

ABSTRACT
Herpesvirus genomes show abundant evidence of past recombination. Its functional importance is unknown. A key question is whether recombinant viruses can outpace the immunity induced by their parents to reach higher loads. We tested this by coinfecting mice with attenuated mutants of murid herpesvirus 4 (MuHV-4). Infection by the natural olfactory route routinely allowed mutant viruses to reconstitute wild-type genotypes and reach normal viral loads. Lung coinfections rescued much less well. Attenuated murine cytomegalovirus mutants similarly showed recombinational rescue via the nose but not the lungs. These infections spread similarly, so route-specific rescue implied that recombination occurred close to the olfactory entry site. Rescue of replication-deficient MuHV-4 confirmed this, showing that coinfection occurred in the first encountered olfactory cells. This worked even with asynchronous inoculation, implying that a defective virus can wait here for later rescue. Virions entering the nose get caught on respiratory mucus, which the respiratory epithelial cilia push back toward the olfactory surface. Early infection was correspondingly focused on the anterior olfactory edge. Thus, by concentrating incoming infection into a small area, olfactory entry seems to promote functionally significant recombination.
IMPORTANCE All organisms depend on genetic diversity to cope with environmental change. Small viruses rely on frequent point mutations. This is harder for herpesviruses because they have larger genomes. Recombination provides another means of genetic optimization. Human herpesviruses often coinfect, and they show evidence of past recombination, but whether this is rare and incidental or functionally important is unknown. We showed that herpesviruses entering mice via the natural olfactory route meet reliably enough for recombination routinely to repair crippling mutations and restore normal viral loads. It appeared to occur in the first encountered olfactory cells and reflected a concentration of infection at the anterior olfactory edge. Thus, natural host entry incorporates a significant capacity for herpesvirus recombination.
KEYWORDS: herpesviruses, host entry, olfactory, recombination
INTRODUCTION
Viruses transmitting between outbred hosts face serial changes in selection. Those with small genomes can diversify solely by point mutation. Those with more essential genes must have lower mutation rates to avoid most virions being defective, and herpesviruses, with relatively large genomes, have proofreading polymerases that are slow to make new mutations (1). Nonetheless, over time, they have accumulated marked diversity in some genes (2–4). Established diversity and present stability give herpesvirus genes allelic forms, which interact with host gene alleles to influence infection outcomes. For example, murine cytomegalovirus (MCMV) m157 can inhibit attack by NK cells (5) but disadvantageously activates them in Ly49H+ mice (6, 7).
Just as meiosis shuffles host alleles, recombination between coinfecting viruses can potentially yield useful new genetic mixes. Coinfection of hosts by different viral strains seems common (2–4), and many circulating strains appear to be recombinants (8–11). However, the complexity of serial infections makes the selective pressures operating on herpesvirus alleles hard to gauge. Thus, the frequency and functional significance of recombination remain speculative. To recombine, herpesviruses must coinfect single cells. Longitudinal analysis has identified a likely instance of recombination only in immunodeficiency, when viral loads are unusually high (12). A key unanswered question is whether normal infection allows herpesvirus strains to form functional gene pools or whether recombination only rarely alters outcomes.
Recombination early in infection, before host immunity limits all spread, is likely to have the most impact. The difficulty of sampling early human herpesvirus infections makes their routes and recombination opportunities hard to know. The presence of Epstein-Barr virus (EBV) in tonsils during infectious mononucleosis led to an assumption that gammaherpesviruses enter orally (13, 14). However, tonsillitis postdates EBV acquisition by at least a month (15), and EBV DNA is found in blood before saliva (16), so tonsillar infection looks more like host exit. Analysis of the related murid herpesvirus 4 (MuHV-4) (17) found oral virions to be noninfectious (18) and submucosal B cell colonization to postdate systemic spread (19). MuHV-4 enters alert mice via the olfactory epithelium (20). Lung infection is a popular experimental model but requires large volume inoculation under anesthesia. The only other known natural route of MuHV-4 uptake is genital (21). After entry, MuHV-4 spreads to lymph nodes (LN) via dendritic cells (22) and then to the spleen and beyond via B cells (23).
Olfactory entry is evident also for MCMV, including spontaneous transmission (24), and for herpes simplex virus 1 (HSV-1) (25). These viruses diverged long before primates split from rodents (26), and heparan binding, a key determinant of olfactory entry (17), is shared by all human herpesviruses. On most differentiated epithelia, heparan is solely basal (27) and therefore inaccessible to incoming virions. However, olfactory neurons display heparan on their apical cilia (20). Human CMV also has a specific olfactory receptor (28). We tested in this study whether olfactory entry offers a significant opportunity for coinfecting herpesvirus strains to recombine.
RESULTS
Nasal infection with latency-deficient MuHV-4 mutants.
Gammaherpesviruses colonize their hosts by lytic spread and by latency-associated lymphoproliferation. MuHV-4 mutants lacking stable latency have been made by overexpressing the open reading frame 50 (ORF50) viral transactivator (20, 29) and by disrupting the ORF73 episome maintenance protein (30, 31). For example, M50 MuHV-4 has an MCMV IE1 promoter fragment inserted in the 5′ untranslated region of ORF50, deregulating its transcription (32), and ORF73FS MuHV-4 has a frameshift mutation in ORF73 (30) (Fig. 1a). Both mutants replicate lytically in the lungs after a 30-μl inoculation under anesthesia, but they poorly colonize lymphoid tissue. Nasal inoculation (5 μl without anesthesia) similarly led to local lytic spread but significantly less LN infection than for the wild type and no detectable splenic infection after 18 days (Fig. 1b).
FIG 1.

Nasal infection by MuHV-4 mutants lacking normal latency. (a) A schematic sketch of MuHV-4 mutations shows the linear genome flanked by terminal repeats (TR), with expanded views below. Replication-deficient ORF50DEL MuHV-4 has most of ORF50 exon 2 replaced by luciferase plus a polyadenylation site. ORF73FS MuHV-4 has a frameshift in ORF73, which encodes the viral episome maintenance protein. The ΔL virus mutation additionally deletes 11 kb from the genome left end, removing ORFs M1-M4. M50 MuHV-4 has an MCMV IE1 promoter inserted in the 5′ untranslated region of ORF50. p1 to p6 show the locations of primers used to identify the ORF50DEL, M50, and ORF73FS mutations. (b) C57BL/6 mice were infected nasally (105 PFU in 5 μl without anesthesia) with wild-type, M50, or ORF73FS virus. Titers in noses were determined by plaque assay. Titers in superficial cervical lymph nodes (SCLN) and spleens were determined by infectious-center assay. Symbols show individual mice; bars show means. The dashed line shows the detection limit. Significant differences in virus recovery relative to the wild type are shown.
Recombinational rescue of latency-deficient mutants after nasal coinfection.
To test rescue by recombination, we coinfected mice nasally with ORF73FS and M50 MuHV-4 and 25 days later determined latent loads by infectious-center assay (Fig. 2). A total of 11/12 coinfected mice showed significant splenomegaly (Fig. 2a) and splenic infection (Fig. 2b). No singly infected mice did so, ruling out spontaneous reversion (or wild-type contamination) as an explanation. PCR genome analysis of viruses cloned from splenic infectious centers of 4/4 coinfected mice showed M50 and ORF73 PCR products of wild-type size (Fig. 2c and d), and the DNA sequences of these PCR products exactly matched the wild type, supporting the idea that they resulted from recombination.
FIG 2.

Recovery of latency after nasal coinfection with latency-deficient MuHV-4 mutants. (a) C57BL/6 mice were infected nasally (105 PFU) with wild-type, M50, or ORF73FS virus or a 1:1 M50/ORF73FS mix. Weights of spleens 25 days later are shown, with significant splenomegaly for the wild type and mixed infections. Symbols show individual mice; bars show means. (b) Titers in the spleens in panel a were determined for latent virus by infectious-center assay. Symbols show individuals; bars show means. The dashed line shows the lower limit of assay detection. Mixed M50/ORF73FS infection yielded significantly more virus than either single infection. No preformed infectious virus was recovered by parallel titer of freeze-thawed spleen samples. (c) Viruses cloned (c1 to c4) from spleens of mixed-infection mice were genotyped by PCR across the M50 insertion site (primers p1 and p2 in Fig. 1a). PCR products were resolved by agarose gel electrophoresis and stained with ethidium bromide. WT and M50, wild-type and M50 input viruses; Mw, molecular weight markers. Sequencing of the main (268 bp) product of cloned virus DNA confirmed identity with the wild type. (d) DNA from the cloned viruses in panel c was genotyped by PCR across the ORF73 frameshift (primers p5 and p6 in Fig. 1a). PCR products were digested or not with BstEII, resolved by agarose gel electrophoresis, and stained with ethidium bromide. 73, ORF73FS input viruses. As the viruses were cloned prior to analysis, we interpret the minor residual 0.43-kb product for BstEII-digested c3 DNA as incomplete digestion. DNA sequencing of the main (428 bp) undigested product of the cloned viruses confirmed identity with the wild type. (e) C57BL/6 mice were infected as for panel a. Three months later, latent virus was detected by infectious-center assay. M50/ORF73FS coinfection yielded significantly more virus than either single infection and was indistinguishable from the wild type. Symbols show individual mice; bars show means. The dashed line shows the detection limit. (f) The spleens in panel e were assayed for viral genomes by quantitative PCR of extracted DNA. Viral copies are expressed relative to cellular β-actin copies amplified in parallel. M50/ORF73FS coinfection yielded significantly more viral genomes than either single infection and was indistinguishable from the wild type.
Recombinant infections necessarily start at a very low dose, so it was unsurprising that the early viral loads of coinfected mice were lower than those of wild-type infection controls. After 3 months, the splenic loads of coinfected mice were indistinguishable from those of the wild type by infectious-center assay (Fig. 2e) and by quantitative PCR of viral DNA (Fig. 2f). Thus, although the parental M50 and ORF73FS infections elicited enough immunity to protect against a later wild-type challenge (33–36), they did not do so quickly enough to stop recombinant outgrowth.
Dual recombinational rescue.
Herpesvirus genomes comprise conserved blocks, between which lie more varied loci. The genome ends also vary. Exchanging an end would require just one recombination, but exchanging a central gene would require two. To test this setting, we coinfected mice with M50 MuHV-4 and an ORF73FS mutant that also lacked 10 kb from its left end (ORF73FSΔL) (36) (Fig. 1a). The ΔL mutation deletes ORFs M1 to M4, which also impairs the establishment of a normal latent viral load (37), and infectious-center assays confirmed the attenuating effect of this mutation (Fig. 3a). However, nasal coinfection again gave rescue, with no detectable splenic infection in the single-inoculum controls. To test for full repair rather than just complementation in trans, we inoculated virus cloned from the spleens of M50/ORF73FSΔL-coinfected mice into the lungs of new naive mice (Fig. 3b). They colonized the spleens of these mice as effectively as the wild type. Thus, there was no barrier to recombinational rescue of a parental virus with more than one attenuating mutation.
FIG 3.
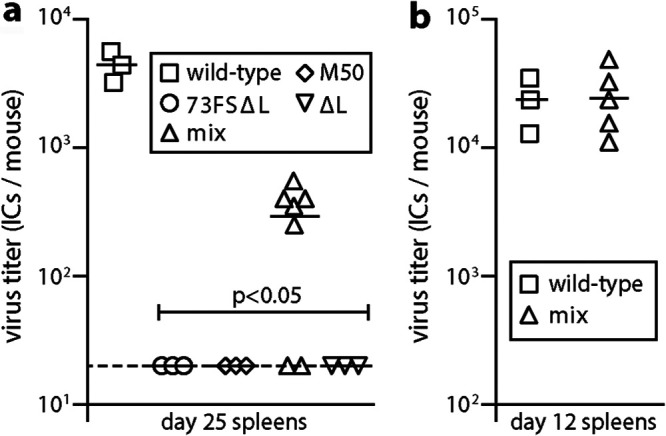
Recovery of latency after coinfection by M50 and ORF73FSΔL mutants. (a) C57BL/6 mice were infected nasally (105 PFU) with wild-type, M50, or ORF73FSΔL virus or a 1:1 M50/ORF73FSΔL mix. We also infected mice with a control ΔL single mutant. Spleens were infectious-center assayed for latent infection 25 days later. Symbols show individual mice; bars show means. The dashed line shows the detection limit. M50/ORF73FSΔL coinfection yielded significantly more latency than either virus alone. No preformed infectious virus was recovered by parallel titer of freeze-thawed spleen samples. (b) To test the viruses recovered from spleens of individual coinfected mice for full rescue, they were inoculated into the lungs of naive C57BL/6 mice (103 PFU). Splenic infectious-center assays 12 days later showed titers equivalent to those of the wild type.
Lung coinfection with latency-deficient MuHV-4 mutants.
To determine whether MuHV-4 coinfection generally provides genetic rescue, or whether this was a particular feature of olfactory entry, we tested lung inoculation, giving mice M50 and ORF73FS MuHV-4 as before but in 30 μl under anesthesia. Exact comparison of lung and olfactory infections is difficult, as they involve different organs. For example, virus inoculation into the lungs is much more efficient, as alert mice retain very little free fluid in their upper airways (38): at least 90% of a 5-μl nasal inoculum is swallowed. Thus, for lung inoculations we gave 104 rather than 105 PFU of virus. MuHV-4 also replicates more extensively in lungs than in noses (18). This leads to LN and spleen infections peaking earlier than after olfactory infection, typically at day 12 to 17 rather than day 18 to 25 (18). This makes day 25 is a relatively insensitive time point for detecting systemic spread via the lungs, so instead we tested day 17 (Fig. 4a), the peak time point for spleen colonization after a low-dose lung inoculum (18). Low levels of latent infection were found in the spleens of 3/6 coinfected mice, with none in single-mutant controls. An additional experiment (Fig. 4b) found no sign of late (day 100) splenic colonization after coinoculation of lungs.
FIG 4.

Little evidence of MuHV-4 recombination after lung coinfection. (a) Wild-type, M50, or ORF73FS virus or a 1:1 M50/ORF73FS mix was inoculated into the lungs of C57BL/6 mice (104 PFU in 30 μl under anesthesia). Spleens were infectious-center assayed for latent virus 17 days later. Symbols show individual mice; bars show means. The dashed line is the detection limit. (b) Mice were infected as for panel a. One hundred days later, DNA from spleens was assayed for viral DNA by quantitative PCR. Values are expressed relative to the cellular DNA load assayed in parallel for each sample. Symbols show individual mice; bars show means. (c) Mice were infected as for panel a. Twelve days later, SCLN and MLN were infectious-center assayed for latent virus.
While nasal inocula do not reach the lungs, lung inocula must navigate the nose, so the low-level splenic latency in Fig. 4a possibly came from contaminating olfactory infection. To test this, we sacrificed mice at 12 days after coinfection and separately determined the virus titers in the superficial cervical LN (SCLN), which drain the nose, and the mediastinal LN (MLN), which drain the lungs (Fig. 4c). At this time after low-dose infection, LN colonization reflects primary viral replication in peripheral sites rather than systemic spread, so we could distinguish lung and nose origins. While 4/7 coinfected mice had a low level of SCLN latency, only 1/7 had MLN latency, arguing that virus capable of lymphoid colonization was coming primarily from the nose rather than the lungs. Wild-type controls showed more virus in MLN than in SCLN—as primary lung infection is more extensive—and the sole positive MLN of the coinfected cohort correspondingly yielded more virus than any SCLN. Thus, we could be confident of not missing recombinational rescue in the lungs. It seemed that viruses coinfecting the lungs rarely recombined—1/7 mice, versus 11/12 for olfactory infection in Fig. 2b (P < 0.002)—and that the low-level spleen infections in Fig. 4a had likely come from the nose.
Recombination via olfactory infection but not lungs applies also to MCMV.
To explore whether recombinational rescue was specific to MuHV-4, we tested MCMV, coinfecting BALB/c mice with mutants lacking M33 or M78 (Fig. 5). MCMV needs both of these G protein-coupled receptor homologs to colonize the salivary glands (39, 40). We used BALB/c mice because K181 strain MCMV is attenuated in C57BL/6 mice. After lung inoculation, day 18 salivary gland plaque assays were positive for only the wild-type control (Fig. 5a). After nose inoculation, day 18 salivary glands were positive for all coinfected mice and no M33 or M78 single controls (Fig. 5b). We cloned viruses from the salivary glands of nasally coinfected mice and checked their M33 and M78 loci by PCR (Fig. 5c and d). A total of 4/4 showed a K181 wild-type-sized M33 locus, and DNA sequencing showed an exact match with the wild type. One clone retained a mutant M78 locus (Fig. 5d), reflecting that M78-deficient viruses do not always completely lack salivary gland infection (40, 41). However, the other M78 loci matched the K181 wild-type size and DNA sequence. Thus, as with MuHV-4, recombination appeared to provide frequent and functionally significant MCMV rescue after olfactory entry but not after lung entry.
FIG 5.

Rescue of MCMV mutants after coinfection in the nose but not the lungs. (a) Wild-type, M33, or M78 MCMV or a 1:1 M33/M78 mix was inoculated into the lungs of BALB/c mice (104 PFU in 30 μl under anesthesia). Eighteen days later, salivary glands were plaque assayed for infectious virus. Symbols show individual mice; bars show means. The dashed line is the detection limit. Only wild-type infection yielded recoverable virus. (b) BALB/c mice were infected nasally (105 PFU) with M33 or M78 MCMV or a 1:1 M33/M78 mix. Eighteen days later, salivary glands were plaque assayed for infectious virus. Symbols show individual mice; bars show means. Only the mixed infection yielded recoverable virus. (c) DNA of virus cloned from mixed infection salivary glands in panel b was checked for M33 mutation by PCR. 33 and 78, M33 and M78 input viruses. The predicted wild-type band is 572 bp. The upper 33 sample band corresponds to the expected size for the lacZ cassette insertion of the mutant (4.4 kb). The source of the lower (2.2 kb) 33 sample band is unclear, but the 33 sample is clearly free of wild-type DNA, and no recovered clone shows a mutant M33 locus. (d) The same DNA samples were checked for M78 mutation. Clone 2 appeared to be a parental M78 virus. The other 3 clones had a wild-type M78 locus, indicating recombination.
Recombination occurs early in olfactory infection.
MuHV-4 lung and olfactory infections both reach B cells via dendritic cells (17), and MCMV lung and olfactory infections both spread via dendritic cell recirculation (42, 43). Therefore, inefficient rescue of either virus via the lungs argued that recombination occurred early after olfactory entry, before spread to LN. Supporting this supposition, SCLN infectious-center assays at day 8 detected more latent MuHV-4 after nasal M50/ORF73FS coinfection than after single infections (Fig. 6a). However, latency-deficient mutants can still reach LN—they just fail to amplify—and with little time for recombinant viruses to grow out, the titers of coinfected mice were not significantly higher than those of M50 alone. Thus, we sought further evidence of recombinational rescue, by inoculating virus clones recovered from SCLN in Fig. 6a into the lungs of naive mice (Fig. 6b). At day 14 after reinoculation, no virus clones from M50 or ORF73FS single infections yielded infectious centers, while 8/9 of those from mixed infections had titers indistinguishable from the wild type. Therefore, olfactory coinfections appeared to recombine before leaving LN.
FIG 6.

Early recombinational rescue of MuHV-4 after nasal coinfection. (a) C57BL/6 mice were infected nasally (105 PFU) with wild-type, M50, or ORF73FS MuHV-4 or a 1:1 M50/ORF73FS mix. Eight days later, latent virus in SCLN was infectious-center assayed. Symbols show individual mice; bars show means. The dashed line is the detection limit. Mixed infections gave higher titers than either component single infection, although the wide spread meant that as a group they were not significantly higher than M50 alone. (b) Viruses cloned from SCLN for each mouse in panel a were given i.n. to naive mice under anesthesia to inoculate the lungs (103 PFU in 30 μl). One clone from each positive mouse was given to one naive mouse. Fourteen days later, spleens were infectious-center assayed for latent infection. Symbols show individual mice; bars show means. (c) C57BL/6 mice were infected nasally (105 PFU in 5 μl) with wild-type, ORF50DEL, or ORF73FS MuHV-4 or a 1:1 ORF50DEL/ORF73FS mix. Eighteen days later, SCLN and spleens were infectious-center assayed for latent virus. Symbols show individual mice; bars show means. Mixed infections gave significantly higher titers than either component single infection. (d) Mice infected as for panel c were infectious-center assayed for splenic virus after 25 days. Mixed infections gave significantly higher titers than either component single infection. (e) DNA of viruses cloned (c1 to c4) from mixed-infection spleens for panel d was PCR amplified across the ORF50 deletion site (primers p3 and p4 in Fig. 1a). The PCR products were resolved by agarose gel electrophoresis and stained with ethidium bromide. 50, ORF50DEL input viruses. DNA sequencing of cloned virus PCR products confirmed identity with the wild type. (f) DNA from the mixed-infection clones in panel e was PCR amplified across the ORF73 frameshift site (primers p5 and p6 in Fig. 1a). The PCR products were digested or not with BstEII, resolved by agarose gel electrophoresis, and stained with ethidium bromide. As in Fig. 2d, as c1 to c4 were cloned prior to analysis; the minor residual 0.43-kb band after incubation with BstEII digestion presumably reflected incomplete digestion rather than mutant DNA. Sequencing of the undigested PCR products confirmed identity with the wild type.
We could not recover recombinant virus clones in the same way from coinfected noses, presumably because without the selective pressure of having to establish latency, the proportion of recombinants remained low. Therefore, to locate more precisely the in vivo recombination site, we used ORF50DEL MuHV-4, which lacks all lytic replication, due to a large deletion in ORF50 (Fig. 1a), coinfecting mice with this virus and the ORF73FS mutant (Fig. 6c). ORF50DEL MuHV-4 requires complementing ORF50+ cells for propagation (18), and in vivo it is confined to the first cells infected (23). At 18 days after nasal coinfection, spleens and SCLN yielded recoverable virus for 3/3 coinfected mice, and at 25 days spleens did so for 5/5 coinfected mice, with none from single-mutant controls. Viruses cloned from day 25 spleens of 4/4 coinfected mice further showed wild-type ORF50 and ORF73 loci by PCR (Fig. 6d and e); DNA sequencing confirmed their identity with the wild type. Therefore, coinfection must have occurred in the first encountered olfactory cells, specifically olfactory neurons or sustentacular cells (20).
Recombination does not require simultaneous coinfection.
ORF50DEL MuHV-4 cannot replicate lytically, and no infectivity could be recovered from noses at 1 day after inoculation, but it can establish latent infection (20). Thus, we used it to test whether olfactory entry might also allow a superinfecting virus to rescue a defective mutant. We infected mice nasally with ORF50DEL MuHV-4, then 5 days later gave the same mice nasal ORF73FS MuHV-4, and assayed spleens for recoverable virus after another 18 days (Fig. 7). A total of 5/6 coinfected mice showed splenic infection, while no singly infected controls did so. Therefore, olfactory entry allowed rescue by superinfection.
FIG 7.
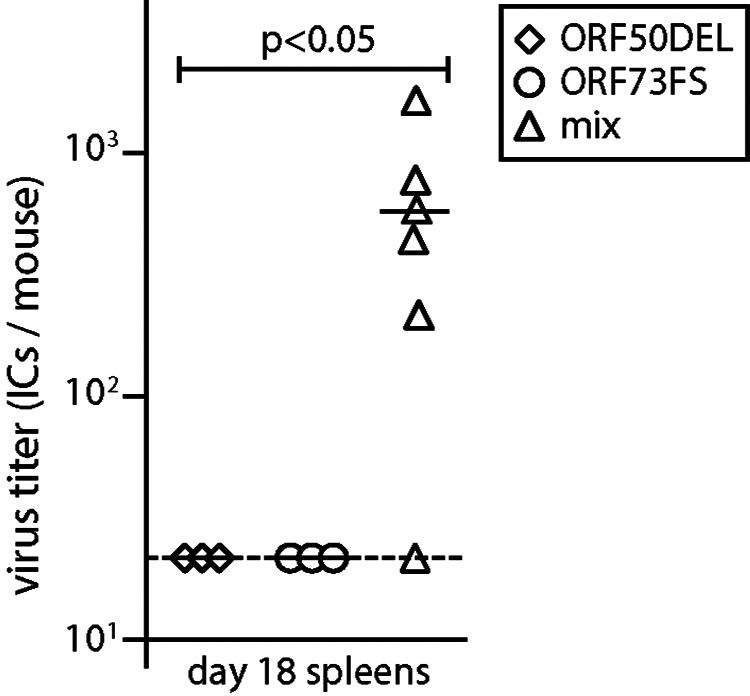
Recombinational rescue after serial mixed infection. C57BL/6 mice were infected nasally (105 PFU) with ORF50DEL MuHV-4. Five days later, they were infected nasally (105 PFU) with ORF73FS MuHV-4. Controls were given either just ORF50DEL or just ORF73FS. Eighteen days after ORF73FS infection, spleens were infectious-center assayed for latent virus. Symbols show individual mice; bars show means. The dashed line shows the detection limit. Serial mixed infection yielded significantly more recoverable virus than either single infection.
A possible site of olfactory recombination.
Olfactory cells far outnumber the number of virions we inoculated, and previous analysis has shown that while MuHV-4 can establish extensive olfactory infections in pups, only a small proportion of adult olfactory cells ever seem to be infected (20). For MCMV, the number of infected cells is lower still (24). Thus, it was a puzzle how coinfecting strains could so often meet. However, a survey of MuHV-4-infected noses showed that the anterior olfactory epithelium, where it borders the respiratory epithelium, is a conspicuous site of early infection (Fig. 8). Anatomical complexity (44) makes this border hard to survey entirely, but even on limited sampling we found evidence of its involvement in 23/30 early infections (1 to 3 days postinoculation). Thus, frequent infection of this site seemed a plausible explanation for efficient rescue after nasal entry.
FIG 8.
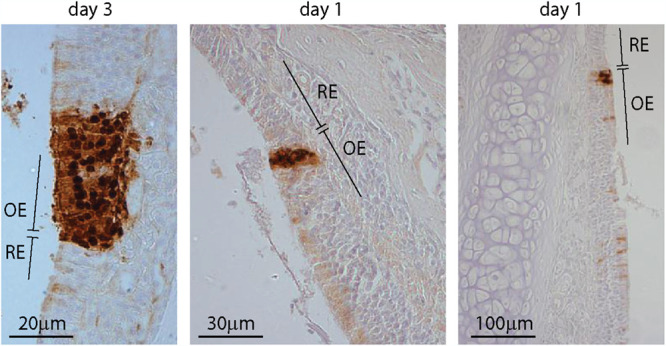
Infection at the olfactory/respiratory border. C57BL/6 mice were infected nasally with wild-type MuHV-4 (105 PFU). One to 3 days later, nose sections were stained for viral lytic antigens with a polyclonal rabbit serum (brown) and counterstained with hemalum (blue). OE, olfactory epithelium; RE, respiratory epithelium. Representative sections show foci of infection on the olfactory side of the olfactory/respiratory border.
DISCUSSION
Herpesviruses interact with polymorphic host genes and so face significant environmental change in each new infection. Coping with such change requires genetic diversity. Herpesvirus mutation rates are constrained by their large complements of essential genes, and the identification of many circulating strains as recombinants suggests that gene shuffling could provide significant additional diversity. However, the in vivo fitness of human herpesvirus strains is hard to compare, and serial selection in an outbred host soon gets unfathomably complicated. Thus, genome sequences alone cannot tell us the frequency or functional importance of viral recombination. We addressed a key functional question: whether a higher titer and therefore more transmissible recombinant infection can arise from attenuated parents entering via a natural route. M50 and ORF73FS MuHV-4 infections elicit strong protection against a later wild-type challenge (33–35). However, olfactory entry allowed recombination early enough to outpace this immune response and so restored wild-type levels of host colonization.
It might be considered that coinfecting herpesviruses could meet anywhere. Yet once infection breaches a tissue barrier, the need to outpace immunity favors rapid spread, which is divergence rather than the convergence needed for recombination. Hence, MuHV-4 recombinants readily escaped the dominant negative impact on latency of the M50 mutation. Systemic infection in immunocompetent hosts remains dilute, with most replication at the point of host exit, so after entry there is no obvious point of convergence until progeny virions pool in saliva for shedding. Even the high plaque titers that MCMV reaches in salivary glands come from relatively few infected acinar cells. Thus, normal infection may offer few postentry opportunities for recombination, consistent with the scarcity of recombination after lung inoculation of MuHV-4 or MCMV.
In minimally manipulated mice, herpesviruses have consistently shown olfactory entry. The olfactory epithelium sits above and behind the respiratory epithelium, near the cribriform plate (44).
Tracking inhaled MuHV-4 has shown that many virions get captured onto the respiratory mucus (20), presumably by surface tension. They then disappear without infecting, as respiratory epithelial cells lack the apical heparan required for binding, and their cilia constantly push the overlying mucus back toward the olfactory epithelium. Here, virions accumulate and infect (20). Hence, MuHV-4 entry concentrated at the anterior olfactory edge. The rescue of replication-deficient MuHV-4 argued that recombination occurs in the first infected olfactory cells, and so the experimentally defined process of virion capture onto respiratory mucus and then transport back to the anterior olfactory edge links directly to the high frequency of recombination observed in natural virus isolates.
The main alternative to olfactory entry for herpesviruses appears to be genital (21). Entry here seems to reflect coitus-related epithelial damage, so the recombination opportunities are probably hard to model across species. For lung infection, which provides a useful albeit somewhat artificial experimental standard, the lack of recombination between coinoculated viruses reflected that inocula get diluted in a very large number alveoli (45, 46). Although the is extensive viral replication in inoculated lungs, there is no evidence for spread between alveoli. This may explain the lack of recombination.
Olfactory anatomy is similar across many vertebrate species, as is heparan binding by their herpesviruses, a key determinant of olfactory virion capture (20). Other heparan-binding viruses might use the same route. For example, both adeno-associated virus (AAV) and adenovirus bind heparan (47, 48). AAV requires a coinfecting adenovirus (or herpesvirus) for replication. When spreading within a host, abundant production can keep AAV ahead of its helper. However, interhost transmission involves limiting virus dilution. It is then a problem for AAV to find new complementation. The concentrating effect of olfactory entry could make a meeting more likely, and AAV latent in olfactory cells could wait to ambush its essential aide.
Coentry of herpesvirus strains implies coacquisition from a coshedding host. While EBV strains can coshed acutely (49), long-term infections seem to select one dominant strain (50). Vaccine studies argue that superinfection is inefficient (46). The explanation for nonetheless abundant coinfections and recombinants may be that establishing effective immunity takes time. The antibody response to MuHV-4 takes 3 months to reach its peak (51); EBNA-specific antibodies do not appear until months after EBV acquisition; and EBV loads remain elevated for at least as long (52). Thus, an infant acquiring a single maternal virus strain would have a substantial window to acquire additional strains from other carers (53) and generate recombinants. This might be helped by a carrier inhaling some of their own shed salivary virus.
MATERIALS AND METHODS
Mice.
C57BL/6 or BALB/c mice were infected intransally (i.n.) when 6 to 14 weeks old, either in 30 μl, under isoflurane anesthesia to reach the lungs (104 PFU), or in 5 μl, restrained without anesthesia (105 PFU) to infect only the nose (18). A higher dose was used for nose infections, as most of these inocula are swallowed (38). Statistical comparison was done by Student’s 2-tailed unpaired t test unless stated otherwise. Mice were killed by rising CO2 concentration. Experiments were approved by the University of Queensland Animal Ethics Committee and were performed in accordance with the Australian code for the care and use of animals for scientific purposes, as published by the Australian National Health and Medical Research Council (projects 391/15, 479/15, 196/18, and 207/18).
Cells and viruses.
BHK-21 fibroblasts (American Type Culture Collection [ATCC] CCL-10), NIH 3T3 cells (ATCC CRL-1658), and NIH 3T3-ORF50 cells (18) were grown in Dulbecco’s modified Eagle's medium (Gibco) with 2 mM glutamine, 100 IU/ml of penicillin, 100 μg/ml of streptomycin, and 10% fetal calf serum (complete medium). Murine embryonic fibroblasts were grown in similarly supplemented minimal essential medium. MuHV-4 variants with a frameshift inactivating ORF73 (30), with an additional deletion of 11 kb from the genome left end, which includes ORFs M1, M2, M3, M4, and 4 (36), with a deletion of ORF50 exon 2 (18), and with an MCMV IE1 promoter fragment inserted in the 5′ untranslated region of ORF50 (32) are described. ORF50DEL MuHV-4 was grown and titers were determined in NIH 3T3-ORF50 cells. Other MuHV-4 derivatives were grown and titers were determined in BHK-21 cells. MCMV mutants with a β-galactosidase expression cassette disrupting M33 (39) or M78 (40) were derived from K181 strain Perth, which was used as the wild type. All were grown in NIH 3T3 cells. Infected cell supernatants were cleared of debris by low-speed centrifugation (200 × g, 5 min). Cell-free virions were then concentrated by ultracentrifugation (35,000 × g, 1.5 h) and stored at –80°C.
Infectivity assays.
Infectious virus was quantified by plaque assay. For MuHV-4 (54), virus stocks or freeze-thawed tissue homogenate dilutions were incubated with BHK-21 cells (2 h, 37°C), overlaid with complete medium/0.3 % carboxymethylcellulose, cultured for 4 days, fixed with 1 % formaldehyde, and stained with 0.1 % toluidine blue for plaque counting. Titers of MCMV were determined similarly but on murine embryonic fibroblast monolayers (40), and the virus was adhered by centrifugation (500 × g, 30 min) before the inoculum was discarded. Total recoverable MuHV-4 (latent plus preformed infectious virus) was quantified by infectious-center assay (54). Freshly isolated LN or spleen cells were layered onto BHK-21 cell monolayers and then cultured and processed as for plaque assays. For parallel assays of preformed infectious virus, samples were first frozen and thawed.
Viral genome quantitation.
MuHV-4 genomic coordinates 4163 to 4308 (M2 ORF; primers 4163 to 4183 and 4288 to 4308) were amplified by PCR (Rotor Gene 3000; Corbett Research) from 50 ng of DNA (Nucleospin tissue kit; Macherey-Nagel). PCR products quantified with Sybr green (Thermo Fisher Scientific) were compared to a standard curve of a cloned template amplified in parallel and were distinguished from paired primers by melting-curve analysis. Correct sizing was confirmed by electrophoresis and ethidium bromide staining. Cellular DNA in the same samples was quantified by parallel amplification of a β-actin gene fragment.
Virus genotyping.
M50 MuHV-4 has the proximal 416 bp of the murine cytomegalovirus IE1/IE3 promoter inserted at genomic coordinate 66718, between the ORF50 transcription (66642) and translation (66760) start sites. To detect this insert, we amplified across genomic coordinates 66580 to 66848, using primers 5′-CACATTATCCCACAATGTGCTGC and 5′-GAAATACTGATCTGTCTGCGTGG. This gave a 268-bp wild-type product and a 684-bp M50 product. ORF50DEL MuHV-4 has genomic coordinates 67792 to 69177 deleted from ORF50 exon 2 (67661 to 69376). In place is ligated a 1,961-bp fragment comprising the firefly luciferase coding sequence in frame with ORF50 (1,672 bp) and a simian virus 40 (SV40) polyadenylation signal (289 bp). We amplified across genomic coordinates 67672 to 69240, using primers 5′-GATCGAAGCAGGTCTACTTGAG and 5′-TCAGCAGTGTCCTGGTTTGCC, to give a 1,569-bp wild-type band or a 2,145-bp mutant band. ORF73FS MuHV-4 has a 5-bp insert at genomic coordinate 104379 in ORF73 (104869 to 103925). This disrupts a BstEII restriction site. We amplified across genomic coordinates 104152 to 104580, using primers 5′-TTCACAGTAGGCCAAGACAACC and 5′-CCACCATCACCAGATGTTGATG. This gave a 428-bp wild-type product, cut by BstEII into 228-bp and 200-bp fragments, or a 433-bp mutant product not cut by BstEII. DNA sequencing of PCR products was performed at the Australian Genome Research Facility (St. Lucia, Queensland, Australia). To identify MCMV M33 disruption, we amplified viral DNA with primers 5′-GTGGTGCTGACGACGCAGCTGCTG and 5′-GTGTGGCTGCGCCTGCGGTACGAG for a wild-type band of 572 bp. The M33 mutant has a 3.8-kb β-galactosidase expression cassette inserted without deletion to give a 4.4-kb band. To identify M78 disruption, we amplified viral DNA with primers 5′-TCGTCTGCCCCTCTAAGGTCA and 5′-CAGACGGTGGGGATCTTGTCG for a wild-type band of 919 bp. The M78 mutant has an equivalent 3.8-kb cassette insertion to give a 4.7-kb band.
Immunostaining of tissue sections.
Organs were fixed in 1% formaldehyde/10 mM sodium periodate/75 mM l-lysine (18 h, 4°C). Noses were then decalcified in 150 mM NaCl/50 mM Tris-Cl (pH 7.2)/270 mM EDTA for 2 weeks at 23°C, with changing of the solution every 3 days, and then washed twice in phosphate-buffered saline (PBS). Samples were dehydrated in graded ethanol solutions, embedded in paraffin, and cut with a microtome. Sections were then dewaxed in xylene, rehydrated, washed 3 times in PBS, and air dried. Endogenous peroxidase activity was quenched in PBS/3% H2O2 for 10 min. Sections were blocked (1 h, 23°C) with 0.3% Triton X-100/5% normal donkey serum and incubated (18h, 4°C) with anti-MuHV-4 rabbit polyclonal antibody (pAb), which recognizes a range of virion proteins by immunoblotting, including the products of ORF4 (gp70), M7 (gp150), and ORF65 (p20) (54). Sections were additionally blocked with an avidin/biotin blocking kit (Vector Laboratories). Detection was done with biotinylated goat anti-rabbit IgG pAb (1 h, 23°C; Vector Laboratories), the Vectastain Elite ABC peroxidase system, and ImmPACT diaminobenzidine substrate (Vector Laboratories). Stained sections were counterstained with Mayer’s hemalum (Merck), dehydrated, and mounted in dibutylphthalate polystyrene xylene (DPX; BDH Chemicals, UK).
ACKNOWLEDGMENTS
We thank Stacey Efstathiou for providing the original ORF73FS and ORF50DEL MuHV-4 mutants.
This work was supported by grants from the National Health and Medical Research Council (project grants 1122070 and 1140169), the Australian Research Council (grant DP190101851), and Queensland Health.
The funders played no part in decisions about the paper.
We report no conflict of interest.
Contributor Information
Helen E. Farrell, Email: h.farrell1@uq.edu.au.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suárez NM, Wilkie GS, Hage E, Camiolo S, Holton M, Hughes J, Maabar M, Vattipally SB, Dhingra A, Gompels UA, Wilkinson GWG, Baldanti F, Furione M, Lilleri D, Arossa A, Ganzenmueller T, Gerna G, Hubáček P, Schulz TF, Wolf D, Zavattoni M, Davison AJ. 2019. Human cytomegalovirus genomes sequenced directly from clinical material: variation, multiple-strain infection, recombination, and gene loss. J Infect Dis 220:781–791. 10.1093/infdis/jiz208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palser AL, Grayson NE, White RE, Corton C, Correia S, Ba Abdullah MM, Watson SJ, Cotten M, Arrand JR, Murray PG, Allday MJ, Rickinson AB, Young LS, Farrell PJ, Kellam P. 2015. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. J Virol 89:5222–5237. 10.1128/JVI.03614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbotts J, Nishiyama Y, Yoshida S, Loeb LA. 1987. On the fidelity of DNA replication: herpes DNA polymerase and its associated exonuclease. Nucleic Acids Res 15:1185–1198. 10.1093/nar/15.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pyzik M, Dumaine A, Dumaine AA, Charbonneau B, Fodil-Cornu N, Jonjic S, Vidal SM. 2014. Viral MHC class I-like molecule allows evasion of NK cell effector responses in vivo. J Immunol 193:6061–6069. 10.4049/jimmunol.1401386. [DOI] [PubMed] [Google Scholar]
- 6.Smith HRC, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA 99:8826–8831. 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corbett AJ, Coudert JD, Forbes CA, Scalzo AA. 2011. Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J Immunol 186:1713–1722. 10.4049/jimmunol.1003308. [DOI] [PubMed] [Google Scholar]
- 8.Pfaff F, Groth M, Sauerbrei A, Zell R. 2016. Genotyping of herpes simplex virus type 1 by whole-genome sequencing. J Gen Virol 97:2732–2741. 10.1099/jgv.0.000589. [DOI] [PubMed] [Google Scholar]
- 9.Cudini J, Roy S, Houldcroft CJ, Bryant JM, Depledge DP, Tutill H, Veys P, Williams R, Worth AJJ, Tamuri AU, Goldstein RA, Breuer J. 2019. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc Natl Acad Sci USA 116:5693–5698. 10.1073/pnas.1818130116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGeoch DJ, Gatherer D. 2007. Lineage structures in the genome sequences of three Epstein-Barr virus strains. Virology 359:1–5. 10.1016/j.virol.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Kakoola DN, Sheldon J, Byabazaire N, Bowden RJ, Katongole-Mbidde E, Schulz TF, Davison AJ. 2001. Recombination in human herpesvirus-8 strains from Uganda and evolution of the K15 gene. J Gen Virol 82:2393–2404. 10.1099/0022-1317-82-10-2393. [DOI] [PubMed] [Google Scholar]
- 12.Hage E, Wilkie GS, Linnenweber-Held S, Dhingra A, Suárez NM, Schmidt JJ, Kay-Fedorov PC, Mischak-Weissinger E, Heim A, Schwarz A, Schulz TF, Davison AJ, Ganzenmueller T. 2017. Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole-genome sequencing directly from clinical specimens. J Infect Dis 215:1673–1683. 10.1093/infdis/jix157. [DOI] [PubMed] [Google Scholar]
- 13.Hoagland RJ. 1955. The transmission of infectious mononucleosis. Am J Med Sci 229:262–272. 10.1097/00000441-195503000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Rickinson AB, Yao QY, Wallace LE. 1985. Epstein-Barr virus as a model of virus-host interactions. Br Med Bull 41:75–79. 10.1093/oxfordjournals.bmb.a072030. [DOI] [PubMed] [Google Scholar]
- 15.Hoagland RJ. 1964. The incubation period of infectious mononucleosis. Am J Public Health Nations Health 54:1699–1705. 10.2105/ajph.54.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunmire SK, Grimm JM, Schmeling DO, Balfour HH, Hogquist KA. 2015. The incubation period of primary Epstein-Barr virus infection: viral dynamics and immunologic events. PLoS Pathog 11:e1005286. 10.1371/journal.ppat.1005286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillet L, Frederico B, Stevenson PG. 2015. Host entry by γ-herpesviruses—lessons from animal viruses? Curr Opin Virol 15:34–40. 10.1016/j.coviro.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Milho R, Smith CM, Marques S, Alenquer M, May JS, Gillet L, Gaspar M, Efstathiou S, Simas JP, Stevenson PG. 2009. In vivo imaging of murid herpesvirus-4 infection. J Gen Virol 90:21–32. 10.1099/vir.0.006569-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frederico B, Milho R, May JS, Gillet L, Stevenson PG. 2012. Myeloid infection links epithelial and B cell tropisms of murid herpesvirus-4. PLoS Pathog 8:e1002935. 10.1371/journal.ppat.1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milho R, Frederico B, Efstathiou S, Stevenson PG. 2012. A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog 8:e1002986. 10.1371/journal.ppat.1002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.François S, Vidick S, Sarlet M, Desmecht D, Drion P, Stevenson PG, Vanderplasschen A, Gillet L. 2013. Illumination of murine gammaherpesvirus-68 cycle reveals a sexual transmission route from females to males in laboratory mice. PLoS Pathog 9:e1003292. 10.1371/journal.ppat.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaspar M, May JS, Sukla S, Frederico B, Gill MB, Smith CM, Belz GT, Stevenson PG. 2011. Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog 7:e1002346. 10.1371/journal.ppat.1002346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frederico B, Chao B, May JS, Belz GT, Stevenson PG. 2014. A murid γ-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 15:457–470. 10.1016/j.chom.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Farrell HE, Lawler C, Tan CSE, MacDonald K, Bruce K, Mach M, Davis-Poynter N, Stevenson PG. 2016. Murine cytomegalovirus exploits olfaction to enter new hosts. mBio 7:e00251-16. 10.1128/mBio.00251-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shivkumar M, Milho R, May JS, Nicoll MP, Efstathiou S, Stevenson PG. 2013. Herpes simplex virus 1 targets the murine olfactory neuroepithelium for host entry. J Virol 87:10477–10488. 10.1128/JVI.01748-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGeoch DJ, Rixon FJ, Davison AJ. 2006. Topics in herpesvirus genomics and evolution. Virus Res 117:90–104. 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi K, Hayashi M, Jalkanen M, Firestone JH, Trelstad RL, Bernfield M. 1987. Immunocytochemistry of cell surface heparan sulfate proteoglycan in mouse tissues. A light and electron microscopic study. J Histochem Cytochem 35:1079–1088. 10.1177/35.10.2957423. [DOI] [PubMed] [Google Scholar]
- 28.E X, Meraner P, Lu P, Perreira JM, Aker AM, McDougall WM, Zhuge R, Chan GC, Gerstein RM, Caposio P, Yurochko AD, Brass AL, Kowalik TF. 2019. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc Natl Acad Sci USA 116:7043–7052. 10.1073/pnas.1814850116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rickabaugh TM, Brown HJ, Martinez-Guzman DAnn, Wu T-T, Tong L, Yu F, Cole S, Sun R. 2004. Generation of a latency-deficient gammaherpesvirus that is protective against secondary infection. J Virol 78:9215–9223. 10.1128/JVI.78.17.9215-9223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fowler P, Marques S, Simas JP, Efstathiou S. 2003. ORF73 of murine herpesvirus-68 is critical for the establishment and maintenance of latency. J Gen Virol 84:3405–3416. 10.1099/vir.0.19594-0. [DOI] [PubMed] [Google Scholar]
- 31.Moorman NJ, Willer DO, Speck SH. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol 77:10295–10303. 10.1128/jvi.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May JS, Coleman HM, Smillie B, Efstathiou S, Stevenson PG. 2004. Forced lytic replication impairs host colonization by a latency-deficient mutant of murine gammaherpesvirus-68. J Gen Virol 85:137–146. 10.1099/vir.0.19599-0. [DOI] [PubMed] [Google Scholar]
- 33.Boname JM, Coleman HM, May JS, Stevenson PG. 2004. Protection against wild-type murine gammaherpesvirus-68 latency by a latency-deficient mutant. J Gen Virol 85:131–135. 10.1099/vir.0.19592-0. [DOI] [PubMed] [Google Scholar]
- 34.Fowler P, Efstathiou S. 2004. Vaccine potential of a murine gammaherpesvirus-68 mutant deficient for ORF73. J Gen Virol 85:609–613. 10.1099/vir.0.19760-0. [DOI] [PubMed] [Google Scholar]
- 35.Lawler C, Stevenson PG. 2020. Limited protection against γ-herpesvirus infection by replication-deficient virus particles. J Gen Virol 101:420–425. 10.1099/jgv.0.001391. [DOI] [PubMed] [Google Scholar]
- 36.Lawler C, Simas JP, Stevenson PG. 2020. Vaccine protection against murid herpesvirus-4 is maintained when the priming virus lacks known latency genes. Immunol Cell Biol 98:67–78. 10.1111/imcb.12299. [DOI] [PubMed] [Google Scholar]
- 37.Clambey ET, Virgin HW, Speck SH. 2002. Characterization of a spontaneous 9.5-kilobase-deletion mutant of murine gammaherpesvirus 68 reveals tissue-specific genetic requirements for latency. J Virol 76:6532–6544. 10.1128/jvi.76.13.6532-6544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan CS, Frederico B, Stevenson PG. 2014. Herpesvirus delivery to the murine respiratory tract. J Virol Methods 206:105–114. 10.1016/j.jviromet.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Farrell HE, Bruce K, Lawler C, Oliveira M, Cardin R, Davis-Poynter N, Stevenson PG. 2017. Murine cytomegalovirus spreads by dendritic cell recirculation. mBio 8:e01264-17. 10.1128/mBio.01264-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yunis J, Farrell HE, Bruce K, Lawler C, Sidenius S, Wyer O, Davis-Poynter N, Stevenson PG. 2018. Murine cytomegalovirus degrades MHC class II to colonize the salivary glands. PLoS Pathog 14:e1006905. 10.1371/journal.ppat.1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis-Poynter N, Yunis J, Farrell HE. 2016. The cytoplasmic C-tail of the mouse cytomegalovirus 7 transmembrane receptor homologue, M78, regulates endocytosis of the receptor and modulates virus replication in different cell types. PLoS One 11:e0165066. 10.1371/journal.pone.0165066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farrell HE, Stevenson PG. 2019. Cytomegalovirus host entry and spread. J Gen Virol 100:545–553. 10.1099/jgv.0.001230. [DOI] [PubMed] [Google Scholar]
- 43.Farrell HE, Bruce K, Lawler C, Stevenson PG. 2019. murine cytomegalovirus spread depends on the infected myeloid cell type. J Virol 93:e00540-19. 10.1128/JVI.00540-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrios AW, Núñez G, Sánchez Quinteiro P, Salazar I. 2014. Anatomy, histochemistry, and immunohistochemistry of the olfactory subsystems in mice. Front Neuroanat 8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawler C, Milho R, May JS, Stevenson PG. 2015. Rhadinovirus host entry by co-operative infection. PLoS Pathog 11:e1004761. 10.1371/journal.ppat.1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glauser DL, Milho R, Lawler C, Stevenson PG. 2019. Antibody arrests γ-herpesvirus olfactory super-infection independently of neutralization. J Gen Virol 100:246–258. 10.1099/jgv.0.001183. [DOI] [PubMed] [Google Scholar]
- 47.Dechecchi MC, Tamanini A, Bonizzato A, Cabrini G. 2000. Heparan sulfate glycosaminoglycans are involved in adenovirus type 5 and 2-host cell interactions. Virology 268:382–390. 10.1006/viro.1999.0171. [DOI] [PubMed] [Google Scholar]
- 48.Summerford C, Samulski RJ. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 72:1438–1445. 10.1128/JVI.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sitki-Green DL, Edwards RH, Covington MM, Raab-Traub N. 2004. Biology of Epstein-Barr virus during infectious mononucleosis. J Infect Dis 189:483–492. 10.1086/380800. [DOI] [PubMed] [Google Scholar]
- 50.Weiss ER, Lamers SL, Henderson JL, Melnikov A, Somasundaran M, Garber M, Selin L, Nusbaum C, Luzuriaga K. 2018. Early Epstein-Barr virus genomic diversity and convergence toward the B95.8 genome in primary infection. J Virol 92:e01466-17. 10.1128/JVI.01466-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stevenson PG, Doherty PC. 1998. Kinetic analysis of the specific host response to a murine gammaherpesvirus. J Virol 72:943–949. 10.1128/JVI.72.2.943-949.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson KH, Webb CH, Schmeling DO, Brundage RC, Balfour HH. 2016. Epstein-Barr virus dynamics in asymptomatic immunocompetent adults: an intensive 6-month study. Clin Trans Immunol 5:e81. 10.1038/cti.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lang DJ, Garruto RM, Gajdusek DC. 1977. Early acquisition of cytomegalovirus and Epstein-Barr virus antibody in several isolated Melanesian populations. Am J Epidemiol 105:480–487. 10.1093/oxfordjournals.aje.a112407. [DOI] [PubMed] [Google Scholar]
- 54.de Lima BD, May JS, Stevenson PG. 2004. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J Virol 78:5103–5112. 10.1128/jvi.78.10.5103-5112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]