

Abstract
Azoospermia patients who carry a monogenetic mutation that causes meiotic arrest may have their biological child through genetic correction in spermatogonial stem cells (SSCs). However, such therapy for infertility has not been experimentally investigated yet. In this study, a mouse model with an X-linked testis-expressed 11 (TEX11) mutation (Tex11PM/Y) identified in azoospermia patients exhibited meiotic arrest due to aberrant chromosome segregation. Tex11PM/Y SSCs could be isolated and expanded in vitro normally, and the mutation was corrected by clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated endonuclease 9 (Cas9), leading to the generation of repaired SSC lines. Whole-genome sequencing demonstrated that the mutation rate in repaired SSCs is comparable with that of autonomous mutation in untreated Tex11PM/Y SSCs, and no predicted off-target sites are modified. Repaired SSCs could restore spermatogenesis in infertile males and give rise to fertile offspring at a high efficiency. In summary, our study establishes a paradigm for the treatment of male azoospermia by combining in vitro expansion of SSCs and gene therapy.
Keywords: azoospermia, gene therapy, male infertility, meiotic arrest, spermatogonial stem cells, testis-expressed 11
INTRODUCTION
Male infertility is a complex pathological condition affecting around 7% of men of reproductive age.1 Usually, infertile men can have their biological child through assisted reproductive technology (ART), such as intracytoplasmic sperm injection (ICSI), because these patients still have the ability to produce a small number of functional sperms for ART.2 In contrast, it is impossible to restore fertility in men with nonobstructive azoospermia (NOA, approximately 10%–15% of all infertile males),3 the most clinically severe form of infertility which is defined by a complete absence of spermatozoa in the ejaculate and testis,4 through the currently available ARTs. Genetic studies reveal that NOA patients are at the highest risk of being carriers of genetic anomalies.1 However, only a small fraction of NOA patients (around 20%) have established causal genetic diagnoses, including chromosomal aberrations (15%) and Y-chromosome azoospermia factor deletions.5
Recent efforts on unraveling the genetic causes for NOA through genome-wide approaches have led to the identification of single-gene mutations that account for spermatogenetic failure.5–13 NOA patients carrying monogenic mutations causing spermatogenic failure14,15 may have substantial amount of functional spermatogonial stem cells (SSCs) in their testis.16 SSCs can self-renew and differentiate into mature sperm in the seminiferous tubules, and can also be long-term expanded in vitro without losing their capacity to produce functional gametes in vivo after transplantation into recipient testis.16,17 Most importantly, the availability of SSCs offers the prospect for correction of a disease-causing genetic mutation by clustered regularly interspaced short palindromic repeats–CRISPR-associated endonuclease 9 (CRISPR-Cas9)-mediated homology-directed repair (HDR),18–20 which has been shown to be effective in repairing a cataract-causing genetic mutation in the SSCs isolated from a cataract mouse model.21 We thus reasoned that successful gene editing in SSCs carrying an azoospermia-causing mutation may resume the spermatogenesis of NOA patients. Moreover, ex vivo gene therapy in SSCs allows for selection of SSC lines with repaired gene and without unexpected mutations for transplantation through derivation of SSC lines from single-cell expansion followed by whole-genome sequencing.21 Therefore, gene therapy in cultured SSCs may potentially provide a chance for NOA patients with an identified single mutation to have their genetically related child without any unwanted genomic changes. However, the feasibility of infertility treatment by combined application of in vitro expansion of SSCs and gene therapy has not been investigated yet. In this study, we establish a strategy of patient-derived mutation xenocorrection (PDMX) to verify and correct a monogenic mutation that causes human azoospermia in mice.
We chose TEX11 gene to develop the PDMX model because TEX11 mutations are recently revealed in NOA patients9,10,22 and Tex11-mutant mice resemble the patients’ phenotypes of spermatogenic failure induced by meiotic arrest.23,24 The PDMX model basically includes the following steps: (1) generation of mouse models carrying NOA patient-originated TEX11 mutations, (2) analysis of testicular phenotypes of males, (3) isolation and in vitro culture of SSCs, (4) correction of the genetic defect by CRISPR-Cas9-mediated HDR, (5) derivation of repaired SSC lines through single-cell expansion, (6) off-target effect analysis by whole-genome sequencing, (7) transplantation of the repaired SSCs into recipient testis to restore spermatogenesis, and (8) isolation of haploid gametes for fertility analysis. Here, we report the successful correction of an azoospermia patient-originated TEX11 mutation and restoration of spermatogenesis in mice.
MATERIALS AND METHODS
Animal use and care
All animal procedures were performed under the ethical guidelines of the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
Derivation of SSCs
To derive SSC lines, we sorted cells from testes of 8- to 10-day-old male mice using CD146. CD146+ cells were seeded on the mouse embryonic fibroblast (MEF) feeder cells and cultured with the modified SSC medium according to a previously reported protocol.
The culture medium consisted of StemPro-34 SFM (Invitrogen, Carlsbad, CA, USA) supplemented with StemPro supplement (Invitrogen), 25 μg ml-1 insulin, 100 μg ml-1 transferrin, 60 μmol l-1 putrescine, 30 nmol l-1 sodium selenite, 6 mg ml-1 D-(+)-glucose, 30 μg ml-1 pyruvic acid, 1 μl ml-1 dl-lactic acid (Sigma, Saint Louis, MO, USA), 5 mg ml-1 bovine serum albumin (Sigma), 2 mmol l-1 L-glutamine (Millipore, Bedford, MA, USA), β-mercaptoethanol (Millipore), minimal essential medium vitamin solution (Invitrogen), nonessential amino acid solution (Millipore), penicillin/streptomycin (Invitrogen), 0.1 mmol l-1 ascorbic acid, 10 μg ml-1 d-biotin (Sigma), 20 ng ml-1 recombinant human epidermal growth factor (Invitrogen), 10 ng ml-1 human basic fibroblast growth factor (Invitrogen), 10 ng ml-1 recombinant human glial cell line-derived neurotrophic factor (GDNF; Invitrogen), and 1% fetal bovine serum (FBS; ES cell-qualified, Invitrogen). The cells were maintained at 37°C in an atmosphere of 5% CO2 in air. The culture medium was changed every 2–4 days. All the cultured cells were tested to ensure that they are mycoplasma free.
Plasmid construction
The CMV-mCherry-pA cassette was inserted into pX330 to get the pX330-mcherry as described previously.18 The single-guide RNAs (sgRNAs) targeting Tex11 genes were synthesized, annealed, and ligated to the pX330-mcherry plasmid that was digested with Bbs I. All the inserted sequences of plasmids were validated by Sanger sequencing. Primers used in this study are listed in Supplementary Table 1.
Supplementary Table 1.
Primers used in this study
| Primer name | Sequence (5’- 3’) | Application |
|---|---|---|
| GAPDH-QF | CACTCTTCCACCTTCGATGC | Real-time PCR |
| GAPDH-QR | CTCTTGCTCAGTGTCCTTGC | |
| Tex11-QF | TCCTACACATGTTTTCACTCGG | |
| Tex11-QR | CTGACCACCTGTAAAGCTCTG | |
| Sohlh2-QF | TCAAGGGGAGGAAGAGCGAT | |
| Sohlh2-QR | TGTAACCTGGGCCATAATGG | |
| Id4-QF | GCCGTGAACAAGCAGGGTGA | |
| Id4-QR | CTTGGAATGACAAGACGAGACG | |
| Stra8-QF | ACAACCTAAGGAAGGCAGTTTAC | |
| Stra8-QR | GACCTCCTCTAAGCTGTTGGG | |
| Kit-QF | CTAGCCAGAGACATCAGGA | |
| Kit-QR | CCATAGGACCAGACATCAC | |
| SgRNA-IVT-R | AAAAGCACCGACTCGGTGCCAC | Amplification of sgRNA transcription templates from PX330-mCherry |
| Tex11-IVT-F | TAATACGACTCACTATAGGGAATATGCTGATGCCCTACACGTTTTAGAGCTAGAAATAG | |
| Tex11-check F | ACTTAGGCTTCAAGTAAGTAGGCA | Genotyping of Tex11 in mice and SSCs |
| Tex11-check R | TCACCTAAGTGCCACAGCAA | |
| Tex11-HDR-F | GACAGGAAGCCTCAACAGGG | Amplification of repair donor for SSCs transfection |
| Tex11-HDR-R | TGCTTCAGTGACAGGCCATT | |
| Tex11 sgRNA-1F | CACC TTAGGTCCAAAAATATGCTT | Construction of sgRNA plasmid for SSCs transfection |
| Tex11 sgRNA-1R | AAACAAGCATATTTTTGGACCTAA | |
| Tex11 sgRNA-2F | CACC TATGCTTTGGTACCCTACAC | |
| Tex11 sgRNA-2R | AAACGTGTAGGGTACCAAAGCATA |
SSCs: spermatogonial stem cells; HDR: homology-directed repair; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; sgRNA: single-guide RNA; Tex11: testis-expressed 11.
Lipofectamine transfection of SSCs and cell sorting
Before lipofectamine transfection, SSCs were trypsinized, centrifuged, and re-suspended with Opti-MEM (Invitrogen) to a new plate. Thirty minutes later, the Opti-MEM was gently replaced with SSC medium. The pX330-mcherry plasmids harboring the corresponding sgRNAs and oligo DNA were transfected into SSCs with lipofectamine 3000 according to the manufacturer’s instruction. Ten hours to 12 h later, the supernatant was aspirated and fresh SSCs were added.
Twenty-four hours after transfection, the SSCs expressing mCherry were sorted with flow cytometry (Beckman Moflo Astrios, Fort Collins, CO, USA), and one mCherry-positive cell was deposited into each well of the 96-well plates. After 3–4 weeks, SSC clones were trypsinized and expanded for genotyping.
Transplantation of SSCs
For SSC transplantation, busulfan (40 mg kg-1)-treated 4-week-old mT/mG (B6D2F1 background) or 14–21-day-old Kitw/Kitwv male mice were used as the recipient mice. Approximately 8 μl (2 × 104 cells per μl) of the donor cell suspensions was injected into the seminiferous tubules of an mT/mG testis, and 2 μl was introduced into the Kitw/Kitwv testis through the efferent duct. The mice were sacrificed 2 months later, and the testes were examined for further analysis.
Round spermatid injection (ROSI)
Metaphase II-arrested oocytes were collected from superovulated B6D2F1 females (8–10 weeks) and cumulus cells were removed using hyaluronidase. After decapsulation and digestion, the testicular cell suspensions were filtered through a 40-m filter (Millipore). Then, the suspensions were suspended with PBS containing 8% FBS. The haploid cells from the testes were sorted according to a reported protocol. First, the cell suspensions were incubated with the Hoechst 33342 (5 μg ml-1) at 37°C for 10 min. After washing with PBS/FBS twice, haploid cells were sorted with flow cytometry. The round spermatids were then injected into the oocytes in a droplet of HEPES-CZB medium containing 5 μg ml-1 cytochalasin B (CB) using a blunt Piezo-driven pipette. The injected oocytes were activated by treatment with SrCl2 for 5–6 h and maintained in the potassium simplex optimization medium (KSOM) medium at 37°C under 5% CO2 in air. Two-cell embryos were transferred into the oviducts of pseudopregnant Institute of Cancer Research (ICR) females. Live fetuses were born on day 19.5 of gestation.
Purification and Sanger sequencing of genomic DNA
Genomic DNA was isolated using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China), followed by PCR amplification for Sanger sequencing. For blunting cloning sequencing, the targeted sequence was amplified from the genome by high-fidelity DNA polymerase and then purified by the Universal DNA Purification Kit (TIANGEN). The editing efficiency was calculated through blunting cloning (TransGen Biotech, Beijing, China) by picking up more than 20 Escherichia coli clones for analysis. Primers used in this study are listed in Supplementary Table 1.
Bisulfite sequencing
Bisulfite sequencing was performed using the EZ DNA methylation kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s manual. The PCR products were cloned into pMD18T TA cloning vector (Takara, Tokyo, Japan), and individual clones were then sequenced for subsequent analysis. Primers used in this study are listed in Supplementary Table 1.
RNA extraction and real-time quantitative PCR
Total RNA was isolated from the cells using TRIzol reagent (Invitrogen). The cDNA was obtained from about 1 μg RNA with reverse transcription reaction by the ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). Real-time quantitative PCR reactions (rtqPCR) were performed on a Bio-Rad CFX96 using the SYBR Green Mix (TOYOBO) in triplicate. All the gene expression levels were normalized to the internal standard gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Primers used in this study are listed in Supplementary Table 1.
Histological and immunostaining analysis
The testis, epididymis, and ovary were fixed in 4% paraformaldehyde overnight at room temperature; dehydrated with different concentration gradients of alcohol; and embedded in paraffin and sectioned. Paraffin sections on slides were dewaxed in xylene and rehydrated in different concentration gradients of alcohol, followed by staining with hematoxylin and eosin.
Library preparation for whole-genome sequencing (WGS)
A total amount of 0.2 μg DNA per sample was used as input material for the DNA library preparations. Sequencing library was generated using NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) following the manufacturer’s recommendations, and index codes were added to each sample. Briefly, genomic DNA sample was fragmented by sonication to a size of 350 bp. Then, DNA fragments were end polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing, followed by further PCR amplification. After PCR products were purified by AMPure XP system (Beckman Coulter, Beverly, MA, USA), DNA concentration was measured by Qubit® 3.0 Fluorometer (Invitrogen), and libraries were analyzed for size distribution by NGS3K/Caliper and quantified by real-time PCR (3 nmol l-1).
Library preparation for transcriptome sequencing
A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB) following the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample.
Briefly, mRNA was purified from total RNA using poly-T-oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First-Strand Synthesis Reaction Buffer (×5). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 250–300 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Brea, CA, USA). Then, 3 μl USER Enzyme (NEB) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. Then, PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. At last, PCR products were purified and library quality was assessed on the Agilent Bioanalyzer 2100 system.
WGS data analysis
Raw reads were filtered by fastp (version 0.20.0) to acquire clean reads, and then mapped to mouse genome (mm10) using BWA tool with default parameters. For single-nucleotide polymorphism (SNP) and Indel calling, data preparation was executed according to the GATK best practices with GATK version 4.1.5.0, and well-prepared bam files were subjected to Mutect2 for SNP calling. Then, the resultant SNPs were filtered by hard filters with parameters, namely DP <10, QD <2.0, QUAL <30.0, SOR >3.0, FS >60.0, Q <40.0, MQRankSum <-12.5, and ReadPosRankSum <-8.0. Indels were filtered with parameters such as DP <10, QD <2.0, QUAL <30.0, ReadPosRankSum <-20.0, and FS >200.0. Passed SNPs and Indels were filtered by allele DP >10, minor allele frequency >0.35 for heterozygous SNPs, allele frequency >0.85 for homozygous SNPs. All SNPs and Indels were annotated by ANNOVAR (October 24, 2019). All primers used to confirm called SNPs and Indels are generated by Primer3 program. All predicted off-target sites were calculated by Cas-OFFinder (mouse genome: mm10) with at most five mismatches. And overlapped all called SNPs within predicted off-target sites (within ±20 bp) using Excel. The top predicted sites were examined by PCR (primers used were generated by Primer3) and sequencing.
Production of Cas9 mRNA and sgRNAs
First, pX330 plasmid including T7 promoter was linearized by Not I. The linearized templates were purified and in vitro transcribed with mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies, Carlsbad, CA, USA). SgRNAs with T7 promoter were amplified by PCR and in vitro transcribed using MEGAshortscript™ T7 kit (Life Technologies). After transcription, the Cas9 mRNA and the sgRNAs were purified with MEGAclear™ kit (Life Technologies) according to the manufacturer’s instructions.
Zygote injection
C57BL/6 female mice were superovulated and mated with the male C57BL/6 mice. For embryo injection, donor oligo (100 ng μl-1) was mixed with Cas9 mRNAs (100 ng μl-1) and sgRNA (50 ng μl-1) and injected into the embryos. The injected embryos were cultured in KSOM until 2-cell embryonic stage and transferred into pseudopregnant female mice.
The sequences of injected sgRNA were as follows: AATATGCTGATGCCCTACAC, oligo donor: GTGAAT TTTGGAAAAATCTATGACTTTGTTGTTTTTTTTTAACTTTA GGTCCAAAAATATGCTTTGGTACCCTACACTGGTACAGTTAT TCTCTGAAGTTGTATGAGTATGATAAAGCAGATCTGGATTT.
Material availability
Plasmids and mouse strains generated in this study are available from the Lead Contact.
Data accession
The raw data of RNA-seq and processed data of bulk RNA-seq and WGBS in this study were deposited at Gene Expression Omnibus (GEO) with accession numbers GSE158977, 158978, and 158979.
RESULTS
Male mice carrying an azoospermia patient-originated TEX11 mutation display meiotic arrest
TEX11 mutations have been identified in patients with azoospermia9,10,22 recently. To test the PDMX model, we chose a frame shift mutation in exon 16 (1258Ins [TT]) revealed in an azoospermic family.10 This mutation results in a stop codon at the 431st amino acid (Supplementary Figure 1a (355.4KB, tif) ), leading to a truncated protein TEX11 without the final 948 amino acids. Histological analysis of the patient’s testis biopsy indicated meiotic arrest at the pachytene stage,10 suggesting the possibility of existing SSCs in the testes. We first introduced the patient-derived mutation into the mouse genome through co-injection of Cas9 mRNA, sgRNA targeting exon 16, and exogenous single-stranded DNA oligo carrying the mutation into zygotes (C57BL/6 background) according to a previous report18 (Supplementary Figure 1b (355.4KB, tif) ). Of 150 injected zygotes, a total of 31 live pups were born (Supplementary Table 2). DNA sequencing analysis revealed that six of them were carrying knockin mutation at the targeted site in exon 16 (Supplementary Figure 1c (355.4KB, tif) ), which were used as founders for further analysis. All founders displayed normal development. However, after long-term breeding of founders with wild-type mice, males failed to give rise to any births, while female heterozygous-mutant founders displayed reproductive ability and could have progeny normally. We dissected ovaries and testes from founders and observed a similar size in mutant ovary but significant smaller size in mutant testes (Supplementary Figure 1d (355.4KB, tif) and 1f (355.4KB, tif) ). Histological analysis indicated normal gametogenesis in mutant ovary (Supplementary Figure 1e (355.4KB, tif) ) but meiotic arrest in mutant testis and no sperm in epididymis (Supplementary Figure 1g (355.4KB, tif) ). These results recapitulate the observations in azoospermic family, in which the patient carried TEX11 mutation inherited from his heterozygous-mutated mother.10
Supplementary Table 2.
Generation of mice carrying Tex11 mutations via direct clustered regularly interspaced short palindromic repeat/associated endonuclease 9 cytoplasmic injection
| Gene | Cas9/sgRNA (ng µl−1) | Oligo Donor (ng µl−1) | Number of injected zygotes | Number of 2-cell embryos | Number of newborn pups (dead) | Gender | Alive pups | Homo | Het |
|---|---|---|---|---|---|---|---|---|---|
| Tex11 | 100/50 | 100 | 150 | 131 | 33 (2) | Male | 17 | 2 | - |
| Female | 14 | 0 | 4 |
sgRNA: single-guide RNA; Tex11: testis-expressed 11
We thus set up a breeding system to obtain Tex11-mutant males (hereafter Tex11PM/Y) for further analysis through using female Tex11PM/X individuals backcrossed with wild-type males. As expected, male offspring carrying mutant Tex11 normally grew up to adult but displayed sterility (Supplementary Figure 2a (355.5KB, tif) ). The weights of the mutant testes were significantly lighter than that of wild-type controls (Figure 1a and 1b). Histological analysis of testes from adult Tex11PM/Y mice showed that Tex11 mutation causes spermatogenic failure, leading to rare germ cells in Tex11PM/Y seminiferous tubules and epididymis (Supplementary Figure 2b (355.5KB, tif) and 2c (355.5KB, tif) ). To check the effect of Tex11 mutation on SSCs, we analyzed the expression of promyelocytic leukemia zinc finger (PLZF), the specific marker of SSCs25 in juvenile testes. The results showed comparable SSC populations in Tex11PM/Y to littermate controls (Figure 1c and 1d), suggesting that TEX11 is not required for the maintenance of undifferentiated spermatogonia. Previous studies have shown that Tex11-/Y exhibited defects in chromosomal synapsis.23,24 We thus performed the co-immunostaining of spermatocyte spreads prepared from Tex11PM/Y for mutl homolog 1 (MLH1, crossover marker) and synaptonemal complex protein 3 (SCP3, synaptonemal complex marker). The results indicated that patient-derived mutation caused aberrant chromosomal synapsis in mouse testes (Figure 1e and 1f). Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) analysis showed that apoptosis increased dramatically in Tex11PM/Y seminiferous epithelium (Figure 1g and 1h), especially in cells at stages of IV and XII (Supplementary Figure 2d (355.5KB, tif) and 2e (355.5KB, tif) ), consistent with previous observations about increased apoptosis in Tex11-/Y testes.24 Taken together, these data show that patient-derived TEX11 mutation disrupts chromosomal synapsis, leading to meiotic arrest, followed by spermatocyte apoptosis in mouse seminiferous tubules, which resembles the phenotypes observed in patients with azoospermia.9
Figure 1.

Male mice carrying an azoospermia patient-derived TEX11 mutation display meiotic arrest. (a) Testis morphology of WT and Tex11PM/Y mice. Scale bar = 100 mm. (b) Relative testis/body weight of WT and Tex11PM/Y mice. The average values of three separate experiments are plotted. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test. (c) Immunostaining of 10 dpp testis sections with an anti-PLZF antibody. Green: PLZF; Blue: Hoechst 33342. Scale bar = 50 μm. (d) Quantification of PLZF+ cells in 10 dpp testis sections. A total of 237 and 222 tubules from three WT and Tex11PM/Y mice were scored. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test. NS: no significance. (e) Immunostaining of MLH1/SCP3 in spermatocyte spreads prepared from 8-week-old WT and Tex11PM/Y mice. Green: MLH1; red: SYCP3. Note the absence of MLH1 foci on three synapsed chromosomes (arrowhead) in the Tex11PM/Y sample shown in the bottom image. (f) Number of MLH1 foci in Tex11PM/Y and WT spermatogonia. A dramatic reduction in the number of MLH1 foci (green) in Tex11PM/Y spermatogonia (approximately 15 foci per cell) relative to WT (22 foci per cell). Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test;*** P < 0.001. (g) TUNEL analysis of Tex11PM/Y and WT testis. Massive apoptosis (green) shown in Tex11PM/Y tubules. Scale bar = 100 μm. (h) Analysis of apoptotic cells in Tex11PM/Y and WT testis. The average values of three separate experiments are plotted. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test;*** P < 0.001. TEX11: testis-expressed 11; WT: wild-type; PLZF: promyelocytic leukemia zinc finger; MLH1: mutl homolog 1; SYCP3: synaptonemal complex protein 3; TUNEL: terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling; DAPI: 4’,6-diamidino-2-phenylindole; s.d.: standard deviation; dpp: days postpartum.
SSCs isolated from Tex11PM/Y mice are normal
Having demonstrated that SSCs were unaffected in the testes of mutant males, we next attempted to derive SSC lines from newborn Tex11PM/Y males according to well-established protocols.21,26,27,28 Briefly, CD146-positive cells were enriched from testicular cells dissociated from mutant testes and cultured in a standard SSC culture medium29 (Figure 2a and 2b), leading to a stable SSC line carrying Tex11PM/Y (Figure 2c). The mutant SSCs showed typical SSC morphology, normal proliferation, and expression of SSC markers at similar levels as wild-type SSCs (Figure 2d–2f). Next, we compared the whole-gene expression of Tex11PM/Y SSCs with those of normal SSCs by performing RNA sequencing (RNA-seq). A similar expression pattern was observed for global genes in both cells (Figure 2g and Supplementary Figure 3a (452.5KB, tif) ). Further analysis showed an unaffected transcriptional level among SSC self-renewal, meiotic, and imprinted genes upon Tex11 mutation (Figure 2h, Supplementary Figure 3b (452.5KB, tif) and 3c (452.5KB, tif) ). Only two genes, including Tex11, were found to be differentially expressed between WT and Tex11PM/Y samples, suggesting a highly similar expression pattern (Supplementary Figure 3d (452.5KB, tif) and 3e (452.5KB, tif) ). Taken together, these results indicated that Tex11PM/Y SSCs are generally normal, implying that these cells after genetic correction may resume spermatogenesis.
Figure 2.

SSCs isolated from Tex11PM/Y mice are normal. (a) Flow cytometric analysis of the ratio of SSCs using CD146 antibody in 10-day-old WT and Tex11PM/Y testicular cells. Small black boxes represent the positive population of SSCs. (b) The ratio of CD146-positive cells in WT and Tex11PM/Y testicular cells (three mice per group) was scored. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test; NS: no significance. (c) Morphology of cultured WT and Tex11PM/Y SSCs. P2: passage 2. Scale bar = 100 μm. (d) Proliferation rate of WT and Tex11PM/Y SSCs during in vitro culture. The average values of three separate experiments are plotted. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test. (e) Expression of marker genes in WT and Tex11PM/Y SSCs. Id4 and Sohlh2 were used as undifferentiated marker genes, while Kit and Stra8 were used as differentiated marker genes. The av?erage values of three separate experiments are plotted; error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test; ***P < 0.001. (f) Immunostaining of PLZF in WT and Tex11PM/Y SSCs. Scale bar = 200 μm. (g) Scatter plot of log10-transformed average gene expression profiles. Global gene expression profiles of WT and Tex11PM/Y SSCs were obtained from RNA-seq analysis (r is the Pearson’s correlation coefficient; yellow lines indicate two-fold upregulation and downregulation). (h) Heatmap correlation of SSC self-renewal genes in WT and Tex11PM/Y SSCs (r is the Pearson’s correlation coefficient). WT: wild-type; Tex11: testis-expressed 11; SSCs: spermatogonial stem cells; s.d.: standard deviation; PLZF: promyelocytic leukemia zinc finger; SSC-A: Side Scatter-area; Id4: inhibitor of DNA binding 4; Sohlh2: spermatogenesis and oogenesis specific basic helix-loop-helix 2; Kit: Kit proto-oncogene; Stra8: stimulated by retinoic acid 8; Ret: ret proto-oncogene; Zbtb16: zinc finger and BTB domain containing 16; Pou5f1: POU class 5 homeobox 1; Etv5: ETS variant transcription factor 5; Gfra1: GDNF family receptor alpha 1; Foxo1: forkhead box O1; Taf4b: TATA-box binding protein associated factor 4b; T: brachyury; Lhx1: LIM homeobox 1; Bcl6b: BCL6B transcription repressor; Cxcr4: C-X-C motif chemokine receptor 4.
Gene therapy in Tex11PM/Y SSCs
To correct the mutation, we designed two sgRNAs targeting upstream and downstream of the mutant site (termed sgRNA-1 and sgRNA-2, respectively) and transfected pX330-mCherry plasmids18 expressing Cas9 and sgRNA-1 or sgRNA-2 and exogenous plasmid carrying wild-type sequence into Tex11PM/Y SSCs (Figure 3a). One day later, the SSCs expressing mCherry were enriched, in which the CRISPR-Cas9 system was successfully transfected, and single mCherry-positive SSCs were deposited into each well of the 96-well plates (Supplementary Figure 4a (356KB, tif) and 4b (356KB, tif) ) according to a previously reported protocol.29 From a total of 580 single SSCs for each sgRNA, we established 17 and 16 clonal SSC lines for sgRNA-1 and sgRNA-2, respectively (Supplementary Table 3. Further analysis showed that sgRNA-1 efficiently targeted the mutant Tex11 and led to HDR-mediated repair of the Tex11 gene in three SSC lines (Figure 3c). By contrast, no HDR-based repair was detected in SSC clones transfected with sgRNA-2 (Supplementary Figure 4c (356KB, tif) and Supplementary Table 3).
Figure 3.
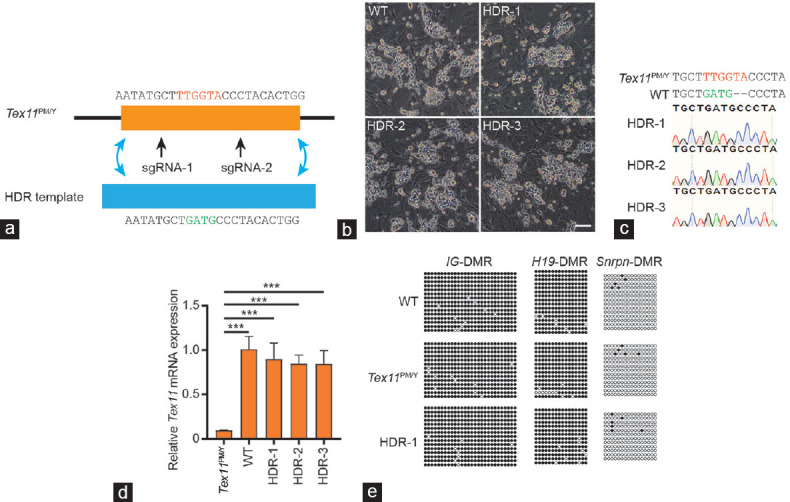
Correction of Tex11 mutation via CRISPR-Cas9-mediated gene editing in SSCs. (a) Schematic for Tex11 gene correction via HDR induced by CRISPR-Cas9 system and HDR template with WT sequence of Tex11. The mutant nucleotides were marked in red, while the WT nucleotides were in green. (b) Morphology of the three established SSC lines with correct Tex11 sequence (termed HDR-1, 2, and 3). Scale bar = 100 μm. (c) DNA sequencing analysis of SSCs from HDR-1/2/3. Note that the sequences of PCR products amplified from the Tex11 gene show that HDR-1/2/3 carry corrected Tex11 gene. (d) Transcriptional analysis of Tex11 in the corrected SSCs lines. The expression values were normalized to that of WT. The average values of three separate experiments are plotted; error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test; ***P < 0.001. (e) DNA methylation analysis of the DMRs of H19, IG, and Snrpn in Tex11PM/Y and HDR-1 SSCs. Open, filled, and gray circles represent unmethylated, methylated, and uncertain CpG sites, respectively. WT: wild-type; Tex11: testis expressed 11; SSCs: spermatogonial stem cells; s.d.: standard deviation; sgRNA: single-guide RNA; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; DMR: differentially methylated regions; H19: H19 imprinted maternally expressed transcript; IG: intergenic differentially methylated region; Snrpn: small nuclear ribonucleoprotein polypeptide N; CpG: cytosine-guanine; HDR: homology-directed repair.
Supplementary Table 3.
Summary of spermatogonial stem cell lines from single-cell expansion via clustered regularly interspaced short palindromic repeat-associated endonuclease 9-mediated gene correction of Tex11PM/Y spermatogonial stem cells
| SgRNA | Number of single cells used for expansion | Number of derived clones | Number of unedited clones (%) | Number of NHEJ clones (%) | Number of HDR clones (%) |
|---|---|---|---|---|---|
| SgRNA-1 | 580 | 17 | 4 (22.5) | 10 (58.8) | 3 (17.6) |
| SgRNA-2 | 580 | 16 | 11 (68.8) | 5 (31.2) | 0 |
HDR: homology-directed repair; SgRNA: single-guide RNA
Three repaired SSC lines (termed HDR-1, 2, and 3) derived from single SSC expansion after CRISPR-Cas9-mediated HDR correction of the patient-derived mutation in Tex11 gene restored the expression of Text11 to the level similar to that of wild-type SSCs (Figure 3d). These SSCs proliferated in vitro normally, showed typical SSC morphology, and expressed SSC marker genes (Figure 3b, Supplementary Figure 5a (144.9KB, tif) and 5b (144.9KB, tif) ). To further assess epigenetic inheritance, we performed bisulfite sequencing of two paternally imprinted regions, H19 imprinted maternally expressed transcript (H19)-DMR and intergenic differentially methylated region (IG)-DMR, and one maternally imprinted region, small nuclear ribonucleoprotein polypeptide N (Snrpn)-DMR in mutant and wild-type SSCs, and confirmed that both cell lines stably sustained typical paternally imprinted state (Figure 3e). One major challenge of CRISPR-Cas9-based therapeutic application is the occurrence of unintended off-target mutation at certain genome site depending on the specificity of the chosen sgRNA and intrinsic properties of Cas9. This becomes even more serious when germ cells are the target cells for gene therapy, because the unwanted modifications produced in the germ cells will pass on to the offspring genome, which might not only give rise to other genetic diseases in current individuals but also will pass on to next generations.30–34 To evaluate the risk of our therapy, we analyzed off-target effects in the three SSC lines derived from single SSCs that harbor corrected Tex11. We first searched all possible off-target sites with at most five mismatches for the sgRNA-1 against the whole mouse genome using Cas-OFFinder35 and found a total of 1166 possible off-target sites with no sites that show 0/1/2 mismatches, which indicates that the specificity of our sgRNA is sufficient. We then amplified and sequenced all possible sites within three mismatches but found that none of the sites were affected by our method (Supplementary Table 4). Next, to dive in the specificity of our method, we analyzed the genomic mutation between one cured SSC line (HDR-1) and its parental Tex11PM/Y SSCs using WGS (×40; Supplementary Table 5). The results indicated normal karyotype features and no significant genome copy number variations in both SSCs (Supplementary Figure 6a (417.6KB, tif) ). We found no Tex11 mutation in the HDR-1 genome sites, confirming that HDR-1 SSC line was derived from a single cell (Supplementary Figure 6b (417.6KB, tif) ). Thirty-six single-nucleotide variations (SNVs) and six indels were called using Mutect2 (GATK toolkit) between Tex11PM/Y SSCs and corrected HDR-1 SSCs (Supplementary Table 6). At the time point of performing WGS, both cell lines have already passaged eight times, individually, leading to an average four SNVs or indels per cell division in vitro, with the mutation rate in our system being comparable with that of spontaneous mutation rate (4–5 per cell division) in vivo.36 None of these SNVs or indels were annotated within the exonic gene region (Supplementary Table 6). Among all the detected SNVs and indels between those two cell lines, none of the mutations resided on any of the predicted off-target sites. Taken together, these results demonstrate that CRISPR-Cas9-mediated gene correction in Tex11PM/Y SSCs does not induce obvious off-target effects in SSCs, consistent with our previous observations in gene-treated SSCs carrying a disease-causing homozygous mutation.21
Supplementary Table 4.
The off-target analysis in repaired spermatogonial stem cell lines
| Potential off-target site | Sequence | Mismatch bases | Mutant SSC lines (total) |
|---|---|---|---|
| chr1:66249826 | TTAaGTaCAAAAATATGCaTTGG | 3 | 0 (3) |
| chr1:102316146 | TTAttTCaAAAAATATGCTTTGG | 3 | 0 (3) |
| chr10:26854499 | TaAaGTCCAAAAAcATGCTTCGG | 3 | 0 (3) |
| chr11:30465584 | TcAGGTCCAAAcATtTGCTTAGG | 3 | 0 (3) |
| chr12:42482925 | TcAGGTCCAAAAATtTGCcTAGG | 3 | 0 (3) |
| chr12:79861136 | caAGGTtCAAAAATATGCTTTGG | 3 | 0 (3) |
| chr12:111449864 | TcAGGTCCAAAAATAgGCTcTGG | 3 | 0 (3) |
| chr14:114782600 | TTAGtTtCcAAAATATGCTTTGG | 3 | 0 (3) |
| chr2:73095017 | TTAGtatCAAAAATATGCTTAGG | 3 | 0 (3) |
| chr4:72841982 | TTAGGTCtAAAAAgATGtTTTGG | 3 | 0 (3) |
| chr6:59018691 | TTAGGaCCAAAAAgATcCTTAGG | 3 | 0 (3) |
| chrY: 20249774 | ccAGGTCCAAAAATtTGCTTAGG | 3 | 0 (3) |
The mismatch nucleotides were shown in lower cases. PAM is marked in red. No off-target nucleotides were detected in the potential off-target sites. SSC: spermatogonial stem cell
Supplementary Table 5.
Summary of whole-genome sequencing analysis
| Raw reads | Clean reads | Clean bases | Rate (%) | Mapped reads | Properly paired | Properly paired reads | Base | Ratio (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Tex11 PM/Y | 763192270 | 747324824 | 109520937180 | 97.921 | 746464276 | 717938106 | 96.1 | 109393973559 | 99.884 |
| HDR-1 | 836485138 | 824798254 | 121183737241 | 98.603 | 822262433 | 809857802 | 98.2 | 120809378500 | 99.691 |
HDR: homology-directed repair
Supplementary Table 6.
Summary of genomic mutations in homology-directed repair-1
| Region | SNV | Indels |
|---|---|---|
| Exon | 0 | 0 |
| Intron | 15 | 3 |
| Intergenic | 21 | 3 |
| Total | 36 | 6 |
SNV: single-nucleotide variatio
Corrected SSCs restore spermatogenesis in the testes of infertility models
Finally, we investigated whether SSCs with corrected Tex11 could restore spermatogenesis in vivo. To this end, we first transplanted HDR-1 SSCs into the testis of Tex11PM/Y mice to mimic autotransplantation situation in patient treatment (Supplementary Figure 7a (149KB, tif) ). However, no spermatogenesis occurred in all the three tested recipient mice (Supplementary Figure 7b (149KB, tif) and 7c (149KB, tif) ), indicating that cured SSCs cannot find the niches when normal Tex11PM/Y SSCs exist in seminiferous tubules. We then attempted to test the ability of the spermatogenesis of cured SSCs by transplantation of HDR-1 SSCs and Tex11PM/Y SSCs as control into the recipient testis of KitW/KitWV (C-Kit double heterozygotes) males, a well-established infertility model37 or busulfan-treated mT/mG males expressing membrane-targeted tdTomato38 (Figure 4a and Supplementary Figure 8a (247.2KB, tif) ). Two months after the transplantation, we observed significantly increased size of testes with transferred HDR-1 SSCs compared to that of control transplanted testes with Tex11PM/Y SSCs (Figure 4b and 4c). Histological analyses indicated that HDR-1 SSCs successfully migrated toward germline niches, recolonized the seminiferous tubules, and restored the spermatogenesis of infertility testes39 (Figure 4d and 4e). Finally, haploid spermatids could be enriched and used for intracytoplasmic round spermatid injection (ROSI; Figure 4f and 4g, and Supplementary Figure 8b (247.2KB, tif) ). The injected oocytes efficiently developed into two-cell embryos, which were then transplanted into the recipient oviducts of pseudopregnant females. We obtained a total of 61 pups on 19.5 days of gestation, at a rate of 16.1% of transferred embryos, similar to that of ROSI with wild-type round spermatids (Figure 4h and Table 1). Genotyping analysis indicated that all of them carry wild-type Tex11 (Supplementary Figure 9a (328.4KB, tif) ). As the resultant pups were derived from a cured SSC line (HDR-1) that carried SNPs detected by WGS, we thus used these SNPs as molecular markers to further demonstrate their paternal origin. As expected, the analysis of five SNPs indicated that two tested pups were originated from HDR-1 (Supplementary Figure 9b (328.4KB, tif) and Supplementary Table 7). The pups grew to adulthood and males could give rise to progeny normally, demonstrating the successful transmission of corrected Tex11 in HDR-1 to next generations (Supplementary Table 8).
Figure 4.

Corrected SSCs restore spermatogenesis in the testes of KitW /KitWV mice. (a) Diagram showing the repaired SSCs derived from single SSCs without off-target mutations which are selected for transplantation into infertile KitW /KitWV male mice. Two months later, spermatids are isolated from reconstituted testes and injected into mature oocytes to produce healthy mice carrying corrected Tex11 gene. (b) Testis morphology of Tex11PM/Y and HDR-1 SSC-transplanted KitW /KitWV mice. Scale bar = 100 mm. (c) Weight of Tex11PM/Y and HDR-1 SSC-transplanted KitW /KitWV testes. The average values of three separate experiments are plotted. Error bar: s.d. Significance was determined by two-tailed, unpaired Student’s t-test;* P < 0.05. (d) Histological analyses of the Tex11PM/Y and HDR-1 SSC-transplanted KitW /KitWV testis. Note successful spermatogenesis in the HDR-1 SSC-transplanted testis. Scale bar = 40 μm. (e) Immunostaining of MVH in Tex11PM/Y and HDR-1-transplanted KitW /KitWV testis. Green: MVH; blue: Hoechst. Scale bar = 100 μm. (f) Round spermatids are sorted from testicular cells of reconstituted testes according to DNA content. (g) Morphology of round sperm sorted from HDR-1-transplanted testis. Scale bar = 50 μm. (h) Pups (black coat color) on 12 dpp developed from transferred two-cell embryos generated after the injection of round spermatids from HDR-1 SSCs into mature oocytes. WT: wild-type; Tex11: testis expressed 11; SSCs: spermatogonial stem cells; ROSI: round spermatid injection; s.d.: standard deviation; HDR: homology-directed repair; dpp: days postpartum.
Table 1.
Mice produced through injection of round spermatids derived from transplanted spermatogonial stem cells into mature oocytes
| Round spermatid source | Recipient mice for SSC transplantation | Types of transplanted SSCs | Genetic background of SSCs | Number of injected oocytes | Number of transferred two-cell embryos | Number of live-born pup, n (%) | Number of pups with corrected Tex11 |
|---|---|---|---|---|---|---|---|
| Recipient mice | KitW/KitWV | HDR-1 | C57×DBA | 161 | 134 | 21 (15.67) | 20 |
| Recipient mice | KitW/KitWV | Tex11 PM/Y | C57×DBA | 155 | 36 | 0 (0) | 0 |
| Recipient mice | Busulfan-treated mT/mG | HDR-1 | C57×DBA | 140 | 120 | 22 (18.33) | 22 |
| Recipient mice | Busulfan-treated mT/mG | HDR-1 | C57×DBA | 145 | 124 | 18 (14.51) | 18 |
| Control | - | - | - | 150 | 130 | 24 (18.46) | 0 |
Tex11: testis-expressed 11; SSCs: spermatogonial stem cells; HDR: homology-directed repair; DBA: Dilute Brown Non-Agouti
Supplementary Table 7.
Summary of single-nucleotide polymorphism sequence analysis of homology-directed repair-1 spermatogonial stem cell-derived pups
| Chr | POS | REF | ALT | Mut (REF, ALT) | HDR-1 (REF, ALT) | C57 | DBA | ROSI-1 | ROSI-2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| SNP-1 | chr6 | 132136304 | G | A | 14, 0 | 1, 17 | G | G | G/A | G |
| SNP-2 | chr5 | 5040740 | A | T | 35, 0 | 6, 17 | A | A | A/T | A |
| SNP-3 | chr15 | 31181191 | C | T | 24, 0 | 9, 22 | C | C | C/T | C/T |
| SNP-4 | chr17 | 21447226 | TATA | T | 31, 0 | 10, 23 | TATA | TATA | TATA | T/TATA |
| SNP-5 | chr12 | 89104845 | T | C | 36, 0 | 11, 26 | T | T | C/T | T |
As Tex11PM/Y and HDR-1 SSCs were derived from C57 and DBA mixed background, unique HDR-1 SNP sites obtained during in vitro culture different from both Tex11PM/Y SSCs and C57/DBA sequence were selected for analysis. For each SNP, genome of WT C57/DBA tails, Tex11PM/Y and HDR-1 SSCs were subjected to PCR and sanger sequence. SNP-1/2/3/5 was detected in the HDR-1 SSC-derived pup ROSI-1, while SNP3/4 in the ROSI-2. SNP: single-nucleotide polymorphism; HDR: homology-directed repair; SSCs: spermatogonial stem cells; ROSI: round spermatid injection
Supplementary Table 8.
Summary of offspring obtained from 4 male mice derived from homology-directed repair-1 spermatogonial stem cells
| 1st litter | 2nd litter | 3rd litter | |
|---|---|---|---|
| RO-M-1 | 3 | 5 | 9 |
| RO-M-2 | 3 | 8 | 7 |
| RO-M-3 | 6 | 7 | - |
| RO-M-4 | 4 | 8 |
DISCUSSION
We have demonstrated the feasibility of infertility treatment by correcting a genetic defect causing azoospermia through a combination of in vitro expansion of SSCs and CRISPR-Cas9-based gene therapy (Figure 5). This PDMX pipeline involved validating the monogenic mutation that causes azoospermia through generation of mouse models by introduction of disease-related TEX11 mutation into mouse genome that recapitulates the phenotype of meiotic arrest. SSCs were isolated from Tex11PM/Y mice and expanded in vitro. CRISPR-Cas9-induced HDR was used to correct the gene defect in SSCs, and SSC lines were established through single-cell expansion. Genotyping, methylation, and off-target analyses enabled the selection of SSCs with the desired genotype, with normal imprinted state, and without off-target mutations for further implementation. To assess whether the genetic correction restored spermatogenesis, we transplanted cured SSCs into the recipient testes of two infertility models. The spermatogenesis was resumed in the seminiferous tubules of these animals, as demonstrated by the enrichment of haploid spermatids 2 months after SSC transplantation. As expected, round spermatids, through ROSI, gave rise to offspring normally. Importantly, the resultant males exhibited normal fertility, indicating the transmission of the corrected gene to next generations. Thus, CRISPR-Cas9-mediated gene therapy in SSCs corrected the genetic defect causing azoospermia.
Figure 5.
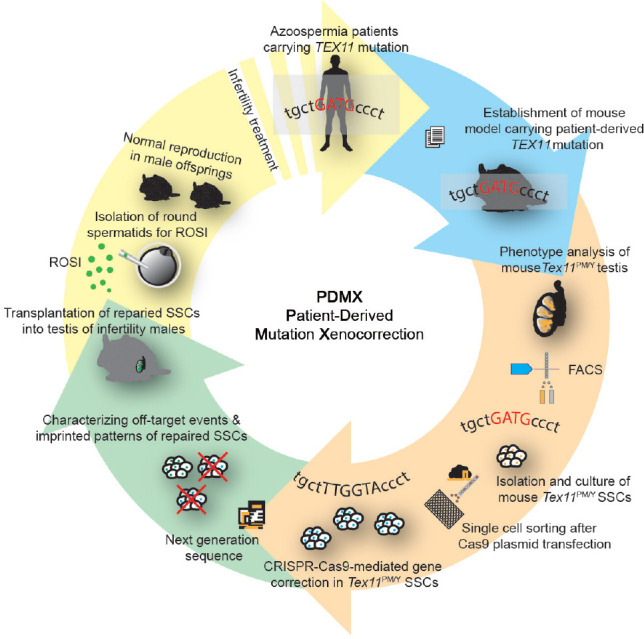
Scheme for PDMX model of male azoospermia therapy. TEX11 mutation has been identified in patients with azoospermia. To validate its pathogenic roles, mouse model with the patient-derived mutation (Tex11PM/Y mice) was derived, which recapitulated patient’s pathological phenotypes and displayed meiotic arrest. SSCs isolated from the testis of the Tex11PM/Y mice were expanded in vitro. CRISPR-Cas9-mediated gene correction was employed in Tex11PM/Y SSCs and repaired SSC lines were derived through single SSC expansion, followed by whole-genome sequence to exclude the off-target mutations. The repaired SSCs were then used for transplantation to restore spermatogenesis in infertility males. PDMX model thus provides proof-of-principle evidence for curing human azoospermia carrying a single mutation that causes meiotic arrest. TEX11: testis-expressed 11; SSCs: spermatogonial stem cells; ROSI: round spermatid injection; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; PDMX: patient-derived mutation xenocorrection.
A critical step of PDMX is the isolation and expansion in vitro of SSCs from infertility individuals. Mouse and rat SSCs can be maintained in long-term culture and still possess the potential to colonize and initiate spermatogenesis in seminiferous tubules when transplanted into the testes of infertility recipients.16 Strikingly, mouse SSCs that were cultured in vitro over 2 years generally sustain normal genetic and epigenetic stability and could be used to produce spermatogenesis and fertile offspring after transplantation.40 Moreover, mouse SSCs after 14-year cryopreservation could still reconstitute the spermatogenesis in the recipient testes.41 These observations imply that SSCs are a promising cell resource for stem cell therapies with regard to male infertility.2 However, while several independent groups reported the in vitro culturing of human SSCs using different protocols, a standard protocol that can be replicated by different groups is still an unmet need.42 Furthermore, transplantation of SSCs to regenerate spermatogenesis, the golden standard assay for SSC functional analysis, needs to be established in humans. Interestingly, the failed spermatogenesis after autotransplantation into meiotic arrest males in this study suggests that it is critical to remove uncorrected SSCs before transplantation of cured SSCs into patient testes, or it may be necessary to establish a protocol of de novo morphogenesis of testis in humans.28 Therefore, a future task is to define appropriate culture conditions that would enable efficient establishment of human SSC lines from seminiferous tubule biopsy samples, which would facilitate the potential applications of these cells in stem cell therapies.
Another critical step of our PDMX model is to identify azoospermia-causing mutations. With the rapid development of high-throughput genome sequencing technologies and bioinformatics methodologies, increasing spermatogenic failure-related mutations are emerging,1 which can be potentially used as targets for gene therapy to treat male infertility. Thus, we first need to validate that the identified mutations can cause azoospermia indeed. The most stringent strategy of validation is to recapitulate the patients’ phenotypes in model organisms, especially in mouse, as demonstrated in this study. Moreover, PDMX model in this study excluded the possibility that the TEX11 mutation disturbs the self-renewing and proliferating abilities of SSCs, thus enabling the stem cell-based gene therapy.
In summary, we have demonstrated that combined application of SSC in vitro expansion and CRISPR-Cas9-mediated gene therapy can be used to cure a genetic disease of male infertility in mice, providing proof-of-principle approach for the treatment of human azoospermia caused by single-gene mutation-induced meiotic arrest.
AUTHOR CONTRIBUTIONS
YHW, MHT, and JSL designed the study. XYL performed mouse embryo microinjection and embryo transplantation. YHW and YFD performed SSC transplantation. YHW performed in vitro culture and gene editing of SSCs; YHW and CPL performed genotyping and gene expression experiments. XZ performed TUNEL analysis and meiotic-related gene immunofluorescence. MY analyzed the whole-exome sequencing and RNA-seq data. YHW and JSL wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
Generation of mice carrying patient-derived Tex11 mutation. (a) Alignment of TEX11 protein sequences. h:, human; m: mouse. (b) Scheme for CRISPR-Cas9-mediated Tex11 mutation in mice. SgRNAs were designed to target exon 16 of Tex11. The sgRNA-targeting sequence is shown in the upper line, and the PAM sequence is labeled in red. The sequence of donor oligo is shown under the gene, with 6 bp changes labeled in green. (c) Genotyping analysis of Tex11-mutant mice. (d) Morphology of WT and Tex11PM/+ female ovary, indicating normal growth and morphology. Scale bar = 25 mm. (e) Histological analysis of the ovary from WT and Tex11PM/+ mice, indicating normal oogenesis in Tex11 heterozygous female. Scale bar = 100 μm. Two independent mutants were analyzed for each group. (f) Morphology of WT and Tex11PM/+ testis. Scale bar = 200 mm. (g) Histological analysis of the testis and epididymis from F0 WT and Tex11PM/+ mice. Scale bar = 50 μm. WT: wild-type; TEX11: testis-expressed 11; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; sgRNA: single-guide RNA.
Meiotic arrest in Tex11PM/Y testis. (a) Image of 8-week-old WT and Tex11PM/Y mice, indicating normal development of Tex11PM/Y mice. (b) Histological analysis of the testis and epididymis of F0 WT and Tex11PM/Y mice. Scale bar = 50 μm. (c) Immunostaining of MVH in testicular sections from 8-week-old WT and Tex11PM/Y mice with nuclei counterstained with Hoechst. Blue: Hoechst; Green: MVH. Scale bar = 50 μm. (d) Histological analysis shows apoptosis cells in stage IV and XII seminiferous tubules. Scale bar = 50 um. (e) TUNEL staining shows apoptosis cells in stage IV and XII seminiferous tubules. Scale bar = 50 um. WT: wild-type; TUNEL: terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling.
Tex11PM/Y SSCs display normal transcriptional profiles. (a) The volcano plot depicting the fold differences in gene expression levels between the WT and Tex11PM/Y SSCs. Colored points refer to the differentially expressed transcripts according to fold change and P-value. (b) Heatmap correlation of log10 genes related to meiotic process in WT and Tex11PM/Y SSCs (r is the Pearson's correlation coefficient). (c) Heatmap correlation of log10 imprinted genes in WT and Tex11PM/Y SSCs. (left, paternal imprinted genes; right, maternal imprinted genes; r is the Pearson's correlation coefficient). (d) A heatmap of log10 gene expression for the two differentially expressed genes. (e) Average gene expression level of the two differentially expressed genes. y axis, TPM from RNA-Seq data. WT: wild-type; SSCs: spermatogonial stem cells; RNA-Seq: RNA sequencing.
Correction of Tex11 mutation via CRISPR-Cas9-mediated gene editing in Tex11PM/Y SSCs. (a) Lipofectamine-mediated CRISPR-Cas9 transfection in Tex11PM/Y SSCs. Two sgRNAs were used for Tex11 gene correction. mCherry indicating that SSCs successfully transfected with CRISPR-Cas9. Scale bar = 100 μm. (b) SSCs with expressed Cas9 (co-expression of mCherry) were enriched through FACS, followed by single-cell expansion to obtain cell clones for Tex11 gene analysis. (c) The sequences of Tex11 gene in the SSCs lines carrying CRISPR-Cas9-induced NHEJ modifications. Small letters represent the inserted nucleotides. TEX11: testis-expressed 11; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; sgRNA: single-guide RNA; SSCs: spermatogonial stem cells.
Characterization of HDR-1 SSCs. (a) Expression of marker genes in Tex11PM/Y and HDR-1 SSCs. The expression values were normalized to that of WT. The average values of three separate experiments are plotted. Significance was determined by two-tailed, unpaired Student's t-test. Id4 and Sohlh2 were used as undifferentiated marker genes, while Kit and Stra8 were used as differentiated marker genes. (b) Flow cytometry analysis of CD146 in cultured Tex11PM/Y and HDR-1 SSCs of passage 14, indicating that the majority of both Tex11PM/Y and HDR-1 SSCs express CD146 during in vitro culture. WT: wild-type; HDR: homology-directed repair; SSCs: spermatogonial stem cells.
WGS analysis of Tex11PM/Y and HDR-1 SSCs. (a) Copy number of chromosomes in Tex11PM/Y and HDR-1 SSCs, indicating normal karyotype of 40 chromosomes (38+X+Y) in both SSC lines. (b) Sequence analysis of Tex11 in Tex11PM/Y and HDR-1 SSCs. Each line represents a single read from WGS. TEX11: testis-expressed 11; SSCs: spermatogonial stem cells; HDR: homology-directed repair; WGS: whole-genome sequencing.
Autotransplantation of HDR-1 SSCs into Tex11PM/Y mice. (a) Diagram showing autotransplantation of HDR-1 SSCs into 3-week-old Tex11PM/Y recipient mice (n = 3). Two months later, the testes were used for FACS and HE analysis. (b) Histological analyses of the Tex11PM/Y-recipient testis, indicating no restoration of spermatogenesis. Scale bar = 40 μm. (c) FACS analysis of Tex11PM/Y-recipient testis according to DNA content. SSCs: spermatogonial stem cells; HDR: homology-directed repair.
Restoration of spermatogenesis following transplantation of HDR-1 SSC into busulfan-treated mT/mG recipients. (a) Diagram showing the repaired SSC lines derived from single SSCs without off-target mutations which are selected for transplantation into busulfan-treated mT/mG mice. Two months later, round spermatids were isolated from reconstituted testes and injected into mature oocytes to produce healthy mice carrying corrected Tex11 gene. (b) Round spermatids are enriched from testicular cells of reconstituted testes. HDR-1 SSC-derived cells represented by RFP negative were enriched based on the expression of fluorescent protein and then subjected to DNA content analysis. SSCs: spermatogonial stem cells; HDR: homology-directed repair; TEX11: testis-expressed 11.
Paternal origin of the ROSI-derived pups from Kitw/Kitwv recipients. (a) Genotyping analysis of C-Kit in the newborn mice. Two peaks can be observed in the sequence of the control Kitw/Kitwv mouse (indicated by arrowheads), while the HDR-1 SSC-derived mice have only one peak. (b) Genotyping analysis of selected SNPs 1–5 in the newborn pups. The SNP sites are indicated by arrowhead. SSCs: spermatogonial stem cells; HDR: homology-directed repair; ROSI: round spermatid injection; SNP: singlenucleotide polymorphism.
ACKNOWLEDGMENTS
This study was supported by Genome Tagging Project and grants from the Chinese Academy of Sciences, the National Key Research and Development Program of China, Shanghai Municipal Commission for Science and Technology, and the National Natural Science Foundation of China (XDB19010204, 2019YFA0109900, OYZDJ-SSW-SMC023, Facility-based Open Research Program, 19411951800, 17JC1420102, 31821004, 32030029, 31730062, 31530048, and 81672117). The research is partly supported by the Fountain-Valley Life Sciences Fund of University of Chinese Academy of Sciences Education Foundation.
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15:369–84. doi: 10.1038/s41585-018-0003-3. [DOI] [PubMed] [Google Scholar]
- 2.Gassei K, Orwig KE. Experimental methods to preserve male fertility and treat male factor infertility. Fertil Steril. 2016;105:256–66. doi: 10.1016/j.fertnstert.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteves SC. Clinical management of infertile men with nonobstructive azoospermia. Asian J Androl. 2015;17:459–70. doi: 10.4103/1008-682X.148719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tournaye H, Krausz C, Oates RD. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. 2017;5:544–53. doi: 10.1016/S2213-8587(16)30040-7. [DOI] [PubMed] [Google Scholar]
- 5.Tuttelmann F, Ruckert C, Ropke A. Disorders of spermatogenesis: perspectives for novel genetic diagnostics after 20 years of unchanged routine. Med Genet. 2018;30:12–20. doi: 10.1007/s11825-018-0181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riera-Escamilla A, Enguita-Marruedo A, Moreno-Mendoza D, Chianese C, Sleddens-Linkels E, et al. Sequencing of a ’mouse azoospermia’ gene panel in azoospermic men: identification of RNF212 and STAG3 mutations as novel genetic causes of meiotic arrest. Hum Reprod. 2019;34:978–88. doi: 10.1093/humrep/dez042. [DOI] [PubMed] [Google Scholar]
- 7.van der Bijl N, Ropke A, Biswas U, Woste M, Jessberger R, et al. Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum Reprod. 2019;34:2112–9. doi: 10.1093/humrep/dez204. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Guo J, Dai L, Zhu Y, Hu H, et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet. 2018;55:628–36. doi: 10.1136/jmedgenet-2017-105145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yatsenko AN, Georgiadis AP, Ropke A, Berman AJ, Jaffe T, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med. 2015;372:2097–107. doi: 10.1056/NEJMoa1406192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang F, Silber S, Leu NA, Oates RD, Marszalek JD, et al. TEX11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol Med. 2015;7:1198–210. doi: 10.15252/emmm.201404967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben Khelifa M, Ghieh F, Boudjenah R, Hue C, Fauvert D, et al. A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family. Hum Reprod. 2018;33:1034–7. doi: 10.1093/humrep/dey073. [DOI] [PubMed] [Google Scholar]
- 12.Gershoni M, Hauser R, Barda S, Lehavi O, Arama E, et al. A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum Reprod. 2019;34:666–71. doi: 10.1093/humrep/dez016. [DOI] [PubMed] [Google Scholar]
- 13.Zou K, Ding G, Huang H. Advances in research into gamete and embryo-fetal origins of adult diseases. Sci China Life Sci. 2019;62:360–8. doi: 10.1007/s11427-018-9427-4. [DOI] [PubMed] [Google Scholar]
- 14.Hann MC, Lau PE, Tempest HG. Meiotic recombination and male infertility: from basic science to clinical reality? Asian J Androl. 2011;13:212–8. doi: 10.1038/aja.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enguita-Marruedo A, Sleddens-Linkels E, Ooms M, de Geus V, Wilke M, et al. Meiotic arrest occurs most frequently at metaphase and is often incomplete in azoospermic men. Fertil Steril. 2019;112:1059–70.e3. doi: 10.1016/j.fertnstert.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Kubota H, Brinster RL. Spermatogonial stem cells. Biol Reprod. 2018;99:52–74. doi: 10.1093/biolre/ioy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulder CL, Zheng Y, Jan SZ, Struijk RB, Repping S, et al. Spermatogonial stem cell autotransplantation and germline genomic editing: a future cure for spermatogenic failure and prevention of transmission of genomic diseases. Hum Reprod Update. 2016;22:561–73. doi: 10.1093/humupd/dmw017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–62. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Zhou H, Fan X, Zhang Y, Zhang M, et al. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. 2015;25:67–79. doi: 10.1038/cr.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sha Y, Zheng L, Ji Z, Mei L, Ding L, et al. A novel TEX11 mutation induces azoospermia: a case report of infertile brothers and literature review. BMC Med Genet. 2018;19:63. doi: 10.1186/s12881-018-0570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adelman CA, Petrini JH. ZIP4H (TEX11) deficiency in the mouse impairs meiotic double strand break repair and the regulation of crossing over. PLoS Genet. 2008;4:e1000042. doi: 10.1371/journal.pgen.1000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang F, Gell K, van der Heijden GW, Eckardt S, Leu NA, et al. Meiotic failure in male mice lacking an X-linked factor. Genes Dev. 2008;22:682–91. doi: 10.1101/gad.1613608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36:653–9. doi: 10.1038/ng1367. [DOI] [PubMed] [Google Scholar]
- 26.Kanatsu-Shinohara M, Morimoto H, Shinohara T. Enrichment of mouse spermatogonial stem cells by melanoma cell adhesion molecule expression. Biol Reprod. 2012;87:139. doi: 10.1095/biolreprod.112.103861. [DOI] [PubMed] [Google Scholar]
- 27.Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod. 2003;69:612–6. doi: 10.1095/biolreprod.103.017012. [DOI] [PubMed] [Google Scholar]
- 28.Zhang M, Zhou H, Zheng C, Xiao J, Zuo E, et al. The roles of testicular c-kit positive cells in de novo morphogenesis of testis. Sci Rep. 2014;4:5936. doi: 10.1038/srep05936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Ding Y, Li J. CRISPR-Cas9-Mediated gene editing in mouse spermatogonial stem cells. Methods Mol Biol. 2017;1622:293–305. doi: 10.1007/978-1-4939-7108-4_20. [DOI] [PubMed] [Google Scholar]
- 30.Lander ES, Baylis F, Zhang F, Charpentier E, Berg P, et al. Adopt a moratorium on heritable genome editing. Nature. 2019;567:165–8. doi: 10.1038/d41586-019-00726-5. [DOI] [PubMed] [Google Scholar]
- 31.The Royal Society; National Academy of Sciences; National Academy of Medicine; International Commission on the Clinical Use of Human Germline Genome Editing. Heritable Human Genome Editing. Washington: National Academies Press (US); 2020. [PubMed] [Google Scholar]
- 32.Wen L, Liu Q, Xu J, Liu X, Shi C, et al. Recent advances in mammalian reproductive biology. Sci China Life Sci. 2020;63:18–58. doi: 10.1007/s11427-019-1572-7. [DOI] [PubMed] [Google Scholar]
- 33.Adashi EY, Cohen IG. Heritable Human Genome Editing: The International Commission Report. JAMA. 2020;324:1941–2. doi: 10.1001/jama.2020.19059. [DOI] [PubMed] [Google Scholar]
- 34.Yan M, Li J. Combined application of CRISPR-Cas and stem cells for clinical and basic research. Cell Regen. 2020;9:19. doi: 10.1186/s13619-020-00062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–5. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–52. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–9. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 38.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 39.Brinster RL, Zimmermann JW. Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A. 1994;91:11298–302. doi: 10.1073/pnas.91.24.11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanatsu-Shinohara M, Ogonuki N, Iwano T, Lee J, Kazuki Y, et al. Genetic and epigenetic properties of mouse male germline stem cells during long-term culture. Development. 2005;132:4155–63. doi: 10.1242/dev.02004. [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Goodyear SM, Abramowitz LK, Bartolomei MS, Tobias JW, et al. Fertile offspring derived from mouse spermatogonial stem cells cryopreserved for more than 14 years. Hum Reprod. 2012;27:1249–59. doi: 10.1093/humrep/des077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valli H, Phillips BT, Shetty G, Byrne JA, Clark AT, et al. Germline stem cells: toward the regeneration of spermatogenesis. Fertil Steril. 2014;101:3–13. doi: 10.1016/j.fertnstert.2013.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of mice carrying patient-derived Tex11 mutation. (a) Alignment of TEX11 protein sequences. h:, human; m: mouse. (b) Scheme for CRISPR-Cas9-mediated Tex11 mutation in mice. SgRNAs were designed to target exon 16 of Tex11. The sgRNA-targeting sequence is shown in the upper line, and the PAM sequence is labeled in red. The sequence of donor oligo is shown under the gene, with 6 bp changes labeled in green. (c) Genotyping analysis of Tex11-mutant mice. (d) Morphology of WT and Tex11PM/+ female ovary, indicating normal growth and morphology. Scale bar = 25 mm. (e) Histological analysis of the ovary from WT and Tex11PM/+ mice, indicating normal oogenesis in Tex11 heterozygous female. Scale bar = 100 μm. Two independent mutants were analyzed for each group. (f) Morphology of WT and Tex11PM/+ testis. Scale bar = 200 mm. (g) Histological analysis of the testis and epididymis from F0 WT and Tex11PM/+ mice. Scale bar = 50 μm. WT: wild-type; TEX11: testis-expressed 11; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; sgRNA: single-guide RNA.
Meiotic arrest in Tex11PM/Y testis. (a) Image of 8-week-old WT and Tex11PM/Y mice, indicating normal development of Tex11PM/Y mice. (b) Histological analysis of the testis and epididymis of F0 WT and Tex11PM/Y mice. Scale bar = 50 μm. (c) Immunostaining of MVH in testicular sections from 8-week-old WT and Tex11PM/Y mice with nuclei counterstained with Hoechst. Blue: Hoechst; Green: MVH. Scale bar = 50 μm. (d) Histological analysis shows apoptosis cells in stage IV and XII seminiferous tubules. Scale bar = 50 um. (e) TUNEL staining shows apoptosis cells in stage IV and XII seminiferous tubules. Scale bar = 50 um. WT: wild-type; TUNEL: terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling.
Tex11PM/Y SSCs display normal transcriptional profiles. (a) The volcano plot depicting the fold differences in gene expression levels between the WT and Tex11PM/Y SSCs. Colored points refer to the differentially expressed transcripts according to fold change and P-value. (b) Heatmap correlation of log10 genes related to meiotic process in WT and Tex11PM/Y SSCs (r is the Pearson's correlation coefficient). (c) Heatmap correlation of log10 imprinted genes in WT and Tex11PM/Y SSCs. (left, paternal imprinted genes; right, maternal imprinted genes; r is the Pearson's correlation coefficient). (d) A heatmap of log10 gene expression for the two differentially expressed genes. (e) Average gene expression level of the two differentially expressed genes. y axis, TPM from RNA-Seq data. WT: wild-type; SSCs: spermatogonial stem cells; RNA-Seq: RNA sequencing.
Correction of Tex11 mutation via CRISPR-Cas9-mediated gene editing in Tex11PM/Y SSCs. (a) Lipofectamine-mediated CRISPR-Cas9 transfection in Tex11PM/Y SSCs. Two sgRNAs were used for Tex11 gene correction. mCherry indicating that SSCs successfully transfected with CRISPR-Cas9. Scale bar = 100 μm. (b) SSCs with expressed Cas9 (co-expression of mCherry) were enriched through FACS, followed by single-cell expansion to obtain cell clones for Tex11 gene analysis. (c) The sequences of Tex11 gene in the SSCs lines carrying CRISPR-Cas9-induced NHEJ modifications. Small letters represent the inserted nucleotides. TEX11: testis-expressed 11; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR-associated endonuclease 9; sgRNA: single-guide RNA; SSCs: spermatogonial stem cells.
Characterization of HDR-1 SSCs. (a) Expression of marker genes in Tex11PM/Y and HDR-1 SSCs. The expression values were normalized to that of WT. The average values of three separate experiments are plotted. Significance was determined by two-tailed, unpaired Student's t-test. Id4 and Sohlh2 were used as undifferentiated marker genes, while Kit and Stra8 were used as differentiated marker genes. (b) Flow cytometry analysis of CD146 in cultured Tex11PM/Y and HDR-1 SSCs of passage 14, indicating that the majority of both Tex11PM/Y and HDR-1 SSCs express CD146 during in vitro culture. WT: wild-type; HDR: homology-directed repair; SSCs: spermatogonial stem cells.
WGS analysis of Tex11PM/Y and HDR-1 SSCs. (a) Copy number of chromosomes in Tex11PM/Y and HDR-1 SSCs, indicating normal karyotype of 40 chromosomes (38+X+Y) in both SSC lines. (b) Sequence analysis of Tex11 in Tex11PM/Y and HDR-1 SSCs. Each line represents a single read from WGS. TEX11: testis-expressed 11; SSCs: spermatogonial stem cells; HDR: homology-directed repair; WGS: whole-genome sequencing.
Autotransplantation of HDR-1 SSCs into Tex11PM/Y mice. (a) Diagram showing autotransplantation of HDR-1 SSCs into 3-week-old Tex11PM/Y recipient mice (n = 3). Two months later, the testes were used for FACS and HE analysis. (b) Histological analyses of the Tex11PM/Y-recipient testis, indicating no restoration of spermatogenesis. Scale bar = 40 μm. (c) FACS analysis of Tex11PM/Y-recipient testis according to DNA content. SSCs: spermatogonial stem cells; HDR: homology-directed repair.
Restoration of spermatogenesis following transplantation of HDR-1 SSC into busulfan-treated mT/mG recipients. (a) Diagram showing the repaired SSC lines derived from single SSCs without off-target mutations which are selected for transplantation into busulfan-treated mT/mG mice. Two months later, round spermatids were isolated from reconstituted testes and injected into mature oocytes to produce healthy mice carrying corrected Tex11 gene. (b) Round spermatids are enriched from testicular cells of reconstituted testes. HDR-1 SSC-derived cells represented by RFP negative were enriched based on the expression of fluorescent protein and then subjected to DNA content analysis. SSCs: spermatogonial stem cells; HDR: homology-directed repair; TEX11: testis-expressed 11.
Paternal origin of the ROSI-derived pups from Kitw/Kitwv recipients. (a) Genotyping analysis of C-Kit in the newborn mice. Two peaks can be observed in the sequence of the control Kitw/Kitwv mouse (indicated by arrowheads), while the HDR-1 SSC-derived mice have only one peak. (b) Genotyping analysis of selected SNPs 1–5 in the newborn pups. The SNP sites are indicated by arrowhead. SSCs: spermatogonial stem cells; HDR: homology-directed repair; ROSI: round spermatid injection; SNP: singlenucleotide polymorphism.