

ABSTRACT
Emerging evidence suggests that endothelial activation plays a central role in the pathogenesis of acute respiratory distress syndrome (ARDS) and multiorgan failure in patients with coronavirus disease 2019 (COVID-19). However, the molecular mechanisms underlying endothelial activation in COVID-19 patients remain unclear. In this study, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral proteins that potently activate human endothelial cells were screened to elucidate the molecular mechanisms involved in endothelial activation. It was found that nucleocapsid protein (NP) of SARS-CoV-2 significantly activated human endothelial cells through Toll-like receptor 2 (TLR2)/NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways. Moreover, by screening a natural microbial compound library containing 154 natural compounds, simvastatin was identified as a potent inhibitor of NP-induced endothelial activation. Remarkably, though the protein sequences of N proteins from coronaviruses are highly conserved, only NP from SARS-CoV-2 induced endothelial activation. The NPs from other coronaviruses such as SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), HUB1-CoV, and influenza virus H1N1 did not activate endothelial cells. These findings are consistent with the results from clinical investigations showing broad endotheliitis and organ injury in severe COVID-19 patients. In conclusion, the study provides insights on SARS-CoV-2-induced vasculopathy and coagulopathy and suggests that simvastatin, an FDA-approved lipid-lowering drug, may help prevent the pathogenesis and improve the outcome of COVID-19 patients.
IMPORTANCE Coronavirus disease 2019 (COVID-19), caused by the betacoronavirus SARS-CoV-2, is a worldwide challenge for health care systems. The leading cause of mortality in patients with COVID-19 is hypoxic respiratory failure from acute respiratory distress syndrome (ARDS). To date, pulmonary endothelial cells (ECs) have been largely overlooked as a therapeutic target in COVID-19, yet emerging evidence suggests that these cells contribute to the initiation and propagation of ARDS by altering vessel barrier integrity, promoting a procoagulative state, inducing vascular inflammation and mediating inflammatory cell infiltration. Therefore, a better mechanistic understanding of the vasculature is of utmost importance. In this study, we screened the SARS-CoV-2 viral proteins that potently activate human endothelial cells and found that nucleocapsid protein (NP) significantly activated human endothelial cells through TLR2/NF-κB and MAPK signaling pathways. Moreover, by screening a natural microbial compound library containing 154 natural compounds, simvastatin was identified as a potent inhibitor of NP-induced endothelial activation. Our results provide insights on SARS-CoV-2-induced vasculopathy and coagulopathy, and suggests that simvastatin, an FDA-approved lipid-lowering drug, may benefit to prevent the pathogenesis and improve the outcome of COVID-19 patients.
KEYWORDS: COVID-19, SARS-CoV-2, nucleocapsid protein, endothelial activation, simvastatin, endothelial cells
INTRODUCTION
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the coronavirus disease 2019 (COVID-19), triggered a global pandemic that has led to an unprecedented worldwide public health crisis (1). Before SARS-CoV-2, two other highly pathogenic coronaviruses emerged in the past 2 decades, including severe acute respiratory syndrome coronavirus (SARS-CoV) (2) and Middle East respiratory syndrome coronavirus (MERS-CoV) (3). In addition, four endemic human coronaviruses (i.e., OC43, 229E, NL63, and HKU1) cause common cold respiratory diseases (4). COVID-19 is characterized by progressive respiratory failure resulting from diffuse alveolar damage, inflammatory infiltrates, endotheliitis, and pulmonary and systemic coagulopathy forming obstructive microthrombi with multiorgan dysfunction (5–8). Pathological findings of cell swelling, severe endothelial injury, disruption of intercellular junctions, and basal membrane contact loss in COVID-19 patients imply that the destruction of endothelial cells (ECs) leads to pulmonary vascular endotheliitis and alveolar capillary microthrombi (9–12). Together, emerging evidence suggests that endothelial activation is an early hallmark of multiorgan damage in patients with COVID-19 (13). Moreover, thrombotic complications are a relevant cause of death in patients with COVID-19 (14). Therefore, understanding the molecular mechanisms of the endothelial activation caused by SARS-CoV-2 and pathways involved in the regulation of endothelial dysfunction could lead to new therapeutic strategies against COVID-19.
SARS-CoV-2 infects the host using angiotensin-converting enzyme 2 (ACE2) as its receptor (15). ACE2 is an integral membrane protein that is expressed by airways and lung alveolar epithelial cells, enterocytes, and vascular endothelial cells (16). Though the primary target tissues of SARS-CoV-2 are airways and lungs, there is also evidence of direct viral infection of endothelial cells and diffuse endothelial inflammation in COVID-19 disease (17). Moreover, vulnerable patients with pre-existing endothelial dysfunction, which is associated with male sex, smoking, hypertension, diabetes, obesity, and established cardiovascular disease, are associated with adverse outcomes in COVID-19 (18). Together, these clinical findings suggest that endothelial cells play a central role in the pathogenesis of ARDS and multiorgan failure in patients with COVID-19. Therefore, the vascular system is increasingly being addressed as a major therapeutic target for defeating COVID-19.
The potential molecular mechanisms whereby SARS-CoV-2 induces endothelial activation, dysfunction and injury may contribute to the direct toxic effect of viral proteins. The genome of SARS-CoV-2 encodes 29 viral proteins, including 16 nonstructural proteins (NSP1 to NSP16), 4 structural proteins, including spike (S), membrane (M), nucleocapsid (N), and envelope (E) proteins, and 9 accessory proteins (19, 20). Though their functions in viral life cycle are increasingly being studied, their impacts on host cells are largely unknown. To determine whether distinct viral proteins can induce endothelial activation, recombinant SARS-CoV-2 proteins were evaluated for their potential to activate human endothelial cells. We found that N protein potently induced endothelial activation via Toll-like receptor 2 (TLR2)/NF-κB and mitogen-activated protein kinase (MAPK) signal pathways. To identify potential therapeutic agents targeting N protein (NP)-induced endothelial activation, a natural microbial compound library containing 154 natural compounds was screened, and a single drug, simvastatin, was identified as specific inhibitor of N protein-induced endothelial activation. These results suggest that N protein released from SARS-CoV-2-infected cells may contribute to the broad activation of endothelium and tissue inflammation and that simvastatin may benefit to prevent the viral infection-induced pathogenesis and improve the outcome of COVID-19 patients.
RESULTS
Identification of NP as a potent inducer of human endothelial cell activation.
To understand the molecular mechanisms whereby SARS-CoV-2 induces endothelial activation, we purchased eight recombinant SARS-CoV-2 viral proteins, including three structural proteins (S, N, and E proteins) and five nonstructural proteins (NSP1, NSP3, NSP5, NSP7, and NSP8) from commercial resources. The recombinant proteins are from HEK293 cells or S2 cells. The protein preparation is reported to be endotoxin free, which we confirmed using the chromogenic endpoint Limulus amebocyte lysate (LAL) assay (<0.01 endotoxin units [EU]/ml). The proteins were added to human primary lung microvascular endothelial cells (HLMECs) for 8 h. Western blotting was used to analyze the expression of ICAM-1 and VCAM-1, the markers of endothelial cell activation. As shown in Fig. 1a, N protein significantly induced the expression of ICAM-1 and VCAM-1.
FIG 1.

SARS-CoV2 nucleocapsid protein (NP) is a potent inducer of human endothelial cell activation. (a) HLMECs were incubated with SARS-CoV2 structural proteins (S, N, and E proteins; 1 μg/ml) and five nonstructural proteins (NSP1, NSP3, NSP5, NSP7, and NSP8; 1 μg/ml) for 8 h. (b) HLMECs were treated with 1 μg/ml of NP or 10 ng/ml of TNF-α for different incubation periods as indicated. (c) HLMECs were incubated with indicated concentrations of NP for 8 h. TNF-α at 10 ng/ml served as a positive control. (d) Different cultured cells, including mouse lung vascular endothelial cells (MECs), A549 cells, 293T cells, HUVECs, HAECs, HCAECs, HDMECs, and HLMECs, were treated with NP (1 μg/ml) for 8 h. The expression of ICAM-1, VCAM-1, and VE-cadherin was detected by Western blotting. β-Actin served as a loading control. (e) HLMECs were treated with PBS, NP (1 μg/ml), TNF-α (10 ng/ml), or lipopolysaccharide (LPS) (1 μg/ml) for 8 h. The total RNA was isolated and qPCR was performed for measuring the mRNA levels of TNF-α, ICAM-1, VCAM-1, MCP-1, and IL-6. (f) HLMECs were treated with PBS, NP (1 μg/ml), or TNF-α (10 ng/ml) for 8 h and cocultured with Zombie Red-labeled THP-1 cells for 1 h. After being washed, the adherent cells were imaged and quantitatively analyzed.
Tumor necrosis factor alpha (TNF-α) is the most potent endogenous inducer of endothelial activation, which serves as a positive control. To further confirm the effect of N protein on endothelial cell activation, we incubated HLMECs with different doses of N protein or for different incubation periods. As shown in Fig. 1b, N protein induced ICAM-1 expression at 4 h, and it continued to increase up to 24 h, which was similar to the expression pattern induced by TNF-α. A similar pattern of expression of VCAM-1 was also induced by N protein. However, the expression of VE-cadherin, an EC junction structure protein, was not changed. In addition, N protein induced ICAM-1 expression significantly at 0.05 μg/ml and more potently at 1 μg/ml, which is comparable to the effect of TNF-α at 10 ng/ml (Fig. 1c). Next, we tested whether N protein also induced activation of other endothelial cells. Human umbilical vein endothelial cells (HUVECs), human aortic endothelial cells (HAECs), human coronal artery endothelial cells (HCAECs), human dermal microvascular endothelial cells (HDMECs), HLMECs, 293T cells, A549 cells, and mouse lung vascular endothelial cells (MECs) were incubated with 1 μg/ml of N protein for 8 h. As shown in Fig. 1d, N protein significantly induced activation of all human endothelial cells tested but did not induce expression of ICAM-1 and VCAM-1 in 293T, A549, and mouse endothelial cells. We also probed the same blot using anti-mouse ICAM-1 and VCAM-1 antibodies and confirmed that N protein did not induce mouse EC activation.
To further confirm whether N protein induce the expression of adhesion molecules and proinflammatory cytokines at the transcriptional level, we examined the changes of ICAM-1, VCAM-1, E-selectin, TNF-α, monocyte chemoattractant protein 1 (MCP-1), and interleukin 1β (IL-1β) mRNA levels in NP-treated HLMECs. As shown in Fig. 1e, N protein potently induced the expression of adhesion molecules, inflammatory cytokines, and chemokines, which is similar to the function of TNF-α, a potent endogenous inducer of endothelial activation. The expression of ICAM-1 and VCAM-1 on the surfaces of endothelial cells contributes to leukocyte adherence to endothelial cells (21, 22). To examine if N protein also induces the monocyte adherence to activated endothelial cells, HLMECs were stimulated with N protein (1 μg/ml) or TNF-α (10 ng/ml) for 8 h and then Zombie Red fluorescence-labeled human primary monocytes (Lonza) were cocultured with the activated endothelial cells for 1 h. After being washed, the adherent cells were visualized by fluorescence microscopy and counted in a double-blind manner. As shown in Fig. 1f, both N protein and TNF-α significantly increased monocyte adherence on ECs, compared with that in the control group. Taken together, these results suggest that N protein is a potent inducer of endothelial cell activation and that it may play a key role in SARS-CoV-2-induced lung inflammation and multiorgan failure.
N protein activated NF-κB and MAPK signaling pathways in human endothelial cells.
It is well known that the expression of ICAM-1 and VCAM-1 is controlled by MAPK and NF-κB signaling pathways (23, 24). To test if N protein can activate these signal pathways, HLMECs were incubated with NP (1 μg/ml) for 0, 15, 30, 45, 60, and 120 min, and the cell lysates were analyzed by Western blotting. As shown in Fig. 2, NP treatment induced the phosphorylation of IκB kinases (IKKs), p65, IκBα, JNK, and p38 and led to IκBα degradation. The time patterns of phosphorylation of these signaling molecules were similar to that of TNF-α-induced activation of the signal pathways. It is interesting that S protein did not induce activation of NF-κB signaling but weakly induced activation of MAPK pathways, including ERK1/2, JNK, and p38. These results suggest that N protein activated JNK, p38, and NF-κB signal pathways in human endothelial cells.
FIG 2.

N protein activated NF-κB and MAPK signaling pathways in human endothelial cells. HLMECs were incubated with NP (1 μg/ml), SP (1 μg/ml), TNF-α (10 ng/ml), and LPS (1 μg/ml) for the indicated times. The phosphorylation of IKKs, p65, IκBα, ERK, JNK, and p38, as well as IκBα degradation, was detected by Western blotting. GAPDH served as a loading control.
N protein induced endothelial cell activation via TLR2-mediated signaling pathway.
Next, we investigated how N protein activates NF-κB and MAPK signaling pathways in human endothelial cells. First, we tested if N protein acts on cell surface receptors or intercellular signal proteins. Pretreatment with inhibitors of endocytosis, such as Pitstop2 and Dynasore hydrate, did not affect NP-induced expression of ICAM-1 and VCAM-1 (Fig. 3a), suggesting that internalization of N protein is not required for its action on endothelial activation. Moreover, we transfected the expression plasmid encoding N protein into HLMECs. Though N protein was overexpressed in endothelial cells, intercellular N protein did not induce the expression of ICAM-1 and VCAM-1 (Fig. 3b). Furthermore, incubation of HLMECs with N protein for different times showed that N protein bound to endothelial cells in a time-dependent manner (Fig. 3c). These results suggest that N protein may bind to a kind of receptor on endothelial cells and trigger the NF-κB and MAPK signal pathways. Next, HLMECs were pretreated with antagonists of TLR2 (CU-CPT22), TLR4 (LPS-RS), and IL-1R (IL-1R antagonist) for 1 h and then incubated with 1 μg/ml of N protein for 8 h. The expression of ICAM-1 and VCAM-1 was detected by Western blotting. As shown in Fig. 3a, TLR2 antagonist (CU-CPT22) significantly blocked NP-induced expression of ICAM-1 and VCAM-1 in human endothelial cells, suggesting that N protein may bind to TLR2 to trigger the activation of NF-κB and MAPK and induce endothelial activation. Figure 3d further shows that CU-CPT22 dose-dependently inhibited NP-induced expression of ICAM-1 and VCAM-1. To further confirm that N protein is able to activate TLR2, both wild-type 293T cells (without TLR2 expression) and TLR2-overexpressed 293T cells were treated with or without N protein. As shown in Fig. 3e, N protein did not induce phosphorylation of JNK and p38 in wild-type 293T cells. However, N protein significantly induced phosphorylation of JNK and p38 in 293T-TLR2 cells. Finally, as shown in Fig. 3a, pretreatment with the inhibitors of IKK, JNK and p38 completely blocked NP-induced expression of ICAM-1 and VCAM-1. Taken together, these results suggest that N protein activates endothelial cells via TLR2-mediated NF-κB and MAPK signal pathways.
FIG 3.

N protein induced endothelial cell activation via the TLR2-mediated signaling pathway. (a) HLMECs were pretreated with inhibitors of endocytosis (Pitstop2 [12.5 μM] and Dynasore hydrate [12.5 μM]) and antagonists of TLR4 (LPS-RS [10 μg/ml]), TLR2 (CU-CPT22 [20 μM]), IL-1R (IL-1R antagonist [20 μM]), inhibitors of IKK (IKK16 [20 μM]), ERK (U0126 [20 μM]), JNK (JNK inhibitor V [20 μM]), and p38 (SB203580 [20 μM]) for 1 h followed by treatment with NP (1 μg/ml) for 8 h. (b) The Flag control and Flag-NP expression plasmids were transfected into HLMECs by electroporation. The whole-cell lysate was harvested after 48 h transfection. The expression of ICAM-1, VCAM-1, NP, and Flag was detected by Western blotting. (c) HLMECs were incubated with NP (1 μg/ml) for different time as indicated. After being washed, the cells were harvested, and NP was detected by Western blotting. (d) HLMECs were treated with the indicated concentrations of CU-CPT22 for 1 h followed by treatment with NP (1 μg/ml) for 8 h. (e) Wild-type (left) and TLR2-overexpressing 293T cells were treated with or without NP (1 μg/ml) for 15 min. pJNK and pP38 were detected by Western blotting. GAPDH served as a loading control. All experiments were repeated at least once.
Identification of simvastatin as an effective inhibitor of N protein-induced endothelial activation.
To screen the inhibitors of N protein-induced endothelial activation, 154 chemicals from a microbial natural product library (Target Molecule, Wellesley Hills, MA) were added to individual wells of 48-well plates at 30 μM 1 h before the induction of N protein (1 μg/ml). The effect of chemicals on endothelial activation was measured by Western blotting with anti-ICAM-1 antibody. Among 154 chemicals, we found that 12 chemicals showed significant inhibition of N protein-induced ICAM-1 expression, which include simvastatin, lovastatin, rapamycin, cyclosporine, menadione, 1,4-naphthoquinone, l-thyroxine, thiostrepton, monensin, amphotericin B, gramicidin, and abamectin (Fig. 4). The latter five are antibiotics which are toxic for cells and used in animals, not suitable for human use. Menadione and 1,4-naphthoquinone are vitamin K derivatives which have antibacterial, antiviral, and anti-inflammation activities. Rapamycin and cyclosporine are immunosuppressants. Simvastatin and lovastatin are FDA-approved blood lipid-lowering drugs with anti-inflammatory roles.
FIG 4.

Screening of chemicals for inhibition of N protein-induced endothelial activation. A total of 155 chemicals from microbial natural product library were added to HLMECs at 30 μM 1 h before the induction of N protein (1 μg/ml). The effect of chemicals on endothelial activation was measured by Western blotting with anti-ICAM-1 antibody. β-Actin served as a loading control. Arrows indicate effective compounds; stars indicate toxic compounds causing cell death. The experiments were repeated once.
Simvastatin is an effective inhibitor of N protein-induced endothelial activation in vitro.
To confirm the specific inhibitory role of simvastatin in N protein-induced endothelial activation, we compared the effect of simvastatin, lovastatin, atorvastatin, mevastatin, and rosuvastatin. As shown in Fig. 5a, among the five statins tested, simvastatin potently inhibited N protein-induced expression of ICAM-1 and VCAM-1. Consistent with the screening results, lovastatin showed mild effect on N protein-induced endothelial activation. The other three statins did not affect N protein-induced endothelial activation. We further confirmed the effect of simvastatin on N protein-induced endothelial activation in a dose-dependent manner (Fig. 5b). Moreover, both simvastatin and lovastatin treatments significantly inhibited N protein-induced NF-κB activation (Fig. 5c). Consistently, simvastatin pretreatment also blocked monocyte adhesion to the activated endothelial cells (Fig. 5d). Simvastatin is a member of the class of hexahydronaphthalenes like lovastatin in which the 2-methylbutyrate ester moiety has been replaced by a 2,2-dimethylbutyrate ester group. Simvastatin is derived from lovastatin. Lovastatin is a fatty acid ester that is mevastatin carrying an additional methyl group on the carbobicyclic skeleton. The structures of simvastatin, lovastatin, and mevastatin are very similar. Mevastatin does not have a methyl group; however, both simvastatin and lovastatin have this group, which suggests that the group may be important for its inhibitor effect on N protein-induced endothelial activation (Fig. 6).
FIG 5.

Simvastatin is an effective inhibitor of N protein-induced endothelial activation in vitro. (a) HLMECs were pretreated with simvastatin, lovastatin, atorvastatin, mevastatin, and rosuvastatin at 30 μM for 1 h followed by treatment with NP (1 μg/ml) for 8 h. (b) HLMECs were pretreated with indicated concentrations of lovastatin and simvastatin for 1 h followed by treatment with NP (1 μg/ml) for 8 h. The expression of ICAM-1 and VCAM-1 was detected by Western blotting. (c) HLMECs were pretreated with indicated concentrations of lovastatin and simvastatin for 1 h by treatment with NP (1 μg/ml) for 15 min. The activation of NF-κB and MAPK signal pathways was detected by Western blotting. (d) HLMECs were pretreated with or without lovastatin (Lova) or simvastatin (Simva) followed by treatment with NP (1 μg/ml) for 8 h and then cocultured with Zombie Red-labeled THP-1 cells for 1 h. After washing, the adherent cells were imaged.
FIG 6.

Chemical structures of simvastatin, lovastatin, mevastatin, atorvastatin, and rosuvastatin. Structures were adapted from Wikipedia (simvastatin, https://commons.wikimedia.org/w/index.php?curid=4486633; lovastatin, https://commons.wikimedia.org/wiki/File:Lovastatin.svg; mevastatin, https://commons.wikimedia.org/wiki/File:Mevastatin.svg; atorvastatin, https://commons.wikimedia.org/wiki/File:Atorvastatin.svg; rosuvastatin, https://commons.wikimedia.org/wiki/File:Rosuvastatin_structure.svg; all structures are available under open access licenses or are in the public domain).
The N protein from SARS-CoV-2 but not the other coronaviruses potently induced endothelial cell activation.
There are seven types of coronaviruses infecting humans, including SARS-CoV-2, SARS-CoV, MERS-CoV, OC43-CoV, HKU1-CoV, 229E-CoV, and NL63-CoV. The first three are highly pathogenic and cause severe problems for humans. However, the latter four are less pathogenic and only cause the common cold. Though SARS-CoV and MERS-CoV display high sequence similarity to SARS-CoV-2, patients infected with SARS-CoV-2 are commonly affected by vascular injury and thrombosis formation (25), which was not observed in the patients infected with SARS-CoV and MERS-CoV. To understand the molecular basis of the pathogenesis, we compared the effect of different N proteins from SARS-CoV-2, SARS-CoV, MERS-CoV, HKU1-CoV, and H7N9 on endothelial activation. Interestingly, as shown in Fig. 7, only the N protein from SARS-CoV-2 potently induced endothelial activation. The other N proteins, though their sequence is highly conserved, did not affect endothelial activation (Fig. 7). These results may explain why SARS-CoV-2-infected patients developed severe vascular injury and thrombosis and why the virus affects many organs.
FIG 7.
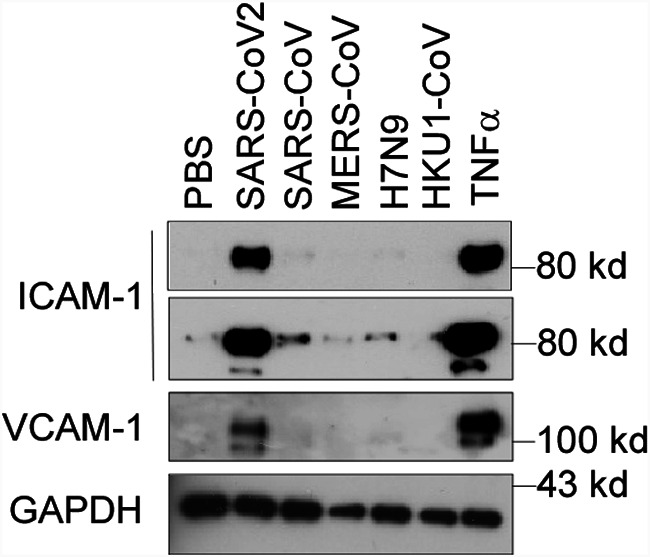
The N protein from SARS-CoV2 but not the other coronaviruses potently induced endothelial cell activation. HLMECs were treated with or without five different recombinant viral N proteins (1 μg/ml), including SARS-CoV2, SARS-CoV, MERS-CoV, H7N9, and HKU1-CoV, for 8 h. The expression of ICAM-1 and VCAM-1 was detected by Western blotting. TNF-α (10 ng/ml) served as a positive control. GAPDH served as the loading control. The experiments were repeated at least once.
DISCUSSION
Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2, is a worldwide challenge for health care systems. The leading cause of mortality in patients with COVID-19 is ARDS and multiorgan failure. Emerging evidence suggests that pulmonary endothelial cells contribute to the initiation and progression of ARDS by altering vessel barrier integrity, promoting a procoagulation state, inducing vascular inflammation (endotheliitis), and mediating inflammatory cell infiltration. However, the molecular mechanisms underlying SARS-CoV-2-induced endothelial activation and vascular injury remain unclear. Though some reports indicated direct infection of endothelial cells by SARS-CoV-2 in human samples, emerging evidence suggests that ACE2 is not highly expressed in human endothelial cells and SARS-CoV-2 is not able to infect human endothelial cells in vitro (26). Thus, the direct damage of endothelial cells by SARS-CoV-2 infection cannot explain the broad endothelial dysfunction in COVID-19 patients. The possible mechanisms by which SARS-CoV-2 infection causes endothelial activation may be attributed to the inflammatory and toxic roles of circulating viral proteins released from infected and lysed cells and inflammatory cytokines secreted from inflammatory and immune cells.
In this study, we screened the recombinant SARS-CoV-2 viral proteins that are able to activate human endothelial cells. We found that nucleocapsid protein (N protein) of SARS-CoV-2 potently activate endothelial cells through TLR2/NF-κB and MAPK signal pathways, by which N protein significantly induced the expression of ICAM-1 and VCAM-1 as well as other inflammatory cytokines and chemokines such as TNF-α, IL-1β, and MCP-1. As ICAM-1 and VCAM-1 are major adhesion molecules expressed on activated endothelial cells and mediate inflammatory cell infiltration into tissues, N protein may play a key role in the development of ARDS and multiorgan injury.
The N protein is highly abundant in the viruses. Its function involves entering the host cells, binding to the viral RNA genome, and forming the ribonucleoprotein core to facilitate its replication and process the virus particle assembly and release (27). Previous reports showed that the N proteins from SARS-CoV and MERS-CoV were highly inflammatory nature to promote the expression of inflammatory cytokines, chemokines, and prothrombinase and were able to induce acute lung inflammation in a mouse model (28–30). The N protein from the hantavirus Andes virus increased basal endothelial cell permeability by activating RhoA signaling (31). The effect of N protein from SARS-CoV-2 on host cells is less studied. In this paper, we report for the first time that the N protein from SARS-CoV-2 acts as a pathogen-associated molecular pattern (PAMP) to directly bind to TLR2 and activate NF-κB and MAPK signaling. In an unbiased survey of the phosphorylation landscape of SARS-CoV-2 infection, SARS-CoV-2 infection truly promotes activation of CK2 and p38 MAPK (32). Moreover, a recent human study showed that the serum levels of soluble ICAM-1 and VACM-1 were elevated in patients with mild COVID-19, dramatically elevated in severe cases, and decreased in the convalescence phase (33). Taken together, the results of the current study identify N protein as a potent factor to induce endothelial activation and provide insights to understand the phenomenon of broad endothelial dysfunction and multiorgan injury that commonly appear in patients with severe COVID-19.
One might ask how the NP protein, a known intracellular protein, binds to the cell surface via TLR2 to activate endothelial cells. Nucleocapsid protein is the most abundant protein in SARS-CoV-2 virions. When the virus-infected host cells lyse, they can be released to extracellular space or circulation. Li et al. recently reported that NP has been detected in the serum from 76% of SARS-CoV-2 positive patients at an early stage (34). Our results suggest that circulating NP may broadly activate microvascular endothelial cells, leading to the development of thrombosis and multiorgan failure in severe SARS-CoV-2 patients.
Our study suggests that targeting of N protein may help prevent or treat pathogenesis and multiorgan injury in COVID-19 patients. By screening a natural microbial compound library containing 154 natural compounds, we identified simvastatin, an FDA-approved lipid-lowering drug, as a potent inhibitor of N protein-induced endothelial activation. Several groups have raised the idea that statins can be used as early therapy to mitigate COVID-19-associated ARDS and cytokine storm syndrome (35–37). Several recent reports showed that in-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19 (38–40). There are more than 20 statins available in clinical use. We tested five different statins. Only simvastatin showed potent inhibitory activity on N protein-induced endothelial activation. Lovastatin also showed a mild inhibitory effect, which may be due to the similarity of its structure and that of simvastatin (Fig. 6). Because many reports showed that simvastatin has an anti-inflammatory role (41), it would be more effective in the treatment of COVID-19 patients by multiple mechanisms. Our results indicate that simvastatin may be of more benefit for the treatment of COVID-19 by potently suppressing endothelial activation.
Many patients with severe COVID-19 show signs of a cytokine storm (42). The high levels of cytokines amplify the destructive process by leading to EC activation, disseminated intravascular coagulation (DIC), inflammation, and vasodilation of the pulmonary capillary bed. This results in alveolar dysfunction, ARDS with hypoxic respiratory failure, and ultimately multiorgan failure and death. EC dysfunction and activation likely codetermine this uncontrolled immune response. This is because ECs promote inflammation by expressing leukocyte adhesion molecules, thereby facilitating the accumulation and extravasation of leukocytes, including neutrophils and monocytes/macrophages, which enhance tissue damage. One recent report showed that SARS-CoV-2 N protein robustly induced proinflammatory cytokines/chemokines in human primary peripheral blood mononuclear cells (PBMCs) (43), suggesting that circulating N protein may also contribute to the initiation and progression of cytokine storm. Remarkably, though the protein sequences of N proteins from coronaviruses are highly conserved, only NP from SARS-CoV-2 induced endothelial activation. The NPs from other coronaviruses, such as SARS-CoV, MERS-CoV, HKU1-CoV, and influenza virus H1N1, did not affect endothelial activation. Thus, these findings are very consistent with the results from clinical investigations. However, it is not clear why other SARS N proteins do not activate endothelial cells while that of SARS-CoV-2 does. We noted that the amino acid sequences of SARS-CoV-2 NP and SARS-CoV NP have 91% homology. Which amino acid sequence difference contributes to their functional difference is under investigation and will be reported in future papers.
In summary, the present study identified SARS-CoV-2 N protein as a potent inducer of human endothelial activation, which can be specifically inhibited by simvastatin. The study provides insights on SARS-CoV-2-induced vasculopathy and coagulopathy and suggests that simvastatin, an FDA-approved lipid-lowering drug, may help to prevent vascular pathogenesis and improve the outcome of COVID-19 patients.
MATERIALS AND METHODS
Reagents.
SARS-CoV-2 nucleocapsid protein (NUN-C5227), S protein (SPN-C52H4), and envelope protein (ENN-C5128) were from Acrobiosystems (Newark, DE). SARS-CoV-2 NSP1 (97-095), NSP5 (10-116), NSP7 (97-096), and NSP8 (97-097) proteins were obtained from Prosci (Poway, CA). SARS-CoV-2 papain-like protease (DB604) was purchased from LifeSensors (Malvern, PA). HCoV-HKU1 coronavirus nucleocapsid protein, H7N9 nucleocapsid protein (40110-V08B), MERS-CoV nucleocapsid protein (40068-V08B), and SARS coronavirus nucleocapsid protein (40143-V08B) were from Sino Biological (Beijing, China). The mammalian expression plasmid for SARS-CoV-2 nucleocapsid protein (152536) was obtained from Addgene (Watertown, MA). ICAM-1 (60299-1-Ig) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase; 60004-1-Ig) antibodies were from Proteintech (Rosemont, IL). VCAM-1 (sc-8304) and β-actin (sc-47778) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). SARS-CoV/SARS-CoV-2 nucleocapsid antibody (40143-MM05) and SARS-CoV-2 nucleocapsid antibody (40588-T62) were from Sino Biological (Beijing, China). The NF-κB and MAPK signal pathway antibodies were all from Cell Signaling Technology (Danvers, MA).
Cells.
Human umbilical vein endothelial cells (HUVECs), human aortic endothelial cells (HAECs), human coronary artery endothelial cells (HCAECs), human dermal microvascular endothelial cells (HDMECs), and human lung microvascular endothelial cells (HLMECs) were purchased from Lonza Bioscience (Houston, TX). Mouse lung microvascular endothelial cells were obtained from Cell Biologics Inc. (Chicago, IL). Cells were cultured in different endothelial cell growth media in a humidified incubator with 5% CO2 at 37°C. Endothelial cells between passages 4 and 8 were grown as a monolayer and were used in all experiments. HLMECs were treated with vehicle (phosphate-buffered saline [PBS]) or various SARS-CoV-2 proteins, including nucleocapsid protein (NP), S protein (SP), envelope protein (EP), NSP1, NSP3, NSP5, NSP7, and NSP8 at 1 μg/ml for 8 h. In another set of experiments, HLMECs were treated with different coronavirus nucleocapsid proteins, including SARS-CoV-2 NP, SARS-CoV NP, MERS-CoV NP, H7N9 NP, and HCoV-HKU1 NP, at 1 μg/ml for 8 h. Different subtypes of endothelial cells were also used to observe the response to NP stimulation. For time course assays, HLMECs were incubated with 1 μg/ml SARS-CoV-2 NP for 2 h, 4 h, 8 h, 16 h, and 24 h, respectively. For dose-dependent assay, HLMECs were treated with various concentrations of SARS-CoV-2 NP ranging from 0.01 to 10 μg/ml. For NF-κB and MAPK signal pathway assay, HLMECs were subject to SARS-CoV-2 NP exposure for 15 min, 30 min, 45 min, 1 h, and 2 h, respectively. TNF-α (10 ng/ml; PeproTech, Cranbury, NJ) was used in all above-described experiments as a positive control for endothelial activation.
Transfection of plasmids.
Flag-NP and Flag-control vectors were transfected to HLMECs by electroporation using Nucleofector device (Lonza) and Nucleofector kits (Lonza, VPB-1002) following the manufacturer’s instructions. The whole-cell lysates were harvested 48 h after electroporation and were analyzed by Western blotting.
Chemical screening.
To screen for inhibitors of NP-induced endothelial activation, HLMECs were pretreated with 30 μM concentrations of individual chemicals from the microbial natural product library (Target Molecule, Wellesley Hills, MA) for 1 h followed by treatment with 1 μg/ml of N protein for 8 h. The effect of chemicals on endothelial activation were measured by ICAM-1 expression. To evaluate the inhibitory effects of statins on NP-induced endothelial activation, HLMECs were pretreated with atorvastatin, lovastatin, mevastatin, rosuvastatin, or simvastatin (all from BioVision, Milpitas, CA) for 1 h and followed by the treatment of N protein (1 μg/ml) for 8 h. The whole-cell lysates were harvested and analyzed by Western blotting.
Inhibitor treatment.
The antagonists or inhibitors involved in the NF-κB and MAPK signal pathways were introduced to verify the action of NP. The endocytosis inhibitors Pitstop2 (12.5 μM, Sigma) and Dynasore hydrate (12.5 μM, Sigma), the TLR4 antagonist LPS-RS (10 μg/ml; InvivoGen, San Diego, CA), the TLR1/TLR2 antagonist CU-CPT22 (20 μM, Millipore, Burlington, MA), the IL-1R antagonist (20 μM; Cayman Chemical Company), IKK-16 (20 μM; Cayman Chemical Company), the JNK inhibitor V (20 μM; Cayman Chemical Company), the ERK1/2 inhibitor U0126 (20 μM, Cell Signal Technology), and the p38 inhibitor SB203580 (20 μM; Enzo Life Sciences, Farmingdale, NY) were added to HLMEC cultures 1 h before NP exposure. The whole-cell lysates were harvested 8 h after NP stimulation and analyzed by Western blotting.
NP-induced NF-κB and MAPK activation in 293T-TLR2 cells.
Human TLR2 overexpressed in 293T cells was use to confirm the interaction between NP and TLR2. Wild-type 293T and 293T-TLR2 (InvivoGen) cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). The cells were treated with or without NP (1 μg/ml) for 15 min. The cells were harvested, and the whole-cell lysates were subjected to detection of pJNK and pP38 by Western blotting.
qPCR.
Total RNA was isolated from HLMEC after 8 h of NP exposure with the RNeasy minikit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. The first-strand cDNAs were synthesized by the high-capacity RNA-to-cDNA kit (Thermo Fisher Scientific, Vilnius, Lithuania). The reaction mixture contained 2×SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA), primer pairs, and cDNAs. The reaction consisted of a 2-step thermocycling protocol (95°C for 15 s and 60°C for 1 min; 40 cycles). The mRNA levels were calculated by using the 2−ΔΔCT method. The primer sequences used in the experiment are listed in Table 1. Results were obtained from at least three biological replicates performed in triplicate.
TABLE 1.
Primers used in qPCRs
| Gene (human) | Forward primer (5′ → 3′) | Reverse primer (5′ → 3′) |
|---|---|---|
| ICAM-1 | AGCTTCGTGTCCTGTATGGC | TTTTCTGGCCACGTCCAGTT |
| VCAM-1 | TGTTTGCAGCTTCTCAAGCTTTT | GATGTGGTCCCCTCATTCGT |
| TNF-α | TCTCGCACCCCGAGTGA | GGAGCTGCCCCTCAGCTT |
| MCP-1 | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT |
| IL-1β | CCACAGACCTTCCAGGAGAATG | ATCCCATGTGTCGAAGAAGATAGG |
| E-selectin | GGCAGTTCCGGGAAAGATCA | GTGGGAGCTTCACAGGTAGG |
Western blotting.
The total protein was collected with ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer after NP stimulation. Equal amounts of protein were loaded into the wells of the SDS-PAGE gel and separated by electrophoresis. Then the proteins were transferred to the polyvinylidene difluoride (PVDF) membrane. Skim milk (5% [wt/vol]) was used to block nonspecific binding. Membranes were incubated with different primary antibodies at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Luminescence was generated after the membranes were exposed to Super Signal West Pico chemiluminescent substrate (Thermo Fisher Scientific) and was detected with X-ray film.
Monocyte adhesion assay.
Monocyte adhesion was analyzed as previously described (44), with some modifications. HLMECs were stimulated with NP or TNF-α for 8 h. The human acute monocytic leukemia cell line THP-1 (Lonza) was prelabeled with Zombie Red fluorescent dye (BioLegend, San Diego, CA) in RPMI 1640 medium for 30 min at 37°C before being added to HLMECs and cocultured for 1 h. Nonadherent cells were removed by gently washing with cold RPMI 1640 medium. The images of adherent THP-1 cells were taken under Cytation 3 cell imaging multimode reader.
Statistical analyses.
Data were obtained from at least three independent experiments and are presented as means and standard deviations (SD). Statistical differences were compared using one-way analysis of variance (ANOVA) followed by a Tukey post hoc test. An unpaired Student's t test was performed to compare data between two independent groups. A difference with a P value of <0.05 was considered statistically significant.
Data availability.
All data are included in the article, and all materials are available upon request.
ACKNOWLEDGMENTS
We thank Peter Klein (University of Pennsylvania) for providing SARS-CoV2 nucleocapsid plasmid via Addgene.com.
This work was supported by National Institutes of Health grant AI138116 (to M.F.). J.Q. was supported by National Institutes of Health grants AI150877 and AI144564. Y.Q. was supported by the China Scholarship Council (20190682503).
M.F. and Y.Q. designed the research and analyzed data; Y.Q., T.L., and P.S.P. did the experiments; C.H.L., P.M.-N., J.Q., and H.-B.X. provided advice and critically read the manuscript; M.F. and Y.Q. wrote the manuscript.
We have no conflict of interests to declare.
Contributor Information
Mingui Fu, Email: fum@umkc.edu.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. 2020. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383:120–128. 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirtipal N, Bharadwaj S, Kang SG. 2020. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect Genet Evol 85:104502. 10.1016/j.meegid.2020.104502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Wit E, van Doremalen N, Falzarano D, Munster VJ. 2016. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534. 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. 2014. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res 109:97–109. 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, Liu L, Shan H, Lei C-L, Hui DSC, Du B, Li L-J, Zeng G, Yuen K-Y, Chen R-C, Tang C-L, Wang T, Chen P-Y, Xiang J, Li S-Y, Wang J-L, Liang Z-J, Peng Y-X, Wei L, Liu Y, Hu Y-H, Peng P, Wang J-M, Liu J-Y, Chen Z, Li G, Zheng Z-J, Qiu S-Q, Luo J, Ye C-J, Zhu S-Y, Zhong N-S, China Medical Treatment Expert Group for Covid-19. 2020. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720. 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carnevale S, Beretta P, Morbini P. 2020. Direct endothelial damage and vasculitis due to SARS-CoV-2 in small bowel submucosa of COVID-19 patient with diarrhea. J Med Virol 10:61–63. 10.1002/jmv.26119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vrints CJM, Krychtiuk KA, Van Craenenbroeck EM, Segers VF, Price S, Heidbuchel H. 2020. Endothelialitis plays a central role in the pathophysiology of severe COVID-19 and its cardiovascular complications. Acta Cardiol 2:1–16. 10.1080/00015385.2020.1846921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser DD, Patterson EK, Slessarev M, Gill SE, Martin C, Daley M, Miller MR, Patel MA, dos Santos CC, Bosma KJ, O’Gorman DB, Cepinskas G. 2020. Endothelial injury and glycocalyx degradation in critically ill coronavirus disease 2019 patients: implications for microvascular platelet aggregation. Crit Care Explor 2:e0194. 10.1097/CCE.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guervilly C, Burtey S, Sabatier F, Cauchois R, Lano G, Abdili E, Daviet F, Arnaud L, Brunet P, Hraiech S, Jourde-Chiche N, Koubi M, Lacroix R, Pietri L, Berda Y, Robert T, Degioanni C, Velier M, Papazian L, Kaplanski G, Dignat-George F. 2020. Circulating endothelial cells as a marker of endothelial injury in severe COVID-19. J Infect Dis 222:1789–1793. 10.1093/infdis/jiaa528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huertas A, Montani D, Savale L, Pichon J, Tu L, Parent F, Guignabert C, Humbert M. 2020. Endothelial cell dysfunction: a major player in SARS-CoV-2 infection (COVID-19)? Eur Respir J 56:2001634. 10.1183/13993003.01634-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, Zhang Y, Yin Q, Cho Y, Andrade L, Shadel GS, Hepokoski M, Lei T, Wang H, Zhang J, Yuan JX, Malhotra A, Manor U, Wang S, Yuan ZY, Shyy JY. 2021. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res 128:1323–1326. 10.1161/circresaha.121.318902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P, Luo R, Zhang M, Wang Y, Song T, Tao T, Li Z, Jin L, Zheng H, Chen W, Zhao M, Zheng Y, Qin J. 2020. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis 11:1042. 10.1038/s41419-020-03252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teuwen LA, Geldhof V, Pasut A, Carmeliet P. 2020. COVID-19: the vasculature unleashed. Nat Rev Immunol 20:389–391. 10.1038/s41577-020-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pons S, Fodil S, Azoulay E, Zafrani L. 2020. The vascular endothelium: the cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit Care 24:353. 10.1186/s13054-020-03062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. 2020. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 16.Albini A, Di Guardo G, Noonan DM, Lombardo M. 2020. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: implications for ACE-inhibitor- and angiotensin II receptor blocker-based cardiovascular therapies. Intern Emerg Med 15:759–766. 10.1007/s11739-020-02364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhatnagar J, Gary J, Reagan-Steiner S, Estetter LB, Tong S, Tao Y, Denison AM, Lee E, deLeon-Carnes M, Li Y, Uehara A, Paden CR, Leitgeb B, Uyeki T, Martines RB, Ritter JM, Paddock CD, Shieh W-J, Zaki SR. 2021. Evidence of SARS-CoV-2 replication and tropism in the lungs, airways and vascular endothelium of patients with fatal COVID-19: an autopsy case-series. J Infect Dis 27:jiab039. 10.1093/infdis/jiab039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korakas E, Ikonomidis I, Kousathana F, Balampanis K, Kountouri A, Raptis A, Palaiodimou L, Kokkinos A, Lambadiari V. 2020. Obesity and COVID-19: immune and metabolic derangement as a possible link to adverse clinical outcomes. Am J Physiol Endocrinol Metab 319:E105–E109. 10.1152/ajpendo.00198.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forster P, Forster L, Renfrew C, Forster M. 2020. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci USA 117:9241–9243. 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wawryk SO, Novotny JR, Wicks IP, Wilkinson D, Maher D, Salvaris E, Welch K, Fecondo J, Boyd AW. 1989. The role of the LFA-1/ICAM-1 interaction in human leukocyte homing and adhesion. Immunol Rev 108:135–161. 10.1111/j.1600-065x.1989.tb00016.x. [DOI] [PubMed] [Google Scholar]
- 22.Yong K, Khwaja A. 1990. Leucocyte cellular adhesion molecules. Blood Rev 4:211–225. 10.1016/0268-960X(90)90001-9. [DOI] [PubMed] [Google Scholar]
- 23.Fusté B, Mazzara R, Escolar G, Merino A, Ordinas A, Díaz-Ricart M. 2004. Granulocyte colony-stimulating factor increases expression of adhesion receptors on endothelial cells through activation of p38 MAPK. Haematologica 89:578–585. [PubMed] [Google Scholar]
- 24.Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. 1992. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J Biol Chem 267:16323–16329. 10.1016/S0021-9258(18)42004-2. [DOI] [PubMed] [Google Scholar]
- 25.Rodríguez C, Luque N, Blanco I, Sebastian L, Barberà JA, Peinado VI, Tura-Ceide O. 2021. Pulmonary endothelial dysfunction and thrombotic complications in COVID-19 patients. Am J Respir Cell Mol Biol 64:407–415. 10.1165/rcmb.2020-0359PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nascimento CJ, Schutt WR, Gorbunova EE, Mackow ER. 2020. Recombinant ACE2 expression is required for SARS-CoV-2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. mBio 11:e03185-20. 10.1128/mBio.03185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan A, Tahir Khan M, Saleem S, Junaid M, Ali A, Shujait Ali S, Khan M, Wei D-Q. 2020. Structural insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput Struct Biotechnol J 18:2174–2184. 10.1016/j.csbj.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. 2014. The SARS coronavirus nucleocapsid protein—forms and functions. Antiviral Res 103:39–50. 10.1016/j.antiviral.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aboagye JO, Yew CW, Ng OW, Monteil VM, Mirazimi A, Tan YJ. 2018. Overexpression of the nucleocapsid protein of Middle East respiratory syndrome coronavirus up-regulates CXCL10. Biosci Rep 38:BSR20181059. 10.1042/BSR20181059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu YG, Qu JM. 2009. Differential characteristics of the early stage of lung inflammation induced by SARS-CoV Nucleocapsid protein related to age in the mouse. Inflamm Res 58:312–320. 10.1007/s00011-009-8062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbunova EE, Simons MJ, Gavrilovskaya IN, Mackow ER. 2016. The Andes virus nucleocapsid protein directs basal endothelial cell permeability by activating RhoA. mBio 7:e01747-16. 10.1128/mBio.01747-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, Batra J, Richards AL, Stevenson E, Gordon DE, Rojc A, Obernier K, Fabius JM, Soucheray M, Miorin L, Moreno E, Koh C, Tran QD, Hardy A, Robinot R, Vallet T, Nilsson-Payant BE, Hernandez-Armenta C, Dunham A, Weigang S, Knerr J, Modak M, Quintero D, Zhou Y, Dugourd A, Valdeolivas A, Patil T, Li Q, Hüttenhain R, Cakir M, Muralidharan M, Kim M, Jang G, Tutuncuoglu B, Hiatt J, Guo JZ, Xu J, Bouhaddou S, Mathy CJP, Gaulton A, Manners EJ, et al. 2020. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182:685–712.E19. 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong M, Jiang Y, Xia D, Xiong Y, Zheng Q, Chen F, Zou L, Xiao W, Zhu Y. 2020. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J Infect Dis 222:894–898. 10.1093/infdis/jiaa349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li T, Wang L, Wang H, Li X, Zhang S, Xu Y, Wei W. 2020. Serum SARS-CoV-2 nucleocapsid protein: a sensitivity and specificity early diagnostic marker for SARS-CoV-2 infection. Front Cell Infect Microb 10:470. 10.3389/fcimb.2020.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon D. 2020. Statins may be a key therapeutic for Covid-19. Med Hypotheses 144:110001. 10.1016/j.mehy.2020.110001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima Martínez MM, Contreras MA, Marín W, D'Marco L. 2020. Statins in COVID-19: is there any foundation? Clin Invest Arterioscler 32:278–281. 10.1016/j.artere.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teoh N, Farrell G. 2020. Statins as early therapy to mitigate COVID-19 (SARS-CoV-2)-associated ARDS and cytokine storm syndrome—time is of the essence. J Clin Transl Res 5:227–229. [PMC free article] [PubMed] [Google Scholar]
- 38.Tan WYT, Young BE, Lye DC, Chew DEK, Dalan R. 2020. Statin use is associated with lower disease severity in COVID-19 infection. Sci Rep 10:17458. 10.1038/s41598-020-74492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X-J, Qin J-J, Cheng X, Shen L, Zhao Y-C, Yuan Y, Lei F, Chen M-M, Yang H, Bai L, Song X, Lin L, Xia M, Zhou F, Zhou J, She Z-G, Zhu L, Ma X, Xu Q, Ye P, Chen G, Liu L, Mao W, Yan Y, Xiao B, Lu Z, Peng G, Liu M, Yang J, Yang L, Zhang C, Lu H, Xia X, Wang D, Liao X, Wei X, Zhang B-H, Zhang X, Yang J, Zhao G-N, Zhang P, Liu PP, Loomba R, Ji Y-X, Xia J, Wang Y, Cai J, Guo J, Li H. 2020. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab 32:176–187.E4. 10.1016/j.cmet.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bifulco M, Gazzerro P. 2020. Statin therapy in COVID-19 infection: much more than a single pathway. Eur Heart J Cardiovasc Pharmacother 6:410–411. 10.1093/ehjcvp/pvaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crespo MJ, Quidgley J. 2015. Simvastatin, atorvastatin, and pravastatin equally improve the hemodynamic status of diabetic rats. World J Diabetes 6:1168–1178. 10.4239/wjd.v6.i10.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Jiang M, Chen X, Montaner LJ. 2020. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol 108:17–41. 10.1002/JLB.3COVR0520-272R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, Kim IS, Silwal P, Kim YJ, Paik S, Chung C, Park C, Kim YS, Jo EK. 2020. COVID-19 patients upregulate Toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J Korean Med Sci 35:e343. 10.3346/jkms.2020.35.e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Huang X, Guo F, Lei T, Li S, Monaghan-Nichols P, Jiang Z, Xin H-B, Fu M. 2020. TRIM65 E3 ligase targets VCAM-1 degradation to limit LPS-induced lung inflammation. J Mol Cell Biol 12:190–201. 10.1093/jmcb/mjz077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the article, and all materials are available upon request.