

Abstract
Multiple morphological abnormalities of the sperm flagella (MMAF) is a specific type of asthenoteratozoospermia, presenting with multiple morphological anomalies in spermatozoa, such as absent, bent, coiled, short, or irregular caliber flagella. Previous genetic studies revealed pathogenic mutations in genes encoding cilia and flagella-associated proteins (CFAPs; e.g., CFAP43, CFAP44, CFAP65, CFAP69, CFAP70, and CFAP251) responsible for the MMAF phenotype in infertile men from different ethnic groups. However, none of them have been identified in infertile Pakistani males with MMAF. In the current study, two Pakistani families with MMAF patients were recruited. Whole-exome sequencing (WES) of patients and their parents was performed. WES analysis reflected novel biallelic loss-of-function mutations in CFAP43 in both families (Family 1: ENST00000357060.3, p.Arg300Lysfs*22 and p.Thr526Serfs*43 in a compound heterozygous state; Family 2: ENST00000357060.3, p.Thr526Serfs*43 in a homozygous state). Sanger sequencing further confirmed that these mutations were segregated recessively in the families with the MMAF phenotype. Semiquantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) was carried out to detect the effect of the mutation on mRNA of the affected gene. Previous research demonstrated that biallelic loss-of-function mutations in CFAP43 accounted for the majority of all CFAP43-mutant MMAF patients. To the best of our knowledge, this is the first study to report CFAP43 biallelic loss-of-function mutations in a Pakistani population with the MMAF phenotype. This study will help researchers and clinicians to understand the genetic etiology of MMAF better.
Keywords: cilia and flagella-associated proteins, male infertility, multiple morphological abnormalities of the sperm flagella, whole-exome sequencing
INTRODUCTION
Multiple morphological abnormalities of the sperm flagellum (MMAF) is one of the more severe forms of sperm defect,1 characterized by bent, coiled, irregular, short, or absent sperm flagella.2,3,4,5,6 The sperm flagellum in MMAF patients often shows ultrastructural abnormalities associated with the “9 + 0” arrangement of dynein microtubules, such as lacking the central pair of microtubules, disorganized axoneme, and mitochondrial sheath, which in turn affects sperm motility and leads to male infertility.4,7,8,9
In the past few years, the development of next-generation sequencing technology has led to identification of a genetic cause in MMAF patients. Various pathogenic mutations have been found in genes encoding cilia and flagella-associated proteins (CFAPs), such as CFAP43, CFAP44, CFAP65, CFAP69, CFAP70, and CFAP251.2,5,10,11,12,13,14,15,16,17,18,19 It has been noted that all these CFAP-associated genes have diverse functions and location. For example, CFAP43, CFAP44, and CFAP65 are associated with the inner dynein arm (IDA) complex tether/tether head (T/TH); CFAP69 is associated with intraflagellar transport (IFT); CFAP70 is related to the outer dynein arm (ODA)-associated complex; and CFAP251 is identified in the calmodulin and spoke-associated complex (CSC).1 In 2017, Tang et al.2 identified biallelic loss-of-function mutations of CFAP43 in Chinese MMAF patients and further confirmed the pathogenicity in knockout mouse models of the Cfap43 ortholog gene. Later, in 2018, Coutton et al.10 also identified CFAP43 biallelic mutations in MMAF patients from different ethnic groups. Biallelic mutations of CFAP43 and CFAP44 have been reported to be account for approximately 8%–31% of studied MMAF cohorts.2,10,11 However, the genetic causes of MMAF among Pakistani patients remain unexplored. Given the existence of a traditional and close-knit society in Pakistan, approximately 65% of the population have consanguineous marriages.20 A high proportion of consanguineous marriage increases the risk of autosomal recessive disorders in offspring. Such kinds of autosomal recessive disease with identified genetic causes have been reported in the Pakistani population including primary microcephaly,21 deafness,22 retinitis pigmentosa,23 and infertility.24,25 Therefore, we wondered, whether CFAP43 mutations could be one of the genetic causes for Pakistani MMAF patients.
CFAP43 (also known as WDR96, ENST00000357060.3) is localized on chromosome 10 and contains 38 exons encoding a predicted 1665-amino-acid protein (Q8NDM7), specifically expressed in the human testis,11 and plays a vital role in the organization of the sperm flagellar axoneme. Animal model studies (knock out of CFAP43 and CFAP44 homologs in mice and Trypanosoma brucei) have produced evidence that mutations in these genes destabilize the entire complex, leading to both periaxonemal and axonemal defects and resulting in aborted flagella.10 However, owing to the absence of a specific antibody for CFAP43, the specific role and localization of the CFAP43 protein in the mouse testis and their molecular and cellular mechanisms are yet to be elucidated.26
We recruited two Pakistani families with three infertile men suffering from MMAF. Through whole-exome sequencing (WES) and Sanger sequencing, we identified novel biallelic loss-of-function mutations in CFAP43 in both families (Family 1: ENST00000357060.3, c.899_900del and c.1577_1578del in a compound heterozygous state; Family 2: ENST00000357060.3, c.1577_1578del in a homozygous state). Mutation (c.1577_1578del) was identified in both families and caused mRNA degradation in spermatozoa of the Family 2 patient.
To our knowledge, this is the first report that CFAP43 biallelic loss-of-function mutations cause MMAF in Pakistani populations. This study will help researchers and clinicians to better understand the genetic etiology of MMAF and would be of high interest for genetic counseling and diagnosis of MMAF.
PARTICIPANTS AND METHODS
Study participants
Two Pakistani families with three interfile men were recruited. Written informed consent was obtained from all the affected and control family members. This study was approved by the Institutional Ethical Committee of University of Science and Technology of China (USTC; Hefei, China) with the approval number of USTCEC202000003.
Semen analysis
All three patients had routine semen analysis performed twice according to the World Health Organization guidelines (2010).27 Sperm morphology was assessed as previously described by Zhang et al.24 The fixed smear slides were sequentially immersed for 30 s in ethanol of 80% and 50% concentration and washed with purified water and then placed in hematoxylin stain (Solarbio, Beijing, China) for 4 min followed by serially immersed for 30 s in purified water, acidic ethanol, running cold tap water, and ethanol of 50%, 80%, and 95% concentration, respectively. These slides were then dipped in Orange-G-6 stain (Solarbio) for 1 min and washed three times with 95% of ethanol. Finally, the slides were forward to Eosin Azure Stain (Solarbio) for 1 min and then washed with 95% and 100% ethanol in each two times for 30 s. These slides were then dipped in xylene:ethanol (1:1 ratio) for 1 min in a fume hood. At least 200 stained spermatozoa per sample were examined by optical microscopy (Nikon Eclipse 80i, Nikon, Tokyo, Japan). According to their characteristic defects, the morphological abnormalities of sperm flagella were divided into five categories: short, coiled, absent, bent, and irregular/caliber.
WES, sequencing data analysis, and Sanger sequencing
Genomic DNA was extracted from the peripheral blood of all available family members by using FlexiGene DNA Kit (QIAGEN, Hilden, Germany) as per the manufacturer’s instructions. For WES, AIExome Enrichment Kit V1 (iGeneTech, Beijing, China)-captured libraries were constructed for family members of Family 1 (I:1, I:2, II:1, and II:2) and Family 2 (III:1, III:2, IV:3, and IV:4) as instructed by the manufacturer. Sequencing was carried out on a Hiseq2000 platform (Illumina, San Diego, CA, USA). Clean reads were mapped to the human reference genome (hg19) by Burrows–Wheeler Alignment tool.28 Variants were discovered and annotated by the Genome Analysis Toolkit (GATK)29 and ANNOVAR.30 After that, specific filtration pipelines for each family are described in Supplementary Figure 1 (884.5KB, tif) and detailed in Supplementary Table 2 and Supplementary Table 3. Sanger sequencing was performed to verify the selected variants in all the available family members. The primers for PCR are listed in Supplementary Table 1.
Supplementary Table 2.
Details of filtered variants from whole-exome sequencing analysis pipeline for family 1
| Gene name | Mutation type | cDNA change | Phenotypes of mutant mice from MGI or literature, or expression in testes |
|---|---|---|---|
| ANKRD36C | Nonsynonymous SNV | C98T | Mutant mice have a mottled retina with photoreceptor degeneration and male infertility associated with oligozoospermia and asthenozoospermia |
| ANKRD36C | Frameshift substitution | 1577_1579G | The same as above |
| CELA3B | Nonsynonymous SNV | G358A | The expression of this gene is not detectable in human testis |
| CELA3B | Frameshift substitution | 2752_2753T | The same as above |
| CFAP43 | Frameshift substitution | 1577_1578G | Mice homozygous for a knock-out allele exhibit complete male sterility, asthenozoospermia, and teratozoospermia characterized by short, thick, and coiled flagella and sperm axonemal defects |
| CFAP43 | Frameshift substitution | 899_901A | The same as above |
| NBPF1 | Nonsynonymous SNV | G1714A | Mice homozygous for a null allele exhibit partial (in utero or perinatal) lethality, hyperactivity, and increased vertical activity |
| NBPF1 | Nonsynonymous SNV | T35G | Mice homozygous for a knock-out allele display delayed mammary tumor progression, impaired intestinal absorption of cholesterol, decreased gastric mucus accumulation, reduced secretion and accumulation of gallbladder mucin, and decreased susceptibility to cholesterol gallstone formation |
| PABPC3 | Frameshift substitution | 232_236T | Homozygotes for a null allele show high brain AEA levels, reduced pain sensation, altered behavioral responses to AEA, and sex-specific changes in ethanol intake and sensitivity. Homozygotes for the C385A variant show enhanced cued fear extinction and reduced anxiety-like behavior |
| PABPC3 | Nonframeshift substitution | The same as above | |
| PABPC3 | Frameshift substitution | 301_309G | Homozygotes for a null allele show high brain AEA levels, reduced pain sensation, altered behavioral responses to AEA, and sex-specific changes in ethanol intake and sensitivity. Homozygotes for the C385A variant show enhanced cued fear extinction and reduced anxiety-like behavior |
| PABPC3 | Nonsynonymous SNV | C17T | The same as above |
| PIK3C2G | Frameshift substitution | 595_596G | Homozygous null mice display hypoplasia of gut-associated lymph tissue due to defects in lymphocyte migration |
| PIK3C2G | Frameshift substitution | 24_25T | The same as above |
| PRIM2 | Frameshift substitution | 899_901A | Mice homozygous for a null allele are viable and fertile with no gross abnormalities |
| PRIM2 | Frameshift substitution | 497_498A | The same as above |
| RRP12 | Splicing | 1657+3A>C | Homozygotes for targeted null mutations exhibit a 1 h shorter circadian period under constant darkness and reduced expression of another circadian gene in the suprachiasmatic nucleus in response to acute light exposure |
| RRP12 | Nonsynonymous SNV | A1178T | The same as above |
| SPTA1 | Splicing | 565-3C>T | Mice homozygous or heterozygous for alleles of this gene exhibit varying degrees of hematopoietic defects |
| SPTA1 | Frameshift substitution | 51_52A | The same as above |
MGI: mouse genome informatic; AEA: anandamide; SNV: single-nucleotide variant
Supplementary Table 3.
Details of filtered variants from whole-exome sequencing analysis pipeline for family 2
| Gene name | Mutation type | cDNA change | Phenotypes of mutant mice from MGI or literature |
|---|---|---|---|
| CFAP43 | Frameshift | 1577_1578G | Mice homozygous for a knock-out allele exhibit complete male sterility, asthenozoospermia, and teratozoospermia characterized by short, thick, and coiled flagella and sperm axonemal defects |
| MYO15A | Nonsynonymous SNV | C10393T | Mutations in this gene result in profound deafness and neurological behavior |
| KRTAP9-9 | Nonsynonymous SNV | G422A | In the hair cortex, hair keratin intermediate filaments are embedded in an interfilamentous matrix, consisting of hair KRTAP, which are essential for the formation of a rigid and resistant hair shaft through their extensive disulfide bond cross-linking with abundant cysteine residues of hair keratins. The matrix proteins include the high-sulfur and high-glycine-tyrosine keratins |
| HTT | Nonsynonymous SNV | A107C | Null mutants gastrulate abnormally and die in utero. Conditional mutants are small with progressive neurodegeneration. Knock-ins of 20–150 CAG repeat units variably mimic Huntington’s with late-onset motor defects, reactive gliosis, and neuronal inclusions |
| KRT25 | Nonsynonymous | A716C | Mutations in this gene have a defect in hair formation resulting in a wavy coat and curly vibrissae |
| DONSON | Nonsynonymous | A752G | Homozygous knockout is early embryonic lethal. Heterozygous knockout causes no observable phenotype |
MGI: mouse genome informatic; SNV: single-nucleotide variant; KRTAP: keratin-associated protein
Supplementary Table 1.
Primers for polymerase chain reaction and Sanger sequencing of CFAP43 variants
| CFAP43 variants | Product size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| c.1577_1578del, p.Thr526Serfs*43 | 442 | ATCAGGAGAATCCCTCATCC | TTACCTCTTCACATGCCAAG |
| c.899_900del, p.Arg300LysfsTer22 | 395 | GCTCCTCTCTCTAATCTAGC | ATGTGACAGATCTGACATCC |
Transmission electron microscopic (TEM) analysis of spermatozoa
TEM analysis was performed according to Zhang et al.31 in 2019. Spermatozoa from the patient and a fertile control individual were taken and fixed in 0.1 mol l−1 phosphate buffer (PB; pH 7.4), comprising 0.2% picric acid, 8% glutaraldehyde, and 4% paraformaldehyde and stored at 4°C overnight. Samples were washed with 0.1 mol l−1 PB, postfixed with 1% osmium tetroxide. Spermatozoa cells were dehydrated through graded alcohol (30%, 60%, 90%, 100%, 100%, and 100%; 10 min for each bath) followed by infiltration of an epon resin and acetone mixture. Ultrathin (70 nm) sections were cut from the samples followed by staining with lead citrate and uranyl acetate. Tecnai 10 or 12 Microscopes (Philips CM10, Philips Electronics, Eindhoven, The Netherlands) at 120 kV or 100 kV were used to capture and examine the ultrastructure of the samples.
RNA extraction and semiquantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total sperm RNA from patient (Family 2-IV:3) and a fertile male was extracted with RNAiso Plus (TAKARA, Beijing, China) and reverse-transcribed into cDNA by PrimeScript RT Reagent Kit (TAKARA) as per the manufacturer’s instructions. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward: 5’-GTCAAGGCTGAGAACGGGAA-3’; reverse: 5’-AAATGAGCCCCAGCCTTCTC-3’) was used as an internal control and CFAP43 (Ensembl transcript ID: ENST00000357060.3) primers used were as follows, forward: 5’-AGCACGTCGTTTATGATCAG-3’; reverse: 5’-TGTGGCAGTAATGTAGGCAG-3’.
RESULTS
Clinical features of patients
This study was performed on two Pakistani families with three infertile men. Family 1-II:1 (57 years), Family 1-II:2 (55 years), and Family 2-IV:3 (39 years) had been married for 31 years, 26 years, and 14 years, respectively, but all were infertile. Detailed information was collected from each patient to exclude the possibility of associated infertility-related disease. All the individuals were healthy, with no previous history of any testicular injury or obstruction, no symptoms of Primary Ciliary Dyskinesia (PCD; disease ID: #MIM 244400). Detailed pedigree charts were constructed on the basis of information provided by their parents (Figure 1). All the physical characteristics and semen parameter values of the patients are presented in Table 1. The semen volumes, pH, and viscosity fell within the normal ranges according to the World Health Organization guidelines (2010).27 However, sperm concentrations were lower than the normal range (Table 1). Sperm morphological analysis reflected severe abnormalities of flagella including bent, short, coiled, irregular, and absent that are typical characteristics of MMAF (Figure 2a).
Figure 1.
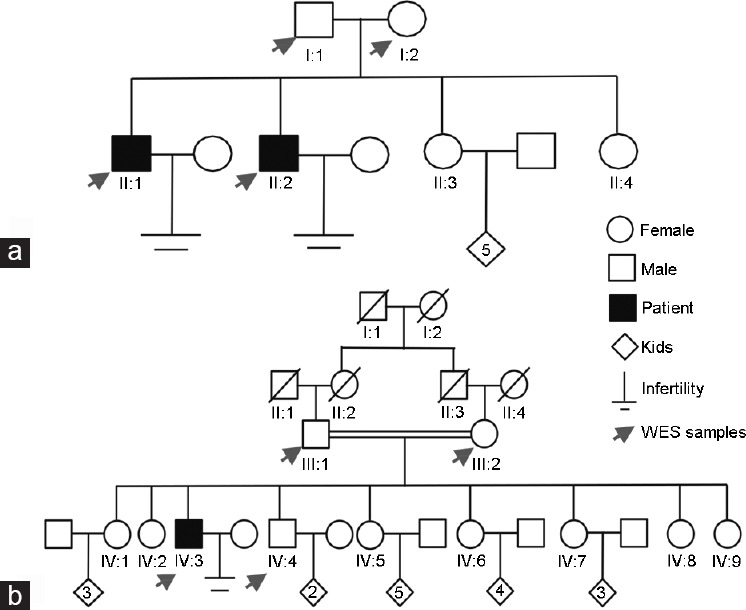
Pedigree of (a) Family 1 and (b) Family 2. Two Pakistani families with three infertile patients were recruited. I, II, III, and IV represent generation 1, 2, 3, and 4, respectively. Squares represent males, circles represent females, diamonds indicate offspring, and the inside numerals indicate the number of offspring. The slashes denote deceased family members. Solid squares indicate patients. Parallel slash lines indicate consanguineous marriage. Red arrows indicate the members selected for WES. WES: whole-exome sequencing.
Table 1.
Characteristics and sperm morphology in the patients
| Characteristic | Reference valuea | Family 1–II:1 | Family 1–II:2 | Family 2–IV:3 |
|---|---|---|---|---|
| Genotype | – | c.899_900del/c.1577_1578del | c.899_900del/c.1577_1578del | c.1577_1578del/c.1577_1578del |
| Age (year)b | – | 57 | 55 | 39 |
| Years of marriagec | – | 31 | 26 | 14 |
| BMI (kg m−2) | – | 37.1 | 31.3 | 23.5 |
| Semen parameters | ||||
| Semen volume (ml) | >1.5 | 2.0 | 3.0 | 3.3 |
| Semen pH | Alkaline | Alkaline | Alkaline | Alkaline |
| Sperm concentration (× 106 ml−1) | >15 | 9 | 6 | 7 |
| Motility (%) | >40 | 0 | 0 | 0 |
| Progressively motility (%) | >32 | 0 | 0 | 0 |
| Sperm morphology | ||||
| Normal flagella (%) | >4.0 | 3.2 | – | 0.8 |
| Abnormal flagella (%) | – | 96.7 | – | 99.1 |
| Short flagella (%) | – | 70.9 | – | 44.5 |
| Absent flagella (%) | – | 17.2 | – | 18.3 |
| Bent flagella (%) | – | 5.4 | – | 14.9 |
| Coiled flagella (%) | – | 5.0 | – | 12.6 |
| Irregular/caliber (%) | – | 0 | – | 8.8 |
| Head defects | ||||
| Normal head (%) | – | 6.8 | – | 4.9 |
| Abnormal head (%) | – | 93.1 | – | 95.2 |
| Tapered head (%) | – | 45.9 | – | 71.1 |
| Pyriform head (%) | – | 25.0 | – | 14.7 |
| Double head (%) | – | 1.4 | – | 0.9 |
| Large/amorphous head (%) | – | 0 | – | 0.9 |
| Round head (%) | – | 10.9 | – | 5.5 |
| Small head (%) | – | 6.5 | – | 0.9 |
| Absent head (%) | – | 3.4 | – | 1.2 |
aReference values were published in WHO (2010). bThe current ages. cThe current years of marriage. –: not available; BMI: body mass index; WHO: World Health Organization; del: deletion
Figure 2.
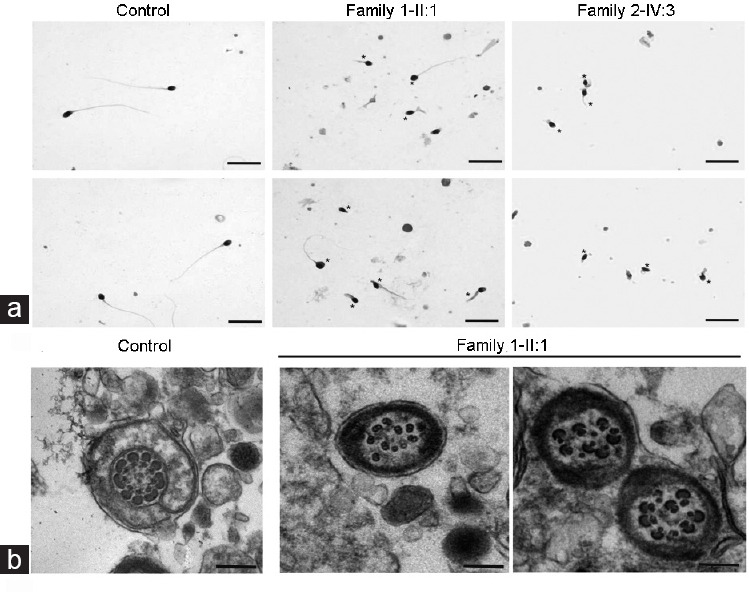
Morphology and transmission electron microscopic analysis of spermatozoa from normal control and infertile patients. (a) Most spermatozoa of patients (middle and right panels) presented abnormal sperm flagella (*), compared with control spermatozoa (left panel). Scale bars = 10 μm. (b) Cross-section of fertile male spermatozoa (left panel). An axoneme of a fertile male’s spermatozoa comprised DMTs circularly arranged around a CPC of microtubules (9 + 2 organization), surrounded by ODFs and FS. Cross-section of the patient II:1 of Family 1 (CFAP43-deficient), see right panel. Spermatozoa display totally disorganized axoneme; outer dense fibers and peripheral microtubules are misarranged. The central pair is displaced. Scale bars = 500 nm. DMTs: doublets of microtubules; CPC: central pair complex; ODF: outer dense fiber; FS: fibrous sheath; CFAP: cilia and flagella-associated protein.
Novel biallelic loss-of-function mutations in CFAP43 are candidate pathogenic variants in the families
To identify the genetic cause of MMAF, WES was performed for all available family members as shown in Figure 1. WES data were filtered according to the detailed pipeline in Supplementary Figure 1 (884.5KB, tif) . As stated in a previous study, MMAF is an autosomal recessive inheritance,17 so as from the family history of Family 1, and the parents in Family 2 were in a consanguineous marriage, we focused on homozygous/compound heterozygous mutations shared by patients. Finally, the filtration pipeline identified novel biallelic loss-of-function mutations in CFAP43 in both families (Family 1: ENST00000357060.3, c.899_900del and c.1577_1578del in a compound heterozygous state; Family 2: ENST00000357060.3, c.1577_1578del in a homozygous state). It is noteworthy that the frameshift mutation (c.1577_1578del) was identified in both families.
CFAP43 mutation induced severe axonemal disorganization
TEM was performed to observe the ultrastructure defects of patient II:1’s spermatozoa of Family 1, as well as normal sperm ultrastructure from a fertile control individual. For TEM, a typical microtubule structure was presented in the spermatozoa of the fertile control that contains a “9 + 2” axonemal arrangement of nine doublets of microtubules (DMTs) and two central pairs (CP), surrounded by a fibrous sheath (FS) and outer dense fibers (ODF) as shown in Figure 2b. In contrast to the fertile male spermatozoa, CFAP43-defecient sperm cross-sections showed axonemal and periaxonemal defects and approximately 82% of the cross-sections were abnormal (Figure 2b). The main defect was severe disorganization of the FS, ODF, and axonemal disassembly, and in some cross-sections the absence of central pair complex (CPC) (9 + 0 conformation).
CFAP43 mutations cosegregated with MMAF phenotype in the families and induced CFAP43 mRNA decay
Sanger sequencing confirmed that the WES-identified CFAP43 mutations cosegregated with MMAF phenotype in both families (Figure 3a and 3b). To determine the effects of the frameshift mutation (c.1577_1578del) on CFAP43 expression, we measured CFAP43 mRNA in spermatozoa of the patient from Family 2, using the sperm sample from a fertile male as control. As shown in Figure 3c, CFAP43 mRNA was detected in the control sample, but not in the patient IV:3. Owing to the unavailability of Family 1 patients’ fresh semen samples for mutant CFAP43 protein/mRNA detection, we compared the mutation c.899_900del with reported CFAP43 mutations that had been confirmed in mRNA or protein level. Our mutation, c.899_900del (predicted truncate protein, p.Arg300Lysfs*22), was close to p.Asn380Lysfs*3, which was previously identified by Wu et al.11 and has been confirmed to cause mRNA decay by quantitative polymerase chain reaction (qPCR), as well as the lack of CFAP43 protein by immunofluorescent staining in patients’ semen samples.
Figure 3.

Sanger sequencing results of CFAP43 mutations in DNA and mRNA levels. Chromatograms of the CFAP43 mutations from (a) Family 1 and (b) Family 2. Red/Blue arrows show the genomic position of CFAP43 mutations. (c) SqRT-PCR analysis of CFAP43 mRNA levels in male control and Family 2-IV:3 sperm samples. SqRT-PCR: semiquantitative reverse-transcriptase polymerase chain reaction; CFAP: cilia and flagella-associated protein; bp: base pair; Ref: reference; Het: heterozygous; chr10: chromosome 10; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; del: deletion.
DISCUSSION
In the current study, we recruited two Pakistani families with MMAF patients. After WES of all available family members, novel biallelic loss-of-function mutations in CFAP43 were identified in both families (Family 1: ENST00000357060.3, c.899_900del and c.1577_1578del in a compound heterozygous state; Family 2: ENST00000357060.3, c.1577_1578del in a homozygous state), as shown in Figure 4. Sanger sequencing further confirmed that these mutations were segregated recessively in the families with MMAF phenotype. Furthermore, the mutation c.1577_1578del has been confirmed to cause mRNA degradation in patient’s spermatozoa from the Family 2. TEM results of the patient II:1’s spermatozoa of Family 1 showed severe disorganization of the axoneme. This is the first report of novel biallelic loss-of-function mutations in CFAP43 causing MMAF in the Pakistani population.
Figure 4.
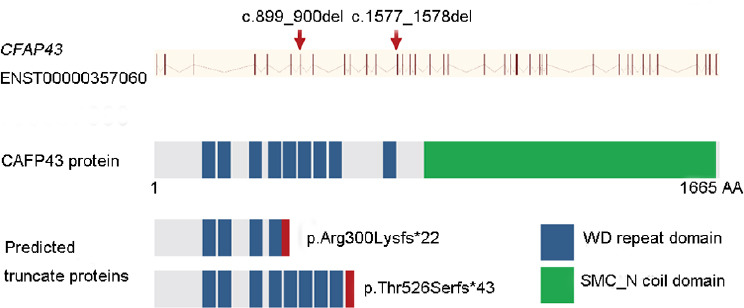
The identified mutations in CFAP43 gene and predicted mutant proteins. CFAP43 gene structure (Ensembl transcript ID: ENST00000357060) is shown with mutations identified in both families. Vertical bars indicate exons and slashed lines represent introns. CFAP43 (1665 AA) comprises two domains: WD (tryptophan-aspartic acid (W-D) repeat domain and SMC_N coil domain. CFAP: cilia and flagella-associated protein; AA: amino acid; SMC_N: N-terminus of structural maintenance of chromosome; del: deletion.
Of all identified CFAP43 mutations, 80% are loss-of-function mutations, which include frameshift, nonsense, and splice-site mutations (Figure 5 and Supplementary Table 4Ref2,10,11,32). These loss-of-function mutations (frameshift and nonsense) might cause mRNA degradation or produce truncate protein. Detailed sperm analyses indicated an increased number of immotile spermatozoa (98%–100%), and all patients’ spermatozoa had typical MMAF characteristics. Furthermore, no significant differences were observed among the semen parameters of the patients harboring CFAP43 mutations in the current study compared with the previously reported patients with other CFAP43 mutations (Supplementary Table 4). Wu et al.11 first examined two CFAP43 mutations’ effects (p.Asn380Lysfs*3 and p.Gln492Arg) on mRNA and protein level in patients’ spermatozoa and found that both mutations cause CFAP43 mRNA degradation. In the current study, we could not obtain fresh semen samples from patients of Family 1 to verify the CFAP43 mutation effects on mRNA and protein level. However, since the mutation (p.Arg300Lysfs*22) is close to the mutations verified by Wu et al.11 (p.Asn380Lysfs*3 and p.Gln492Arg), we speculate that CFAP43 mutations identified in our study have a similar effect on CFAP43 expression, resulting in complete loss of CFAP43 (Figure 4).
Figure 5.

Summary of all reported CFAP43 mutations in MMAF patients. (a) All compound heterozygous mutations are listed above the gene map; horizontal connections represent two mutations identified in one patient. All homozygous mutations are listed below the gene map. Red ones indicate the mutations identified in current study. (b) Statistic of all CFAP43 mutations. CFAP: cilia and flagella-associated protein; MMAF: multiple morphological abnormalities of the sperm flagella; del: deletion; WD: tryptophan-aspartic acid (W-D).
Supplementary Table 4.
Semen characteristics in the subjects carrying CFAP43 mutations
| Patient identified in the study | cDNA change | Effect on protein, or protein alteration | Semen volume (ml) | Sperm count (106 ml) | Motility (%) | Immotile (%) | MMAF phenotype | Reference |
|---|---|---|---|---|---|---|---|---|
| P003 | c.2802T>A | p.Cys934* | 2.2–3.8 | 16.1–39.4 | 0 | 100 | Yes | 2 |
| P028 | c.253C>T | p.Arg85Trp | 1.5–2.5 | 16.1–39.4 | 2 | 98 | Yes | |
| P029 | c.386C>A | p.Ser129Tyr | 2.5–4.0 | 12.2–18.9 | 1 | 99 | Yes | 32 |
| P6 | c.3661-2A> | NA | 3.0 | 15.8 | 0 | 100 | Yes | |
| P1 | c.1140_1143del | p.Asn380Lysfs*3 | 2.3 | 7.6 | 0 | 100 | Yes | 11 |
| P8 | c.739A>T | p.Lys247* | 2.4 | 25.8 | 0 | 100 | Yes | |
| P9 | c.1474G>C | p.Gln492Arg | 3.5 | 32.1 | 0 | 100 | Yes | |
| P10 | c.4600C>G | p.Leu1534Val | 4.1 | 19.2 | 0 | 100 | Yes | |
| P5 | c.4963C>T | p.Arg1655* | 2.9 | 20.1 | 0 | 100 | Yes | |
| P=10 | c.3541−2A>C c.1240_1241delGT c.2658G>A c.2680C>T c.3882delA c.3352C>T c.1302dupT c.1040T>C c.2141+5G>A |
p.Ser1181Lysfs*4 p.Val414LeufsTer46 p.Trp886Ter p.Arg894Ter p.Glu1294AspfsTer47 p.Arg1118Ter p.Leu435SerfsTer26 p.Val347Ala p.Lys714Val*11 |
3.5±1.4 (n=8) | 27.2±23.4 | 0±0 (n=9) | 100 | Yes | 10 |
| P=2 | c.899_900del c.1577_1578del |
p.Arg300Lysfs*22 p.Thr526Serfs*43 |
3.3 | 07 | 0 | 100 | Yes | Current study |
| P1 | c.1577_1578del | p.Thr526Serfs*43 | 2–3 | 6–9 | 0 | 100 | Yes | Current study |
CFAP43 and CFAP44 mutations account for 7.5%–30.8% of MMAF patients from a different study cohort, specified in a recent review.1 Tang et al.2 identified patients harboring CFAP44 or CFAP43 mutations, explaining 7.5% (4/30) of all patients with MMAF. However, Yan et al.32 identified 22.2% of 27 patients carrying CFAP44 or CFAP43 mutations. The most recent study by Wu et al.11 reported 30.8% of all patients Supplementary Table 5Ref2,7,11,12,15,18,32–35 summarized the percentages of involvement of CFAP43 and CFAP44, as well as other MMAF reported genes in different cohorts. Until now, only CFAP43 mutations have been identified in Pakistani MMAF patients in the current study.
Supplementary Table 5.
Percentages of involvement of the different sperm flagellum reported genes in the different cohorts
| Gene | Protein features | Percentage of involvement (%) | Reference |
|---|---|---|---|
| DNAH1 | Dynein heavy chain | 28 | 7 |
| CFAP65 | Coiled-coil domain-containing protein | 6.8 | 12 |
| CFAP43 and CFAP44 | WD repeat domains | 7.5 | 2 |
| 22.22 | 32 | ||
| 30.8 | 11 | ||
| FSIP2 | AKAP4 interacting domain | 5.1 | 33 |
| AK7 | ADK domain, coiled coil domain, DPY30 domain | 1.2 | 34 |
| WDR66 (CFAP251) | calcium regulating EF-hand domain | 9 | 15 |
| CFAP69 | Armadillo-type α-helical repeats | 2.6 | 18 |
| ARMC2 | Armadillo repeat-containing protein 2 | 2.4 | 35 |
According to previous information, good intracytoplasmic sperm injection (ICSI) outcomes are reported for MMAF patients with CFAP43 and CFAP44 mutations. The recorded rates of transferable embryo, implantation, and clinical pregnancy were 80%, 50%, and 100%, respectively, in CFAP43.5 Hence, it is worth mentioning that it would be more interesting for researchers and clinicians to apply ICSI for CFAP43-mutant MMAF patients and improving the prediction of ICSI outcomes for MMAF patients in Pakistan. However, it is very important to know the genetic screening of the wives of male patients carrying CFAP43 mutation before the couple asks for ICSI, to reduce the chances of genetic diseases in the offspring.
In conclusion, our study identified novel loss-of-function mutations in CFAP43 in Pakistani MMAF patients. These findings highlight the significance for genetic counseling and diagnosis for MMAF patients in the Pakistani population, while CFAP43 could be routinely genetic diagnosed. Further studies are needed to identify other pathogenic genes to characterize better MMAF in the Pakistani population.
AUTHOR CONTRIBUTIONS
IK and BS wrote the manuscript and performed semen analysis; SD, NU, AK, HA, XHJ, WS, MZ, and RK collected patients’ samples. JTZ, DRZ, and YWZ performed the WES sequencing and WES data analysis. QHS and HZ conceived and supervised the study, designed and analyzed data, and wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declared no competing interests.
Whole-exome sequencing (WES) analysis pipeline for (a) Family 1 and (b) Family 2.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 32070850), the National Natural Science Foundation of China (No. 31630050, 31890780, and 32061143006), the National Key Research and Developmental Program of China (2018YFC1003900, 2019YFA0802600, and 2016YFC1000600), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB19000000), and the Fundamental Research Funds for the Central Universities (No. YD2070002006).
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Touré A, Martinez G, Kherraf ZE, Cazin C, Beurois J, et al. The genetic architecture of morphological abnormalities of the sperm tail. Hum Genet. 2021;140:21–42. doi: 10.1007/s00439-020-02113-x. [DOI] [PubMed] [Google Scholar]
- 2.Tang S, Wang X, Li W, Yang X, Li Z, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2017;100:854–64. doi: 10.1016/j.ajhg.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chemes HE, Brugo S, Zanchetti F, Carrere C, Lavieri JC. Dysplasia of the fibrous sheath: an ultrastructural defect of human spermatozoa associated with sperm immotility and primary sterility. Fertil Steril. 1987;48:664–9. doi: 10.1016/s0015-0282(16)59482-5. [DOI] [PubMed] [Google Scholar]
- 4.Rawe V, Galaverna G, Acosta A, Olmedo SB, Chemes H. Incidence of tail structure distortions associated with dysplasia of the fibrous sheath in human spermatozoa. Hum Reprod. 2001;16:879–86. doi: 10.1093/humrep/16.5.879. [DOI] [PubMed] [Google Scholar]
- 5.Sha YW, Wang X, Su ZY, Mei LB, Ji ZY, et al. Patients with multiple morphological abnormalities of the sperm flagella harbouring CFAP44 or CFAP43 mutations have a good pregnancy outcome following intracytoplasmic sperm injection. Andrologia. 2019;51:131–51. doi: 10.1111/and.13151. [DOI] [PubMed] [Google Scholar]
- 6.Barthelemy C, Tharanne M, Lebos C, Lecomte P, Lansac J. Tail stump spermatozoa: morphogenesis of the defect. An ultrastructural study of sperm and testicular biopsy. Andrologia. 1990;22:417–25. doi: 10.1111/j.1439-0272.1990.tb02020.x. [DOI] [PubMed] [Google Scholar]
- 7.Khelifa MB, Coutton C, Zouari R, Karaouzène T, Rendu J, et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2014;94:95–104. doi: 10.1016/j.ajhg.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang SM, Li HB, Wang JX, Shi YC, Cheng HB, et al. Morphological characteristics and initial genetic study of multiple morphological anomalies of the flagella in China. Asian J Androl. 2015;17:513. doi: 10.4103/1008-682X.146100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chemes HE, Rawe VY. Sperm pathology: a step beyond descriptive morphology. Origin, characterization and fertility potential of abnormal sperm phenotypes in infertile men. Hum Reprod Update. 2003;9:405–28. doi: 10.1093/humupd/dmg034. [DOI] [PubMed] [Google Scholar]
- 10.Coutton C, Vargas AS, Amiri-Yekta A, Kherraf ZE, Mustapha SF, et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat Commun. 2018;9:1–18. doi: 10.1038/s41467-017-02792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, Li W, He X, Liu C, Fang Y, et al. Novel CFAP43 and CFAP44 mutations cause male infertility with multiple morphological abnormalities of the sperm flagella (MMAF) Reprod Biomed Online. 2019;38:769–78. doi: 10.1016/j.rbmo.2018.12.037. [DOI] [PubMed] [Google Scholar]
- 12.Wang WL, Tu CF, Nie HC, Meng LL, Li Y, et al. Biallelic mutations in CFAP65 lead to severe asthenoteratospermia due to acrosome hypoplasia and flagellum malformations. J Med Genet. 2019;56:750–7. doi: 10.1136/jmedgenet-2019-106031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CY, Lv MR, He XJ, Zhu Y, Amiri-Yekta A, et al. Homozygous mutations in SPEF2 induce multiple morphological abnormalities of the sperm flagella and male infertility. J Med Genet. 2020;57:31–7. doi: 10.1136/jmedgenet-2019-106011. [DOI] [PubMed] [Google Scholar]
- 14.Liu CY, He XJ, Liu WJ, Yang SM, Wang LB, et al. Bi-allelic mutations in TTC29 cause male subfertility with asthenoteratospermia in humans and mice. Am J Hum Genet. 2019;105:1168–81. doi: 10.1016/j.ajhg.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kherraf ZE, Amiri-Yekta A, Dacheux D, Karaouzene T, Coutton C, et al. A homozygous ancestral SVA-insertion-mediated deletion in WDR66 induces multiple morphological abnormalities of the sperm flagellum and male infertility. Am J Hum Genet. 2018;103:400–12. doi: 10.1016/j.ajhg.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Auguste Y, Delague V, Desvignes JP, Longepied G, Gnisci A, et al. Loss of calmodulin- and radial-spoke-associated complex protein CFAP251 leads to immotile spermatozoa lacking mitochondria and infertility in men. Am J Hum Genet. 2018;103:413–20. doi: 10.1016/j.ajhg.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He XJ, Li WY, Wu H, Lv MR, Liu WJ, et al. Novel homozygous CFAP69 mutations in humans and mice cause severe asthenoteratospermia with multiple morphological abnormalities of the sperm flagella. J Med Genet. 2019;56:96–103. doi: 10.1136/jmedgenet-2018-105486. [DOI] [PubMed] [Google Scholar]
- 18.Dong FN, Amiri-Yekta A, Martinez G, Saut A, Tek J, et al. Absence of CFAP69 causes male infertility due to multiple morphological abnormalities of the flagella in human and mouse. Am J Hum Genet. 2018;102:636–48. doi: 10.1016/j.ajhg.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beurois J, Martinez G, Cazin C, Kherraf ZE, Amiri-Yekta A, et al. CFAP70 mutations lead to male infertility due to severe astheno-teratozoospermia. A case report. Hum Reprod. 2019;34:2071–9. doi: 10.1093/humrep/dez166. [DOI] [PubMed] [Google Scholar]
- 20.Manzoor R, Imran W, Maken A, Syed T. Consanguineous marriages: effects on pregnancy outcomes in Pakistan. J Dev Policy Pract. 2018;2:78–105. [Google Scholar]
- 21.Ansar M, Ebstein F, Özkoç H, Paracha SA, Iwaszkiewicz J, et al. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum Mol Genet. 2020;29:1132–43. doi: 10.1093/hmg/ddaa032. [DOI] [PubMed] [Google Scholar]
- 22.Santos-Cortez RL, Faridi R, Rehman AU, Lee K, Ansar M, et al. Autosomal-recessive hearing impairment due to rare missense variants within S1PR2. Am J Hum Genet. 2016;98:331–8. doi: 10.1016/j.ajhg.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan MI, Kersten FF, Azam M, Collin RW, Hussain A, et al. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2011;118:1444–8. doi: 10.1016/j.ophtha.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 24.Zhang B, Ma H, Khan T, Ma A, Li T, et al. A DNAH17 missense variant causes flagella destabilization and asthenozoospermia. J Exp Med. 2020;217:e20182365. doi: 10.1084/jem.20182365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin H, Ma H, Hussain S, Zhang H, Xie X, et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med. 2018;21:62–70. doi: 10.1038/s41436-018-0015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Y, Wang J, Zhou L, Li H, Zheng B, et al. CFAP43-mediated intra-manchette transport is required for sperm head shaping and flagella formation. Zygote. 2021;29:75–81. doi: 10.1017/S0967199420000556. [DOI] [PubMed] [Google Scholar]
- 27.World Health Organization. Laboratory Manual for the Examination and Processing of Human Semen. 5th ed. Geneva: WHO Press; 2010. [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Khan I, Liu C, Ma A, Khan A, et al. Novel loss-of-function variants in DNAH17 cause multiple morphological abnormalities of the sperm flagella in humans and mice. Clin Genet. 2020;99:176–86. doi: 10.1111/cge.13866. [DOI] [PubMed] [Google Scholar]
- 32.Sha YW, Wang X, Xu X, Su ZY, Cui Y, et al. Novel mutations in CFAP44 and CFAP43 cause multiple morphological abnormalities of the sperm flagella (MMAF) Reprod Sci. 2019;26:26–34. doi: 10.1177/1933719117749756. [DOI] [PubMed] [Google Scholar]
- 33.Martinez G, Kherraf ZE, Zouari R, Mustapha SF, Saut A, et al. Whole-exome sequencing identifies mutations in FSIP2 as a recurrent cause of multiple morphological abnormalities of the sperm flagella. Hum Reprod. 2018;33:1973–84. doi: 10.1093/humrep/dey264. [DOI] [PubMed] [Google Scholar]
- 34.Lorès P, Coutton C, El Khouri E, Stouvenel L, Givelet M, et al. Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia. Hum Mol Genet. 2018;27:1196–211. doi: 10.1093/hmg/ddy034. [DOI] [PubMed] [Google Scholar]
- 35.Coutton C, Martinez G, Kherraf ZE, Amiri-Yekta A, Boguenet M, et al. Bi-allelic mutations in ARMC2 lead to severe astheno-teratozoospermia due to sperm flagellum malformations in humans and mice. Am J Hum Genet. 2019;104:331–40. doi: 10.1016/j.ajhg.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole-exome sequencing (WES) analysis pipeline for (a) Family 1 and (b) Family 2.