

Abstract
We used parabiosis to determine whether the central nervous system (CNS)-mediated antidiabetic effects of leptin are mediated by release of brain-derived circulating factors. Parabiosis was surgically induced at 4 weeks of age, and an intracerebroventricular (ICV) cannula was placed in the lateral cerebral ventricle at 12 weeks of age for ICV infusion of leptin or saline vehicle. Ten days after surgery, food intake, body weight, and blood glucose were measured for 5 consecutive days, and insulin-deficiency diabetes was induced in all rats by a single streptozotocin (STZ) injection (40 mg/kg). Five days after STZ injection, leptin or vehicle was infused ICV for 7 days, followed by 5-day recovery period. STZ increased blood glucose and food intake. Chronic ICV leptin infusion restored normoglycemia in leptin-infused rats while reducing blood glucose by ∼27% in conjoined vehicle-infused rats. This glucose reduction was caused mainly by decreased hepatic gluconeogenesis. Chronic ICV leptin infusion also reduced net cumulative food intake and increased GLUT4 expression in skeletal muscle in leptin/vehicle compared with vehicle/vehicle conjoined rats. These results indicate that leptin’s CNS-mediated antidiabetic effects are mediated, in part, by release into the systemic circulation of leptin-stimulated factors that enhance glucose utilization and reduce liver gluconeogenesis.
Introduction
Leptin, a peptide hormone produced by adipose tissue, plays an important role in glucose regulation and metabolism. We (1–3) and others (4–9) showed that leptin infusion has powerful antidiabetic actions in experimental animals, reverses severe insulin resistance in humans with lipodystrophy, and markedly reduces daily insulin requirements in patients with type 1 diabetes (9). Although leptin directly enhances glucose utilization by peripheral tissues (8), long-term increases in circulating leptin also markedly increase insulin sensitivity and whole-body glucose utilization by activating leptin receptors in the central nervous system (CNS). Chronic infusion of leptin directly into the CNS at low doses that do not increase plasma leptin concentration completely restores euglycemia in insulin-deficient diabetic rats (2,10), demonstrating that the CNS effects of leptin on glucose regulation are independent of insulin actions.
Previous acute experiments showed that acute injections of leptin into the brain enhance glucose and lipid utilization in skeletal muscle by increasing muscle sympathetic nervous system activity (11), which can be attenuated by local denervation or administration of the β1/β2 adrenergic receptor antagonist propranolol (6,12). However, combined α1 and β1/β2 adrenergic receptor blockade did not abolish the chronic antidiabetic effects of leptin or alter the ability of leptin to improve insulin sensitivity in normal rats (1,10). Also, the antidiabetic effects of leptin were not impaired by blockade of β3 adrenergic receptors (1). These findings suggest that the chronic CNS-mediated antidiabetic actions of leptin may be independent of adrenergic activation due to sympathetic nervous system activation.
Acute studies also showed that leptin or insulin injected into the CNS reduced liver gluconeogenesis, and this effect was mediated by increased parasympathetic activity via the hepatic branch of the vagus (4). However, selective hepatic vagotomy or ganglionic blockade with hexamethonium did not alter leptin’s ability to restore euglycemia in diabetic rats when chronically administered into the CNS (10). Thus, the mechanisms linking the powerful chronic CNS-mediated antidiabetic actions of leptin are still unknown and cannot be attributed mainly to activation of the parasympathetic nervous system or to sympathetically mediated adrenergic activation.
In the current study, we tested the hypothesis that leptin, acting on the CNS, may trigger release into the systemic circulation of factors that exert antidiabetic effects in the tissues. We conducted parabiosis experiments to examine whether intracerebroventricular (ICV) infusion of leptin into insulin-deficient diabetic rats reduces blood glucose levels in conjoined diabetic rats treated with vehicle. Our results indicate that chronic ICV leptin infusion, at rates that did not increase plasma leptin levels, restored normoglycemia in streptozotocin (STZ)–induced diabetic rats and reduced blood glucose concentration by ∼27% in the conjoined parabiotic rat infused with vehicle. We also found that chronic CNS leptin infusion increased GLUT4 in skeletal muscle and reduced liver gluconeogenesis and liver glucokinase (GK) mRNA expression in the leptin-infused and vehicle-infused parabiotic rats. These findings suggest that leptin acts on the CNS to trigger the release of a factor that is exchangeable in parabiosis and that improves glycemic control in diabetic conjoined rats by increasing muscle GLUT4 and reducing liver gluconeogenesis.
Research Design and Methods
Animals
Experiments were performed in inbred male Lewis rats (n = 26; Charles River Laboratories), beginning at 4 weeks of age. The experimental protocol and procedures were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Surgical Protocols
Parabiosis
Inbred Lewis rats were anesthetized with isoflurane (2–3%) and, using aseptic techniques, were conjoined by skin-to-skin anastomosis from the shoulder to the pelvic girdle as described previously (13) (Fig. 1A). Rats were monitored daily for 5 days for signs of pain and distress. Disharmony between conjoined rats was not observed in any pair. We tested if an established cross-circulation was developed by injecting Evans Blue dye intravenously in one rat of the pair and examining its appearance in the conjoined animal. Three hundred microliters of 1% Evans Blue in Hanks’ salt solution was injected into one counterpart via the penile vein. Blood was collected by tail snip at different time points in both animals of the pair (Fig. 1B). Functional cross-circulation was determined by separating plasma from the blood cells by centrifugation, diluting it, and determining Evans Blue’s absorbance at 620 nm. This procedure resulted in an average blood exchange ratio of 1.45% of total blood volume per minute calculated as previously described (14), leading to no significant differences in optical density for Evans dye between conjoined pairs as early as 2 h postinjection and reaching almost identical values at 19 h and 24 h postinjection.
Figure 1.
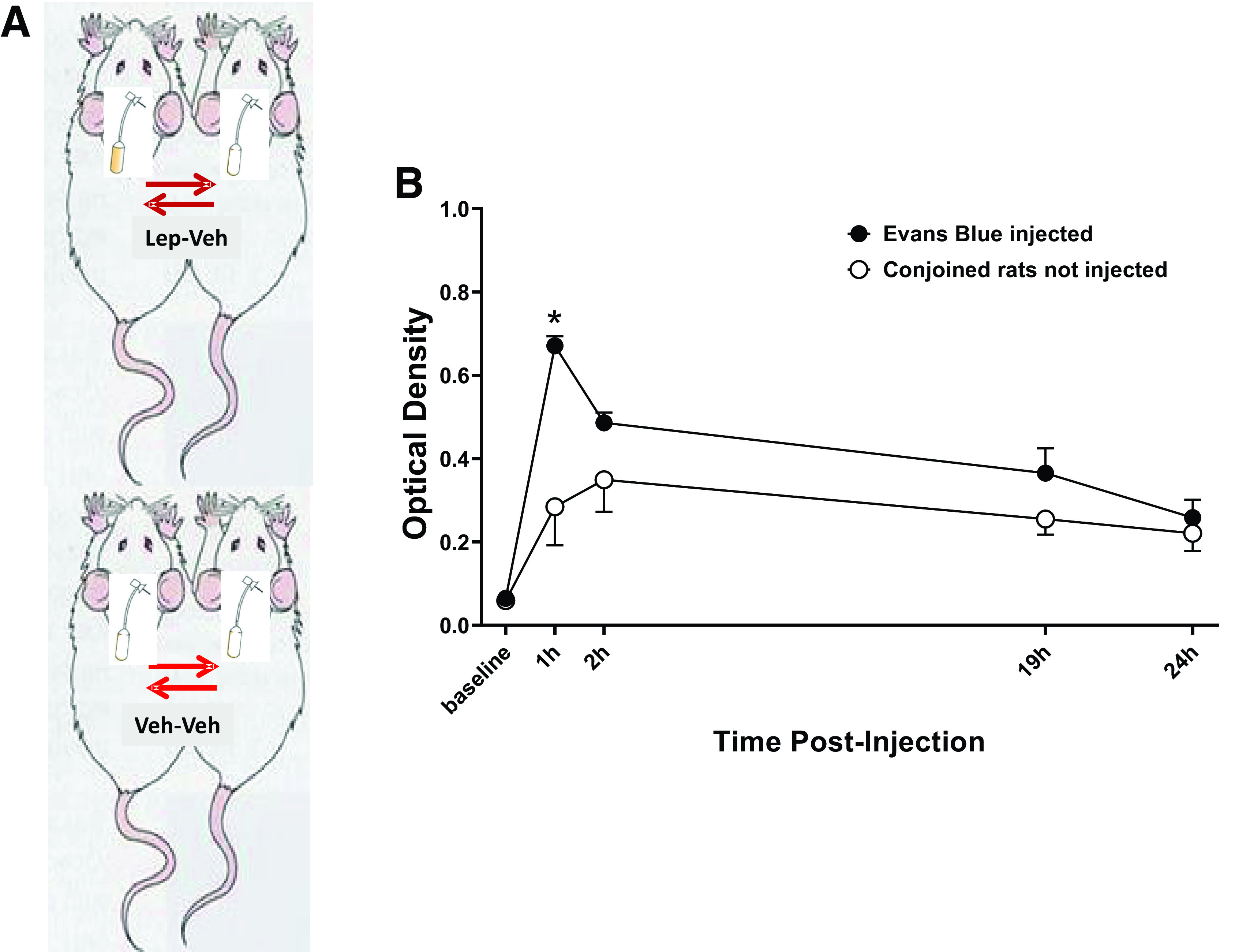
Demonstration of shared circulation between parabiotic animals by using Evans blue. A: Parabiotic experimental model, B: Evans Blue absorbance at 620 nm of plasma samples from parabiotic rats (n = 7). *P < 0.05 compared with noninjected pair using two-way ANOVA.
Implantation of ICV Cannula
Six weeks after the parabiosis surgery, a stainless-steel cannula (21 gauge; 10 mm long) was placed into the right lateral cerebral ventricle in both rats using coordinates previously described (2,3,15). The guide cannula was anchored into place with three stainless-steel machine screws, a metal cap, and dental acrylic, and a stylet was inserted to seal the cannula until use. During stereotaxic manipulation, anesthesia was maintained with 1.5% isoflurane in both rats. Eight days after recovery from surgery, accuracy of the cannula placement was tested by the dipsogenic response (immediate drinking of at least 5 mL of water in 10 min) to an ICV injection of 100 ng of angiotensin II.
Food and Water Intake, Body Weight, and Blood Glucose Measurements
The rats were allowed to recover for 8 to 10 days after ICV surgery before control measurements were recorded. The rats received food and water ad libitum during the study, and food intake and body weight were measured daily. A small amount of blood (10 μL) was collected from a small prick at the end of the tail and used to determine blood glucose levels with glucose strips (ReliOn Ultima).
Experimental Protocols
Induction of Insulin-Deficient Diabetes
After 5 days of stable control measurements, insulin-deficient diabetes was induced by a single penile vein injection of STZ (40 mg/kg, dissolved in 0.5 mL of 0.05 mol/L citrate buffer, pH 4.5; Sigma-Aldrich). Blood glucose concentration was measured each morning between 9:00 and 11:00 a.m. in all experiments.
Chronic ICV Leptin Infusion in Diabetic Rats
After at least 5 days of stable control measurements and 5 days after STZ injection, leptin (0.62 μg/h) or vehicle (1 μL/h of saline) was infused ICV for 7 days via osmotic minipump implanted subcutaneously (Model 2001; Alzet) in the scapular region. We continued to follow the animals for 5 days after leptin or vehicle ICV infusion was stopped. This leptin dose was chosen based on our previous studies after adjustment for the volume and turnover of cerebrospinal fluid (1,3). The following groups of conjoined rats were randomized: 1) ICV leptin in one rat of the pair plus ICV vehicle in the conjoined rat (Lep-Veh group); and 2) ICV vehicle in both rats of the pair (Veh-Veh group).
Whole-Body Gluconeogenesis
At the end of day 7 of leptin infusion, a separate cohort of rats from both groups (n = 5 pairs/group) were fasted for 4 h, arterial and venous (femoral) catheters were implanted under gas anesthesia for blood sampling and intravenous infusions, then the skin connecting the partners of each pair was clamped with large hemostats, and an incision was made to physically separate each conjoined partner before a 24 μCi bolus of [3-3H] glucose was given followed by continuous infusion of 0.2 μCi/min infusion for 90 min, while blood samples were taken at 10-min intervals from 60 to 90 min and processed to determine plasma [3-3H] concentration.
Western Blotting
Skeletal muscle (soleus muscle) and liver from pooled Veh-Veh and each rat of Lep-Veh pairs were dissected and homogenized in lysis buffer (KPO4, pH 7.4) and cleared by centrifugation (1,000g, 5 min at 4°C). After determination of supernatant protein concentration by the Bradford method (Bio-Rad Laboratories, Hercules, CA), 50 μg of protein was separated in a 4–15% precast linear gradient polyacrylamide gel (Bio-Rad Laboratories). After being transferred to a nitrocellulose membrane, blots were rinsed in PBS, blocked in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature, and incubated with rabbit polyclonal anti–phospho-AMPKα (p-AMPKα; Thr172) or AMPKα antibody (1:1,000; Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-phospho–mammalian target of rapamycin (p-mTOR) or mTOR antibody (1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit polyclonal anti–phospho-Akt (p-Akt; Ser473) or Akt (pan) antibody (1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit polyclonal anti–peroxisome proliferator–activated receptor γ coactivator 1α (PGC1α) (1:1,000, overnight at 4°C; Novus Biologicals), anti-mouse Glut4 (1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit polyclonal anti–acetyl CoA carboxylase (ACC) (1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit polyclonal anti–phospho-ACC (Ser79, 1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit monoclonal anti–pyruvate dehydrogenase (PDH) (1:1,000, overnight at 4°C; Cell Signaling Technology), rabbit polyclonal antiphospho-PDH (Ser293, 1:1,000, overnight at 4°C; Cell Signaling Technology), and rabbit monoclonal anti-fructose 1,6-bisphosphatase 1 (FBP1) (1:1,000, overnight at 4°C; Cell Signaling Technology). For GLUT4 protein expression, we used a membrane protein extraction kit (#89842; Thermo Fisher Scientific) to isolate integral plasma membrane proteins in skeletal muscle. The membrane was probed for mouse anti-GAPDH (1:3,000; Abcam) as loading control. The membrane was then incubated with IR700-conjugated donkey anti-rabbit IgG and IR800-conjugated donkey and anti-rabbit antibodies (1:2,000; Rockland Immunochemicals, Pottstown, PA). Antibody labeling was visualized using the Odyssey infrared scanner (LI-COR Biosciences) for simultaneous detection of two probes. Fluorescence intensity analysis was performed using Odyssey software (LI-COR Biosciences). p-AMPKα, AMPK, p-Akt, Akt, PGC1α, p-mTOR, and mTOR levels were normalized to GAPDH after subtraction of background.
RT-PCR
Livers from diabetic Veh-Veh pairs (n = 6), ICV leptin-treated (n = 7), and conjoined pairs treated with ICV vehicle were quickly removed and cleaned on an ice-cold platform. Tissue samples were immediately frozen by immersion in liquid nitrogen and stored at −80°C. Total RNA was extracted from ∼50 mg tissue, cleaned, and quantified. RNA (2.5 μg) was reverse transcribed by SuperScript VILO kit (Thermo Fisher Scientific) and treated with DNase for genomic DNA removal. Quantitative RT-PCR was performed using 4 μL cDNA with the StepOne Plus qRT-PCR system with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific). The following primer pairs were used: GK (NM_012 565), 5′-GTGTACAAGCTGCACCCGA-3′ (forward) and 5′-CAGCATGCAAGCCTTCTTG-3′ (reverse); lipoprotein lipase (LPL) (NM_012598), 5'-CTTAAGTGGAAGAACGACTCCTACT-3′ (forward) and 5′-GTCATGGCATTTCACAAACACTGCA-3′ (reverse); and PEPck (NM_198780.3), 5′-GGCGGAGCATATGCTG-ATCC-3′ (forward) and 5′-CCACAGGCACTAGGGAAGGC-3′ (reverse). Rat 18S rRNA was used as an internal control. Samples were incubated at 50°C for 2 min and then at 95°C for 2 min before cycling 40 times at 95, 60, and 72°C for 15, 30, and 60 s, respectively. A melting curve was conducted to ensure a single product was amplified.
Plasma Hormone Measurements
Blood samples (150 μL) were collected during baseline, day 5 after STZ injection, day 7 of leptin or vehicle infusion, and on the last day of recovery periods, and fasting plasma leptin and insulin concentrations were measured with ELISA kits (R&D Systems and Crystal Chem Inc., respectively). Glucose concentration was measured using ReliOn glucose strips. In rats that underwent whole-body gluconeogenesis measurement, blood samples were also collected at time 0 before [3-3H] glucose infusion for determination of plasma glucagon and corticosterone concentrations using ELISA kits (Crystal Chem Inc.).
Statistical Analyses
The results are expressed as means ± SEM. The data were analyzed by one-way ANOVA with repeated measures followed by Dunnett post hoc test for comparisons between control and experimental values within each group when appropriate. Comparisons between different groups were made by two-way ANOVA followed by Dunnett post hoc test when appropriate. Single time points between two groups were compared using t test. Statistical significance was accepted at P < 0.05.
Data and Resource Availability
The data sets of the current study are available from the corresponding author on reasonable request. No applicable resources were generated during the current study.
Results
Chronic ICV Leptin Infusion Reduced Body Weight but Not Food Intake in Insulin-Deficient Diabetic Rats
We first examined if diabetic Lewis rats exhibited similar food intake, body weight, and glucose-lowering responses to ICV leptin treatment as previously observed in other strains, and we found similar food intake and body weight responses to leptin in diabetic rats (Supplementary Fig. 1). We also found that leptin completely normalized blood glucose levels in these insulin-deficient diabetic rats (baseline: 77 ± 4; STZ: 359 ± 13; last 2 days of leptin infusion: 76 ± 2 mg/dL; P < 0.05). In parabiotic pairs, diabetes induced by STZ injection significantly increased food intake in Lep-Veh and Veh-Veh groups (from 42 ± 1 to 49 ± 3 [P < 0.05] and from 37 ± 3 to 44 ± 2 g/day [P < 0.05], respectively, for combined food consumption by the two rats of each pair) and reduced the combined body weight of the pairs (4–7%). Chronic ICV leptin infusion (only in one partner of the Lep-Veh parabiotic pair) for 7 days caused a small reduction in food intake in Lep-Veh pairs (Fig. 2A), with more pronounced decrease in the combined body weight of the Lep-Veh pair compared with the Veh-Veh–treated group (Fig. 2C). The effect of ICV leptin on the combined food intake in Lep-Veh pairs is more evident when analyzing the net cumulative food intake during the 7 days of leptin infusion (Fig. 2B), which shows a negative net cumulative food intake for Lep-Veh pairs in contrast to the always positive and gradually increasing cumulative food intake in Veh-Veh pairs. After stopping ICV leptin treatment, however, we observed an overshoot of the combined food intake in the Lep-Veh pair (67 ± 4 vs. 53 ± 3 g; P < 0.05), which was not observed after stopping the ICV vehicle infusion in the Veh-Veh group (Fig. 2B).
Figure 2.
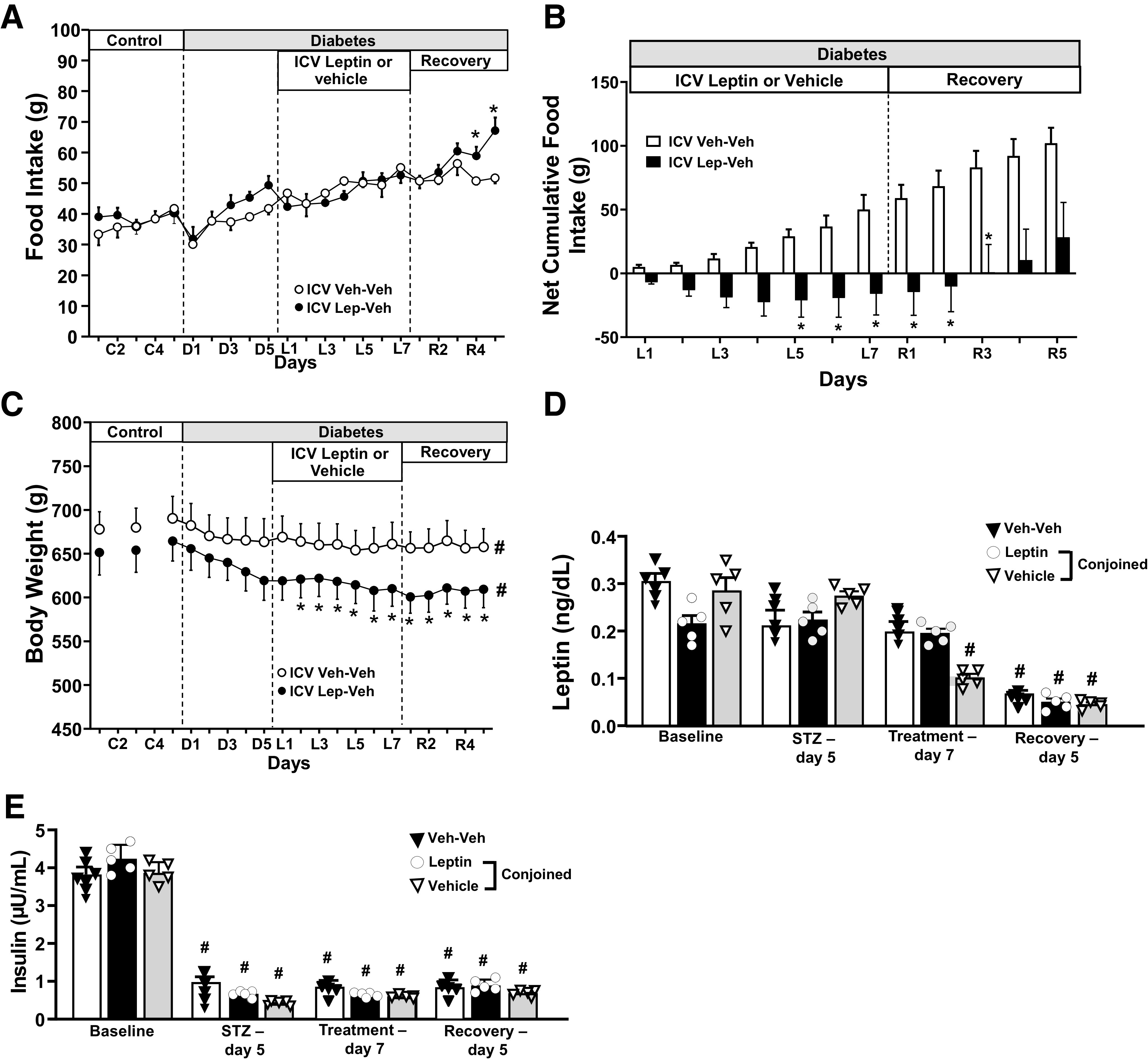
Effects of chronic ICV leptin or vehicle infusion on food intake, net cumulative food intake, body weight, and plasma hormones in parabiotic rats. Food intake (A), net cumulative food intake (B), body weight (C), fasting blood leptin (D), and insulin concentrations (E) measured during baseline, day 5 after STZ injection, day 7 of leptin or vehicle infusion, and last day of recovery periods in parabiotic rats. Values for food intake and body weight represent the sum of the pair and not for each individual rat. *P < 0.05 compared with Veh-Veh group using two-way ANOVA; #P < 0.05 compared with baseline using one-way ANOVA with repeated measures.
Chronic ICV Leptin Infusion Did Not Alter Plasma Insulin Concentration but Reduced Plasma Leptin, Glucagon, and Corticosterone Concentrations in Conjoined Pairs
Plasma leptin and insulin concentrations in the conjoined paired rats were not significantly different during baseline before STZ injection (Fig. 2D and E). Chronic ICV leptin infusion did not significantly alter plasma leptin concentration in leptin-infused rats, whereas reductions in leptin levels were observed in the vehicle-infused conjoined pair (Fig. 2D). We also observed significantly lower plasma leptin concentration in all groups during the recovery period (Fig. 2E), likely due to weight loss (664 ± 20 vs. 609 ± 20 and 690 ± 15 vs. 658 ± 13 g in the last day of the recovery period for Lep-Veh and Veh-Veh groups, respectively). As expected, STZ injection markedly reduced plasma insulin concentration, which was sustained during leptin infusion and recovery periods (Fig. 2E). STZ injection increased plasma glucagon and corticosterone concentrations in Veh-Veh rats (Supplementary Fig. 2). In Lep-Veh conjoined rats, ICV leptin infusion reduced glucagon and corticosterone levels in infused rats (∼50% and ∼35%, respectively) and in conjoined rats treated with vehicle (∼30% and ∼20%, respectively) to values that were not different than in control nondiabetic rats (Supplementary Fig. 2).
Chronic ICV Leptin Infusion Reduced Blood Glucose and Liver Gluconeogenesis in Insulin-Deficient Diabetic Parabiotic Rats
Induction of diabetes was associated with rapid development of hyperglycemia (∼350 mg/dL on day 5 post–STZ injection, (Fig. 3A and B). In Lep-Veh conjoined rats, chronic ICV leptin infusion restored normoglycemia in the leptin-treated rats and reduced glucose by ∼27% in the rats infused with vehicle (Fig. 3A and B). After stopping leptin treatment, blood glucose returned to diabetic values in leptin-treated rats and their conjoined pair. We also measured blood glucose levels in parabiotic Veh-Veh pairs (average values of both rats), which remained very high during the 17-day protocol (5 days post-STZ plus 7 days of treatment plus 5 days of recovery) (Fig. 3A and B).
Figure 3.
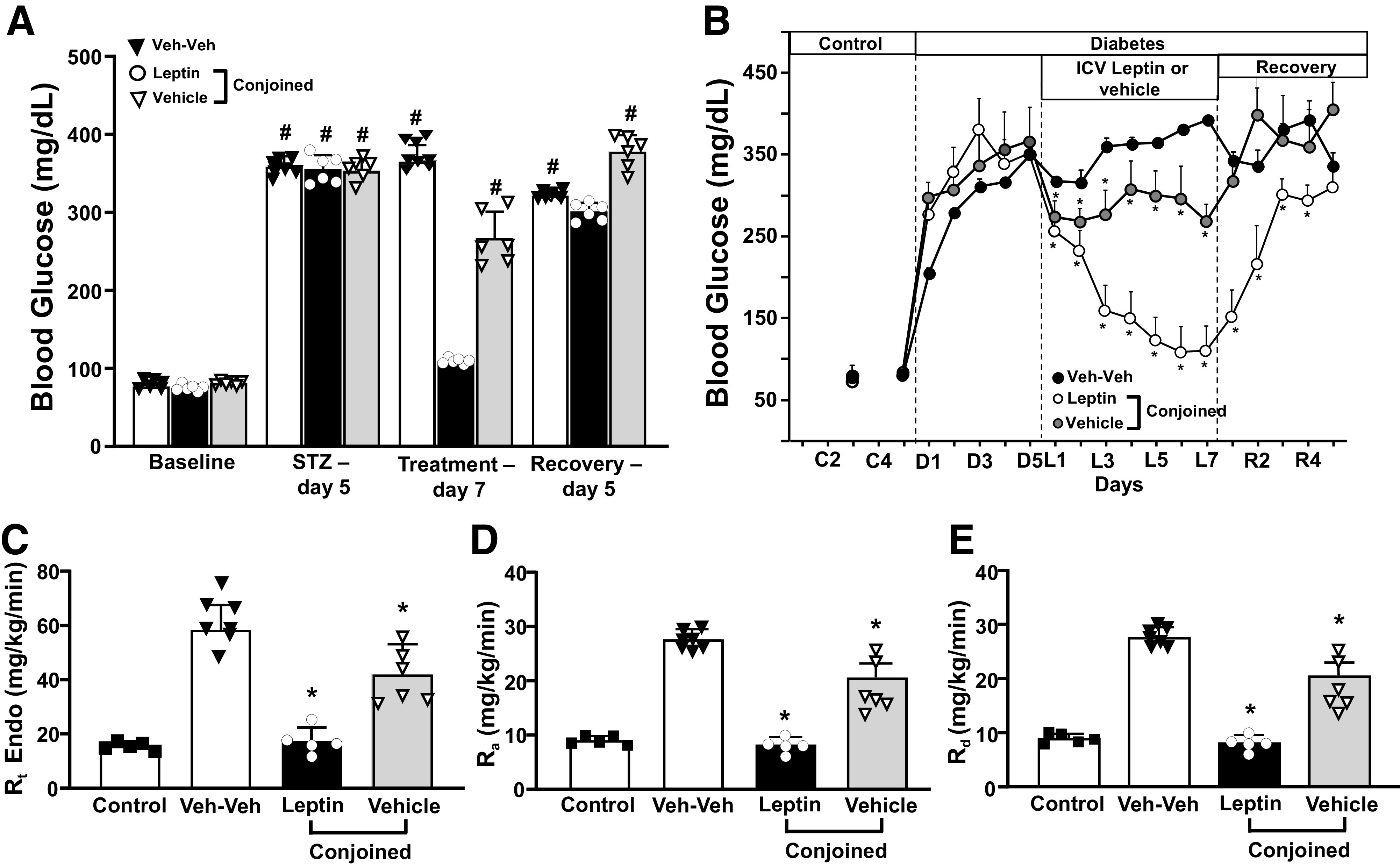
Effects of chronic ICV leptin or vehicle infusion on blood glucose and liver gluconeogenesis in parabiotic rats. A: Average of the last 3 days of blood glucose during control, STZ, leptin, or vehicle treatment and recovery periods. Daily blood glucose concentration (B), liver Rt (C), Ra (D), and Rd (E) in responses to ICV leptin or vehicle in conjoined rats. *P < 0.05 compared with Veh-Veh group using two-way ANOVA or one-way ANOVA as appropriate; #P < 0.05 compared with baseline using one-way ANOVA with repeated measures.
Using tracer dilution techniques to determine whole-body gluconeogenesis (Rt), we found that Rt was completely normalized in diabetic rats receiving ICV leptin infusion and reduced by ∼28% in diabetic rats infused with vehicle and conjoined with ICV leptin-infused rats (Fig. 3C). We further examined the Ra and Rd to determine the effects of ICV leptin infusion on these parameters in diabetic parabiotic rats. Consistent with increased gluconeogenesis, Ra was markedly increased in diabetic vehicle-treated (average of both rats) compared with control rats (Fig. 3D). ICV leptin infusion reduced Ra and Rd to values similar to observed in nondiabetic control rats and markedly reduced Ra in vehicle-infused rats that were conjoined with ICV leptin-infused rats (Fig. 3D and E).
To test if a dilutional effect of glucose “leaking” from the hyperglycemic vehicle-treated partner into its conjoined normoglycemic leptin-treated partner could contribute, at least in part, to the reduction of glucose levels in the vehicle-treated rat of the Lep-Veh pair described above, we used blood glucose levels measured during the whole-body gluconeogenesis protocol as an indirect index of glucose “leakage,” since the larger the leakage, the more glucose levels should increase in the hyperglycemic rat once it is physically separated from its normoglycemic partner. We, however, did not observe an increase in blood glucose levels in the vehicle-treated partner of the Lep-Veh pair after separation of the conjoined partners. In fact, blood glucose levels showed a tendency to fall in both partners during this protocol (Supplementary Fig. 3). This observation suggests that a dilutional effect is unlikely to have contributed importantly to the reduced blood glucose levels observed in vehicle-treated rats of the Lep-Veh pair.
Chronic ICV Leptin Infusion Increased GLUT4, Akt, ACC, and PDH Phosphorylation and Reduced mTOR Signaling in Skeletal Muscle of Insulin-Deficient Diabetic Parabiotic Rats
Chronic ICV leptin infusion increased GLUT4 (approximately threefold) in plasma membrane of leptin-treated rats (Fig. 4A). We also found significantly increased GLUT4 protein expression (∼1.5 fold) in conjoined vehicle-treated rats when compared with Veh-Veh diabetic parabiotic rats (Fig. 4A). Skeletal muscle glucose uptake is mainly regulated by GLUT4 translocation to the cell membrane in response to increased p-Akt levels. Therefore, to test if the Akt pathway is activated in Lep-Veh pairs, we measured p-Akt/Akt ratio in skeletal muscle and found that ICV leptin treatment doubled p-Akt/Akt ratio and markedly increased p-Akt/Akt in conjoined rats infused with vehicle (Fig. 4B), corroborating the increased GLUT4 protein expression in parabiotic Lep-Veh pairs. Since p-Akt can regulate AMPK and mTOR levels, we also measured expression of these proteins in skeletal muscle of parabiotic rats. We found that the p-AMPK/AMPK ratio was similar in the Lep-Veh and Veh-Veh groups (Fig. 4C); however, p-mTOR/mTOR ratio was significantly lower in Lep-Veh rats compared with Veh-Veh group (Fig. 4D). We also found increased PGC1α expression in conjoined vehicle-treated rats of the Lep-Veh group (Fig. 4E).
Figure 4.
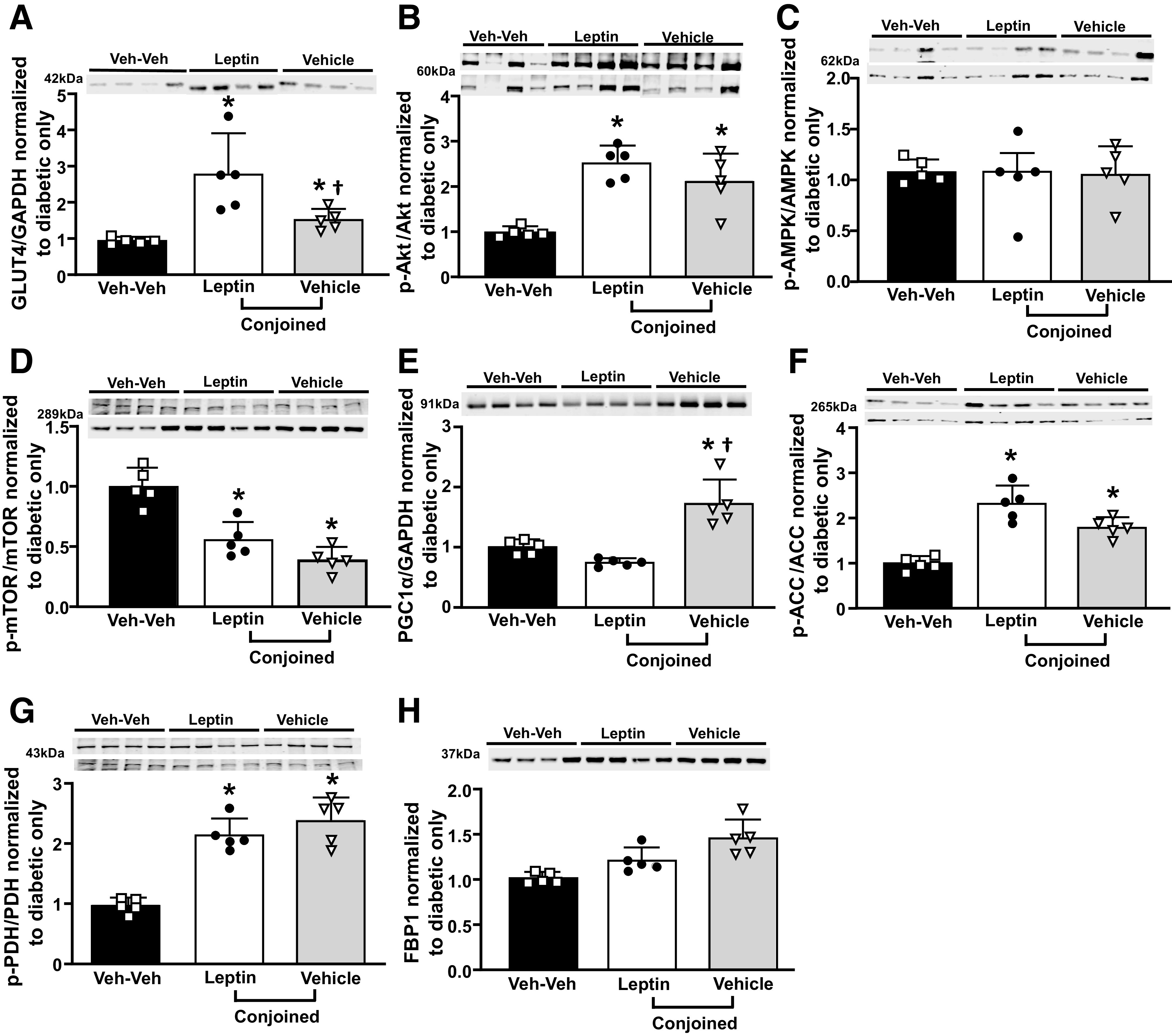
Effects of chronic ICV leptin infusion on GLUT4 plasma membrane expression and key proteins involved in glucose and fatty acid oxidation in skeletal muscle of parabiotic rats. A: GLUT4. B: p-Akt/Akt ratio. C: p-AMPK/AMPK ratio. D: p-mTOR/mTOR ratio. E: PGC1α. F: p-ACC/ACC. G: p-PDH/PDH. H: FBP1 protein expression in diabetic ICV vehicle-treated only, leptin, or vehicle-conjoined treated rats. *P < 0.05 compared with diabetic Veh-Veh using one-way ANOVA; †P < 0.05 compared with ICV leptin-infused partner using one-way ANOVA.
We further examined two key regulatory enzymes that control skeletal muscle mitochondrial fatty acid (ACC) and glucose oxidation (PDH) and found increased p-ACC/ACC and p-PDH/PDH ratios in ICV leptin-treated rats and their conjoined pairs compared with Veh-Veh rats (Fig. 4F and G). We also observed a tendency for increased FBP1 expression in the Lep-Veh group, but this increase did not reach significance (Fig. 4H).
Chronic ICV Leptin Infusion Reduced GK but Did Not Alter PEPCK and LPL mRNA Levels in Livers From Insulin-Deficient Diabetic Parabiotic Rats
We examined the effects of ICV leptin infusion on gene expression of enzymes involved in hepatic gluconeogenesis (GK and PEPCK) and lipid metabolism (LPL). Chronic ICV leptin infusion significantly reduced GK mRNA levels in Lep-Veh rats but did not alter PEPCK or LPL mRNA levels (Fig. 5A–C). Additionally, we found reduced p-ACC/ACC and p-PDH/PDH ratios as well as lower PPARγ protein expression in ICV leptin-treated rats and their conjoined pair (Fig. 5D–F), while FBP1 protein expression was increased in Lep-Veh rats compared with the Veh-Veh group (Fig. 5G).
Figure 5.
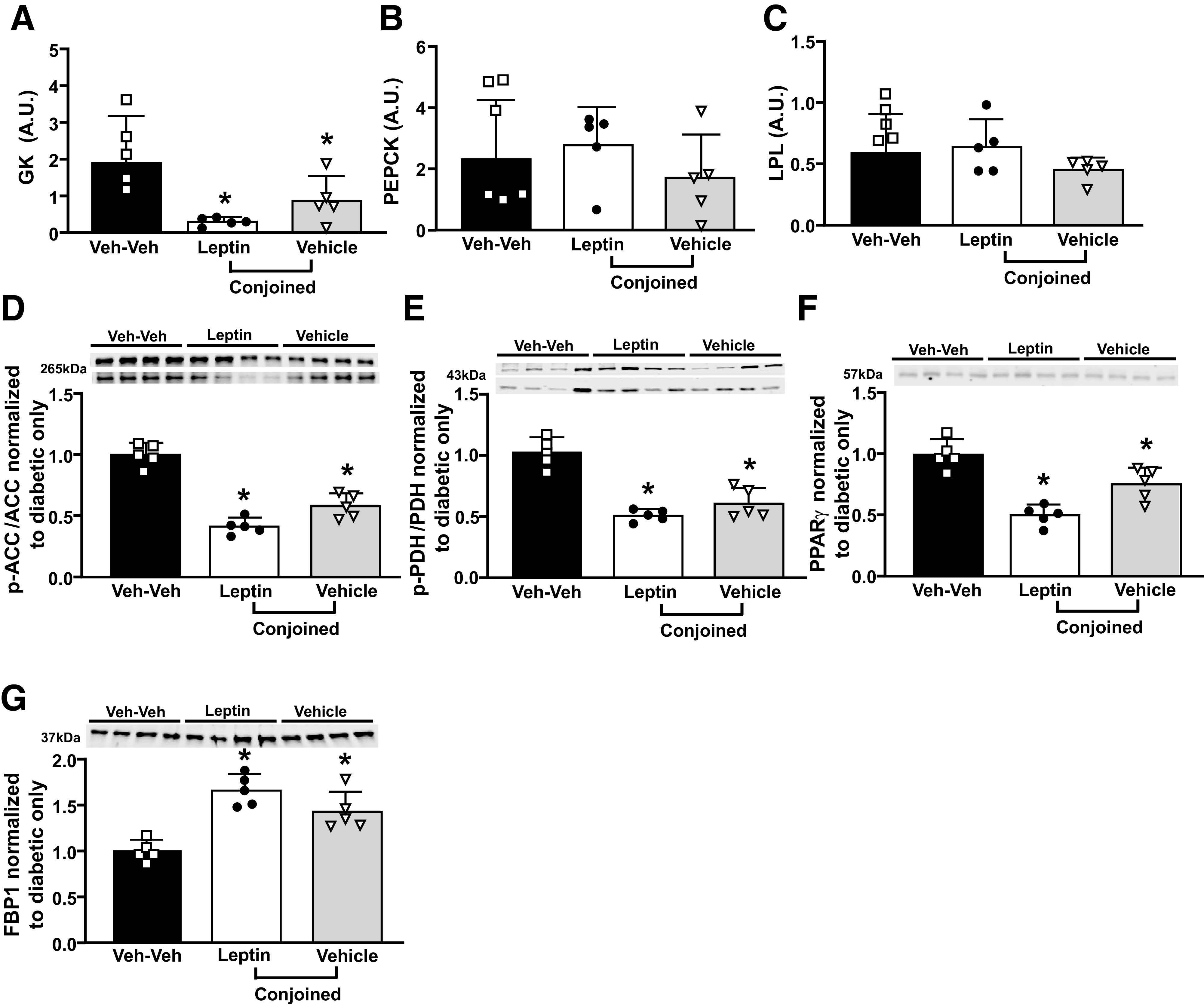
Effects of chronic ICV leptin infusion on mRNA expression of GK, PEPCK, and LPL and key proteins involved in glucose and fatty acid oxidation in livers of parabiotic rats. Quantitative RT-PCR analysis of GK (A), PEPCK (B), and LPL (C) mRNA expression in liver samples. Protein expression of p-ACC/ACC (D), p-PDH/PDH (E), PPARγ (F), and FBP1 (G) (in liver from diabetic ICV vehicle-treated only, leptin, or vehicle-conjoined treated rats. *P < 0.05 compared with diabetic Veh-Veh group using one-way ANOVA. A.U., arbitrary units.
Discussion
In the current study, we show that leptin, via its effects on the CNS, restores euglycemia in insulin-deficient diabetic rats and reduces blood glucose by 27% in parabiotic conjoined rats that were treated with vehicle. We also demonstrate that this powerful glucose-lowering effect of leptin is mediated, to a large extent, by reducing liver glucose production and increasing glucose transport into skeletal muscle and likely other tissues by increased GLUT4 content in plasma membrane. Although these effects were more pronounced in rats infused ICV with leptin, the fact that conjoined rats also showed significant decreases in plasma glucose in the absence of increases in plasma leptin suggests that the chronic CNS-mediated antidiabetic effects of leptin involve production and release of a factor that circulates in the blood to reduce gluconeogenesis and to enhance glucose uptake in peripheral tissues. Our results also support the notion that this circulating factor may have a short half-life and appears to be cleared from the circulation before reaching an equilibrium between the conjoined rats of the Lep-Veh pair. This may help explain the partial glucose-lowering effect in the conjoined rat not receiving leptin but sharing the systemic circulation with the ICV leptin-treated rat of the pair.
Another factor that may have contributed to an attenuated reduction in glucose levels in the conjoined partner not receiving leptin is hyperphagia. Although we did not quantify food intake for each rat of the pair, the fact that only a small reduction in overall food consumption was observed in the Lep-Veh pair and that leptin exerts marked anorexia in STZ-diabetic rats (1–3) suggests the conjoined partner remained hyperphagic during the 7-day treatment period. Thus, the hyperphagia in the conjoined rat not infused with leptin likely blunted the overall impact of the antidiabetic circulating factor.
The mechanisms responsible for the powerful CNS actions of leptin to improve peripheral glucose homeostasis and normalize blood glucose levels in insulin-deficient rats are still poorly understood. However, our previous studies indicate that activation of proopiomelanocortin neurons and subsequent stimulation of melanocortin 4 receptors are required for leptin’s CNS-mediated antidiabetic actions (3). Genetic deletion of leptin receptors specifically in proopiomelanocortin neurons abolished reductions in blood glucose and insulin during chronic leptin infusion (16), and pharmacological blockade of CNS melanocortin 4 receptors abolished the chronic antidiabetic effects of leptin in insulin-deficient diabetic rats (3).
The CNS-mediated antidiabetic effect of leptin appears to be largely independent of insulin. Although leptin, via its CNS actions, significantly improves insulin sensitivity in peripheral tissues, ICV leptin infusion can restore euglycemia even in insulin-deficient diabetic animals, as shown in the current study, as well as in animals with absolute lack of insulin production (17).
The antidiabetic actions of activating the CNS leptin-melanocortin system do not appear to be mediated by reductions in food intake since we previously showed that plasma glucose levels remained very high in insulin-deficient diabetic rats that were pair-fed the same amount of food consumed by leptin-treated rats (1). Also, in the current study, the anorexigenic effects of leptin cannot explain reduction in plasma glucose in the vehicle-infused rat conjoined with the ICV leptin-infused rat since these rats remained hyperphagic. Studies by other investigators also indicate that the CNS-mediated antidiabetic effects of leptin are independent of decreased food intake (18).
Previous acute studies suggest that the autonomic nervous system (ANS) may be an important mediator of leptin’s short-term effects on glucose metabolism. For example, adrenergic receptor blockade attenuated the increase in tissue glucose uptake in response to acute microinjection of leptin into the ventromedial hypothalamus (6). In addition, selective denervation attenuated the effects of leptin to increase glucose uptake in skeletal muscle (4). Leptin-mediated acute suppression of hepatic glucose production in nondiabetic animals was also prevented by hepatic vagal denervation (4), suggesting an important role of the ANS in mediating the CNS acute effects of leptin on glucose homeostasis.
In contrast to these acute studies, we found that combined α1 and β1/β2 adrenergic blockade or β3 adrenergic blockade did not alter the ability of chronic leptin treatment (7–10 days) to improve insulin sensitivity in normal rats (1) or to restore normoglycemia in insulin-deficient diabetic rats (1,10). We also showed that neither ganglionic blockade nor selective hepatic vagotomy attenuated the chronic antidiabetic effects of leptin in insulin-deficient rats (10). Thus, increased ANS activity to peripheral tissues may play an important role in mediating the short-term effects of leptin on glucose utilization, but the long-term, sustained CNS-mediated antidiabetic actions of leptin appear to be largely independent of ANS activation.
Suppression of hyperglucagonemia and of the hypothalamic-pituitary-adrenal axis have also been proposed to mediate the CNS antidiabetic effects of leptin (17,19–21). Previous studies, including our own, have shown, however, that although these effects of leptin may contribute to leptin’s antidiabetic actions, they do not appear to play a major role since removal of the pituitary gland does not prevent STZ-induced hyperglycemia (22), and infusion of glucagon-neutralizing antibody does not prevent hyperglycemia in STZ-diabetes (21). Therefore, the mechanisms responsible for leptin’s CNS-mediated powerful antidiabetic actions, whether autonomic, endocrine, or a combination of the two, are still unclear and deserve further investigation.
Previous studies showed that the total volume of blood exchanged in similar parabiosis experiments is ∼10 times the blood volume of one animal in 24 h (13). We demonstrated that a viable cross-circulation was established in all parabiotic pairs used in this study; thus, lack of effective cross-circulation cannot explain the partial glucose-lowering effect of leptin in the conjoined rats treated with vehicle. Potential explanations for this observation are: 1) lack of equilibrium in the concentration of the circulating factor between the two rats of the Lep-Veh group likely due to rapid clearance of the circulating factor; 2) the rate of blood exchange in this parabiosis model may be slow, limiting the effectiveness of the circulating factor; 3) a combination of these two possibilities; and 4) the hyperphagia of the conjoined partners may have also blunted the antidiabetic effects of the circulating factor in these rats.
It is also possible that some dilutional component of the expanded volume of distribution may have contributed to part of the reduced hyperglycemia in nonleptin-treated animals of the Lep-Veh group. However, we observed no increase in blood glucose concentration in nonleptin-treated rats after they were physically separated from their normoglycemic conjoined partners. This suggests that if leakage of glucose from the hyperglycemic rats to their normoglycemic partners did occur, it could not account significantly for the reduced hyperglycemia in the nonleptin-treated rats of the Lep-Veh pairs during the 7-day treatment period. In addition, if this dilutional effect of glucose leaking from the nonleptin-treated rat into its conjoined partner treated with leptin was a major factor responsible for the reduced hyperglycemia in the nontreated rats, we would expect compensatory stimulation of gluconeogenesis, which contrasts with the ∼25% reduction in gluconeogenesis observed in nonleptin-treated rats of the Lep-Veh group. In fact, in addition to reduced gluconeogenesis observed in nonleptin-treated rats of the Lep-Veh group, these rats also showed increased GLUT4 and p-AKT protein levels in skeletal muscle and reduced plasma glucagon concentration. Moreover, leptin treatment completely restored euglycemia in diabetic leptin-treated rats of the Lep-Veh pair, similar to what we previously observed in single-house diabetic rats undergoing similar ICV leptin treatment (1–3). Any major leakage of glucose from the hyperglycemic vehicle-infused rats into the leptin-infused rats should have attenuated the decrease in plasma glucose in the leptin-infused rats, which did not occur. These results strongly support our conclusion that leptin, via its effects on the CNS, triggers the production/release of antidiabetic factors that are capable of improving glucose homeostasis in conjoined parabiotic animals that were not treated with leptin in addition to any unavoidable dilutional effect that may have occurred between the conjoined partners.
Another important finding of this study is that although we used a higher dose of leptin than reported by some investigators, plasma leptin concentration was not altered in the Lep-Veh pairs. In fact, plasma leptin concentration was reduced in both rats of the Lep-Veh group. This observation rules out potential spillover of ICV leptin into the systemic circulation that could cross into the paired rat and induce its antidiabetic action. It is also important to note that even if some small amount of spillover of leptin into the systemic circulation occurred, this would likely have no significant impact on glucose homeostasis, since we recently demonstrated that when this same dose of leptin was administered peripherally in insulin-deficient diabetic rats for 7 days, it did not reduce blood glucose levels (23). Thus, our current results are in agreement with previous studies demonstrating that this dose of leptin infused ICV does not increase plasma leptin concentration in normal or diabetic rats (1,23). Additionally, our results support previous studies by our group and others showing that the antidiabetic effects of leptin occur independently of insulin actions, as insulin levels remained very low during the treatment period in the current study.
We further examined potential changes in signaling pathways and genes involved in glucose regulation that may contribute to the CNS-mediated antidiabetic effects of leptin in diabetic parabiotic rats. Previous studies showed that Akt and AMPK are key signal transduction pathways for GLUT4 translocation (24,25). In this study, we found significantly increased membrane fraction GLUT4 protein levels and activation of Akt signaling in parabiotic Lep-Veh pairs compared with parabiotic Veh-Veh pairs. We found no major alterations in AMPK phosphorylation and FBP1 protein expression in skeletal muscle. These findings suggest that GLUT4 translocation to the plasma membrane may be mediated by activation of Akt signaling, which, in turn, is induced by a circulating factor released from the brain. mTOR signaling has also been proposed to play an important role in glucose regulation by fine-tuning Akt activation (26,27). Despite significantly increased Akt pathway activation in Lep-Veh conjoined rats, we did not find increased mTOR signaling. In fact, this pathway was downregulated in leptin- and vehicle-treated parabiotic rats.
Although the identity and precise location of the circulating factors that mediate the chronic antidiabetic effects of leptin are still unknown, it is clear that the reductions in blood glucose levels are initiated by a stimulus in the brain. How the brain communicates with the peripheral tissues is still uncertain, but our previous studies indicate that the chronic CNS-mediated antidiabetic effects of leptin are not due to changes in ANS activity (1,10) or by a factor released by the pituitary gland. We found that hypophysectomy did not attenuate the glucose-lowering or appetite-suppressing actions of leptin in diabetic and nondiabetic rats (22). Thus, additional studies are needed to identify the powerful neuroendocrine factors that increase peripheral glucose utilization and reduce hepatic glucose output.
In summary, leptin has powerful CNS-mediated antidiabetic effects in insulin-deficient diabetic parabiotic rats that make it a potential adjunct therapy for type 1 diabetes. We provide evidence that this antidiabetic action of leptin may be mediated by a circulating factor released by the CNS that acts in peripheral tissues to reduce gluconeogenesis and increase glucose transport into the cells. Unraveling the mechanisms responsible for the powerful antidiabetic effects of leptin and how the brain communicates with peripheral tissues to control glucose levels independently of insulin and the ANS offer potentially important new therapeutic targets for diabetes and its metabolic abnormalities.
Article Information
Funding. This study was supported by the National Heart, Lung, and Blood Institute (P01 HL51971), National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK121411), and the National Institute of General Medical Sciences (P20 GM104357 and U54 GM115428) of the National Institutes of Health.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. A.A.d.S., J.E.H., and J.M.d.C. were responsible for study conception and design. A.A.d.S., X.D., Z.W., M.C.S., and J.M.d.C. were responsible for data collection. A.A.d.S., J.E.H., and J.M.d.C. were responsible for draft manuscript preparation. All authors reviewed the results and approved the final version of the manuscript. A.A.d.S. and J.M.d.C. are the guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.15067695.
References
- 1. da Silva AA, Tallam LS, Liu J, Hall JE. Chronic antidiabetic and cardiovascular actions of leptin: role of CNS and increased adrenergic activity. Am J Physiol Regul Integr Comp Physiol 2006;291:R1275–R1282 [DOI] [PubMed] [Google Scholar]
- 2. do Carmo JM, Hall JE, da Silva AA. Chronic central leptin infusion restores cardiac sympathetic-vagal balance and baroreflex sensitivity in diabetic rats. Am J Physiol Heart Circ Physiol 2008;295:H1974–H1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. da Silva AA, do Carmo JM, Freeman JN, Tallam LS, Hall JE. A functional melanocortin system may be required for chronic CNS-mediated antidiabetic and cardiovascular actions of leptin. Diabetes 2009;58:1749–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. German J, Kim F, Schwartz GJ, et al. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology 2009;150:4502–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. German JP, Thaler JP, Wisse BE, et al. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 2011;152:394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes 1999;48:1706–1712 [DOI] [PubMed] [Google Scholar]
- 7. Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 1999;341: 879–884 [DOI] [PubMed] [Google Scholar]
- 8. Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev 2011;91:389–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002;109:1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. da Silva AA, Hall JE, Moak SP, et al. Role of autonomic nervous system in chronic CNS-mediated antidiabetic action of leptin. Am J Physiol Endocrinol Metab 2017;312:E420–E428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci 2011;31: 12189–12197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 1999;48:287–291 [DOI] [PubMed] [Google Scholar]
- 13. Conese M, Carbone A, Beccia E, Angiolillo A. The fountain of youth: a tale of parabiosis, stem cells, and rejuvenation. Open Med (Wars) 2017;12:376–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris RB, Martin RJ. Specific depletion of body fat in parabiotic partners of tube-fed obese rats. Am J Physiol 1984;247:R380–R386 [DOI] [PubMed] [Google Scholar]
- 15. do Carmo JM, Bassi M, da Silva AA, Hall JE. Systemic but not central nervous system nitric oxide synthase inhibition exacerbates the hypertensive effects of chronic melanocortin-3/4 receptor activation. Hypertension 2011;57:428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension 2011;57:918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujikawa T. Central regulation of glucose metabolism in an insulin-dependent and -independent manner. J Neuroendocrinol 2021;33:e12941. [DOI] [PubMed] [Google Scholar]
- 18. Hidaka S, Yoshimatsu H, Kondou S, et al. Chronic central leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. FASEB J 2002;16:509–518 [DOI] [PubMed] [Google Scholar]
- 19. Oberlin D, Buettner C. How does leptin restore euglycemia in insulin-deficient diabetes? J Clin Invest 2017;127:450–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 2011;60:391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meek TH, Dorfman MD, Matsen ME, et al. Evidence that in uncontrolled diabetes, hyperglucagonemia is required for ketosis but not for increased hepatic glucose production or hyperglycemia. Diabetes 2015;64:2376–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. da Silva AA, Hall JE, do Carmo JM. Leptin reverses hyperglycemia and hyperphagia in insulin deficient diabetic rats by pituitary-independent central nervous system actions. PLoS One 2017;12:e0184805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. da Silva AA, Pinkerton MA, Spradley FT, Palei AC, Hall JE, do Carmo JM. Chronic CNS-mediated cardiometabolic actions of leptin: potential role of sex differences. Am J Physiol Regul Integr Comp Physiol 2021;320:R173–R181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem 2008;283:9187–9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J 2008;411:89–95 [DOI] [PubMed] [Google Scholar]
- 26. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 2017;169:381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol 2017;45:62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]