

Abstract

Chiral cis-cyclopropanes are strained rigid analogues of alkyl chains, whose study and application are limited by their difficult synthesis. A modular approach from olefin materials is enabled by the discovery of the electron donor–acceptor (EDA) interaction between 2-substituted benzothiazolines and N-hydroxyphthalimide esters. These complexes are activated by visible light without photocatalysts, and the benzothiazoline reagent plays a triple role as a photoreductant, a stereoselective hydrogen-atom donor, and a Brønsted acid. Beyond the enantioselective synthesis of cis-cyclopropanes, these results introduce benzothiazolines as accessible and easily tunable self-sensitized photoreductants.
Keywords: redox-active carbene, EDA complex, photochemistry, cis-cyclopropanes, stereoselective decarboxylation, benzothiazoline
Cyclopropanes are central motifs in organic synthesis.1 They have been widely used in the field of medicinal chemistry to improve the properties of potential drug candidates due to their resistance toward metabolic degradation and their structural rigidity (Scheme 1A).1c,2 As such, several enantioselective protocols have been developed over the years, mainly targeting the more thermodynamically and kinetically favored trans-cyclopropanes.3 In contrast, the synthesis of cis-cyclopropanes, an important class of stable and conformationally restricted alkyl chain analogues,1c,2a,4 remains a synthetic challenge with only a limited number of protocols being reported.5
Scheme 1. Background and Concept.

The asymmetric syntheses of these products require the preparation and derivatization of enantiopure (Z)-vinylboronates (Scheme 1B, top left)6 or complex catalytic systems employing transition metals7 or engineered proteins8 to obtain cyclopropyl esters. The complexity of these catalysts7,8 highlights the challenge to kinetically favor cis-cyclopropanes over their more stable trans diasteroisomers. Desirable catalytic approaches only offer limited scope9 or low diastereo- and enantioselectivity.10 In particular, the cis-cyclopropanation of alkenes employing benzylidenes is still problematic, due to the instability of the phenyldiazomethane precursors and the difficult taming of the resulting reactive intermediates. Thus, current methodologies are mostly nonenantioselective,11 and the only asymmetric catalytic methods require specific allylic alcohol materials (Scheme 1B, bottom left).12 Seminal studies with chiral iron benzylidenes have also been reported but require stoichiometric chiral complexes and are limited in scope (Scheme 1B, right).13 Also, a diastereoselective approach from the chiral pool has been demonstrated by a single example5f using the decarboxylation of a Barton ester. Nevertheless, this approach has not found further applications due to the long route to access chiral cyclopropyl Barton esters and the large excess of expensive tris(trimethylsilyl)silane required to trap the cis isomer of the cyclopropyl radical intermediate.5f
Recently, our group reported the use of redox-active diazoacetate reagents for the general enantioselective synthesis of cyclopropane building blocks from feedstock olefins.14 We envisioned that redox-active aryldiazoacetates 1 could be used to convert olefins 2 into cis-arylcyclopropanes cis-4, by means of sequential asymmetric cyclopropanation and stereoselective decarboxylative reduction of the redox-active ester (RAE, 3; Scheme 1C). The cyclopropyl radical intermediates cis-A and trans-A are known to be σ-hybridized (pyramidal) and more electrophilic than conventional alkyl σ-radicals.15a Their stereoinversion is rapid even at extremely low temperatures (kinv ≈ 108–109 s–1), and this results in thermodynamically controlled stereoselectivities.15 Thus, the feasibility of this methodology was contingent upon the design of a suitable hydrogen atom transfer (HAT) reagent that would kinetically control the reaction with the less populated (less stable) cis-cyclopropyl radical conformer (cis-A) in the equilibrium. In this respect, the high reactivity of cyclopropyl radicals15a further complicates the challenge to combine chemoselectivity (efficiency) and stereocontrol.
Initially, we evaluated known HAT reagents for the reduction of model substrate 3a (Table 1). It was found that the known nickel-catalyzed protocol,16 although highly diastereoselective, could only provide the desired cyclopropane cis-4a in low yields (entry 1). In contrast, chloroform17 could not afford high stereoselectivity (entry 2). The photoreductions using Hantzsch ester18 or N-butyl dihydronicotinamide (5b)19 recently developed by Shang et al.18a and our group19 were promising (entries 3 and 4), but further attempts to increase the yield or diastereoselectivity by tuning the structure of the dihydropyridines proved unsuccessful (see the Supporting Information for details). On account of these results, we explored the possibility of employing a reductant with a more sterically hindered hydrogen atom to impose a higher kinetic barrier in the HAT toward the undesired diasteroisomer trans-4a. 2-Substituted benzothiazolines (BTA, 6) have been used as alternative hydride sources to Hantzsch esters in transfer hydrogenation reactions.20 More recently, these compounds have been used as hydrogen atom donors in photocatalytic reactions21 requiring auxiliary thiyl radical carriers21b or metal photocatalysts.21a However, benzothiazolines have never been employed as self-sensitized photoreductants or in reductive decarboxylative reactions, as far as we know. We explored several benzothiazolines in this context, finding promising results (entries 5–10). In particular, phenyl- and tert-butylbenzothiazolines 6a,b (entries 5 and 6) provide an optimal performance with the highest diastereoselectivity, whereas other substituents result in either lower yields or lower diastereomeric ratios (entries 7–10). Control experiments with the optimal reagents 6a,b confirmed the need for blue-light irradiation for efficient reduction (entries 11 and 12). These results introduce the benzothiazoline platform for the design of cheap, easy to handle, readily available, and fine-tunable HAT reagents in reductive decarboxylative reactions without any auxiliary light harvesting or chain carrier systems.
Table 1. Optimization of the Stereoselective Decarboxylative Reduction of Redox-Active Ester 3aa.

| entry | HAT reagent | x (equiv) | solvent | 4a (%)b | d.r. (cis:trans)c |
|---|---|---|---|---|---|
| 1d,e | PhSiH3 | 1.5 | footnote e | 30 | 90:10 |
| 2f | CHCl3 | >100 | CHCl3 | 43 | 77:23 |
| 3 | 5a | 1.2 | DMSO | 76 | 90:10 |
| 4 | 5b | 1.2 | DMSO | 60 | 94:6 |
| 5 | 6a | 1.2 | DMSO | 88 | 95:5 |
| 6 | 6b | 1.2 | DMSO | 81 | 95:5 |
| 7 | 6c | 1.2 | DMSO | nd | |
| 8 | 6d | 1.2 | DMSO | 92 | 89:11 |
| 9 | 6e | 1.2 | DMSO | 54 | 88:12 |
| 10 | 6f | 1.2 | DMSO | 44 | 90:10 |
| 11d | 6a | 1.2 | DMSO | <10 | 97:3 |
| 12d | 6b | 1.2 | DMSO | nd |

See the Supporting Information for details.
Yields measured by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard.
Determined by GC-MS.
No light irradiation.
Reaction conditions: PhSiH3 (1.5 equiv), Zn (0.5 equiv), NiCl2(H2O)6 (10 mol %), 4,4′-di-t-Bu-2,2′-bipyridyl (20 mol %), THF:DMF:iPrOH 10:2:1, 40 °C.
Reaction conditions: Et3N (2 equiv), 4CzIPN (2 mol %), CHCl3.
The simplicity of the new photoreduction conditions allowed us to telescope the cyclopropanation and stereoselective reduction into a one-pot method that delivers cis-cyclopropanes cis-4 from olefins 2 and redox-active diazo compounds 1. The latter are modularly synthesized from unsubstituted NHPI-DA (7) and aryl iodides 8 through a method previously developed by our group.14b The scope of the one-pot synthesis of cis-cyclopropanes was explored using the optimal benzothiazoline 6a, which was easily prepared and stored in multigram amounts.
For the initial cyclopropanation step, we adapted the recently reported conditions by our group14b using strictly stoichiometric amounts of the olefin (1.0 equiv) and a shorter reaction time (5 h). As shown in Scheme 2A, electron-rich and electron-poor styrenes were tolerated in this transformation, furnishing cis-diarylcyclopropanes 4b–l in good yields and high enantio- and diastereoselectivities. Substitutions in various positions in the aromatic ring were tolerated. Interesting naphthyl (4i) and indolyl (4j) cyclopropanes could also be generated with this protocol. The slightly lower stereoselectivity observed in the tricyclic indene derivative 4k may be explained by a slower stereoinversion equilibrium or the particular instability of the corresponding trisubstituted cis-cyclopropyl radical intermediate. Divinylbenzene undergoes double cis-cyclopropanation to afford the C2-symmetric product 4l as a single enantiomer in 43% yield over the four reactions performed in one pot. It is important to note that negligible erosion of stereoselectivity was observed for all products relative to the intermediate cyclopropanes,14b indicating that the stereochemical information is conserved throughout the photochemical reduction step. The photodecarboxylation can also proceed with aliphatic substrates, albeit with lower stereoselectivity (82:18)22 likely due to lower facial discrimination in the key HAT process. Further optimization of the benzothiazoline structure to address the limitation in this substrate class is ongoing in our laboratories. The modular nature of the NHPI-aryldiazoacetates allows for the asymmetric transfer of a variety of aromatic fragments. This way, olefin 2a can be transformed into a number of cis-cyclopropane products decorated with different functionalities (4m–u), which include pendant alkyne (4p), nitrile (4r), and ketone (4t) moieties. To further explore the synthetic potential of this system, we obtained a cis-cyclopropane-modified phenylalanine amino acid (4u) in two steps from commercially available 4-iodophenylalanine. Moreover, the asymmetric total synthesis of the combretastatin A4 analogue 4v(6a) was achieved in three steps starting from isovanillin (9) in 39% overall yield (Scheme 2B). To put these results in perspective, twice as many steps (including a resolution) were previously required to obtain this product in <10% overall yield from comparable materials.6a
Scheme 2. Scope Studies and Synthetic Applications.
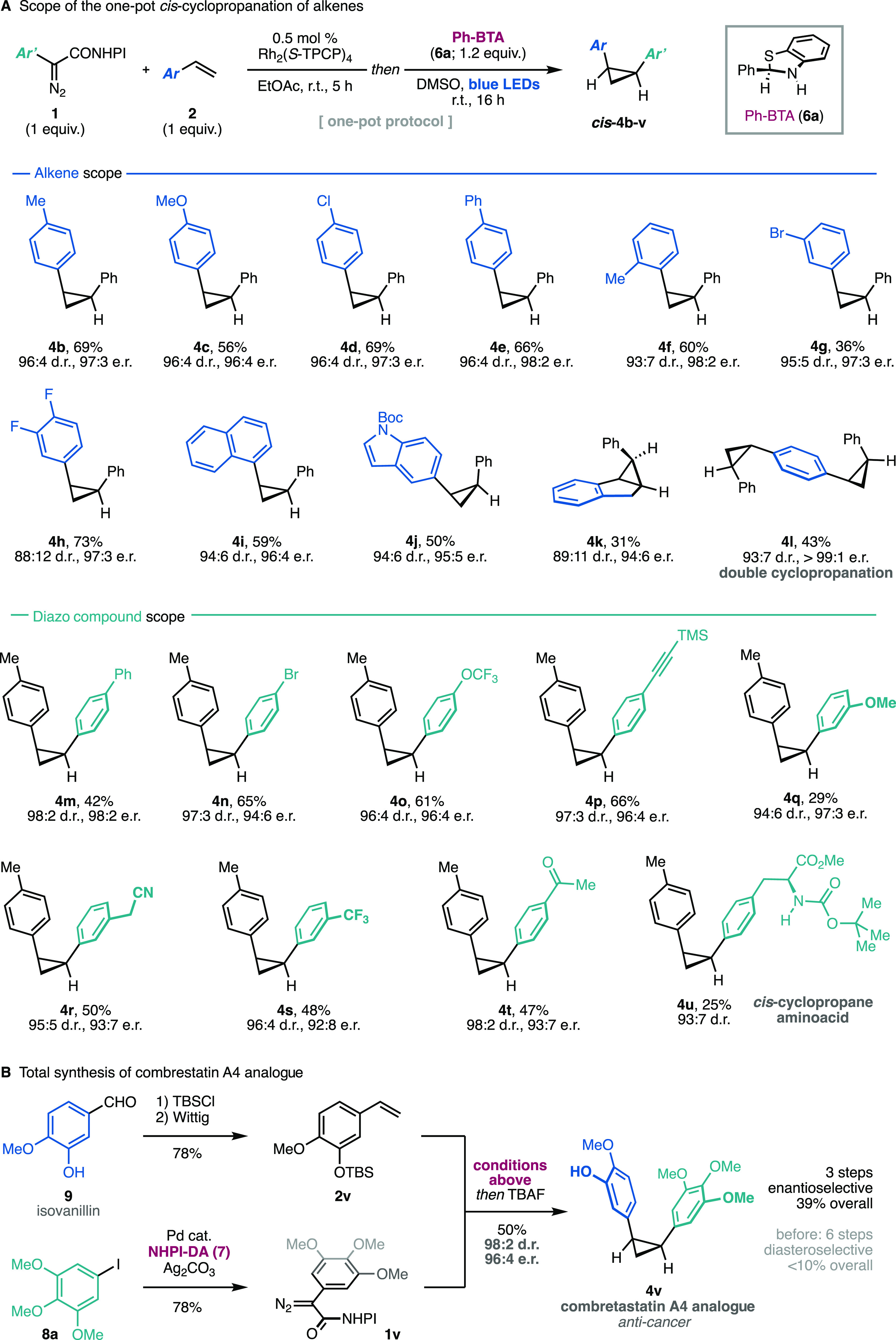
Reaction conditions: 1 (1 equiv), 2 (1 equiv), Rh2(S-TPCP)4 (0.5 mol %), EtOAc (0.05 M), r.t., 5 h; then 6a (1.2 equiv), DMSO (0.1 M), 450 nm LEDs, rt, 16 h. Isolated yields are given. Diasteromeric ratios were determined by HPLC. The photodecarboxylation of aliphatic substrates proceeds with lower stereoselectivity.22
The autonomous photoactivation of benzothiazoline 6a was unexpected on the basis of the previously known reactivity of these systems based on HAT followed by proaromatic radical reduction with auxiliary photosensitization or chain carriers.21 Thus, photochemical studies were performed to investigate the mechanism of the photoreduction. UV–visible spectroscopy revealed that neither 2-phenylbenzothiazoline 6a nor NHPI-ester 3a absorb light effectively in the visible range (Figure 1A). Upon mixing, enhanced absorption in the visible range (450 nm) is observed, and a Job plot (Figure 1B) revealed that it is at a maximum when 3a and 6a are mixed in a 1:1 stoichiometry, suggesting that a bimolecular EDA complex23 absorbing at the LED irradiation wavelength is the dominant species in solution. Clearly defined excitation and emission features (λmax = 435 nm; λem = 490 nm) of the new EDA complex can also be detected by fluorescence (Figure 1C). The formation of this species is further confirmed by time-correlated single photon counting (TCSPC), which allowed us to identify different fluorescence lifetimes for the benzothiazoline 6a (τ0 = 1.7 ns) and the EDA complex (τ = 1.4 ns). Stern–Volmer quenching studies performed by increasing the concentration of redox-active ester 3a revealed an unconventional increase in the steady-state fluorescence intensity (see the Supporting Information), while the corresponding fluorescence lifetime remained constant (Figure 1D). This feature strongly supports a static quenching scenario through the formation of a more emissive bimolecular EDA complex, and it rules out dynamic processes involving the excited state of free benzothiazoline (6a*) that would instead result in a concentration-dependent decrease in the observed fluorescence lifetime.
Figure 1.

Photophysical characterization of the stereoselective photodecarboxylation: (A) UV–visible spectra of NHPI-ester 3a, Ph-BTA (6a), and their 1:1 mixture; (B) Job plot of mixtures 3a/6a measured at 450 nm (ctot = 0.1 M); (C) Normalized excitation and emission spectra of Ph-BTA (6a; 0.02 M) and its EDA complex (0.1 M) with NHPI-ester 3a (EDA emission profile recorded at the excitation wavelength of TCSPC measurements); (D) lifetime Stern–Volmer plot of Ph-BTA (6a; 0.1 M) with NHPI-ester 3a (λex = 450 nm) determined by TCSPC. Legend: (a) λem = 490 nm; (b) λex = 450 nm; (c) λem = 410 nm; (d) λex = 355 nm.
Despite our initial hypothesis, our results could also be explained by a fast stereoretentive hydrogen atom transfer (HAT). To explore this possibility, the diastereoisomer of the redox-active cyclopropane diast-3a was independently synthesized and subjected to the reaction conditions (Scheme 3A). A similar yield and stereoselectivity for the product cis-4a is observed, demonstrating that the stereoinversion equilibrium is faster than the HAT process and that the latter is kinetically controlled. This result opens the door for future stereoconvergent applications. In principle, benzothiazoline radical cations have two hydrogen atoms susceptible to undergo the key HAT transfer. To assess their relative contribution, several deuterium incorporation experiments were carried out (Scheme 3B). A first control experiment with DMSO-d6 ruled out any relevant contribution from the solvent. The monodeuterated benzothiazoline at the benzylic carbon 6a-d1 resulted in 70% deuterium incorporation (56% yield), while the analogue deuterated in the N–H moiety 6a-d1′ led to <5% isotopic labeling and higher efficiency (77% yield). These observations indicate that the benzylic C–H bond is the main hydrogen atom donor but HAT from either the N–H moiety or the imine tautomer24 of 6a may have a secondary role. Indeed, the use of benzothiazoline 6a-d2 increased the degree of deuteration to >90%, thus accounting for the most relevant HAT processes. These results are consistent with the variable diastereoselectivities observed in the benzothiazoline screening (Table 1) with aliphatic (entries 6 and 10) and aromatic substituents (entries 5, 8, and 9) of different sizes at the C2 position, which affect the relative barriers of the HAT step. Furthermore, the quantum yield of the reaction was determined to be 0.09 ± 0.03 (Scheme 3C), disfavoring the possibility of a radical-chain mechanism. This behavior is fundamentally distinct from that of the previously known dihydronicotinamide system 5b, operating through a radical chain reaction.19,25 The formation of the EDA complex was also directly observed by 1H NMR NOE experiments (see the Supporting Information)26 that clearly evidence the spatial proximity of 3a and 6a in their equimolar mixture in DMSO (Scheme 3C).
Scheme 3. Mechanistic Experiments and Model.

See the Supporting Information for details. Diastereomeric ratios were determined by (a) GC-MS or (b) 1H NMR.
The data presented above supports the mechanism presented in Scheme 3C. Redox-active esters 3 and benzothiazoline 6 associate in solution to form the EDA complex 10, which undergoes photoinduced electron transfer (PET) in the excited state to form the radical ion pair 11. After fragmentation of the NHPI moiety with loss of CO2, the resulting cyclopropyl radical abstracts a hydrogen atom primarily from the benzylic C–H bond in the benzothiazoline radical cation (intermediate 12). The alternative HAT process through the N–H bond seems to have a secondary role. Either way, the cis-cyclopropane product 4 is kinetically preferred despite the higher energy of the cis-cyclopropyl radical in comparison to that of the alternative trans conformer (intermediate 12′). The HAT produces benzothiazole (13) and phthalimide (14) after an acid–base reaction of the phthalimidate salt 15. The alternative possibility of the cyclopropyl radical undergoing HAT directly with the benzothiazoline 6 would result in radical chain reactions that can be ruled out on the basis of the quantum yield measurements. Remarkably, the benzothiazoline 6a has a triple role in this system as a self-sensitized single-electron photoreductant to promote the fragmentation of the redox-active ester, a sterically tuned hydrogen atom source to enhance stereoselectivity, and a proton source to neutralize the phthalimidate anion byproduct.
In summary, a general and highly enantioselective method to obtain cis-diarylcyclopropanes from olefins and redox-active carbenes has been developed. This protocol allows for quick and modular access to ring-strained and conformationally strained compounds from available olefin materials, ultimately facilitating the synthesis of interesting bioactive molecules. These advances are bestowed by a new, efficient, and stereoselective photodecarboxylation driven by a novel EDA complex between redox-active esters and benzothiazoline reagents. The photophysical properties of the newly discovered system have been investigated, disclosing a new reactivity manifold of benzothiazolines as single-electron transfer reagents. Beyond enantiopure cis-cyclopropanes, these discoveries open the door for further progress in reductive decarboxylative reactions driven by benzothiazolines as a new platform to develop fine-tuned autonomous photoreductants.
Acknowledgments
We are indebted to the personnel of AstraZeneca Gothenburg and the Department of Organic Chemistry at Stockholm University for unrestricted support. The authors are grateful to Ioannis Athanassiadis (ACES, SciLifeLab) for his technical support with GC-HRMS analyses.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c03949.
Experimental procedures, characterization data, and mechanistic experiments (PDF)
Financial support from the Knut and Alice Wallenberg Foundation (KAW2016.0153) and the European Research Council (714737) is gratefully acknowledged.
The authors declare no competing financial interest.
Notes
NMR, HRMS and HPLC raw data for this article can be downloaded from Zenodo, DOI: 10.5281/zenodo.5575492.
Supplementary Material
References
- a Chen D. Y. K.; Pouwer R. H.; Richard J.-A. Recent advances in the total synthesis of cyclopropane-containing natural products. Chem. Soc. Rev. 2012, 41 (13), 4631–4642. 10.1039/c2cs35067j. [DOI] [PubMed] [Google Scholar]; b Ebner C.; Carreira E. M. Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117 (18), 11651–11679. 10.1021/acs.chemrev.6b00798. [DOI] [PubMed] [Google Scholar]; c Talele T. T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59 (19), 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- a Martin S. F.; Dwyer M. P.; Hartmann B.; Knight K. S. Cyclopropane-Derived Peptidomimetics. Design, Synthesis, and Evaluation of Novel Enkephalin Analogues. J. Org. Chem. 2000, 65 (5), 1305–1318. 10.1021/jo991288h. [DOI] [PubMed] [Google Scholar]; b Wipf P.; Skoda E. M.; Mann A.. Conformational Restriction and Steric Hindrance in Medicinal Chemistry. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth C. G., Aldous D., Raboisson P., Rognan D., Eds.; Academic Press: 2015; Chapter 11, pp 279–299. [Google Scholar]
- a Doyle M. P.; Forbes D. C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98 (2), 911–936. 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; b Lebel H.; Marcoux J.-F.; Molinaro C.; Charette A. B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103 (4), 977–1050. 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; c Pellissier H. Recent developments in asymmetric cyclopropanation. Tetrahedron 2008, 64 (30), 7041–7095. 10.1016/j.tet.2008.04.079. [DOI] [Google Scholar]
- a Shimamoto K.; Ohfune Y. Syntheses and Conformational Analyses of Glutamate Analogs:2 -(2-Carboxy-3-substituted-cyclopropyl)glycinesas Useful Probes for Excitatory Amino Acid Receptors. J. Med. Chem. 1996, 39 (2), 407–423. 10.1021/jm9502908. [DOI] [PubMed] [Google Scholar]; b Sekiyama T.; Hatsuya S.; Tanaka Y.; Uchiyama M.; Ono N.; Iwayama S.; Oikawa M.; Suzuki K.; Okunishi M.; Tsuji T. Synthesis and Antiviral Activity of Novel Acyclic Nucleosides:Discovery of a Cyclopropyl Nucleoside with Potent Inhibitory Activity against Herpesviruses. J. Med. Chem. 1998, 41 (8), 1284–1298. 10.1021/jm9705869. [DOI] [PubMed] [Google Scholar]; c Kazuta Y.; Hirano K.; Natsume K.; Yamada S.; Kimura R.; Matsumoto S.-i.; Furuichi K.; Matsuda A.; Shuto S. Cyclopropane-Based Conformational Restriction of Histamine. (1S,2S)-2-(2-Aminoethyl)-1-(1H-imidazol-4-yl)cyclopropane, a Highly Selective Agonist for the Histamine H3 Receptor, Having a cis-Cyclopropane Structure. J. Med. Chem. 2003, 46 (10), 1980–1988. 10.1021/jm020415q. [DOI] [PubMed] [Google Scholar]; d Watanabe M.; Kazuta Y.; Hayashi H.; Yamada S.; Matsuda A.; Shuto S. Stereochemical Diversity-Oriented Conformational Restriction Strategy. Development of Potent Histamine H3 and/or H4 Receptor Antagonists with an Imidazolylcyclopropane Structure. J. Med. Chem. 2006, 49 (18), 5587–5596. 10.1021/jm0603318. [DOI] [PubMed] [Google Scholar]; e Yonezawa S.; Yamamoto T.; Yamakawa H.; Muto C.; Hosono M.; Hattori K.; Higashino K.; Yutsudo T.; Iwamoto H.; Kondo Y.; Sakagami M.; Togame H.; Tanaka Y.; Nakano T.; Takemoto H.; Arisawa M.; Shuto S. Conformational Restriction Approach to β-Secretase (BACE1) Inhibitors: Effect of a Cyclopropane Ring To Induce an Alternative Binding Mode. J. Med. Chem. 2012, 55 (20), 8838–8858. 10.1021/jm3011405. [DOI] [PubMed] [Google Scholar]
- a Fang G.-H.; Yan Z.-J.; Deng M.-Z. Palladium-Catalyzed Cross-Coupling of Stereospecific Potassium Cyclopropyl Trifluoroborates with Aryl Bromides. Org. Lett. 2004, 6 (3), 357–360. 10.1021/ol036184e. [DOI] [PubMed] [Google Scholar]; b Piou T.; Romanov-Michailidis F.; Ashley M. A.; Romanova-Michaelides M.; Rovis T. Stereodivergent Rhodium(III)-Catalyzed cis-Cyclopropanation Enabled by Multivariate Optimization. J. Am. Chem. Soc. 2018, 140 (30), 9587–9593. 10.1021/jacs.8b04243. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rosenberg M. L.; Vlašaná K.; Gupta N. S.; Wragg D.; Tilset M. Highly cis-Selective Rh(I)-Catalyzed Cyclopropanation Reactions. J. Org. Chem. 2011, 76 (8), 2465–2470. 10.1021/jo102140z. [DOI] [PubMed] [Google Scholar]; d Spencer J. A.; Jamieson C.; Talbot E. P. A. One-Pot, Three-Step Synthesis of Cyclopropylboronic Acid Pinacol Esters from Synthetically Tractable Propargylic Silyl Ethers. Org. Lett. 2017, 19 (14), 3891–3894. 10.1021/acs.orglett.7b01778. [DOI] [PubMed] [Google Scholar]; e Sugawara M.; Yoshida J.-i. Remarkable γ-Effect of Tin:Acid -PromotedCyclopropanation Reactions of α -((Alkoxycarbonyl)oxy)stannaneswith Alkenes. J. Am. Chem. Soc. 1997, 119 (49), 11986–11987. 10.1021/ja973092+. [DOI] [Google Scholar]; f Yamaguchi K.; Kazuta Y.; Abe H.; Matsuda A.; Shuto S. Construction of a cis-Cyclopropane via Reductive Radical Decarboxylation. Enantioselective Synthesis of cis- and trans-1-Arylpiperazyl-2-phenylcyclopropanes Designed as Antidopaminergic Agents. J. Org. Chem. 2003, 68 (24), 9255–9262. 10.1021/jo0302206. [DOI] [PubMed] [Google Scholar]
- a Ty N.; Pontikis R.; Chabot G. G.; Devillers E.; Quentin L.; Bourg S.; Florent J.-C. Synthesis and biological evaluation of enantiomerically pure cyclopropyl analogues of combretastatin A4. Bioorg. Med. Chem. 2013, 21 (5), 1357–1366. 10.1016/j.bmc.2012.11.056. [DOI] [PubMed] [Google Scholar]; b Luithle J. E. A.; Pietruszka J. Synthesis of Enantiomerically Pure cis-Cyclopropylboronic Esters. Eur. J. Org. Chem. 2000, 2000 (14), 2557–2562. . [DOI] [PubMed] [Google Scholar]
- a Niimi T.; Uchida T.; Irie R.; Katsuki T. Co(II)–salen-catalyzed highly cis- and enantioselective cyclopropanation. Tetrahedron Lett. 2000, 41 (19), 3647–3651. 10.1016/S0040-4039(00)00433-0. [DOI] [Google Scholar]; b Suematsu H.; Kanchiku S.; Uchida T.; Katsuki T. Construction of Aryliridium–Salen Complexes: Enantio- and Cis-Selective Cyclopropanation of Conjugated and Nonconjugated Olefins. J. Am. Chem. Soc. 2008, 130 (31), 10327–10337. 10.1021/ja802561t. [DOI] [PubMed] [Google Scholar]; c Uchida T.; Irie R.; Katsuki T. Cis- and Enantio-selective Cyclopropanation with Chiral (ON+)Ru–Salen Complex as a Catalyst. Tetrahedron 2000, 56 (22), 3501–3509. 10.1016/S0040-4020(00)00273-8. [DOI] [Google Scholar]; d Zhu S.; Perman J. A.; Zhang X. P. Acceptor/Acceptor-Substituted Diazo Reagents for Carbene Transfers: Cobalt-Catalyzed Asymmetric Z-Cyclopropanation of Alkenes with α-Nitrodiazoacetates. Angew. Chem., Int. Ed. 2008, 47 (44), 8460–8463. 10.1002/anie.200803857. [DOI] [PubMed] [Google Scholar]
- Knight A. M.; Kan S. B. J.; Lewis R. D.; Brandenberg O. F.; Chen K.; Arnold F. H. Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes. ACS Cent. Sci. 2018, 4 (3), 372–377. 10.1021/acscentsci.7b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bachmann S.; Furler M.; Mezzetti A. Cis-Selective Asymmetric Cyclopropanation of Olefins Catalyzed by Five-Coordinate [RuCl(PNNP)]+ Complexes. Organometallics 2001, 20 (10), 2102–2108. 10.1021/om010020p. [DOI] [Google Scholar]; b Bonaccorsi C.; Mezzetti A. Optimization or Breakthrough? The First Highly cis- and Enantioselective Asymmetric Cyclopropanation of 1-Octene by “Electronic and Counterion” Tuning of [RuCl(PNNP)]+ Catalysts. Organometallics 2005, 24 (21), 4953–4960. 10.1021/om050396t. [DOI] [Google Scholar]
- a Alexander K.; Cook S.; Gibson C. L. cis-Selective cyclopropanations using chiral 5,5-diaryl bis(oxazoline) catalysts. Tetrahedron Lett. 2000, 41 (36), 7135–7138. 10.1016/S0040-4039(00)01232-6. [DOI] [Google Scholar]; b Hu W.; Timmons D. J.; Doyle M. P. Search of High Stereocontrol for the Construction of cis-Disubstituted Cyclopropane Compounds. Total Synthesis of a Cyclopropane-Configured Urea-PETT Analogue That Is a HIV-1 Reverse Transcriptase Inhibitor. Org. Lett. 2002, 4 (6), 901–904. 10.1021/ol017276b. [DOI] [PubMed] [Google Scholar]
- a Verdecchia M.; Tubaro C.; Biffis A. Olefin cyclopropanation with aryl diazocompounds upon catalysis by a dirhodium(II) complex. Tetrahedron Lett. 2011, 52 (10), 1136–1139. 10.1016/j.tetlet.2011.01.002. [DOI] [Google Scholar]; b Solorio-Alvarado C. R.; Wang Y.; Echavarren A. M. Cyclopropanation with Gold(I) Carbenes by Retro-Buchner Reaction from Cycloheptatrienes. J. Am. Chem. Soc. 2011, 133 (31), 11952–11955. 10.1021/ja205046h. [DOI] [PubMed] [Google Scholar]; c Solorio-Alvarado C. R.; Echavarren A. M. Gold-Catalyzed Annulation/Fragmentation: Formation of Free Gold Carbenes by Retro-Cyclopropanation. J. Am. Chem. Soc. 2010, 132 (34), 11881–11883. 10.1021/ja104743k. [DOI] [PubMed] [Google Scholar]; d Ringger D. H.; Chen P. Rational Design of a Gold Carbene Precursor Complex for a Catalytic Cyclopropanation Reaction. Angew. Chem., Int. Ed. 2013, 52 (17), 4686–4689. 10.1002/anie.201209569. [DOI] [PubMed] [Google Scholar]; e Lévesque É.; Goudreau S. R.; Charette A. B. Improved Zinc-Catalyzed Simmons–Smith Reaction: Access to Various 1,2,3-Trisubstituted Cyclopropanes. Org. Lett. 2014, 16 (5), 1490–1493. 10.1021/ol500267w. [DOI] [PubMed] [Google Scholar]; f Carden R. G.; Widenhoefer R. A. Gold Sulfonium Benzylide Complexes Undergo Efficient Benzylidene Transfer to Alkenes. Chem. - Eur. J. 2019, 25 (47), 11026–11030. 10.1002/chem.201902845. [DOI] [PubMed] [Google Scholar]; g Aggarwal V. K.; de Vicente J.; Bonnert R. V. Catalytic Cyclopropanation of Alkenes Using Diazo Compounds Generated in Situ. A Novel Route to 2-Arylcyclopropylamines. Org. Lett. 2001, 3 (17), 2785–2788. 10.1021/ol0164177. [DOI] [PubMed] [Google Scholar]
- Goudreau S. R.; Charette A. B. In Situ Generation of Zinc Carbenoids from Diazo Compounds and Zinc Salts: Asymmetric Synthesis of 1,2,3-Substituted Cyclopropanes. J. Am. Chem. Soc. 2009, 131 (43), 15633–15635. 10.1021/ja9074776. [DOI] [PubMed] [Google Scholar]
- a Wang Q.; Mayer M. F.; Brennan C.; Yang F.; Hossain M. M.; Grubisha D. S.; Bennett D. A New Approach to Diastereoselective and Enantioselective Cyclopropane Syntheses Using the Chiral Iron Carbene Complexes S- and R-[(η5-C5H5)(CO)2FeCH[(η6-o-CH3OC6H4)Cr(CO)3]]+. Tetrahedron 2000, 56 (28), 4881–4891. 10.1016/S0040-4020(00)00203-9. [DOI] [Google Scholar]; b Wang Q.; Försterling F. H.; Hossain M. M. Enantiospecific Cis-Cyclopropane Synthesis Using the Chiral Iron Carbene Complexes S- and R-(η5-C5H5)(CO)2FeCH[(η6-o-CH3C6H4)Cr(CO)3]+. Organometallics 2002, 21 (13), 2596–2598. 10.1021/om020336c. [DOI] [Google Scholar]; c Theys R. D.; Hossain M. M. Asymmetric cyclopropanation reactions via iron carbene complexes having chirality at the carbene ligand. Tetrahedron Lett. 1995, 36 (29), 5113–5116. 10.1016/00404-0399(50)0962C-. [DOI] [Google Scholar]; d Seitz W. J.; Hossain M. M. Iron Lewis acid catalyzed reactions of phenyldiazomethane and olefins: Formation of cyclopropanes with very high cis selectivity. Tetrahedron Lett. 1994, 35 (41), 7561–7564. 10.1016/S0040-4039(00)78343-2. [DOI] [Google Scholar]; e Brookhart M.; Liu Y.; Goldman E. W.; Timmers D. A.; Williams G. D. Enantioselective cyclopropane syntheses using the chiral carbene complexes (SFe)- and (RFe)-C5H5(CO)(PR3)Fe:CHCH3+. A mechanistic analysis of the carbene transfer reaction. J. Am. Chem. Soc. 1991, 113 (3), 927–939. 10.1021/ja00003a028. [DOI] [Google Scholar]; f Brookhart M.; Buck R. C. Enantioselective benzylidene transfer reactions using the chiral-at-iron benzylidene complexes (SFeSc)- and (RFeSc)-Cp(CO)(Ph2R★P_Fe = CH6H5+ (R★ = (S-2-methylbutyl) and SFe- and (RFe)-Cp(CO)PEt3Fe = CHC6H5+. J. Organomet. Chem. 1989, 370 (1-3), 111–127. 10.1016/0022-328X(89)87279-1. [DOI] [Google Scholar]
- a Montesinos-Magraner M.; Costantini M.; Ramírez-Contreras R.; Muratore M. E.; Johansson M. J.; Mendoza A. General Cyclopropane Assembly by Enantioselective Transfer of a Redox-Active Carbene to Aliphatic Olefins. Angew. Chem., Int. Ed. 2019, 58 (18), 5930–5935. 10.1002/anie.201814123. [DOI] [PubMed] [Google Scholar]; b Yu Z.; Mendoza A. Enantioselective Assembly of Congested Cyclopropanes using Redox-Active Aryldiazoacetates. ACS Catal. 2019, 9 (9), 7870–7875. 10.1021/acscatal.9b02615. [DOI] [Google Scholar]
- a Walborsky H. M. The cyclopropyl radical. Tetrahedron 1981, 37, 1625–1651. 10.1016/S0040-4020(01)98924-0. [DOI] [Google Scholar]; b Boche G.; Schneider D. R. Configurational stability of cyclopropyl radicals in electron-transfer reactions with naphthalene radical anion. Tetrahedron Lett. 1978, 19, 2327–2330. 10.1016/S0040-4039(01)91527-8. [DOI] [Google Scholar]
- Qin T.; Malins L. R.; Edwards J. T.; Merchant R. R.; Novak A. J. E.; Zhong J. Z.; Mills R. B.; Yan M.; Yuan C.; Eastgate M. D.; Baran P. S. Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem., Int. Ed. 2017, 56 (1), 260–265. 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ko E. J.; Savage G. P.; Williams C. M.; Tsanaktsidis J. Reducing the Cost, Smell, and Toxicity of the Barton Reductive Decarboxylation: Chloroform as the Hydrogen Atom Source. Org. Lett. 2011, 13 (8), 1944–1947. 10.1021/ol200290m. [DOI] [PubMed] [Google Scholar]; b Patra T.; Mukherjee S.; Ma J.; Strieth-Kalthoff F.; Glorius F. Visible-Light-Photosensitized Aryl and Alkyl Decarboxylative Functionalization Reactions. Angew. Chem., Int. Ed. 2019, 58, 10514–10520. 10.1002/anie.201904671. [DOI] [PubMed] [Google Scholar]
- a Zheng C.; Wang G.-Z.; Shang R. Catalyst-free Decarboxylation and Decarboxylative Giese Additions of Alkyl Carboxylates through Photoactivation of Electron Donor-Acceptor Complex. Adv. Synth. Catal. 2019, 361 (19), 4500–4505. 10.1002/adsc.201900803. [DOI] [Google Scholar]; b Buzzetti L.; Prieto A.; Roy S. R.; Melchiorre P. Radical-Based C–C Bond-Forming Processes Enabled by the Photoexcitation of 4-Alkyl-1,4-dihydropyridines. Angew. Chem., Int. Ed. 2017, 56 (47), 15039–15043. 10.1002/anie.201709571. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang W.; Cheng X. Hantzsch Esters as Multifunctional Reagents in Visible-Light Photoredox Catalysis. Synlett 2017, 28 (02), 148–158. 10.1055/s-0036-1588129. [DOI] [Google Scholar]; d Milligan J. A.; Phelan J. P.; Badir S. O.; Molander G. A. Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem., Int. Ed. 2019, 58 (19), 6152–6163. 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]; e van Leeuwen T.; Buzzetti L.; Perego L. A.; Melchiorre P. A Redox-Active Nickel Complex that Acts as an Electron Mediator in Photochemical Giese Reactions. Angew. Chem., Int. Ed. 2019, 58 (15), 4953–4957. 10.1002/anie.201814497. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wang P.-Z.; Chen J.-R.; Xiao W.-J. Hantzsch esters: an emerging versatile class of reagents in photoredox catalyzed organic synthesis. Org. Biomol. Chem. 2019, 17 (29), 6936–6951. 10.1039/C9OB01289C. [DOI] [PubMed] [Google Scholar]
- Chowdhury R.; Yu Z.; Tong M. L.; Kohlhepp S. V.; Yin X.; Mendoza A. Decarboxylative Alkyl Coupling Promoted by NADH and Blue Light. J. Am. Chem. Soc. 2020, 142 (47), 20143–20151. 10.1021/jacs.0c09678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chikashita H.; Miyazaki M.; Itoh K. 2-Phenylbenzothiazoline as a Reducing Agent in the Conjugate Reduction of α,β-Unsaturated Carbonyl Compounds. Synthesis 1984, 1984 (04), 308–310. 10.1055/s-1984-30819. [DOI] [Google Scholar]; b Chikashita H.; Miyazaki M.; Itoh K. Lewis acid-promoted conjugate reduction of α,β-unsaturated carbonyl compounds by 2-phenylbenzothiazoline (2-phenyl-2,3-dihydrobenzothiazole). J. Chem. Soc., Perkin Trans. 1 1987, 1 (0), 699–706. 10.1039/P19870000699. [DOI] [Google Scholar]; c Enders D.; Liebich J. X.; Raabe G. Organocatalytic Asymmetric Synthesis of trans-1,3-Disubstituted Tetrahydroisoquinolines via a Reductive Amination/Aza-Michael Sequence. Chem. - Eur. J. 2010, 16 (32), 9763–9766. 10.1002/chem.201001623. [DOI] [PubMed] [Google Scholar]; d Henseler A.; Kato M.; Mori K.; Akiyama T. Chiral Phosphoric Acid Catalyzed Transfer Hydrogenation: Facile Synthetic Access to Highly Optically Active Trifluoromethylated Amines. Angew. Chem., Int. Ed. 2011, 50 (35), 8180–8183. 10.1002/anie.201103240. [DOI] [PubMed] [Google Scholar]; e Saito K.; Akiyama T. Enantioselective organocatalytic reductive amination of aliphatic ketones by benzothiazoline as hydrogen donor. Chem. Commun. 2012, 48 (38), 4573–4575. 10.1039/c2cc31486j. [DOI] [PubMed] [Google Scholar]; f Zhu C.; Akiyama T. Benzothiazoline: Highly Efficient Reducing Agent for the Enantioselective Organocatalytic Transfer Hydrogenation of Ketimines. Org. Lett. 2009, 11 (18), 4180–4183. 10.1021/ol901762g. [DOI] [PubMed] [Google Scholar]; g Zhu C.; Saito K.; Yamanaka M.; Akiyama T. Benzothiazoline: Versatile Hydrogen Donor for Organocatalytic Transfer Hydrogenation. Acc. Chem. Res. 2015, 48 (2), 388–398. 10.1021/ar500414x. [DOI] [PubMed] [Google Scholar]
- a Tarantino K. T.; Liu P.; Knowles R. R. Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2013, 135 (27), 10022–10025. 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]; b Chen J.; Huang W.; Li Y.; Cheng X. Visible-Light-Induced Difluoropropargylation Reaction with Benzothiazoline as a Reductant. Adv. Synth. Catal. 2018, 360 (7), 1466–1472. 10.1002/adsc.201800066. [DOI] [Google Scholar]; c Bhunia A.; Studer A. Recent advances in radical chemistry proceeding through pro-aromatic radicals. Chem. 2021, 7, 2060. 10.1016/j.chempr.2021.03.023. [DOI] [Google Scholar]
- The RAE derived from 2-phenethyl-1-phenylcyclopropanecarboxylate can be decarboxylated using 6a (75% yield; 67:33 d.r.) or 6b (82% yield; 82:18 d.r.).
- a Hilinski E. F.; Masnovi J. M.; Amatore C.; Kochi J. K.; Rentzepis P. M. Charge-transfer excitation of electron donor-acceptor complexes. Direct observation of ion pairs by time-resolved (picosecond) spectroscopy. J. Am. Chem. Soc. 1983, 105 (19), 6167–6168. 10.1021/ja00357a042. [DOI] [Google Scholar]; For reviews on EDA complexes, see:; b Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41 (5), 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]; c Lima C. G. S.; de M. Lima T.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6 (3), 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]; d Mori T.; Inoue Y. Charge-transfer excitation: unconventional yet practical means for controlling stereoselectivity in asymmetric photoreactions. Chem. Soc. Rev. 2013, 42 (20), 8122–8133. 10.1039/c3cs60117j. [DOI] [PubMed] [Google Scholar]; e Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other EDA complexes involving redox-active esters, see:; f Bosque I.; Bach T. 3-Acetoxyquinuclidine as catalyst in electron donor–acceptor complex-mediated reactions triggered by visible light. ACS Catal. 2019, 9, 9103–9109. 10.1021/acscatal.9b01039. [DOI] [Google Scholar]; g Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]; h Fu M.-C.; Wang J.-X.; Shang R. Triphenylphosphine-catalyzed Alkylative Iododecarboxylation with Lithium Iodide under Visible Light. Org. Lett. 2020, 22 (21), 8572–8577. 10.1021/acs.orglett.0c03173. [DOI] [PubMed] [Google Scholar]; i Wang G.-Z.; Fu M.-C.; Zhao B.; Shang R. Photocatalytic decarboxylative alkylations of C(sp3)-H and C(sp2)-H bonds enabled by ammonium iodide in amide solvent. Sci. China: Chem. 2021, 64, 439–444. 10.1007/s11426-020-9905-1. [DOI] [Google Scholar]; j Correia J. T. M.; Piva da Silva G.; Kisukuri C. M.; André E.; Pires B.; Carneiro P. S.; Paixão M. W. Metal-Free Photoinduced Hydroalkylation Cascade Enabled by an Electron-Donor–Acceptor Complex. J. Org. Chem. 2020, 85 (15), 9820–9834. 10.1021/acs.joc.0c01130. [DOI] [PubMed] [Google Scholar]; k Kammer L. M.; Badir S. O.; Hu R.-M.; Molander G. A. Photoactive electron donor–acceptor complex platform for Ni-mediated C(sp3)–C(sp2) bond formation. Chem. Sci. 2021, 12, 5450–5457. 10.1039/D1SC00943E. [DOI] [PMC free article] [PubMed] [Google Scholar]; l de Pedro Beato E.; Spinnato D.; Zhou W.; Melchiorre P. A General Organocatalytic System for Electron Donor–Acceptor Complex Photoactivation and Its Use in Radical Processes. J. Am. Chem. Soc. 2021, 143 (31), 12304–12314. 10.1021/jacs.1c05607. [DOI] [PMC free article] [PubMed] [Google Scholar]; m McClain E. J.; Monos T. M.; Mori M.; Beatty J. W.; Stephenson C. R. J. Design and Implementation of a Catalytic Electron Donor-Acceptor Complex Platform for Radical Trifluoromethylation and Alkylation. ACS Catal. 2020, 10 (21), 12636–12641. 10.1021/acscatal.0c03837. [DOI] [Google Scholar]
- a Goetz F. J. Heterocyclic tautomerisms. I. An investigation of the 2-arylbenzothiazoline-2-(benzylideneamino)thiophenol tautomerism. Part 1. J. Heterocycl. Chem. 1967, 4 (1), 80–84. 10.1002/jhet.5570040114. [DOI] [Google Scholar]; b Goetz F. J. Heterocyclic tautomerisms. III. An investigation of the 2-arylbenzothiazoline-2-(benzylideneamino)thiophenol tautomerism. Part 3. J. Heterocycl. Chem. 1968, 5 (4), 509–512. 10.1002/jhet.5570050411. [DOI] [Google Scholar]; c Mashraqui S. H.; Kellogg R. M. A ring expansion method for the preparation of 2,3-dihydro-1,4-benzothiazines from 2-aryl-2,3-dihydrobenzothiazoles. Tetrahedron Lett. 1985, 26 (11), 1457–1460. 10.1016/S0040-4039(00)99070-1. [DOI] [Google Scholar]
- For related radical chain reactions promoted by heterocyclic reductants, see:; a Fukuzumi S.; Hironaka K.; Tanaka T. Photoreduction of alkyl halides by an NADH model compound. An electron-transfer mechanism. J. Am. Chem. Soc. 1983, 105 (14), 4722–4727. 10.1021/ja00352a034. [DOI] [Google Scholar]; b Fukuzumi S.; Mochizuki S.; Tanaka T. Photoreduction of phenacyl halides by NADH analogues. Origins of different mechanistic pathways. J. Chem. Soc., Perkin Trans. 2 1989, 10, 1583–1589. 10.1039/p29890001583. [DOI] [Google Scholar]; c Emmanuel M. A.; Greenberg N. R.; Oblinsky D. G.; Hyster T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417. 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]
- Aganda K. C. C.; Kim J.; Lee A. Visible-light-mediated direct C3-arylation of 2H-indazoles enabled by an electron-donor–acceptor complex. Org. Biomol. Chem. 2019, 17 (45), 9698–9702. 10.1039/C9OB02074H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.