

Abstract

Radical addition to chiral N-acylhydrazones has generated unusual amino acids tubuphenylalanine (Tup) and tubuvaline (Tuv) that are structural components of the tubulysin family of picomolar antimitotic agents and previously led to a tubulysin tetrapeptide analog with a C-terminal alcohol. To improve efficiency in this synthetic route to tubulysins, and to address difficulties in oxidation of the C-terminal alcohol, here we present two alternative routes to Tuv that (a) improve step economy, (b) provide modified conditions for Mn-mediated radical addition in the presence of aromatic heterocycles, and (c) expose an example of double diastereocontrol in radical addition to a β-benzyloxyhydrazone with broader implications for asymmetric amine synthesis via radical addition. An efficient coupling sequence affords 11-O-benzyltubulysin V benzyl ester.
1. Introduction
Stereocontrolled Mn-Mediated Radical Additions to Chiral Hydrazones
Radical additions to imino compounds offer a useful carbon–carbon bond construction approach to chiral amine synthesis.1 Seminal efforts to generalize this reaction for intermolecular coupling led to stereocontrolled alkylborane-, zinc-, and tin-mediated additions by Naito,2 Bertrand,3 and our group.4 A significant early limitation on this chemistry restricted the scope of radicals to simple 2° and 3° alkyls, usually from reagents in large excess, due to unfavorable halogen atom transfer or competitive reduction of the radicals. Such concerns make it difficult to apply this chemistry to more complex synthetic targets, prompting our search for alternative methods to generate the radicals for this purpose. With this in mind, we initiated a long-standing effort to develop the scope of a photochemical method to initiate radical addition to imino compounds via halogen atom abstraction by •Mn(CO)5 (Figure 1a).5 Meanwhile, the portfolio of radical generation conditions continues to widen6 and now includes a variety of photoredox catalysis methods7 as well as very recent approaches to Mn-mediated radical chemistry that render the processes catalytic in Mn.8
Figure 1.

(a) Methodology for Mn-mediated radical generation and stereocontrolled addition to C=N bonds using chiral N-acylhydrazones. (b) Key bond constructions (blue highlights) in synthetic applications to various chiral amines.
Our focus has been on establishing reliable and versatile modes of stereocontrol for radical addition to imino compounds, so that this type of transformation can be applied for synthesis design even when both the radical and acceptor bear structural complexity.9,10 Chiral N-acylhydrazones11 (Figure 1a) emerged as excellent radical acceptors that afford >95:5 diastereomer ratios for a host of intermolecular radical additions to the C=N bond. Together with the Mn-mediated conditions, this approach has been successfully applied to functionalized chiral amines (Figure 1b) such as γ-amino acids12 and α,α-disubstituted α-amino acids,13 to radical–polar crossover annulations,14 and to a formal synthesis of quinine.15 Broad vetting showed compatibility with multifunctional radical acceptors as well as multifunctional radicals, where large excesses of either component would normally be prohibitive. This feature makes Mn-mediated radical addition to chiral N-acylhydrazones well-suited for target-oriented synthesis and for preparing unusual amino acids and other chiral amine building blocks in the context of medicinal chemistry. These considerations drew our attention to application toward synthesis of tubulysins and analogues.
Tubulysins
Naturally occurring peptides containing unusual amino acids offer bioactivities of potential utility in drug discovery as exemplified by dolastatin 10, plitidepsin, and didemnin B, all of which have reached Phase 2 clinical trials as cancer chemotherapeutic candidates.16 Similarly, the tubulysin family of antimitotic agents (Figure 2),17 the first examples of which were reported by Höfle et al. in 2000,18 are tetrapeptides of myxobacterial origin and are composed of d-N-methylpipecolic acid (Mep), l-isoleucine (Ile), tubuvaline (Tuv), and a C-terminal γ-amino acid, either tubuphenylalanine (Tup) or tubutyrosine (Tut).19 These extraordinarily active antimitotic agents rival dolastatin 10 and epothilone, with some members of the family reaching picomolar potency through a mechanism involving noncompetitive binding at the vinca domain of β-tubulin.20 Early evaluation of tubulysin A indicated selective cytotoxicity and potential antiangiogenic activity,21 although more in vivo studies indicated a limited therapeutic window.22 More recently, targeting strategies involving folate–tubulysin and antibody–tubulysin conjugates have attracted attention to address the toxicity problem.23
Figure 2.

Structures of selected tubulysins, highlighting the unusual amino acids tubuvaline (green) and tubuphenylalanine (blue).
As a consequence of their potent antimitotic effects, much effort has been devoted to chemical synthesis of tubulysins,24 especially 1–4 (Figure 2), leading to several approaches to the natural products as well as numerous analogues.25 Creative strategies for stereocontrol have appeared in preparations of Tuv and Tup,26 and the N,O-acetal functionality in certain tubulysins (e.g., 3) has also inspired new methodology.27 In the diverse slate of creative strategies to prepare Tuv, the main innovations tend to focus on stereocontrol issues (known to impact activity34) at the hydroxyl- or acetoxy-bearing center at C11, stereocontrol at the C13 chiral amine, and introduction of the thiazole. The C11 configuration has been addressed by stereocontrolled enolate oxidation,28,29 metalloenamine aldol addition,30 thiazolyl anion addition,31 hydride reduction,32−35 hetero-Diels–Alder reactions,36 and proline-catalyzed aldol reactions.37 An interesting multicomponent coupling reaction (MCR) rapidly built both C11 and the thiazole, albeit with modest stereocontrol38,39 that was later improved via a catalytic asymmetric Passerini reaction.40 Most Tuv syntheses begin with precursors having the C13 chiral amine already established from various valine derivatives and their homologues. Asymmetric induction at C13 has been accomplished by kinetic resolution of racemic aza-Michael or Mannich adducts,33,41 hydride reduction,30 and additions of various C-nucleophiles (enolate,42 organomagnesium,43 and allylindium reagents44) to imines. A nitrone cycloaddition approach established both C11 and C13 stereogenic centers.45 The breadth of the innovative synthetic route designs directed toward Tuv over many years attest to the challenge of this unusual multifunctional amino acid as well as the continuing interest in the tubulysins as potential cancer chemotherapeutics.46
Our own efforts toward Tup and Tuv were initially published in 2004,47 highlighting the utility of radical addition to chiral N-acylhydrazones in control of the chiral amine configurations of both Tup and Tuv. Subsequent optimizations led to an efficient seven-step route to Tuv (Scheme 1).48 Beginning with known alcohol 5,49 a three-step sequence to N-acylhydrazone 6 was followed by photolysis with isopropyl iodide in the presence of InCl3 and Mn2(CO)10. This key step efficiently furnished 7 with complete stereocontrol at the C13 chiral amine (dr >98:2, minor isomer not detected). Three functional group transformations led to γ-amino acid 8.
Scheme 1.

We later disclosed a high-yielding but step-intensive route to convert 8 to N-TFA tubuvaline methyl ester (10, Scheme 2) in six steps and its application in a synthesis of a tubulysin analogue with a C-terminal alcohol (11, Figure 3).48 Attempts at oxidation of the C-terminus of 11 led to complicated mixtures; a putative C-terminal aldehyde intermediate appeared to have engaged in a cyclization with the γ-amido group as a nucleophile. Thus, we considered more conventional strategies that introduced the C-terminus at the carboxylate oxidation state, along with improvements to synthesis of Tuv derivative 10.
Scheme 2.

Figure 3.

Structures of selected tubulysin analogues.
Despite the excellent overall yield of the prior route to 10, undesirable step economics prompted development of two alternative plans to access 10 (Scheme 3). First, because constructing the tubuvaline thiazole added six steps after introduction of the chiral amine, we envisioned an improved step economy if the thiazole could be tolerated in the radical acceptor such as 13 (route A). Second, we also envisioned merging our previous Mn-mediated radical addition with a less step-intensive thiazole construction that introduced sulfur via condensation of cysteine with aldehyde 14 (route B).50 Here, we present the results of investigating these two routes, the former of which led to greater versatility of Mn-mediated radical addition51 and uncovered new insights concerning 1,3-diastereocontrol in a C=N radical acceptor and the latter of which culminated the synthesis of a dibenzyl analogue of tubulysin V (12, Figure 3).
Scheme 3.

2. Results and Discussion
Mn-Mediated Radical Addition Compatibility with Aromatic Heterocycles
An improved step-economic synthesis of 10 according to route A (Scheme 3) required solving a problem that had emerged during prior synthetic studies on quinine.52 Preliminary studies in that venture revealed an incompatibility of the Mn-mediated radical addition with the basic nitrogen of a quinoline aromatic N-heterocycle present in the N-acylhydrazone radical acceptor. With this in mind, we anticipated that further modification of the Mn-mediated radical addition conditions would be needed in order to accommodate other N-heteroaromatics such as a thiazole.
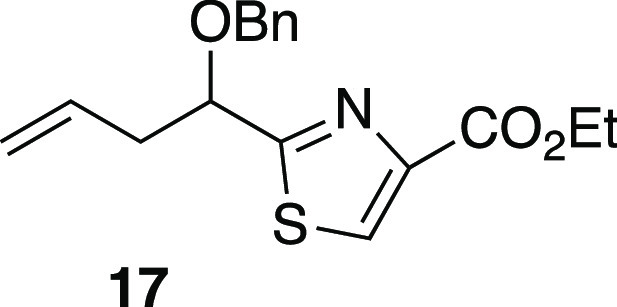
We began this study with a screen of the effects of various aromatic heterocyclic additives on the Mn-mediated isopropyl addition to hydrazone 15 (Table 1). According to our established method, the reaction conditions include a Lewis acid (InCl3) to activate the acceptor toward nucleophilic radical addition.5 The Lewis acid coordinates in bidentate fashion to N-acylhydrazones, as evidenced by spectroscopic data.12 Various N-heterocycles were placed into the control reaction as additives to assess their effects on conversion and yield of isopropyl adduct 16. The control reaction under the usual conditions (conditions A) without any additive gave 16 with a normal yield of 86% (Table 1, entry 1). In separate runs, the presence of four different aromatic N-heterocycles (pyridine, imidazole, benzothiazole, and thiazole 17) in equimolar quantity each diminished the yields of the control reaction to an average of 40% (entries 2–5).
Table 1.

| yield
(%) |
|||
|---|---|---|---|
| entry | additive | Aa | Bb |
| 1 | none | 86 | 72 |
| 2 | pyridine | 44 | 54 |
| 3 | imidazole | 29 | 61 |
| 4 | benzothiazole | 24 | 32 |
| 5 | 17 | 64 | 70 |
Conditions A: 0.11 M (hydrazone), 10 equiv of i-PrI, 2.2 equiv of InCl3.
Conditions B: 0.017 M (hydrazone), 3 equiv of i-PrI, 3.5 equiv of InCl3.
We hypothesized that diminished yields in the presence of aromatic heterocycles was attributable to basic sites of aromatic heterocycles interfering with the desired bidentate binding of InCl3. Modifying the conditions to obviate this problem, we found that increasing the Lewis acid loading to 3.5 equiv and diluting the reaction mixture to one-sixth of the usual concentration generally improved the results. The isolated yields of 16 in the presence of the N-heterocycles using these modifications (conditions B) increased to an average of 54% (Table 1). The hypothesis suggests that more basic N-heterocycles should interfere to a greater degree; indeed, pyridine and imidazole (entries 2 and 3) exhibited more substantial improvement with the modified conditions. Reactions were also cleaner under the modified conditions, with recovery of unreacted 15 accounting for much of the mass balance.
To apply these improved conditions to a more step-economical second-generation approach to tubuvaline, a series of N-acylhydrazone radical acceptors were required. From formyl thiazole 18(53) (Scheme 4), treatment with allylzinc bromide afforded homoallylic alcohol 19 (racemic). Keck allylation54 was identified as a suitable enantioselective counterpart affording 19 with er 10:1, but most of the subsequent route was initially developed using racemic 19. After O-benzylation to afford 17, reduction of the ester gave primary alcohol 20. Oxidative cleavage55 and subsequent condensation of the racemic aldehydes with enantiopure N-aminooxazolidinone 21 then furnished N-acylhydrazone 22 as a diastereomeric mixture. The primary alcohol was acylated to furnish the pivaloate derivative 23. A variety of related hydrazone acceptors with various replacements of the Piv ester with other groups, such as silyl or benzyl ethers, were prepared in a similar fashion (see the Supporting Information). However, these were not as effective in subsequent radical additions.
Scheme 4.

When 23 (dr 1:1, Scheme 4) was subjected to Mn-mediated addition of isopropyl radical, there was exceptionally clean reactivity under the modified conditions (conditions B of Table 1). Although the reaction was incomplete, with 48% recovery of unreacted hydrazone (23′), it furnished 47% yield of isopropyl adduct 24 (90% yield based on conversion). Surprisingly, radical adduct 24 produced in this reaction appeared to be a single diastereomer; it gave only one set of signals in the 13C NMR spectrum, in contrast to the starting 1:1 11R/11S mixture 23 which had shown two sets of signals as expected.56 In attempting to secure complete conversion of N-acylhydrazone 23, an unexpected phenomenon was observed; resubjection of recovered 23′ to the Mn-mediated radical addition did not provide any of the desired adduct 24, and with longer reaction times, gradual decomposition was instead observed. Closer comparison of analytical data for original reactant 23 and recovered reactant 23′ showed that recovered 23′ had only one set of 13C NMR resonances. This contrasted with the two sets of 13C NMR peaks observed for reactant 23, as expected for the C11 epimer mixture. Thus, both 24 and recovered 23′ were single diastereomers; one diastereomer of 23 had produced 24 via radical addition, while the other afforded no radical adduct.
Elaboration to Tubuvaline
Conversion of radical adduct 24 to a protected form of tubuvaline (Scheme 4) entailed TFA installation and reductive N–N bond cleavage (SmI2), removal of the Piv ester (K2CO3, MeOH), and oxidation to the C-terminal carboxylate. The latter conversion involved telescoped oxidation and esterification, as the intermediate carboxylic acid suffered significant material loss upon attempted isolation. Thus, following oxidation of the primary alcohol by catalytic TEMPO in the presence of water, direct esterification by MeI gave the desired methyl ester. This sequence smoothly proceeded to afford N-TFA-O-benzyltubuvaline methyl ester (10) with spectroscopic properties that matched those of 10 from our previously published route.48 This confirmed the configurational assignment of (11R,13R)-24. Overall, the yield of 10 was 8.6% over 10 steps via route A (Scheme 3), a three-step improvement in step economy versus our prior approach.
An Unexpected Kinetic Resolution Reveals 1,3-Diastereocontrol
The isolation of 23′ and 24 as single diastereomers indicates a kinetic resolution process via double diastereoselection; the 1:1 diastereomeric mixture of 23 would present matched and mismatched stereocontrol involving the two stereocontrol elements. The lower relative rate of the mismatched case could permit isolation of one diastereomeric product 24 and recovery of unreacted 23′, also with diastereomeric enrichment. This kinetic resolution via radical addition is unprecedented, prompting further analysis.
For allylmetal addition to chiral β-alkoxyimines, the seminal studies of Yamamoto addressed double diastereoselection involving the β stereogenic center of 25 with alternate configurations of a chiral auxiliary at the imine nitrogen (Figure 4a).57 The chelated (allylMgCl) and nonchelated (allylborane) stereocontrol models discussed by Yamamoto have relevance to various other nonradical additions to β-substituted imino compounds.58,59 However, despite considerable literature searches, we have identified no previously documented cases of double diastereoselection in intermolecular radical additions to β-alkoxyimino compounds.60 In the closest precedent, Lin et al. reported a SmI2-mediated aza-pinacol coupling with β-alkoxysulfinimine 27 (Figure 4b) that incorporates two stereocontrol elements, but stereocontrol was attributed to the N-sulfinyl group and no comparison was made of matched and mismatched control.61 Similarly, we had observed complete stereocontrol in our Mn-mediated radical addition to β-alkoxyhydrazone 6(47) (Scheme 1) and related compounds,12 but without examining matched/mismatched cases, we lacked a basis to expect strong contributions from 1,3-diastereocontrol.
Figure 4.

Examples of 1,3-diastereocontrol in additions to β-alkoxyimines. (a) Chelation-controlled antiselective Grignard addition with matched and mismatched α-phenethyl imines. (b) Pinacol-type radical addition to a β-alkoxysulfinimine.
Because these closest precedents were of questionable relevance to our radical addition, we explored our 1,3-diastereocontrol further. First, to determine the C11 configuration of 24, N-acylhydrazone 23 was prepared with diastereomeric enrichment from a precursor of known configuration at C11. Asymmetric Keck allylation54 of formyl thiazole 18 in the presence of (R)-BINOL afforded (R)-19 (er 10:1, Scheme 4) as judged by Mosher ester analysis, with a configuration consistent with the Keck precedent. Benzylation, carboxylate reduction, oxidative cleavage, and condensation with N-aminooxazolidinone afforded (11R)-23 by the same sequence described in Scheme 4. The 13C NMR spectrum of this compound did not match that of 23′ recovered from incomplete reactions; instead, it matched the spectrum of the 23 diastereomer that was consumed during the radical addition. Thus, the recovered 23′ was assigned the 11S configuration, and the product 24 was assigned the 11R configuration.
The configurational assignments of 23′ and 24 allow for structural hypotheses regarding modes of stereocontrol via chelated structures A and B (Scheme 5). In structure A that can reasonably be presumed to form upon mixing (11S)-23 with InCl3,62 both re and si faces of the C=N acceptor carbon are blocked by the chiral N-acylhydrazone and the thiazole, respectively. This is consistent with poor reactivity due to mismatched double diastereocontrol. In alternative structure B with the thiazole coordinating to InCl3, the si face of the C=N in B appears to be unhindered; thus, we favor structure A to explain the mismatched double diastereocontrol with (11S)-23.
Scheme 5.

If the hypothetical structure A effectively blocks both faces as proposed, an analogous structure bearing the C11 stereogenic center as the only sterecontrol element would also be predicted to provide control of the relative configuration in the radical addition. A simple deletion of the N-acylhydrazone stereocontrol element tested this prediction. Racemic N-acylhydrazone 31 (Scheme 6) was prepared from alkene rac-20 and achiral N-aminooxazolidinone 29 through oxidative cleavage, condensation, and esterification (i.e., the same sequence used to convert 20 to 23 in Scheme 4). Isopropyl radical addition, under the same Mn-mediated conditions of Scheme 4, gave rac-32 with excellent diastereoselectivity, with dr >95:5 as judged by 1H NMR and 13C NMR spectra. Trifluoroacetylation and reductive cleavage of the N–N bond gave rac-33 (dr >95:5), with spectroscopic data matching the previous nonracemic sample obtained during conversion of 24 to 10 (Scheme 4). All these data confirm that auxiliary and substrate stereocontrol were mutually reinforcing in N-acylhydrazone (11R)-23, both favoring si face radical addition to the imino carbon, and that the β-alkoxy substituent is indeed a viable stereocontrol element for such radical additions. Thus, this route to Tuv yielded important new insights into 1,3-diastereocontrol and double differentiation in the context of the radical addition approach to asymmetric amine synthesis.
Scheme 6.

Alternative Elaboration of the Tubuvaline Thiazole
Although we had some success in our plan to improve the step efficiency by conducting radical addition in the presence of the thiazole (route A, Scheme 3), the overall chemical yield of 8.6% was less than desired. Our attention had also been drawn to a tactic from the tubulysin synthetic studies of Zanda33 and Chandrasekhar,29 in which tubuvaline assembly included cyclocondensation of an aldehyde with cysteine methyl ester followed by oxidation to forge the thiazole ring. We hypothesized that this thiazole construction could be applied as an alternative way to streamline our original preparation of the tubuvaline fragment. Thus, from alcohol 34(48) (Scheme 7), Swern oxidation gave the corresponding aldehyde; cysteine methyl ester hydrochloride was directly added to the Swern oxidation mixture to furnish thiazoline 35 in a one-pot operation. Oxidation with manganese dioxide then completed route B to N-TFA-O-benzyltubuvaline methyl ester (10). This material gave spectral data matching that produced from route A as well as our earlier published route.48 Importantly, the efficiency as judged by both step count and overall yield were excellent, with 45% yield over eight steps from alcohol 5.
Scheme 7.

Modified Route to Tubulysins
Our prior studies of tubulysin tetrapeptide assembly yielded a tetrapeptide C-terminal alcohol analog 11 (Figure 3),48 but oxidation to the corresponding carboxylic acid proved difficult. To avoid the late-stage oxidation, the tetrapeptide was assembled with the C-terminal amino acid Tup already in the carboxylate oxidation state. In hopes that an endgame debenzylation would deprotect both C11–OH and the C-terminal carboxylate concurrently, the Tup unit was introduced as a benzyl ester. This material was obtained from primary alcohol 36 (Scheme 8), available in stereochemically pure form via our previously published Mn-mediated radical addition route to Tup.48 Exchange of the N-TFA for N-Boc and oxidation with PDC proceeded via known compounds 37(28) and 38,34 and the known sequence of basic hydrolysis and benzyl esterification gave N-Boc-γ-amino ester 39.34 Removal of the Boc group with trifluoroacetic acid furnished Tup ester 40 in the free amine form, suitable for peptide assembly.
Scheme 8.

Our previously reported tetrapeptide assembly48 required modification of the sequence of couplings. Basic conditions hydrolyzed both N-TFA and methyl ester functions of Tuv derivative 10 (Scheme 9); the crude material was taken up in methanolic HCl to reinstall the methyl ester, providing amino ester 41. Next, attachment of the Mep-Ile dipeptide 42 via its mixed anhydride gave tripeptide 43. Ester saponification at the C-terminus and DECP-mediated coupling with Tup benzyl ester 40 then completed the tetrapeptide assembly to furnish 12 in excellent yield.
Scheme 9.

The final step en route to tubulysin V was envisioned to be a convenient hydrogenolysis of both benzyl groups. To our dismay, all efforts to hydrogenate the di-O-benzyl tetrapeptide 12 led to destruction of this material or no reaction at all.63 After considerable experimentation, a surprisingly effective alternative was finally identified: In a model study, a mixture of 10 and stannic chloride in boiling dichloromethane cleanly removed the benzyl ether to furnish 45 (eq 1).
 |
1 |
Exploratory experiments on debenzylation of the intact tetrapeptide 12 offer some evidence of its feasibility. Exposure of 12 to SnCl4 in refluxing CH2Cl2 indeed furnished a compound that had been twice debenzylated, as judged by high-resolution mass spectrometry (HRMS). The chemical behavior of this compound was consistent with the known reactivity of tubulysin V; it reacted with Ac2O and pyridine to give an O-acetyl derivative (tubulysin U) as judged by mass spectrometry. These results provide supporting evidence for the structure of 11-O-benzyltubulysin V benzyl ester (12), prepared in 43% overall yield via a 12-step longest linear sequence.
3. Conclusion
Building on prior data establishing Mn-mediated radical addition as an excellent means of stereocontrol for tubuphenylalanine (Tup) and tubuvaline (Tuv) synthesis, two alternative routes are presented toward the goal of improved efficiency in synthesis of Tuv. One of these routes required modification of Mn-mediated radical addition conditions to accommodate a thiazole in the radical acceptor, allowing broader versatility of the radical addition approach to asymmetric amine synthesis. While this route did not meet the efficiency goals, it uncovered a case of 1,3-diastereocontrol in radical addition to β-alkoxyimino compounds: in combination with the chiral N-acylhydrazone as a second stereocontrol element, a kinetic resolution occurred, separating two diastereomeric radical acceptors according to their differential reactivity in radical addition. The second route merged our radical addition chemistry with a rapid cyclocondensation and oxidation sequence to afford the thiazole of Tuv, with greater improvement to the overall efficiency, facilitating a high-yielding assembly of the 11-O-benzyltubulysin V benzyl ester.
4. Experimental Section
Materials and Methods
Reactions employed oven- or flame-dried glassware under nitrogen unless otherwise noted. Toluene, tetrahydrofuran (THF), and CH2Cl2 were purchased inhibitor-free, deoxygenated by sparging with argon, and passed through columns of activated alumina under an argon atmosphere prior to use. Nitrogen was passed successively through columns of anhydrous CaSO4 and R3-11 catalyst for removal of water and oxygen, respectively. All other materials were used as received from commercial sources unless otherwise noted. Thin-layer chromatography (TLC) employed glass 0.25 mm silica gel plates with a UV indicator. Flash chromatography columns were packed with 230–400 mesh silica gel as a slurry in the initial elution solvent. Gradient flash chromatography was conducted by adsorption of product mixtures on silica gel, packing over a short pad of clean silica gel as a slurry in hexane, and eluting with a continuous gradient as indicated. Radial chromatography refers to centrifugally accelerated thin-layer chromatography performed using commercially supplied rotors. Nuclear magnetic resonance (NMR) data were obtained at operating frequencies indicated in the text and are reported in units of ppm. Infrared spectra were recorded using a single beam FT-IR spectrophotometer by standard transmission method. Low- and high-resolution mass spectra (TOF) were obtained from local instrumentation facilities services.
Conditions for Compatibility of N-Heteroaromatics with Mn-Mediated Radical Addition
General Procedure A (Table 1, Footnote a)
A Schlenk flask charged with N-acylhydrazone 15 (1 equiv), N-heteroaromatic additive (1 equiv), indium chloride (2.2 equiv), and CH2Cl2 (0.11 M) was stirred for 30 min. Isopropyl iodide (10 equiv) was added via syringe, followed by dimanganese decacarbonyl (1 equiv). The Schlenk flask was sealed and irradiated (300 nm, Rayonet photoreactor) for 6 h. Concentration and flash chromatography or gradient flash chromatography furnished the N-acylhydrazine 16. Preparation and characterization of hydrazine 16 have been previously reported.5
General Procedure B (Table 1, Footnote b)
A Schlenk flask charged with N-acylhydrazone 15 (1 equiv), additive (1 equiv), indium chloride (3.5 equiv), and CH2Cl2 (0.017 M) was stirred for 30 min. Isopropyl iodide (3 equiv) was added via syringe, followed by dimanganese decacarbonyl (1 equiv). The Schlenk flask was sealed and irradiated (broad spectrum with maximum at 300 nm, Rayonet photoreactor)64 for 6 h. Concentration and flash chromatography or gradient flash chromatography furnished the N-acylhydrazine 16.5
Preparation and Characterization Data for New Compounds.65
Ethyl 2-(1-Hydroxybut-3-enyl)thiazole-4-carboxylate (rac-19)
To a mixture of ZnCl2 (5.90 g, 43.3 mmol) and THF (120 mL) was added allylmagnesium
chloride
(2 M in THF, 19.7 mL, 39.4 mmol) slowly via syringe. After being stirred
for 0.5 h, this mixture was transferred via cannula into a solution
of formyl thiazole 18(53) (7.20
g, 39.4 mmol) in THF (150 mL). After 3 h, the reaction was quenched
with water (20 mL) and extracted with EtOAc. The organic phase was
dried over Na2SO4. Concentration and gradient
flash chromatography (25% EtOAc in petroleum ether to 50% EtOAc in
petroleum ether) afforded homoallylic alcohol rac-19 (5.27 g, 60% yield) as a pale yellow solid: mp 46.0–46.5
°C; IR (film) 3411, 2983, 1725, 1489, 1215 cm–1; 1H NMR (300 MHz, CDCl3) δ 8.13 (s,
1H), 5.90–5.76 (m, 1H), 5.27–5.20 (m, 2H), 5.11 (dd, J = 7.9, 4.0 Hz, 1H), 4.42 (q, J = 7.1
Hz, 2 H), 2.91–2.82 (m, 1H), 2.79 (br s, 1H), 2.66–2.55
(m, 1H), 1.40 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ 175.9, 161.5, 146.9,
132.8, 127.5, 119.7, 71.0, 61.6, 42.4, 14.4; HRMS (ESI) m/z [M + H]+ calcd for C10H14NO3S 228.0694; found 228.0686.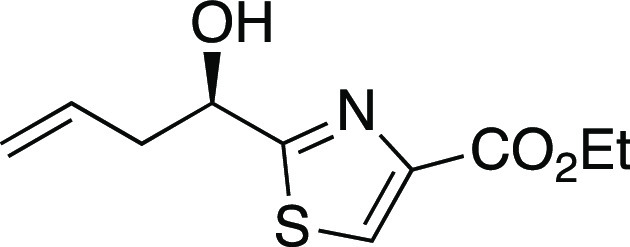
Ethyl 2-(1R-Hydroxybut-3-enyl)thiazole-4-carboxylate ((R)-19)
To activated 4 Å
molecular sieves (23 mg) were added (R)-BINOL (6.2
mg, 0.022 mmol). CH2Cl2 (0.6 mL) and Ti(O-i-Pr)4 (12 μL, 0.040 mmol) were added via
syringe. After 1 h, formyl thiazole 18(53) (20 mg, 0.11 mmol) was added. After cooling to −78
°C, allyltributylstannane (0.10 mL, 0.32 mmol) was added via
syringe. The mixture was stirred for 10 min at −78 °C
and then stored in a freezer (ca. −20 °C) for 24 h. The
mixture was partitioned between saturated aqueous sodium bicarbonate
solution (2 mL) and EtOAc, and the organic phase was dried over Na2SO4. Concentration and flash chromatography (17%
EtOAc in hexane to 33% EtOAc in hexane to 50% EtOAc in hexane) afforded
alcohol (R)-19 (22.7 mg, 93% yield)
as a pale yellow solid: [α]D20.6 = +70.6
(c 1.14, CHCl3).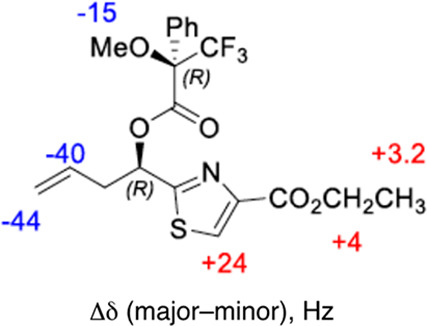
Configuration Assignment: Mosher Ester of (R)-19 (S1)
To a solution of (R)-19 (1 mg, 4.4 μmol) in CH2Cl2 (0.25
mL) was added pyridine (1 μL, 0.0124 mmol) via syringe. (S)-Mosher’s acid chloride (5 mg, 0.020 mmol) was
added. After 24 h, reaction mixture was concentrated. Flash chromatography
afforded Mosher’s ester S1 (1.6 mg, 82% yield)
as a colorless oil: 1H NMR (400 MHz, CDCl3)
δ 8.14 (s, 1H), 8.09 (s, minor diastereomer peak), 7.58–7.51
(m, 2H), 7.45–7.36 (m, 3H), 6.43 (dd, J =
7.5, 4.7 Hz, 1H), 5.80- 5.70 (m, minor diastereomer peak), 5.70–5.60
(m, 1H), 5.19–5.11 (m, minor diastereomer peak), 5.08–5.00
(m, 2H), 4.43 (dd, J = 14.3, 7.0 Hz, 2H), 3.58 (s,
minor diastereomer peak), 3.54 (s, 3H), 2.95–2.76 (m, 2H),
1.40 (t, minor diastereomer peak), 1.41 (t, J = 7.1
Hz, 3H).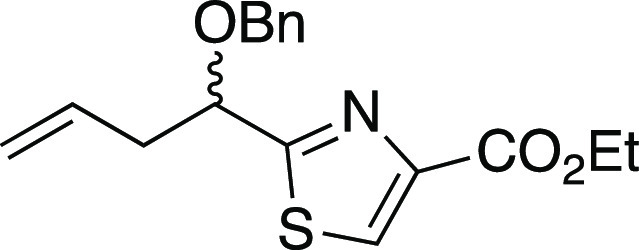
Ethyl 2-(1-(Benzyloxy)but-3-enyl)thiazole-4-carboxylate) (S2)
To a solution of homoallylic alcohol rac-19 (1.16 g, 5.12 mmol) in THF (100 mL)
was added KH (30 wt % in mineral oil, 821 mg, 6.14 mmol) in one portion.
After 5 min, benzyl bromide (0.73 mL, 6.14 mmol) was added via syringe.
After 4 h, the reaction mixture was quenched with water (30 mL), concentrated
to remove THF, and extracted with EtOAc. The organic phase was dried
over Na2SO4. Concentration and flash chromatography
(9% EtOAc in petroleum ether) afforded benzyl ether S2 (884 mg, 54% yield) as a pale yellow oil: IR (film) 3015, 2910,
1725, 1487, 1315, 1217, 1096 cm–1; 1H
NMR (300 MHz, CDCl3) δ 8.17 (s, 1H), 7.36–7.28
(m, 5H), 5.90–5.77 (m, 1H), 5.15–5.05 (m, 2H), 4.91
(dd, J = 7.0, 5.4 Hz, 1H), 4.58 (ABq, Δν = 23.9 Hz, J = 11.6 Hz, 2H), 4.44 (q, J = 7.1 Hz, 2H), 2.74–2.63 (m, 2H), 1.41 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ 174.8, 161.5, 147.1, 137.4, 133.0, 128.6, 128.1,
128.0 (2C), 118.5, 78.8, 72.3, 61.6, 41.4, 14.5; HRMS (ESI) m/z [M + H]+ calcd for C17H20NO3S 318.1164, found 318.1154.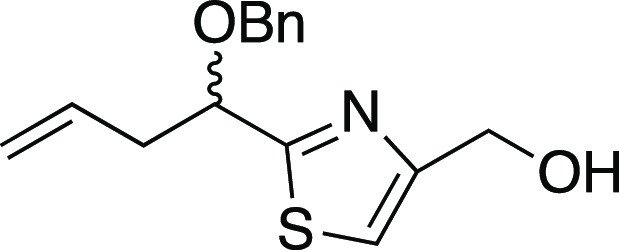
(2-(1-(Benzyloxy)but-3-enyl)thiazol-4-yl)methanol (rac-20)
A solution of ester S2 (1.56
g, 4.91 mmol) in THF (98 mL) was cooled to −78 °C, and
lithium aluminum hydride (2.0 M in THF, 4.91 mL, 9.82 mmol) was added
slowly via syringe. After 1 h, the reaction was warmed to room temperature,
quenched by slow addition of water (50 mL), and extracted with EtOAc.
The organic phase was dried over Na2SO4 and
concentrated. Flash chromatography (67% EtOAc in petroleum ether)
afforded alcohol rac-20 (1.26 g, 93%
yield) as a pale yellow oil: IR (film) 3339, 3078, 3031, 2978, 2869,
1642, 1454, 1324, 1075 cm–1; 1H NMR (300
MHz, CDCl3) δ 7.40–7.25 (m, 5H), 7.20 (s,
1H), 5.93–5.74 (m, 1H), 5.19–5.02 (m, 2H), 4.83–4.74
(m, 3H), 4.58 (ABq, Δν = 36.5 Hz, J = 11.6 Hz, 2H), 3.36 (br s, 1H), 2.80–2.57 (m,
2H); 13C{1H} NMR (75 MHz, CDCl3)
δ 174.2, 156.2, 137.6, 133.2, 128.5, 128.0 (2C), 118.2, 115.3,
78.7, 72.0, 60.8, 41.4; HRMS (ESI) m/z [M + H]+ calcd for C15H18NO2S 276.1058; found 276.1053.
(S)-3-(3-(Benzyloxy)-3-(4-(hydroxymethyl)thiazol-2-yl)propylideneamino)-4-benzyloxazolidin-2-one (22)
To a solution of alkene rac-20 (86 mg, 0.31 mmol) in THF (21 mL) and water (21
mL) was added osmium tetraoxide (2.5 wt % in tert-butyl alcohol, 0.12 mL, 0.012 mmol). After 5 min, sodium periodate
(265 mg, 1.24 mmol) was added. After 5.5 h, the reaction was quenched
with saturated aqueous sodium thiosulfate solution (50 mL) and extracted
with EtOAc. The organic phase was dried over Na2SO4, concentrated, then filtered through a short column of silica
gel, eluting with EtOAc. Concentration furnished the crude aldehyde,
which was dissolved in CH2Cl2 (31 mL) along
with (S)-3-amino-4-phenylmethyl-2-oxazolidinone (21, 119 mg, 0.62 mmol). After 12.5 h, concentration and gradient
flash chromatography (83% EtOAc in petroleum ether to EtOAc to 5%
methanol in EtOAc) afforded 22 (106 mg, 75% yield, inseparable
1:1 mixture of diastereomers) as a colorless wax: IR (film) 3404,
3013, 1763, 1405, 1217, 1092 cm–1; 1H
NMR (300 MHz, CDCl3) δ 7.91–7.84 (m, 1H),
7.38–7.19 (m, 9H), 7.09–7.04 (m, 2H), 5.08–5.02
(m, 1H), 4.75–4.52 (m, 4H), 4.33–4.23 (m, 1H), 4.20–4.15
(m, 1H), 4.10–4.03 (m, 1H), 3.73 (br s, 1H), 3.15–2.93
(m, 3H), 2.77–2.63 (m, 1H), (diastereomer peaks were unresolved); 13C{1H} NMR (75 MHz, CDCl3) δ 172.5,
156.8, 154.0, 149.6 and 149.4, 137.0, 134.9, 129.2, 128.8, 128.43
and 128.42, 127.9 (2C), 127.2, 115.4, 76.5 and 76.4, 71.9 and 71.8,
65.6, 60.6, 56.4, 40.04 and 39.97, 36.1, and 36.0 (some diastereomer
peaks were unresolved); HRMS (ESI) m/z [M + Na]+ calcd for C24H25N3O4SNa 474.1463; found 474.1451.
(S)-3-(3-(Benzyloxy)-3-(4-((pivaloyloxymethyl)thiazol-2-yl)propylideneamino)-4-benzyloxazolidin-2-one (23)
To a solution of alcohol 22 (220 mg, 0.487 mmol) and pyridine (0.20 mL, 2.4 mmol) in CH2Cl2 (4.9 mL) was added pivaloyl chloride (0.09
mL, 0.7 mmol) via syringe. After 16 h, concentration and flash chromatography
(50% EtOAc in petroleum ether) afforded pivaloate 23 (209
mg, 80% yield, inseparable 1:1 mixture of diastereomers) as a colorless
oil: IR (film) 3019, 2980, 2939, 1742, 1208, 1144, 1047 cm–1; 1H NMR (300 MHz, CDCl3) δ 8.02–7.95
(m, 1H), 7.38–7.23 (m, 9H), 7.14–7.04 (m, 2H), 5.22–5.19
(m, 2H), 5.09–5.04 (m, 1H), 4.74–4.56 (m, 2H), 4.37–4.18
(m, 2H), 4.14–4.08 (m, 1H), 3.20–3.08 (m, 1H), 3.08–3.03
(m, 2H), 2.80–2.65 (m, 1H), 1.23 (s, 9H) (diastereomer peaks
were unresolved); 13C{1H} NMR (75 MHz, CDCl3) δ 178.1, 172.53 and 172.48, 154.1, 152.0, 150.0 and
149.8, 137.2, 135.1, 129.4, 129.0, 128.6, 128.1 (2C), 127.4, 117.21
and 117.17, 76.6 and 76.5, 72.1 and 72.0, 65.7, 61.9, 57.0, 40.24
and 40.16, 38.9, 36.5 and 36.4, 27.2 (some diastereomer peaks were
unresolved); HRMS (ESI) m/z [M +
H]+ calcd for C29H34N3O5S 536.2219; found 536.2208.
(1S,1′R,3′R)-3-(1′-(Benzyloxy)-1′-(4″-(pivaloyloxymethyl)thiazol-2″-yl)-4′-methylpentan-3′-ylamino)-4-benzyloxazolidin-2-one (24)
From N-acylhydrazone 23 (104 mg, 0.194 mmol, 1:1 mixture of diastereomers) via
general procedure B was obtained amine 24 (53 mg, 47%
yield) as a colorless oil: IR (film) 3290, 2963, 2872, 1755, 1604,
1497, 1398, 1368, 1281, 1216, 1160, 1095 cm–1; 1H NMR (300 MHz, CDCl3) δ 7.40–7.22
(m, 9H), 7.12–7.05 (m, 2H), 5.22 (s, 2H), 5.08 (dd, J = 7.7, 3.9 Hz, 1H), 4.64 (ABq, Δν = 36.2 Hz, J = 11.4 Hz, 2H), 4.35 (br s, 1H), 4.05
(dd, apparent t, J = 8.2 Hz, 1H), 3.95 (dd, J = 8.8, 4.5 Hz, 1H), 3.91–3.80 (m, 1H), 3.21 (dd, J = 13.4, 3.4 Hz, 1H), 3.17–3.08 (m, 1H), 2.49 (dd, J = 13.4, 9.9 Hz, 1H), 2.14–1.78 (m, 3H), 1.24 (s,
9H), 0.94 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ 178.1, 174.2, 158.5, 151.8, 137.3, 135.7, 129.1,
128.8, 128.5, 128.0, 127.9, 127.0, 116.7, 77.6, 72.4, 65.5, 61.9,
59.8, 57.8, 38.8, 36.4, 35.8, 28.7, 27.2, 18.9, 15.9; HRMS (ESI) m/z [M + H]+ calcd for C32H42N3O5S 580.2845; found
580.2840.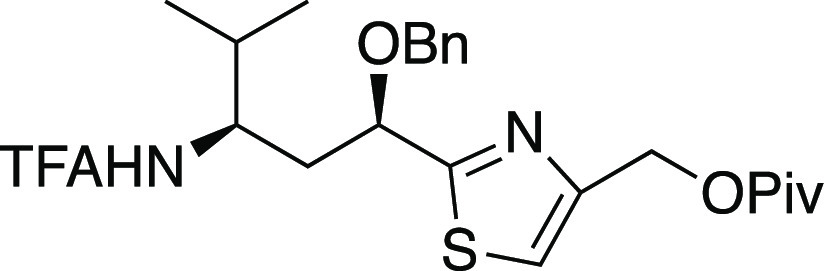
(2-((1′R,3′R)-3′-(Trifluoroacetamido)-1′-(benzyloxy)-4′-methylpentyl)thiazol-4-yl)methyl pivaloate (33), from Chiral Oxazolidinone Precursor 24
To the solution of amine 24 (31 mg, 0.053 mmol) and DMAP (130 mg, 1.06 mmol) in CH2Cl2 (0.5 mL) was added TFAA (0.5 mL). After 15 h, mixture was filtered and concentrated. Flash chromatography (25% EtOAc in hexane) afforded intermediate (34 mg, 94% yield) as colorless oil. Part of this intermediate (6 mg, 0.0089 mmol) was dissolved in THF (0.045 mL) and MeOH (0.045 mL, dried over activated 4 Å molecular sieves), to which samarium iodide solution (0.2 M in THF) was added until the blue color persisted for 1 s. After quenching by exposure to air, concentration and flash chromatography (25% EtOAc in hexane) afforded TFA protected amine 33 (3.3 mg, 70% yield over two steps) as a colorless oil. IR (film) 3336, 3020, 2963, 2927, 2853, 1720, 1457 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.39–7.30 (m, 5H), 7.25 (s, 1H), 6.88 (d, J = 9.2 Hz, 1H), 5.21 (s, 2H), 4.81 (dd, J = 8.9, 4.3 Hz, 1H), 4.55 (ABq, Δν = 58.4 Hz, J = 10.7 Hz, 2H), 4.17–4.07 (m, 1H), 2.15–1.99 (m, 2H), 1.83–1.73 (m, 1H), 1.25 (s, 9H), 0.94 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 178.2, 173.8, 157.2 (q, J = 36 Hz), 152.0, 136.8, 128.8, 128.7, 128.5, 117.2, 116.1 (t, J = 288 Hz), 76.5, 72.9, 61.9, 52.7, 39.0, 38.9, 31.7, 27.3, 19.0, 18.4; HRMS (ESI) m/z [M + H]+ calcd for C24H32F3N2O4S 501.2035; found 501.2031.
(2-((1′R*,3′R*)-3-(2,2,2-Trifluoroacetamido)-1-(benzyloxy)-4-methylpentyl)thiazol-4-yl)methyl Pivalate (rac-33), from Achiral Oxazolidinone Precursor rac-32
To the solution
of amine rac-32 (10 mg, 0.020 mmol)
and DMAP (49 mg, 0.4 mmol) in CH2Cl2 (0.2 mL)
was added TFAA (0.2 mL). After 27 h, the mixture was filtered and
concentrated. Flash chromatography (17% EtOAc in hexane) afforded
a colorless oil: HRMS (ESI) m/z [M
+ H]+ calcd for C27H35F3N3O6S 586.2199; found 586.2185. To a solution
of this material in THF (0.05 mL) and MeOH (0.05 mL, dried over activated
4 Å molecular sieves) was added samarium iodide solution (0.2
M) until the blue color remained for 1 s. After quenching by exposure
to air, concentration and flash chromatography (17% EtOAc in hexane)
afforded TFA protected amine rac-33 (9.4
mg, 92% yield over two steps) as colorless oil.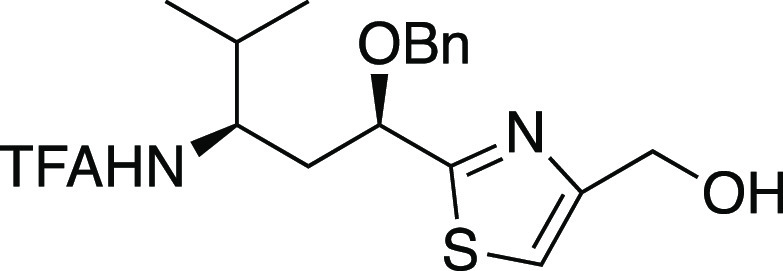
(2-((1′R,3′R)-3′-(Trifluoroacetamido)-1′-(benzyloxy)-4′-methylpentyl)thiazol-4-yl)methanol (S3)
To a solution of pivaloate 33 (9.0 mg, 0.018 mmol) in MeOH (1.8 mL) was added potassium
carbonate (5.0 mg, 0.036 mmol). After 16 h, saturated aqueous ammonium
chloride solution (0.5 mL) was added. This mixture was extracted with
EtOAc, and the organic layer was dried over Na2SO4. Concentration and gradient flash chromatography (17% EtOAc in hexane
to 67% EtOAc in hexane) afforded 24% recovery of reactant 33 and alcohol S3 (5.7 mg, 76% yield) as a colorless oil:
[α]D20 + 22.4 (c 0.285, CHCl3); IR (film) 3279, 3018, 2966,
2400, 1720, 1215 cm–1; 1H NMR (400 MHz,
CDCl3) δ 7.40–7.31 (m, 5H), 7.22 (s, 1H),
6.72 (d, J = 8.8 Hz, 1H), 4.81 (dd, J = 9.3, 3.7 Hz, 1H), 4.77 (s, 2H), 4.55 (ABq, Δν = 64.5 Hz, J = 10.7 Hz, 2H), 4.18–4.07 (m,
1H), 2.63 (br s, 1H), 2.15–1.96 (m, 2H), 1.83–1.72 (m,
1H), 0.93 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 174.1, 157.2 (q, J = 36 Hz), 156.2,
136.7, 128.6, 128.5, 128.4, 116.2 (q, J = 290 Hz),
115.4, 76.4, 72.9, 60.9, 52.5, 38.9, 31.7, 18.8, 18.3; HRMS (ESI) m/z [M + Na]+ calcd for C19H23F3N2O3SNa
439.1279; found 439.1270.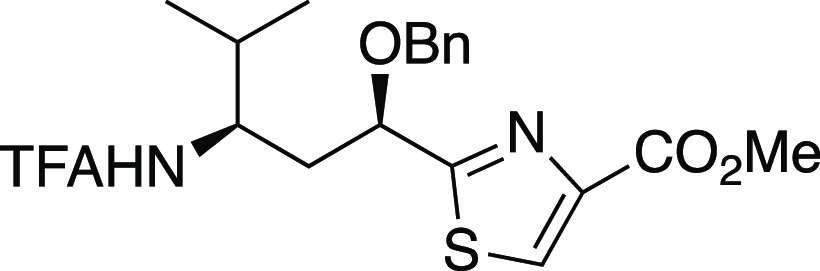
Methyl (2-((1′R,3′R)-1′-Benzyloxy-4′-methyl-3′-(trifluoroacetamido)pent-1′-yl)-thiazol-4-yl)carboxylate (N-TFA-11-O-benzyltubuvaline methyl ester, 10). Procedure via oxidation of alcohol S3
A solution of TEMPO (0.7 mg, 5 μmol), BAIB (30 mg, 0.092 mmol) and CH2Cl2 (0.6 mL) was prepared. A portion (ca. 10%) of this solution was added into alcohol S3 (1.9 mg, 0.0046 mmol), then water (0.012 mL) was added. After 30 min, an additional portion of TEMPO (0.3 mg, 2 μmol) was added. After 20 h, the reaction mixture was partitioned between diethyl ether and saturated sodium bicarbonate aqueous solution. The aqueous phase was acidified with 2 M HCl, then extracted with EtOAc. The organic phase was dried over Na2SO4 and concentrated to afford the crude carboxylic acid as colorless oil. To a solution of this carboxylic acid in DMF (0.2 mL) was added potassium carbonate (0.7 mg, 5.2 μmol) and iodomethane (0.6 mg, 3.9 μmol; measured via syringe weight before and after addition). After 21 h, flash chromatography (25% EtOAc in hexane) afforded methyl ester 10 as colorless oil (1.3 mg, 64% yield).
Procedure via oxidation of thiazolidine 35
To a solution of thiazolidine 35 (10 mg, 0.02 mmol)
in anhydrous acetonitrile was added MnO2 (20 mg, 0.2 mmol).
After 9 h, another portion of MnO2 (20 mg, 0.2 mmol) was
added. After 12 h, the reaction mixture was filtered through Celite
and concentrated. Flash chromatography (2:1 petroleum ether/ethyl
acetate) afforded 10 (8 mg, 80% yield) as a pale yellow
oil: [α]D23 +12.0 (c 0.26,
CHCl3); IR (film) 3583, 3315, 2962, 2924, 1722, 1710, 1207,
1180; 1H NMR (400 MHz, CDCl3) δ 8.21 (s,
1H), 7.48–7.29 (m, 5H), 6.63 (d, J = 9.7 Hz,
1H), 4.89 (dd, J = 9.7, 3.8 Hz, 1H), 4.54 (ABq, J = 10.6 Hz, Δν = 35 Hz, 2H),
4.29–4.12 (m, 1H), 3.97 (s, 3H), 2.12–1.98 (m, 2H),
1.78 (dq, J = 13.4, 6.7 Hz, 1H), 0.94 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) 174.8, 161.6,
157.0 (q, J = 36 Hz), 146.9, 136.4, 128.6, 128.6,
128.4, 128.2, 115.9 (q, J = 288 Hz), 76.3, 73.2,
52.6, 52.1, 39.2, 31.8, 18.9, 18.0; MS (ESI) m/z (rel intensity) 467 ([M + Na]+, 100), 445 [M
+ H]+, 26); HRMS (ESI) m/z [M + Na]+ calcd for C20H23F3N2O4SNa 467.1228; found: 467.1224.
3-(3-(Benzyloxy)-3-(4-(hydroxymethyl)thiazol-2-yl)propylideneamino)oxazolidin-2-one (rac-30)
To a solution of alkene rac-20 (170 mg, 0.62 mmol) in THF (31 mL) and
water (31 mL) was added osmium tetraoxide (2.5 wt % in tert-butyl alcohol, 0.06 mL, 0.006 mmol). After 5 min, sodium periodate
(535 mg, 2.5 mmol) was added. After 13 h, the reaction was quenched
with saturated aqueous sodium thiosulfate solution (100 mL) and extracted
with EtOAc. The organic phase was dried over Na2SO4, concentrated, and filtered through a short column of silica
gel, eluting with EtOAc. Concentration furnished the crude aldehyde
(180 mg), part of which (150 mg) was dissolved in CH2Cl2 (50 mL) along with 3-amino-2-oxazolidinone (120 mg, 1.18
mmol). After 12 h, concentration and flash chromatography (5% MeOH
in CH2Cl2) afforded hydrazone rac-30 (98 mg, 53% yield over two steps, racemic) as a
colorless wax: IR (film) 3400, 3018, 2252, 1772, 1407, 1215, 1091
cm–1; 1H NMR (600 MHz, CDCl3) δ 7.37–7.27 (m, 5H), 7.22 (s, 1H), 6.94 (t, J = 5.5 Hz, 1H), 4.98 (dd, J = 6.8, 5.5
Hz, 1H), 4.74 (s, 2H), 4.61 (ABq, Δν =
85.0 Hz, J = 11.7 Hz, 2H), 4.44 (dd, apparent t, J = 8.0 Hz, 2H), 3.69–3.62 (m, 2H), 3.28 (br s, 1H),
3.06–2.95 (m, 2H); 13C{1H} NMR (150 MHz,
CDCl3) δ 172.5, 156.7, 154.6, 143.6, 137.2, 128.6,
128.19, 128.16, 115.5, 76.6, 72.0, 61.5, 60.8, 42.1, 39.4; HRMS (ESI) m/z [M + H]+ calcd for C17H20N3O4S 362.1174; found
362.1167.
3-(3-(Benzyloxy)-3-(4-((pivaloyloxymethyl)thiazol-2-yl)propylideneamino)oxazolidin-2-one (rac-31)
To a solution of alcohol rac-30 (88 mg, 0.24 mmol) in CH2Cl2 (2.4 mL) was added pivaloyl chloride (44 μL,
0.36 mmol) and pyridine (58 μL, 0.72 mmol) via syringe. After
7 h, gradient flash chromatography (9% EtOAc in hexane to 17% EtOAc
in petroleum ether) afforded pivaloate rac-31 (90 mg, 83% yield) as a colorless oil: IR (film) 3017,
2976, 1772, 1728, 1479, 1407, 1216, 1152 cm–1; 1H NMR (600 MHz, CDCl3) δ 7.38–7.29
(m, 5H), 7.24 (s, 1H), 6.97 (dd, apparent t, J =
5.5 Hz, 1H), 5.21 (s, 2H), 4.98 (dd, J = 6.8, 5.7
Hz, 1H), 4.62 (ABq, Δν = 84.2 Hz, J =
11.7 Hz, 2H), 4.47 (t, J = 7.9 Hz, 2 H), 3.71–3.64
(m, 2H), 3.09–2.97 (m, 2H), 1.24 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 178.2, 172.3, 154.5,
152.1, 143.7, 137.3, 128.7, 128.3, 128.2, 117.3, 76.6, 72.0, 61.9,
61.4, 42.2, 39.4, 39.0, 27.3; HRMS (ESI) m/z [M + H]+ calcd for C22H28N3O5S 446.1750; found 446.1743.
(1′R*,3′R*)-3-(1′-(Benzyloxy)-1′-(4″-(pivaloyloxymethyl)thiazol-2″-yl)-4′-methylpentan-3′-ylamino)oxazolidin-2-one (rac-32)
From N-acylhydrazone rac-31 (49 mg, 0.11 mmol) via general procedure
B was obtained amine rac-31 (24 mg,
45% yield) as a colorless oil: IR (film) 3440, 3293, 2960, 2872, 1731,
1479, 4555, 1397, 1368, 1281, 1150 cm–1; 1H NMR (600 MHz, CDCl3) δ 7.40–7.25 (m, 5H),
7.22 (s, 1H), 5.21 (s, 2H), 5.01 (dd, J = 8.2, 3.6
Hz, 1H), 4.62 (ABq, Δν = 83.0 Hz, J = 11.5 Hz, 2H), 4.37 (br s, 1H), 4.25–4.18 (m,
2H), 3.50 (dd, J = 14.9, 8.0 Hz, 1H), 3.42 (dd, J = 16.1, 8.1 Hz, 1H), 3.03 (d, J = 9.42
Hz, 1H), 2.00–1.93 (m, 1H), 1.92–1.84 (m, 1H), 1.78–1.70
(m, 1H), 1.24 (s, 9H), 0.89 (d, J = 6.8 Hz, 3H),
0.87 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (150 MHz, CDCl3) δ 178.2, 174.5, 159.2, 151.8,
137.6, 128.6, 128.2, 128.0, 116.8, 77.3, 72.4, 62.0, 61.3, 60.5, 47.9,
38.9, 35.9, 29.1, 27.3, 19.0, 16.0; HRMS (ESI) m/z [M + H]+ calcd for C25H36N3O5S 490.2376; found 490.2363.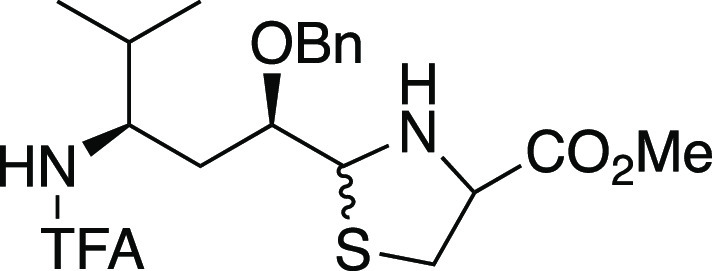
Methyl (2-((1′R,3′R)-3′-(Trifluoroacetamido)-1′-(benzyloxy)-4′-methylpentyl)thiazolidine-4-yl)carboxylate (35)
A solution of DMSO (0.02 mL, 0.27 mmol)
in 0.128 mL CH2Cl2 was cooled to −78
°C followed by addition of oxalyl chloride (0.013 mL, 0.15 mmol).
The mixture was stirred at −78 °C for 20 min followed
by addition of alcohol 34(48) (23 mg, 0.06 mmol) as a solution in 0.057 mL CH2Cl2. After 2 h at −78 °C, triethylamine (0.086 mL,
0.63 mmol) was added, and the temperature was allowed to rise to 0
°C over a period of 2 h. The reaction mixture at 0 °C was
then diluted using 0.3 mL anhydrous ethyl alcohol followed by addition
of cysteine methyl ester hydrochloride (18 mg, 0.1 mmol). After 12
h, concentration and flash chromatography (1:1 petroleum ether/ethyl
acetate) afforded 35 (24 mg, 77% yield, mixture of diastereomers)
as pale yellow oil: [α]D22 −32.5
(c 0.25, CHCl3); IR (film) 3583, 3314,
2960, 2925, 1742, 1721, 1710, 1203, 1181, 1160; 1H NMR
(CDCl3, 400 MHz) δ 7.41–7.27 (m, 10H), 6.74
(d, J = 9.0 Hz, 1H), 6.63 (d, J =
9.2 Hz, 1H), 5.02 (d, J = 10.6 Hz, 1H), 4.81–4.72
(m, 2H), 4.67–4.53 (m, 3H), 4.10–3.91 (m, 3H), 3.87–3.81
(m, 1H), 3.80 (s, 3H), 3.78 (s, 3H), 3.43–3.34 (m, 1H), 3.30–3.23
(m, 2H), 2.93 (dd, J = 10.6, 7.6 Hz, 1H), 2.83 (dd, J = 10.1, 9.4 Hz, 2H), 2.02–1.95 (m, 2H), 1.89–1.77
(m, 4H), 1.75–1.69 (m, 2H), 0.92 (d, J = 6.8
Hz, 3H), 0.91–0.86 (m, 9H), some diastereomer peaks were not
resolved; 13C{1H} NMR (100 MHz, CDCl3) 171.8, 171.3, 156.9 (q, J = 36 Hz), 137.70, 137.67,
128.5, 128.3, 128.2, 128.02, 127.99, 115.9 (q, J =
288 Hz), 111.6, 80.5, 76.7, 75.2, 73.9, 73.2, 71.7, 65.4, 64.3, 53.3,
52.9, 52.6, 52.5, 37.53, 37.45, 35.9, 34.3, 31.9, 31.8, 19.0, 18.5,
18.2, some diastereomer peaks were not resolved; MS (ESI) m/z (rel intensity) 471 ([M + Na]+, 12), 449 ([M + H]+, 100); HRMS (ESI) m/z [M + H]+ calcd for C20H28F3N2O4S 449.1722; found:
449.1714.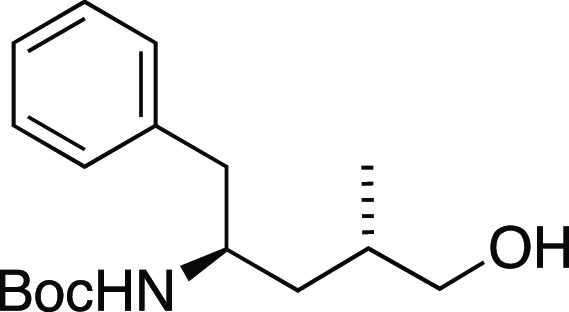
(2S,4R)-4-(tert-Butoxycarbonylamino)-2-methyl-5-phenyl-1-pentanol (37)
A solution of trifluoroacetyl amino alcohol 36(48) (57 mg, 0.02 mmol) in a suspension
of methanol–water (9.5 mL, 5:1) was cooled to 0 °C followed
by addition of Ba(OH)2·8H2O (124 mg, 0.39
mmol). The reaction mixture was allowed to warm to room temperature
over 3 h and then heated at 40 °C with an oil bath. After 12
h, the mixture was cooled to room temperature and concentrated. To
a solution of the residual material in diethyl ether (2.7 mL) at 0
°C were added triethylamine (0.033 mL, 0.23 mmol) and Boc anhydride
(17 mg, 0.078 mmol), and the reaction was allowed to reach ambient
temperature. After 12 h, the reaction was quenched with saturated
aqueous NaHCO3 and partitioned between water and CH2Cl2. The organic phase was washed with brine, dried
over Na2SO4, and concentrated. Flash chromatography
(petroleum ether to 1:1 petroleum ether/ethyl acetate) afforded the
known alcohol 37(28) (25 mg,
44%).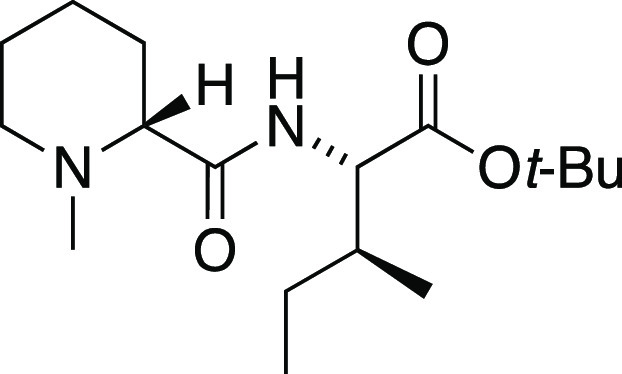
N-Methyl-d-pipecolyl-l-isoleucine tert-Butyl Ester (S4)
A solution of N-methyl-d-pipecolic
acid66 (100 mg, 0.55 mmol) in DMF (9.1
mL) was cooled to 0 °C followed
by addition of diisopropylethylamine (0.387 mL, 2.22 mmol). To this
mixture was added l-isoleucine tert-butyl
ester hydrochloride (56 mg, 0.35 mmol) followed by addition of diethyl
cyanophosphate (DECP, 0.101 mL, 0.66 mmol). The reaction was allowed
to reach ambient temperature. After 12 h, the reaction was quenched
with water and partitioned between saturated aqueous NaHCO3 and EtOAc. The organic phase was washed with brine, dried over Na2SO4, and concentrated. Flash chromatography (1:1
petroleum ether/ethyl acetate to 1:4 petroleum ether/ethyl acetate)
afforded S4 (144 mg, 83% yield) as a pale yellow oil:
[α]D26 +70.5 (c 0.96, CHCl3); IR (film) 3388, 2965, 2936, 2791, 1736, 1727, 1677, 1502, 1147,
847 cm–1; 1H NMR (CDCl3, 400
MHz) δ 6.96 (d, J = 9.5 Hz, 1H), 4.38 (dd, J = 9.2, 4.5 Hz, 1H), 2.83 (br d, J = 11.6
Hz, 1H), 2.41 (dd, J = 11.2, 3.4 Hz, 1H), 2.15 (s,
3H), 1.99 (ddd, J = 12.7, 11.6, 3.1 Hz, 1H), 1.88–1.80
(m, 2H), 1.71–1.62 (m, 1H), 1.60–1.46 (m, 2H), 1.46–1.36
(m, 2H), 1.38 (s, 9H), 1.28–1.03 (m, 2H), 0.92 (t, J = 6.7 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) 174.0, 170.7,
81.4, 69.6, 56.1, 55.3, 44.7, 37.7, 30.6, 27.9, 25.2, 25.1, 23.2,
15.6, 11.6; MS (ESI) m/z (rel intensity)
335 ([M + Na]+, 4), 313 ([M + H]+, 100); HRMS
(ESI) m/z [M + H]+ calcd
for C17H33N2O3 313.2491;
found 313.2498.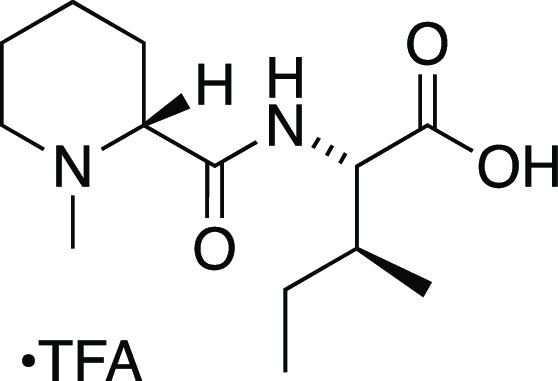
N-Methyl-d-pipecolyl-l-isoleucine Trifluoroacetate Salt (42)
A solution of tert-butyl ester S4 (50 mg, 0.16 mmol) in dichloromethane
(1.6 mL) was treated with trifluoroacetic acid (0.204 mL, 2.66 mmol)
followed by reflux for 4 h with heat applied by oil bath. The mixture
was then cooled to room temperature and concentrated to afford the
carboxylic acid 42(62) as the
TFA salt, which was used without further purification. MS (ESI) m/z (rel intensity) 257 ([M + H]+, 100).
N-Methyl-d-pipecolyl-l-isoleucinyl-(O-benzyl)tubuvaline Methyl Ester (43)
A solution of trifluoroacetyl amino ester 10 (12 mg, 0.02 mmol) in methanol–water (1 mL, 5:1) was cooled to 0 °C followed by addition of Ba(OH)2·8H2O (42 mg, 0.16 mmol). The reaction mixture was allowed to warm ambient temperature over 3 h and then maintained at 40 °C overnight with heat applied by oil bath. The mixture was then cooled to room temperature and concentrated to afford the crude amino acid: MS (ESI) m/z (rel intensity) 357 ([M + Na]+, 5), 335 ([M + H]+, 100). The crude amino acid was treated with methanolic HCl (2 mL of a stock solution prepared from 5 mL methanol and 0.71 mL acetyl chloride) at 0 °C for 10 min and then heated at reflux for 2.5 h. The reaction mixture was cooled to 0 °C and concentrated. The residue was taken up in toluene and concentrated to ensure azeotropic removal of traces of acetic acid. Flash chromatography (2:1 petroleum ether/ethyl acetate to 9:1 dichloromethane/methanol) afforded the Tuv methyl ester 41 (9 mg, 96% yield) as a pale yellow oil; MS (ESI) m/z (rel intensity) 371 ([M + Na]+, 3), 349 ([M + H]+, 100). This material was used immediately in the coupling reaction.
To a
solution of the Mep-Ile dipeptide 42 (12 mg, 0.03 mmol)
in anhydrous ethyl acetate (0.212 mL) was added N-methyl morpholine (0.004 mL, 0.03 mmol). The mixture was cooled
to −10 °C and isobutyl chloroformate (0.005 mL, 0.034
mmol) was added via syringe, followed by a solution of Tuv amino ester 41 (6 mg, 0.01 mmol) in anhydrous ethyl acetate (0.085 mL)
via cannula. The reaction mixture was allowed to warm slowly to ambient
temperature. After 12 h, the reaction mixture was partitioned between
water and EtOAc. The organic phase was washed with brine, dried over
MgSO4, and concentrated. Flash chromatography (petroleum
ether to dichloromethane to 9:1 dichloromethane/methanol) afforded
the tripeptide 43 (10 mg, > 99%) as a pale yellow
oil;
[α]D25 −2.7 (c 0.07, CDCl3); IR (film) 3583, 2959, 2925, 1737, 1726,
1691, 1678; 1H NMR (CDCl3, 400 MHz) δ
8.17 (s, 1H), 7.55–7.28 (m, 5H), 7.00 (d, J = 8.5 Hz, 1H), 6.18 (d, J = 9.9 Hz, 1H), 4.85 (dd, J = 9.2, 4.2 Hz, 1H), 4.56 (ABq, J = 10.3
Hz, Δν = 38 Hz, 2H), 4.33–4.23
(m, 1H), 4.09 (dd, J = 8.6, 8.4 Hz, 1H), 3.96 (s,
3H), 2.91 (br d, J = 11.7 Hz, 1H), 2.50 (dd, J = 11.2, 3.4 Hz, 1H), 2.22 (s, 3H), 2.09–1.86 (m,
6H), 1.81–1.69 (m, 2H), 1.57–1.47 (m, 2H), 1.44–1.33
(m, 1H), 1.24–1.13 (m, 2H), 0.97 (d, J = 6.7
Hz, 3H), 0.96–0.88 (m, 3H), 0.87 (d, J = 2.4
Hz, 3H), 0.86 (d, J = 2.7 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) 175.9, 174.9, 171.0, 161.8,
146.9, 137.1, 128.6, 128.5, 128.2, 128.0, 77.2, 73.5, 69.7, 57.8,
55.4, 52.5, 50.5, 45.1, 40.2, 35.3, 32.2, 30.9, 25.2, 24.9, 23.3,
19.0, 17.8, 16.2, 10.7; MS (ESI) m/z (rel intensity) 609 ([M + Na]+, 100), 587 ([M + H]+, 15); HRMS (ESI) m/z [M
+ H]+ calcd for C31H47N4O5S 587.3267; Found: 587.3255.
11-O-Benzyltubulysin V Benzyl Ester (12)
To a solution of tripeptide 43 (5 mg, 8 μmol) in THF (0.22 mL) was added an aqueous solution of LiOH·H2O (1 N, 0.021 mL). The reaction mixture was stirred for 72 h and then acidified using TFA to pH 1–2. The mixture was partitioned between water and ethyl acetate, and the organic phase was dried over Na2SO4. Concentration afforded carboxylic acid 44 that was used without further purification; MS (ESI) m/z (rel intensity) 595 ([M + Na]+, 20), 573 ([M + H]+, 100).
To a solution of N-Boc amine 39(34) (6 mg, 0.015 mmol) in CH2Cl2 (0.02 mL) was added TFA (0.02 mL) at ambient temperature. After 2 h the mixture was concentrated to furnish the trifluoroacetate salt of Tup benzyl ester (40) that was used without further purification.
To a solution of carboxylic acid 44 (5 mg, 7 μmol)
and i-Pr2NEt (5.2 μL, 0.029 mmol)
in DMF (0.01 mL) at 0 °C was added a solution of 40 (4.5 mg, 0.015 mmol) in DMF (0.110 mL) followed by diethyl cyanophosphonate
(DECP, 1.2 μL, 8 μmol). The mixture was allowed to warm
to ambient temperature. After 12 h, the reaction was quenched with
water and partitioned between saturated aqueous NaHCO3 and
EtOAc. The organic phase was washed with brine and dried over Na2SO4. Flash chromatography (CH2Cl2 to 24:1 CH2Cl2/MeOH) afforded tetrapeptide 12 (7 mg, > 99%) as a pale yellow oil: 1H NMR
(CDCl3, 400 MHz) δ 8.02 (s, 1H), 7.66–7.28
(m, 15H),
7.03 (d, J = 8.8 Hz, 1H), 6.27–6.08 (d, J = 9.8 Hz, 1H), 5.09 (q, J = 12.3 Hz,
2H), 4.68 (dd, J = 10.6, 2.6 Hz, 1H), 4.61–4.42
(m, 3H), 4.33–4.21 (m, 1H), 4.20–4.07 (m, 2H), 3.02–2.83
(m, 3H), 2.76–2.63 (m, 1H), 2.52 (dd, J =
11.6, 3.5 Hz, 1H), 2.23 (s, 3H), 2.11–1.98 (m, 11H), 1.97–1.70
(m, 4H), 1.20 (d, J = 7.1 Hz, 3H), 0.96 (d, J = 6.7 Hz, 3H), 0.93–0.87 (m, apparent overlap of
doublets, 9H); MS (ESI) m/z (rel
intensity) 874 ([M + Na]+, 100), 852 ([M + H]+, 75); HRMS (ESI) m/z [M + H]+ calcd for C49H65N5O6S 852.4734; found 852.4723. Due to material loss, 13C NMR data were unavailable for this compound. Treatment of 12 with SnCl4, using the procedure given for 45, furnished a yellow oil with mass spectral data consistent
with 2-fold debenzylation: HRMS (ESI) m/z [M + H]+ calcd for C35H53N5O6S 672.3795; found 672.3801.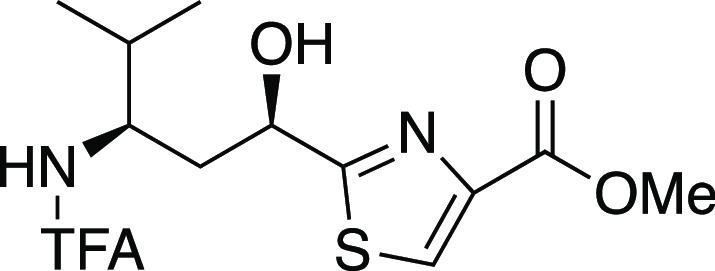
Methyl (2-((1′R,3′R)-1′-Hydroxy-4′-methyl-3′-(trifluoroacetamido)pent-1′-yl)thiazol-4-yl)carboxylate (45)
To a solution of N-TFA amino ester 10 (23 mg, 0.05 mmol) in CH2Cl2 (0.25 mL) was added tin(IV) chloride (0.03 mL, 0.25 mmol) at ambient temperature. The reaction mixture was then heated at reflux for 25 h. After cooling to 0 °C the reaction was quenched by dropwise addition of satd NaHCO3 and extracted with CH2Cl2. The organic phase was washed with brine and dried over Na2SO4. Concentration and flash chromatography afforded alcohol 45(67) (15 mg, 83% yield) as a pale yellow oil: 1H NMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 6.46 (d, J = 9.5 Hz, 1H), 4.96 (ddd, J = 11.1, 4.8, 2.4 Hz, 1H), 4.24 (d, J = 5.0 Hz, 1H), 4.08–4.16 (m, 1H), 3.93 (s, 3H), 2.26 (ddd, J = 14.1, 11.6, 2.4 Hz, 1H), 2.02–1.83 (m, 2H), 1.00 (d, J = 7 Hz, 3H), 0.99 (d, J = 7 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) 175.8, 161.8, 158.6 (q, J = 37 Hz), 146.6, 127.8, 115.8 (q, J = 287 Hz), 68.7, 52.6, 52.4, 40.2, 31.8, 19.2, 18.2; MS (ESI) m/z (rel intensity) 377 ([M + Na]+, 100), 355 ([M + H]+, 8).
Acknowledgments
We thank the University of Iowa (Graduate College Fellowship to M.L.) and the National Science Foundation (NSF, CHE-1362111) for generously supporting this research. The Thermo Q-Exactive mass spectrometer used in this research was acquired through the National Science Foundation Major Research Instrumentation and the Chemical Instrumentation Programs (CHE-1919422).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01798.
Comparisons of tubulysin syntheses, 1H and 13C NMR spectra for new compounds, enantiomer ratio and configuration assignment of (R)-19 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Friestad G. K.Control of Asymmetry in the Radical Addition Approach to Chiral Amine Synthesis. In Topics In Current Chemistry: Stereoselective Formation of Amines; Li W., Zhang X., Eds.; Springer-Verlag: Berlin, 2014; Vol. 343, pp 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miyabe H.; Ushiro C.; Naito T. Highly diastereoselective radical addition to glyoxylic oxime ether: asymmetric synthesis of alpha-amino acids. Chem. Commun. 1997, 1789–1790. 10.1039/a704562j. [DOI] [PubMed] [Google Scholar]; b Miyabe H.; Ushiro C.; Ueda M.; Yamakawa K.; Naito T. Asymmetric synthesis of alpha-amino acids based on carbon radical addition to glyoxylic oxime ether. J. Org. Chem. 2000, 65, 176–185. 10.1021/jo991353n. [DOI] [PubMed] [Google Scholar]
- a Bertrand M. P.; Coantic S.; Feray L.; Nouguier R.; Perfetti P. Et3B- and Et2Zn-mediated radical additions to glyoxylate imines, compared stereoinductions. Tetrahedron 2000, 56, 3951–3961. 10.1016/S0040-4020(00)00327-6. [DOI] [Google Scholar]; b Bertrand M. P.; Feray L.; Nouguier R.; Perfetti P. Diethylzinc mediated radical additions to glyoxylate imines. Synlett 1999, 1999, 1148–1150. 10.1055/s-1999-2754. [DOI] [Google Scholar]; c Bertrand M. P.; Feray L.; Nouguier R.; Stella L. 1,3-stereoinduction in radical additions to glyoxylate imines. Synlett 1998, 1998, 780–782. 10.1055/s-1998-1763. [DOI] [Google Scholar]
- Friestad G. K.; Qin J. Highly Stereoselective Intermolecular Radical Addition to Aldehyde Hydrazones from a Chiral 3-Amino-2-oxazolidinone. J. Am. Chem. Soc. 2000, 122, 8329–8330. 10.1021/ja002173u. [DOI] [Google Scholar]
- a Friestad G. K.; Qin J.; Suh Y.; Marie J.-C. Mn-Mediated Coupling of Alkyl Iodides and Chiral N-Acylhydrazones: Optimization, Scope, and Evidence for a Radical Mechanism. J. Org. Chem. 2006, 71, 7016–7027. 10.1021/jo061158q. [DOI] [PubMed] [Google Scholar]; b Friestad G. K.; Qin J. Intermolecular Alkyl Radical Addition to Chiral N-Acylhydrazones Mediated by Manganese Carbonyl. J. Am. Chem. Soc. 2001, 123, 9922–9923. 10.1021/ja011312k. [DOI] [PubMed] [Google Scholar]
- For selected examples involving radical addition to C=N, see:; a Lo J. C.; Kim D.; Pan C.-M.; Edwards J. T.; Yabe Y.; Gui J.; Qin T.; Gutiérrez S.; Giacoboni J.; Smith M. W.; Holland P. L.; Baran P. S. Fe-Catalyzed C–C Bond Construction from Olefins via Radicals. J. Am. Chem. Soc. 2017, 139, 2484–2503. 10.1021/jacs.6b13155. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Saladrigas M.; Loren G.; Bonjoch J.; Bradshaw B. Hydrogen Atom Transfer (HAT)-Triggered Iron-Catalyzed Intra- and Intermolecular Coupling of Alkenes with Hydrazones: Access to Complex Amines. ACS Catal. 2018, 8, 11699–11703. 10.1021/acscatal.8b03794. [DOI] [Google Scholar]; c Matos J. L. M.; Vásquez-Céspedes S.; Gu J.; Oguma T.; Shenvi R. A. Branch-Selective Addition of Unactivated Olefins into Imines and Aldehydes. J. Am. Chem. Soc. 2018, 140, 16976–16981. 10.1021/jacs.8b11699. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Uraguchi D.; Kinoshita N.; Kizu T.; Ooi T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]
- Review:; a Cullen S. T. J.; Friestad G. K. Synthesis of Chiral Amines by C–C Bond Formation with Photoredox Catalysis. Synthesis 2021, 53, 2319–2341. 10.1055/a-1396-8343. [DOI] [Google Scholar]; Selected examples:; b Cullen S. T. J.; Friestad G. K. Alkyl radical addition to aliphatic and aromatic N-acylhydrazones using an organic photoredox catalyst. Org. Lett. 2019, 21, 8290–8294. 10.1021/acs.orglett.9b03053. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pantaine L. R. E.; Milligan J. A.; Matsui J. K.; Kelly C. B.; Molander G. A. Photoredox Radical/Polar Crossover Enables Construction of Saturated Nitrogen Heterocycles. Org. Lett. 2019, 21, 2317–2321. 10.1021/acs.orglett.9b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jindakun C.; Hsieh S.-Y.; Bode J. W. Iridium-catalyzed Synthesis of Saturated N-Heterocycles from Aldehydes and SnAP Reagents with Continuous Flow Photochemistry. Org. Lett. 2018, 20, 2071–2075. 10.1021/acs.orglett.8b00611. [DOI] [PubMed] [Google Scholar]; e Wang J. C.; Shao Z. Y.; Tan K.; Tang R.; Zhou Q. L.; Xu M.; Li Y. M.; Shen Y. Synthesis of Amino Acids by Base-Enhanced Photoredox Decarboxylative Alkylation of Aldimines. J. Org. Chem. 2020, 85, 9944–9954. 10.1021/acs.joc.0c01246. [DOI] [PubMed] [Google Scholar]; f Jia J.; Lefebvre Q.; Rueping M. Reductive Coupling of Imines with Redox-Active Esters by Visible Light Photoredox Organocatalysis. Org. Chem. Front. 2020, 7, 602–608. 10.1039/C9QO01428D. [DOI] [Google Scholar]; g Weigel W. K. III; Dang H. T.; Yang H.-B.; Martin D. B. C. Synthesis of amino-diamondoid pharmacophores via photocatalytic C-H aminoalkylation. Chem. Commun. 2020, 56, 9699–9702. 10.1039/D0CC02804E. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Garrido-Castro A. F.; Choubane H.; Daaou M.; Maestro M. C.; Alemán J. Asymmetric Radical Alkylation of N-Sulfinimines Under Visible Light Photocatalytic Conditions. Chem. Commun. 2017, 53, 7764–7767. 10.1039/C7CC03724D. [DOI] [PubMed] [Google Scholar]; i Han B.; Li Y.; Yu Y.; Gong L. Photocatalytic enantioselective alpha-aminoalkylation of acyclic imine derivatives by a chiral copper catalyst. Nat. Commun. 2019, 10, 3804–3813. 10.1038/s41467-019-11688-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rafferty S. M.; Rutherford J. E.; Zhang L.; Wang L.; Nagib D. A. Cross-Selective Aza-Pinacol Coupling via Atom Transfer Catalysis. J. Am. Chem. Soc. 2021, 143, 5622–5628. 10.1021/jacs.1c00886. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang X.; Zhu B.; Dong J.; Tian H.; Liu Y.; Song H.; Wang Q. Visible-light-mediated multicomponent reaction for secondary amine synthesis. Chem. Commun. 2021, 57, 5028–5031. 10.1039/D1CC01560E. [DOI] [PubMed] [Google Scholar]; c Weng W.-Z.; Liang H.; Liu R.-Z.; Ji Y.-X.; Zhang B. Visible-Light-Promoted Manganese-Catalyzed Atom Transfer Radical Cyclization of Unactivated Alkyl Iodides. Org. Lett. 2019, 21, 5586–5590. 10.1021/acs.orglett.9b01918. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Shen Y.; Ruggles E. L. Enantioselective Radical Addition to N-Acylhydrazones Mediated by Chiral Lewis Acids. Angew. Chem., Int. Ed. 2003, 42, 5061–5063. 10.1002/anie.200352104. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Massari S. E. A Silicon Tether Approach for Addition of Functionalized Radicals to Chiral alpha-Hydroxyhydrazones: Diastereoselective Additions of Hydroxymethyl and Vinyl Synthons. J. Org. Chem. 2004, 69, 863–875. 10.1021/jo035405r. [DOI] [PubMed] [Google Scholar]
- Friestad G. K. Chiral N-Acylhydrazones: Versatile Imino Acceptors for Asymmetric Amine Synthesis. Eur. J. Org. Chem. 2005, 2005, 3157–3172. 10.1002/ejoc.200500232. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Banerjee K. Synthesis of gamma-Amino Esters via Mn-Mediated Radical Addition to Chiral gamma-Hydrazonoesters. Org. Lett. 2009, 11, 1095–1098. 10.1021/ol802932v. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Ji A. Mn-Mediated Coupling of Alkyl Iodides and Ketimines: A Radical Addition Route to alpha,alpha-Disubstituted alpha-Aminoesters. Org. Lett. 2008, 10, 2311–2313. 10.1021/ol800733b. [DOI] [PubMed] [Google Scholar]
- Slater K. A.; Friestad G. K. Mn-Mediated Radical-Ionic Annulations of Chiral N-Acylhydrazones. J. Org. Chem. 2015, 80, 6432–6440. 10.1021/acs.joc.5b00863. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Ji A.; Baltrusaitis J.; Korapala C. S.; Qin J. Scope of Stereoselective Mn-Mediated Radical Addition to Chiral Hydrazones and Application in a Formal Synthesis of Quinine. J. Org. Chem. 2012, 77, 3159–3180. 10.1021/jo2026349. [DOI] [PubMed] [Google Scholar]
- a Vaishampayan U.; Glode M.; Du W.; Kraft A.; Hudes G.; Wright J.; Hussain M. Phase II Study of Dolastatin-10 in Patients with Hormone-refractory Metastatic Prostate Adenocarcinoma. Clin. Cancer Res. 2000, 6, 4205–4208. [PubMed] [Google Scholar]; b Alonso-Álvarez S.; Pardal E.; Sánchez-Nieto D.; Navarro M.; Caballero M. D.; Mateos M. V.; Martín A. Plitidepsin: design, development, and potential place in therapy. Drug Des., Dev. Ther. 2017, 11, 253–264. 10.2147/DDDT.S94165. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chun H. G.; Davies B.; Hoth D.; Suffness M.; Plowman J.; Flora K.; Grieshaber C.; Leyland-Jones B. Didemin B: The first marine compound entering clinical trials as an antineoplastic agent. Invest. New Drugs 1986, 4, 279–284. 10.1007/BF00179597. [DOI] [PubMed] [Google Scholar]; d Nuijen B.; Bouma M.; Manada C.; Jimeno J. M.; Schellens J. H. M.; Bult A.; Beijnen J. H. Pharmaceutical development of anticancer agents derived from marine sources. Anti-Cancer Drugs 2000, 11, 793. 10.1097/00001813-200011000-00003. [DOI] [PubMed] [Google Scholar]
- Murray B. C.; Peterson M. T.; Fecik R. A. Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat. Prod. Rep. 2015, 32, 654–662. 10.1039/C4NP00036F. [DOI] [PubMed] [Google Scholar]
- Sasse F.; Steinmetz H.; Heil J.; Höfle G.; Reichenbach H. Tubulysins, New Cytostatic Peptides from Myxobacteria Acting on Microtubuli Production, Isolation, Physico-chemical and Biological Properies. J. Antibiot. 2000, 53, 879–885. 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]
- a Höfle G.; Glaser N.; Leibold T.; Karama U.; Sasse F.; Steinmetz H. Semisynthesis and degradation of the tubulin inhibitors epothilone and tubulysin. Pure Appl. Chem. 2003, 75, 167–178. 10.1351/pac200375020167. [DOI] [Google Scholar]; b Steinmetz H.; Glaser N.; Herdtweck E.; Sasse F.; Reichenbach H.; Höfle G. Isolation, Crystal and Solution Structure Determination, and Biosynthesis of Tubulysins—Powerful Inhibitors of Tubulin Polymerization from Myxobacteria. Angew. Chem., Int. Ed. 2004, 43, 4888–4892. 10.1002/anie.200460147. [DOI] [PubMed] [Google Scholar]
- Khalil M. W.; Sasse F.; Lünsdorf H.; Elnakady Y. A.; Reichenbach H. Mechanism of Action of Tubulysin, an Antimitotic Peptide from Myxobacteria. ChemBioChem 2006, 7, 678–683. 10.1002/cbic.200500421. [DOI] [PubMed] [Google Scholar]
- Kaur G.; Hollingshead M.; Holbeck S.; Schauer-Vukašinović V.; Camalier; Richard F.; Dömling A.; Agarwal S. Biological evaluation of tubulysin A: a potential anticancer and antiangiogenic natural product. Biochem. J. 2006, 396, 235–242. 10.1042/BJ20051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reddy J. A.; Dorton R.; Dawson A.; Vetzel M.; Parker N.; Nicoson J. S.; Westrick E.; Klein P. J.; Wang Y.; Vlahov I. R.; Leamon C. P. In Vivo Structural Activity and Optimization Studies of Folate–Tubulysin Conjugates. Mol. Pharmaceutics 2009, 6, 1518–1525. 10.1021/mp900086w. [DOI] [PubMed] [Google Scholar]; b Schluep T.; Gunawan P.; Ma L.; Jensen G. S.; Duringer J.; Hinton S.; Richter W.; Hwang J. Polymeric Tubulysin-Peptide Nanoparticles with Potent Antitumor Activity. Clin. Cancer Res. 2009, 15, 181–189. 10.1158/1078-0432.CCR-08-1848. [DOI] [PubMed] [Google Scholar]
- a Szigetvari N. M.; Dhawan D.; Ramos-Vara J. A.; Leamon C. P.; Klein P. J.; Ruple A. A.; Heng H. G.; Pugh M. R.; Rao S.; Vlahov I. R.; Deshuillers P. L.; Low P. S.; Fourez L. M.; Cournoyer A. M.; Knapp D. W. Phase I/II clinical trial of the targeted chemotherapeutic drug, folate-tubulysin, in dogs with naturally-occurring invasive urothelial carcinoma. Oncotarget 2018, 9, 37042–37053. 10.18632/oncotarget.26455. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reddy J. A.; Dorton R.; Bloomfield A.; Nelson M.; Dircksen C.; Vetzel M.; Kleindl P.; Santhapuram H.; Vlahov I. R.; Leamon C. P. Pre-clinical evaluation of EC1456, a folate-tubulysin anti-cancer therapeutic. Sci. Rep. 2018, 8, 8943–8943. 10.1038/s41598-018-27320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tumey L. N.; Leverett C. A.; Vetelino B.; Li F.; Rago B.; Han X.; Loganzo F.; Musto S.; Bai G.; Sukuru S. C. K.; Graziani E. I.; Puthenveetil S.; Casavant J.; Ratnayake A.; Marquette K.; Hudson S.; Doppalapudi V. R.; Stock J.; Tchistiakova L.; Bessire A. J.; Clark T.; Lucas J.; Hosselet C.; O’Donnell C. J.; Subramanyam C. Optimization of Tubulysin Antibody–Drug Conjugates: A Case Study in Addressing ADC Metabolism. ACS Med. Chem. Lett. 2016, 7, 977–982. 10.1021/acsmedchemlett.6b00195. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Parker J. S.; McCormick M.; Anderson D. W.; Maltman B. A.; Gingipalli L.; Toader D. The Development and Scale-Up of an Antibody Drug Conjugate Tubulysin Payload. Org. Process Res. Dev. 2017, 21, 1602–1609. 10.1021/acs.oprd.7b00232. [DOI] [Google Scholar]
- a Xiangming X.; Friestad G.; Lei Y. Recent Advances in the Synthesis of Tubulysins. Mini-Rev. Med. Chem. 2013, 13, 1572–1578. 10.2174/13895575113139990063. [DOI] [PubMed] [Google Scholar]; b See ref (17).
- a Ullrich A.; Chai Y.; Pistorius D.; Elnakady Y. A.; Herrmann J. E.; Weissman K. J.; Kazmaier U.; Müller R. Pretubulysin, a Potent and Chemically Accessible Tubulysin Precursor from Angiococcus disciformis. Angew. Chem., Int. Ed. 2009, 48, 4422–4425. 10.1002/anie.200900406. [DOI] [PubMed] [Google Scholar]; b Toader D.; Wang F.; Gingipalli L.; Vasbinder M.; Roth M.; Mao S.; Block M.; Harper J.; Thota S.; Su M.; Ma J.; Bedian V.; Kamal A. Structure–Cytotoxicity Relationships of Analogues of N14-Desacetoxytubulysin H. J. Med. Chem. 2016, 59, 10781–10787. 10.1021/acs.jmedchem.6b01023. [DOI] [PubMed] [Google Scholar]
- Strategies to assemble Tup (or Tut) include chiral enolate alkylation by enantioenriched aziridine (Domling and Wessjohann, ref (38)), Ru-catalyzed asymmetric reductive amination (Domling, ref (40)), α-aminoalkyl Michael addition (Ellman, ref (30)), diastereoselective hydrogenation of α,β-unsaturated ester (Zanda, ref (33); Wipf, ref (28); Kazmaier/Muller, ref (25a)), Evans aldol addition to α-aminoaldehyde followed by reductive deoxygenation (Tamura, ref (45)), diastereoselective alkylation of chiral lactam (Fecik, ref (34)), Grignard addition to sulfinylimine (Chen, ref (42)), chiral pool approach from (−)-citronellol (Chandresekhar, ref (29)), and radical addition to chiral N-acylhydrazones (Friestad, refs (47). (48))
- a Vlahov I. R.; Wang Y.; Vetzel M.; Hahn S.; Kleindl P. J.; Reddy J. A.; Leamon C. P. Acid mediated formation of an N-acyliminium ion from tubulysins: A new methodology for the synthesis of natural tubulysins and their analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6778–6781. 10.1016/j.bmcl.2011.09.041. [DOI] [PubMed] [Google Scholar]; b Ibrahim S. M. S.; Banerjee K.; Slater K. A.; Friestad G. K. A Tamao-Fleming oxidation route to dipeptides bearing N,O-acetal functionality. Tetrahedron Lett. 2017, 58, 4864–4866. 10.1016/j.tetlet.2017.11.036. [DOI] [Google Scholar]
- Wipf P.; Takada T.; Rishel M. J. Synthesis of the Tubuvaline-Tubuphenylalanine (Tuv-Tup) Fragment of Tubulysin. Org. Lett. 2004, 6, 4057–4060. 10.1021/ol048252i. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S.; Mahipal B.; Kavitha M. Toward Tubulysin: Gram-Scale Synthesis of Tubuvaline-Tubuphenylalanine Fragment. J. Org. Chem. 2009, 74, 9531–9534. 10.1021/jo9015503. [DOI] [PubMed] [Google Scholar]
- a Peltier H. M.; McMahon J. P.; Patterson A. W.; Ellman J. A. The Total Synthesis of Tubulysin D. J. Am. Chem. Soc. 2006, 128, 16018–16019. 10.1021/ja067177z. [DOI] [PubMed] [Google Scholar]; b Patterson A. W.; Peltier H. M.; Sasse F.; Ellman J. A. Design, Synthesis, and Biological Properties of Highly Potent Tubulysin D Analogues. Chem. - Eur. J. 2007, 13 (34), 9534–9541. 10.1002/chem.200701057. [DOI] [PubMed] [Google Scholar]; c Patterson A. W.; Peltier H. M.; Ellman J. A. Expedient Synthesis of N-Methyl Tubulysin Analogues with High Cytotoxicity. J. Org. Chem. 2008, 73, 4362–4369. 10.1021/jo800384x. [DOI] [PubMed] [Google Scholar]
- Wipf P.; Wang Z. Total Synthesis of N14-Desacetoxytubulysin H. Org. Lett. 2007, 9, 1605–1607. 10.1021/ol070415q. [DOI] [PubMed] [Google Scholar]
- Colombo R.; Wang Z.; Han J.; Balachandran R.; Daghestani H. N.; Camarco D. P.; Vogt A.; Day B. W.; Mendel D.; Wipf P. Total Synthesis and Biological Evaluation of Tubulysin Analogues. J. Org. Chem. 2016, 81, 10302–10320. 10.1021/acs.joc.6b01314. [DOI] [PubMed] [Google Scholar]
- Sani M.; Fossati G.; Huguenot F.; Zanda M. Total Synthesis of Tubulysins U and V. Angew. Angew. Chem., Int. Ed. 2007, 46, 3526–3529. 10.1002/anie.200604557. [DOI] [PubMed] [Google Scholar]
- a Balasubramanian R.; Raghavan B.; Begaye A.; Sackett D. L.; Fecik R. A. Total Synthesis and Biological Evaluation of Tubulysin U, Tubulysin V, and Their Analogues. J. Med. Chem. 2009, 52, 238–240. 10.1021/jm8013579. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Raghavan B.; Balasubramanian R.; Steele J. C.; Sackett D. L.; Fecik R. A. Cytotoxic Simplified Tubulysin Analogues. J. Med. Chem. 2008, 51, 1530–1533. 10.1021/jm701321p. [DOI] [PubMed] [Google Scholar]
- a Nicolaou K. C.; Yin J.; Mandal D.; Erande R. D.; Klahn P.; Jin M.; Aujay M.; Sandoval J.; Gavrilyuk J.; Vourloumis D. Total Synthesis and Biological Evaluation of Natural and Designed Tubulysins. J. Am. Chem. Soc. 2016, 138, 1698–1708. 10.1021/jacs.5b12557. [DOI] [PubMed] [Google Scholar]; b Nicolaou K. C.; Erande R. D.; Yin J.; Vourloumis D.; Aujay M.; Sandoval J.; Munneke S.; Gavrilyuk J. Improved Total Synthesis of Tubulysins and Design, Synthesis, and Biological Evaluation of New Tubulysins with Highly Potent Cytotoxicities against Cancer Cells as Potential Payloads for Antibody–Drug Conjugates. J. Am. Chem. Soc. 2018, 140, 3690–3711. 10.1021/jacs.7b12692. [DOI] [PubMed] [Google Scholar]
- Park Y.; Lee J. K.; Ryu J.-S. Synthesis of a Cyclic Analogue of Tuv N-Methyl Tubulysin. Synlett 2015, 26, 1063–1068. 10.1055/s-0034-1379900. [DOI] [Google Scholar]
- Paladhi S.; Das J.; Samanta M.; Dash J. Asymmetric Aldol Reaction of Thiazole-Carbaldehydes: Regio- and Stereoselective Synthesis of Tubuvalin Analogues. Adv. Synth. Catal. 2014, 356, 3370–3376. 10.1002/adsc.201400640. [DOI] [Google Scholar]
- Dömling A.; Beck B.; Eichelberger U.; Sakamuri S.; Menon S.; Chen Q.-Z.; Lu Y.; Wessjohann L. A. Total Synthesis of Tubulysin U and V. Angew.. Angew. Chem., Int. Ed. 2006, 45, 7235–7239. 10.1002/anie.200601259. [DOI] [PubMed] [Google Scholar]
- Pando O.; Dörner S.; Preusentanz R.; Denkert A.; Porzel A.; Richter W.; Wessjohann L. First Total Synthesis of Tubulysin B. Org. Lett. 2009, 11, 5567–5569. 10.1021/ol902320w. [DOI] [PubMed] [Google Scholar]
- Vishwanatha T. M.; Giepmans B.; Goda S. K.; Dömling A. Tubulysin Synthesis Featuring Stereoselective Catalysis and Highly Convergent Multicomponent Assembly. Org. Lett. 2020, 22, 5396–5400. 10.1021/acs.orglett.0c01718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S. P.; Jagodzinska M.; Malpezzi L.; Lazzari P.; Manca I.; Greig I. R.; Sani M.; Zanda M. Synthesis and structure–activity relationship studies of novel tubulysin U analogues – effect on cytotoxicity of structural variations in the tubuvaline fragment. Org. Biomol. Chem. 2013, 11, 2273–2287. 10.1039/c3ob27111k. [DOI] [PubMed] [Google Scholar]
- Yang X.-D.; Dong C.-M.; Chen J.; Ding Y.-H.; Liu Q.; Ma X.-Y.; Zhang Q.; Chen Y. Total Synthesis of Tubulysin U and Its C-4 Epimer. Chem. - Asian J. 2013, 8, 1213–1222. 10.1002/asia.201300051. [DOI] [PubMed] [Google Scholar]
- Tao W.; Zhou W.; Zhou Z.; Si C.-M.; Sun X.; Wei B.-G. An enantioselective total synthesis of tubulysin V. Tetrahedron 2016, 72, 5928–5933. 10.1016/j.tet.2016.08.038. [DOI] [Google Scholar]
- Wang R.; Tian P.; Lin G. Stereoselective Total Synthesis of Tubulysin V. Chin. J. Chem. 2013, 31, 40–48. 10.1002/cjoc.201200984. [DOI] [Google Scholar]
- a Shibue T.; Hirai T.; Okamoto I.; Morita N.; Masu H.; Azumaya I.; Tamura O. Stereoselective synthesis of tubuvaline methyl ester and tubuphenylalanine, components of tubulysins, tubulin polymerization inhibitors. Tetrahedron Lett. 2009, 50, 3845–3848. 10.1016/j.tetlet.2009.04.046. [DOI] [Google Scholar]; Shibue T.; Hirai T.; Okamoto I.; Morita N.; Masu H.; Azumaya I.; Tamura O. Total Syntheses of Tubulysins. Chem. - Eur. J. 2010, 16, 11678–11688. 10.1002/chem.201000963. [DOI] [PubMed] [Google Scholar]
- Further details on Tuv and Tup syntheses are briefly reviewed in the Supporting Information
- Friestad G. K.; Marié J.-C.; Deveau A. M. Stereoselective Mn-Mediated Coupling of Functionalized Iodides and Hydrazones: A Synthetic Entry to the Tubulysin γ-Amino Acids. Org. Lett. 2004, 6, 3249–3252. 10.1021/ol048986v. [DOI] [PubMed] [Google Scholar]
- Friestad G. K.; Banerjee K.; Marié J.-C.; Mali U.; Yao L. Stereoselective access to tubuphenylalanine and tubuvaline: improved Mn-mediated radical additions and assembly of a tubulysin tetrapeptide analog. J. Antibiot. 2016, 69, 294–298. 10.1038/ja.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins M. T.; Emmitte K. A.; Katz J. D. Diastereoselective Alkylations of Oxazolidinone Glycolates: A Useful Extension of the Evans Asymmetric Alkylation. Org. Lett. 2000, 2, 2165–2167. 10.1021/ol006091m. [DOI] [PubMed] [Google Scholar]
- The aldehyde–cysteine cyclocondensation tactic was also employed in the tubulysin synthetic studies of Zanda (ref (33)) and Chandrasekhar (ref (29)).
- Initial efforts toward route A have appeared in a prior communication; Li M.; Goff L.; Friestad G. K. Aromatic N-heterocycles in Mn-mediated radical additions to N-acylhydrazones. Tetrahedron Lett. 2020, 61, 152058. 10.1016/j.tetlet.2020.152058. [DOI] [Google Scholar]
- See ref (15).
- Inami K.; Shiba T. Total Synthesis of Antibiotic Althiomycin. Bull. Chem. Soc. Jpn. 1985, 58, 352–360. 10.1246/bcsj.58.352. [DOI] [Google Scholar]
- Keck G. E.; Tarbet K. H.; Geraci L. S. Catalytic Asymmetric Allylation of Aldehydes. J. Am. Chem. Soc. 1993, 115, 8467–8468. 10.1021/ja00071a074. [DOI] [Google Scholar]
- a See ref (12).; b Yu W.; Mei Y.; Kang Y.; Hua Z.; Jin Z. Improved Procedure for the Oxidative Cleavage of Olefins by OsO4–NaIO4. Org. Lett. 2004, 6, 3217–3219. 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- Aldehyde N-acylhydrazones are typically formed with a >98:2 E/Z ratio, and we have found no evidence for C=N isomerism in recovered starting materials in Mn-mediated radical additions to aldehyde N-acylhydrazones.
- Yamamoto Y.; Komatsu T.; Maruyama K. Enantiodivergent 1,2- and 1,3-Asymmetric Induction in a- and b-AIkoxyimines via Metal Tuning and Double Stereodifferentiation. J. Chem. Soc., Chem. Commun. 1985, 814–816. 10.1039/C39850000814. [DOI] [Google Scholar]
- Selected examples of double diastereoselection have been reported in nonradical additions to β-alkoxyimines.; a Grignard addition:Zhou Q.-R.; Wei X.-Y.; Li Y.-Q.; Huang D.; Wei B.-G. An efficient method for the preparation of 3-hydroxyl-5-substituted 2-pyrrolidones and application in the divergent synthesis of (−)-preussin and its analogues. Tetrahedron 2014, 70, 4799–4808. 10.1016/j.tet.2014.05.037. [DOI] [Google Scholar]; b Nitrone cycloaddition:Stecko S.; Pasniczek K.; Jurczak M.; Urbanczyk-Lipkowska Z.; Chmielewski M. Double asymmetric induction in 1,3-dipolar cycloaddition of five-membered cyclic nitrones to 2-(5H)-furanones. Tetrahedron: Asymmetry 2006, 17, 68–78. 10.1016/j.tetasy.2005.11.027. [DOI] [Google Scholar]; c Gilbertson S. R.; Lopez O. D. Kinetic Resolution of Diiron Acyl Complexes—An Approach to Asymmetric Bicyclic β-Lactams. Angew. Chem., Int. Ed. 1999, 38, 1116–1119. . [DOI] [PubMed] [Google Scholar]; d Allyltitanium addition:Okamoto S.; Teng X.; Fujii S.; Takayama Y.; Sato F. An Allyltitanium Derived from Acrolein 1,2-Dicyclohexylethylene Acetal and (η2-propene)Ti(O-i-Pr)2 as a Chiral Propionaldehyde Homoenolate Equivalent that Reacts with Imines with Excellent Stereoselectivity. An Efficient and Practical Access to Optically Active γ-Amino Carbonyl Compounds. J. Am. Chem. Soc. 2001, 123, 3462–3471. 10.1021/ja004140k. [DOI] [PubMed] [Google Scholar]
- Chelation models may allow for 1,3-diastereocontrolled additions to β-alkoxyimines.; a Organozinc addition: CeCl3-catalyzed AllylZnBr addition of β-alkoxyimine with a chelation model for acyclic stereocontrol:Jain R. P.; Williams R. M. Asymmetric Synthesis of (+)-Negamycin. J. Org. Chem. 2002, 67, 6361–6365. 10.1021/jo025636i. [DOI] [PubMed] [Google Scholar]; b Allylstannane addition:Shimizu M.; Morita A.; Fujisawa T. AlCl3-promoted addition of allylstannane to b-alkoxyimine with chelation model: Stereocontrol in the Addition of Allyl Metal Reagents to an Optically Active Imine Derived from Malic Acid, Leading to a Formal Synthesis of (+)-Negamycin. Chem. Lett. 1998, 27, 467–468. 10.1246/cl.1998.467. [DOI] [Google Scholar]; c Strecker reaction:Makino K.; Jiang H.; Suzuki T.; Hamada Y. The efficient synthesis of (3R,5R)-5-hydroxypiperazic acid and its diastereomer using Lewis acid-promoted diastereoselective Strecker synthesis. Tetrahedron: Asymmetry 2006, 17, 1644–1649. 10.1016/j.tetasy.2006.06.004. [DOI] [Google Scholar]
- A few cases of radical cyclizations upon β-alkoxyimino compounds have been reported:; a Cronjé J.J.; Holzapfel C. W. Samarium (II) iodide mediated transformations of carbohydrate derived iodo oxime ethers into stereodefined alkoxy aminocyclopentanes. Tetrahedron Lett. 1997, 38, 7429–7432. 10.1016/S0040-4039(97)01748-6. [DOI] [Google Scholar]; b Godineau E.; Landais Y. Synthesis of Piperidinones through a Radical Cascade. Synthesis 2009, 2009, 2646–2649. 10.1055/s-0029-1216860. [DOI] [Google Scholar]; c Godineau E.; Schenk K.; Landais Y. Synthesis of Fused Piperidinones through a Radical-Ionic Cascade. J. Org. Chem. 2008, 73, 6983–6993. 10.1021/jo801308j. [DOI] [PubMed] [Google Scholar]; d Xu J.; Liu Y.; Christelle Dupouy C.; Chattopadhyaya J. Synthesis of Conformationally Locked Carba-LNAs through Intramolecular Free-Radical Addition to C=N. Electrostatic and Steric Implication of the Carba-LNA Substituents in the Modified Oligos for Nuclease and Thermodynamic Stabilities. J. Org. Chem. 2009, 74, 6534–6554. 10.1021/jo901009w. [DOI] [PubMed] [Google Scholar]
- Wang R.; Fang K.; Sun B.-F.; Xu M.-H.; Lin G.-Q. Concise Asymmetric Synthesis of Antimalarial Alkaloid (+)-Febrifugine. Synlett 2009, 2009, 2301–2304. 10.1055/s-0029-1217713. [DOI] [Google Scholar]
- Previously published 1H and 13C NMR studies of N-acylhydrazones with InCl3 provided evidence of chelation by the N-acylhydrazone. See ref (12).
- Poisoning of the catalyst by sulfur-containing heterocycles could explain this result.
- Recent studies suggest LED light sources can be effective in the Mn-mediated radical addition to C=N. See ref (8).
- a Li M.Synthesis of Tubuvaline Utilizing Mn-Mediated Radical Addition Conditions Modified for Compatibility with Heteroaromatics. Ph.D. Thesis, University of Iowa, 2021. [Google Scholar]; b Banerjee K. B.Mn-Mediated Radical Addition Approach Toward gamma-Amino Esters and Synthetic Studies of the Tubulysins. Ph.D. Thesis, University of Iowa, 2011. [Google Scholar]
- Patrick K. S.; Singletary J. L. Relative configuration of thioridazine enantiomers. Chirality 1991, 3, 208–211. 10.1002/chir.530030312. [DOI] [Google Scholar]
- Further confirmation of the relative configuration of 45 was obtained via 13C NMR chemical shift comparison with literature data on both diastereomers of a closely related Tuv derivative. See ref (33).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.