (Affymetrix). See Supplementary Material for further details.
Statistical Analysis
Data are expressed as medians for dot plot or mean and SD for bar plots. Data were analyzed using Prism version 8.0 for Windows (GraphPad Software, San Diego, CA). Mann-Whitney test and Kruskal-Wallis test or two-way ANOVA for multiple comparisons was performed. Results were considered statistically significant when P was <0.05; asterisks indicate significant differences (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001) between two groups according to statistical analysis performed.
Data and Resource Availability
All original microarray data were deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus database (accession number GSE166899). Unprocessed 16S rRNA gene sequencing data are deposited in the Genome Sequence Archive in the National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA003914.
Results
DChBcl-2 Mice Demonstrate Resistance to HFD-Induced Metabolic Alterations
To decipher how DCs orchestrate DIO and metabolic alterations, we used a mouse model enriched for DCs, where the antiapoptotic human gene hBcl2 is expressed under the control of CD11c, a DC-related promoter.
We challenged adult WT and DChBcl-2 mice with a 60% HFD or CCD. Although there was no difference in weight between WT and DChBcl-2 mice on CCD, DChBcl-2 mice gained 10% less weight than WT mice after 12 weeks of HFD, a difference that reached 15% after 24 weeks of HFD, when we observed a weight stabilization (Fig. 1A and B).
Figure 1.
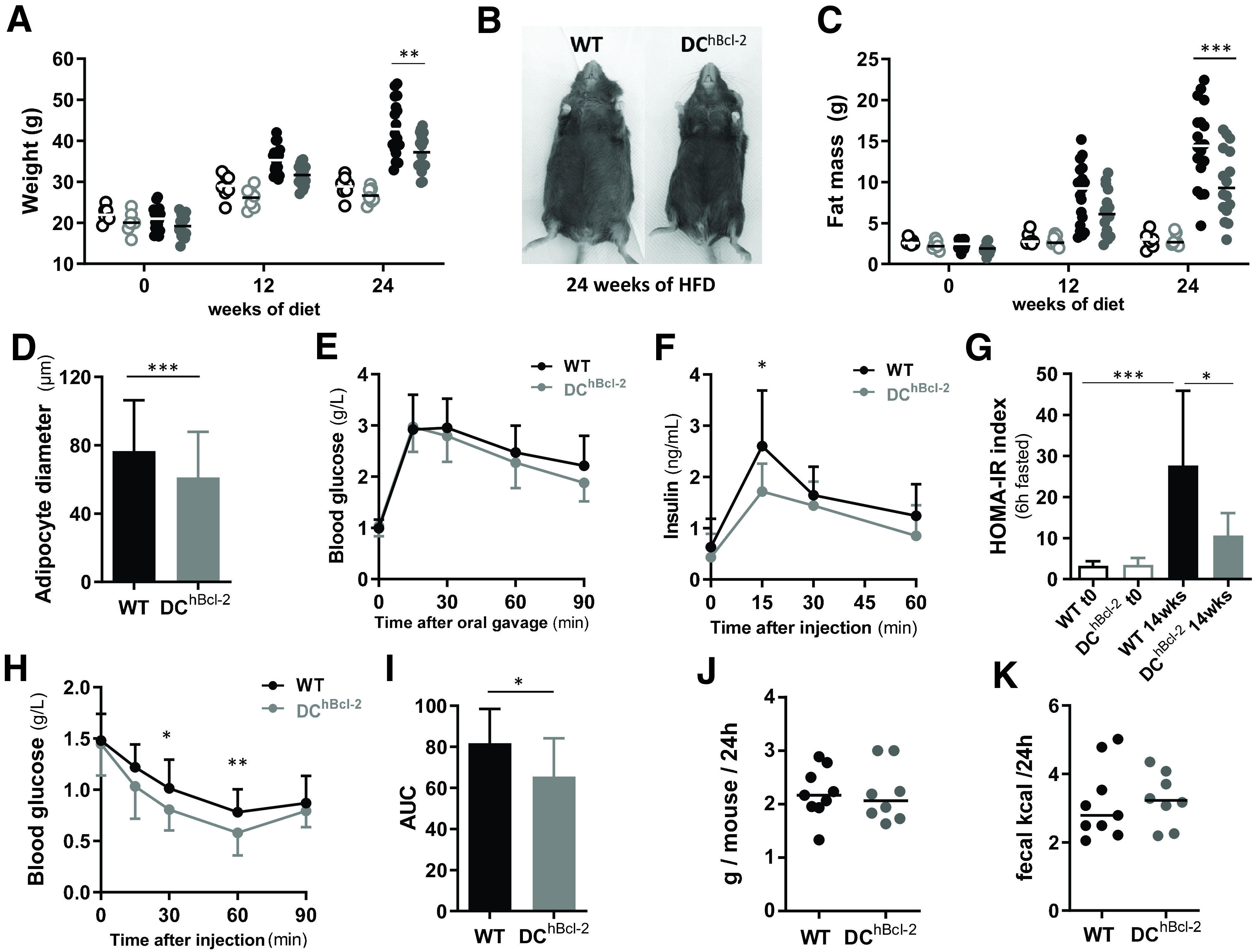
DChBcl-2 mice are resistant to HFD-induced obesity and associated metabolic alterations. Body weight (A) and fat mass (C) monitoring of WT (black circles) and DChBcl-2 (gray circles) at day 0 and 12 and 24 weeks after starting CCD (open circles) or HFD (filled circles). B: Abdominal photographs of representative WT and DChBcl-2 mice HFD-fed for 24 weeks. D: Average adipocyte diameter in the epididymal adipose tissue of WT and DChBcl-2 HFD-fed for 24 weeks (N = 7–9 mice/group). Blood glucose (g/L) (E) and insulin (ng/mL) (F) levels during oral glucose tolerance test after 13 weeks of HFD (N = 10–14 mice/group). G: Insulin resistance index (HOMA-IR) before and after 14 weeks of HFD (N = 10 to 14 mice/group). H: Blood glucose level (g/L) during an ITT after 14 weeks of HFD (N = 10–14 mice/group). I: Area under the curve (AUC) of the glucose profile during the ITT (N = 10–14 mice/group). Mean of daily food intake (J) and fecal energy (K) monitored for 1 week after 12 weeks of HFD. Data are presented as median for dot plots and mean ± SD for others. *P < 0.05; **P < 0.01; ***P < 0.001.
Body mass composition analysis during the entirety of the HFD indicated that despite the same increase in lean mass (Supplementary Fig. 1A), DChBcl-2 mice gained significantly less fat mass than their WT counterparts (Fig. 1C), which was associated with reduced adipocyte size (Fig. 1D). Looking at AT monocytes/macrophages that share the CD11c marker and that are known to contribute to tumor necrosis factor-α (TNF-α)–mediated obesity-induced inflammation and insulin resistance (15,16), we observed no difference in the percentage and total numbers. Although complementary analysis of AT inflammatory parameters is required to conclude on AT inflammation state between the two groups, we observed no difference in terms of total SVF-derived TNF-α secretion (Supplementary Fig. 1B–D).
To investigate further the metabolic phenotype, we looked at glucose metabolism after 13 weeks of HFD, a current timing described in the literature for the C57BL/6J mice background (17). Although blood glucose levels were comparable (Fig. 1E), we observed significant differences in circulating insulin levels following oral glucose administration (Fig. 1F). HOMA-IR indicated that DChBcl-2 mice were significantly more sensitive to insulin than WT mice (Fig. 1G and Supplementary Fig. 1E). Insulin tolerance test (ITT) confirmed that DChBcl-2 mice displayed enhanced insulin sensitivity compared with WT mice (Fig. 1H and I).
We next investigated the DChBcl-2 mice DIO-resistant phenotype, evaluating their daily energy expenditure. Daily food intake (Fig. 1J), as well as intestinal absorptive capacity looking at loss of energy in the feces by bomb calorimetry, demonstrated no significant differences in both groups of mice with no difference observed in terms of transit time (Fig. 1K and Supplementary Fig. 1F) and feces production (Supplementary Fig. 1G). Indirect gas calorimetry and locomotor activity assessment indicated that there were no changes between WT and DChBcl-2 mice on HFD (Supplementary Fig. 1H–K).
All of these results suggested that differences in weight gain, adiposity, and insulin sensitivity in HFD-fed WT and DChBcl-2 mice occurred, although we observed similar food intake, caloric intestinal absorptive capacity, and energy expenditure.
Intestinal Barrier Integrity in DChBcl-2 Mice Is Associated With Increased CD103+CD11b+ DCs
Long-term interactions between HFD and microbiota have been shown to be required to promote microbial-derived metabolic alterations in a chronicled manner (18). We further evaluated how the IBF, known to be impacted by diet-mediated microbial disturbances, could be involved in the respective discrepant metabolic phenotypes after 24 weeks of HFD. We first investigated intestinal permeability parameters and observed that DChBcl-2 mice displayed a lower increase in paracellular intestinal permeability than WT mice (Fig. 2A and B). Paracellular permeability kinetic study highlighted that the distal part of the intestine appeared more affected by the HFD in WT mice compared with DChBcl-2 mice on HFD (Fig. 2B). This was associated with lower fecal albumin levels (Supplementary Fig. 2A), used as a marker for gut vascular barrier leakage. We determined how those differences in intestinal permeability relate to the overall intestinal inflammatory tone. We quantified fecal lipocalin-2 (Lcn-2), known as an early biomarker for intestinal inflammation (19). Under HFD, DChBcl-2 mice displayed lower levels of Lcn-2 compared with WT mice (Fig. 2C).
Figure 2.
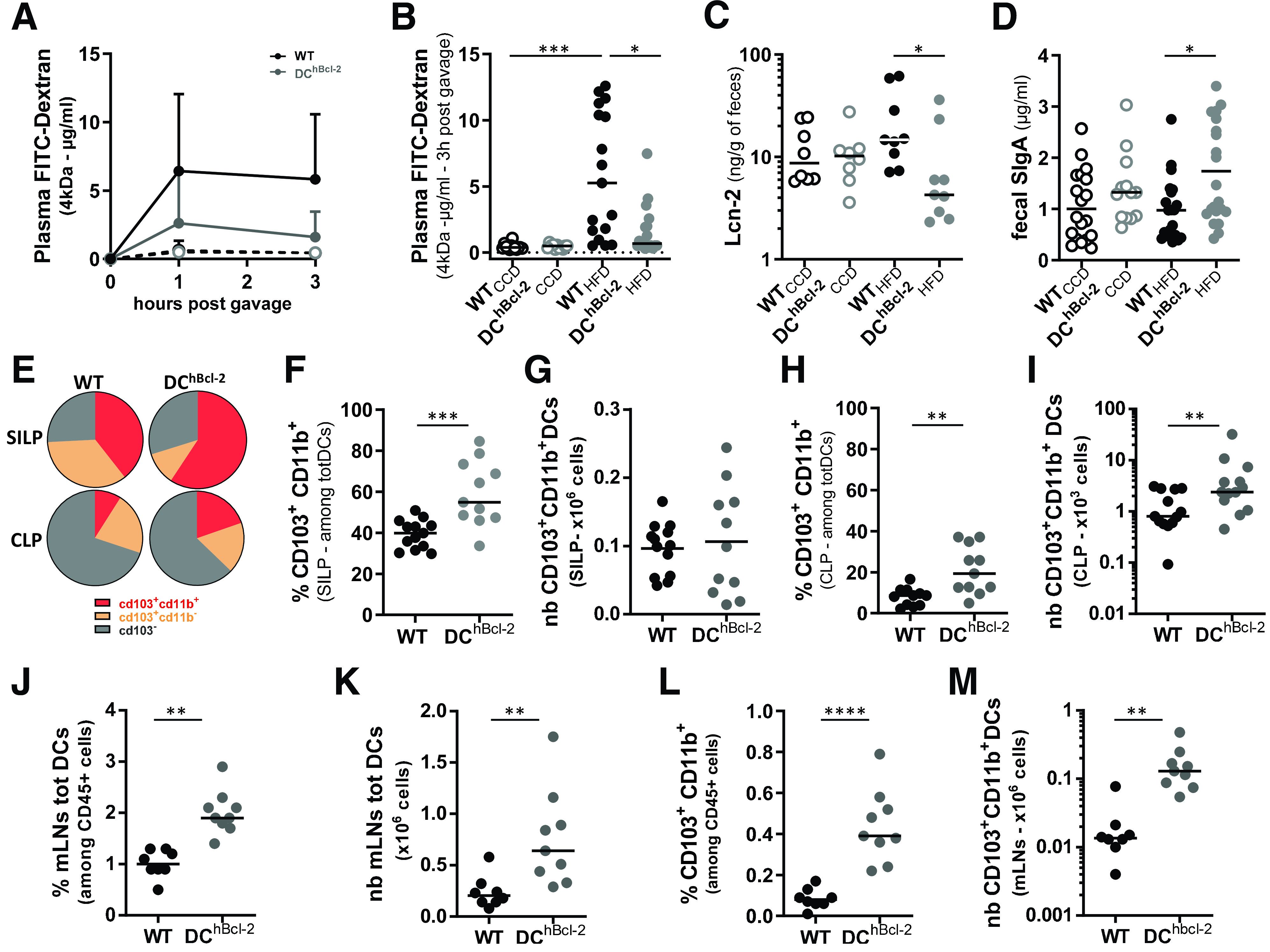
Maintenance of the IBF is associated with a strong increase in tolerogenic DCs. Open circles and filled circles are representative of mice fed a CCD or an HFD, respectively, for 24 weeks. A: Kinetics of plasma levels of dextran-FITC (FD 4 kDa) (600 mg/kg body wt) (N = 10–14 mice/subcutaneous group). FD4 plasma levels 3 h after oral gavage (B) and Lcn-2 (C) and sIgA (D) levels in the feces determined by ELISA. E: Mean proportions of CD103+CD11b+, CD103+CD11b−, and CD103− among totDCs in the SILP and the CLP. Proportions (F) and total numbers (nb) (G) of CD103+CD11b+ DCs in the SILP. Proportions (H) and total numbers (I) of CD103+CD11b+ DCs in the CLP. Percentage (J) and total numbers (K) of totDCs among CD45+ cells in the mLNs. Percentage (L) and total numbers (M) of CD103+CD11b+ DCs in the mLNs. Data are presented as mean for circle graphs, median for dot plots, and mean ± SD for others. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
IBF also comprises fecal secreted IgA (sIgA) that counteracts several antigens to access the intestinal wall (bacteria, viruses, food, etc.) (20). Although we observed no difference on CCD, fecal sIgA titers were 1.8 times higher in DChBcl-2 than in WT mice on 24 weeks of HFD (Fig. 2D).
All of these results suggested that DChBcl-2 mice displayed enhanced IBF compared with WT mice after 24 weeks of HFD.
We further assessed how manipulating DC life span impacted intestinal DC populations, looking at global markers and subsets of intestinal DCs by flow cytometry (Supplementary Fig. 2B). We observed no significant differences in the percentages and total numbers of DCs (totDCs) in both SILP and CLP (Supplementary Fig. 2C–F). Based on CD103 and CD11b surface markers discriminating the three subsets of intestinal conventional DCs (cDCs) (21,22), we found that the SILP and CLP CD103+CD11b+ cDCs were significantly increased in the proportion of totDCs in DChBcl-2 compared with WT mice (Fig. 2E, F, and H). Although we observed no difference in the SILP (Fig. 2G), total numbers of CD103+CD11b+ cDCs in the CLP were threefold increased in DChBcl-2 mice relative to WT mice (Fig. 2I). Since hBcl-2 is expressed under the CD11c promoter also shared by monocytes/macrophages, we looked at CD64+CD11c+MHC class II+ intestinal monocytes/macrophages. We observed no difference in the percentages and total numbers in both groups of mice (Supplementary Fig. 2G–J).
Those results demonstrated that the hbcl2 insertion strongly enhanced the CD103+CD11b+ cDC subpopulation.
One important feature of DCs after antigenic stimuli is their ability to migrate from the intestinal lamina propria to the mLNs. After 24 weeks of HFD, we observed a marked increase in totDCs in both percentage and total numbers (Fig. 2J and K) in the mLNs of DChBcl-2 mice compared with WT mice. The CD103+CD11b+ cDC subset in DChBcl-2 mice was even more increased in this compartment, representing 4 times more in percentage than in WT mice and 10 times more in total numbers (Fig. 2L and M).
These observations highlighted that the maintenance of DChBcl-2 mice IBF is associated with increased CD103+CD11b+ cDCs.
hBcl2 Transgene Promotes DC Tolerogenic Properties With Enhanced RALDH Activity
The first screen of mLN DC subpopulations indicated that the hBcl2 transgene was significantly more expressed in the CD103+ DCs than in CD103− DCs regardless of the diet (Fig. 3A). Among the CD103+ DCs, CD11b+ DCs were the most enriched in transgenic mice compared with WT mice (Supplementary Fig. 3A). Therefore, we performed global transcriptomic analysis on mLN sorted CD103+CD11b+ DCs in both groups of mice before starting the HFD to avoid any other environmental effect.
Figure 3.
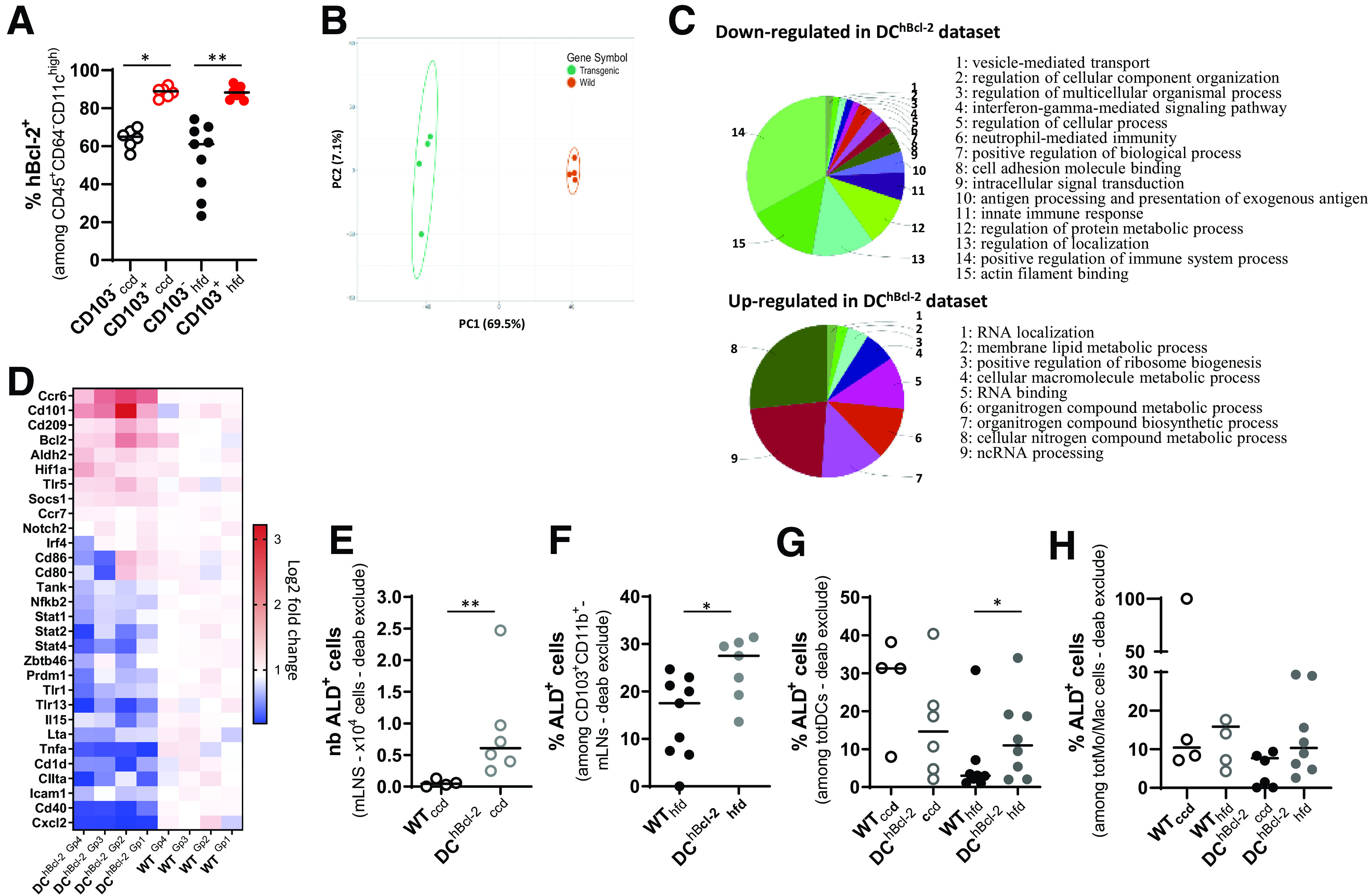
hBcl2 transgene modulates tolerogenic DC function that impacts RALDH activity. A: Proportion of hBcl-2+ DCs among CD103+ and CD103− cDC subpopulations in the mLNs after 24 weeks of CCD or HFD. B–D: Microarray gene expression analysis of sorted CD103+CD11b+ DCs from mLNs of WT and DChBcl-2 before starting the diet. B: Principal component (PC) analysis showing separation of sample groups. C: Biological enrichment and annotation of pathways downregulated and upregulated in DChBcl-2 data set using ClueGo plugin. D: Heat map of log twofold-change value of key gene expression related to DC maturation or activation and DC tolerogenic markers. E: Total numbers (nb) of ALD+ in CD11b+CD103+ cDC subpopulation in the mLNs of mice under CCD. F: Percentage of ALD+ cells among CD11b+CD103+ cDC subpopulation in the mLNs of mice under HFD. Percentage of ALD+ cells among totDCs (G) or monocytes/macrophages (totMo/Mac) (H) in the CLP of mice after 24 weeks of CCD or HFD. Data are presented as median for dot plots. *P < 0.05; **P < 0.01. deab, diethylaminobenzaldehyde.
Principal component analysis of CD103+CD11b+ gene sets discriminated the sample genotypes on the high axis percentages (69.5% and 7.1% for x- and y-axis, respectively), confirming the major impact of the transgene expression in this cDC subset (Fig. 3B).
Differential expression of genes showed that 2,774 genes discriminated DChBcl-2 and WT samples, with a marked downregulation of immune-related pathways in DCs sorted from DChBcl-2 mice (Fig. 3C). Expression of genes related to the maturation or activation status of DCs were downregulated in DChBcl-2CD103+CD11b+ DCs (CXCL2, CD40, CIITA, and CD1D) (Fig. 3D). These results were confirmed looking at predictive signaling pathways involved in DC maturation using Ingenuity Pathway Analysis (Supplementary Fig. 3B). We observed that the transgene may prevent DCs from acquiring antigen sensing through TLRs 2/3/4/9, antigen-presenting properties through MHC class II/I or cell adhesion markers, such as the intercellular adhesion molecule 1 (ICAM1), as well as markers for costimulation of adaptive immune cells (CD40/CD86) (Supplementary Fig. 3B). Functionally, the immaturity of DCs relates to their inability to mount proinflammatory responses after stimulation (23). The immature status of transgenic-sorted DCs was in line with a downregulation of immune-related inflammatory signaling pathways such as nuclear factor κ-light-chain enhancer of activated B-cell pathway (nuclear factor-κB, TANK, and NFKB2) and STAT pathways (STAT1, 2, and 4) (Fig. 3D and Supplementary Fig. 3B). This global downregulation of proinflammatory responses was associated with a decreased capacity for proinflammatory cytokines release, such as TNF-α, interferon-γ, and interleukin-15 (IL-15) (Fig. 3D and Supplementary Fig. 3B).
All of these results strongly suggest that the hBcl2 transgene prompted CD103+CD11b+ DCs to keep an immature phenotype, altering their capacity to elicit the proinflammatory immune responses.
The immature immune stage in DCs has been associated with increased tolerogenic capacity (23,24). Expression levels of gene-sustaining tolerogenic functions, such as Cd101, Socs1, Hif1-a, as well as Aldh2, were upregulated in DChBcl-2CD103+CD11b+ DCs compared with WT (Fig. 3D).
Since the tolerogenic ability of mucosal DCs has been related to their capacity to process vitamin A into retinoic acid through the enzymatic activity of RALDHs (25), we next analyzed this function (Supplementary Fig. 3C). We observed enhanced RALDH activity (aldefluor-positive [ALD+] cells) in CD103+CD11b+ DC subpopulations isolated from mLNs of DChBcl-2 mice. ALD+ cells represented nearly a threefold increase in percentage among CD103+ CD11b+ cells, relative to WT mice, that resulted in a 13-fold increase in cDC CD103+CD11b+ALD+ total cell numbers (Fig. 3E and Supplementary Fig. 3D).
We next wondered how this DC function was impacted upon HFD, and we observed no impact of the diet on the RALDH activity in both groups, with DChBcl-2 mice significantly harboring their higher rate of RALDH activity (Fig. 3F and Supplementary Fig. 3E). Studies have highlighted the importance of DCs RALDH activity in mouse models of colitis and in patients with inflammatory bowel disease (26,27). They demonstrated that upon inflammation, tolerogenic DCs were losing their RALDH activity, promoting the subsequent loop of chronic inflammation. Although we observed no difference in intestinal RALDH activity in the SILP (data not shown), DC RALDH activity was significantly decreased in the colon of HFD-fed WT mice compared with CCD-fed WT (Fig. 3G). On the contrary, DChBcl-2 mice maintained their RALDH activity (Fig. 3G and Supplementary Fig. 3F). We did not observe any effect of the hBcl-2 transgene on monocytes/macrophages’ colonic RALDH activity regard-less of diet, demonstrating that the hBcl-2 transgene targeted particularly DCs’ tolerogenic RALDH activity (Fig. 3H and Supplementary Fig. 3G).
Those results indicated that modulating DCs through hBcl2 insertion promotes tolerogenic DC function with increased capacity to process vitamin A through RALDH activity.
Tolerogenic DCs Strongly Impact Colonic Adaptive Immunity in the Context of DIO
The important role of DCs in shaping the appropriate immune responses through adaptive immune system activation has been widely documented (22,28). After 24 weeks of HFD, we observed no significant differences in the percentages and total numbers of T and B lymphocytes in the mLNs of both groups of mice (Supplementary Fig. 4A–C and data not shown). Focusing on Th subsets, we detected significantly higher proportions and total numbers of CD4+IL-17+ cells (Th17) as well as significantly higher total numbers of CD4+Foxp3+ (regulatory T [Treg] cells) in the mLNs of DChBcl-2 compared with WT mice (Fig. 4A and B and Supplementary Fig. 4D–F). In the same compartment, we also observed significantly higher percentages and total numbers of CD19−sIgA+ plasmablasts in DChBcl-2 mice (Fig. 4C and D).
Figure 4.
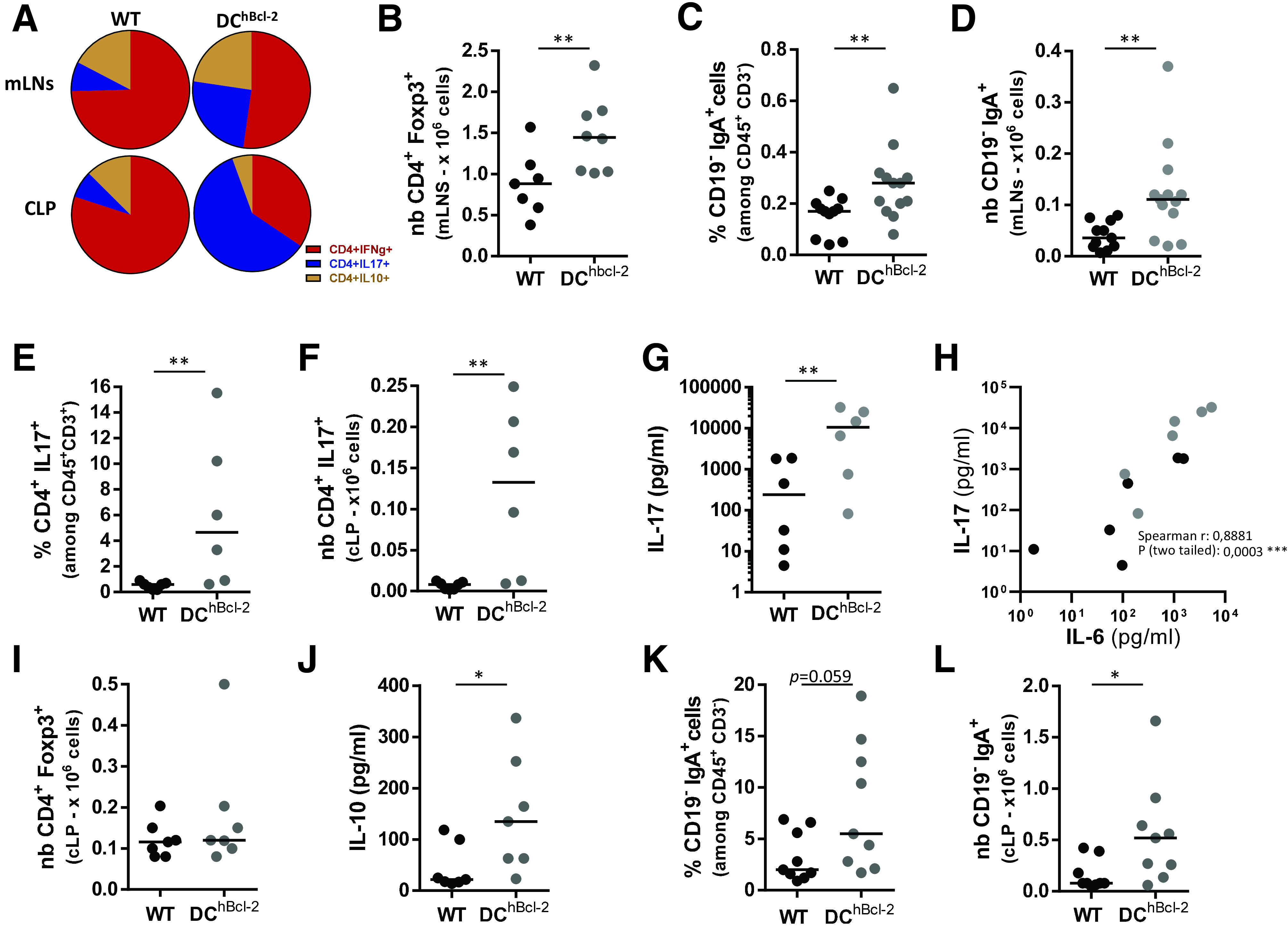
HFD-fed DChBcl-2 mice showed enhanced Treg, Th17, and sIgA+ B-cells that established in the colon. All data are representative of mice fed an HFD for 24 weeks. A: Circle graphs representing the mean proportions of interferon-γ (IFN-γ–), IL-17–, and IL-10–producing CD4+ T cells in the mLNs or CLP after intracellular staining of cytokines. Total numbers (nb) of CD4+Foxp3+ T lymphocytes in the mLNs (B) or in the CLP (I). Proportions of CD19−IgA+ plasmablasts in the mLNs (C) or in the CLP (K). Total numbers of CD19−IgA+ plasmablasts in the mLNs (D) or the CLP (L). Percentages (E) and total numbers (F) of IL-17–producing CD4+ T cells in the CLP after intracellular staining of cytokines. IL-17 (G) and IL-10 (J) secretion in the supernatants of ex vivo anti-CD3/CD28–stimulated CLP cells for 72 h. H: Correlation graph of IL-17 and IL-6 cytokines secreted in the supernatants of ex vivo anti-CD3/CD28–stimulated CLP cells for 72 h. Data are presented as mean for circle graphs or median for dot plots. *P < 0.05; **P < 0.01.
Altogether, these results demonstrated that a higher number of tolerogenic CD103+CD11b+ cDCs were associated with enhanced priming of Th17, Treg, and sIgA+ B-cell responses in DChBcl-2 after 24 weeks of HFD.
We next assessed where those adaptive immune responses established throughout the intestinal compartment. We observed no significant differences in CD4+ T-cell responses or sIgA+ B-cell responses in the SILP compartment (Supplementary Fig. 4G and H). Conversely, we observed a marked and significant skewing toward Th17 responses in the CLP of DChBcl-2 mice upon HFD. While Th1 cells were the predominant T-cell population in the CLP of WT mice, Th17 cells represented 60% of the total CD4+ T cells in DChBcl-2 mice (Fig. 4A). DChBcl-2 mice Th17 colonic responses displayed a 4-fold increase in percentage and a 10-fold increase in total number relative to WT mice (Fig. 4E and F). Cellular ex vivo experiments with a nonspecific T-cell receptor stimulation confirmed that colonic T cells isolated from DChBcl-2 mice displayed a higher capacity to secrete IL-17 than those from WT mice (Fig. 4G). CD103+CD11b+ cDCs have been shown to promote Th17 differentiation through IL-6 secretion, a capacity that appeared impaired on HFD (4,29). IL-17 secretions and IL-6 levels correlated positively, suggesting a potent mechanism involving an increased capacity of DChBcl-2-derived DCs to produce IL-6 (Fig. 4H). Despite equivalent levels of CD4+Foxp3+ Treg cells, ex vivo–stimulated colonic T cells isolated from DChBcl-2 mice secreted higher levels of IL-10 (Fig. 4I and J). Th17 and Treg cell responses that established in the colon of DChBcl-2 mice were associated with a significant increase in the percentage and the total number of CD19−sIgA+ plasmablasts (Fig. 4K and L).
Altogether, those results suggested that on HFD, increased tolerogenic DCs strongly impact the colonic immunity through enhanced Th17, Treg, and sIgA+ B-cell responses.
DChBcl-2 Intestinal Microbiota Displays Lower Inflammatory Signatures After DIO
The immune system strongly influences intestinal microbiota composition that, in turn, is a major determinant of the metabolic response to HFD (30,31). We wondered how the strong colonic immunological differences observed could impact the intestinal microbiota. We analyzed the fecal microbiota of WT and DChBcl-2 mice by 16S rRNA gene sequencing before and after 12 weeks of HFD challenge, just before we observed differences in glucose metabolism. 16S rRNA gene analyses first indicated that the fecal microbiota of WT and DChBcl-2 mice were not distinguishable before starting the diet (Fig. 5A and B). Conversely, we observed marked differences in WT and DChBcl-2 microbiota after 12 weeks of HFD (Fig. 5C and D). Linear discriminant analysis (LDA) effect size (LEfSe) revealed that specific taxa were enriched in either the microbiota of WT or the DChBcl-2 mice. WT mice harbored enrichment of the Epsilonproteobacteria family members, whereas the Mogibacteriaceae members appeared increased in DChBcl-2 mice (Fig. 5E and F). Those results demonstrated that upon HFD, microbiota composition has differentially shifted on HFD in the two groups of mice.
Figure 5.
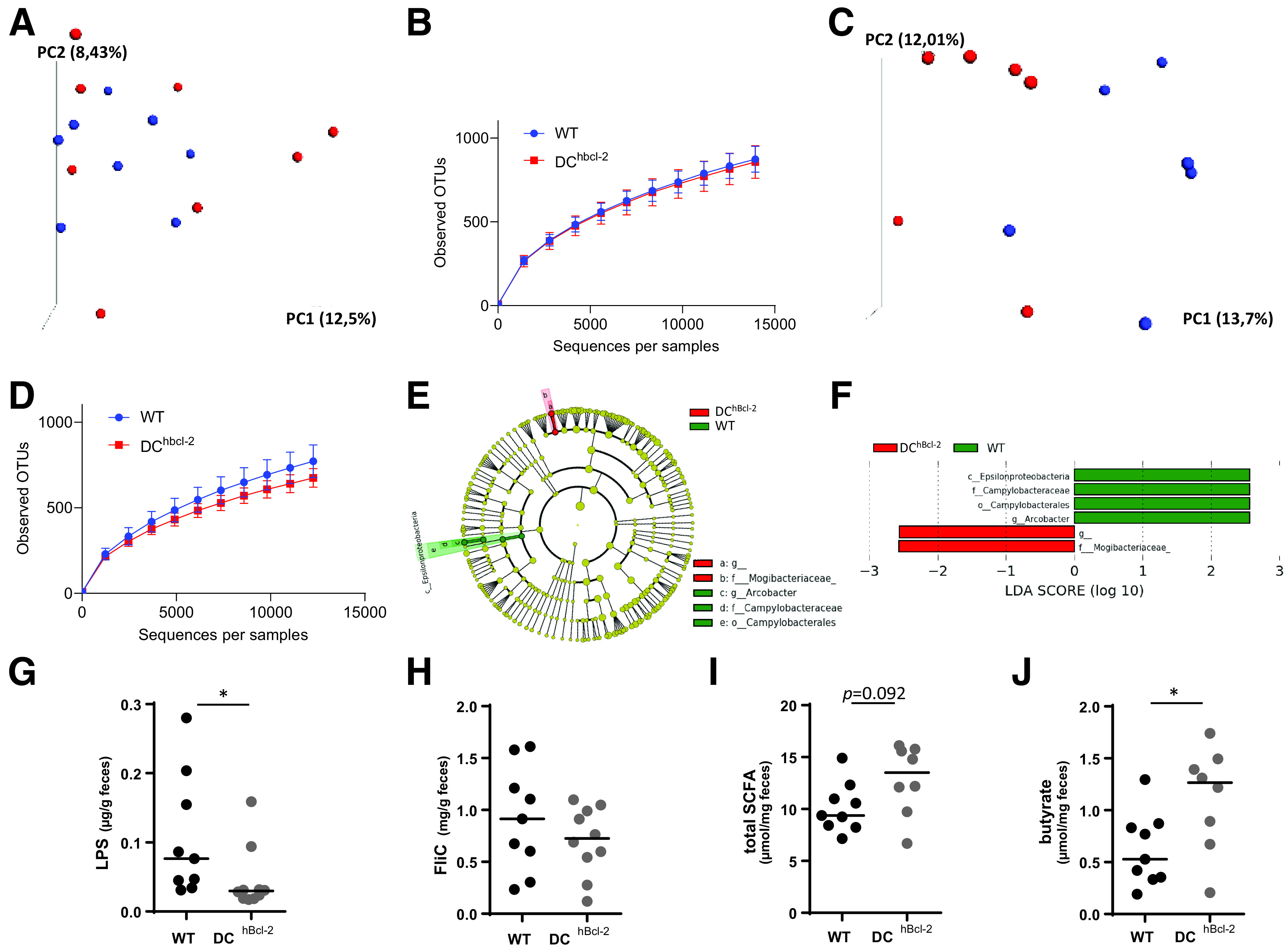
HFD-fed DChBcl-2 mice shape a gut microbiota characterized by lower inflammatory signatures. Principal component (PC) analysis of the unweighted UniFrac distance matrix (A and C) and α diversity assessment (B and D) of fecal WT and DChBcl-2 microbiota at baseline (A and B) and after 12 weeks of HFD (C and D). E and F: LEfSe was used to investigate bacterial members that drive the differences between the fecal microbiota of WT and DChBcl-2 mice. E: Taxonomic cladogram obtained from LEfSe analysis. Red indicates taxa significantly more abundant in WT mice; green indicates taxa significantly more abundant in DChBcl-2 mice. F: LDA scores for the differentially altered taxa. Green indicates taxa significantly more abundant in WT mice; red indicates taxa significantly more abundant in DChBcl-2 mice. Only taxa meeting an LDA significance threshold >2.0 are presented. G and H: Fecal LPS and FliC levels in WT and DChBcl-2 mice assessed by HEK reporter cell lines. Fecal total SCFA (I) and fecal butyrate concentrations (J) in WT and DChBcl-2 mice. Data are represented as median for dot plots. OTU, operational taxonomic unit. *P < 0.05.
Epsilonproteobacteria are members of Gram-negative bacteria. Their motility as well as their LPS, major components of their outer membrane, could trigger proinflammatory immunoreactivity. Since long-term interactions between diet and microbiota have been shown to elicit microbiota-derived host metabolic dysregulations (18), we assessed the immunogenic properties of each type of microbiota through the quantification of fecal bioactive LPS and flagellin (FliC), after 24 weeks of HFD, when the discrepant body mass compositions stabilized in each group. Upon HFD, DChBbcl-2 mice displayed a lower amount of both fecal bioactive LPS and FliC, and this was more particularly statistically significant for the bioactive LPS (Fig. 5G and H).
Pattern recognition receptor–independent signals can also orientate DC function, and bacterial fermentation products have been shown to participate in the immunoregulatory function of DCs, contributing to intestinal immune tolerance and intestinal homeostasis (32). We quantified fecal SCFAs using gas-liquid chromatography. Although we observed no significant differences in the amount of total fecal SCFA concentration (Fig. 5I), DChBcl-2 harbored particular SCFA profiles with a marked enrichment in fecal butyrate concentration, representing 2.3-fold increased levels relatively to WT mice (Fig. 5J).
Altogether, those results demonstrated that under HFD, DChBcl-2 microbiota behave differently in terms of bacterial composition and functions, leading to less immunogenicity as well as sustained immune tolerance.
DChBcl-2 Intestinal Microbiota Drives Resistance to HFD-Induced Metabolic Alterations
Horizontal transmission of the microbiota by cohousing different mouse populations has been shown to nearly normalize the fecal microbiota later on, especially when starting at weaning period when the introduction of solid food strongly modified the microbiota composition (33). To unravel the respective role of WT and DChBcl-2 microbiota in modulating their metabolic phenotype, we first compared HFD-treated cohoused WT (WT CoH) and DChBcl-2 (DCh CoH) mice versus single-housed WT or single-housed DChBcl-2 mice. Looking at body mass composition after 24 weeks of HFD, we observed that cohousing transmitted the DIO-resistant phenotype to WT mice (Supplementary Fig. 5A–D).
To assess whether the microbiota was driving the HFD-resistant DChBcl-2 phenotype, we transferred fecal microbiota from both groups of mice into GF recipients. We colonized 8-week-old GF recipients with the microbiota of 24 weeks of HFD-fed WT and DChBcl-2 (Supplementary Fig. 5E). We strikingly observed that recipient mice followed the same kinetic of body mass composition evolution as the donors. Indeed, 24 weeks after starting the diet, DChBcl-2 microbiota recipient mice (i.e., FT-DChBcl-2) gained significantly less weight than WT microbiota recipient mice (i.e., FT-WT) (39.2 ± 3.9 g and 44.2 ± 3.9 g, respectively) (Fig. 6A). We observed that although mice developed the same lean mass (Supplementary Fig. 5F), FT-DChBcl-2 mice displayed significantly lower adiposity than FT-WT mice (10.7 ± 2.3 g and 15.6 ± 3.2 g, respectively) (Fig. 6B).
Figure 6.
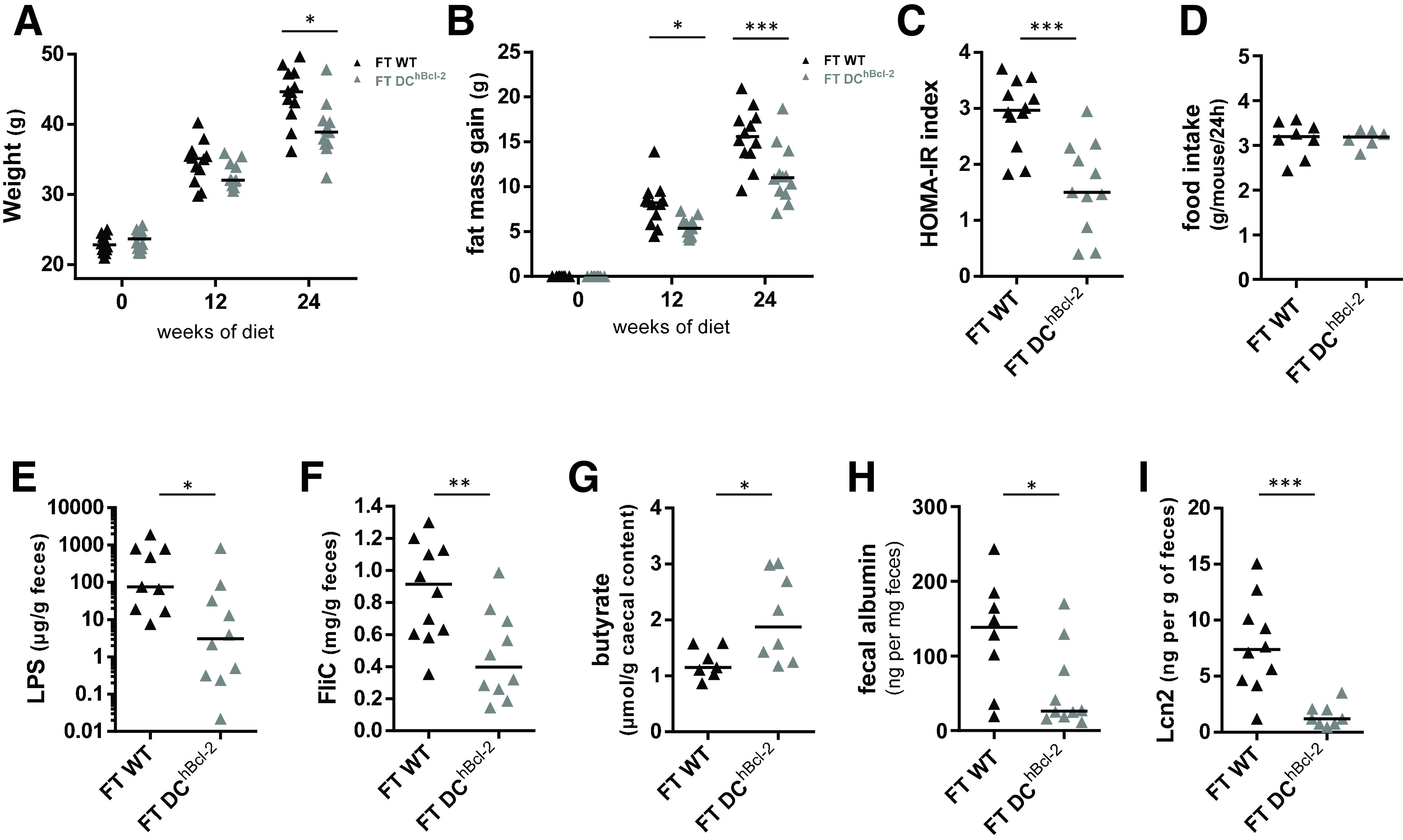
DChBcl-2 gut microbiota triggers the DIO-resistant phenotype. A: Body weight monitoring of WT-microbiota recipients (FT-WT) and DChBcl-2-microbiota recipients (FT-DChBcl-2) at day 0 and 12 and 24 weeks after both fecal microbial transplant (FT) and starting the HFD. B: Fat mass gain at day 0 and 12 and 24 weeks after both FT and HFD. C: Insulin resistance index (HOMA-IR) after 12 weeks of both FT and HFD. D: Mean of daily food intake of individually housed mice monitored for 1 week after 12 weeks of both FT and HFD. Fecal bioactive LPS (E) and FliC (F) load after 24 weeks of both FT and HFD. G: Cecal butyrate concentration after 24 weeks of both FT and HFD. Albumin (H) and Lcn-2 (I) levels in the feces after 24 weeks of both FT and HFD. Data are presented as median for dot plots. *P < 0.05; **P < 0.01; ***P < 0.001.
We further investigated glucose metabolism and observed marked differences in insulin sensitivity. FT-DChBcl-2 recipient mice had a significantly lower HOMA-IR index than FT-WT mice (1.6 ± 0.8 and 2.9 ± 0.6, respectively) (Fig. 6C). As monitored in donors, we never observed any variations in food intake in the recipient groups (Fig. 6D).
Those results were in line with cohousing experiments and demonstrated that DChBcl-2 microbiota by itself was able to drive the HFD-resistant phenotype. This overall demonstrated that DChBcl-2 tolerogenic DCs are shaping a transmissible DIO-protective intestinal microbiota.
We next assessed whether the immunogenic properties of each type of microbiota have been transmitted through fecal transplantation. FT-DChBcl-2 recipient mice displayed a significant lower amount of both fecal bioactive LPS and FliC (Fig. 6E and F). We quantified cecal SCFA (Supplementary Fig. 5G) and observed that FT-DChBcl-2 recipients harbored a significant enrichment in cecal butyrate concentration, representing 1.6-fold increased levels compared to FT-WT recipients (Fig. 6G). We wondered if those discrepant microbial properties transferred from donors to recipient mice have impacted intestinal permeability by looking at fecal albumin content. We observed a threefold increase in fecal albumin content in FT-WT mice relative to FT-DChBcl-2 mice (Fig. 6H). After 24 weeks of HFD, FT-DChBcl-2 recipients demonstrated a lower level of fecal Lcn-2 than FT-WT recipients, arguing for a lower intestinal inflammatory tone (Fig. 6I).
Altogether, those results demonstrated that an increase in tolerogenic DCs is associated with a DIO-resistant microbiota that is sufficient to drive by itself the DIO-resistant phenotype. This transmissible DIO-resistant microbiota is characterized by less immunogenicity and enhanced butyrate-producing capability related to decreased intestinal inflammatory tone (Fig. 7).
Figure 7.

Scheme representing how tolerogenic DCs sustain the DIO-resistant microbiota characterized by lower immunogenicity and enhanced butyrate production.
Discussion
The Bcl-2–regulated apoptosis pathway has been shown to act as a molecular regulator of both DC life span and immunogenicity (11,12,34,35). The functional importance of this survival pathway tested in vivo revealed that Bcl-2 regulates the accumulation of DCs associated with enhanced T-cell activation, which, in turn, enables resistance to lethal septic shock in mice (12). Here, we questioned what could be the impact of such DC-mediated Th polarization in the context of HFD-induced metabolic endotoxemia, where LPS is playing a central role in driving the deleterious metabolic effect (36,37). The DChBcl-2 DIO-resistant phenotype depicted healthier indexes of IBF together with a lower inflammatory tone. Since CD11c is not a DC-restrictive promoter, we might have expected the same impact of the hBcl-2 transgene on both monocytes/macrophages and DCs populations. Although we observed a minor impact on monocyte/macrophage populations, a deeper characterization of these cells would be necessary to ascertain their possible role. However, since it has been shown that, depending on the immune cell type, different antiapoptotic BCL-2 family members could be required for survival pathway (38,39), we might expect that a strategy that targets bcl2 could differentially affect the different CD11c-bearing immune cells. Indeed, we observed that the hBcl-2 transgene was affecting not all DCs but more particularly a cDC subset, since we observed a marked enrichment toward CD103+CD11b+ cDCs. This enrichment, especially in the colon of DChBcl-2 mice, is of particular interest since the same CD103+CD11b+ cDCs subset has been recently shown to be reduced in the colon upon HFD (40). Although we did not observe any change in AT and intestinal monocyte/macrophage populations, we cannot exclude residual impact in particular subsets as well as in other compartments that we did not investigate in our study. Additional experiments are needed to clearly demonstrate these aspects.
Nevertheless, the accumulation of CD103+CD11b+ cDCs observed in the intestinal compartment of DChBcl-2 mice is particularly interesting since this subpopulation has been shown to induce the differentiation of Th17 cells in the gut at steady state (29), and we indeed observed a strong colonic Th17 polarization in the intestinal draining lymph nodes as well as in the CLP of DChBcl-2 compared with WT mice. Previous studies have shown that DIO triggers an increase of intestinal Th1 immune response associated with a decrease of intestinal Th17 response (3,4,41). In Garidou et al. (4) and Hong et al. (41), the authors even demonstrated the important role of intestinal Th17 cells in mediating DIO resistance. Our data are in line with what has been published, reinforcing the hypothesis that intestinal Th17 responses play a major role in counteracting DIO and metabolic alterations.
The mechanism by which intestinal Th17 responses are decreased in DIO is still questioned. One possible explanation could be a lack of a proper antigen stimulation of T cells by APCs (4,41). Indeed, it has been reported that after 3 weeks of HFD, intestinal APCs’ ability to prompt Th17 differentiation is reduced (4). Since DCs are mainly involved in this process, we investigated how the hBcl-2–targeted CD103+CD11b+ cDCs subpopulation may have induced Th17 polarization. We found out that RALDH tolerogenic DC function, converting the vitamin A into retinoic acid, is increased in DChBcl-2 mice relative to WT mice. The importance of such DC tolerogenic activity has been previously demonstrated at steady state (42–45). In a context of inflammation and, more precisely, in the colon of patients with ulcerative colitis, the RALDH DCs activity is impaired (27). In the context of HFD, a vitamin A–deficient diet worsens the metabolic phenotype and is associated with a more severe decrease of intestinal Th17 cells (41). The overall enhanced tolerogenic DC activity in the intestinal compartment, including draining lymph nodes, could explain the discrepant intestinal adaptive immunity that established in DChBcl-2 and WT mice. Indeed Treg, Th17, and sIgA+ B cells, all increased in DChBcl-2 compared with WT mice, are major components of intestinal homeostasis (46). Understanding how Bcl-2 survival pathway translates into increased tolerogenic cDCs needs further investigation, but Hif1-a is one interesting candidate revealed through our transcriptomic analysis. This molecular regulator that could be stabilized by Bcl-2 (47) has been shown to sustain DC tolerogenic function and associated Treg cell expansion (48).
Several research groups demonstrated how the adaptive immunity is impacting the systemic metabolism through intestinal microbiota modulations (4,5,41). Th17-mediated DIO resistance involves their ability to control microbiota composition (4,41). We demonstrated here, through cohousing and fecal microbiota transplantation approaches, that DChBcl-2 microbiota is sufficient to transmit the DIO-resistance phenotype. Analysis of fecal microbial composition revealed that WT mice depicted an enrichment toward the Epsilonproteobacteria members under HFD. Those Gram-negative bacteria represent an important source of immunogenic LPS, known to trigger metabolic endotoxemia (36). The respective microbial immunogenic property was confirmed by looking at the fecal load of bioactive LPS, which was increased in the WT fecal microbiota compared with DChBcl-2. This particular immunogenic trait of DIO-sensible WT microbiota was transmitted to the recipients, suggesting that such a discrepant immunogenic load of fecal LPS plays a role in the different phenotypes resulting from DIO treatment.
Another interesting bacterial component increased in DIO-resistant DChBcl-2 mice compared with WT mice is butyrate, an SCFA involved in many metabolic processes, promoting host fitness and shaping the intestinal immune system (32,49,50). Although butyrate has been related to impact feeding behavior and/or energy expenditure, we never noticed any differences in term of food intake or any other parameters using metabolic cages. The increased butyrate content could sustain the immunoregulatory responses that were enhanced in DIO-resistant DChBcl-2 mice. Several researchers have indeed highlighted the important role of butyrate in downregulating the expression of proinflammatory immune responses as well as promoting immunoregulatory ones (51,52). DChBcl-2 mice harbored increased polarization of Treg cells in their gut draining lymph nodes, and HFD-treated DChBcl-2 colonic Treg cells displayed an enhanced capacity to produce IL-10 compared with WT mice. Another potent mechanism of a butyrate-mediated immunoregulatory process could have directly impacted the local tolerogenic capacity of DCs, which in turn have been shown to sustain Treg cell activity (53). It has been more particularly demonstrated that butyrate-conditioned human DCs are able to prime Treg cells through the induction of RALDH function (9). This latter observation overall reinforces the importance of RALDH tolerogenic DC function and demonstrates how microbial-derived metabolites may sustain these DC-mediated immunoregulatory activities. Furthermore, with the observation that butyrate was also increased in the FT-DChBcl-2 recipients, our results strongly suggest that such discrepant microbiota compositions and functions can trigger metabolic phenotype, even in the absence of the transgene.
Article Information
Acknowledgments. The authors thank the PreclinICAN and CytoICAN platforms from IHU-ICAN, the “plate-forme de Génomique, Institut Cochin, Inserm 1016-CNRS 8104-Paris Descartes,” and the animal facility of “Centre d’expérimentation fonctionnelle, Equipe du 105B, La Pitié-Salpêtrière, Paris” for the excellent technical support. The authors also thank François Déjardin (Microenvironment and Immunity Unit - Inserm U1224, Pasteur Institute, Paris, France) and Julien Verdier (Center of Research in Myology, Institute of Myology, Institute of Myology Team 7 - Myasthenia Gravis, Paris-Sorbonne University (UPMC)/INSERM UMRS 974/AIM, Hôpital de La Pitié-Salpétriére, France) for fruitful discussions and careful reading of the manuscript P.L. and B.C. research groups are members of the French INSERM National Program on Microbiota.
Funding. This study was supported by INSERM, Sorbonne Université, the Fondation de France (00029519), and the Institute of Cardiometabolism and Nutrition (IHU-ICAN, ANR-10-IAHU-05). E.L. was supported by the Fondation Lefoulon Delalande/Institut de France and the Region Ile-de-France CORDDIM.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. E.L., T.L.R., A.G., A.L., N.V., M.G., and P.L. were responsible for conceptualization. E.L., T.L.R., A.G., A.L., C.P., J.-B.H., M.F., F.I., S.B., M.R., E.M., N.K., P.G., B.C., and P.L. were responsible for investigation and resources. E.L., T.L.R., A.G., A.L., C.P., M.P., J.-B.H., N.K., and B.C. were responsible for formal analysis. E.L., T.L.R., M.P., and B.C. wrote the original draft of the manuscript. E.L., T.L.R., M.F., F.I., T.H., E.G., N.K., P.G., N.V., B.C., and P.L. reviewed and edited the manuscript. E.L., M.G., and P.L. were responsible for funding acquisition. E.L. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.14687523.
References
- 1. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep 2018;20:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ding S, Chi MM, Scull BP, et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 2010;5:e12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luck H, Tsai S, Chung J, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 2015;21:527–542 [DOI] [PubMed] [Google Scholar]
- 4. Garidou L, Pomié C, Klopp P, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab 2015;22:100–112 [DOI] [PubMed] [Google Scholar]
- 5. Petersen C, Bell R, Klag KA, et al. T cell-mediated regulation of the microbiota protects against obesity. Science 2019;365:eaat9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawano Y, Nakae J, Watanabe N, et al. Colonic pro-inflammatory macrophages cause insulin resistance in an intestinal Ccl2/Ccr2-dependent manner. Cell Metab 2016;24:295–310 [DOI] [PubMed] [Google Scholar]
- 7. Zlotnikov-Klionsky Y, Nathansohn-Levi B, Shezen E, et al. Perforin-positive dendritic cells exhibit an immuno-regulatory role in metabolic syndrome and autoimmunity. Immunity 2015;43:776–787 [DOI] [PubMed] [Google Scholar]
- 8. Sun C-M, Hall JA, Blank RB, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 2007;204:1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory T cells via both histone deacetylase inhibition and G protein-coupled receptor 109A signaling. Front Immunol 2017;8:1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qiang Y, Xu J, Yan C, et al. Butyrate and retinoic acid imprint mucosal-like dendritic cell development synergistically from bone marrow cells. Clin Exp Immunol 2017;189:290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nopora A, Brocker T. Bcl-2 controls dendritic cell longevity in vivo. J Immunol 2002;169:3006–3014 [DOI] [PubMed] [Google Scholar]
- 12. Gautier EL, Huby T, Saint-Charles F, Ouzilleau B, Chapman MJ, Lesnik P. Enhanced dendritic cell survival attenuates lipopolysaccharide-induced immunosuppression and increases resistance to lethal endotoxic shock. J Immunol 2008;180:6941–6946 [DOI] [PubMed] [Google Scholar]
- 13. Winer DA, Luck H, Tsai S, Winer S. The intestinal immune system in obesity and insulin resistance. Cell Metab 2016;23:413–426 [DOI] [PubMed] [Google Scholar]
- 14. Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl Environ Microbiol 1997;63:2802–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cho KW, Morris DL, Lumeng CN. Flow cytometry analyses of adipose tissue macrophages. Methods Enzymol 2014;537:297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91 [DOI] [PubMed] [Google Scholar]
- 17. Burcelin R, Crivelli V, Dacosta A, Roy-Tirelli A, Thorens B. Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am J Physiol Endocrinol Metab 2002;282:E834–E842 [DOI] [PubMed] [Google Scholar]
- 18. Foley KP, Zlitni S, Denou E, et al. Long term but not short term exposure to obesity related microbiota promotes host insulin resistance. Nat Commun 2018;9:4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 2012;7:e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol 2011;4:603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013;31:563–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev 2014;260:86–101 [DOI] [PubMed] [Google Scholar]
- 23. Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol 2002;80:477–483 [DOI] [PubMed] [Google Scholar]
- 24. Tisch R. Immunogenic versus tolerogenic dendritic cells: a matter of maturation. Int Rev Immunol 2010;29:111–118 [DOI] [PubMed] [Google Scholar]
- 25. Cassani B, Villablanca EJ, De Calisto J, Wang S, Mora JR. Vitamin A and immune regulation: role of retinoic acid in gut-associated dendritic cell education, immune protection and tolerance. Mol Aspects Med 2012;33:63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laffont S, Siddiqui KRR, Powrie F. Intestinal inflammation abrogates the tolerogenic properties of MLN CD103+ dendritic cells. Eur J Immunol 2010;40:1877–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Magnusson MK, Brynjólfsson SF, Dige A, et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol 2016;9:171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol 2008;8:435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Persson EK, Uronen-Hansson H, Semmrich M, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 2013;38:958–969 [DOI] [PubMed] [Google Scholar]
- 30. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell 2014;157:121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Roy T, Llopis M, Lepage P, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013;62:1787–1794 [DOI] [PubMed] [Google Scholar]
- 32. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology 2018;154:28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caruso R, Ono M, Bunker ME, Núñez G, Inohara N. Dynamic and asymmetric changes of the microbial communities after cohousing in laboratory mice. Cell Rep 2019;27:3401–3412.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hou W-S, Van Parijs L. A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat Immunol 2004;5:583–589 [DOI] [PubMed] [Google Scholar]
- 35. Gautier EL, Huby T, Saint-Charles F, et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation 2009;119:2367–2375 [DOI] [PubMed] [Google Scholar]
- 36. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–1772 [DOI] [PubMed] [Google Scholar]
- 37. Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008;57:1470–1481 [DOI] [PubMed] [Google Scholar]
- 38. Carrington EM, Zhang JG, Sutherland RM, et al. Prosurvival Bcl-2 family members reveal a distinct apoptotic identity between conventional and plasmacytoid dendritic cells. Proc Natl Acad Sci USA 2015;112:4044–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol 2003;21:71–105 [DOI] [PubMed] [Google Scholar]
- 40. Luck H, Khan S, Kim JH, et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun 2019;10:3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hong C-P, Park A, Yang BG, et al. Gut-specific delivery of T-helper 17 cells reduces obesity and insulin resistance in mice. Gastroenterology 2017;152:1998–2010 [DOI] [PubMed] [Google Scholar]
- 42. Mora JR, Iwata M, Eksteen B, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 2006;314:1157–1160 [DOI] [PubMed] [Google Scholar]
- 43. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004;21:527–538 [DOI] [PubMed] [Google Scholar]
- 44. Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007;317:256–260 [DOI] [PubMed] [Google Scholar]
- 45. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med 2007;204:1757–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang L, Zhu L, Qin S. Gut microbiota modulation on intestinal mucosal adaptive immunity. J Immunol Res 2019;2019:4735040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trisciuoglio D, Gabellini C, Desideri M, Ziparo E, Zupi G, Del Bufalo D. Bcl-2 regulates HIF-1α protein stabilization in hypoxic melanoma cells via the molecular chaperone HSP90. PLoS One 2010;5:e11772. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Flück K, Breves G, Fandrey J, Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol 2016;9:379–390 [DOI] [PubMed] [Google Scholar]
- 49. Gao Z, Yin J, Zhang J, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009;58:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Vadder F, Kovatcheva-Datchary P, Goncalves D, et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014;156:84–96 [DOI] [PubMed] [Google Scholar]
- 51. Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013;504:451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li M, van Esch BCAM, Wagenaar GTM, Garssen J, Folkerts G, Henricks PAJ. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur J Pharmacol 2018;831:52–59 [DOI] [PubMed] [Google Scholar]
- 53. Singh N, Gurav A, Sivaprakasam S, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014;40:128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]