

Abstract
T cells exhibit heterogeneous functional states, which correlate with responsiveness to immune checkpoint blockade and prognosis of tumor patients. However, the molecular regulatory mechanisms underlying the dynamic process of T cell state transition remain largely unknown. Based on single-cell transcriptome data of T cells in non-small cell lung cancer, we combined cell states and pseudo-times to propose a pipeline to construct dynamic regulatory networks for dissecting the process of T cell dysfunction. Candidate regulators at different stages were revealed in the process of tumor-infiltrating T cell dysfunction. Through comparing dynamic networks across the T cell state transition, we revealed frequent regulatory interaction rewiring and further refined critical regulators mediating each state transition. Several known regulators were identified, including TCF7, EOMES, ID2, and TOX. Notably, one of the critical regulators, TSC22D3, was frequently identified in the state transitions from the intermediate state to the pre-dysfunction and dysfunction state, exerting diverse roles in each state transition by regulatory interaction rewiring. Moreover, higher expression of TSC22D3 was associated with the clinical outcome of tumor patients. Our study embedded transcription factors (TFs) within the temporal dynamic networks, providing a comprehensive view of dynamic regulatory mechanisms controlling the process of T cell state transition.
Keywords: T cell dysfunction, state transition trajectory, dynamic regulatory network, pseudo-times, critical regulators
Graphical abstract
Antigen recognition in the tumor microenvironment drives T cell dysfunction, which is governed by a complex regulatory network. We constructed dynamic regulatory networks and identified critical regulators during state transition processes along T cell dysfunction. TSC22D3 was frequently identified in different state transition stages, and it exhibited prognostic significance for NSCLC patients.
Introduction
T cell immune checkpoint blockade (ICB) therapy has revolutionized cancer treatment to induce durable responses in patients through reactivating tumor-specific T cell responses.1 However, only a subset of patients could achieve long-term effective clinical benefit, and our understanding of the mechanisms underlying response or resistance to these therapies is still incomplete. Recent studies of single-cell analysis have provided evidence for heterogeneous compositions of tumor-infiltrating CD8+ T cells.2, 3, 4 The main components of intratumoral CD8+ T cells comprise “naive-like” cells marked by CCR7, LEF1, and TCF7, “cytotoxic effector” cells characterized by CX3CR1, PRF1, and KLRG1,5 and “dysfunction” or “exhaustion” T cells, which occupy a higher proportion in the tumor.6 Dysfunctional T cells, which are characterized by the increased co-inhibitory receptors, such as PD1, LAG3, TIM3, CTLA4, and TIGIT, have limited effector function compared with effector CD8+ T cells. Dysfunctional T cells may be further divided into subgroups, such as pre-dysfunctional, progenitor exhausted, and terminally exhausted T cells.7 Moreover, these heterogeneous state compositions exhibit different intrinsic effector functions and reinvigoration potential,8 indicating the likelihood to form a determining factor in therapy outcome.
Indeed, the composition and relative proportions of different states of CD8+ T cells in tumors are related to the clinical outcome, such as the tumor stage,6 response to immune therapy,3 and survival probability of patients.9 Zheng et al.6 demonstrated increased proportions of dysfunction CD8+ T cells in hepatocellular carcinoma (HCC) patients at a late stage than at an early stage. Single-cell profiling of T cells in breast cancer revealed a tissue-resident memory subset, which expressed signature genes of dysfunction and was associated with improved prognosis.10 Importantly, a subset of TCF1-expressing PD1+ pre-dysfunction T cells were revealed to be critical for tumor control on single agent anti-PD1 therapy or anti-PD1 and anti-CTLA4 combination therapy in melanoma.11 In addition, the pre-dysfunction subset of T cells has also been observed in human non-small cell lung cancer (NSCLC) and colorectal cancer (CRC), and their cell proportions were increased in tumors treated with ICB and might indicate better survival of tumor patients.3,11, 12, 13 Given the distinct functions and relationship to clinical outcome and response to ICB therapy of different states of T cells, there is an urgent need to identify regulators that modulate the transition among T cell states in cancer.
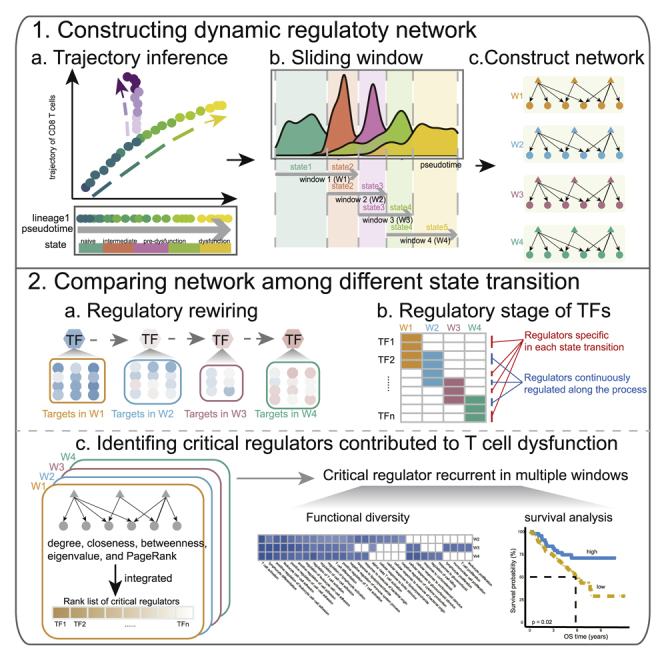
In this study, we proposed a comprehensive analysis of the dynamic network during the T cell dysfunction process and dissected the critical transcription factors (TFs) for each state transition stage. First, we constructed the development trajectory of CD8+ T cells and identified dynamic genes differentially expressed along the dysfunction lineage induced by the tumor environment. Next, we assigned cells into state transition stages along the trajectory, and then constructed a dynamic regulatory network for each cell state transition stage. Then, by comparing the regulatory networks among different stages, we found high-degree rewiring and dissected diverse rewiring patterns of TFs. Furthermore, critical regulators for each stage were pinpointed by using centrality metrics.
Results
Constructing the trajectory of state transition of CD8+ T cells in the tumor microenvironment
We obtained 3,700 CD8+ T cells from 14 NSCLC patients.13 These cells were isolated from adjacent normal lung tissues (NTC), tumor tissues (TTC), or peripheral blood (PTC) (Figure S1A). In the original study, all T cells underwent unsupervised clustering and were well annotated as different states, including naive T cells, intermediate T cells, effector T cells, two clusters of pre-dysfunction T cells marked with GZMK and ZNF683, respectively, and dysfunction cells (Figure S1B).
In order to depict the relationship across CD8+ T cells in different states, we performed pseudo-time analysis to establish trajectories across cell states using the Slingshot algorithm.14 The inferred state transition trajectory contains two lineages, presenting a bifurcated structure from the naive state to dysfunction and to the effect state, respectively (Figure 1A). Both lineages started from the naive state and diverged after the intermediate state. Then, pre-dysfunction_GZMK and pre-dysfunction_ZNF683 states were sequentially located in lineage 1 (i.e., dysfunction lineage), which ended with the dysfunction state (Figure 1B). In contrast, lineage 2 (i.e., effect lineage) ended directly with the terminal effect state (Figure 1C). The two lineages were also observed in individual patients, indicating that the trajectory of T cell state transition was prevailing across different patients (Figures S2A and S2B).
Figure 1.

scRNA-seq revealed a bifurcated trajectory of CD8+ T cells
(A) Pseudo-time analysis of CD8+ T cells using Slingshot algorithm, resulting in two lineages (colored lines) in the trajectory. Cells on the trajectory are labeled with cell states, revealing a dysfunction lineage (red line) versus effect lineage (blue line). (B and C) Pseudo-time density distribution of each state on the dysfunction lineage (B) and on the effect lineage (C). (D) Expression levels of co-inhibitory receptors along pseudo-time of the dysfunction lineage. (E) Expression levels of effector molecules along pseudo-time of the effect lineage. (F and G) Tissues of origin for cells on the dysfunction lineage (F) and on the effect lineage (G). Points shaped by ellipse, triangle, and rectangle denote PTC, NTC, and TTC, which were short for CD8+ T cells from peripheral blood, adjacent normal tissues, and tumor tissues, respectively. (H and J) Pseudo-time distribution (H) and state composition (J) of cells from different origins of tissues on the dysfunction lineage. The lineage is divided evenly into 10 bins according to pseudo-time. The height of bars represents the number of cells from various tissues in each bin. (I and K) Pseudo-time distribution (I) and state composition (K) of cells from different origins of tissues on the effect lineage.
For each lineage, cells in different states showed different pseudo-time distributions as expected5 (Figures 1B and 1C), indicating the accuracy of the pseudo-time calculation. Moreover, genes that act at the early and late stages of T cell dysfunction showed pseudo-temporal kinetics along the dysfunction lineage that were highly consistent with expectations, with naive state markers (CCR7, SELL, and IL7R) active early in pseudo-time and co-inhibitory receptors (PDCD1, CTLA4, TIGIT, LAG3, and HAVCR2) active later (Figure 1D; Figure S2F), which confirmed the accuracy of the dysfunction lineage. Likewise, the expression of effector molecules, including GNLY, PRF1, GZMB, GZMA, and NKG7, increased along the pseudo-time of the effect lineage, further confirming the accuracy of the cell ordering along the effect lineage (Figure 1E; Figure S2G).
Projection of the tissue origin to the state transition trajectory showed the clear differences of tissue distribution in the two lineages (Figures 1F and 1G). To further clearly dissect the relationship of tissue origin and trajectory, we evenly split cells into 10 groups according to the pseudo-time and compared their tissue origin/cell state compositions. For the dysfunction lineage, peripheral blood-derived naive and intermediate T cells were dominant in the first four groups of cells, then sharply declined (Figures 1H and 1J). Meanwhile, pre-dysfunction T cells from the tumor and normal tissues gradually increased. Normal tissue-derived cells were decreased until cell group 6. In contrast, tumor-infiltrating T cells were further increased and dominant in the subsequent cell groups. For the effect lineage, blood-derived naive and intermediate T cells also dominated the start groups, and normal tissue-derived and tumor-infiltrating effect T cells were gradually increased along pseudo-time, without obvious bias to any tissue origin (Figures 1I and 1K). Combining tissue distribution and cell state composition along the trajectory, these results suggested a tendency that blood circulating naive T cells are recruited to the tumor, and persistent antigen stimulation drives intermediate state cells to pre-dysfunction cells and further differentiate to the dysfunctional state, which was similar to the CD8+ T cell differentiation model proposed by Andreatta et al.15 Another lineage differentiated from intermediate state cells directly to normal effect cells or intratumoral bystander cells.
Characterizing the dynamic genes specific to T cell dysfunction trajectory
To determine which genes regulated the progression of T cell dysfunction, we first attempted to identify genes that were differentially expressed along the dysfunction lineage by applying the tradeSeq algorithm.16 As a result, a total of 1,787 genes were identified to be dynamically regulated (false discovery rate [FDR] < 0.05; Figure 2A). In addition to well-known co-inhibitory receptors such as PDCD1, CTLA4, and HAVCR2, a lot of genes involved in T cell differentiation were identified (Figures 2B–2F). For instance, we observed a continuous increase in effector molecules, such as IFNG, PRF1, GZMB, and GZMH (Figure 2D), and the tumor necrosis factor family (Figure 2E), indicating that dysfunction T cells may not have completely lost their anti-tumor effector potential.17 Furthermore, genes related to lymphocyte migration were dynamically expressed along the lineage, including chemokines CXCL13, CCL3, XCL1, XCL2, CCL4, and CCL5 (Figure 2F).
Figure 2.

Dynamic gene analysis on the dysfunction lineage
(A) (Left) Heatmap showing average expression of dynamic genes in 10 bins of cells evenly divided according to pseudo-time. All of the dynamic genes were grouped into three clusters. (Middle) Expression trends along the dysfunction lineage for all genes in each cluster (gray lines) and for the cluster average (red lines). (Right) Representative enriched Gene Ontology (GO) terms for each cluster. (B–F) Dynamic changes in the expression of representative genes contained in dynamic genes, including known TFs (B), co-inhibitory receptors (C), effector molecules (D) tumor necrosis factor family (TNF family) (E), and chemokines (F). (G–I) Dynamic changes in the activities of function entities for cluster 2 (G), cluster 1 (H), and cluster 3 (I). The top 10 enriched terms for each cluster were displayed.
We next grouped genes with similar expression trends. Three distinct trends were observed according to the clustering results, with implications in different biological functions (Figure 2A). Genes in the cluster 1, which were downregulated early and gradually upregulated late along the pseudo-time, were highly enriched for biological processes related to T cell cytotoxicity, including cytokine signaling in the immune system and interferon signaling (Figure 2A; Figure S3A). Genes in cluster 2 with early activation and late downregulation along the pseudo-time were involved in ribosome, translational initiation protein targeting to membrane, and peptide chain elongation (Figure 2A; Figure S3B). However, the third cluster included many genes with no obvious consistent dynamic trend and fluctuated randomly over time, which played broad roles in lymphocyte activation, leukocyte cell-cell adhesion, and lymphocyte differentiation (Figure 2A; Figure S3C).
In order to further dissect temporal relationships of biological functions throughout the dysfunction process, we performed a high-resolution functional activity analysis (see Materials and methods). Signaling pathways related to cell translation were highly active in the naive stage and rapidly decreased from the intermediate active stage (Figure 2G), consistent with the recent proposal that immediate translation accelerated the T cell activation process.18 The decline in translation-related functions was accompanied by a continuous increase in cytotoxic effect and leukocyte cell migration-related functions. The cytotoxic function continued to be upregulated and reached the highest activity in the latest dysfunction stage (Figure 2H). However, the activity of T cell activation and cell migration reached the peak in the pre-dysfunction stage, and then slightly decreased in the subsequent stages (Figure 2I). Taken together, gene expression analysis along the tumor microenvironment-induced dysfunction lineage revealed the molecular dynamics of the T cell dysfunction process and depicted the sequential dynamics of biological events.
Constructing dynamic networks along the T cell dysfunction trajectory
State transition of T cells during the dysfunction process are governed by TFs and their associated cofactors, which work together to regulate target expression dynamically. To better understand the molecular mechanisms that drive the T cell dysfunction, we proposed a pipeline for dynamic regulatory network construction, with a variable-length sliding-window approach to split cells (see Materials and methods; Figure S4). In brief, we binned cells on the dysfunction lineage into four overlapped windows based on the pseudo-time distribution of cell states, with each window mainly composed of cells in two consecutive states. For instance, window 1 (W1) was mainly composed of cells in the naive and intermediate states, and W2 was mainly composed of cells in the intermediate and pre-dysfunction_GZMK states (Figure 3A; Figure S5A). Then, gene regulatory networks (GRNs) were constructed for each window separately to reflect the regulatory links controlling transition between the corresponding two states. Overall, 7,117 interactions between 82 regulators and 1,641 targets were inferred in at least one network (Figure 3A), and the size of each regulon varied from 1 to 229 genes (Figure S5B). W1, W2, and W3 had similar numbers of nodes and interactions, while the network of the last window, W4, contained relatively fewer nodes (1,143) and fewer interactions (1,886) (Figure S5C).
Figure 3.

Constructing and validating dynamic networks along the dysfunction lineage
(A) (Upper) Pseudo-time densities for cells in different states (naive, intermediate, pre-dysfunction_GZMK, pre-dysfunction_ZNF683, dysfunction). The cells in two consecutive adjacent states were windowed to construct the gene regulatory network (GRN), which reflected the transition between the two states. (Middle) The sub-networks of TFs in each window. For each network, nodes are colored by average expression in the corresponding window. The sizes of nodes were scaled by their numbers of target genes. (Lower) The lollipop plot displays the number of target genes for the top 10 regulators. (B) Overlap of regulators identified in different datasets for each state transition. The pie plot displays the proportions of regulators identified only in the core dataset (light blue) and identified in at least one independent dataset (blue). The significance was evaluated by a hypergeometric test. (C) The heatmap displays the scaled Jaccard index estimating overlap of targets for each TF in each window and that in the network without windowing. The Venn diagram displays the overlap of target genes of the TOX in network W4 and network without windowing. The number of target genes for top regulators in the two networks were listed, and curated TFs related to T cell dysfunction were colored red. (D) Receiver operating characteristic (ROC) curve and area under the ROC (AUC) depicting the performance on prediction of dysfunction-related regulatory factors for six GRN construction algorithms.
We also applied the dynamic GRN construction pipeline to another eight single-cell RNA sequencing (scRNA-seq) datasets (Table S1), involving NSCLC, melanoma, breast cancer, head and neck cancer, HCC, Merkel cell carcinoma, and squamous cell carcinoma. We used the SingleR19 method to map cell states across these datasets, and then applied the variable-length sliding window approach and GENIE3 to construct dynamic networks for each dataset (Figure S6; see Materials and methods). We compared the regulators of each state transition among these datasets. We found that there was significant overlap between the regulators in the core dataset and those in the independent datasets, with 85% (from naive to intermediate state), 74% (from intermediate to pre-dysfunction), and 63% (from pre-dysfunction to dysfunction) of the regulators having been identified in at least one additional dataset (Figure 3B, p = 2.10e−06, 2.69e−07, and 5.27e−08, respectively).
To illustrate advantages of using the variable-length sliding window approach in the dynamic GRN construction pipeline, we implemented the GENIE3 algorithm for all cells in the dysfunction lineage without windowing cells. We demonstrated that TFs identified without windowing displayed overlap of target genes with those of our approach (Figure 3C, left panel), indicating that the GRN constructed using all cells (GRN_all) could capture certain information across the whole lineage. However, GRN_all could be biased toward large cell subpopulations, with some loss of information from other cells. Indeed, these TFs mainly capture target genes in W3 (Figure 3C, left panel), indicating information bias from cells in W3, which had the largest number of cells. Moreover, we could not distinguish the actional stages of these TFs based solely on GRN_all. For instance, we identified 50 targets for TOX in GRN_all, among which 30 target genes were shared with those identified in W4 (Figure 3C, upper right panel). TOX is a key transcriptional regulator in the process from the pre-dysfunctional state to a late dysfunctional state,5 which could not be determined to play roles in the dysfunctional stage in GRN_all. In addition, TOX had lower rank in GRN_all than in W4 (Figure 3C, lower right panel). These results demonstrated advantages and necessity of the variable-length sliding-window approach.
To further estimate the influence of different GRN construction algorithms on the resulting GRNs, we chose the late dysfunction window, W4, to perform various algorithms for the reason that we could curate TFs regulating T cell dysfunction from the literature (Table S2). These algorithms included LEAP,20 PIDC,21 SCENIC,22 SCODE,23 and SINCERITIES.24 We did not observe performance promotion when choosing other algorithms (Figure 3D; Figure S7A) than GENIE3, which was used in this study. Although the SCENIC algorithm also implemented GENIE3 plus considering motif enrichment, it could reveal fewer regulators than other algorithms (Figure S7B), potentially resulting in a higher false negative. For instance, the well-known dysfunction-related regulators ID2 and TOX were totally missed in the GRN constructed using SCENIC. Thus, we finally chose the GENIE3 algorithm to construct GRNs.
Next, we extracted candidate regulators with significant differences of regulon activities between the two consecutive adjacent states in the window (Figure S7C; Table S3; see Materials and methods). Many well-known regulators of T cell state transition (Figure S7D) were detected. For instance, in the early regulatory network of W1, LEF1, MYC, PRDM1, and TCF7 were among the top differential TFs in the state transition. Previous studies demonstrated implications of LEF1, TCF7, and MYC in cell cycle regulation, transcription activation, and metabolic reprogramming upon antigen encounter,25 confirming their critical roles in early activation of T cells. Taken together, our computational analysis revealed the temporal dynamics of complex regulatory interactions during the dysfunction of T cells and highlighted the usefulness of our pipeline in characterizing the regulatory mechanisms during the process of state transition.
Substantial regulatory rewiring controls the state transition
To sophisticatedly characterize dynamics of regulatory interactions step by step during T cell dysfunction, we systematically compared the regulatory networks at different transition processes. We observed substantial overlap of nodes among different networks, with 44.4% present in all four networks (Figure 4A) and even higher (63.4%) for TFs (Figure 4B). However, through inspecting the edge overlaps across the four networks, we found a large number of edges specific in each window (Figure 4C). Furthermore, we calculated the Jaccard index (JI) of each TF present in any two networks to measure the overlap of its regulons. The average JI values between two consecutive windows (W1_W2, W2_W3, W3_W4) were 0.11, 0.14, and 0.12 (Figure 4D), respectively, indicating that TFs had substantially reconnected to different targets (i.e., “rewiring”) along the T cell trajectory. PRDM1 was upregulated in the first window and expressed sustainedly in the subsequent states (Figures 3B–3E). However, PRDM1 regulated specific genes in different windows, and it participated in window-specific functions. For example, PRDM1 played roles in interferon-γ-related functions in W1, while it participated in T cell activation and dephosphorylation for W3 and W4, respectively (Figure 4E). Another example was ID2, which was an important regulator for the formation of terminally differentiated T cells.26 We showed that ID2 was also rewiring across the four networks and exerted diverse roles (Figure 4F).
Figure 4.

Large-scale network rewiring during T cell dysfunction
(A and B) Venn diagram showing the overlap of nodes (A) or TFs (B) across the four windows. (C) UpSetR plot showing intersection of network edges across the four windows. The number of window-specific edges is marked in red. (D) Boxplots showing JI of TFs in any two windows. Significance of differences across different groups were assessed with an ANOVA test. (E and F) Regulatory rewiring of transcription factor PRDM1 (E) and ID2 (F). The first four groups of targets represent window-specific targets in W1, W2, W3, and W4, respectively, and the last group of genes contained targets that were shared by at least two windows. Representative functions of window-specific groups are shown below the genes. (G) Subnetworks of co-inhibitory receptors (TIGIT, LAG3, PDCD1, HAVCR2, and CTLA4) in each window.
T cell exhaustion is manifested by the high expression of co-inhibitory molecules. We extracted the subnetworks of co-inhibitory receptors in each window to investigate their regulatory rewiring (Figure 4G). The initial event was the activation of TIGIT by LYAR, KLF6, JUNB, and PRDM1 in the early stage (Figure 4G left panel), which was expressed significantly higher than other co-inhibitory receptors (Figures S8A and S8B). Meanwhile, LAG3 was activated by KLF6, SP100, and HOPX (Figure 4G, second panel), resulting in upregulation in the intermediate state (Figures S8A and S8C). Subsequently, PDCD1 and HAVCR2 were sequentially activated by PRDM1, IKZF3, STAT1, and ID2 in the networks of W2 and W3 (Figure 4G, second and third panels; Figure S8A). At last, CTLA4 was upregulated by TOX and RBPJ in the late dysfunction stage (Figure 4G, fourth panel; Figure S8A). The dynamic rewiring of co-inhibitory receptors showed that they were regulated by different TFs during the T cell dysfunction process, resulting in an activation cascade.
Dissecting the dynamic rewiring patterns of TFs
There were a large number of specific edges among the state transition networks, which was further confirmed by the Spearman correlation of TF-target pairs in four windows. We observed the highest correlation for regulatory pairs in their corresponding windows as expected (Figure 5A), and we also noted the gradual changes of the correlation across the windows. Fluctuations and rewiring of regulatory interactions among the windows during state transitions indicated that TFs might play dominant regulatory roles in certain stages. In order to elucidate the dominant regulating stages for each TF, we assigned it to the windows in which the number of targets of the TF was higher than the average number in all windows. A total of 11 regulation patterns were obtained, including 4 window-specific and 7 sustained regulatory patterns (Figures 5B and 5C). For instance, naive state-related TFs BACH2, LEF1, and MYC27, 28, 29 played specific roles in W1, while dysfunction-related TFs TOX and BATF30 were identified to play dominant roles in W4. In addition, some TFs played sustained dominant roles in two or three windows. For instance, TCF7 sustained to play roles in W1 and W2, consistent with its critical role in transition from naive to pre-dysfunction state.31 SP140, which was related to T cell dysfunction by regulating co-inhibitory receptors LAG3 and TIGIT in viral infection,32 played a regulatory role continuously from W1 to W3.
Figure 5.

Dynamic regulatory pattern of transcription factors
(A) Median Spearman correlation of TF-target pairs within each window calculated in the four windows respectively. The whiskers denoted the 25th percentile and 75th percentile values. The significance was evaluated by ANOVA test. (B) Heatmap showing numbers of targets for each dynamic TF normalized by its maximum number of targets. (C) Sankey diagram showing the relationship between networks W1–W4 and 11 regulatory patterns of TFs. TFs corresponding to each regulatory pattern are listed in the right table. (D–G) TF modules identified based on the Simpson index for each window. (Left) Heatmaps showing the Simpson index for TF pairs. (Right) Subnetworks of the corresponding TF modules with nodes colored by expression levels.
In order to explore the synergistic effects of TFs, we estimated regulatory similarities between TFs in each window based on the Simpson index33 and identified two modules for W1 and W2, respectively, and one module for W3 and W4, respectively (Figures 5D–5G; Figures S9A and S9B). Strikingly, TFs in the same module tended to have the same regulation pattern, especially the window-specific pattern. The module M1 in W1 contained TCF7, LEF1, MYC, SATB1, and BACH2, all of which played dominant roles in W1, except for TCF7 (Figure 5D), and this module was involved in eukaryotic translation elongation (Figure S9C). M2 in W2 contained W2-specific regulators ZBTB38, EOMES, and ZEB2 (Figure 5E), playing roles in cell killing and leukocyte activation involved in immune response (Figure S9D). M1 in W3 contained W3-specific regulators HOPX, ZNF683, and ID2, as well as two sustained regulators, which only regulated a small number of targets (Figure 5F), relating to cytokine signaling in the immune system and leukocyte activation involved in the immune response (Figure S9E). M1 in W4 contained TOX, ETV1, and RBPJ, which played dominant roles in W4 (Figure 5G), which was enriched in lymphocyte activation and T cell co-stimulation (Figure S9F).
Critical regulators contribute to T cell dysfunction
In order to further measure the importance of diverse TFs in the state transition networks, we established an integrated centrality metric by combining degree, closeness, betweenness, eigenvalue, and PageRank (see Materials and methods). The top 10 TFs with the highest centrality metrics in the four state transition networks are displayed in Figures 6A–6D. The set of critical regulators included some well-known regulators of mediating T cell state transition, including TCF7,31 EOMES,34 ID2,26 and TOX.35 Notably, TSC22D3 constantly played regulatory roles in W2, W3, and W4 (Figure 5C), and it was among the most important regulators in all three windows (Figures 6B–6D). Although TSC22D3 regulated different targets in different windows (Figure 6E), it could regulate some biological functions constantly over time, such as T cell activation and cell-cell adhesion (Figure 6F). Ayroldi et al.36 had reported that induction of TSC22D3 expression could contribute to the modulation of T cell activation and apoptosis. Nonetheless, we also observed window-specific functions for TSC22D3. For instance, it could regulate the cellular response to biotic stimulus specifically in W2, while regulating T cell proliferation specifically in W3 (Figure 6F).
Figure 6.

Critical regulators of the four networks during T cell dysfunction
(A–D) Ranks for TFs in the four networks based on the integrated centrality measure. For each network, the top 10 TFs are labeled and colored by their regulatory patterns. The arrow marks the critical TF TSC22D3 present in W2, W3, and W4. (E) Venn diagram denoting overlap of targets of TSC22D3 in W2, W3, and W4. (F) Enriched GO terms of TSC22D3-regulated genes in each window. The color showing the −log10 transformed p values of enrichment significance. (G) Violin plots showing expression levels of TSC22D3 in each cell state. Only cells with expression greater than 5 were used for visualization. (H–J) Boxplots displaying expression levels of TSC22D3 in pre-dysfunction and dysfunction states for hepatocellular carcinoma (H), melanoma (I), and colorectal cancer (J). The significance was evaluated by Wilcoxon test. Similarly, cells with low expression were not included for visualization, with a threshold of 5 for Smart-Seq2 datasets (GEO: GSE98638 and GSE108989) and 0.1 for the MARS-seq2 dataset (GEO: GSE123139). (K and L) Kaplan-Meier survival curves showing significant differences of survival probabilities for patient stratification based on TSC22D3 expression in GEO: GSE30219 (K) and GEO: GSE50081 (L).
Along the state transition timeline, we found that TSC22D3 showed gradually increased and then decreased expression, with highest expression in the pre-dysfunction state (Figure 6G). We further investigated the expression of TSC22D3 in another three scRNA-seq datasets of CRC,17, HCC,6 and melanoma37 and observed consistently significantly higher expression in the pre-dysfunction state than in the dysfunction state (Figures 6H and 6I), indicating its critical roles in pre-dysfunction of T cells. Previous studies reported that a higher proportion of pre-dysfunction state T cells was of benefit to patient survival.8,11 Here, we attempted to explore whether the expression of TSC22D3, a potential critical regulator of pre-dysfunction T cell state, was sufficient to predict prognosis of NSCLC patients. Based on two cohorts of NSCLC patients in GEO: GSE30219 and GSE50081, we found that higher expression of TSC22D3 was significantly associated with overall survival (Figures 6K and 6L). These results suggested that TSC22D3 could act as a critical regulator of T cell pre-dysfunction and was associated with clinical outcome of NSCLC patients.
Discussion
In this study, we combined trajectory inference with GRN construction based on single-cell transcriptomics to map the gene regulatory landscape of the cell state transition during the T cell dysfunction process. Through constructing the T cell state transition trajectory, we detected a high degree of transcriptional heterogeneity and expression dynamics specific in the lineage induced by the tumor microenvironment. Dynamic network analysis revealed candidate regulators and substantial network rewiring that control the state transition. We further optimized critical genes in each state transition and demonstrated that TSC22D3, with high centrality in multiple networks, was highly expressed in the pre-dysfunction state and correlated with the clinical outcome of tumor patients.
Under different cellular contexts, TFs may perform dramatic differential functions by changing their targets.38 The rewiring of regulatory interactions has been found to be a hallmark in state transition, and the altered regulatory programs generated by network rewiring have been reported to have strong phenotypic impacts.39 Along the T cell dysfunction process, we also observed substantial regulatory rewiring and revealed the altered regulatory programs. For instance, EOMES (Figure S5D) and PRDM1 (Figure 4E), which have been reported to modulate different targets with important context-specific function in acute and chronic infection,40 showed substantial rewiring across different T cell states. We also observed distinct functions for different targets of rewired TFs in specific states, suggesting the importance of network rewiring for controlling state transition. Importantly, our study dissected the rewiring of co-inhibitory receptors and depicted their activation cascade, which may facilitate the future identification of stage-specific targets in immunotherapy.8
Our analysis allowed a broad, unbiased investigation into dynamics regulatory mechanisms along the T cell dysfunction process in tumors. For example, consistent with published works,41 we identified TOX and BATF as critical regulators of the transition from the pre-dysfunction state to the dysfunction state. Our data identified RBPJ as a new potential regulator of CD8+ dysfunction cells, as RBPJ is upregulated after T cell activation, particularly at the peak of dysfunction, and RBPJ was synergistically targeting co-inhibitory receptors HAVCR2 and CTLA4 with TOX in the late stage of T cell dysfunction (Figure 4G). Moreover, another potential regulator, TSC22D3, was sustained to exert diverse functions after the pre-dysfunction state until the dysfunction state. TSC22D3 participated in the cell response to corticosteroid and glucocorticoid stimuli, consistent with a previous study,42 which demonstrated that endogenous glucocorticoid signaling can shape T cell differentiation from the naive to dysfunction state, suggesting that a regulatory cascade from glucocorticoid stimulation to the dysfunction state was mediated by TSC22D3. Survival analysis of TSC22D3 demonstrated its prognostic significance in NSCLC patients. Although we noticed wide expression of TSC22D3 in various immune cell types using the TISCH web resource43 (Figure S10A), we have normalized the expression levels of TSC22D3 by the geometric mean of CD3 gene expression levels (CD3D, CD3E, and CD3G) to only consider the relative expression of TSC22D3 in T cells in tumor samples. We also noticed the expression of TSC22D3 in CD4+ T cells, which could potentially confound the results. However, if the expression levels of TSC22D3 in T cells were positively correlated with those in CD8+ T cells/pre-dysfunction T cells, the expression level of TSC22D3 in the total T cells would be proportional to those in CD8+ T cells/pre-dysfunction T cells. Indeed, we observed a strong positive correlation between TSC22D3 expression levels in the total T cells and in CD8+ T cells/pre-dysfunction T cells (Figure S10B). Thus, the prognostic relationship of the relative expression of TSC22D3 in T cells could reflect the clinical impact of its expression in CD8+ T cells/pre-dysfunction T cells. In addition, ID2, ZNF683, HOPX, and IRF9 were among the most significantly upregulated TFs of network W3 (Figure 3A; Figures S7C and S7D). The dynamic expression of these TFs indicated that cells gradually acquire a long-term cytotoxicity and memory phenotype in the dysfunction process of transition,26,44, 45, 46 allowing pre-dysfunctional T cells to play sustained effector roles in tumors. Taken together, we think that this strategy for identifying potential regulators of cell trajectory holds promise and will facilitate the elucidation of complex transcriptional networks that control the differentiation of dysfunction T cells at various stages.
Materials and methods
Data collection and processing
We downloaded the scRNA-seq data of 14 patients from the GEO database under GEO: GSE99254.13 We extracted the count expression profile of CD8+ T cells except mucosal-associated invariant T (MAIT) cells, which had distinct T cell receptors (TCRs) and development processes relative to other CD8+ cells.13 After removing genes with a mean count less than 1, count normalization was performed by first dividing counts by the total counts in each cell, followed by multiplication with the median of the total counts across cells. Then, we performed log transformation for the expression profile. Finally, we retained a total of 12,306 protein-coding genes and 3,700 CD8+ T cells with well-annotated cell states, including 303 naive cells (CD8_C1-LEF1), 206 intermediate cells (CD8_C2-CD28), 1,192 effector T cells (CD8_C3-CX3CR1), 674 GZMK marked pre-dysfunction cells (CD8_C4-GZMK), 832 ZNF683 marked pre-dysfunction cells (CD8_C5-ZNF683), and 439 dysfunction cells (CD8_C6-LAYN).
CD8+ T cell state transition trajectory inference
To infer CD8+ T cell developmental trajectories, we used the Slingshot algorithm.14 Slingshot is a fast and robust method for branching trajectories inference, and it was shown to be among the top-performance methods in a recent benchmarking study.47 In short, Slingshot uses pre-exiting clusters to infer lineage hierarchies based on a minimal spanning tree, and align cells on a pseudo-time trajectory. Specifically, we first downloaded the cluster-specific signature genes13 and performed principal-component analysis (PCA) on all cells to keep the major biological variation among states. Then, we ran Slingshot on the top three principal components, with the naive state as the starting cluster. Using the Slingshot pipeline, we obtained the pseudo-time values and assigned branches of cells (Figure S2E). In addition, we performed the same processing flow of trajectory inference for each patient. After trajectory inference, we obtained three branches with branch 1 consisting of almost all pre-dysfunction and dysfunction T cells, branch 2 enriched with effector T cells, and branch “1,2” composed of naive and intermediate T cells (Figures S2F and S2G). In order to ensure the accuracy of the trajectory, we removed the ambiguous cells that did not belong to the enriched states from the corresponding branches.
Identification of dynamic genes along the trajectory of the state transition
We used tradeSeq16 version 1.2.1 to identify genes dynamically expressed along the trajectories of T cell dysfunction induced by the tumor environment. The differential analysis was restricted to genes with high quality, which passed three filtering criteria, that is, (1) average expression greater than 0.5, (2) ratio of expressed cells greater than 0.05, and (3) detected in more than 100 cells. For each gene, we fit a general additive model (GAM) with parameter K (number of knots) of six to model the relationships between gene expression and pseudo-time and tested for the significance of their associations using the associationTest function. We picked out the significant genes with FDR-corrected p values <0.05 as dynamic genes. Function enrichment analyses of dynamic genes were performed using Metascape.48
Functional activity analysis of gene sets along pseudo-time
We first determined the on/off binary state for each gene throughout pseudo-time using an hidden Markov model (HMM) approach as previously described.49 In brief, we divided pseudo-time into 40 bins and averaged the expression level of each gene within each bin. We assigned the averaged expression values to observed variables for HMM and extracted the most possible emission probabilities and transition probabilities using a Baum-Welch algorithm. Then, the Viterbi algorithm was applied to predict the on/off binary state for each gene. For each functional gene set, we averaged the binary states of all expressed genes within it in each bin, which were further normalized to represent the proportion of turned-on genes. Finally, the proportions of turned-on genes along pseudo-time were used to estimate activity transition of functional gene sets.
Construction of dynamic regulatory network for state transition
A number of computational methods have been developed to predict GRNs from single-cell gene expression data,50,51 but most of them ignore the time sequence of cells. In addition, the distribution of cells along the trajectory is not uniform, which makes the regulatory network biased toward cells in the high-density areas of pseudo-time. To avoid these biases, we combined the cell states and pseudo-times to re-divide the cells with a variable-length sliding-window approach and constructed regulatory networks across state transition stages. First, based on the pseudo-time density of cells in different states, we calculated the intersection of the density curves between two adjacent states, which was used as the boundary to split cells into multiple pseudo-time intervals. Then, every two consecutive intervals were regarded as a window. Therefore, cells on the dysfunction lineage were divided into 4 windows to represent different state transition stages. Each window is mainly composed of cells in two consecutive states. Then, we constructed a GRN for each window separately.
We first filtered genes in each window to remove genes that are expressed either at very low levels (average expression less than 1) or in too few cells (the number of detected cells less than 10 and the proportion of detected cells less than 0.05). Then, unsupervised GRNs were constructed using GENIE3,52 which was the top-performance method in the DREAM4 and DREAM5 GRN reconstruction challenges. GENIE3 takes advantage of the random forest (RF) machine learning algorithm, can deal with combinatorial and non-linear interactions, and is suitable for single-cell data.50 Briefly, it trains random forest models to predict the expression variance of each target gene and uses as input the expression of the TFs. Each model was then used to derive weights for the TFs. We extract weights higher than a pre-defined threshold, which was determined as the mean of all weights plus twice the standard deviation, as the high-confident regulatory links. Then, the rankings of all target models were aggregated to get a global ranking of all regulatory links. The links with weights greater than 0.02 were used to construct the GRN. Furthermore, we split the targets into positive- and negative-correlated targets (according to the Spearman correlations) to separate likely activated and repressed targets. Finally, only activated links were kept for the following step. The code of the pipeline is available at https://github.com/MinYan19940/DynamicGRNPipe.
Constructing networks for independent datasets
We downloaded eight datasets from GEO and ArrayExpress databases, involving seven cancer types. For GEO: GSE120575, ArrayExpress: E-MTAB-6149, and GEO:GSE123813, we extracted CD8+ T cells based on the original cell annotation. For GEO: GSE110868, GSE140228, GSE118056, GSE103322, and GSE127471, we extracted CD8+ T cells based on the expression of T cell markers. After quality control, we obtained 53,842 CD8+ T cells in total (Table S1). To obtain consistent cell states, we used a unified pipeline to annotate cells and construct the state transition trajectory. First, the Seurat R package was used to identify major cell clusters. The top 2,000 highly variable genes were generated and used to perform PCA. The first five principal components (PCs) were used for graph-based clustering (with parameter res = 0.3) to identify different clusters. Second, the SingleR R package was employed to map the clusters to the six cell states used in this study. In brief, we calculated the average expression of genes in each cluster. Then, taking the average expression profile of cell clusters as input, we used the core dataset (GEO: GSE99254) as the reference to map the clusters to a known cell state using SingleR (Figure S6A). Next, based on the mapped states of cells, we applied the Slingshot algorithm to construct the state transition trajectory and calculate the pseudo-time of cells (Figure S6B). Since the pre-dysfunction_GZMK state is missing in the ArrayExpress: E-MTAB-6149 and GEO: GSE110686 data and the pre-dysfunction_ZNF683 state is missing in the GEO: GSE103322 data, the pre-dysfunction_GZMK and pre-dysfunction_ZNF683 are not distinguished when constructing the regulatory network from pre-dysfunction to dysfunction or the intermediate to pre-dysfunction state. Finally, we used the cell states, pseudo-time, and dynamic gene expression profile as input for the pipeline to construct a dynamic regulatory network for each dataset. A hypergeometric test was used to evaluate the significance of overlap of regulators in the independent datasets and the core dataset.
Comparing with other GRN reconstruction algorithms
To assess the impact of different GRN reconstruction algorithms on the identification of dysfunction-related TFs, we also implemented other algorithms to construct GRNs in the fourth window, including LEAP,20 PIDC,21 SCENIC,22 SCODE,23 and SINCERITIES.24 For SCENIC, we employed the Docker image of pySCENIC, and for the others, we employed the corresponding Docker images from the BEELINE pipeline.53 To compare the performance of these algorithms, we performed receiver operating characteristic (ROC) analyses by curating T cell dysfunction-related TFs from the literature (Table S2). ROC analyses were performed utilizing the plotROC package.54
Refining candidate regulators during state transition
To refine candidate regulators in each window, we first estimated the average expression of each TF regulon in cells, corresponding to the two states in each window. Then, we examined the differential significance of regulon activity between the two states using a Wilcoxon rank-sum test. p values were then corrected for the multiple testing problem according to the FDR approach. TFs with FDR <0.05 were considered as candidate regulators during state transition in the window.
Measuring node centrality
The node centrality metrics were employed to measure the importance of TFs in the GRNs. Here, we used degree, closeness, betweenness, eigenvalue, and PageRank to evaluate the centrality of nodes.55 Then, a rank aggregation method (RRA)56 was applied to integrate all centrality metrics. Finally, the rank integration score for each TF is calculated as –log10(p value) to indicate the final centrality measure.
Survival analysis of TSC22D3 in NSCLC patients
We collected two datasets from GEO (GEO: GSE30219 and GSE50081) to assess association of TSC22D3 expression with clinical outcome of NSCLC patients. To correct for the effect of T cell infiltration levels in tumor samples, we normalized the expression of TSC22D3 by geometric mean of the expression levels of CD3D, CD3E, and CD3G.35 Then, we stratified NSCLC patients into two groups according to the third quartile of the normalized TSC22D3 expression levels. Kaplan-Meier survival curves were plotted using the survminer package.57 A log-rank test was performed to examine the significance of difference between survival probabilities between the two groups.
Statistical analysis
A hypergeometric test was used to identify significantly enriched T cell states on each branch. A Wilcoxon rank-sum test was used to examine the significance of differences between the expression of co-inhibitory receptors, as well as the differences between the expression of critical regulators in the pre-dysfunction and dysfunction states. A one-way ANOVA was used to test the dynamic of the Spearman correlation of TF-target pairs across four windows.
Acknowledgments
This work was supported in part by the National Key R&D Program of China (2018YFC2000100), the National Natural Science Foundation of China (61873075, 32070673, 31871336, and 31900478), the Heilongjiang Provincial Natural Science Foundation (YQ2019C012), the Heilongjiang Postdoctoral Foundation (LBH-Q18099), and the Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2017059).
Author contributions
X.L., Y.X., and Y.Z. conceived and designed the study. M.Y. and J.H. designed the framework. M.Y., J.H., H.Y., and L.X. analyzed the data and implemented the methodology. X.L. and Y.X. revised the manuscript. G.L. and Z.J. acquired the data. J.Z. and B.P. organized figures. B.P. and Y.P. provided constructive discussions. Y.Z. and Y.P. helped in interpreting the results. M.Y., J.H., and H.Y. drafted the manuscript. All authors read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.011.
Contributor Information
Yunpeng Zhang, Email: zhangyp@hrbmu.edu.cn.
Yun Xiao, Email: xiaoyun@ems.hrbmu.edu.cn.
Xia Li, Email: lixia@hrbmu.edu.cn.
Supplemental information
References
- 1.Pauken K.E., Wherry E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Leun A.M., Thommen D.S., Schumacher T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer. 2020;20:218–232. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 7.Blank C.U., Haining W.N., Held W., Hogan P.G., Kallies A., Lugli E., Lynn R.C., Philip M., Rao A., Restifo N.P. Defining “T cell exhaustion”. Nat. Rev. Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kallies A., Zehn D., Utzschneider D.T. Precursor exhausted T cells: Key to successful immunotherapy? Nat. Rev. Immunol. 2020;20:128–136. doi: 10.1038/s41577-019-0223-7. [DOI] [PubMed] [Google Scholar]
- 9.Clemente C.G., Mihm M.C., Jr., Bufalino R., Zurrida S., Collini P., Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Savas P., Virassamy B., Ye C., Salim A., Mintoff C.P., Caramia F., Salgado R., Byrne D.J., Teo Z.L., Dushyanthen S., Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 11.Miller B.C., Sen D.R., Al Abosy R., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiqui I., Schaeuble K., Chennupati V., Fuertes Marraco S.A., Calderon-Copete S., Pais Ferreira D., Carmona S.J., Scarpellino L., Gfeller D., Pradervand S. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 13.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 14.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreatta M., Corria-Osorio J., Müller S., Cubas R., Coukos G., Carmona S.J. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 2021;12:2965. doi: 10.1038/s41467-021-23324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van den Berge K., Roux de Bézieux H., Street K., Saelens W., Cannoodt R., Saeys Y., Dudoit S., Clement L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020;11:1201. doi: 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 18.Wolf T., Jin W., Zoppi G., Vogel I.A., Akhmedov M., Bleck C.K.E., Beltraminelli T., Rieckmann J.C., Ramirez N.J., Benevento M. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 2020;21:927–937. doi: 10.1038/s41590-020-0714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Specht A.T., Li J. LEAP: Constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics. 2017;33:764–766. doi: 10.1093/bioinformatics/btw729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan T.E., Stumpf M.P.H., Babtie A.C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017;5:251–267.e3. doi: 10.1016/j.cels.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto H., Kiryu H., Furusawa C., Ko M.S.H., Ko S.B.H., Gouda N., Hayashi T., Nikaido I. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-seq during differentiation. Bioinformatics. 2017;33:2314–2321. doi: 10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papili Gao N., Ud-Dean S.M.M., Gandrillon O., Gunawan R. SINCERITIES: Inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics. 2018;34:258–266. doi: 10.1093/bioinformatics/btx575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willinger T., Freeman T., Herbert M., Hasegawa H., McMichael A.J., Callan M.F. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J. Immunol. 2006;176:1439–1446. doi: 10.4049/jimmunol.176.3.1439. [DOI] [PubMed] [Google Scholar]
- 26.Kaech S.M., Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsukumo S., Unno M., Muto A., Takeuchi A., Kometani K., Kurosaki T., Igarashi K., Saito T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA. 2013;110:10735–10740. doi: 10.1073/pnas.1306691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preston G.C., Sinclair L.V., Kaskar A., Hukelmann J.L., Navarro M.N., Ferrero I., MacDonald H.R., Cowling V.H., Cantrell D.A. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 2015;34:2008–2024. doi: 10.15252/embj.201490252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing S., Li F., Zeng Z., Zhao Y., Yu S., Shan Q., Li Y., Phillips F.C., Maina P.K., Qi H.H. Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol. 2016;17:695–703. doi: 10.1038/ni.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leavy O. Exhaustion through BATF. Nat. Rev. Immunol. 2010;10:747. doi: 10.1038/nri2881. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z., Ji Z., Ngiow S.F., Manne S., Cai Z., Huang A.C., Johnson J., Staupe R.P., Bengsch B., Xu C. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. 2019;51:840–855.e5. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumida T.S., Dulberg S., Schupp J., Stillwell H.A., Axisa P.P., Comi M., Lincoln M., Unterman A., Kaminski N., Madi A. Type I interferon transcriptional network regulates expression of coinhibitory receptors in human T cells. bioRxiv. 2020 doi: 10.1101/2020.10.30.362947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuxman Bass J.I., Diallo A., Nelson J., Soto J.M., Myers C.L., Walhout A.J. Using networks to measure similarity between genes: association index selection. Nat. Methods. 2013;10:1169–1176. doi: 10.1038/nmeth.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J., He Y., Hao J., Ni L., Dong C. High levels of Eomes promote exhaustion of anti-tumor CD8+ T cells. Front. Immunol. 2018;9:2981. doi: 10.3389/fimmu.2018.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim K., Park S., Park S.Y., Kim G., Park S.M., Cho J.W., Kim D.H., Park Y.M., Koh Y.W., Kim H.R. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12:22. doi: 10.1186/s13073-020-00722-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayroldi E., Migliorati G., Bruscoli S., Marchetti C., Zollo O., Cannarile L., D’Adamio F., Riccardi C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor κB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- 37.Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J.B.A.G., Blank C.U. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lou S., Li T., Kong X., Zhang J., Liu J., Lee D., Gerstein M. TopicNet: A framework for measuring transcriptional regulatory network change. Bioinformatics. 2020;36(Suppl_1):i474–i481. doi: 10.1093/bioinformatics/btaa403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H.J., Osteil P., Humphrey S.J., Cinghu S., Oldfield A.J., Patrick E., Wilkie E.E., Peng G., Suo S., Jothi R. Transcriptional network dynamics during the progression of pluripotency revealed by integrative statistical learning. Nucleic Acids Res. 2020;48:1828–1842. doi: 10.1093/nar/gkz1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doering T.A., Crawford A., Angelosanto J.M., Paley M.A., Ziegler C.G., Wherry E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mann T.H., Kaech S.M. Tick-TOX, it’s time for T cell exhaustion. Nat. Immunol. 2019;20:1092–1094. doi: 10.1038/s41590-019-0478-y. [DOI] [PubMed] [Google Scholar]
- 42.Cannarile L., Delfino D.V., Adorisio S., Riccardi C., Ayroldi E. Implicating the role of GILZ in glucocorticoid modulation of T-cell activation. Front. Immunol. 2019;10:1823. doi: 10.3389/fimmu.2019.01823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun D., Wang J., Han Y., Dong X., Ge J., Zheng R., Shi X., Wang B., Li Z., Ren P. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021;49(D1):D1420–D1430. doi: 10.1093/nar/gkaa1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braun J., Frentsch M., Thiel A. Hobit and human effector T-cell differentiation: The beginning of a long journey. Eur. J. Immunol. 2015;45:2762–2765. doi: 10.1002/eji.201545959. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y., Zhang W. The role of HOPX in normal tissues and tumor progression. Biosci. Rep. 2020;40 doi: 10.1042/BSR20191953. BSR20191953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huber M., Suprunenko T., Ashhurst T., Marbach F., Raifer H., Wolff S., Strecker T., Viengkhou B., Jung S.R., Obermann H.L. IRF9 prevents CD8+ T cell exhaustion in an extrinsic manner during acute lymphocytic choriomeningitis virus infection. J. Virol. 2017;91:e01219-17. doi: 10.1128/JVI.01219-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saelens W., Cannoodt R., Todorov H., Saeys Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019;37:547–554. doi: 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin J., Berg D.A., Zhu Y., Shin J.Y., Song J., Bonaguidi M.A., Enikolopov G., Nauen D.W., Christian K.M., Ming G.L., Song H. Single-cell RNA-seq with Waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S. ppcor: An R package for a fast calculation to semi-partial correlation coefficients. Commun. Stat. Appl. Methods. 2015;22:665–674. doi: 10.5351/CSAM.2015.22.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pratapa A., Jalihal A.P., Law J.N., Bharadwaj A., Murali T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods. 2020;17:147–154. doi: 10.1038/s41592-019-0690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sachs M.C. plotROC: A tool for plotting ROC curves. J. Stat. Softw. 2017;79:2. doi: 10.18637/jss.v079.c02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou S., Huang Y.E., Liu H., Zhou X., Yuan M., Hou F., Wang L., Jiang W. Single-cell RNA-seq dissects the intratumoral heterogeneity of triple-negative breast cancer based on gene regulatory networks. Mol. Ther. Nucleic Acids. 2021;23:682–690. doi: 10.1016/j.omtn.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolde R., Laur S., Adler P., Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kassambara A., Kosinski M., Biecek P. 2021. survminer: Drawing survival curves using “ggplot2”. R package version 0.4.9.https://cran.r-project.org/web/packages/survminer/index.html [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.