

Abstract

Secondary phosphine oxides incorporating various aryl and alkyl groups were synthesized in racemic form, and these products formed the library reported in this study. TADDOL derivatives were used to obtain the optical resolution of these P-stereogenic secondary phosphine oxides. The developed resolution method showed a good scope under the optimized reaction conditions, as 9 out of 14 derivatives could be prepared with an enantiomeric excess (ee) ≥ 79% and 5 of these derivatives were practically enantiopure >P(O)H compounds (ee ≥ 98%). The scalability of this resolution method was also demonstrated. Noncovalent interactions responsible for the formation of diasteromeric complexes were elucidated by single-crystal XRD measurements. (S)-(2-Methylphenyl)phenylphosphine oxide was transformed to a variety of P-stereogenic tertiary phosphine oxides and a thiophosphinate in stereospecific Michaelis–Becker, Hirao, or Pudovik reactions.
Introduction
P-Stereogenic phosphines, phosphine oxides, or phosphonium salts represent an important class among organophophorus compounds, as these chiral derivatives have found widespread application as ligands,1 organocatalysts,2 or even biologically active compounds.3 The preparation of enantiopure P-compounds still represents a challenge, which inhibits their more diverse use. Most of the synthetic methods give the organophosphorus compound of interest in optically active form.4 Syntheses involving the preparation of P-chiral precursors followed by their incorporation in the desired scaffold represent a modular strategy carrying high synthetic potential. Bench stability, low toxicity, odorless property, and the stereospecific functionalization of their P–H bond make secondary phosphine oxides (SPOs) desired P-stereogenic precursors.5 Moreover, when the >P(O)H–>P(OH) tautomerization is utilized,6 secondary phosphine oxides can be regarded as (pre)ligands, which can be used in transition metal catalyzed asymmetric transformations.7
These properties make P-stereogenic SPOs valuable targets, and a variety of stereoselective syntheses or enantioseparation methods have been developed in recent years.8 Most of the stereoselective syntheses rely on nucleophilic substitution of enantiopure H-phosphinates (Figure 1A). The mode of preparation of the chiral H-phosphinate is a key difference between the given methods. Menthyl and later adamantyl phenyl-H-phosphinate were among the first simple optically active H-phosphinates used for stereoselective synthesis of chiral SPOs. Separation of the corresponding racemic compounds was used for the preparation of optically active H-phosphinate starting materials, and these studies also highlighted that undesired racemization may happen during nucleophilic substitution when certain organometallic reagents are used.9 Another strategy utilizes amino-alcohols as chiral templates, and hydrolysis of the chiral oxazaphospholidine intermediate gives the corresponding H-phosphinates, which are then reacted with organometallic reagents.10 Despite the advantages of the stereoselective syntheses, carefully controlled reaction conditions were required for each of the three key steps in order to avoid partial racemization and to obtain secondary phosphine oxides with high enantiomeric purity. In contrast, racemic secondary phosphine oxides can be prepared in just two steps considering the most straightforward synthetic path.11
Figure 1.

Selected examples showing the main methods for the preparation of P-stereogenic secondary phosphine oxides in optically active form (yields were calculated on the basis of the full amount of the racemate).
Thus, various separation methods of the corresponding racemates represent alternative methods for the preparation of optically active SPOs. In recent years, a few kinetic resolutions were developed employing chiral catalysts to give various P-stereogenic tertiary phosphine oxides with excellent enantiomeric excesses (ee’s).12 In theory, such reactions would give the unreacted portion of the SPO with high enanantiomeric purity (Figure 1B). However, optically active SPOs were seldom isolated from the corresponding reaction mixtures,12c−12e and a few mechanistic studies revealed that optically active SPOs may racemize to some extent under the reaction conditions.12b−12d In a recent publication, Zhang and co-workers demonstrated an organocatalytic kinetic resolution using chiral phosphine catalysts. Besides the TPO products, the corresponding optically active SPOs were also isolated in good to excellent ee-s, showing a wide scope among P-stereogenic diaryl secondary phosphine oxides.13
Classical resolutions, which are based on the formation and separation of crystalline diastereomeric salts or complexes, were among the first few methods developed for the enantioseparation of P-stereogenic secondary phosphine oxides. It is noteworthy that t-butyl-phenylphosphine oxide was the main focus of those studies. One type of resolution method involves the transformation of SPOs to the corresponding thiophosphinic acids or phosphinous acid-boranes. The enantioseparation of these acidic derivatives was elaborated with chiral bases, and optically active SPOs were obtained after the removal of the sulfur or borane. Haynes et al. used this reaction sequence for the preparation of enantiopure t-Bu(Ph)P(O)H in multigram quantities via a thiophosphinic intermediate,14 whereas the phosphinous acid-borane route was explored by Stankevič and Pietrusiewicz.15 There are a few other reports detailing the resolution of the acidic derivatives of other SPOs, but deprotection is generally omitted in those studies.16 These optical resolution methods comprise two additional steps, which potentially lower the yield and increase the risk of undesired racemization (Figure 1C).15,17 Enantiomeric separation without derivatization is a more straightforward approach to obtain optically active secondary phosphine oxides. Chromatographic separation using chiral stationary phases was used to prepare the enantiomers of chiral SPOs on a small scale (Figure 1D).18 Considering classical resolutions of secondary phosphine oxides, enantiopure t-butyl-phenylphosphine oxide could be prepared using mandelic acid or O,O′-dibenzoyltartaric acid (DBTA) as the resolving agents,11a,19 and partial enantiomeric separation was achieved with BINOL19 or chalcone sulfonic acid.11a Minnaard and co-workers found that the radical racemization of t-Bu(Ph)P(O)H could be promoted using a catalytic amount of iodine. When this racemization process was implemented into the optical resolution with O,O′-dibenzoyltartaric acid, a dynamic resolution procedure was developed, which afforded nearly full conversion of the racemate to (R)-t-Bu(Ph)P(O)H with high enantiomeric purity.20 Despite these promising results, such direct resolutions have never been extended to other secondary phosphine oxides, leaving the enantiomers of t-butyl-phenylphosphine oxide as the sole example prepared by this method (Figure 1E).
This summary shows that renewed interest in P-stereogenic SPOs populated especially the stereoselective or kinetic resolution methods, whereas the number of classical resolutions still remains low with limited scope. One of our ongoing interests involves the preparation of chiral organophosphorus compounds, and the high synthetic potential of P-stereogenic SPOs prompted us to develop enantiomeric separation methods for this compound class. To date, only tertiary phosphine oxides were made available in optically active form by our resolution methods.21 The elaboration of such a method for P-stereogenic SPOs is not straightforward, as one of the TPO functional groups capable of the formation of secondary interactions is replaced by a proton, which makes the enantiomeric recognition and the separation of SPOs challenging.
The aim of this research was to develop a versatile enantioseparation method for the preparation of optically active secondary phosphine oxides (1). On the one hand, diaryl secondary phosphine oxides (1a–i) containing substituents in the ortho-, meta-, or para-position were our focus in order to investigate the effect of the substitution pattern on the efficiency of the enantiomeric separation and to increase the number of separation methods for such SPOs. On the other hand, alkyl-aryl derivatives (1j–n) were also included in this study in order to investigate the general scope of our resolution method. The secondary interactions within the diastereomers were investigated by single crystal X-ray crystallography. Starting from a selected secondary phosphine oxide, the synthesis of various optically active P-stereogenic tertiary phosphine oxides and thiophosphinates were attempted using stereospecific transformations (Figure 2).
Figure 2.

Outline of this research project.
Results and Discussion
A synthetic strategy employing the diethylamino protecting group was selected for the synthesis of the racemic diaryl phosphine oxides (1a–i) (Scheme 1). First, P,P-dichlorophenylphosphine (4) was reacted with diethylamine, and pure N,N-diethylamino-chloro-phenylphosphine (6) could be isolated by vacuum distillation. N,N-Diethylamino-chloro-phenylphosphine (6) was the key intermediate in our divergent synthetic strategy, and it was reacted with aryllithiums to give the corresponding aminophosphines (7). Then, the toluene solution of the protected intermediate (7) was treated with cc. HCl (aq.) in order to remove the diethylamino group and to facilitate hydrolysis to give the corresponding secondary phosphine oxides (1a–i). The overall yield of this reaction sequence (1a–i) was in between 42% and 77% after a final purification by column chromatography. This one pot strategy for the aminophosphine SPO transformation was first demonstrated by Hoge et al.,22 and it has advantages over other literature procedures in which the removal of the protecting group and the hydrolysis of the intermediate are performed in two separate steps.23 The racemic alkyl-aryl secondary phosphine oxides (1j–n) were prepared according to a literature procedure by reacting PhPCl2 with ethanol and treating the in situ formed ethyl phenyl-H-phosphinate with the corresponding organometallic reagent.12b,24
Scheme 1. Preparation of Racemic Diaryl Secondary Phosphine Oxides (1a–i).

With the racemic secondary phosphine oxides (1) in hand, we began our investigation to find a suitable resolving agent. Our previous research21a−21c suggested that TADDOL derivatives might be good candidates for the optical resolution of the target secondary phosphine oxides (1). From our SPO library, the (2-methylphenyl)-phenylphosphine oxide (1a) was selected as the model racemic compound, and its resolution was attempted by various TADDOL derivatives and in a variety of solvents or solvent mixtures selected on the basis of our previous study (see the Supporting Information for details) (Scheme 2).21c Even the first few experiments suggested that spiro-TADDOL [(R,R)-2] is the most suitable resolving agent, as a change either in the aromatic groups or in the dioxolane ring of the TADDOL scaffold led to a significant decrease in the enantioseparation of secondary phosphine oxide 1a. Considering the solvents and the crystallization techniques, a precipitation-mediated crystallization from a mixture of toluene and hexane and a “classical” crystallization from 2-PrOH were the two methods that showed the most promising results affording the (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] in an ee above 95%. The other solvents investigated gave significantly lower ee values and yields (see the Supporting Information for details).
Scheme 2. Summary of the Best Results for the Resolution of P-Stereogenic Secondary Phosphine Oxides (1) with (R,R)-spiro-TADDOL [(R,R)-2].

Considering the scope of the secondary phosphine oxides (1), diaryl SPOs incorporating either a moderately electron donating methyl group or a strong electron withdrawing trifluoromethyl group in the ortho-, meta-, or para-position were investigated (1a–f). Moreover, 1-naphthyl-, 2-methoxyphenyl, and (2-phenylphenyl)-phenylphosphine oxides (1g–i) were also included in the study. Considering the alkyl-phenylphospine oxide derivatives (1j–n), normal, branched, and cycloalkyl groups along with the benzyl group were selected in order to represent a variety of alkyl chains.
On the basis of the results of the preliminary studies, the optical resolution of all target secondary phosphine oxides (1) was attempted with spiro-TADDOL [(R,R)-2] using either 2-propanol or a mixture of toluene and hexane as the solvent. It was found that even traces of water in the solvent cause inconsistent results; for this, dry solvents were used during the crystallization of the diastereomers. In all resolution experiments, the diastereomeric complexes were purified by recrystallization(s) in order to increase the diastereomeric purity. The given enantiomerically enriched secondary phosphine oxide (1) was liberated from the diastereomeric complexes by column chromatography, and the resolving agent [(R,R)-2] could also be recycled. Our previous study showed that organic solvent nanofiltration could be a scalable alternative process of such decompositions and recycling.21c Most of the resolution experiments were attempted according to half-equivalent and equivalent methods in order to investigate which separation method is the more effective one. Thus, the amount of spiro-TADDOL [(R,R)-2] varied between 0.5 and 2 equiv, depending on the composition of the diastereomeric complex formed (vide infra). Results are summarized in Scheme 2 (see the Supporting Information for the complete set of experimental data).
The results indicated that the position and the nature of the substituent significantly influence the efficiency of the enantiomeric separation. Beside (2-methylphenyl)-phenylphosphine oxide (1a), the (4-methylphenyl) and (4-trifluoromethylphenyl) derivatives (1c and 1f) could be prepared in enantiopure form (ee: 99%) in yields of 40% or 29%, respectively. Interestingly, the enantiomers of (2-methylphenyl)-phenylphosphine oxide (1a) could be separated effectively (ee > 95%, S > 0.52) either in 2-propanol or in a mixture of toluene and hexane. On the other hand, only one of the two crystallization methods afforded pure enantiomers for the para-substituted derivatives (1c and 1f). One may conclude that the substituent at the para-position has the least effect on the outcome of the resolution. A para-substituent is not in the proximity of the >P(O)H function; hence, it does not interfere with the mode of binding and noncovalent interactions formed between the given secondary phosphine oxide (1c or 1f) and spiro-TADDOL [(R,R)-2].
In contrast, ortho- and meta-substituents in a phenyl ring have a more decisive role on the overall efficiency of the resolution. 3-Methyl- or (3-trifluoromethyl-phenyl)phenylphosphine oxide (1b and 1e) could be prepared with a maximum ee of 62% or 79%, respectively. The yields were rather low (14–16%), and consequently, the resolving capability values fell in the range of 0.08–0.13. Considering the ortho-substituted derivatives (1a, 1d, 1g, and 1h), the change of the methyl group of the racemic compound to a trifluoromethyl, methoxy, or phenyl group led to a decrease in enantiomeric excess (0–67%) and in resolving capability values (0.00–0.23). The (2-trifluoromethylphenyl)-phenylphosphine oxide (1d) was the only derivative when the enantiomeric separation was completely unsuccessful, as no diastereomeric complex was formed. One may assume that the trifluoromethyl group being close to the P(O)H group lowers the Lewis basicity of the P=O function; thus, the guest molecule (1d) loses its ability to form a stable H bond with the resolving agent [(R,R)-2] (vide infra). The methoxy group is another H-bond acceptor, whereas the 2-phenylphenyl group significantly increases the steric bulk in the ortho-position. Each effect has a negative impact on the enantiomeric recognition and, consequently, on the efficiency of the resolution (ee = 67% and S = 0.12 for 1g; ee = 40% and S = 0.10 for 1h).
Moderate results were obtained for (1-naphthyl)-phenylphosphine oxide (1i) (ee = 38% and S = 0.16 or ee = 83% and S = 0.12), which was another indication that increased steric demand has a negative impact on the host–guest interactions and, consequently, on the overall efficiency of the resolution. Considering the alkyl-aryl secondary phosphine oxides (1j–n), the selected derivatives could be resolved with good overall efficiency. Four (1j, 1k, 1m, and 1n) out of the five derivatives could be prepared with an ee greater than 87%, and the resolving capability values fell in the range of 0.11–0.63. The (S)-methyl- or (t-butyl)-phenylphoshine oxide [(S)-1k or (S)-1m] were the two derivatives that could be prepared in practically enantiopure form (ee > 98%). These results indicated that the optical resolution with (R,R)-spiro-TADDOL [(R,R)-2] tolerated normal, cyclo, and aralkyl chains. The butyl-phenylphosphine oxide (1l) was the only derivative that could not be separated effectively (ee ≤ 45%; S ≤ 0.10). One underlying reason might be that the normal butyl chain cannot form stable second order interactions, whereas these contacts might be more pronounced for the more compact or sterically more demanding (cyclo)alkyl groups. The resolution of a few selected SPOs of special interest, such as the 2-methoxyphenyl-, 1-naphthyl-, and t-butyl-derivative (1g, 1i, and 1m), was also attempted with two additional spiro-TADDOL derivatives. The partial separation of the enantiomers was only successful in the case of the (1-naphthyl)-phenylphosphine oxide (1i), but a lower enantiomeric excess value could be obtained with 1i (ee: 70%) than with the spiro-TADDOL [(R,R)-2] (ee: 83%). These results show that better enantioseparation in this SPO library may not be obtained by changing the structure of the resolving agent (see Table S4 for details).
1H NMR studies revealed that the composition of the diastereomeric complexes was dependent on the solvent used. Diastereomers incorporating the given secondary phosphine oxide enantiomer (1) and (R,R)-spiro-TADDOL [(R,R)-2] in a 1:1 ratio were formed in a mixture of toluene and hexane, whereas this SPO–spiro-TADDOL [1–[(R,R)-2] ratio changed predominantly to 1:2 when 2-propanol was used as solvent. This change in the composition of the host–guest complexes indicates that the mode of binding and, consequently, the ratio of 1 and [(R,R)-2] changes when the protic 2-propanol or the aprotic toluene–hexane mixture is used as solvent. The enantiopreference of the resolving agent [(R,R)-2] was not solvent dependent,21a and the same secondary phosphine enantiomer [(R)-1 or (S)-1] was incorporated into the diastereomeric complex under both crystallization conditions. The absolute configuration of the secondary phosphine oxides 1a and 1g–n was assigned according to literature data.9b,10b,14b Moreover, a single crystal XRD measurement has also confirmed the absolute configuration of (S)-1a (vide infra).
After the evaluation of the substrate scope of our resolution method, the scalability was assessed. A submillimolar scale (ca. 0.10 g) was typically used for optimization, and the resolution of (2-methylphenyl)- and (t-butyl)-phenylphosphine oxide (1a and 1m) were also elaborated on a gram scale (ca. 2 g of 1a or 1m) using (R,R)-spiro-TADDOL [(R,R)-2] as the resolving agent and 2-PrOH as the solvent (Scheme 3). In both cases, the enantioseparation could be elaborated successfully on a gram scale affording (S)-1a and (R)-1m with an ee of 98%. Similarly to the optimization experiments, three crystallizations of the corresponding diastereomer (R)-1m·(spiro-TADDOL) were necessary to reach a high optical purity of (R)-1m, and the yield improved from 36% to 60%. Intriguingly, the (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] was obtained with an ee of 98% and in a yield of 92% (S: 0.90) even after one crystallization and decomposition of the diastereomeric complex (S)-1a·(spiro-TADDOL)2 by column chromatography. These results are somewhat better than the ones obtained after the first crystallization and decomposition of the corresponding diastereomer [(S)-1a·(spiro-TADDOL)2] during the preliminary studies (ee: 79%; yield: 83%; S: 0.65). The larger scale allowed a more efficient crystallization and a better separation of the crystalline diastereomer from the mother liquor, which consequently led to a better yield in both cases and a more simple procedure for (S)-1a. It is noteworthy that the mother liquor of the optical resolutions contained the other SPO antipode (R)-1a in excess. Enantiopure (R)-1a could be obtained by the optical resolution of this mother liquor with (S,S)-spiro-TADDOL under the same crystallization conditions, as it was demonstrated in our previous studies.21a,21c
Scheme 3. Gram-Scale Resolution of (2-Methylphenyl)-phenylphosphine Oxide (1a) and (t-Butyl)-phenylphosphine Oxide (1m) with (R,R)-spiro-TADDOL [(R,R)-2].

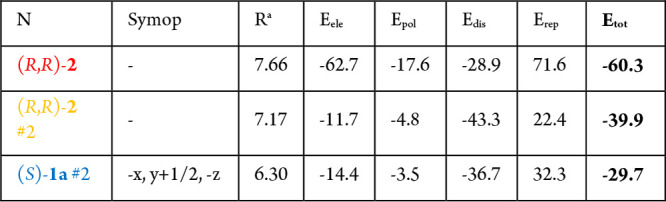
The diastereomeric complex (S)-1a·(spiro-TADDOL) (diastereomeric excess (de): 95%) prepared from a mixture of toluene and hexane was selected for X-ray analysis, and the single crystals were grown from the same solvent mixture. The absolute configuration of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] was determined using the known absolute configuration of (R,R)-spiro-TADDOL [(R,R)-2] as reference. The crystal lattice comprises the SPO (S)-1a and the resolving agent (R,R)-2 in a 1:1 ratio, which is in accordance with our NMR measurements. A characteristic intramolecular H bond is present between the two OH groups of (R,R)-spiro-TADDOL [(R,R)-2],21a,21c and the (S)-1a and (R,R)-2 host–guest molecules are held together by an O–H···O bridge (Figure 3).25 Energy calculations were conducted with the Crystal Explorer program using the B3LYP/6-31G(d,p) level of theory (Table 1).26 Total energies (Etot) were the sum of the Coulomb interactions (Eele), polar interactions (Epol), dispersion interactions (Edis), and repulsive interactions (Erep). The four energy components were scaled in the total energy (Etot = 1.019Eele + 0.651Epol + 0.901Edis + 0.811Erep). Interaction energies were investigated for a 3.8 Å cluster around the selected secondary phosphine oxide (S)-1a. These DFT calculations also confirmed that the two O–H···O interactions are the strongest ones in the crystal lattice (Table 1 and Figure 3). The second and third strongest interaction come from another neighboring (R,R)-spiro-TADDOL [(R,R)-2] or (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] molecule, respectively. The O–H···O hydrophilic contact turns the (R,R)-2 and (S)-1a molecules toward each other, creating a tight fit in between the host and guest molecules and creating a hydrophobic outer surface for this associate. Hirshfeld surface analysis also showed a high number of H contacts of the associated molecules. The H atom interactions corresponded to 84% of the intermolecular interactions, while the O atoms had a contribution of only 2.7% (see the Supporting Information for details).27
Figure 3.

(a) Hydrogen bonds formed in the diastereomeric complex (S)-1a·(spiro-TADDOL). Nonhydrogen atom ellipsoids are drawn on a 30% probability level. (b) The neighboring molecules with the strongest interactions of (S)-1a for the calculation of interaction energies (DFT calculation details can be found in Table 1).
Table 1. Calculated Total Energies for the Neighboring Molecules of (S)-1ab.
As the last step of this study, a few typical SPO transformations were also elaborated in order to prepare the corresponding tertiary phosphine oxides [(S)-3a–d], hydroxyphosphine-oxides [(S)-3f and (SP,RC)-3g], and a tiophosphonate [(R)-3e] (Scheme 4). Previously, such transformations were described using mainly the t-butyl-phenylphosphine oxide (1m) as the benchmark starting material. As the nature of the substituents may influence the P-inversion barrier,28 the (S)-(2-methylphenyl)phenylphosphine oxide [(S)-1a] with an ee of 98% was chosen as a diaryl-SPO model compound for these stereoselective transformations. One method for the preparation of P-stereogenic tertiary phosphine oxides is the alkylation of metalated secondary phosphine oxides (Michaelis–Becker reaction).14b,29 First, (S)-1a was added to the THF suspension of NaH at 0 °C to prepare (2-MePh)PhP(ONa), and it was treated with the corresponding primary alkyl halide to give (S)-methyl-, ethyl- or benzyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3a–c] in yields of 76–87%. The enantiomeric purity of the tertiary phosphine oxides (S)-3a–c was in the range of 95–98%, indicating high configurational stability of the secondary phosphine oxides and their deprotonated derivatives under the reaction conditions (Scheme 4A). Literature data suggested that this reaction proceeds with a retention at the P-stereogenic center.14b,29
Scheme 4. Stereoselective Synthesis of Various P-Stereogenic Tertiary Phosphine Oxides and a Thiophosphinate (3) from (S)-(2-Methylphenyl)phenylphosphine Oxide [(S)-1a].

Hirao coupling of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] with 1-bromonaphtalene was also elaborated. Despite the popularity of this method for the formation of P–Csp2 bonds,30 only a few reports can be found on the stereospecific arylation of P-stereogenic secondary phosphine oxides,10c,13,31 and the chiral SPOs might be prone to (partial) racemization under the reaction conditions.12b The P–C cross coupling of (S)-1a was performed in toluene under reflux conditions in the presence of Pd(PPh3)4 using K2CO3 as the base, and these conditions were successfully applied for t-BuPhP(O)H (1m).31b (S)-(1-Naphthyl)-(2-methylphenyl)-phenylphosphine oxide [(S)-3d] could be prepared with a yield of 85% and in an ee of 88%. The high reaction temperature might be responsible for the partial racemization observed (ee0 = 98% → ee = 88%). This cross-coupling is expected to take place with retention of the P absolute configuration (Scheme 4B).31
The addition of (S)-1a to formaldehyde or benzaldehyde was also studied in the presence of aq. NaOH. The (S)-hydroxymethyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3f] could be prepared in nearly quantitative yield (98%) and with an ee of 96%. The addition of benzaldehyde to P-stereogenic SPOs involves the formation of a new carbon stereogenic center. 31P NMR spectra indicated that this reaction was highly diastereoselective, as the ratio of the two diastereomers was 97.5:2.5 in the crude product. Moreover, the (SP,RC) enantiomer could be prepared with excellent enantiomeric purity (ee > 98%) and in a yield of 80% (Scheme 4D). We believe that a crystallization induced asymmetric transformation is the underlying reason for this high diastereoselectivity.20,32 Crystalline product (3g) was immediately formed when (S)-1a was reacted with benzaldehyde at room temperature. The subsequent stirring of the aqueous suspension at 80 °C for 16 h allows the dissolution of the more soluble diastereomer and its transformation to the less soluble stereoisomer via an equilibrium cascade, which involves a retro-Pudovik reaction in the supernatant. This hypothesis was supported by a few control experiments. When the reaction was performed either at a lower temperature or with shorter reaction times, a significant decrease in the diastereomeric excess of 3g could be observed. As in the literature,33 X-ray crystallography was used for characterization. The XRD measurement of (SP,RC)-3g confirmed that the reaction proceeds with retention of the P-configuration, which is in accordance with the literature.20,34 Moreover, the (R) absolute configuration of the formed carbon stereogenic center could also be established. The neighboring hydroxy(phenyl)methyl-(2-methylphenyl)-phenylphosphine oxides [(SP,RC)-3g] were held together by H bonds formed between OH and P=O functional groups of two different (SP,RC)-3g molecules (see the Supporting Information for details). (S)-1a was also reacted with elemental sulfur to prepare the corresponding thiophosphonic acid 3e with retention of the P-configuration (Scheme 4C).9b,16d Interestingly, the enantiomeric excess values of 3g could be determined by 31P NMR using (S)-1-naphthylethylamine as a chiral solvating agent, as no chiral chromatographic method could be developed for 3g.
Conclusions
In conclusion, a series of P-stereogenic secondary phosphine oxides (1) was prepared in racemic form, and their enantiomeric separation was elaborated with (R,R)-spiro-TADDOL [(R,R)-2]. The scope of the SPOs comprised several diaryl-derivatives containing a phenyl group and another aryl moiety with various substitution patterns (1a–i) and alkyl-phenylphospine oxides (1j–n) incorporating normal, branched, or cycloalkyl groups or a benzyl function. According to the preliminary experiments, the optical resolution of the target secondary phosphine oxides (1) was elaborated with (R,R)-spiro-TADDOL [(R,R)-2] in 2-propanol or in a mixture of toluene and hexane. Nine derivatives (1a, 1c, 1e, 1f, 1i–k, 1m, and 1n) could be prepared with an ee > 79% (yield: 12–65%), and five of these derivatives (1a, 1c, 1f, 1k, and 1m) were practically enantiopure secondary phosphine oxides (ee > 98%, yield 27–65%). To date, this current method has the widest scope among classical resolution methods developed for P-chiral secondary phosphine oxides. Our results indicated that substituents in the para-position of a phenyl group has the least effect on the overall efficiency of the resolution. On the contrary, an increased steric bulk, especially in the ortho-position, had a negative effect on the enantiomeric separation. Several alkyl-phenylphospine oxides (1j, 1k, 1m, and 1n) containing various normal, cyclo, or aralkyl chains could be prepared with good enantiomeric purity and in acceptable yields (ee: 87–99%; yield: 12–41%). A gram-scale resolution of (2-methylphenyl)-phenylphosphine oxide (1a) was performed, indicating the scalability of this resolution method. X-ray quality crystals were prepared from (S)-1a·(spiro-TADDOL), which was complemented with the DFT calculation to study the main interaction between the secondary phosphine oxide and the resolving agent.
Starting from (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] (ee: 98%), several P-stereogenic phosphine oxides and a thiophosphonate (3) were prepared with excellent enantiomeric excess values (ee > 95%). Only the Hirao coupling of secondary phosphine oxide (S)-1a with 1-bromonaphtalene afforded (S)-(1-naphthyl)-(2-methylphenyl)-phenylphosphine oxide [(S)-3d] with a somewhat lower optical purity (ee: 88%), which could be attributed to the partial racemization of (S)-1a at the elevated reaction temperature.
Experimental Section
General Information
The commercially available reagents were purchased from commercial sources, and they were used without further purification unless otherwise stated. The TADDOL derivatives [(R,R)-2, (R,R)-SI-1, (R,R)-SI-3, and (R,R,R,R)-SI-4],35,36 benzyl-phenylphosphine oxide (1j), methyl-phenylphosphine oxide (1k), butyl-phenylphosphine oxide (1l), tert-butyl-phenylphosphine oxide (1m), and cyclohexyl-phenylphosphine oxide (1n) were synthesized as described in the literature, and their analytical data were identical to the ones reported in the literature.12b,24 The solvents were purchased from Merck Chemical Ltd., and they were used without further purification. Solvents used in moisture sensitive reactions were purified and dried according to the standard procedures.37 Dry solvents were stored over molecular sieves of 3 or 4 Å. The 31P, 19F, 13C, and 1H NMR spectra were taken on a Bruker AV-300 or DRX-500 spectrometer operating at 121.5, 282.4, 75.5, and 300 MHz or 202.5, 470.6, 125.8, and 500 MHz, respectively. Chemical shifts (δ) are given in parts per million (ppm). Chemical shifts (δ) were for 1H and 13C in CDCl3 and referenced to 7.26 and 77.16 ppm, respectively. An 85% solution of H3PO4 was the external reference for 31P NMR chemical shifts. Coupling constants are expressed in Hertz (Hz). The following abbreviations are used: s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet, and dd = doublet of doublets. The exact mass measurements were performed using an Agilent 6230C TOF LCMS System with Agilent Jet Stream source in positive ESI mode (buffer: ammonium-formate in water/acetonitrile; drying gas: 325 °C; capillary: 3000 V; Fragmentor 100 V). LCMS measurements were performed using an Agilent 1100 and Agilent 6130 LCMS system in positive and negative electrospray mode. For the single crystal structure determination, the intensity data were collected on a Rigaku RAXIS-RAPID II diffractometer (using a graphite monochromator; Mo Kα radiation, λ = 0.71075 Å). The crystals were measured with fiber. Crystal Clear (developed by Rigaku Company) software was used for data collection and refinement.38 Numerical and empirical absorption corrections were applied to the data.39 The structures were solved by direct methods. Anisotropic full-matrix least-squares refinements were performed on F2 for all non-hydrogen atoms. Hydrogen atoms bonded to C atoms were placed in calculated positions and refined in a riding-model approximation. The computer programs used for the structure solution, refinement, and analysis of the structures were Shelx,40 Sir2014,41 Wingx,42 Platon,43 and Crystal Explorer.44 Melting points were obtained on a melting point apparatus and are uncorrected. The preparation of the air and moisture sensitive intermediates (6 and 7) or racemic secondary phosphine oxides (1a–n) was carried out under a nitrogen atmosphere in Schlenk-type reaction vessels using standard Schlenk techniques.45 Thin layer chromatography (TLC) was performed on Merck precoated Silica gel 60 F254 or neutral Al2O3 60 F254 aluminum plates with realization by UV irradiation, iodine, and phosphomolybdic acid. Column chromatography was performed on Silica gel 60 or neutral Al2O3 90 with a particle size of 0.063–0.200 mm supplied by Merck. Flash column chromatography was performed using a Combi-Flash (Teledyne ISCO) with gradient elution on Silica gel 60 or neutral Al2O3 90 columns. The enantiomeric excess (ee) value of 3e was determined by 31P NMR using 5.0 mg (20 μmol) of the analyte, 4.8 μL (30 μmol) of (S)-naphthyl-ethyl-amine as CSA, and 750 μL of CDCl3 as solvent. Enantiomeric excess (ee) values of secondary and tertiary phosphine oxides 1a–n, 3a–d, and 3f–g were determined by chiral HPLC on a PerkinElmer Series 200 instrument equipped with Phenomenex Lux 5 μm Cellulose-1, Phenomenex Lux 5 μm Cellulose-2, Phenomenex Lux 5 μm Amylose-2 column, or Kromasil 5-Amycoat (250 × 4.6 mm ID, a mixture of hexane/ethanol was used as the eluent with a flow rate of 0.8 mL/min (T = 20 °C, UV detector α = 254 nm). Exact chromatographic parameters are detailed in Table S7. Optical rotations were determined on a Perkin–Elmer 341 polarimeter.
Preparation of Racemic Diaryl Secondary Phosphine Oxides (1a–i)
N,N-Diethylamino-chloro-phenylphosphine (6)
Under nitrogen atmosphere, 21 mL (260 mmol) of anhydrous pyridine was added dropwise over 5 min to a solution of 17 mL (130 mmol) of P,P-dichlorophenylphosphine (4) in 100 mL of anhydrous hexane, and then, the opaque solution was stirred for 10 min at 25 °C. To this reaction mixture, 27 mL (260 mmol) of anhydrous diethyl-amine (5) was added dropwise over 20 min. The resulting white suspension was refluxed in an oil bath for 3 h, and it was cooled to 25 °C. The precipitated salt was filtered through a sintered glass funnel under nitrogen atmosphere, and it was washed 2× with 50 mL of anhydrous hexane. The filtrate was concentrated on the rotary evaporator. Distillation under reduced pressure yielded 24 g (84%) of N,N-diethylamino-chloro-phenylphosphine (6) as colorless oil. bp: 95 °C @ 0.5 mbar; (lit. bp: 68–70 °C @ 0.13 mbar).46 The distillate was used immediately.
(2-Methylphenyl)-phenylphosphine oxide (1a) (Representative Procedure I)
Under nitrogen atmosphere, a solution of 4.3 g (20 mmol) N,N-diethylamino-chloro-phenylphosphine (6) in 10 mL of anhydrous THF was added dropwise over 1 h to a solution of 20 mmol of 2-methylphenyllithium at −78 °C [2-methylphenyllithium was prepared by adding 13 mL (20 mmol) of n-butyllithium (1.6 M hexane solution) to a solution of 2.4 mL (20 mmol) of 2-bromotoluene and 24 mL of anhydrous THF over 30 min at −78 °C followed by an additional 30 min of stirring at the same temperature]. The reaction mixture was stirred for 2 h at −78 °C. Then, it was allowed to warm to 25 °C, and it was stirred overnight. The solution was concentrated under vacuum, and the residue was redissolved in 20 mL of toluene. Fifteen mL of cc. HCl solution was added over 10 min at 0 °C, and the resulting emulsion was stirred for 15 min. Then, the pH was adjusted to 8 by adding 20% aqueous NaOH solution. The phases were separated, and the aqueous layer was extracted with DCM (3 × 40 mL). The organic layers were combined, dried (Na2SO4), and evaporated. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 4.0 g (92%) of (2-methylphenyl)-phenylphosphine oxide (1a) as a white solid. mp: 115–117 °C (mplit: 121.5–123 °C);9b31P{1H} NMR (121.5, MHz, CDCl3) δ 21.9 (δlit 21.9);9b1H NMR (500 MHz, CDCl3) δ 8.09 (d, 1H, J = 480.1), 7.71–7.60 (m, 3H), 7.53–7.50 (m, 1H), 7.46–7.43 (m, 3H), 7.33–7.30 (m, 1H), 7.23–7.20 (m, 1H), 2.35 (bs, 3H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 141.5 (d, J = 9.2), 132.8 (d, J = 2.6), 132.3 (d, J = 3.0), 132.3 (d, J = 13.6), 131.5 (d, J = 100.4), 131.4 (d, J = 10.4), 130.8 (d, J = 11.4), 129.6 (d, J = 101.0), 128.9 (d, J = 12.7), 126.0 (d, J = 13.6), 20.2 (d, J = 6.8); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14OP 217.0782; Found 217.0786.
(3-Methylphenyl)-phenylphosphine oxide (1b)
The (3-methylphenyl)-phenylphosphine oxide (1b) was prepared according to Representative Procedure I by reacting 4.3 g (20 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 10 mL of anhydrous THF with 20 mmol of 3-methylphenyllithium at −78 °C. 3-Methylphenyllithium was prepared from 2.4 mL (20 mmol) of 3-bromotoluene and 13 mL (20 mmol) of n-butyllithium (1.6 M hexane solution) in 24 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 3.0 g (70%) of (3-methylphenyl)-phenylphosphine oxide (1b) as a clear oil. 31P{1H} NMR (121.5 MHz, CDCl3) δ 21.8; 1H NMR (500 MHz, CDCl3) δ 8.04 (d, 1H, J = 479.6), 7.72–7.67 (m, 2H), 7.58–7.44 (m, 5H), 7.38–7.36 (m, 2H), 2.38 (s, 3H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 139.0 (d, J = 12.7), 133.5 (d, J = 2.9), 132.6 (d, J = 3.0), 131.8 (d, J = 101.1), 131.3 (d, J = 11.1), 130.8 (d, J = 11.4), 130.0 (d, J = 100.8), 129.0 (d, J = 12.8), 128.9 (d, J = 13.7), 127.8 (d, J = 11.8), 21.5; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14OP 217.0782; Found 217.0786.
(4-Methylphenyl)-phenylphosphine oxide (1c)
The (4-methylphenyl)-phenylphosphine oxide (1c) was prepared according to Representative Procedure I by reacting 4.3 g (20 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 10 mL of anhydrous THF with 20 mmol of 4-methylphenyllithium at −78 °C. The 4-methylphenyllithium was prepared from 2.5 mL (20 mmol) of 4-bromotoluene and 13 mL (20 mmol) of n-butyllithium (1.6 M hexane solution) in 24 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 2.9 g (68%) of (4-methylphenyl)-phenylphosphine oxide (1c) as white solid. mp: 70–72 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 21.6 (δlit 21.9);471H NMR (500 MHz, CDCl3) δ 8.04 (d, 1H, J = 479.5), 7.71–7.66 (m, 2H), 7.61–7.46 (m, 5H), 7.31–7.29 (m, 2H), 2.40 (s, 3H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 143.4 (d, J = 2.6), 132.6 (d, J = 3.0), 131.9 (d, J = 101.7), 130.9 (d, J = 11.9), 130.9 (d, J = 96.5), 130.8 (d, J = 11.4), 129.8 (d, J = 13.3), 129.0 (d, J = 12.8), 21.8; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14OP 217.0782; Found 217.0782.
(2-Trifluoromethylphenyl)-phenylphosphine oxide (1d)
The (2-trifluoromethylphenyl)-phenylphosphine oxide (1d) was prepared according to the Representative Procedure I by reacting 5.2 g (24 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 12 mL of anhydrous THF with 24 mmol of 2-trifluoromethylphenyllithium at −78 °C. The 2-trifluoromethylphenyllithium was prepared from 3.3 mL (24 mmol) of 2-bromobenzotrifluoride and 15 mL (24 mmol) of n-butyllithium (1.6 M hexane solution) in 28 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 3.8 g (59%) of (2-trifluoromethylphenyl)-phenylphosphine oxide (1d) as white solid. mp: 45 °C; 31P{1H} NMR (121.5 MHz, CDCl3) δ 15.4 (q, J = 7.5); 1H NMR (500 MHz, CDCl3) δ 8.33 (dq, 1 H, J = 510.4, 3.1), 8.21–8.17 (m, 1H), 7.80–7.78 (m, 1H), 7.76–7.69 (m, 2H), 7.68–7.61 (m, 2H), 7.57–7.53 (m, 1H), 7.49–7.45 (m, 2H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 134.2 (d, J = 6.9), 132.8 (d, J = 3.0), 132.6 (d, J = 2.5), 132.3 (d, J = 10.7), 131.5 (d, J = 105.0), 131.4 (dd, J = 39.1, 6.4), 130.8 (d, J = 11.7), 130.6 (d, J = 101.4), 129.0 (d, J = 13.2), 126.8 (m), 123.8 (dd, J = 274.3, 2.6); 19F NMR (282.4 MHz, CDCl3) δ −56.9 (d, J = 7.4); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H11F3OP 271.0500; Found 271.0499.
(3-Trifluoromethylphenyl)-phenylphosphine oxide (1e)
(3-Trifluoromethylphenyl)-phenylphosphine oxide (1e) was prepared according to Representative Procedure I by reacting 5.2 g (24 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 12 mL of anhydrous THF with 24 mmol of 3-trifluoromethylphenyllithium at −78 °C. 3-Trifluoromethylphenyllithium was prepared from 3.3 mL (24 mmol) of 3-bromobenzotrifluoride and 15 mL (24 mmol) of n-butyllithium (1.6 M hexane solution) in 28 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 3.2 g (50%) of (3-trifluoromethylphenyl)-phenylphosphine oxide (1e) as clear oil. 31P{1H} NMR (121.5 MHz, CDCl3) δ 19.8; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, 1H, J = 486.2), 7.99 (d, 1H, J = 13.5), 7.89–7.81 (m, 2H), 7.73–7.69 (m, 2H), 7.66–7.59 (m, 2H), 7.55–7.51 (m, 2H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 134.1 (d, J = 11.1), 133.2 (d, J = 3.0), 133.2 (d, J = 99.9), 131.7 (dd, J = 32.1, 13.6), 130.8 (d, J = 11.7), 130.6 (d, J = 102.9), 129.7 (d, J = 12.5), 129.4 (d, J = 6.2), 129.3 (d, J = 13.1), 127.7 (m), 123.6 (dd, J = 272.8, 1.7); 19F NMR (282.4 MHz, CDCl3) δ −62.8; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H11F3OP 271.0500; Found 271.0500.
(4-Trifluoromethylphenyl)-phenylphosphine oxide (1f)
(4-Trifluoromethylphenyl)-phenylphosphine oxide (1f) was prepared according to Representative Procedure I by reacting 5.2 g (24 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 12 mL of anhydrous THF with 24 mmol of 4-trifluoromethylphenyllithium at −78 °C. 4-Trifluoromethylphenyllithium was prepared from 3.3 mL (24 mmol) of 4-bromobenzotrifluoride and 15 mL (24 mmol) of n-butyllithium (1.6 M hexane solution) in 28 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 3.3 g (51%) of (4-trifluoromethylphenyl)-phenylphosphine oxide (1f) as clear oil. 31P{1H} NMR (121.5 MHz, CDCl3) δ 19.8 (δlit 19.6);481H NMR (500 MHz, CDCl3) δ 8.11 (d, 1H, J = 486.5), 7.87–7.81 (m, 2H), 7.76–7.68 (m, 4H), 7.63–7.58 (m, 1H), 7.55–7.50 (m, 2H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 135.8 (d, J = 97.9), 134.5 (dd, J = 32.8, 3.1), 133.2 (d, J = 2.9), 131.4 (d, J = 11.7), 130.8 (d, J = 11.7), 129.9 (d, J = not visible), 129.3 (d, J = 13.1), 125.9 (dq, J = 12.8, 3.6), 123.6 (dd, J = 272.9, 1.0); 19F NMR (282.4 MHz, CDCl3) δ −63.2; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H11F3OP 271.0500; Found 271.0502.
(2-Methoxyphenyl)-phenylphosphine oxide (1g)
(2-Methoxyphenyl)-phenylphosphine oxide (1g) was prepared according to Representative Procedure I by reacting 4.3 g (20 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 10 mL of anhydrous THF with 20 mmol of 2-methoxyphenyllithium at −78 °C. 2-Methoxyphenyllithium was prepared from 2.5 mL (20 mmol) of 2-bromoanisole and 13 mL (20 mmol) of n-butyllithium (1.6 M hexane solution) in 24 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 3.0 g (65%) of (2-methoxyphenyl)-phenylphosphine oxide (1g) as pale yellow solid. mp: 94–96 °C (mplit: 101–103 °C);4931P{1H} NMR (121.5 MHz, CDCl3) δ 12.5 (δlit 14.4);491H NMR (500 MHz, CDCl3) δ 8.16 (d, 1H, J = 499.0), 7.81–7.70 (m, 3H), 7.55–7.43 (m, 4H), 7.11–7.08 (m, 1H), 6.92–6.89 (m, 1H), 3.77 (s, 3H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 160.8 (d, J = 3.8), 134.5 (d, J = 1.9), 133.2 (d, J = 7.1), 132.3 (d, J = 104.5), 132.1 (d, J = 3.0), 130.6 (d, J = 11.8), 128.6 (d, J = 13.1), 121.2 (d, J = 12.1), 119.6 (d, J = 101.9), 110.9 (d, J = 6.1), 55.6; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14O2P 233.0731; Found 233.0733.
(2-Phenylphenyl)-phenylphosphine oxide (1h)
(2-Phenylphenyl)-phenylphosphine oxide (1h) was prepared according to Representative Procedure I by reacting 4.3 g (20 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 10 mL of anhydrous THF with 20 mmol of 2-phenylphenyllithium at −78 °C. 2-Phenylphenyllithium was prepared from 3.4 mL (20 mmol) of 2-bromobiphenyl and 13 mL (20 mmol) of n-butyllithium (1.6 M hexane solution) in 24 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 4.5 g (81%) of (2-phenylphenyl)-phenylphosphine oxide (1h) as clear oil. 31P{1H} NMR (202.5 MHz, CDCl3) δ 18.4 (δlit 18.4);10b1H NMR (500 MHz, CDCl3) δ 7.95 (dd, 1H, J = 14.1, 7.6), 7.88 (d, 1H, J = 493.3), 7.60 (t, 1H, J = 7.5), 7.51 (t, 1H, J = 7.6), 7.43–7.21 (m, 11H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 146.1 (d, J = 10.1), 139.3 (d, J = 5.2), 132.9 (d, J = 10.4), 132.4 (d, J = 2.7), 132.0 (d, J = 2.9), 131.7 (d, J = 102.5), 130.8 (d, J = 9.3), 130.6 (d, J = 11.5), 130.4 (d, J = 105.7), 129.5, 128.5 (d, J = 13.1), 128.3, 128.1, 127.6 (d, J = 12.0); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C18H16OP 279.0939; Found 279.0934.
(1-Naphthyl)-phenylphosphine oxide (1i)
(1-Naphthyl)-phenylphosphine oxide (1i) was prepared according to Representative Procedure I by reacting 5.2 g (24 mmol) of N,N-diethylamino-chloro-phenylphosphine (6) in 12 mL of anhydrous THF with 24 mmol of 1-naphthyllithium at −78 °C. 1-Naphthyllithium was prepared from 3.4 mL (24 mmol) of 1-bromonaphthalene and 15 mL (24 mmol) of n-butyllithium (1.6 M hexane solution) in 28 mL of anhydrous THF at −78 °C. The crude product was purified by flash column chromatography (Al2O3, gradient elution, hexane to EtOAc) to give 4.5 g (74%) of (1-naphthyl)-phenylphosphine oxide (1i) as clear oil. 31P{1H} NMR (121.5 MHz, CDCl3) δ 23.2 (δlit 23.2);9b1H NMR (500 MHz, CDCl3) δ 8.43 (d, 1H, J = 484.1), 8.28 (d, 1H, J = 8.13), 8.08 (d, 1H, J = 8.31), 8.00–7.90 (m, 2H), 7.75–7.70 (m, 2H), 7.60–7.43 (m, 6H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 133.9 (d, J = 3.0), 133.8 (d, J = 9.0), 132.8 (d, J = 8.7), 132.5 (d, J = 3.0), 132.3 (d, J = 13.4), 131.7 (d, J = 102.8), 130.9 (d, J = 11.4), 129.2 (d, J = 1.6), 129.0 (d, J = 12.8), 127.9, 127.6 (d, J = 100.3), 127.0, 125.5 (d, J = 7.5), 124.9 (d, J = 15.4); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C16H14OP 253.0782; Found 253.0781.
Preparation of ((2R,3R)-1,4-Dioxaspiro[4.5]decane-2,3-diyl)bis(bis(4-(tert-butyl)phenyl)methanol) [(R,R)-SI-2]
(R,R)-SI-2 was synthesized according to a modified procedure of Beck et al.35 To a solution of 44 mmol of (4-(tert-butyl)phenyl)magnesium bromide in 35 mL of anhydrous THF was added 2.5 g (8.7 mmol) of diethyl (2R,3R)-1,4-dioxaspiro[4.5]decane-2,3-dicarboxylate in 15 mL of anhydrous THF over 30 min at 0 °C under nitrogen atmosphere. [(4-(tert-butyl)phenyl)magnesium bromide was prepared from 7.6 mL (44 mmol) of 1-bromo-4-(tert-butyl)benzene and 1.2 g (48 mmol) of Mg in 35 mL of anhydrous THF]. The resulting solution was heated at reflux in an oil bath for 4 h; then, it was allowed to cool to room temperature, and it was stirred overnight. 40 mL of saturated NH4Cl and 20 mL of water were added. The phases were separated, and the aqueous layer was extracted with DCM (3 × 40 mL). The organic layers were combined, dried (Na2SO4), and evaporated. The crude product was purified by flash column chromatography (silica gel, gradient elution, hexane to CHCl3) to give 4.3 g (67%) of ((2R,3R)-1,4-dioxaspiro[4.5]decane-2,3-diyl)bis(bis(4-(tert-butyl)phenyl)methanol) [(R,R)-SI-2] as a white solid. mp: 160–163 °C; [α]D25 = −60.9 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.47–7.29 (m, 16H), 4.59 (s, 2H), 3.97 (s, 2H), 1.46–1.03 (m, 46H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 150.0, 149.7, 143.6, 139.9, 128.1, 127.3, 124.9, 124.0, 109.9, 80.9, 77.9, 36.5, 34.4, 31.4, 31.3, 25.2, 24.1; HRMS (ESI/TOF) m/z: [M – H]− Calcd for C50H65O4P 729.4888; Found 729.4885.
Representative Resolution Procedures
Over the course of this research project, it was observed that even the residual water content of the solvent may influence the outcome of the resolution in a negative manner. Thus, all the solvents used for resolution were dried according to the standard procedures. Dry solvents were stored over molecular sieves of 3 or 4 Å.
Resolution of (2-Methylphenyl)-phenylphosphine oxide (1a) with spiro-TADDOL [(R,R)-2] Using the Crystallization Method (Representative Procedure II)
0.10 g (0.46 mmol) of racemic (2-methylphenyl)-phenylphosphine oxide (1a) and 0.23 g (0.46 mmol) of spiro-TADDOL [(R,R)-2] were dissolved in 1.4 mL of hot 2-PrOH using a hot plate as the heat source. The colorless crystalline diastereomeric complex of (S)-1a·(spiro-TADDOL)2 appeared after cooling the mixture to 25 °C. After standing at 25 °C for 3 h, the crystals were separated by filtration and washed with 0.46 mL of 2-PrOH to give 0.25 g (89%) of (S)-1a·(spiro-TADDOL)2 with a de of 79%. The diastereomeric complex (S)-1a·(spiro-TADDOL)2 was purified by two recrystallizations from 1.4 mL of 2-PrOH according to the procedure described above to afford 0.18 g (65%) of the (S)-1a·(spiro-TADDOL)2 with a de of 98% (Scheme 2; Table S2, Entry 1). (S)-(2-Methylphenyl)-phenylphosphine oxide [(S)-1a] was recovered from the diastereomer by flash column chromatography (silica gel, gradient elution, DCM to DCM–MeOH 95:5) to give 0.031 g (61%) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] with an ee of 98%.
The resolution of secondary phosphine oxides (1) with TADDOL derivatives was performed according to Representative Procedure II when i-Pr2O, acetone, MeOH, EtOH, or 2-PrOH was used as solvent. All conditions and results can be found in Tables S1, S2, and S4. Scheme 2 shows the selected results.
Resolution of (2-Methylphenyl)-phenylphosphine oxide (1a) with spiro-TADDOL [(R,R)-2] Using the Precipitation Method (Representative Procedure III)
0.10 g (0.46 mmol) of racemic (2-methylphenyl)-phenylphosphine oxide (1a) and 0.12 g (0.23 mmol) of spiro-TADDOL [(R,R)-2] were dissolved in 0.58 mL of hot toluene using a hot plate as the heat source, and then, 0.58 mL of hexane was added. The colorless crystalline diastereomeric complex of (S)-1a·(spiro-TADDOL) appeared immediately. After standing at 25 °C for 3 h, the crystals were separated by filtration and washed with 0.20 mL of hexane to give 0.13 g (75%) of (S)-1a·(spiro-TADDOL) with a de of 77%. The diastereomeric complex (S)-1a·(spiro-TADDOL) was purified by two recrystallizations from a mixture of 0.58 mL of toluene and 0.58 mL of hexane according to the procedure described above to afford 0.093 g (55%) of the (S)-1a·(spiro-TADDOL) with a de of 95% (Scheme 2; Table S3, Entry 1). (S)-(2-Methylphenyl)-phenylphosphine oxide [(S)-1a] was recovered from the diastereomer by flash column chromatography (silica gel, gradient elution, DCM to DCM–MeOH 95:5) to give 0.025 g (50%) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] with an ee of 95%.
The resolution of secondary phosphine oxides (1) with TADDOL derivatives was performed according to Representative Procedure III when a toluene/hexane or EtOAc/hexane mixture was used as solvent. All conditions and results can be found in Tables S1, S3, and S4. Scheme 2 shows the selected results.
(S)-(2-Methylphenyl)-phenylphosphine Oxide [(S)-1a]
[α]D25 = −44.2 (c = 1.07, CHCl3, ee = 98%), Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (85:15), tR1 20.8 min (R), tR2 22.5 min (S).
(−)-(3-Methylphenyl)-phenylphosphine Oxide [(−)-1b]
[α]D25 = −1.3 (c = 1.35, CHCl3, ee = 62%), Chiral HPLC: Phenomenex Lux 5 μm Cellulose-2 column, hexane/ethanol (50:50), tR1 11.1 min (−), tR2 12.8 min (+).
(−)-(4-Methylphenyl)-phenylphosphine Oxide [(−)-1c]
[α]D25 = −7.2 (c = 0.72, CHCl3, ee = 99%), Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 10.3 min (−), tR2 11.2 min (+).
(2-Trifuoromethylphenyl)-phenylphosphine Oxide (1d)
[α]D25 = not measured, ee = 0%, Chiral HPLC: Phenomenex Lux 5 μm Cellulose-2 column, hexane/ethanol (50:50), tR1 7.3 min, tR2 7.9 min.
(+)-(3-Trifuoromethylphenyl)-phenylphosphine Oxide [(+)-1e]
[α]D25 = +10.0 (c = 0.78, CHCl3, ee = 79%); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 6.7 min (+), tR2 8.7 min (−).
(+)-(4-Trifuoromethylphenyl)-phenylphosphine Oxide [(+)-1f]
[α]D25 = +13.0 (c = 0.68, CHCl3, ee = 99%); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 6.4 min (+), tR2 8.3 min (−).
(R)-(2-Methoxylphenyl)-phenylphosphine Oxide [(R)-1g]
[α]D25 = +40.9 (c = 0.91, CHCl3, ee = 67%), Chiral HPLC: Phenomenex Lux 5 μm Cellulose-1 column, hexane/ethanol (85:15), tR1 10.8 min (S), tR2 15.4 min (R).
(S)-(2-Phenylphenyl)-phenylphosphine Oxide [(S)-1h]
[α]D25 = −30.4 (c = 0.58, CHCl3, ee = 40%); Chiral HPLC: Phenomenex Lux 5 μm Cellulose-2 column, hexane/ethanol (50:50), tR1 10.0 min (R), tR2 12.0 min (S).
(R)-(1-Naphthyl)-phenylphosphine Oxide [(R)-1i]
[α]D25 = −9.7 (c = 1.79, CHCl3, ee = 83%); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 8.9 min (R), tR2 9.8 min (S).
(S)-Benzyl-phenylphosphine Oxide [(S)-1j]
[α]D25 = +47.8 (c = 0.49, CHCl3, ee = 87%); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 9.7 min (S), tR2 16.1 min (R); 31P{1H} NMR (202.5 MHz, CDCl3) δ 29.5 (δlit 29.6);9b1H NMR (500 MHz, CDCl3) δ 7.56–7.41 (m, 5H), 7. 47 (dt, J = 474.7, 3.6, 1H), 7.27–7.22 (m, 3H), 7.06–7.04 (m, 2H), 3.48 (ddd, J = 17.3, 14.5, 3.0, 1H), 3.35 (td, J = 14.9, 4.2, 1H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 132.7 (d, J = 3.0), 130.6 (d, J = 7.4), 130.2 (d, J = 10.8), 130.1 (d, J = 97.0), 129.9 (d, J = 5.7), 129.0 (d, J = 3.1), 128.8 (d, J = 12.4), 127.4 (d, J = 3.6), 38.9 (d, J = 62.6); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14OP 217.0782; Found 217.0778.
(R)-Methyl-phenylphosphine Oxide [(R)-1k]
[α]D25 = +11.4 (c = 0.70, CHCl3, ee = 99%); Chiral HPLC: Phenomenex Lux 5 μm Cellulose-2 column, hexane/ethanol (50:50), tR1 9.1 min (S), tR2 10.8 min (R); 31P{1H} NMR (202.5 MHz, CDCl3) δ 20.4 (δlit 20.3);9b1H NMR (500 MHz, CDCl3) δ 7.69–7.64 (m, 2H), 7.59 (dq, J = 472.4, 3.8, 1H), 7.54–7.44 (m, 3H), 1.75 (dd, J = 14.0, 3.8 Hz, 3H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 132.5 (d, J = 2.9), 132.0 (d, J = 99.9), 129.6 (d, J = 11.3), 129.0 (d, J = 12.7), 16.3 (d, J = 69.0); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C7H10OP 141.0469; Found 141.0464.
(S)-Butyl-phenylphosphine Oxide [(S)-1l]
[α]D25 = −11.2 (c = 0.51, CHCl3, ee = 45%); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (50:50), tR1 7.1 min (S), tR2 7.8 min (R); 31P{1H} NMR (202.5 MHz, CDCl3) δ 28.0 (δlit 28.0);9b1H NMR (500 MHz, CDCl3) δ 7.71–7.67 (m, 2H), 7.58–7.48 (m, 3H), 7.47 (dtd, J = 463.1, 3.4, 1.6, 1H), 2.04–1.95 (m, 2H), 1.64–1.52 (m, 2H), 1.48–1.37 (m, 2H), 0.90 (td, J = 7.3, 1.6, 3H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 132.5 (d, J = 2.9), 131.3 (d, J = 96.4), 130.0 (d, J = 10.9), 129.0 (d, J = 12.3), 30.2 (d, J = 68.1), 23.8 (d, J = 14.7), 23.7 (d, J = 3.7), 13.7; HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C10H16OP 183.0939; Found 183.0934.
(R)-tert-Butyl-phenylphosphine Oxide [(R)-1m]
[α]D25 = +34.4 (c = 1.32, CHCl3, ee = 98%); Chiral HPLC: Kromasil 5-Amycoat column, hexane/ethanol (85:15), tR1 8.8 min (S), tR2 11.9 min (R); 31P{1H} NMR (202.5 MHz, CDCl3) δ 47.5 (δlit 47.6);9b1H NMR (500 MHz, CDCl3) δ 7.71–7.67 (m, 2H), 7.60–7.57 (m, 1H), 7.52–7.50 (m, 2H), 7.04 (d, J = 452.8, 1H), 1.16 (d, J = 16.6, 9H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 132.6 (d, J = 2.7), 131.0 (d, J = 10.0), 129.1 (d, J = 90.1), 128.6 (d, J = 11.8), 32.1 (d, J = 69.2), 23.6 (d, J = 2.1); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C10H16OP 183.0939; Found 183.0936.
(S)-Cyclohexyl-phenylphosphine Oxide [(S)-1n]
[α]D25 = −25.3 (c = 1.06, CHCl3, ee = 92%); Chiral HPLC: Phenomenex Lux 5 μm Cellulose-2 column, hexane/ethanol (50:50), tR1 10.0 min (S), tR2 19.7 min (R); 31P{1H} NMR (202.5 MHz, CDCl3) δ 36.6 (δlit 36.7);10b1H NMR (500 MHz, CDCl3) δ 7.68–7.64 (m, 2H), 7.58–7.47 (m, 3H), 7.18 (d, J = 446.6, 1H), 1.93–1.79 (m, 5H), 1.70–1.68 (m, 1H), 1.39–1.17 (m, 5H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 132.5 (d, J = 2.8), 130.4 (d, J = 10.4), 130.0 (d, J = 92.9), 128.9 (d, J = 12.0), 38.7 (d, J = 69.7), 26.1 (d, J = 5.3), 26.0 (d, J = 4.9), 25.9 (d, J = 1.7), 25.4 (d, J = 1.9), 24.7 (d, J = 2.6); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C12H18OP 209.1095; Found 209.1093.
Gram-Scale Resolution of (2-Methylphenyl)-phenylphosphine Oxide (1a) or tert-Butyl-phenylphosphine Oxide (1m) with spiro-TADDOL [(R,R)-2]
(2-Methylphenyl)-phenylphosphine Oxide (1a)
2.0 g (9.3 mmol) of racemic (2-methylphenyl)-phenylphosphine oxide (1a) and 4.7 g (9.3 mmol) of spiro-TADDOL [(R,R)-2] were dissolved in 28 mL of hot 2-PrOH using an oil bath as the heat source. The colorless crystalline diastereomeric complex of (S)-1a·(spiro-TADDOL)2 appeared after cooling the mixture to 25 °C. After standing at 25 °C for 3 h, the crystals were separated by filtration and washed 2× with 2.8 mL of 2-PrOH to give 5.3 g (94%) of (S)-1a·(spiro-TADDOL)2 with a de of 98%. (S)-(2-Methylphenyl)-phenylphosphine oxide [(S)-1a] was recovered from the diastereomer by flash column chromatography (silica gel, gradient elution, DCM to DCM–MeOH 95:5) to give 0.92 g (92%) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a] with an ee of 98% (Scheme 3).
tert-Butyl-phenylphosphine Oxide (1m)
2.2 g (12.1 mmol) of racemic tert-butyl-phenylphosphine oxide (1m) and 3.1 g (6.1 mmol) of spiro-TADDOL [(R,R)-2] were dissolved in 18 mL of hot 2-PrOH using an oil bath as the heat source. The colorless crystalline diastereomeric complex of (R)-1m·(spiro-TADDOL) appeared after cooling the mixture to 25 °C. After standing at 25 °C for 3 h, the crystals were separated by filtration and washed 2× with 1.8 mL of 2-PrOH to give 3.6 g (87%) of (R)-1m·(spiro-TADDOL) with a de of 78%. The diastereomeric complex (R)-1m·(spiro-TADDOL) was purified by two recrystallizations from 18 mL of 2-PrOH according to the procedure described above to afford 2.6 g (63%) of the (R)-1m·(spiro-TADDOL) with a de of 98%. (R)-tert-Butyl-phenylphosphine oxide [(R)-1m] was recovered from the diastereomer by flash column chromatography (silica gel, gradient elution, DCM to DCM–MeOH 95:5) to give 0.66 g (60%) of (R)-tert-butyl-phenylphosphine oxide [(R)-1m] with an ee of 98% (Scheme 3).
Preparation of (S)-Methyl-(2-methylphenyl)-phenylphosphine Oxide [(S)-3a] via Stereospecific Alkylation (Representative Procedure IV)
Under argon atmosphere, 20 mg of 60% (w/w%) NaH dispersion (12 mg of NaH, 0.48 mmol) was washed with anhydrous THF (3 × 1.0 mL); then, 1.0 mL of anhydrous THF was added. To this suspension, 80 mg (0.37 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] in 1.0 mL of anhydrous THF was added dropwise over 10 min at 0 °C, and the reaction mixture was stirred for 45 min at this temperature. Then, 28 μL (0.44 mmol) of MeI in 1.0 mL of anhydrous THF was added dropwise over 30 min at 0 °C. The reaction mixture was allowed to warm to 25 °C, and it was stirred overnight. Then, 1 mL of saturated NH4Cl solution and 1 mL of water were added. Phases were separated, and the aqueous layer was extracted with EtOAc (3 × 1 mL). The organic layers were combined, dried (Na2SO4), and evaporated. The crude product was purified by flash column chromatography (silica gel, gradient elution, CHCl3 to CHCl3–MeOH 97:3) to give 74 mg (87%) of (S)-methyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3a] with an ee of 98% as a white solid. mp: 104 °C (mplit: 114 °C);50 [α]D25 = −33.0 (c = 1.00, CHCl3, ee = 98%, SP) ([α]lit = −28.2 (c = 1.0, CHCl3, ee = 96%, SP));50 Chiral HPLC: Kromasil 5-Amycoat column, hexane/ethanol (85:15), tR1 10.9 min (S), tR2 13.5 min (R); 31P{1H} NMR (121.5 MHz, CDCl3) δ 31.8 (δlit 31.4);501H NMR (500 MHz, CDCl3) δ 7.68–7.61 (m, 3H), 7.52–7.40 (m, 4H), 7.30–7.21 (m, 2H), 2.36 (s, 3H), 2.03 (d, 3H, J = 13.1); 13C{1H} NMR (75.5 MHz, CDCl3) δ 142.2 (d, J = 8.3), 134.8 (d, J = 100.1), 132.2 (d, J = 2.8), 132.0 (d, J = 10.3), 131.6, 131.6 (d, J = 14.6), 130.9 (J = not visible), 130.5 (d, J = 10.0), 128.7 (d, J = 12.0), 125.6 (d, J = 12.3), 21.4 (d, J = 4.7), 17.3 (d, J = 74.1); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C14H16OP 231.0939; Found 231.0937.
(S)-Ethyl-(2-methylphenyl)-phenylphosphine Oxide [(S)-3b]
(S)-Ethyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3b] was prepared according to Representative Procedure IV by reacting 12 mg (0.48 mmol) of NaH in 1.0 mL of anhydrous THF with 80 mg (0.37 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] in 1.0 mL of anhydrous THF and 36 μL (0.44 mmol) of EtI in 1.0 mL of anhydrous THF at 0 °C. The crude product was purified by flash column chromatography (silica gel, gradient elution, CHCl3 to CHCl3–MeOH 97:3) to give 72 mg (80%) of (S)-ethyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3b] with an ee of 95% as a white solid. mp 90–92 °C (mplit: 94–95 °C);21b [α]D25 = −31.0 (c = 1.00, CHCl3, ee = 95%, SP) ([α]lit = +32.6 (c = 1.2, CHCl3, ee = 99%, RP));21b Chiral HPLC: Kromasil 5-Amycoat column, hexane/ethanol (85:15), tR1 8.4 min (S), tR2 12.6 min (R). 31P{1H} NMR (121.5 MHz, CDCl3) δ 36.2 (δlit 35.5);511H NMR (500 MHz, CDCl3) δ 7.67–7.60 (m, 3H), 7.50–7.39 (m, 4H), 7.30–7.26 (m, 1H), 7.22–7.20 (m, 1H), 2.43–2.19 (m, 5H), 1.19 (dt, 3H J = 16.9, 7.6); 13C{1H} NMR (75.5 MHz, CDCl3) δ 142.7 (d, J = 7.6), 133.7 (d, J = 96.5), 132.1 (d, J = 10.2), 132.0 (d, J = 2.6), 131.6 (d, J = 11.1), 131.5 (d, J = 3.1), 130.9 (d, J = 9.5), 130.2 (J = not visible), 128.6 (d, J = 11.6), 125.5 (d, J = 11.8), 22.7 (d, J = 73.4), 21.5 (d, J = 4.3), 5.8 (d, J = 4.9); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C15H18OP 245.1095; Found 245.1085.
(S)-Benzyl-(2-methylphenyl)-phenylphosphine Oxide [(S)-3c]
(S)-Benzyl-(2-methylphenyl)phosphine oxide [(S)-3c] was prepared according to Representative Procedure IV by reacting 12 mg (0.48 mmol) of NaH in 1.0 mL of anhydrous THF with 80 mg (0.37 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] in 1.0 mL of anhydrous THF and 53 μL (0.44 mmol) of BnBr in 1.0 mL of anhydrous THF at 0 °C. The crude product was purified by flash column chromatography (silica gel, gradient elution, CHCl3 to CHCl3–MeOH 95:5) to give 86 mg (76%) of (S)-benzyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3c] with an ee of 96% as a white solid. mp: 167–170 °C; [α]D25 = −84.9 (c = 1.01, CHCl3, ee = 96%, SP) ([α]lit = +29.8 (c = 1, CHCl3, RP));52 Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (85:15), tR1 17.2 min (S), tR2 24.3 min (R). 31P NMR (121.5 MHz, CDCl3) δ 31.0; 1H NMR (500 MHz, CDCl3) δ 7.70–7.66 (m, 1H), 7.51–7.35 (m, 6H), 7.29–7.17 (m, 5H), 7.09–7.07 (m, 2H), 3.80–3.61 (m, 2H), 2.35 (s, 3H); 13C{1H} NMR (75.5 MHz, CDCl3) δ 143.1 (d, J = 7.7), 133.2 (d, J = 98.6), 132.2 (d, J = 8.7), 132.1, 131.7, 131.6 (d, J = 7.9), 131.3 (d, J = 7.8), 131.1 (d, J = 9.5), 130.6 (d, J = 97.6), 130.4 (d, J = 5.2), 128.5 (d, J = 11.6), 128.4, 126.8 (d, J = 3.0), 125.5 (d, J = 12.0), 37.8 (d, J = 66.7), 21.5 (d, J = 4.2); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C20H20OP 307.1252; Found 307.1252.
Preparation of (S)-(1-Naphthyl)-(2-methylphenyl)-phenylphosphine Oxide [(S)-3d]
A solution of 43 μL (0.41 mmol) of 1-bromonaphthalene, 0.10 g (0.74 mmol) of anhydrous K2CO3, and 22 mg (0.019 mmol) of Pd(PPh3)4 in 1.0 mL of anhydrous toluene was stirred at 25 °C under argon atmosphere for 30 min. Then, 80 mg (0.37 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] in 1.0 mL of anhydrous toluene was added dropwise. The reaction mixture was stirred at 110 °C for 22 h using an oil bath as the heat source. After cooling to 25 °C, it was filtered through a short plug of silica gel, washed with EtOAc, and evaporated. The crude product was purified by flash column chromatography (silica gel, gradient elution, hexane to hexane–EtOAc 20:80) to give 107 mg (85%) of (S)-(1-naphthyl)-(2-methylphenyl)-phenylphosphine oxide [(S)-3d] with an ee of 88% as a white solid. mp 87–92 °C; [α]D25 = −7.8 (c = 1.00, CHCl3, ee = 88%, SP) ([α]lit = −8 (c = 1, CHCl3, SP));52 Chiral HPLC: Kromasil 5-Amycoat column, hexane/ethanol (80:20), tR1 8.7 min (R), tR2 11.1 min (S); 31P{1H} NMR (121.5 MHz, CDCl3) δ 35.6; 1H NMR (500 MHz, CDCl3) δ 8.72 (d, 1H, J = 8.4), 8.00 (d, 1H, J = 8.1), 7.88 (d, 1H, J = 7.9), 7.68–7.64 (m, 2H), 7.56–7.35 (m, 7H), 7.32–7.26 (m, 2H), 7.07 (dt, 1H, J = 7.7, 3.9), 7.03–6.98 (m, 1H), 2.56 (s, 3H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 143.5 (d, J = 7.9), 134.1 (d, J = 1.8), 134.0, 133.6 (d, J = 97.2), 133.4 (d, J = 12.9), 133.2 (d, J = 2.9), 133.1 (d, J = 11.9), 132.4 (d, J = 9.8), 132.1 (d, J = 5.5), 132.0 (d, J = 7.6), 131.9 (d, J = 2.8), 131.0 (d, J = 103.2), 129.1 (d, J = 101.2), 128.8, 128.6 (d, J = 12.1), 127.9 (d, J = 5.3), 127.4, 126.6, 125.4 (d, J = 12.9), 124.4 (d, J = 14.3), 22.0 (d, J = 4.4); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C23H20OP 343.1252; Found 343.1238.
Preparation of (R)-(2-Methylphenyl)-phenylphosphothioic Acid [(R)-3e]
To a solution of 58 mg (0.27 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] in 0.54 mL of anhydrous THF was added 8.6 mg (0.27 mmol) of sulfur. Using an oil bath, the reaction mixture was heated at 67 °C under argon atmosphere for 1.5 h; then, the solvent was evaporated. The residue was crystallized from 1.1 mL of hexane to afford 53 mg (80%) of (R)-(2-methylphenyl)-phenylphosphothioic acid [(R)-3e] with an ee of >98% as a white solid. mp: 103–104 °C; [α]D25 = +48.3 (c = 0.97, CHCl3, ee = 99%, RP); 31P{1H} NMR (202.5 MHz, CDCl3) δ 75.3; 1H NMR (500 MHz, CDCl3) δ 7.98 (dd, 1H, J = 15.2, 7.7), 7.80–7.76 (m, 2H), 7.48 (dt, 1H, J = 7.4, 4.5), 7.42–7.38 (m, 3H), 7.29–7.27 (m, 1H), 7.17 (t, 1H, J = 6.4), 2.32 (s, 3H); 13C{1H} NMR (125.8 MHz, CDCl3) δ 141.0 (d, J = 11.7), 135.4 (d, J = 108.3), 132.5 (d, J = 11.6), 132.5 (d, J = 109.3), 132.3 (d, J = 3.0), 132.0 (d, J = 5.9), 131.9 (d, J = 3.4), 131.0 (d, J = 12.2), 128.5 (d, J = 13.7), 125.7 (d, J = 13.4), 21.7 (d, J = 4.9); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C13H14OPS 249.0503; Found 249.0497.
Preparation of (S)-Hydroxymethyl-(2-methylphenyl)-phenylphosphine Oxide [(S)-3f]
To 2.6 mL of NaOH solution (0.05 M) were added 61 mg (0.28 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] and 25 μL (0.34 mmol) of 37% formaldehyde solution at 25 °C. The reaction mixture was heated in an oil bath at 80 °C for 16 h and then cooled to 25 °C. Two mL of DCM was added; the phases were separated, and the aqueous layer was extracted with DCM (3 × 2 mL). The organic layers were combined, dried (Na2SO4), and evaporated to give 72 mg (98%) of (S)-hydroxymethyl-(2-methylphenyl)-phenylphosphine oxide [(S)-3f] with an ee of 96% as a white solid. mp: 140–142 °C; [α]D25 = −42.7 (c = 1.00, CHCl3, ee = 96%, SP); Chiral HPLC: Phenomenex Lux 3 μm Cellulose-4 column, hexane/ethanol (50:50), tR1 8.6 min (S), tR2 9.2 min (R); 31P{1H} NMR (202.5 MHz, DMSO-d6) δ 29.9; 1H NMR (500 MHz, DMSO-d6) δ 7.78–7.74 (m, 1H), 7.64–7.44 (m, 6H), 7.35–7.26 (m, 2H), 5.76 (s, 1H), 4.34–4.22 (m, 2H), 2.30 (s, 3H); 13C{1H} NMR (125.8 MHz, DMSO-d6) δ 141.9 (d, J = 7.2), 132.7 (d, J = 92.9), 131.9, 131.8 (d, J = 9.0), 131.5 (d, J = 6.0), 131.6, 131.0 (d, J = 9.0), 130.3 (d, J = 93.1), 128.4 (d, J = 11.0), 125.5 (d, J = 11.8), 60.2 (d, J = 87.3), 20.6 (d, J = 4.2); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C14H16O2P 247.0888; Found 247.0884.
Preparation of (SP)-[(RC)-Hydroxy(phenyl)methyl]-(2-methylphenyl)-phenylphosphine oxide [(SP,RC)-3g]
To 2.3 mL of NaOH solution (0.05 M) were added 54 mg (25 mmol) of (S)-(2-methylphenyl)-phenylphosphine oxide [(S)-1a, ee = 98%] and 30 μL (0.30 mmol) of benzaldehyde at 25 °C. The reaction mixture was heated in an oil bath at 80 °C for 16 h and then cooled to 25 °C. The resulting white suspension was filtered and washed with 2.3 mL of water and 2.3 mL of hexane to give 64 mg (80%) of (SP)-[(RC)-hydroxy(phenyl)methyl]-(2-methylphenyl)-phenylphosphine oxide [(SP,RC)-3g] with an ee of 99% and de of 95% as a white solid. mp: 178–179 °C; [α]D25 = −83.3 (c = 0.28, MeOH, ee = 99%, SP,RC); Chiral HPLC: Phenomenex Lux 5 μm Amylose-2 column, hexane/ethanol (85:15), tR1 10.2 min (RP,SC), tR2 24.5 min (SP,RC); 31P{1H} NMR (121.5 MHz, DMSO-d6) δ 31.6 (major diastereomer, 97.5%), 31.3 (minor diastereomer, 2.5%); 1H NMR (500 MHz, DMSO-d6) δ 7.96–7.92 (m, 1H), 7.55–7.18 (m, 13H), 6.49 (dd, 1H, J = 17.2, 5.9), 5.62–5.60 (m, 1H), 2.18 (s, 3H); 13C{1H} NMR (75.5 MHz, DMSO-d6) δ 143.4 (d, J = 6.8), 138.5, 133.8 (d, J = 90.2), 133.1 (d, J = 8.7), 132.1, 132.0 (d, J = 7.8), 131.8 (d, J = 2.5), 131.6 (d, J = 8.8), 129.7 (d, J = 91.5), 128.6 (d, J = 11.1), 128.4 (d, J = 4.5), 127.8 (d, J = 1.9), 127.8 (d, J = 2.4), 125.5 (d, J = 11.7), 72.7 (d, J = 85.9), 21.3 (d, J = 3.7); HRMS (ESI/TOF) m/z: [M + H]+ Calcd for C20H20O2P 323.1201; Found 323.1197.
X-ray Crystallography
Diastereomeric Complex (S)-1a·(spiro-TADDOL)
X-ray quality crystals were prepared by slowly diffusing hexane to the toluene solution of the (S)-1a·(spiro-TADDOL) diastereomeric complex.
Crystal data: C34H34O4, C13H13OP, Fwt.: 722.81, colorless, needle, size: 0.500 × 0.040 × 0.040 mm, monoclinic, space group P21, a = 10.553(3) Å, b = 9.719(3) Å, c = 18.977(6) Å, α = 90°, β = 93.918(7)°, γ = 90°, V = 1941.8(11) Å3, T = 294(2) K, Z = 2, F(000) = 768, Dx = 1.236 Mg/m3, μ 0.118 mm–1. A crystal of (S)-1a·(spiro-TADDOL) was mounted on a fiber. Cell parameters were determined by least-squares using 10 345 (2.99° ≤ θ ≤ 25.275°) reflections. Intensity data were collected on a Rigaku RAXIS-RAPID II diffractometer (monochromator; Mo Kα radiation, λ = 0.71075 Å) at 293(2) K in the range of 2.991° ≤ θ ≤ 21.967°. A total of 20 355 reflections were collected of which 5047 were unique [R(int) = 0.2604, R(σ) = 0.2178]; intensities of 2392 reflections were greater than 2σ (I). Completeness to θ = 0.997. A NUMABS absorption correction was applied to the data (the minimum and maximum transmission factors were 0.965396 and 0.995849). The structure was solved by direct methods (and subsequent difference syntheses). Anisotropic full-matrix least-squares refinement on F2 for all non-hydrogen atoms yielded R1 = 0.0889 and wR2 = 0.1329 for 1332 [I > 2σ (I)] and R1 = 0.1869 and wR2 = 0.1668 for all (5047) intensity data (number of parameters = 481, goodness-of-fit = 0.960, the maximum and mean shifts/esd are 0.001 and 0.000). The absolute structure parameter is 0.0(3) (Friedel coverage: 0.867; Friedel fraction max.: 0.999; Friedel fraction full: 0.999). The maximum and minimum residual electron densities in the final difference map were 0.222 and −0.219 e·Å–3. The weighting scheme applied was w = 1/[σ2(Fo2) + (0.00390.0000P)2 + 0.0000P] where P = (Fo2 + 2Fc2)/3.
(SP)-[(RC)-Hydroxy(phenyl)methyl]-(2-methylphenyl)-phenylphosphine Oxide [(SP,RC)-3g]
X-ray quality crystals were prepared by the slow evaporation of the solvent from the dichloromethatne solution of (SP,RC)-3g.
Crystal data: C20H19O2P, Fwt.: 322.32, colorless, platelet, size: 0.300 × 0.100 × 0.100 mm, orthorhombic, space group P212121, a = 8.1680(7) Å, b = 9.9573(9) Å, c = 20.1721(17) Å, α = 90°, β = 90°, γ = 90°, V = 1640.6(2) Å3, T = 294(2) K, Z = 4, F(000) = 680, Dx = 1.305 Mg/m3, μ 0.175 mm–1. A crystal of (SP,RC)-3g was mounted on a fiber. Cell parameters were determined by least-squares using 25 498 (3.21° ≤ θ ≤ 27.475°) reflections. Intensity data were collected on a Rigaku RAXIS-RAPID II diffractometer (monochromator; Mo Kα radiation, λ = 0.71075 Å) at 293(2) K in the range of 3.209° ≤ θ ≤ 25.350°. A total of 32 784 reflections were collected of which 2996 were unique [R(int) = 0.0807, R(σ) = 0.0367]; intensities of 2614 reflections were greater than 2σ (I). Completeness to θ = 0.998. A numerical absorption correction was applied to the data (the minimum and maximum transmission factors were 0.993616 and 0.998669). The structure was solved by direct methods (and subsequent difference syntheses). Anisotropic full-matrix least-squares refinement on F2 for all non-hydrogen atoms yielded R1 = 0.0500 and wR2 = 0.0854 for 1332 [I > 2σ(I)] and R1 = 0.0620 and wR2 = 0.0893 for all (2996) intensity data (number of parameters = 210, goodness-of-fit = 1.122, the maximum and mean shifts/esd are 0.000 and 0.000). The absolute structure parameter is 0.05(4) (Friedel coverage: 0.725; Friedel fraction max.: 1.000; Friedel fraction full: 1.000). The maximum and minimum residual electron densities in the final difference map were 0.209 and −0.196 e·Å–3. The weighting scheme applied was w = 1/[σ2(Fo2) + (0.03550.2831P)2 + 0.2831P] where P = (Fo2 + 2Fc2)/3. The crystallographic data are in Table S6.
Acknowledgments
This research was funded by the New National Excellence Program of the Ministry of Human Capacities (Grant No.: ÚNKP-20-3-II-BME-282). T.H. is grateful for the support of the National Research, Development and Innovation Office-NKFIH (Grant Nos. PD 128504 and K 124544).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01364.
Conditions and results of the resolution experiments; optical rotation values [α] of secondary phosphine oxides (1) and the assignation of their absolute configuration; X-ray measurements; NMR spectra; HPLC traces (PDF)
Accession Codes
CCDC 2081817 and 2081818 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Börner A.Phosphorus Ligands in Asymmetric Catalysis; Wiley-VCH: Weinheim, 2008. [Google Scholar]; b Grabulosa A.P-Stereogenic Ligands in Enantioselective Catalysis; The Royal Society of Chemistry: Cambridge, 2010. [Google Scholar]; c Kamer P. C. J., Van Leeuwen P. W. N. M., Eds. Phosphorus(III)Ligands in Homogeneous Catalysis: Design and Synthesis; New York: John Wiley & Sons, 2012. [Google Scholar]; d Imamoto T. Searching for Practically Useful P-Chirogenic Phosphine Ligands. Chem. Rec. 2016, 16, 2659–2673. 10.1002/tcr.201600098. [DOI] [PubMed] [Google Scholar]
- a Benaglia M.; Rossi S. Chiral Phosphine Oxides in Present-Day Organocatalysis. Org. Biomol. Chem. 2010, 8, 3824–3830. 10.1039/c004681g. [DOI] [PubMed] [Google Scholar]; b Golandaj A.; Ahmad A.; Ramjugernath D. Phosphonium Salts in Asymmetric Catalysis: A Journey in a Decade’s Extensive Research Work. Adv. Synth. Catal. 2017, 359, 3676–3706. 10.1002/adsc.201700795. [DOI] [Google Scholar]; c Guo H.; Fan Y. C.; Sun Z.; Wu Y.; Kwon O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. 10.1021/acs.chemrev.8b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ni H.; Chan W.-L.; Lu Y. Phosphine-Catalyzed Asymmetric Organic Reactions. Chem. Rev. 2018, 118, 9344–9411. 10.1021/acs.chemrev.8b00261. [DOI] [PubMed] [Google Scholar]; e Ayad T.; Gernet A.; Pirat J.-L.; Virieux D. Enantioselective Reactions Catalyzed by Phosphine Oxides. Tetrahedron 2019, 75, 4385–4418. 10.1016/j.tet.2019.06.042. [DOI] [Google Scholar]
- a Pradere U.; Garnier-Amblard E. C.; Coats S. J.; Amblard F.; Schinazi R. F. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev. 2014, 114, 9154–9218. 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mehellou Y.; Rattan H. S.; Balzarini J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018, 61, 2211–2226. 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bellingham R.; Borrett G.; Bret G.; Choudary B.; Colclough D.; Hayes J.; Hayler J.; Hodnett N.; Ironmonger A.; Ochen A.; Pascoe D.; Richardson J.; Vit E.; Alexandre F. R.; Caillet C.; Amador A.; Bot S.; Bonaric S.; Da Costa D.; Lioure M. P.; Roland A.; Rosinovsky E.; Parsy C.; Dousson C. B. Development and Scale-Up of a Manufacturing Route for the Non-Nucleoside Reverse Transcriptase Inhibitor GSK2248761A (IDX-899): Synthesis of an Advanced Key Chiral Intermediate. Org. Process Res. Dev. 2018, 22, 200–206. 10.1021/acs.oprd.7b00356. [DOI] [Google Scholar]
- a Dutartre M.; Bayardon J.; Jugé S. Applications and Stereoselective Syntheses of P-Chirogenic Phosphorus Compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. 10.1039/C6CS00031B. [DOI] [PubMed] [Google Scholar]; b Chrzanowski J.; Krasowska D.; Drabowicz J. Synthesis of Optically Active Tertiary Phosphine Oxides: A Historical Overview and the Latest Advances. Heteroat. Chem. 2018, 29, e21476 10.1002/hc.21476. [DOI] [Google Scholar]; c Lemouzy S.; Giordano L.; Hérault D.; Buono G. Introducing Chirality at Phosphorus Atoms: An Update on the Recent Synthetic Strategies for the Preparation of Optically Pure P-Stereogenic Molecules. Eur. J. Org. Chem. 2020, 2020, 3351–3366. 10.1002/ejoc.202000406. [DOI] [Google Scholar]; d Cui Y. M.; Lin Y.; Xu L. W. Catalytic Synthesis of Chiral Organoheteroatom Compounds of Silicon, Phosphorus, and Sulfur via Asymmetric Transition Metal-Catalyzed C–H Functionalization. Coord. Chem. Rev. 2017, 330, 37–52. 10.1016/j.ccr.2016.09.011. [DOI] [Google Scholar]; e Lin Y.; Ma W. Y.; Sun Q. Y.; Cui Y. M.; Xu L. W. Catalytic Synthesis of Chiral Phosphole Oxides via Desymmetric C-H Arylation of o -Bromoaryl Phosphine Oxides. Synlett 2017, 28 (12), 1432–1436. 10.1055/s-0036-1588983. [DOI] [Google Scholar]; f Ye X.; Peng L.; Bao X.; Tan C.-H.; Wang H. Recent Developments in Highly Efficient Construction of P-Stereogenic Centers. Green Synth. Catal. 2021, 2 (1), 6–18. 10.1016/j.gresc.2020.12.002. [DOI] [Google Scholar]
- Ackermann L. Air- and Moisture-Stable Secondary Phosphine Oxides as Preligands in Catalysis. Synthesis 2006, 2006, 1557–1571. 10.1055/s-2006-926427. [DOI] [Google Scholar]
- a Christiansen A.; Li C.; Garland M.; Selent D.; Ludwig R.; Spannenberg A.; Baumann W.; Franke R.; Börner A. On the Tautomerism of Secondary Phosphane Oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741. 10.1002/ejoc.201000037. [DOI] [Google Scholar]; b Janesko B. G.; Fisher H. C.; Bridle M. J.; Montchamp J.-L. P(=O)H to P–OH Tautomerism: A Theoretical and Experimental Study. J. Org. Chem. 2015, 80, 10025–10032. 10.1021/acs.joc.5b01618. [DOI] [PubMed] [Google Scholar]; c Vincze D.; Ábrányi-Balogh P.; Bagi P.; Keglevich G. A Mechanistic Study on the Tautomerism of H-Phosphonates, H-Phosphinates and Secondary Phosphine Oxides. Molecules 2019, 24, 3859. 10.3390/molecules24213859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shaikh T. M.; Weng C.-M.; Hong F.-E. Secondary Phosphine Oxides: Versatile Ligands in Transition Metal-Catalyzed Cross-Coupling Reactions. Coord. Chem. Rev. 2012, 256, 771–803. 10.1016/j.ccr.2011.11.007. [DOI] [Google Scholar]; b Achard T. Advances in Homogeneous Catalysis Using Secondary Phosphine Oxides (SPOs): Pre-Ligands for Metal Complexes. Chimia 2016, 70, 8–19. 10.2533/chimia.2016.8. [DOI] [PubMed] [Google Scholar]; c Gallen A.; Riera A.; Verdaguer X.; Grabulosa A. Coordination Chemistry and Catalysis with Secondary Phosphine Oxides. Catal. Sci. Technol. 2019, 9, 5504–5561. 10.1039/C9CY01501A. [DOI] [Google Scholar]
- Glueck D. Asymmetric Synthesis of P-Stereogenic Secondary Phosphine Oxides (SPOs). Synthesis 2021, 10.1055/a-1582-0169. [DOI] [Google Scholar]
- a Leyris A.; Bigeault J.; Nuel D.; Giordano L.; Buono G. Enantioselective Synthesis of Secondary Phosphine Oxides from (RP)-(−)-Menthyl Hydrogenophenylphosphinate. Tetrahedron Lett. 2007, 48, 5247–5250. 10.1016/j.tetlet.2007.05.140. [DOI] [Google Scholar]; b Xu Q.; Zhao C.-Q.; Han L.-B. Stereospecific Nucleophilic Substitution of Optically Pure H-Phosphinates: A General Way for the Preparation of Chiral P-Stereogenic Phosphine Oxides. J. Am. Chem. Soc. 2008, 130, 12648–12655. 10.1021/ja804412k. [DOI] [PubMed] [Google Scholar]; c Gatineau D.; Nguyen D. H.; Hérault D.; Vanthuyne N.; Leclaire J.; Giordano L.; Buono G. H-Adamantylphosphinates as Universal Precursors of P-Stereogenic Compounds. J. Org. Chem. 2015, 80, 4132–4141. 10.1021/acs.joc.5b00548. [DOI] [PubMed] [Google Scholar]
- a Leyris A.; Nuel D.; Giordano L.; Achard M.; Buono G. Enantioselective Synthesis of Both Enantiomers of Tert-Butylphenylphosphine Oxide from (S)-Prolinol. Tetrahedron Lett. 2005, 46, 8677–8680. 10.1016/j.tetlet.2005.10.058. [DOI] [Google Scholar]; b Copey L.; Jean-Gérard L.; Andrioletti B.; Framery E. Synthesis of P-Stereogenic Secondary Phosphine Oxides Using α-d-Glucosamine as a Chiral Precursor. Tetrahedron Lett. 2016, 57, 543–545. 10.1016/j.tetlet.2015.12.074. [DOI] [Google Scholar]; c Han Z. S.; Wu H.; Xu Y.; Zhang Y.; Qu B.; Li Z.; Caldwell D. R.; Fandrick K. R.; Zhang L.; Roschangar F.; Song J. J.; Senanayake C. H. General and Stereoselective Method for the Synthesis of Sterically Congested and Structurally Diverse P -Stereogenic Secondary Phosphine Oxides. Org. Lett. 2017, 19, 1796–1799. 10.1021/acs.orglett.7b00568. [DOI] [PubMed] [Google Scholar]; d Li S.-G.; Yuan M.; Topic F.; Han Z. S.; Senanayake C. H.; Tsantrizos Y. S. Asymmetric Library Synthesis of P-Chiral t -Butyl-Substituted Secondary and Tertiary Phosphine Oxides. J. Org. Chem. 2019, 84, 7291–7302. 10.1021/acs.joc.9b00945. [DOI] [PubMed] [Google Scholar]
- a Holt J.; Maj A. M.; Schudde E. P.; Pietrusiewicz K. M.; Sieroń L.; Wieczorek W.; Jerphagnon T.; Arends I. W. C. E.; Hanefeld U.; Minnaard A. J. On the Resolution of Secondary Phosphine Oxides via Diastereomeric Complex Formation: The Case of Tert-Butylphenylphosphine Oxide. Synthesis 2009, 2009, 2061–2065. 10.1055/s-0029-1216811. [DOI] [Google Scholar]; b Rajendran K. V.; Gilheany D. G. Simple Unprecedented Conversion of Phosphine Oxides and Sulfides to Phosphine Boranes Using Sodium Borohydride. Chem. Commun. 2012, 48, 817–819. 10.1039/C1CC14856G. [DOI] [PubMed] [Google Scholar]
- a Zhang Y.; He H.; Wang Q.; Cai Q. Asymmetric Synthesis of Chiral P-Stereogenic Triaryl Phosphine Oxides via Pd-Catalyzed Kinetic Arylation of Diaryl Phosphine Oxides. Tetrahedron Lett. 2016, 57, 5308–5311. 10.1016/j.tetlet.2016.10.048. [DOI] [Google Scholar]; b Beaud R.; Phipps R. J.; Gaunt M. J. Enantioselective Cu-Catalyzed Arylation of Secondary Phosphine Oxides with Diaryliodonium Salts toward the Synthesis of P-Chiral Phosphines. J. Am. Chem. Soc. 2016, 138, 13183–13186. 10.1021/jacs.6b09334. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dai Q.; Li W.; Li Z.; Zhang J. P-Chiral Phosphines Enabled by Palladium/Xiao-Phos-Catalyzed Asymmetric P–C Cross-Coupling of Secondary Phosphine Oxides and Aryl Bromides. J. Am. Chem. Soc. 2019, 141, 20556–20564. 10.1021/jacs.9b11938. [DOI] [PubMed] [Google Scholar]; d Liu X.-T.; Zhang Y.-Q.; Han X.-Y.; Sun S.-P.; Zhang Q.-W. Ni-Catalyzed Asymmetric Allylation of Secondary Phosphine Oxides. J. Am. Chem. Soc. 2019, 141, 16584–16589. 10.1021/jacs.9b08734. [DOI] [PubMed] [Google Scholar]; e Dai Q.; Liu L.; Qian Y.; Li W.; Zhang J. Construction of P-Chiral Alkenylphosphine Oxides through Highly Chemo-, Regio-, and Enantioselective Hydrophosphinylation of Alkynes. Angew. Chem., Int. Ed. 2020, 59, 20645–20650. 10.1002/anie.202009358. [DOI] [PubMed] [Google Scholar]
- Qiu H.; Dai Q.; He J.; Li W.; Zhang J. Access to P -Chiral Sec - and Tert -Phosphine Oxides Enabled by Le-Phos-Catalyzed Asymmetric Kinetic Resolution. Chem. Sci. 2020, 11, 9983–9988. 10.1039/D0SC04041J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Haynes R. K.; Freeman R. N.; Mitchell C. R.; Vonwiller S. C. Preparation of Enantiomerically Pure Tertiary Phosphine Oxides from, and Assay of Enantiomeric Purity with, (Rp)- and (Sp)-Tert-Butylphenylphosphinothioic Acids. J. Org. Chem. 1994, 59, 2919–2921. 10.1021/jo00090a003. [DOI] [Google Scholar]; b Haynes R. K.; Au-Yeung T.-L. L.; Chan W.-K. K.; Lam W.-L. L.; Li Z.-Y. Y.; Yeung L.-L. L.; Chan A. S. C. C.; Li P.; Koen M.; Mitchell C. R.; Vonwiller S. C. Reaction of Metallated Tert-Butyl(Phenyl)Phosphane Oxide with Electrophiles as a Route to Functionalized Tertiary Phosphane Oxides: Alkylation Reactions. Eur. J. Org. Chem. 2000, 2000, 3205–3216. . [DOI] [Google Scholar]
- Stankevič M.; Pietrusiewicz K. M. Resolution and Stereochemistry of Tert-Butylphenylphosphinous Acid-Borane. J. Org. Chem. 2007, 72, 816–822. 10.1021/jo061896e. [DOI] [PubMed] [Google Scholar]
- a Harger M. J. P. Chemical Shift Non-Equivalence of Enantiomers in the Proton Magnetic Resonance Spectra of Partly Resolved Phosphinothioic Acids. J. Chem. Soc., Perkin Trans. 2 1978, (4), 326–331. 10.1039/p29780000326. [DOI] [Google Scholar]; b Skrzypczynski Z.; Michalski J. Stereoselective Synthesis and Stereochemistry of Optically Active Tert-Butylphenylphosphine Sulfide. J. Org. Chem. 1988, 53, 4549–4551. 10.1021/jo00254a024. [DOI] [Google Scholar]; c Kobayashi Y.; Handa H.; Maeda J.; Saigo K. Factors Determining the Pattern of a Hydrogen-Bonding Network in the Diastereomeric Salts of 1-Arylethylamines with Enantiopure P-Chiral Acids. Chirality 2008, 20, 577–584. 10.1002/chir.20530. [DOI] [PubMed] [Google Scholar]; d Drabowicz J.; Pokora-Sobczak P.; Zając A.; Wach-Panfiłow P. A New Procedure for the Synthesis of Optically Active t -Butylphenylphosphinothioic Acid. Heteroat. Chem. 2014, 25, 674–677. 10.1002/hc.21206. [DOI] [Google Scholar]; e Stankevič M.; Pietrusiewicz K. M. An Efficient Resolution of Phosphinous Acid-Boranes. Tetrahedron: Asymmetry 2007, 18, 552–556. 10.1016/j.tetasy.2007.02.020. [DOI] [Google Scholar]
- Gallen A.; Orgué S.; Muller G.; Escudero-Adán E. C.; Riera A.; Verdaguer X.; Grabulosa A. Synthesis and Coordination Chemistry of Enantiopure: T -BuMeP(O)H. Dalton Trans. 2018, 47, 5366–5379. 10.1039/C8DT00897C. [DOI] [PubMed] [Google Scholar]
- a Jiang X.; Minnaard A. J.; Hessen B.; Feringa B. L.; Duchateau A. L. L.; Andrien J. G. O.; Boogers J. A. F.; de Vries J. G. Application of Monodentate Secondary Phosphine Oxides, a New Class of Chiral Ligands, in Ir(I)-Catalyzed Asymmetric Imine Hydrogenation. Org. Lett. 2003, 5, 1503–1506. 10.1021/ol034282u. [DOI] [PubMed] [Google Scholar]; b Jiang X.; van den Berg M.; Minnaard A. J.; Feringa B. L.; de Vries J. G. The Application of Monodentate Secondary Phosphine Oxide Ligands in Rhodium- and Iridium-Catalyzed Asymmetric Hydrogenation. Tetrahedron: Asymmetry 2004, 15, 2223–2229. 10.1016/j.tetasy.2004.05.002. [DOI] [Google Scholar]; c Bigeault J.; Giordano L.; Buono G. [2 + 1] Cycloadditions of Terminal Alkynes to Norbornene Derivatives Catalyzed by Palladium Complexes with Phosphinous Acid Ligands. Angew. Chem., Int. Ed. 2005, 44, 4753–4757. 10.1002/anie.200500879. [DOI] [PubMed] [Google Scholar]
- Drabowicz J.; Łyżwa P.; Omelańczuk J.; Pietrusiewicz K. M.; Mikołajczyk M. New Procedures for the Resolution of Chiral Tert-Butylphenylphosphine Oxide and Some of Its Reactions. Tetrahedron: Asymmetry 1999, 10, 2757–2763. 10.1016/S0957-4166(99)00264-5. [DOI] [Google Scholar]
- Kortmann F. A.; Chang M.-C.; Otten E.; Couzijn E. P. A.; Lutz M.; Minnaard A. J. Consecutive Dynamic Resolutions of Phosphine Oxides. Chem. Sci. 2014, 5, 1322–1327. 10.1039/c3sc52913d. [DOI] [Google Scholar]
- a Bagi P.; Ujj V.; Czugler M.; Fogassy E.; Keglevich G. Resolution of P-Stereogenic P-Heterocycles via the Formation of Diastereomeric Molecular and Coordination Complexes (a Review). Dalton Trans. 2016, 45, 1823–1842. 10.1039/C5DT02999F. [DOI] [PubMed] [Google Scholar]; b Bagi P.; Varga B.; Szilágyi A.; Karaghiosoff K.; Czugler M.; Fogassy E.; Keglevich G. The Resolution of Acyclic P -Stereogenic Phosphine Oxides via the Formation of Diastereomeric Complexes: A Case Study on Ethyl-(2-Methylphenyl)-Phenylphosphine Oxide. Chirality 2018, 30, 509–522. 10.1002/chir.22816. [DOI] [PubMed] [Google Scholar]; c Varga B.; Herbay R.; Székely G.; Holczbauer T.; Madarász J.; Mátravölgyi B.; Fogassy E.; Keglevich G.; Bagi P. Scalable Enantiomeric Separation of Dialkyl-Arylphosphine Oxides Based on Host-Guest Complexation with TADDOL-Derivatives, and Their Recovery. Eur. J. Org. Chem. 2020, 2020, 1840–1852. 10.1002/ejoc.202000035. [DOI] [Google Scholar]; d Varga B.; Bagi P. Preparation of Enantiomerically Enriched P -Stereogenic Dialkyl-Arylphosphine Oxides via Coordination Mediated Optical Resolution. Symmetry 2020, 12, 215. 10.3390/sym12020215. [DOI] [Google Scholar]
- a Hoge B.; Garcia P.; Willner H.; Oberhammer H. Bis(Trifluoromethyl)Phosphinous Acid (CF3)2P-O-H: An Example of a Thermally Stable Phosphinous Acid - Synthesis, Gas-Phase Structure, and Rotational Isomers. Chem. - Eur. J. 2006, 12, 3567–3574. 10.1002/chem.200501066. [DOI] [PubMed] [Google Scholar]; b Kurscheid B.; Wiebe W.; Neumann B.; Stammler H.-G.; Hoge B. Investigations of the Tautomeric Equilibria between Phosphane Oxides and Their Corresponding Phosphinous Acids Bearing Electron-Withdrawing Perfluoroaryl Groups. Eur. J. Inorg. Chem. 2011, 2011, 5523–5529. 10.1002/ejic.201100984. [DOI] [Google Scholar]
- Petit C.; Favre-Réguillon A.; Mignani G.; Lemaire M. A Straightforward Synthesis of Unsymmetrical Secondary Phosphine Boranes. Green Chem. 2010, 12, 326–333. 10.1039/b920324a. [DOI] [Google Scholar]
- Trost B. M.; Spohr S. M.; Rolka A. B.; Kalnmals C. A. Desymmetrization of Phosphinic Acids via Pd-Catalyzed Asymmetric Allylic Alkylation: Rapid Access to P-Chiral Phosphinates. J. Am. Chem. Soc. 2019, 141, 14098–14103. 10.1021/jacs.9b07340. [DOI] [PubMed] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spackman M. A.; Jayatilaka D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. 10.1039/B818330A. [DOI] [Google Scholar]
- McKinnon J. J.; Spackman M. A.; Mitchell A. S. Novel Tools for Visualizing and Exploring Intermolecular Interactions in Molecular Crystals. Acta Crystallogr., Sect. B: Struct. Sci. 2004, 60, 627–668. 10.1107/S0108768104020300. [DOI] [PubMed] [Google Scholar]
- Holz J.; Jiao H.; Gandelman M.; Börner A. About the Inversion Barriers of P-Chirogenic Triaryl-Substituted Phosphanes. Eur. J. Org. Chem. 2018, 2018, 2984–2994. 10.1002/ejoc.201701734. [DOI] [Google Scholar]
- Nie S.-Z.; Zhou Z.-Y.; Wang J.-P.; Yan H.; Wen J.-H.; Ye J.-J.; Cui Y.-Y.; Zhao C.-Q. Nonepimerizing Alkylation of H–P Species to Stereospecifically Generate P-Stereogenic Phosphine Oxides: A Shortcut to Bidentate Tertiary Phosphine Ligands. J. Org. Chem. 2017, 82, 9425–9434. 10.1021/acs.joc.7b01413. [DOI] [PubMed] [Google Scholar]
- a Jablonkai E.; Keglevich G. P-C Bond Formation by Coupling Reactions Utilizing > P(O)H Species as the Reagents. Curr. Org. Synth. 2014, 11, 429–453. 10.2174/15701794113109990066. [DOI] [Google Scholar]; b Henyecz R.; Keglevich G. New Developments on the Hirao Reactions, Especially from “Green” Point of View. Curr. Org. Synth. 2019, 16, 523–545. 10.2174/1570179416666190415110834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bloomfield A. J.; Herzon S. B. Room Temperature, Palladium-Mediated P-Arylation of Secondary Phosphine Oxides. Org. Lett. 2012, 14, 4370–4373. 10.1021/ol301831k. [DOI] [PubMed] [Google Scholar]; b Chrzanowski J.; Krasowska D.; Urbaniak M.; Sieroń L.; Pokora-Sobczak P.; Demchuk O. M.; Drabowicz J. Synthesis of Enantioenriched Aryl- Tert -Butylphenylphosphine Oxides via Cross-Coupling Reactions of Tert -Butylphenylphosphine Oxide with Aryl Halides. Eur. J. Org. Chem. 2018, 2018, 4614–4627. 10.1002/ejoc.201800698. [DOI] [Google Scholar]
- Zhang H.; Sun Y. M.; Yao L.; Ji S. Y.; Zhao C. Q.; Han L. B. Stereogenic Phosphorus-Induced Diastereoselective Formation of Chiral Carbon during Nucleophilic Addition of Chiral H-P Species to Aldehydes or Ketones. Chem. - Asian J. 2014, 9, 1329–1333. 10.1002/asia.201301650. [DOI] [PubMed] [Google Scholar]
- Nolla-Saltiel R.; Geer A. M.; Taylor L. J.; Churchill O.; Davies E. S.; Lewis W.; Blake A. J.; Kays D. L. Hydrophosphination of Activated Alkenes by a Cobalt(I) Pincer Complex. Adv. Synth. Catal. 2020, 362 (15), 3148–3157. 10.1002/adsc.202000514. [DOI] [Google Scholar]
- Haynes R. K.; Lam W. W.-L.; Yeung L. Stereoselective Preparation of Functionalized Tertiary P-Chiral Phosphine Oxides by Nucleophilic Addition of Lithiated Tert-Butylphenylphosphine Oxide to Carbonyl Compounds. Tetrahedron Lett. 1996, 37, 4729–4732. 10.1016/0040-4039(96)00952-5. [DOI] [Google Scholar]
- Beck A. K.; Bastani B.; Plattner D. A.; Petter W.; Seebach D.; Braunschweiger H.; Gysi P.; Lavecchia L. Large-Scale Preparation of Alpha,Alpha,Alpha,Alpha-Tetraaryl-1,3-Dioxolane-4,5-Dimethanols (TADDOL) - Versatile Auxiliaries for EPC Synthesis and Its Solid-State Structure. Chimia 1991, 45, 238–244. [Google Scholar]
- Legrand S.; Luukinen H.; Isaksson R.; Kilpeläinen I.; Lindström M.; Nicholls I. A.; Unelius C. R. Synthesis, NMR Conformational Studies and Host-Guest Behaviour of New (+)-Tartaric Acid Derivatives. Tetrahedron: Asymmetry 2005, 16 (3), 635–640. 10.1016/j.tetasy.2004.11.025. [DOI] [Google Scholar]
- Armarego W. L. F.Purification of Laboratory Chemicals, 8th ed.; Butterworth-Heinemann: Oxford, 2017. [Google Scholar]
- CrystalClear SM 1.4.0; Rigaku/MSC Inc., 2008.
- Higashi T.NUMABS, rev. 2002; Rigaku/MSC Inc., 1998.
- a Sheldrick G. M. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]; b Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burla M. C.; Caliandro R.; Carrozzini B.; Cascarano G. L.; Cuocci C.; Giacovazzo C.; Mallamo M.; Mazzone A.; Polidori G. Crystal Structure Determination and Refinement via SIR2014. J. Appl. Crystallogr. 2015, 48 (1), 306–309. 10.1107/S1600576715001132. [DOI] [Google Scholar]
- Spek A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65 (2), 148–155. 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Macrae C. F.; Edgington P. R.; McCabe P.; Pidcock E.; Shields G. P.; Taylor R.; Towler M.; van de Streek J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39 (3), 453–457. 10.1107/S002188980600731X. [DOI] [Google Scholar]; b van der Sluis P.; Spek A. L. BYPASS: An Effective Method for the Refinement of Crystal Structures Containing Disordered Solvent Regions. Acta Crystallogr., Sect. A: Found. Crystallogr. 1990, 46 (3), 194–201. 10.1107/S0108767389011189. [DOI] [Google Scholar]; c Kitaigorodsky A. I.Molecular Crystals and Molecules; Academic Press: New York, 2012. [Google Scholar]
- a Spackman M. A.; Jayatilaka D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11 (1), 19–32. 10.1039/B818330A. [DOI] [Google Scholar]; b Thomas S. P.; Spackman P. R.; Jayatilaka D.; Spackman M. A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14 (3), 1614–1623. 10.1021/acs.jctc.7b01200. [DOI] [PubMed] [Google Scholar]
- Shriver D. F.; Drezdzon M. A.. The Manipulation of Air-Sensitive Compounds, 2nd ed.; Wiley & Sons: New York, 1986. [Google Scholar]
- Oliana M.; King F.; Horton P. N.; Hursthouse M. B.; Hii K. K. M. Practical Synthesis of Chiral Vinylphosphine Oxides by Direct Nucleophilic Substitution. Stereodivergent Synthesis of Aminophosphine Ligands. J. Org. Chem. 2006, 71, 2472–2479. 10.1021/jo052575q. [DOI] [PubMed] [Google Scholar]
- Jablonkai E.; Keglevich G. Catalyst-Free P–C Coupling Reactions of Halobenzoic Acids and Secondary Phosphine Oxides under Microwave Irradiation in Water. Tetrahedron Lett. 2015, 56, 1638–1640. 10.1016/j.tetlet.2015.02.015. [DOI] [Google Scholar]
- Castoldi L.; Rajkiewicz A. A.; Olofsson B. Transition Metal-Free and Regioselective Vinylation of Phosphine Oxides and H -Phosphinates with VBX Reagents. Chem. Commun. 2020, 56, 14389–14392. 10.1039/D0CC05992G. [DOI] [PubMed] [Google Scholar]
- Dziuba K.; Lubańska M.; Pietrusiewicz K. M. Enantiodivergent Synthesis of Both PAMPO Enantiomers Using l -Menthyl Chloroacetate and Stereomutation at P in Classical Quaternisation Reactions. Synthesis 2020, 52, 909–916. 10.1055/s-0039-1691531. [DOI] [Google Scholar]
- Adams H.; Collins R. C.; Jones S.; Warner C. J. A. Enantioselective Preparation of P -Chiral Phosphine Oxides. Org. Lett. 2011, 13, 6576–6579. 10.1021/ol202916j. [DOI] [PubMed] [Google Scholar]
- Nikitin K.; Rajendran K. V.; Müller-Bunz H.; Gilheany D. G. Turning Regioselectivity into Stereoselectivity: Efficient Dual Resolution of P-Stereogenic Phosphine Oxides through Bifurcation of the Reaction Pathway of a Common Intermediate. Angew. Chem., Int. Ed. 2014, 53, 1906–1909. 10.1002/anie.201309556. [DOI] [PubMed] [Google Scholar]
- Chookiewicz W.; Jore D.; Wodzki W. Optically Active Phosphines: New Synthetic Approach. Tetrahedron Lett. 1979, 20, 1069–1072. 10.1016/S0040-4039(01)87194-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.