

Abstract
The RAT1 gene of Saccharomyces cerevisiae encodes a 5′→3′ exoribonuclease which plays an essential role in yeast RNA degradation and/or processing in the nucleus. We have cloned a previously uncharacterized gene (YGL246c) that we refer to as RAI1 (Rat1p interacting protein 1). RAI1 is homologous to Caenorhabditis elegans DOM-3 and human DOM3Z. Deletion of RAI1 confers a growth defect which can be complemented by an additional copy of RAT1 on a centromeric vector or by directing Xrn1p, the cytoplasmic homolog of Rat1p, to the nucleus through the addition of a nuclear targeting sequence. Deletion of RAI1 is synthetically lethal with the rat1-1ts mutation and shows genetic interaction with a deletion of SKI2 but not XRN1. Polysome analysis of an rai1 deletion mutant indicated a defect in 60S biogenesis which was nearly fully reversed by high-copy RAT1. Northern blot analysis of rRNAs revealed that rai1 is required for normal 5.8S processing. In the absence of RAI1, 5.8SL was the predominant form of 5.8S and there was an accumulation of 3′-extended forms but not 5′-extended species of 5.8S. In addition, a 27S pre-rRNA species accumulated in the rai1 mutant. Thus, deletion of RAI1 affects both 5′ and 3′ processing reactions of 5.8S rRNA. Consistent with the in vivo data suggesting that RAI1 enhances RAT1 function, purified Rai1p stabilized the in vitro exoribonuclease activity of Rat1p.
In the yeast Saccharomyces cerevisiae, two 5′→3′ exoribonucleases, Rat1p and Xrn1p (23, 35), are functionally interchangeable and are localized to the nucleus and cytoplasm, respectively (19). These enzymes are conserved throughout eukaryotes but are apparently absent from prokaryotic cells based on analysis of protein sequences predicted from prokaryotic genomes. The mammalian counterparts of Rat1p and Xrn1p function in yeast and complement their respective mutants (8, 33, 34). These exoribonucleases processively degrade RNA containing a 5′-monophosphate (37, 38). RNA molecules with 7mG cap, 5′-triphosphate, or 5′-hydroxyl are resistant to degradation by these enzymes (38). The role of Xrn1p in general deadenylation-dependent mRNA degradation and in nonsense-mediated degradation is well established (18, 26, 27). Xrn1p also degrades the 5′ fragment of internal transcribed spacer 1, resulting from cleavage at sites D and A2 (36; reviewed in reference 40). Xrn1p is not an essential protein, probably reflecting its partial redundancy with factors required for a 3′ degradation pathway (6, 21).
On the other hand, Rat1p activity in the nucleus is essential. The enzyme acts in numerous RNA 5′ processing reactions important for ribosome biogenesis. These include maturation of 5.8S rRNA (16) and 5′ trimming of intron-encoded and polycistronic small nucleolar RNAs (snoRNAs) (30, 32). Interestingly, many of the known Rat1p-mediated reactions can be accomplished to some extent by Xrn1p, and thus, the observation of processing intermediates requires double mutants of XRN1 and RAT1 (30, 32).
RAT1 has been identified from a variety of genetic and biochemical approaches. The temperature-sensitive rat1-1 allele was identified in a screen for factors involved in the export of poly(A) mRNA from the nucleus (5). The gene was also identified as TAP1, mutations in which activated polymerase III transcription of a mutated tRNA gene (12). However, this activation was dependent on the sequence context of the tRNA gene (2), suggesting that RAT1 was not a general factor in polymerase III transcription. RAT1 was identified as HKE1 from a combination of a screen for temperature-sensitive mutants and a reverse genetic approach based on the sequence of the purified exonuclease (23). More recently, the rsf11 allele of RAT1 was identified as synthetically lethal with a deletion of SWI4 (39). The double mutant could be rescued by high-copy expression of CLN1, CLN2, or RME1. However, rsf11-specific suppressors were not reported. How mutations in RAT1 account for these various phenotypes is not presently understood.
Although numerous substrates for Rat1p activity have been identified, the essential function of Rat1p remains to be demonstrated. The rat1-1 mutation results in accumulation of poly(A) RNA in the nucleus within 1 h after a shift to a restrictive temperature (5), and the hke1 temperature-sensitive allele leads to inhibition of protein synthesis within 4 h of a shift to a restrictive temperature (23). The known substrates of Rat1p are required for ribosome biogenesis; however, ribosomes are stable and are only slowly depleted from cells not actively synthesizing ribosomes. Consequently, many hours of growth under conditions inhibiting ribosome synthesis would be required to sufficiently deplete yeast cells of ribosomes and inhibit protein synthesis to the degree observed for hke1ts mutants after 4 h at a restrictive temperature. Thus, the kinetics of inhibition of poly(A) mRNA export and inhibition of protein synthesis in rat1-1 conditional mutants are not consistent with the idea that a defect in ribosome biogenesis is responsible for the growth arrest observed for a rat1 mutant.
Previously, highly purified preparations of Rat1p contained two polypeptides, the 116-kDa polypeptide encoded by the RAT1 gene and a second, uncharacterized 45-kDa polypeptide (38). To gain further insight into the function of Rat1p, we have cloned the gene encoding this associated protein. We have named this gene RAI1 for Rat1p interacting protein 1. This protein is conserved throughout eukaryotes, and its human homolog is DOM3Z (43), the gene for which maps adjacent to the human homolog of yeast SKI2 (11), encoding a putative RNA helicase required for 3′-mRNA degradation and repression of translation of poly(A)− mRNA. Here we show that Rai1p enhances the activity of Rat1p in vivo and in vitro and is required for normal 5.8S rRNA processing.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The yeast strains used in this study are listed in Table 1. Rich medium (yeast extract-peptone-dextrose) and dropout media (synthetic complete [SC]) containing 2% dextrose or 1% raffinose as the carbon source were as described previously (22). Yeast transformations were carried out using a lithium acetate method as described previously (15), except that cells to be transformed were grown as light lawns on plates.
TABLE 1.
Strains used in this work
| Strain | Genotype | Source or reference |
|---|---|---|
| AJY667 | MATα ade2 ade3 lys2-801 ura3-52 rai1::leu2::hisG::URA3::hisG | This study |
| AJY903 | MATa ade2 ade3 lys2-801 ura3-52 rai1::LEU2 | This study |
| AJY917 | MATα ade2 ade3 trp1Δ63 rai1::LEU2 | This study |
| AJY942 | MATα ura3-52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1 rai1::LEU2 | This study |
| BJ5464 | MATα ura3-52 trp1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1 | E. Jones |
| CH1305 | MATa ade2 ade3 leu2 lys2-801 ura3-52 | Connie Holm |
| RKY1973 | MATα ski2::LEU2 ade2 ade3 leu2 ura3-52 | 21 |
| RDKY1978 | MATα ade2 ade3 ura3-52 leu2 xrn1::URA3 | 21 |
| Dat1-1 | MATα ura3-52 leu2Δ1 his3Δ200 trp1Δ63 rat1-1ts | D. C. Amberg |
Enzymes, chemicals, and other reagents.
Enzymes were purchased from New England Biolabs and Promega. Tobacco acid pyrophosphatase (TAP) was purified as described previously (14) from tobacco BY2 cells (kindly provided by Kathleen Dana) and was free of detectable RNase activity. Single-stranded DNA (ssDNA) cellulose, Source 15Q, and glutathione-Sepharose 4B were from Pharmacia.
Plasmids.
Plasmids used in this work are listed in Table 2. pAJ246 contained a fusion of glutathione S-transferase (GST) to the first 815 nucleotides of RAI1 (GST-RAI1Δ115). It was prepared by PCR amplification of the RAI1 genomic locus using the oligonucleotide primers AJO154 (5′-CGCGGATCCATGGGTGTTAGTGCAAATTTGTTCG), which introduced a BamHI site, and AJO155 (5′-CGCGGATCCAAGCTTAGATACAAAATCGGTTCGCC), which introduced a HindIII site. The PCR product was digested with BamHI, and the fragment containing the 5′ 815 nucleotides of the RAI1 gene was ligated into BamHI-digested pEG(KT) downstream of the GST-encoding region. pAJ245, containing a GST-RAI1(wt) gene fusion, was constructed by replacing the SalI-to-HindIII fragment of pAJ246 with the SalI-to-HindIII fragment of the PCR fragment obtained using primers AJO154-AJO155 (described above).
TABLE 2.
Plasmids used in this work
| Plasmid | Relevant markersa | Source or reference |
|---|---|---|
| pAJ202 | CEN URA3 RAT1 (wt) | 19 |
| pAJ226 | CEN URA3 RAT1 (wt) GFP | 19 |
| pAJ236 | 2μm URA3 XRN1-SV40 NLS | 19 |
| pAJ245 | 2μm URA3 GAL10-GST-RAI1 (wt) | This study |
| pAJ246 | 2μm URA3 GAL10-GST-RAI1Δ115 | This study |
| pAJ501 | pUC18 RAI1 | This study |
| pAJ502 | pUC18 rai1::LEU2 | This study |
| pAJ503 | CEN URA3 RAI1 | This study |
| pAJ504 | CEN TRP1 RAT1 (NLSΔ) | This study |
| pAJ505 | CEN TRP1 RAT1 (wt) | This study |
| pAJ506 | 2μm LEU2 GAL10-RAT1 (5′-2208 nt) | This study |
| pAJ507 | 2μm LEU2 GAL10-RAT1 (wt) | This study |
| pAJ508 | 2μm TRP1 GAL10-RAT1 (wt) | This study |
| pAJ509 | 2μm URA3 RAT1 | This study |
| pEG(KT) | 2μm URA3 GAL10-GST | B. Deschenes |
| pNKY85 | LEU2::hisG::URA3::hisG | 1 |
| pRDK242 | 2μm URA3 XRN1 | 20 |
| pRDK304 | 2μm LEU2 GAL10::XRN1 | 29 |
| pSP6A0 | MFA2 | A. Stevens |
wt, wild type.
Cloning and disruption of RAI1.
The RAI1 gene and flanking sequence were amplified from wild-type genomic DNA by PCR using primers AJO155, described above, and AJO181 (5′-CCGTGAGCTCTTTATCACCATTATCGAAG), which introduced a SacI site. The PCR product was digested with HindIII and SacI and ligated into the HindIII and SacI sites of pUC18 to create pAJ501. The HindIII-SacI-digested PCR fragment was also cloned into the respective sites of pRS416 to create pAJ503. An rai1::LEU2 disruption was made as follows. pAJ501 was digested with SalI and BamHI to remove a 967-bp internal fragment from the RAI1 gene. The plasmid was then blunt ended with T4 DNA polymerase and ligated to a LEU2-containing HpaI fragment to give pAJ502. For disruption of RAI1, pAJ502 was digested with SacI and HindIII to release a ∼2.7-kb fragment which was transformed into the haploid yeast strains CH1305 and BJ5464 to give strains AJY903 and AJY942, respectively. The rai1::LEU2 disruption was in turn disrupted with a hisG::URA3::hisG cassette by transforming AJY903 with pNKY85 to give strain AJY667. All integrants were confirmed by PCR analysis (data not shown).
Construction of a RAT1 overexpression vector.
The 5′ end of RAT1 was amplified by PCR with primers AJO150 and AJO208 using pAJ202 as the template. AJO150 (5′-CGCTATGCCTTGCCAGG) was complementary to the sense strand of RAT1 at nucleotides 2192 to 2208. AJO208 (5′-CCTCGCCATGGGTGTTCCGTCATTT) was complementary to the antisense strand of RAT1 and introduced an NcoI site at −1 relative to the translation start site of RAT1. The PCR product was digested with NcoI, and the fragment corresponding to the 5′ end of RAT1 was then cloned into pRDK304 downstream of the GAL10 promoter, replacing the ∼1.2-kb NcoI segment of pRDK304 to create pAJ506. The correct orientation was determined by restriction digest. A 3.4-kb SalI-NheI digestion fragment from pAJ202 containing the 3′ end of RAT1 was subcloned into pAJ506, replacing the 4.6-kb SalI-NheI segment of pAJ506 to yield pAJ507. Finally, the BamHI-NheI fragment containing GAL10-RAT1 from pAJ507 was cloned into BamHI-SpeI-digested pRS424, a 2μm-TRP1 vector, to yield pAJ508.
Purification of Rat1p. (i) Cell growth.
Strain BJ5464 containing plasmid pAJ508 was grown at 30°C in SC Trp− medium supplemented with 1% (wt/vol) raffinose to a density of 1.8 × 107 cells/ml. Galactose was added to 1% (wt/vol), and the cells were grown for an additional 6.5 h. Cells were collected and washed with 20 mM Tris-HCl (pH 7.6)–10% (wt/vol) glycerol–20 mM KCl–1 mM EDTA–0.5 mM dithiothreitol (DTT)–0.5 mM phenylmethylsulfonyl fluoride (PMSF), and the cell pellet (∼20 g) was stored at −80°C until use.
(ii) Preparation of crude extract.
All purification steps were carried out at 0 to 4°C. Rat1p purification was monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. The cell pellet was thawed on ice and resuspended in 40 ml of extraction buffer (buffer A; 20 mM Tris-HCl [pH 7.6], 10% [wt/vol] glycerol, 1 mM EDTA, and 0.5 mM PMSF supplemented with 20 mM KCl, 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml). The cell suspension was broken in a Bead Beater (Biospec Co.) by agitating it 10 times for 0.5 min each. The cell lysate was recovered and clarified by centrifugation at 20,000 × g for 2.5 h, and the supernatant was collected (fraction I, 30 ml, 30 mg/ml).
(iii) PEI precipitation.
Fraction I was diluted with extraction buffer to give a protein concentration of 20 mg/ml. Polyethyleneimine (PEI) (5% [wt/vol]) was slowly added to the diluted fraction I with stirring to give a final concentration of 0.075% (wt/vol) PEI. After 30 min of stirring, the suspension was centrifuged for 15 min at 20,000 × g. The supernatant was discarded, and the pellet was washed once with extraction buffer. Proteins were eluted by mixing the pellet with 20 ml of buffer A supplemented with 200 mM KCl, 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml. The sample was clarified by centrifugation, and the supernatant was recovered (fraction II, 20 ml, 3.5 mg/ml).
(iv) ssDNA cellulose column chromatography.
A 2-ml column of ssDNA cellulose was equilibrated with buffer A supplemented with 200 mM KCl, 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml. Fraction II was loaded onto the column at 10 ml/h. The column was washed with 15 ml of the same buffer, and then proteins were eluted with a 30-ml linear KCl gradient (200 to 500 mM) in buffer A supplemented with 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml. Fractions of 0.5 ml were collected, and fractions containing Rat1p, which eluted at ∼280 mM KCl, were pooled (fraction III, 10 ml, 0.14 mg/ml).
(v) Source 15Q fast-phase liquid chromatography.
A 2-ml Source 15Q column was equilibrated with buffer B (20 mM Tris-HCl [pH 8.5], 10% [wt/vol] glycerol, 1 mM EDTA, and 0.5 mM PMSF) supplemented with 70 mM KCl, 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml. Fraction III was diluted with buffer B supplemented with 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml to a conductivity corresponding to 70 mM KCl in buffer B and then loaded onto the column at 1 ml per min. After the column was washed with 3 column volumes of the same buffer, proteins were eluted with a 10-ml linear KCl gradient (70 to 500 mM) in buffer B supplemented with 0.5 mM DTT, 0.5 μg of leupeptin per ml, and 0.7 μg of pepstatin A per ml. Fractions of 0.5 ml were collected, and Rat1p-containing fractions (fractions 13 to 17) were saved. Fraction 14 (0.5 ml, 0.2 mg/ml) was used for all subsequent in vitro exoribonuclease assays.
Purification of GST fusion proteins. (i) Cell growth.
Strain BJ5464 containing plasmid pEG(KT), pAJ245, or pAJ246 and AJY917 containing pAJ245 and pAJ508 were grown at 30°C in SC Ura− medium supplemented with 1% (wt/vol) raffinose to a density of 1.5 × 107 cells/ml. Galactose was added to 1% (wt/vol), and the cells were grown for an additional 6 h. Cells were harvested and washed once with phosphate-buffered saline (PBS) buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH7.4), and the cell pellets (∼3 g) were stored at −80°C.
(ii) Preparation of crude extracts.
All operations were performed at 0 to 4°C unless otherwise specified. The cell pellet was thawed on ice and resuspended in 5 ml of PBS buffer supplemented with 1 mM EDTA, 0.5 mM DTT, 0.5 μg of leupeptin per ml, 0.7 μg of pepstatin A per ml, and 0.5 mM PMSF. Cold, acid-washed glass beads (0.4 mm; Sigma) were added to the cell suspension. Cells were disrupted by vortexing them six times for 0.5 min with 1-min rests on ice in between vortexings. The cell lysate was recovered, and the beads were washed with 1 ml of the same buffer. The cell lysate and wash were combined and centrifuged at 20,000 × g for 30 min, and the supernatant was recovered as the crude extract (3.7 ml, 10 mg/ml).
(iii) Glutathione-Sepharose 4B chromatography.
A portion of the crude extract (1.7 ml) was mixed gently with 0.25 ml of a 50% slurry of glutathione-Sepharose 4B for 1 h at 4°C. The glutathione-Sepharose 4B was then transferred to a small column (0.4 by 1.5 cm). After the column was washed with 10 ml of PBS buffer supplemented with 1 mM EDTA, 0.5 mM DTT, 0.5 μg of leupeptin per ml, 0.7 μg of pepstatin A per ml, and 0.5 mM PMSF, the proteins were eluted with 1 ml of glutathione elution buffer containing 50 mM Tris-HCl, 10 mM reduced glutathione, and 140 mM KCl. Eluted proteins were stored at −80°C. All proteins were quantitated by the Bradford assay using bovine serum albumin as a standard (9). The amount of Rat1p in the Rat1p-GST-Rai1p complex was quantitated by comparison of Coomassie blue staining of Rat1p in the complex with that of purified free Rat1p.
SDS-PAGE and Western blot analysis.
For Western blot analysis, proteins were transferred from SDS-polyacrylamide gels to nitrocellulose membranes as described previously (24). Rabbit polyclonal antiserum raised against Rat1p (kindly provided by M. MacCammon) or mouse anti-GST (Zymed Laboratories Inc.) was used as the primary antibody, and blots were developed as described by the manufacturer (Promega ProtoBlot Kit).
RNA substrate.
Plasmid pSP6A0 DNA bearing the yeast MFA2 gene (13) was digested with EcoRI and then transcribed using the Riboprobe in vitro transcription system (Promega). The reaction mixture (100 μl) contained 40 mM Tris-HCl (pH 7.9), 10 mM NaCl, 6 mM MgCl2, 2 mM spermidine, 10 mM DTT, 0.5 mM ATP, 0.5 mM GTP, 0.5 mM CTP, 0.5 mM UTP, 50 μCi of [α-32P]CTP, 10 μg of linearized pSP6A0 template DNA, 100 U of RNase inhibitor, and 100 U of SP6 RNA polymerase. After incubation at 37°C for 1 h, RNase-free DNase was added to the reaction mixture and incubation was continued at 37°C for 15 min to remove the DNA template. The transcript was extracted with phenol-chloroform (pH 4.5) and separated from unincorporated nucleotides by chromatography through Sephadex G-50 (Boehringer Mannheim). The 5′-triphosphate was hydrolyzed to 5′-monophosphate with TAP. The TAP reaction mixture (100 μl) contained 50 mM sodium acetate (pH 5.0), 1 mM EDTA, 0.1% β-mercaptoethanol, 0.01% Triton X-100, 42 pmol of MFA2 transcript, and 10 U of TAP. This amount of TAP was found to be optimal for the activation of in vitro transcripts for degradation by Xrn1p, which requires 5′-phosphate for efficient degradation. Higher levels of TAP, either commercially prepared or prepared as described here, lead to lower levels of activation of in vitro transcripts for degradation by Xrn1p, probably due to hydrolysis of the 5′-phosphate at saturating TAP concentrations. After incubation at 37°C for 15 min, the reaction mixture was extracted with phenol-chloroform (pH 4.5) and ethanol precipitated, and the RNA was resuspended in H2O.
In vitro exoribonuclease assays.
The assay was based on the release of acid-soluble radioactivity from 32P-labeled TAP-treated MFA2 transcript. The reaction mixture (30 μl) contained 0.05 pmol of TAP-treated 32P-MFA2 transcript, 20 mM Tris-HCl (pH 8.5), 75 mM NaCl, 5 mM MgCl2, 0.1 mg of bovine serum albumin per ml, 0.5 mM DTT, and purified protein as indicated. After incubation at 30°C for 10 min, the reactions were stopped on ice by the addition of 10 μl of stop buffer (1 mg of ssDNA per ml and 20 mM EDTA). Sixty microliters of 0.6 M trichloroacetic acid was then added, reaction mixtures were incubated on ice for 10 min and centrifuged for 10 min at 15,000 × g, and the radioactivity in the supernatant was measured by liquid scintillation counting. A reaction mixture lacking the purified protein was used as a control.
Polysome analysis.
Sucrose gradient analyses for polysomes and for quantitation of total 40S and 60S subunits under conditions to dissociate subunits were performed as described in reference 17 with the modification that gradients were analyzed by continuous monitoring of absorbance at 254 nm using an ISCO model 640 gradient density fractionator.
Northern blotting.
Total RNA was prepared as described previously (17) from mid-log-phase cultures grown in SC Ura− medium. RNA was fractionated on 1% agarose-formaldehyde gels for large rRNA species or on 7 M urea–8% polyacrylamide gels as described previously (17). The RNAs in the gels were then transferred to nylon membranes (Zetaprobe; Bio-Rad) and probed with the appropriate radiolabeled oligonucleotide probes. Probes were as follows: a, 5′-GGTCTCTCTGCTGCCGGAAATG; b, 5′-TCTTGCCCAGTAAAAGCTCTCATGC; c, 5′-TCCAGTTACGAAAATTCTTGTTTTTGACAA; d, 5′-GGCCAGCAATTTCAAGTTA; e, 5′-GTTCGCCTAGACGCTCTCTTC; and f, 5′-CGCTGCGTTCTTCATCGATGCG. The probes were end labeled with [γ-32P]ATP using T4 polynucleotide kinase.
Primer extension.
Reactions were carried out as described previously (7) using probe f (above) end labeled with [γ-32P]ATP and 12 μg of total RNA prepared from the indicated strains. Reaction products were separated by electrophoresis through an 8% polyacrylamide–7 M urea gel. The gel was dried and autoradiographed.
RESULTS
Cloning of the RAI1 gene.
During purification of Rat1p from wild-type cells, a 45-kDa protein was found to copurify with Rat1p (31). The N-terminal sequence of this protein was determined by automated Edman degradation to be GVSANLSVKQRGSTXTA, where X was an unidentified residue. This sequence unambiguously identified a previously uncharacterized gene in yeast, YGL246c. We have named this gene RAI1 (Rat1p interacting protein 1).
Sequence analysis of Rai1p.
Based on a BLAST search of the GenBank database, Rai1p homologs are found throughout eukaryotes (Fig. 1). This protein family does not show significant similarity to other proteins of known function. A second open reading frame in S. cerevisiae, YDR370c, is predicted to encode a protein with similarity to Rai1p (data not shown). YDR370c is not an essential gene (42), and high-copy YDR370c did not suppress the growth defect of an rai1 deletion mutant, suggesting that these proteins are not functionally redundant (data not shown).
FIG. 1.

Amino acid alignment of Rai1p with homologs. Proteins (Rai1p, from S. cerevisiae; Schizosaccharomyces pombe predicted protein [GenBank accession no. 2440182]; DOM-3 from C. elegans [GenBank accession no. U34893]; mouse predicted protein [GenBank accession no. AAC05281]; and DOM3Z from humans [GenBank accession no. 2347139]) were aligned using CLUSTALW at http://dot.imgen.bcm.tmc.edu:9331/multi-align/multi-align.html. Shading was done using BOXSHADE at http://ulrec3.unil.ch/software/BOX_form.html. Positions of at least 60% identity are shaded in black, and positions with at least 60% similarity are shaded in gray.
RAI1 is not an essential gene.
In order to examine the function of Rai1p in vivo, we created a null mutant by targeted gene disruption by replacing a 967-bp SalI-BamHI fragment, beginning at codon 13 of the genomic RAI1 gene, with the yeast LEU2 selectable marker. The resulting haploid disruptants were viable, indicating that the gene is not essential. However, they displayed a slow-growth phenotype (Fig. 2) but were not temperature sensitive or cold sensitive (data not shown).
FIG. 2.

Complementation of rai1Δ growth defect. Strain AJY903 (rai1::LEU2) was transformed with pRS416 (empty vector), pAJ503 (RAI1), pAJ202 (RAT1), pRDK242 (XRN1), or pAJ236 (XRN1-SV40 NLS). The XRN1 vectors were high copy number, whereas all other plasmids were single-copy centromeric vectors. Transformants were streaked for single colonies on SC Ura− plates and incubated for 2 days at 30°C.
RAT1 and nucleus-targeted XRN1 complement the growth defect of an rai1 deletion mutant.
A screen for high-copy-number suppressors of the rai1::LEU2 slow-growth phenotype identified multiple isolates of RAI1 and RAT1 but not additional genes. Since high-copy RAT1 complemented the growth defect, we asked if RAT1 expressed from a low-copy-number centromeric vector could do the same. Indeed, low-copy-number RAT1 suppressed the growth defect of the rai1 disruption mutant (Fig. 2). Since Rat1p and Xrn1p are functionally interchangeable exonucleases that are restricted to the nucleus and cytoplasm, respectively (19), we also tested if Xrn1p directed to the nucleus by the presence of the simian virus 40 large-T antigen nuclear localization signal (NLS) could complement the growth defect of an rai1 mutant. High-copy-number XRN1-SV40 NLS complemented the growth defect of the rai1 deletion strain (Fig. 2). These results suggest that Rai1p acts primarily to enhance the activity of Rat1p in vivo.
Genetic interaction of rai1Δ with rat1-1ts and ski2Δ.
Rai1p was initially identified based on its copurification with Rat1p. Since temperature-sensitive mutations in proteins that form complexes are often destabilized in the absence of their binding partners, we asked if deletion of RAI1 affected Rat1p function in the temperature-sensitive rat1-1 mutant. An rai1::LEU2 mutant strain was crossed with a rat1-1 mutant. The resultant diploid was sporulated, and tetrads were dissected. From this cross, no viable Leu+ and temperature-sensitive spore clones were recovered from 27 tetrads, indicating that the double mutant was inviable (data not shown). Spore clones containing the single mutations were recovered at the expected frequency. No genetic interaction was observed between rai1::LEU2 and a deletion of XRN1 (data not shown). In the human genome, DOM3Z, the human homolog of RAI1, is adjacent to SKI2W, encoding a protein homologous to yeast Ski2p. This striking physical proximity of the human genes prompted us to examine the possible genetic interaction of rai1 with ski2 mutations. A heterozygous diploid (RAI1/rai1::hisG::URA3::hisG ski2::LEU2/SKI2) made by mating AJY667 and RKY1973 was sporulated, and the resulting spore clones were scored for Ura+ and Leu+. The double mutants were recovered at the expected frequency; however, all double mutants displayed severely impaired growth compared to the single mutants (Fig. 3). Since the ski2 mutation alone does not affect growth, the enhanced growth defect that a ski2 mutation causes in an rai1 deletion background indicates genetic interaction between these two mutations. Yeast Ski2p is similar in sequence to Mtr4p, a putative RNA helicase that is required for mRNA export from the nucleus. Whereas xrn1 is synthetically lethal with ski2 mutations, we observed no genetic interaction between mtr4-1 and rai1::LEU2 mutations (data not shown).
FIG. 3.
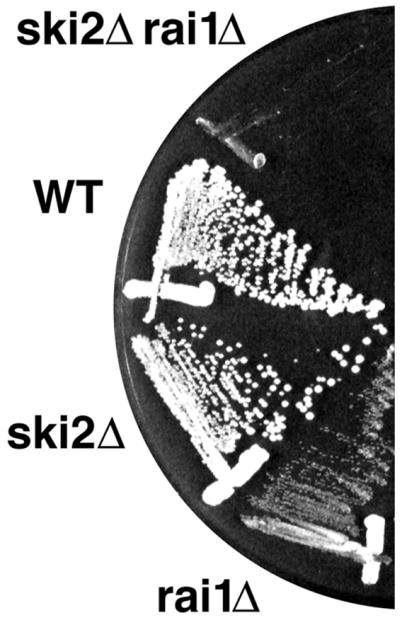
Genetic interaction between rai1Δ and ski2Δ. Strains AJY667 and RKY1973 were mated and sporulated, and the resulting tetrads were dissected. A representative tetrad was restreaked for single colonies on yeast extract-peptone-dextrose, incubated at room temperature for 3 days, and photographed. Similar results were obtained at 30 and 37°C (data not shown). WT, wild type.
Rai1p is not required for the nuclear localization of Rat1p.
Deletion of the Rat1p bipartite NLS sequence (KKHRLEKDNEEEEIAKDSKKVK) causes mislocalization of the Rat1p protein throughout the cell (19) but does not completely block the nuclear function of Rat1p (19). We tested if Rai1p was needed for the residual nuclear localization of Rat1p lacking an NLS (RAT1-NLSΔ). We made a rat1-1ts rai1Δ double mutant rescued by plasmid pAJ202 harboring the RAT1 gene and a URA3 selectable marker. We then determined in a plasmid shuffle assay if RAT1-NLSΔ could replace the RAT1 plasmid. The RAT1-NLSΔ plasmid allowed growth on 5-fluoroorotic acid, which is toxic if the RAT1 URA3 plasmid cannot be lost. Thus, Rai1p is not required for the nuclear function of RAT1-NLSΔ.
An rai1 mutant displays a deficit of free 60S ribosomal subunits.
The slow growth of the rai1 deletion mutant and the requirement of Rat1p for numerous RNA processing reactions involved in rRNA biogenesis led us to ask if an rai1 mutant had a translation defect. We did this by sucrose gradient sedimentation. Extracts were prepared and sedimented through 7 to 47% sucrose gradients, and the gradients were analyzed by continuously monitoring the absorbance at 254 nm. As shown in Fig. 4, the rai1 mutant (rai1/vector) had a severely reduced ratio of free 60S to free 40S subunits and showed halfmers, polysomes with 43S initiation complexes not yet joined with 60S. Halfmers are often observed under conditions in which 60S levels are limiting (28). Similar results were obtained with wild-type and deletion mutant strains (strains CH1305 and AJY903, respectively) not carrying vectors and grown in rich medium (data not shown). Quantitation of total 40S and 60S levels after sedimentation in sucrose gradients under conditions that dissociate polysomes and 80S couples indicated that 60S levels were reduced 40% in the rai1 mutant (data not shown). Since we found that RAT1 was a dosage suppressor of the rai1 deletion, we also examined the polysome profiles of rai1 mutants carrying RAT1 on a low-copy-number vector (CEN) or a high-copy-number vector (hc). Figure 4 shows that high-copy-number RAT1 restored the polysome profile to nearly wild-type levels. Halfmers were no longer evident, and there was only a slight imbalance in the free subunit ratio. RAT1 on a centromeric vector also improved the polysome profile but to a lesser extent than high-copy-number RAT1. Similarly, high-copy-number XRN1 containing an NLS restored polysome profiles similar to those obtained with single-copy-number RAT1 (data not shown).
FIG. 4.

Sucrose gradient analysis. Extracts were prepared from mid-log-phase cultures and sedimented through 7 to 47% sucrose gradients. Gradients were fractionated, and the absorbance at 254 nm was monitored. The positions of 40S and 60S subunits, 80S monosomes, and halfmers, where evident, are indicated. The strain used was AJY903 (rai1::LEU2) containing pAJ503 (RAI1), pRS416 (vector), single-copy pAJ202 (RAT1 CEN), or high-copy-number pAJ509 (RAT1 hc).
RAI1 is required for normal processing of 5.8S rRNA.
The reduction in 60S levels in an rai1 mutant suggested a defect in the rRNA biogenesis. We examined this by Northern blotting using total RNA prepared from the strains used in the polysome analysis. 5.8S rRNA is generated by two different and redundant pathways in yeast to give rise to two species, 5.8SL and 5.8SS, which differ in length at their 5′ ends (16). In wild-type cells, 5.8SS is the predominant species and is generated by cleavage at site A3 followed by exonucleolytic trimming by Rat1p and Xrn1p to site B1S. The 5′ end of 5.8SL results from sequential cleavages at A2 and B1L (Fig. 5A). In the absence of 5′ exonucleolytic trimming, the production of 5.8SS is inhibited and 5.8SL becomes the predominant form (16). This was also observed for an rai1 deletion mutant (Fig. 5B, lane 2). This defect was reversed by increasing the gene dosage of RAT1 (Fig. 5B, lanes 3 and 4). The percentage of total 5.8S present as 5.8SL in Fig. 5B, lanes 1 through 4, was 29, 64, 40, and 32%, respectively. This restoration of normal 5.8S levels by increasing RAT1 copy number closely paralleled the restoration of normal polysome profiles shown in Fig. 4. Using probe c, specific for the 3′ region of ITS1 between sites A3 and B1L (Fig. 5A), we did not detect any aberrant 5′-extended forms of 5.8S (Fig. 5C, lane 8, and data not shown), although this probe readily hybridized with 35S and 27S rRNAs, intermediates that contain this region of RNA (Fig. 5C, lane 8). Unexpectedly, for the rai1 deletion mutant we observed accumulation of 3′-extended species in the position expected for the 7S precursor rRNA (Fig. 5B, lane 6). Such 3′-extended forms suggest that the 3′ processing pathway, which depends on the exonucleolytic activities of the exosome (3, 25), is also partially inhibited in an rai1 mutant. As seen in Fig. 5B, these 3′ defects were reversed by ectopically expressing RAI1 or RAT1.
FIG. 5.

Northern blot analysis of rRNAs. (A) 35S rRNA precursor with the 5.8S region enlarged. Mature rRNAs are labeled and indicated as thick lines, external transcribed spacers (ETS) and internal transcribed spacers (ITS) are shown as thin lines, and the major processing sites are labeled. Oligonucleotide probes used for Northern blot analysis are indicated as short bars (labeled a to f) below the primary transcript. (B) Northern blot analysis of 5.8S rRNA. Total RNA was prepared from AJY903 (rai1Δ) containing pAJ503 (RAI1), pRS416 (vector), pAJ202 (RAT1), or high-copy-number pAJ509 (RAT1 hc) and separated by electrophoresis through a 8% polyacrylamide–7 M urea gel. RNAs were blotted to membrane and hybridized with 32P-labeled oligonucleotide probes (f and d as described for panel A). The identities of bands are indicated. (C) Northern blot analysis of the large rRNA intermediates. Total RNA was prepared from AJY903 (rai1Δ) containing pAJ503 (RAI1), pRS416 (vector), or pAJ202 (RAT1) and separated by electrophoresis through a 1% agarose-formaldehyde gel. RNAs were blotted to membrane and hybridized with 32P-labeled oligonucleotide probes (a to e as described for panel A). The identities of bands are indicated to the right.
Northern analysis of the larger rRNA species showed higher steady-state levels of 35S and 27S pre-rRNAs, indicating that the processing of these species was slowed in an rai1 mutant (Fig. 5C, lane 8). This defect also was reversed by ectopically expressing RAI1 or RAT1. Mature 25S and 18S rRNAs were observed in the rai1 mutant (data not shown), although the ratio of 25S rRNA relative to 18S rRNA was reduced based on our observation of a reduced ratio of total 60S to 40S subunits. No defect was observed for 5S rRNA (data not shown). Pulse-chase analysis of rRNA processing using [methyl-3H]methionine (17) confirmed that cleavage of 27S rRNA was slowed in the rai1 deletion mutant (data not shown).
Primer extension analysis was carried out using probe f (Fig. 5A), which hybridized to 5.8S rRNA. As shown in Fig. 6, there was a significant increase in a primer extension product with a 5′ end at site A3 in the rai1 mutant (Fig. 6, lane 6). The accumulation of this product was reversed by increasing the gene dosage of RAT1 (Fig. 6, lanes 7 and 8). A3 is the processing site that is used for the generation of 5.8SS by 5′ trimming by Rat1p (16). Since we were unable to detect 5′-extended 5.8S species, this primer extension product to site A3 indicates the accumulation of a 27S species extended to site A3.
FIG. 6.
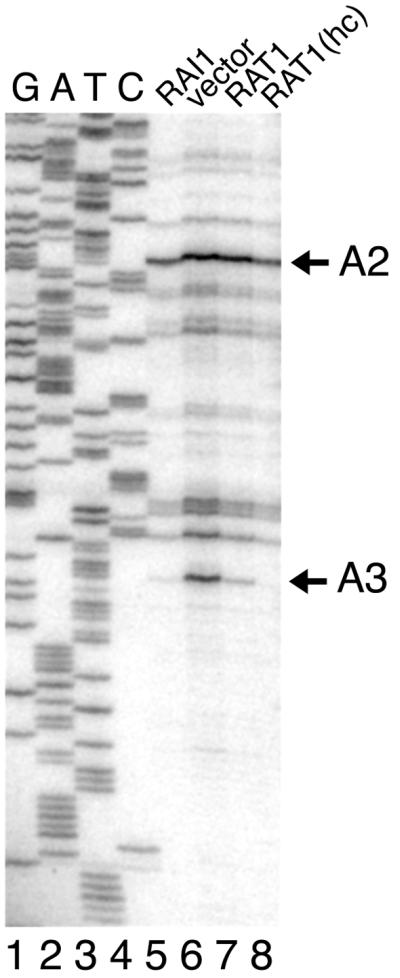
Primer extension analysis. Probe f (see Fig. 5A legend), complementary to 5.8S rRNA, was used for primer extension on the RNAs used in Fig. 5B. Extension products (lanes 5 to 8) were compared to a sequencing ladder (lanes 1 to 4) using probe f to prime DNA synthesis on plasmid p518 containing the ribosomal DNA locus. Reaction products were separated by urea-polyacrylamide gel electrophoresis and visualized by autoradiography after drying. The positions of species resulting from cleavage at sites A2 and A3 (Fig. 5A) are indicated. Lanes 5 to 8 are labeled as in Fig. 5B.
Rai1p binds to Rat1p.
A GST-RAI1 fusion was constructed which complemented the slow-growth phenotype of an rai1 disruption strain (data not shown), indicating that it was functional. Extracts prepared from cells overexpressing the GST-Rai1p fusion protein were incubated with glutathione-Sepharose beads, and the bound proteins were subsequently analyzed by SDS-PAGE and Western blotting. As shown in Fig. 7, Rat1p copurified with the GST-Rai1p fusion (lane 6) but was not retained by the glutathione-Sepharose beads when extracts contained GST protein alone (lane 3). These results confirmed that Rai1p binds Rat1p in vivo.
FIG. 7.

Rai1p binds Rat1p in vivo. Crude extracts from BJ5464 containing plasmid pEG(KT) (GST), pAJ245 (GST-RAI1), or pAJ246 (GST-RAI1Δ115) were incubated with glutathione-Sepharose beads. Bound proteins were eluted by heating in the presence of Laemmli sample buffer, separated by SDS-PAGE, and analyzed for the presence of Rat1p by Western blotting. T, total extract; ft, flowthrough; E, eluted proteins.
A GST fusion to a truncated Rai1p, GST-rai1Δ115p, lacking the C-terminal 115 amino acids of Rai1p, did not bind Rat1p (Fig. 7, lane 9), suggesting that the C-terminal portion of Rai1p is important for its physical association with Rat1p. In addition, the GST-RAI1Δ115 construct did not complement the growth defect of an rai1 disruption mutant (data not shown), consistent with the idea that binding of Rai1p to Rat1p is important for Rai1p function in vivo.
In vitro analysis of purified Rat1p and Rai1p.
Our results thus far suggested that Rai1p stimulates Rat1p function in vivo. We sought to understand the mechanism of stimulation by purifying the individual proteins to assay the activities separately and in combination in vitro in exonuclease assays.
Since Rat1p had previously been purified and characterized in a complex with Rai1p and not as a free protein, we purified free Rat1p from an rai1 disruption strain to examine the in vitro characteristics of free Rat1p compared with the protein complexed with Rai1p. Although a purification protocol for the Rat1p-Rai1p complex had been established, it was necessary to develop a new procedure for free Rat1p. As described in Materials and Methods, the purification of Rat1p entailed three steps: PEI precipitation, ssDNA cellulose column chromatography, and Source 15Q fast-phase liquid chromatography. The use of PEI at the first stage was critical to the purification procedure. Some proteins, most of which are nucleic acid-binding proteins, are precipitated by PEI to form the PEI-nucleic acid-protein complex under low-ionic-strength conditions. We found that Rat1p was precipitated at extremely low PEI concentrations (quantitatively precipitated at 0.075% PEI) and in fact was not precipitated at intermediate PEI concentrations. Precipitation of Rat1p by very low PEI concentrations at which most proteins remain in solution gave a significant purification. The PEI eluate was subsequently run on an ssDNA cellulose column and finally purified by Source 15Q fast-phase liquid chromatography. We found that use of other ion-exchange columns before or in place of ssDNA cellulose resulted in large losses of Rat1p due to contaminating proteases (data not shown). Figure 8A shows the SDS-PAGE of purified Rat1p. The lower-molecular-weight species in the Rat1p lane were due to degradation of Rat1p, demonstrated by Western blotting using polyclonal antibody against Rat1p (data not shown). The contaminating protease eluted from ssDNA cellulose slightly earlier but partially overlapping with Rat1p. The preparation was stable with respect to proteolysis after Source 15Q chromatography.
FIG. 8.

Purification of proteins for in vitro analysis. Proteins were separated by SDS-PAGE and visualized by staining with Coomassie blue. (A) Rat1p was purified from AJY942 (rai1Δ) containing pAJ508 as described in Materials and Methods. Lane 1, crude extract; lane 2, PEI fraction; lane 3, ssDNA cellulose fraction; lane 4, Source 15Q fraction. (B) GST-Rai1p and GST-Rai1p complexed with Rat1p were overexpressed and purified as described in Materials and Methods. Lane 1, GST-Rai1p; lane 2, GST-Rai1p with Rat1p. The positions of molecular mass markers are indicated.
GST-Rai1p fusion protein was also overexpressed and purified from yeast (Fig. 8B). Although GST-Rai1p was purified from wild-type yeast containing Rat1p, the high level of overexpression allowed the purification of GST-Rai1p that was free of significant exonuclease activity (see below), suggesting that contamination with endogenous Rat1p was minimal and would not affect subsequent analysis. GST-Rai1Δ115p and free GST as a control were purified similarly.
We were unable to reconstitute the Rat1p-Rai1p complex from purified Rat1p and GST-Rai1p, even though these proteins formed a stable complex in vivo. Consequently, we purified the Rat1p-GST-Rai1p complex from yeast cooverexpressing both proteins (Fig. 8B).
Rai1p enhances the exoribonuclease activity of Rat1p in vitro.
We carried out in vitro exoribonuclease assays of the purified proteins. GST-Rai1p alone showed no significant exoribonuclease activity (0.5% of the activity of free Rat1p) and demonstrated that it was essentially free of contaminating Rat1p (Fig. 9). Rat1p alone displayed significant 5′→3′ exoribonuclease activity with a maximum rate of reaction of 650 nucleotides released per Rat1p molecule in a standard 10-min reaction. The Rat1p-GST-Rai1p complex displayed a more robust exonuclease activity, with a maximum rate of 940 nucleotides released per Rat1p molecule in a standard 10-min reaction. The addition of excess free GST-Rai1p to an exonuclease assay with purified free Rat1p did not significantly affect the exonuclease activity of free Rat1p (data not shown), indicating that the excess GST-Rai1p in the purified complex did not affect Rat1p activity.
FIG. 9.

Rai1p enhances Rat1p exonuclease activity in vitro. Standard exonuclease assays were performed using varying amounts of Rat1p, GST-Rai1p, and Rat1p-GST-Rai1p complex. Picomoles of nucleotide released is plotted against picomoles of enzyme in each reaction. Protein amounts are indicated, and in the case of Rat1p-GST-Rai1p complex, the protein amounts are those of Rat1p in the complex.
Rai1p stabilizes the exoribonuclease activity of Rat1p in vitro.
The enhancement of Rat1p exonuclease activity by Rai1p could be explained by several mechanisms including increased RNA binding and increased stability. We examined the effect of Rai1p on the stability of Rat1p exonuclease activity. Similar amounts of Rat1p or Rat1p in a complex with GST-Rai1p were preincubated at 30°C for increasing times and then assayed for remaining exonuclease activity under standard conditions. The first time point taken was after 2 min of preincubation to allow for temperature equilibration. The activities remaining at subsequent times are shown relative to that at 2 min. As seen in Fig. 10, the exonuclease activity of Rat1p was significantly more stable when in a complex with Rai1p than as free Rat1p protein. The reason for the initial rapid drop in activity observed between 2 and 5 min for free protein and complexed protein is not known, but this effect was observed in multiple experiments. Western blotting for Rat1p during the course of this experiment showed no change in Rat1p, indicating that the loss of activity was not due to loss of the protein by proteolysis (data not shown). Thus, Rai1p stabilizes Rat1p exonuclease activity in vitro.
FIG. 10.

Rai1p stabilizes Rat1p exonuclease activity in vitro. Similar amounts of Rat1p and Rat1p complexed with GST-Rai1p were assayed for stability of the exonuclease activity at 30°C. Samples were prepared in exonuclease assay buffer lacking substrate and incubated at 30°C. At the indicated times after incubation at 30°C, aliquots were removed and assayed for exonuclease activity in a standard reaction. Activity relative to that at 2 min of incubation is plotted against time. The amount of enzyme used in these assays was in the linear range of the assay.
DISCUSSION
Rai1p defines a new family of proteins that includes human DOM3Z and Caenorhabditis elegans DOM-3. Since Rai1p binds Rat1p in yeast, it is likely that the homologs of Rai1p in other organisms bind their respective homologs of Rat1p. This protein family has no motifs common to other proteins of known function, and like the Xrn1p and Rat1p family of proteins, there are no proteins of similar sequence predicted in prokaryotic cells.
The growth defect of an rai1 deletion mutant was complemented by increasing gene dosage of RAT1 and was partially complemented by Xrn1p directed to the nucleus. Since Rat1p is an essential nuclear exoribonuclease, these results suggest that the low growth rate observed for an rai1 deletion mutant is due to impaired nuclear RNA turnover or processing. Because Rai1p itself does not apparently encode a nuclease activity, Rai1p appears to function to enhance Rat1p activity in vivo. This is supported by our in vitro demonstration that Rai1p binding to Rat1p stabilizes the exonuclease activity of Rat1p. In addition, deletion of the C-terminal 115 amino acids abolishes Rat1p binding and complementation of the slow-growth phenotype, suggesting that direct interaction is necessary for function. These results do not indicate a specific function for Rai1p apart from its interaction with Rat1p. However, the Rai1p protein family shows conservation of amino acid sequence throughout the length of the protein and is not restricted to the presumed Rat1p binding domain. We assayed GST-Rai1p for RNA binding activity. GST-Rai1p purified from Escherichia coli showed no RNA binding (data not shown).
The human homolog of Rai1p is encoded by DOM3Z, a gene identified within the HLA class III locus (43). The 3′ end of the DOM3Z coding sequence is remarkably only 59 nucleotides from the 3′ end of the coding sequence of SKI2W, the human homolog of the yeast antiviral gene SKI2 (43). SKI2 encodes a putative RNA helicase and is required for repression of translation of poly(A) mRNA and for normal 3′ mRNA degradation. The physical proximity of the human genes prompted us to examine the potential genetic interaction of the homologous yeast genes. We identified mutations in SKI2 from a screen for mutations synthetically lethal with a deletion of XRN1, encoding the cytoplasmic exoribonuclease Xrn1p (21). Subsequently, we showed that mislocalization of Rat1p to the cytoplasm suppressed the lethality of xrn1 ski2 double mutants (19). We have recently shown that Ski2p forms a heterotrimeric complex with Ski3p and Ski8p and is predominantly located in the cytoplasm (10). It has been suggested that Ski2p acts to recruit the 3′ exoribonucleolytic exosome to the 3′ ends of mRNAs for degradation. It is also possible that Ski2p affects ribosome function directly, perhaps acting during ribosome biogenesis. The localization of human Ski2w to the nucleolus (as well as the cytoplasm) supports such a notion (32a). Genetic interaction between mutations in RAI1 and SKI2, but not XRN1, suggests that there is overlap in function between Ski2p and Rai1p. Thus, Ski2p may have an additional role in the nucleus or Rai1p may have an additional cytoplasmic function. Using conditional rai1 ski2 double mutants should now allow us to examine a potential role of Ski2p in nuclear RNA processing events.
Rat1p is required for the normal processing of the 5′ end of 5.8S rRNA as well as processing various snoRNAs. Because Rai1p acts to enhance Rat1p activity, it was not surprising to find that deletion of RAI1 inhibited the Rat1p-dependent pathway responsible for generating 5.8SS. Consequently, in an rai1 deletion mutant, the majority of 5.8S rRNA is 5.8SL and is made by an alternate pathway. However, blocking the 5′ exonucleolytic trimming from site A3 to B1S should result in accumulation of a 5′-extended form of 5.8S (16). We did not detect this species. On the other hand, our data show the accumulation of a 27S species extending to site A3, suggesting that cleavage at site C2, which releases 7S rRNA, is inefficient. In addition, an rai1 mutant accumulates 7S rRNA, the 3′-extended precursor of 5.8S. The 3′ processing of 5.8S rRNA depends on the multisubunit 3′ exonucleolytic complex, the exosome (4, 25).
These results suggest that RAI1 is also needed for efficient processing of the 3′ end of 5.8S rRNA. Since Rai1p itself is physically associated with the 5′ processing enzyme Rat1p, the effect on 3′ processing could be indirect. For example, Rat1p is required for 5′ processing of various small RNA species including snoRNAs (30, 32, 41). It is possible that the function or assembly of a 3′ processing complex depends on a substrate of Rat1p. Alternatively, Rai1p may be required directly for efficient 3′ processing, perhaps physically interacting with factors required for 3′ processing and thereby coordinating 5′ and 3′ processing events. Because an rai1 deletion mutant is viable and behaves as a constitutive partial loss-of-function rat1 mutant, we believe that it will provide a useful genetic background to examine the role of Rat1p in additional nuclear RNA turnover and processing reactions.
ACKNOWLEDGMENTS
We thank M. MacCammon for providing anti-Rat1p antiserum, J. Woolford for providing plasmid p518, and K. Dana for providing tobacco BY2 cultures.
REFERENCES
- 1.Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldrich T L, Di Segni G, McConaughy B L, Keen N J, Whelen S, Hall B D. Structure of the yeast TAP1 protein: dependence of transcription activation on the DNA context of the target gene. Mol Cell Biol. 1993;13:3434–3444. doi: 10.1128/mcb.13.6.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999;18:5399–5410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, Mitchell P. The yeast exosome and human PM-Scl are related complexes of 3′→5′ exonucleases. Genes Dev. 1999;13:2148–2158. doi: 10.1101/gad.13.16.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amberg D C, Goldstein L A, Cole C N. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- 6.Anderson J, Parker R. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998;17:1497–1506. doi: 10.1093/emboj/17.5.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ausubel F M, editor. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1988. [Google Scholar]
- 8.Bashkirov V I, Scherthan H, Solinger J A, Buerstedde J M, Heyer W D. A mouse cytoplasmic exoribonuclease (mXRN1p) with preference for G4 tetraplex substrates. J Cell Biol. 1997;136:761–773. doi: 10.1083/jcb.136.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Brown J T, Bai X, Johnson A W. The yeast antiviral proteins Ski2p, Ski3p and Ski8p form a heterotrimeric complex. RNA. 2000;6:449–457. doi: 10.1017/s1355838200991787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dangel A W, Shen L, Mendoza A R, Wu L C, Yu C Y. Human helicase gene SKI2W in the HLA class III region exhibits striking structural similarities to the yeast antiviral gene SKI2 and to the human gene KIAA0052: emergence of a new gene family. Nucleic Acids Res. 1995;23:2120–2126. doi: 10.1093/nar/23.12.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Segni G, McConaughy B L, Shapiro R A, Aldrich T L, Hall B D. TAP1, a yeast gene that activates the expression of a tRNA gene with a defective internal promoter. Mol Cell Biol. 1993;13:3424–3433. doi: 10.1128/mcb.13.6.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doktycz M J, Larimer F W, Pastrnak M, Stevens A. Comparative analyses of the secondary structures of synthetic and intracellular yeast MFA2 mRNAs. Proc Natl Acad Sci USA. 1998;95:14614–14621. doi: 10.1073/pnas.95.25.14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efstratiadis A, Vournakis J N, Donis K H, Chaconas G, Dougall D K, Kafatos F C. End labeling of enzymatically decapped mRNA. Nucleic Acids Res. 1977;4:4165–4174. doi: 10.1093/nar/4.12.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gietz D, St. John A, Woods R A, Schiestl R H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henry Y, Wood H, Morrissey J P, Petfalski E, Kearsey S, Tollervey D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 1994;13:2452–2463. doi: 10.1002/j.1460-2075.1994.tb06530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho J, Johnson A W. NMD3 encodes an essential cytoplasmic protein required for stable 60S ribosomal subunits in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2389–2399. doi: 10.1128/mcb.19.3.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu C L, Stevens A. Yeast cells lacking 5′→3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson A W. Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol Cell Biol. 1997;17:6122–6130. doi: 10.1128/mcb.17.10.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson A W, Kolodner R D. Strand exchange protein 1 from Saccharomyces cerevisiae. A novel multifunctional protein that contains DNA strand exchange and exonuclease activities. J Biol Chem. 1991;266:14046–14054. [PubMed] [Google Scholar]
- 21.Johnson A W, Kolodner R D. Synthetic lethality of sep1 (xrn1) ski2 and sep1 (xrn1) ski3 mutants of Saccharomyces cerevisiae is independent of killer virus and suggests a general role for these genes in translation control. Mol Cell Biol. 1995;15:2719–2727. doi: 10.1128/mcb.15.5.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 23.Kenna M, Stevens A, McCammon M, Douglas M G. An essential yeast gene with homology to the exonuclease-encoding XRN1/KEM1 gene also encodes a protein with exoribonuclease activity. Mol Cell Biol. 1993;13:341–350. doi: 10.1128/mcb.13.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsudaira P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem. 1987;262:10035–10038. [PubMed] [Google Scholar]
- 25.Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- 26.Muhlrad D, Decker C J, Parker R. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Genes Dev. 1994;8:855–866. doi: 10.1101/gad.8.7.855. [DOI] [PubMed] [Google Scholar]
- 27.Muhlrad D, Parker R. Premature translational termination triggers mRNA decapping. Nature. 1994;370:578–581. doi: 10.1038/370578a0. [DOI] [PubMed] [Google Scholar]
- 28.Ohtake Y, Wickner R B. Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol Cell Biol. 1995;15:2772–2781. doi: 10.1128/mcb.15.5.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Page A M, Davis K, Molineux C, Kolodner R D, Johnson A W. Mutational analysis of exoribonuclease I from Saccharomyces cerevisiae. Nucleic Acids Res. 1998;26:3707–3716. doi: 10.1093/nar/26.16.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petfalski E, Dandekar T, Henry Y, Tollervey D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol Cell Biol. 1998;18:1181–1189. doi: 10.1128/mcb.18.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poole T L, Stevens A. Comparison of features of the RNase activity of 5′-exonuclease-1 and 5′-exonuclease-2 of Saccharomyces cerevisiae. Nucleic Acids Symp Ser. 1995;33:79–81. [PubMed] [Google Scholar]
- 32.Qu L H, Henras A, Lu Y J, Zhou H, Zhou W X, Zhu Y Q, Zho J, Henry Y, Caizergues F M, Bachellerie J P. Seven novel methylation guide small nucleolar RNAs are processed from a common polycistronic transcript by Rat1p and RNase III in yeast. Mol Cell Biol. 1999;19:1144–1158. doi: 10.1128/mcb.19.2.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a.Qu X, Yang Z, Zhang S, Shen L, Dangel A W, Hughes J H, Redman K L, Wu L C, Yu C Y. The human DEVH-box protein SkiZw from the HLA is localized in nucleoli and ribosomes. Nucleic Acids Res. 1998;26:4068–4077. doi: 10.1093/nar/26.17.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shobuike T, Sugano S, Yamashita T, Ikeda H. Characterization of cDNA encoding mouse homolog of fission yeast dhp1+ gene: structural and functional conservation. Nucleic Acids Res. 1995;23:357–361. doi: 10.1093/nar/23.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shobuike T, Sugano S, Yamashita T, Ikeda H. Cloning and characterization of mouse Dhm2 cDNA, a functional homolog of budding yeast SEP1. Gene. 1997;191:161–166. doi: 10.1016/s0378-1119(97)00053-x. [DOI] [PubMed] [Google Scholar]
- 35.Stevens A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5′-mononucleotides by a 5′→3′ mode of hydrolysis. J Biol Chem. 1980;255:3080–3085. [PubMed] [Google Scholar]
- 36.Stevens A, Hsu C L, Isham K R, Larimer F W. Fragments of the internal transcribed spacer 1 of pre-rRNA accumulate in Saccharomyces cerevisiae lacking 5′→3′ exoribonuclease 1. J Bacteriol. 1991;173:7024–7028. doi: 10.1128/jb.173.21.7024-7028.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens A, Maupin M K. A 5′→3′ exoribonuclease of Saccharomyces cerevisiae: size and novel substrate specificity. Arch Biochem Biophys. 1987;252:339–347. doi: 10.1016/0003-9861(87)90040-3. [DOI] [PubMed] [Google Scholar]
- 38.Stevens A, Poole T L. 5′-Exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J Biol Chem. 1995;270:16063–16069. doi: 10.1074/jbc.270.27.16063. [DOI] [PubMed] [Google Scholar]
- 39.Toone W M, Johnson A L, Banks G R, Toyn J H, Stuart D, Wittenberg C, Johnston L H. Rme1, a negative regulator of meiosis, is also a positive activator of G1 cyclin gene expression. EMBO J. 1995;14:5824–5832. doi: 10.1002/j.1460-2075.1995.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venema J, Tollervey D. Processing of pre-ribosomal RNA in Saccharomyces cerevisiae. Yeast. 1995;11:1629–1650. doi: 10.1002/yea.320111607. [DOI] [PubMed] [Google Scholar]
- 41.Villa T, Ceradini F, Presutti C, Bozzoni I. Processing of the intron-encoded U18 small nucleolar RNA in the yeast Saccharomyces cerevisiae relies on both exo- and endonucleolytic activities. Mol Cell Biol. 1998;18:3376–3383. doi: 10.1128/mcb.18.6.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winzeler E A, Shoemaker D D, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke J D, Bussey H, Chu A M, Connelly C, Davis K, Dietrich F, Dow S W, El B M, Foury F, Friend S H, Gentalen E, Giaever G, Hegemann J H, Jones T, Laub M, Liao H, Davis R W, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z, Shen L, Dangel A W, Wu L C, Yu C Y. Four ubiquitously expressed genes, RD (D6S45)-SKI2W (SKIV2L)-DOM3Z-RP1 (D6S60E), are present between complement component genes factor B and C4 in the class III region of the HLA. Genomics. 1998;53:338–347. doi: 10.1006/geno.1998.5499. [DOI] [PubMed] [Google Scholar]