

Abstract
Frontotemporal dementia accounts for a significant fraction of dementia cases. Frontotemporal dementia with parkinsonism linked to chromosome 17 is associated with either exonic or intronic mutations in the tau gene. This highlights the involvement of aberrant pre-mRNA splicing in the pathogenesis of neurodegenerative disorders. Little is known about the molecular mechanisms of the splicing defects underlying these diseases. To establish a model system for studying the role of pre-mRNA splicing in neurodegenerative diseases, we have constructed a tau minigene that reproduces tau alternative splicing in both cultured cells and in vitro biochemical assays. We demonstrate that mutations in a nonconserved intronic region of the human tau gene lead to increased splicing between exon 10 and exon 11. Systematic biochemical analyses indicate the importance of U1 snRNP and, to a lesser extent, U6 snRNP in differentially recognizing wild-type versus intron mutant tau pre-mRNAs. Gel mobility shift assays with purified U1 snRNP and oligonucleotide-directed RNase H cleavage experiments support the idea that the intronic mutations destabilize a stem-loop structure that sequesters the 5′ splice site downstream of exon 10 in tau pre-mRNA, leading to increases in U1 snRNP binding and in splicing between exon 10 and exon 11. Thus, mutations in nonconserved intronic regions that increase rather than decrease alternative splicing can be an important pathogenic mechanism for the development of human diseases.
tau is a major microtubule (MT)-binding protein that promotes MT assembly and stabilizes the MT tracks (for reviews, see references 26, 27, 43, and 44). The tau gene is expressed in developing and mature neurons and is especially enriched in the axon. A low level of tau expression is also found in glial cells. In addition to posttranslational regulation by phosphorylation, expression and function of the tau gene are under complex regulation by alternative splicing. In the human brain, alternative splicing produces six isoforms with variations in the amino-terminal region and in the carboxyl domain, containing either three or four MT-binding repeats (MT1 to MT4) (1, 19, 35, 40). This expression pattern results from a combination of alternative inclusion of exon 2 or exons 2 and 3 in the amino-terminal region and of exon 10 in the MT-binding domain of the carboxyl-terminal region. Exclusion or inclusion of exon 10 leads to the formation of tau proteins containing either three or four MT-binding repeats. In the normal human adult cerebral cortex, the ratio of four- to three-MT-binding repeat-containing tau transcripts (Tau4R/Tau3R) is approximately 1 (see Fig. 2) (15, 16, 18, 29, 30).
FIG. 2.

tau alternative splicing in tissues and cell lines. (A) Expression of endogenous tau exon 10+ (Tau4R) and exon 10− (Tau3R) mRNAs in the adult human brain, rat brain, and human (HeLa, 293T, HEK293, HeLa RB, and SH-SY5Y) and murine (N2a and GT1-7) cell lines as determined by RT-PCR with specific primers in exon 9 and exon 11 of human or rat and mouse tau. (B) Alternative splicing of exon 10 from transfected wild-type (WT) and mutant tau minigenes (AusI and DDPAC). The tau minigene constructs (TauEx9+11d5) with wild-type and mutant intronic sequences were transfected into HeLa RB and N2a cells. Alternatively spliced tau products expressed from the transfected minigene were detected by RT-PCR with primers specific to the transfected plasmids (T7 and SP6 primers). The percentage of Tau4R (exon 10 inclusion) in the total splicing products was measured using a PhosphorImager.
A large number of studies have suggested that changes in the tau protein play a critical role in neurodegeneration (reviewed in references 17, 35, 41, 61, 62). tau-containing neurofibrillary lesions are found in myotonic dystrophy, Pick's disease, corticobasal degeneration and progressive supranuclear palsy. In addition, several other neurodegenerative diseases have intraneuronal lesions containing aberrantly processed tau protein, including Niemann-Pick disease type C, Gerstmann-Straussler-Scheinker disease with tangles, prion protein amyloid angiopathy, amyotrophic lateral sclerosis-parkinsonism-dementia complex of Guam, Down syndrome, and familial presenile dementia with tangles (reference 61 and references therein). Recent genetic studies have placed the tau gene at the center of pathogenesis of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), an autosomal dominant condition characterized by prominent atrophy of the frontal and temporal cortex with later involvement of subcortical structures (14). The brain atrophy is usually accompanied by neuronal cell death, gliosis, and formation of intraneuronal deposits containing tau protein. Missense mutations in the tau gene have been identified in FTDP-17 cases, and several of these mutations lead to a reduction in the ability of tau to bind MT and to promote MT assembly (24, 29–31, 50). In addition to mutations found in the coding region of the tau gene, several intronic mutations were identified, accounting for a significant fraction of FTDP-17 cases (Fig. 1A) (for reviews, see references 16 and 61). All of the intronic mutations reported so far are associated with increases in the ratio of four- to three-repeat-containing isoforms of tau (Tau4R/Tau3R). This suggests that a proper ratio of Tau4R to Tau3R is essential for normal function of tau in the human brain and that disturbance of this delicate balance may lead to deleterious effects on neuronal survival and function. A change in the Tau4R/Tau3R ratio appears to be sufficient for development of the filamentous tauopathy (16, 32, 61). Discovery of these intronic mutations in the tau gene in dementia patients highlights the importance for normal brain function of controlling the balance of different tau isoforms by alternative splicing.
FIG. 1.

(A) Positions of reported mutations in exon 10 or the downstream intron region of the human tau gene. The first nucleotide in the intron is defined as position +1. 5′ SS, 5′ splice site. (B) Schematic of a series of tau minigene constructs. The genomic DNA fragments containing exons 9, 10, and 11 as well as intronic sequences (wild type [wt], AusI, or DDPAC) flanking exon 10 were inserted in mammalian expression vector pcDNA3 under the control of the cytomegalovirus promoter (PCMV). The chimeric minigene constructs Ad-TauEx10+11wt and -DDPAC were made by replacing the upstream exon 9 and associated intronic sequences with the first exon region (L1) of the adenovirus (Ad) major late transcription unit. The sizes of the corresponding exons and introns are indicated above the respective regions (in base pairs).
Based on analyses of the nucleotide sequence of the tau gene and the observation that intronic mutations increase inclusion of exon 10 in the tau transcripts, a hypothesis was proposed that a stem-loop-type secondary structure may form in the tau pre-mRNA around the exon-intron junction that could regulate tau exon 10 alternative splicing (6, 30, 63). In this study, we have established a minigene model system to examine tau alternative splicing using both cell culture and in vitro biochemical assays. We have tested the splicing efficiency of wild-type and mutant tau pre-mRNAs containing either DDPAC+14 or AusI+16 intronic mutations and demonstrated that the intronic mutations enhance splicing of exon 10 without affecting RNA stability. Systematic analyses using biochemical approaches indicate that among the different spliceosomal U snRNPs, U1 snRNP plays a prominent role in differentially recognizing wild-type versus intronic mutant tau pre-mRNAs and that U6 snRNP also appears to be involved in this process. A gel mobility shift assay with purified U1 snRNP preparations demonstrates a significantly higher level of U1 snRNP binding to the mutant tau than to the wild-type tau pre-mRNA. Our data support the model in which base-pairing interactions between the intronic sequence downstream of exon 10 and the splice junction itself prevent maximal recognition of this 5′ splice site. Partial disruption of these base-pairing interactions by intronic mutations leads to enhanced U1 snRNP interaction with the 5′ splice site of exon 10 and therefore higher splicing efficiency. To our knowledge, this is the first study with systematic biochemical characterization of a human disease-causing alternative splicing event in which intronic mutations increase rather than decrease splicing efficiency.
MATERIALS AND METHODS
Plasmids.
The tau genomic DNA fragments containing exons 9, 10, and 11 as well as intronic sequences (wild type or mutant) flanking exon 10 were amplified by PCR from normal adult human or FTDP-17 patient brain genomic DNA. tau minigene constructs were made by inserting the genomic fragments into mammalian expression vector pcDNA3 (Invitrogen) between the HindIII and XhoI sites under the control of the cytomegalovirus promoter. DNA sequence analysis of tau genomic fragments and different expression plasmids was carried out on an ABI 373A automatic sequencer using the PRISM Ready Reaction DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems).
Cell culture and transient transfection.
For transfection experiments, all plasmid DNA samples were purified by double cesium chloride centrifugation. HeLa RB or N2a cells were grown in 3.5-cm dishes in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. At the time of transfection, HeLa RB cells were approximately 50% confluent and N2a cells were approximately 60% confluent. Transfection was performed by the calcium phosphate precipitation method with 2 to 3 μg of tau minigene construct DNA. Cells were harvested 48 h after transfection. Under these conditions, the transfection efficiency in these experiments was approximately 60% for HeLa RB cells and 50% for N2a cells.
RT-PCR assay.
RNA was prepared from different tissues or transfected HeLa RB or N2a cells with Trizol reagent (Gibco-BRL), and reverse transcription (RT)-PCR was carried out in the presence of [α-32P]dCTP (Amersham Life Sciences) as previously described (68). Human or murine and rat tau-specific primers in exon 9 and exon 11 were used to detect the endogenous tau mRNA. Alternatively spliced tau products expressed from the transfected minigene were detected by RT-PCR with primers specific to the transfected plasmid (T7 and SP6 primers). The PCR cycle number was kept to a minimum (20 cycles) to maintain linearity. The ratio of tau exon 10+ (Tau4R) to tau exon 10− (Tau3R) was measured with a PhosphorImager (Molecular Dynamics).
Transcription and splicing.
Splicing substrates were synthesized using T7 RNA polymerase (Promega) from corresponding linear templates, with catabolite gene activator protein analog (Pharmacia Biotech) and [α-32P]UTP (Amersham Life Sciences). RNA purification was performed as described before (5). HeLa cell nuclear extracts were prepared as described before (8). Some preparations yielded very low splicing activity for uncharacterized reasons and allowed only the first step of the splicing reaction to occur when incubated for 2 h at 30°C. It was observed previously that these extracts could be rendered fully active by the addition of 2 U of creatine kinase per 25-μl splicing reaction mixture (7). Splicing mixtures were set up and processed as described before (45). Splicing products were separated on 7 or 8% acrylamide (acrylamide-bisacrylamide, 38:2)–8 M urea gels. The identity of lariat intermediates was confirmed by performing debranching reactions in S100 extracts followed by gel migration.
Oligonucleotide-targeted RNase H cleavage assay.
Minigene constructs TauEx10+11 and TauEx10+11d5 were linearized with EcoRI (Promega) and transcribed into RNA with T7 RNA polymerase. [α-32P]UTP-labeled wild-type and mutant RNAs were incubated at 37°C for 20 min in standard splicing buffer in the presence of 0.5 U of RNase H (Gibco-BRL) in 12.5-μl reaction mixtures with various concentrations of an oligonucleotide (5′-GAAGGTACTCACACTGCC-3′) complementary to the exon 10 splice donor site. When assays were performed in the presence of nuclear extracts, RNAs were incubated with nuclear extract for the indicated times in the presence of RNase H and 0.2 pmol of oligonucleotide. These concentrations of oligonucleotide and RNase H were used so that differences in cleavage between the wild-type and mutant tau RNAs were not masked by excessive amounts of oligonucleotide or RNase H. Cleaved RNA products were then separated on a 6% polyacrylamide–8 M urea gel. The cleavage ratios were measured with a PhosphorImager (Molecular Dynamics).
U snRNP inactivation and blocking assays.
U1 snRNA was either inactivated using oligonucleotide-targeted RNase H cleavage as described above or blocked by incubation with 2′-O-methyl-oligoribonucleotides. Specifically, 2′-O-methyl-oligonucleotides (U1, 3 to 12 μM; U2, 0.3 μM; U5, 12 μM; U6, 13 μM) were added to splicing reaction mixtures and incubated at 30°C for 10 min prior to the addition of RNA substrates (55). Incubations were then carried on for 1.5 h at 30°C in the presence or absence of creatine kinase (1 U). In the absence of creatine kinase, splicing reactions with this particular HeLa nuclear extract did not efficiently proceed through the second step of the splicing reaction. As a control, a 2′-O-methyl-oligonucleotide against U7 was used (6 μM). HeLa cell nuclear extracts depleted of U1 snRNP activity were produced by incubation at 30°C for 1 h in the presence of RNase H and the oligonucleotide CCAGGTAAGTAT, complementary to the 5′ end of U1 snRNA (3). As a control, a mock-treated nuclear extract was obtained by incubation with RNase H and an unrelated oligonucleotide. Splicing products were processed as before and resolved on 8% denaturing gels. Aliquots of the mock- and U1 oligonucleotide-treated nuclear extracts were incubated with proteinase K (1 mg/ml) for 30 min at 37°C, extracted with phenol, and ethanol precipitated. The extracted RNAs were then run on an 8% polyacrylamide–8 M urea gel and visualized by ethidium bromide staining to assess the efficiency of RNase H cleavage.
U1 snRNP protection assay.
Splicing reactions were carried out with 2 fmol of 32P-labeled-tau pre-mRNA transcripts with U1-depleted or mock-treated nuclear extracts at 30°C for 0, 10, or 20 min. RNase H (0.4 U) and 20 pmol (molar excess) of the oligonucleotide complementary to the 5′ splice site of exon 10 (GAAGGTACTCACACTGCC) were then added to 12.5-μl splicing reaction mixtures (11), and incubation was continued for 15 min at 37°C to completely cleave the pre-mRNA transcripts that were not protected. U1 snRNP- or mock-depleted extracts were prepared as described above.
U1 binding assay.
The U1 snRNP preparation was a kind gift from A. Kramer, and its purification and characterization were described previously (37, 38). The major snRNA species detected in the preparation is U1 snRNA. 32P-labeled pre-mRNA transcripts from constructs TauEx10+11d5wt and -DDPAC were incubated under splicing conditions with various amounts of purified U1 snRNP at 30°C for 5 min except that no ATP or creatine phosphate was added. Heparin was then added to a final concentration of 0.5 mg/ml, and reaction mixtures were incubated at 30°C for an additional 5 min. The RNA-U1 snRNP complexes were resolved on 4% nondenaturing polyacrylamide gels (acrylamide-bisacrylamide, 80:1), and electrophoresis was carried out in Tris-glycine buffer (50 mM Tris, 50 mM glycine) (34).
RESULTS
Alternative splicing of tau exon 10.
To understand alternative splicing regulation of genes involved in neuronal function and neurodegeneration, we have initiated a study of tau pre-mRNA alternative splicing. Because all of the intronic mutations identified so far in the tau gene are in the intron following exon 10 and many exonic mutations are also located in exon 10 (6, 25, 30, 63), we have focused our efforts on this region of the human tau gene (Fig. 1A). Based on our previous experience (33, 68) we established a specific and sensitive RT-PCR protocol to examine tau alternative splicing in tissues or cultured cells. This allowed us to reliably measure the level of inclusion or exclusion of tau exon 10 in human brain tissues and in cultured cell lines. A number of human or rodent cell lines of neural as well as nonneural origin were screened. The tau alternative splicing pattern detected in these cell lines was compared with that in adult human and rat brain tissues. Consistent with previous studies (30, 35), endogenous tau expression was mostly restricted to cell lines derived from neural tissues. tau expression was also detected in embryonic kidney cell lines HEK293 and 293T in addition to neural cell lines such as SH-SY5Y, N2a, and GT1-7. In the adult human brain, the ratio of Tau3R to Tau4R is approximately 1 (Fig. 2A, lane 6) (30). However, in the adult rat brain, Tau4R is the predominant tau RNA transcript (Fig. 2A, lane 9). This suggests that alternative splicing regulation of the tau gene in human brain is different from that in rat brain.
Intronic mutations affect exon 10 alternative splicing.
tau minigenes were constructed by using genomic DNA fragments containing intact exons 9, 10, and 11 with shortened flanking intronic sequences (Fig. 1B, TauEx9-11wt and TauEx9-11d5wt). These tau minigenes, when introduced into either HeLa RB or N2a cells, underwent alternative splicing similar to that seen in the human adult brain, with the formation of both Tau3R and Tau4R (data not shown) (Fig. 2B, lanes 1 and 2 and 7 and 8). We therefore concluded that the minimal TauEx9-11d5wt minigene with a total of 800 bp of tau genomic DNA sequence contains cis elements essential for tau exon 10 alternative splicing. We noticed that transfected tau minigenes in these cells produced a slightly higher level of Tau4R compared to the endogenous tau expression pattern (Fig. 2), suggesting that overexpression of the tau minigene may titrate certain limiting factors controlling the ratio of Tau3R to Tau4R.
Derivatives of tau minigenes containing the FTDP-17-associated mutations AusI+16 and DDPAC+14 were then constructed (Fig. 1B, TauEx9-11AusI and -DDPAC and TauEx9-11d5AusI and -DDPAC, respectively). These intronic mutant tau minigenes were introduced into either HeLa RB or N2a cells. The tau splicing pattern in these transfected cells was examined by RT-PCR. Both AusI+16 and DDPAC+14 derivatives led to predominantly Tau4R production when transiently expressed in these cell lines (data not shown) (Fig. 2B, lanes 3 to 6 and lanes 9 to 12), as detected in brain tissues of FTDP-17 patients (30; Z. Jiang and J. Y. Wu, unpublished data). This observation suggests that the AusI+16 and DDPAC+14 intronic mutations in the tau gene most likely mediate their effect by affecting exon 10 alternative splicing and that cis elements required to observe the mutant profile were present in our TauEx9-11d5 minigene. However, these results, similar to previous studies (6, 8, 23, 30, 63), could not rule out the possibility that differences in RNA stability contributed to the alternative splicing difference. Therefore, we tested whether the AusI+16 and DDPAC+14 mutations affected the RNA stability of tau pre-mRNA transcripts both in transfected cells and in an in vitro biochemical assay. Specifically, following transfections, cells were treated with actinomycin D to block transcription, and levels of tau pre-mRNA and mRNA transcripts were then quantified at different time points by RT-PCR. No significant difference in RNA stability was detected between wild-type and AusI+16 or DDPAC+14 pre-mRNAs (Fig. 3). We also compared the stability of tau wild-type and mutant pre-mRNAs using in vitro-synthesized and radiolabeled transcripts. Following incubation with HeLa nuclear extracts for different periods of time, the tau pre-mRNA and splicing products were separated by gel electrophoresis. The levels of the RNA transcripts were then quantified with a PhosphorImager. Again, no difference was detected in the stability of wild-type and mutant tau transcripts (Fig. 4 and 5). These results demonstrated that the effects of these FTDP-17 intronic mutations on tau alternative splicing were not due to differential RNA stability.
FIG. 3.

Intronic mutations do not affect the RNA stability of tau transcripts. Wild-type (WT) and mutant (AusI+16 and DDPAC+14) tau minigenes were transfected into N2a cells. Following transfections, cells were treated with actinomycin D to block transcription, and tau pre-mRNA and mRNA transcripts were then detected at different time points by RT-PCR (A). The tau transcripts were quantified using a PhosphorImager. RNA stability was expressed as the ratio of total tau transcripts at different time points to that at time zero (B).
FIG. 4.
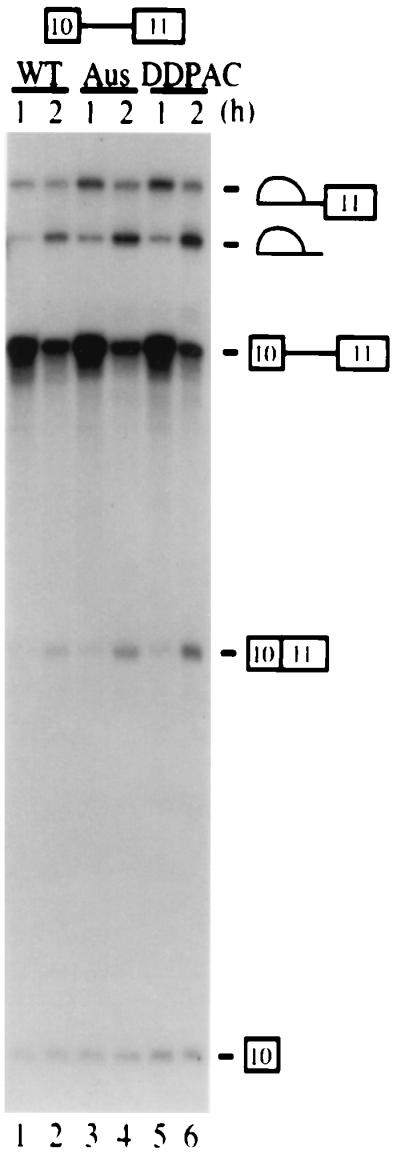
Intronic mutations affect exon 10 alternative splicing. In vitro splicing of TauEx10+11d5wt and mutant substrates. Labeled pre-mRNA substrates were incubated in HeLa nuclear extracts (approximately 6 μg/μl) under splicing conditions for the times indicated.
FIG. 5.

Oligonucleotide-targeted RNase H cleavage assays support a potential secondary structure around the 5′ splice site of exon 10. (A) The stem-loop structure formed with exon 10 downstream intronic sequences. The mutations AusI+16 and DDPAC+14 are indicated. (B, C, and D). Oligonucleotide-targeted RNase H cleavage assays. Minigene constructs of TauEx10+11 (B) and TauEx10+11d5 (C and D) were linearized with EcoRI and transcribed into RNA with T7 RNA polymerase. Labeled wild-type (WT) and mutant (AusI and DDPAC) tau RNAs were incubated with RNase H at 37°C for 20 min under splicing conditions in the presence of various concentrations of the oligonucleotide complementary to the 5′ splice site. The line with an arrowhead over the stem-loop shows the position of the oligonucleotide used (A). TauEx10+11 constructs contain a cryptic 5′ splice site, as indicated by the asterisk (B). (D) RNase H cleavage assay carried out under splicing conditions with 0.2 pmol of the oligonucleotide in the presence of HeLa nuclear extract (1.6 μg/μl). In this experiment, tau transcripts, the oligonucleotide, the nuclear extract, and RNase H were mixed together prior to incubation at 37°C for the time indicated in minutes). The uncut RNA and products of cleavage at the exon 10 splice donor site are shown on the right, while the products resulting from the cryptic site are marked on the left (B). Quantification of the RNA cleavage products in panel B is shown in panels E (cleavage at the authentic site) and F (cleavage at the cryptic site). Panels G and H show the quantification of RNA cleavage products in panels C and D, respectively. The efficiency of cleavage was expressed as the ratio of total cleavage products to the corresponding input transcript as measured using a PhosphorImager.
To investigate the biochemical mechanism by which the intronic mutations affected tau exon 10 alternative splicing, we set up an in vitro splicing system using tau minigenes. When incubated under splicing conditions in HeLa nuclear extracts, the TauEx9-11d5wt and mutant substrates both yielded only the Tau3R (exon 10 skipping) mRNAs, with no detectable Tau4R (exon 10 inclusion) mRNAs (data not shown). This could be explained by the fact that the 3′ splice site upstream of exon 10 as well as the 5′ splice site of exon 9 are both divergent from consensus sequences, and therefore these splicing signals are too weak to be recognized by the splicing machinery in vitro. Therefore, we replaced the upstream exon 9 and associated intronic sequences for the first exon region (L1) of the adenovirus (Ad) major late transcription unit (Fig. 1B, Ad-TauEx10+11wt and -DDPAC). Even with this chimeric substrate, we were able to detect only a low level of exon 10 inclusion in the wild-type tau substrate and a slight enhancement of exon 10 inclusion with the DDPAC+14 derivative (data not shown). We then used single-intron substrates to determine which tau splicing unit (9-10 or 10-11) was specifically affected by these intronic mutations (Fig. 1B, TauEx9-10d5 and TauEx10+11d5). Neither DDPAC+14 nor AusI+16 intronic mutations affected splicing between exon 9 and exon 10 (data not shown). However, splicing between exons 10 and 11 was significantly enhanced by AusI+16 and DDPAC+14 mutations, because significantly higher levels of splicing intermediates or products can be detected at various time points (Fig. 4, compare lanes 3 and 5 with lane 1 or lanes 4 and 6 with lane 2). This result suggests that AusI+16 and DDPAC+14 intronic mutations increase specific recognition of the sequence around the 5′ splice site of exon 10 by the splicing machinery.
Oligonucleotide-directed RNase H cleavage experiments support the presence of a secondary structure around the 5′ splice site of exon 10.
It has been proposed in previous studies that the tau exon 10 splice donor region could form a stem-loop structure with downstream intronic sequences. This stem-loop model was first proposed based on sequence analysis and exon-trapping experiments where exon 10 was inserted with minimal intronic sequence into a heterologous splicing cassette (6, 23, 30, 63). In this model (Fig. 5A), there are base-pairing interactions between the nucleotide residues surrounding the 5′ splice site of exon 10 in the region that extends from positions −2 to +16 (with the first G nucleotide in the intron being +1). In wild-type tau pre-mRNA, this structure would contain at least six uninterrupted base pairs, whereas in either AusI+16 or DDPAC+14 mutant tau pre-mRNA, a G-C interaction is disrupted in the stem region, presumably leading to a less stable structure. More recently, nuclear magnetic resonance spectroscopy was employed to demonstrate that short RNA oligonucleotides corresponding to this region of tau pre-mRNA could form a stable, folded stem-loop structure in the absence of protein factors (65). However, it is not clear whether such a secondary structure exists in the longer tau pre-mRNA transcripts under splicing conditions or whether the splicing machinery indeed differentially recognizes wild-type versus mutant tau pre-mRNA transcripts.
We probed the potential secondary structure of the wild-type and mutant tau gene in the region around the exon 10 5′ splice site using an oligonucleotide-directed RNase H cleavage assay with a specific DNA oligonucleotide (illustrated in Fig. 5A) complementary to the 5′ splice site region. To avoid complication of splicing products (when the assay was performed in the presence of nuclear extract) and nonspecific cleavage, we used shorter RNA transcripts. With tau transcripts containing exon 10 and 261 nucleotides of downstream intronic sequences, both DDPAC+14 and AusI+16 mutant tau consistently showed significantly more cleavage products than wild-type tau. Consistently, RNase H cleavage at a cryptic splice site downstream of the authentic 5′ splice site occurred at a lower level in the mutant than in the wild-type transcripts (Fig. 5B and F). These data are consistent with an increased accessibility of the sequence around the 5′ splice site to the oligodeoxynucleotide-mediated RNase H cleavage in the mutant tau transcripts (Fig. 5B). These results support the idea that a secondary structure forms around the 5′ splice site of exon 10, reducing the binding of the oligonucleotide and, therefore, the efficiency of RNase H cleavage. Destabilization of this secondary structure by the DDPAC+14 or AusI+16 mutations increases cleavage by RNase H. To further define this potential secondary structure, we made additional truncations in the intron. Shortening this intronic sequence downstream of the 5′ splice site to 53 nucleotides did not affect the differential RNase H cleavage of wild-type and mutant tau transcripts (Fig. 5C and G), consistent with the observation that these shortened tau minigene constructs behave similarly to those with longer intron sequences (data not shown). Finally, the RNase H cleavage assay was carried out in the presence of HeLa nuclear extract. Again, as was observed in the absence of nuclear extract, there were significantly more cleavage products in the reactions with DDPAC+14 or AusI+16 tau transcripts than in those with the wild-type tau transcript (Fig. 5D and H). Quantification of the cleavage products at different oligonucleotide concentrations and at different time points clearly indicates that wild-type and intronic mutant (DDPAC+14 as well as AusI+16) tau RNAs have distinct susceptibilities to the RNase H cleavage. Because the oligonucleotide used in the RNase H cleavage assay does not extend to the position of these mutations (+14 for DDPAC and +16 for AusI; Fig. 5A) and because the difference in cleavage efficiency between the wild-type and mutant RNAs was detectable in the absence of other protein factors or spliceosomal components, these data are best explained by the difference in the RNA secondary structure. Taken together, these results support the hypothesis that at the U1 snRNP binding site around the 5′ splice site downstream of exon 10 there exists a secondary structure involving 53 or fewer intronic nucleotides. This secondary structure could form on naked tau pre-mRNA transcripts as well as with tau transcripts in the presence of splicing-competent nuclear extracts. This structure is altered in the DDPAC+14 and AusI+16 mutants so that base-pairing interaction in the stem is weakened and the accessibility of the oligonucleotide is increased, leading to enhanced RNase H cleavage in the reaction mixtures containing the mutant tau transcripts.
U1 and U6 snRNPs are important players in differential recognition of tau wild-type and mutant pre-mRNAs.
To dissect the mechanism by which intronic mutations affect tau exon 10 splicing, we employed the in vitro splicing assay using tau pre-mRNA substrates containing exon 10 and exon 11 with the shortened intron in between. Because the effect of the DDPAC+14 mutation was consistently stronger than that of the AusI+16 mutation in enhancing exon 10 splicing both in vitro and in transfected cells, we used the DDPAC+14 mutant for further characterization.
We first carried out titration experiments to examine whether certain splicing factors were limiting for tau exon 10 splicing. Serial dilution of the HeLa nuclear extract led to a general reduction in splicing efficiency for both wild-type and DDPAC+14 mutant tau pre-mRNAs (Fig. 6A). However, at medium to low concentrations of HeLa nuclear extracts, the difference in splicing efficiency between the DDPAC+14 and wild-type tau became even more obvious (Fig. 6A, compare lanes 2 with 5 and 3 with 6, respectively). This suggested the involvement of trans-acting factors in differential recognition of wild-type and mutant splicing substrates. We then investigated which spliceosomal U snRNPs were involved in differentially recognizing wild-type versus mutant tau pre-mRNAs. U1, U2, U5, or U6 snRNPs were partially blocked by using specific individual 2′-O-methyl-oligoribonucleotides at appropriate concentrations to treat HeLa nuclear extracts prior to the splicing reactions (55). In the mock-treated or control (U7) oligoribonucleotide-treated nuclear extracts, splicing of either wild-type or DDPAC mutant tau pre-mRNAs was the same as that in the untreated nuclear extract, with an approximately twofold increase in the splicing products in the reactions with DDPAC tau compared with wild-type tau constructs (Fig. 6B, lanes 1 and 2 and lanes 11 and 12). When either U2 snRNP or U5 snRNP was partially blocked, both wild-type tau splicing and DDPAC+14 tau splicing were partially inhibited to a similar extent. The ratio of DDPAC+14 splicing products to wild-type tau splicing products remained approximately the same (Fig. 6B, lanes 5 and 6 and lanes 7 and 8). With U6 snRNP partially blocked, splicing of wild-type tau was more severely affected than that of DDPAC+14 tau (Fig. 6B, lanes 9 and 10). However, when U1 snRNP was partially blocked, splicing of wild-type tau was drastically decreased, whereas DDPAC+14 tau splicing was only slightly affected. Thus, the ratio of DDPAC+14 splicing products to wild-type tau splicing products was increased from 2-fold in mock-treated nuclear extract to approximately 10-fold when U1 snRNP was partially blocked. This is consistent with the idea that the secondary structure on the wild-type tau pre-mRNA renders the 5′ splice site of exon 10 less accessible for recognition by U1 snRNP at the early stage and by other factors, such as U6, at a later stage during spliceosome assembly.
FIG. 6.

(A) Presence of a limiting factor(s) for tau splicing in HeLa nuclear extracts. Splicing reactions were set up as previously described except that decreasing amounts of HeLa nuclear extract (NE) were used, specifically: lanes 1 and 5, 6 μg/μl; lanes 2 and 6, 4 μg/μl; lanes 3 and 7, 2 μg/μl; and lanes 4 and 8, 0.8 μg/μl. Positions of pre-mRNA, splicing intermediates, and products are indicated. (B) U snRNP inactivation differentially affects wild-type (WT) and mutant tau splicing. 2′-O-Methyl-oligoribonucleotides (2′O-MeU) complementary to U1, U2, U5, U6, and U7 snRNAs were added individually to HeLa nuclear extracts, and the splicing reaction mixtures were preincubated at 30°C for 10 min. The concentration of individual 2′-O-methyl-oligonucleotides was titrated to give partial inhibition of splicing (U1, 8 μM; U2, 0.3 μM; U5, 12 μM; and U6, 13 μM). The U7 2′-O-methyl-oligonucleotide (6 μM), which has been shown not to affect splicing, was used as a control. TauEx10+11d5wt and the DDPAC+14 mutant pre-mRNAs (WT and DD, respectively) were then added, and the incubation was continued for 2 h. Splicing reaction products were analyzed by gel electrophoresis.
To further demonstrate that the involvement of U1 snRNP in differentially recognizing wild-type and DDPAC+14 tau substrates was an early event, we made use of HeLa nuclear extracts that would not proceed to the second step of splicing without the addition of exogenous creatine kinase (see Materials and Methods). In the presence of 8 μM 2′-O-methyl U1 oligonucleotide and creatine kinase, wild-type tau splicing was remarkably reduced but DDPAC+14 tau splicing was only slightly decreased (Fig. 7A, lanes 1 to 4). When exogenous creatine kinase was omitted from the splicing reaction and the splicing reaction was blocked at the first step, wild-type tau splicing was almost completely blocked in the presence of 3 to 16 μM 2′-O-methyl U1 oligonucleotide (Fig. 7A, lanes 7 and 8). However, under the same conditions, DDPAC tau splicing was hardly affected (Fig. 7A, compare lanes 10 to 12 with lanes 6 to 8). This observation clearly indicates that U1 snRNP is crucial for wild-type tau splicing, especially during the earliest step of the splicing reaction, and that the DDPAC+14 intronic mutation allows tau splicing to occur even when functional U1 snRNP is at a very low level. To rule out the possibility of artifacts related to the use of 2′-O-methyl-oligonucleotides, we depleted the extract of U1 snRNAs by using a DNA oligonucleotide and RNase H (Fig. 7B and C). Under these conditions, more than 90% of U1 snRNA in the nuclear extract was cleaved (Fig. 7C, lane 2). When splicing reactions were carried out using this U1-inactivated nuclear extract, wild-type tau splicing was completely blocked (Fig. 7B, lane 2), whereas significant amounts of splicing intermediates were still detected in the reactions with DDPAC+14 tau pre-mRNA (Fig. 7B, lane 4). This demonstrates that a low level of U1 snRNP (less than 10% of that in the untreated nuclear extract) was sufficient to support splicing of DDPAC+14 but not wild-type tau pre-mRNA. This result also suggests that the DDPAC+14 mutation may affect tau splicing through increasing initial recognition of the 5′ splice site of exon 10 by U1 snRNP.
FIG. 7.

Wild-type (WT) tau pre-mRNA is more sensitive to U1 snRNP inactivation than DDPAC mutant pre-mRNA. (A) Effect of U1 snRNP 2′-O-methyl-oligonucleotide inactivation on TauEx10+11d5 splicing. The same experiment was done using a different preparation of HeLa nuclear extract in the presence (lanes 1 to 4) and absence (lanes 5 to 12) of creatine kinase. This extract specifically needs the addition of creatine kinase to proceed through the second step of the splicing reaction. Incubation was for 1.5 h at 30°C. Addition of the 2′-O-methyl U1 (2′O-MeU1) oligonucleotide is shown above each lane. The concentration used was 8 μM for lanes 2 and 4 and 0, 4, 8, or 16 μM for lanes 5 and 9, 6 and 10, 7 and 11, and 8 and 12, respectively. (B) Inactivation of U1 snRNA by RNase H cleavage affects wild-type (WT) and DDPAC tau splicing. Splicing reactions with tau transcripts were performed with either mock-treated HeLa nuclear extract (lanes 1 and 3) or nuclear extract after treatment of oligonucleotide-targeted RNase H cleavage of U1 snRNA (lanes 2 and 4). Splicing reactions were incubated at 30°C for 1.5 h. An asterisk marks the RNA species produced by a cleavage induced by nonspecific hybridization of the U1-specific oligonucleotide to the tau pre-mRNA transcripts. (C) Efficiency of oligonucleotide-targeted RNase H cleavage of U1 snRNA. An aliquot of mock-treated (lane 1) or U1 snRNA-depleted (lane 2) nuclear extracts as described for panel B was analyzed by gel electrophoresis for the presence of different snRNA species (as indicated on the right). U1∗, U1 snRNA molecules containing a shortened 5′ end after RNase H cleavage. Under these conditions, more than 90% of U1 snRNA in the nuclear extract was cleaved.
DDPAC+14 intronic mutation promotes more efficient assembly of U1-dependent complexes on the 5′ splice site of exon 10.
To examine whether the DDPAC+14 intronic mutation could influence the formation of U1 snRNP-dependent early complexes on the 5′ splice site, a specific RNase H protection assay (Fig. 8) was performed as described before (11). Wild-type or DDPAC+14 tau splicing substrates were first incubated with HeLa nuclear extract under splicing conditions for 0 to 20 min. Then, 20 pmol (in molar excess to tau pre-mRNA transcripts) of the oligodeoxynucleotide complementary to the 5′ splice site and corresponding to the U1 snRNP binding region was added together with RNase H, and the incubation was continued for another 15 min. The RNase H-cleaved and -protected fragments were then resolved by denaturing polyacrylamide gel electrophoresis and quantified. An increasing level of protection was observed for both substrates upon incubation at 30°C (Fig. 8A, lanes 2 to 4 and 6 to 8), which likely reflects early 5′ splice site recognition by the spliceosomal commitment complexes. This is consistent with the previous observation with other pre-mRNAs that U1 snRNP binding to the 5′ splice site is detectable at 0°C but requires incubation with nuclear extract at 30°C to be stabilized (11). The DDPAC+14 tau mutant transcript reproducibly gave rise to twofold more protected product than wild-type tau (Fig. 8A, lanes 2 to 4 and lanes 6 to 8, and Fig. 7B, 10- and 20-min time points). Finally, when the nuclear extract was depleted of U1 snRNP by oligonucleotide-targeted RNase H cleavage, no protected tau pre-mRNA was detectable in the reactions with either wild-type or DDPAC+14 tau transcripts (Fig. 8A, lanes 1 and 5). The requirement for preincubation with nuclear extract to observe formation of the U1 snRNP protected band also suggests that certain trans-acting factors in the nuclear extract may play a role in making the 5′ splice site accessible for U1 snRNP binding. These results demonstrate that the DDPAC+14 intronic mutation enhances the formation of U1 snRNP-dependent complexes on the 5′ splice site of exon 10 in a dynamic fashion during splicing. Supporting this result, we also observed greater accumulation of spliceosomal complexes A and B at these same time points, as monitored by native gel electrophoresis (data not shown).
FIG. 8.

Disruption of the putative secondary structure by the DDPAC+14 intronic mutation promotes the assembly of U1-dependent complexes on the 5′ splice site of exon 10. (A) TauEx10+11d5wt and DDPAC+14 RNAs were incubated with nuclear extract at 0°C (lanes 2 and 6) or 30°C for 10 min (lanes 3 and 7) or 20 min (lanes 4 and 8) or with U1 snRNP-depleted (U1−) HeLa nuclear extracts for 20 min (lanes 1 and 5). Following incubation, the oligonucleotide complementary to the 5′ splice site of exon 10 (Fig. 1A) was added along with RNase H (0.4 U), and the incubation was continued for another 15 min at 37°C. The RNA cleavage products were then analyzed by gel electrophoresis. Positions of the U1-protected pre-mRNA and cleavage products are indicated on the right. The asterisk indicates the position of an artifactual cleavage product generated by hybridization of the U1 snRNA-specific oligonucleotide on the tau pre-mRNAs. After a longer exposure, the U1 snRNP-protected band was visible in lanes 2 and 6 but not detectable in the lanes 1 and 5. (B) Histogram representation of the ratio of protected tau pre-mRNA to digested products for wild-type (WT) and mutant substrates. Error bars are derived from five independent experiments.
FIG. 10.

Model explaining enhanced splicing of exon 10 in DDPAC+14 and AusI+16 intronic tau mutations via increased U1 snRNA interaction. In the wild-type tau, a putative stem-loop structure forms around the 5′ splice site of exon 10, as depicted in panel A. The base-pairing interaction between U1 snRNA and the 5′ splice site is shown in panel B. The stem-loop structure prevents efficient interaction of U1 snRNA with the 5′ splice site and leads to partial skipping of exon 10. Such a stem-loop structure is less stable in the presence of the DDPAC+14 and AusI+16 mutations (or other likely mutations, including those at positions −2, +3, and +13) because of reduced base-pairing interactions, resulting in increased recognition by U1 snRNP and therefore increased exon 10 splicing. The Watson-Crick base-pairing interactions are depicted as thick black bars. The G-to-A mutations at the +3 (MSTD) and −2 (S305N) positions not only disrupt the base-pairing interactions in the stem (A) but also lead to the formation of additional base-pairing interactions between the tau pre-mRNA and U1 snRNA (••). Thus, it is possible that such mutations at +3 or −2 positions have a more severe effect on exon 10 splicing.
To test directly whether DDPAC+14 intronic mutation promotes U1 snRNP binding, we used a gel mobility shift assay with purified U1 snRNP as described in Materials and Methods. As shown in Fig. 9, incubation of the purified U1 snRNP preparation with 32P-labeled tau pre-mRNA transcripts led to the formation of a complex that migrated more slowly than the free RNA, with significantly more complex detected in the DDPAC+14 tau reaction than in the wild-type tau reaction.
FIG. 9.

Tau DDPAC+14 intronic mutation increases U1 snRNP binding to tau pre-mRNA. TauEx10+11d5wt (lanes 1 to 5) and -DDPAC+14 (lanes 6 to 10) RNAs were incubated with purified U1 snRNP, 0 μl for lanes 1 and 6, 0.35 μl for lanes 2 and 7, 0.7 μl for lanes 3 and 8, and 1.4 μl for lanes 4 and 9, or with depleted U1 snRNP (U1−) (lanes 5 and 10, 1.4 μl of protein used). The RNA-U1 snRNP complexes were resolved in a nondenaturing 4% polyacrylamide gel. In lanes 5 and 10, the U1 snRNP preparation was treated with oligonucleotide-targeted RNase H cleavage as described above to inactivate U1 snRNA.
The difference in the amount of complex formed with the DDPAC+14 and wild-type tau pre-mRNAs is more obvious at the lower concentration of U1 snRNP used (Fig. 9, compare lanes 7 and 8 with lanes 2 and 3). Quantification with the PhosphorImager revealed that there is an approximately a seven- to ninefold increase in the level of the complex detected with the DDPAC+14 mutant compared with the wild-type tau transcript. The detected complex was U1 snRNP dependent, because inactivation of U1 snRNP in the preparation with RNase H in the presence of the U1-specific oligonucleotide abolished the formation of this complex (Fig. 9, lanes 5 and 10). These results demonstrate that the DDPAC+14 intronic mutation enhances the formation of U1 snRNP-dependent complexes on the 5′ splice site of exon 10.
DISCUSSION
We have established a minigene system to dissect the molecular mechanism underlying alternative splicing of human tau pre-mRNA exon 10, an event important for pathogenesis of neurodegenerative disorder FTDP-17. Our systematic biochemical analyses of wild-type and intronic mutant tau pre-mRNAs demonstrated that single-nucleotide mutations in an evolutionarily nonconserved intronic region enhance splicing between exon 10 and exon 11. Experiments using RNase H and an oligonucleotide complementary to positions −6 to +12 at the splice junction suggest that the intronic mutant tau transcripts harbor a more “open” RNA structure in this region than wild-type tau in the presence or absence of HeLa nuclear extracts. This is consistent with the presence of an RNA stem-loop structure forming around the exon 10 5′ splice site in wild-type tau pre-mRNA. Comparison of the splicing efficiency of wild-type and mutant tau transcripts when various spliceosomal U snRNPs were made limiting by specific 2′-O-methyl oligonucleotides demonstrated that wild-type tau splicing was most sensitive to a reduction in the level of functional U1 snRNP. Finally, the U1 snRNP protection assay and the gel mobility shift experiment with purified U1 snRNP revealed an increase in the binding of U1 snRNP to the 5′ splice site of exon 10 in the DDPAC+14 mutant, correlating well with the increase in splicing efficiency observed in our in vitro splicing assay. These results strongly support the model depicted in Fig. 10B, where the stem-loop structure in the wild-type tau pre-mRNA is destabilized by FTDP-17 intronic mutations, leading to enhanced recognition of this 5′ splice site by the U1 snRNP-containing early splicing complex and increased formation of Tau4R transcripts.
This stem-loop structure model could also explain the behavior of several other mutations found in a number of patients with tau exon 10 aberrant splicing, including +3 and +13 mutations (6, 23, 30, 63) in addition to the DDPAC+14 and AusI+16 mutations (Fig. 10A). It is also consistent with the observation that in the rat (or mouse), the predominant isoform of tau is the exon 10-containing isoform. Exon 10 splicing may be enhanced in these species because of the destabilization of the stem structure caused by the naturally occurring G at position +13 in the rat tau gene (G at both +13 and +16 positions in mouse; see Fig. 2); (23; Jiang and Wu, unpublished data). Thus, a single-nucleotide change in this nonconserved intronic region can have a significant impact on alternative splicing of exon 10.
While this paper was in preparation, two studies were published in which tau exon 10 alternative splicing was examined more extensively using exon-trapping assays (9, 23). The study by Grover and colleagues lends further support for the stem-loop model. On the other hand, D'Souza and colleagues suggested that the stem-loop structure was not supported by a mutation at +12 that should restore base-pairing in the stem structure (9). However, results from compensatory-mutation analyses have to be carefully interpreted. First, A-U base pairing (in the +12 compensatory mutant) is expected to be weaker than G-C base-pairing (in the wild-type tau). Second, multiple RNA-RNA and RNA-protein interactions are known to influence splice site selection and splicing efficiency. The stem-loop structure has to be viewed in the context of these multiple interactions. It is likely that events other than the U1 snRNA-pre-mRNA interaction also play important roles in regulating tau exon 10 splicing. In fact, we have found that certain non-U1 snRNP splicing regulators affect tau exon 10 splicing, and we are currently characterizing the differential recognition of the wild-type versus intronic mutant pre-mRNAs by these splicing regulators (Jiang and Wu, unpublished). The single-nucleotide changes around the 5′ splice site may have multiple effects on the RNA-RNA interactions (intramolecular and intermolecular) as well as on RNA-protein interactions. These effects may not be necessarily in the same direction. For example, “compensatory mutations” for the DDPAC+14 and AusI+16 mutants (at positions +1 and −2, respectively) that “restore” the base-pairing interactions in the stem will also affect U1 snRNP binding at the same time. Therefore, the compensatory mutation strategy that we and other groups used may not be optimal for testing the stem-loop model in the presence of the spliceosome.
The role of U1 snRNP in mammalian pre-mRNA splicing has been well established (for reviews, see references 2, 36, 46, and 52). It has been demonstrated that differential binding of U1 snRNP could affect 5′ splice site recognition in both yeast and mammalian cells (10, 20–22, 39, 49, 57, 58, 66). It is worth noting that these previous studies have all focused on either the upstream exonic region or the intronic region at the U1 snRNP binding site less than 10 nucleotides from the splice junction. Sequences further downstream in the intron have been found to be less conserved (47, 56). Our results demonstrate that a nonconserved intronic region outside the U1 snRNP binding site can also influence U1 snRNP binding via formation of a secondary structure(s) that masks the U1 snRNP binding site. The formation of secondary structures that potentially sequester 5′ splice sites has been proposed as a mechanism to regulate alternative 5′ splice site selection (12, 13, 59, 60). However, there has been little direct biochemical evidence demonstrating differential U1 snRNP binding in the pre-mRNA with proposed secondary structures (4, 58). In yeast cells, a systematic analysis to examine the effects of secondary structures on U1 snRNP binding using artificial hairpins to sequester the 5′ splice site of the yeast RP51A intron has been carried out (22). Pre-mRNAs containing hairpin structures with longer than 9 consecutive base pairs began to show a reduction in splicing in vivo, whereas structures with up to 6 consecutive base pairs had little effect on splicing efficiency (22). Our study demonstrates that even a single-nucleotide change at position +14 or +16, which potentially disrupts one base-pairing in a 6-bp stem involving the 5′ splice site, leads to a significant increase in splicing of tau exon 10 both in vitro and in vivo. In addition, these mutations are located in a nonconserved region of the intron. This is significant and prompts revised strategies for identifying potentially important genes for human diseases, since many have focused only on evolutionarily conserved regions.
It should be pointed out that this secondary-structure model is not inconsistent with the potential involvement of factors other than U1 snRNP. It is possible that the intronic mutations also disrupt the interaction of certain splicing-repressing factors (either protein or RNA). The potential involvement of splicing factors other than U1 snRNP in differentiating wild-type and intronic mutant tau pre-mRNAs was suggested by several observations. In human brain tissues and in cells transfected with tau minigenes, the increase in exon 10 splicing in both AusI+16 and DDPAC+14 mutant tau pre-mRNAs, compared with wild-type tau, appeared more dramatic than the difference observed in the RNase H cleavage assay in the absence of nuclear extract. In this study (Fig. 2) and in a previous study (30), the ratio of Tau4R to Tau3R was increased from 1 in wild-type tau to 3 or higher in AusI+16 tau and 4 or higher in DDPAC+14 tau. SR proteins have been shown to bind directly to 5′ splice sites (69) and recruit and/or stabilize the binding of the U1 snRNP to pre-mRNA (11, 34). In our previous studies, we have demonstrated interaction between SR proteins and U1 70K, a U1 snRNP protein (67), and distinct functional activities of SR proteins in alternative 5′ splice site selection (68) as well as in alternative exon inclusion (33). Recently, a yeast U1 snRNP protein, Nam8p, was shown to interact with nonconserved intronic sequences and affect 5′ splice site selection (51). U5 PRP8 yeast and human homologs have been shown to interact with 5′ and 3′ splice sites (53, 54, 64), although their role in regulating alternative splicing is not yet clear. It is possible that some of these proteins, or other novel or known alternative splicing regulators, also play a role in tau alternative splicing.
Abnormal pre-mRNA splicing has been implicated in the pathogenesis of a large number of human diseases, including neurodegenerative disorders such as amyotrophic lateral sclerosis (42). Almost all splicing mutations reported in human diseases either weaken recognition by spliceosomal snRNPs or cause activation of cryptic splice sites, leading to exon skipping, intron retention, or usage of cryptic splice sites (28, 48). The FTDP-17-associated intronic mutations analyzed in this study represent the first case in which single-nucleotide mutations cause increased rather than decreased splicing of an alternatively spliced exon, thereby altering the balance between different isoforms of normal gene products and leading to neurodegeneration. Our study provides strong evidence that enhanced U1 snRNP binding to a normal alternative splice site, as a result of single-nucleotide mutations in the nonconserved intronic region outside of the U1 snRNP binding site, can be a pathogenic mechanism. Such aberrant splicing can cause alteration in the delicate balance of different alternative splicing products. Considering the size of introns compared with exons and the complexity of alternative splicing regulation in mammalian genes, it is likely that simple alterations in the balance of different isoforms of critical genes as a result of aberrant splicing could be a more important mechanism for pathogenesis of human diseases than previously appreciated.
ACKNOWLEDGMENTS
We thank A. Kramer, W.-Y. Tarn, and M. McNally for generous gifts of purified U1 snRNP preparation and 2′-O-methyl-oligonucleotides and Y. Rao, A. Strauss, and members of the Wu laboratory for critical reading of the manuscript.
This work is supported by grants from the National Institute of Health (RO1 GM53945/AG17518 to J.Y.W. and P50 AG05681 to A.M.G.), by the Leukemia Society of America Scholarship to J.Y.W., by a postdoctoral fellowship from Natural Sciences and Engineering Research Council of Canada to J.C., by NSADA to J.M.K., and by an NIH career development award to A.M.G. (AG000634).
REFERENCES
- 1.Andreadis A, Brown W M, Kosik K S. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 2.Black D L. Finding splice sites within a wilderness of RNA. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 3.Black D L, Chabot B, Steitz J A. U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell. 1985;42:737–750. doi: 10.1016/0092-8674(85)90270-3. [DOI] [PubMed] [Google Scholar]
- 4.Blanchette M, Chabot B. A highly stable duplex structure sequesters the 5′ splice site region of hnRNP A1 alternative exon 7B. RNA. 1997;3:405–419. [PMC free article] [PubMed] [Google Scholar]
- 5.Chabot B. Synthesis and purification of RNA substrates. In: Hames D, Higgins S, editors. RNA processing. I. Oxford, United Kingdom: Oxford University Press; 1994. pp. 1–29. [Google Scholar]
- 6.Clark L N, Poorkaj P, Wszolek Z, Geschwind D H, Nasreddine Z S, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee V M, Reed L, Trojanowski J Q, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen K C. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Côté J, Chabot B. Natural base-pairing interactions between 5′ splice site and branch site sequences affect mammalian 5′ splice site selection. RNA. 1997;3:1248–1261. [PMC free article] [PubMed] [Google Scholar]
- 8.Dignam J D, Lebovitz R M, Roeder R G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee V M, Bird T D, Schellenberg G D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng F J, Warner J R. Structural basis for the regulation of splicing of a yeast messenger RNA. Cell. 1991;65:797–804. doi: 10.1016/0092-8674(91)90387-e. [DOI] [PubMed] [Google Scholar]
- 11.Eperon I C, Ireland D C, Smith R A, Mayeda A, Krainer A R. Pathways for selection of 5′ splice sites by U1 snRNPs and SF2/ASF. EMBO J. 1993;12:3607–3617. doi: 10.1002/j.1460-2075.1993.tb06034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eperon L P, Estibeiro J P, Eperon I C. The role of nucleotide sequences in splice site selection in eukaryotic pre-messenger RNA. Nature. 1986;324:280–282. doi: 10.1038/324280a0. [DOI] [PubMed] [Google Scholar]
- 13.Eperon L P, Graham I R, Griffiths A D, Eperon I C. Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell. 1988;54:393–401. doi: 10.1016/0092-8674(88)90202-4. [DOI] [PubMed] [Google Scholar]
- 14.Foster N L, Wilhelmsen K, Sima A A, Jones M Z, D'Amato C J, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol. 1997;41:706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 15.Goedert M. Neurofibrillary pathology of Alzheimer's disease and other tauopathies. Prog Brain Res. 1998;117:287–306. doi: 10.1016/s0079-6123(08)64022-4. [DOI] [PubMed] [Google Scholar]
- 16.Goedert M, Crowther R A, Spillantini M G. Tau mutations cause frontotemporal dementias. Neuron. 1998;21:955–958. doi: 10.1016/s0896-6273(00)80615-7. [DOI] [PubMed] [Google Scholar]
- 17.Goedert M, Spillantini M G, Crowther R A. Tau proteins and neurofibrillary degeneration. Brain Pathol. 1991;1:279–286. doi: 10.1111/j.1750-3639.1991.tb00671.x. [DOI] [PubMed] [Google Scholar]
- 18.Goedert M, Spillantini M G, Davies S W. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol. 1998;8:619–632. doi: 10.1016/s0959-4388(98)80090-1. [DOI] [PubMed] [Google Scholar]
- 19.Goedert M, Spillantini M G, Jakes R, Rutherford D, Crowther R A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 20.Goguel V, Liao X L, Rymond B C, Rosbash M. U1 snRNP can influence 3′-splice site selection as well as 5′-splice site selection. Genes Dev. 1991;5:1430–1438. doi: 10.1101/gad.5.8.1430. [DOI] [PubMed] [Google Scholar]
- 21.Goguel V, Rosbash M. Splice site choice and splicing efficiency are positively influenced by pre-mRNA intramolecular base pairing in yeast. Cell. 1993;72:893–901. doi: 10.1016/0092-8674(93)90578-e. [DOI] [PubMed] [Google Scholar]
- 22.Goguel V, Wang Y, Rosbash M. Short artificial hairpins sequester splicing signals and inhibit yeast pre-mRNA splicing. Mol Cell Biol. 1993;13:6841–6848. doi: 10.1128/mcb.13.11.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 24.Hasegawa M, Smith M J, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 25.Hasegawa M, Smith M J, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- 26.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 27.Hirokawa N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 28.Hodges D, Bernstein S I. Genetic and biochemical analysis of alternative RNA splicing. Adv Genet. 1994;31:207–281. doi: 10.1016/s0065-2660(08)60399-5. [DOI] [PubMed] [Google Scholar]
- 29.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller B I, Geschwind D H, Bird T D, McKeel D, Goate A, Morris J C, Wilhelmsen K C, Schellenberg G D, Trojanowski J Q, Lee V M. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 30.Hutton M, Lendon C L, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen R C, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon J M, Nowotny P, Heutink P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 31.Iijima M, Tabira T, Poorkaj P, Schellenberg G D, Trojanowski J Q, Lee V M, Schmidt M L, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 32.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee M K, Trojanowski J Q, Lee V M. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Z, Zhang W, Rao Y, Wu J Y. Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc Natl Acad Sci USA. 1998;95:9155–9160. doi: 10.1073/pnas.95.16.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohtz J D, Jamison S F, Will C L, Zuo P, Luhrmann R, Garcia-Blanco M A, Manley J L. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 35.Kosik K S. Tau protein and neurodegeneration. Mol Neurobiol. 1990;4:171–179. doi: 10.1007/BF02780339. [DOI] [PubMed] [Google Scholar]
- 36.Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 37.Kramer A, Frick M, Keller W. Separation of multiple components of HeLa cell nuclear extracts required for pre-messenger RNA splicing. J Biol Chem. 1987;262:17630–17640. [PubMed] [Google Scholar]
- 38.Kramer A, Keller W. Preparation and fractionation of mammalian extracts active in pre-mRNA splicing. Methods Enzymol. 1990;181:3–19. doi: 10.1016/0076-6879(90)81107-6. [DOI] [PubMed] [Google Scholar]
- 39.Kuo H C, Nasim F H, Grabowski P J. Control of alternative splicing by the differential binding of U1 small nuclear ribonucleoprotein particle. Science. 1991;251:1045–1050. doi: 10.1126/science.1825520. [DOI] [PubMed] [Google Scholar]
- 40.Lee G, Neve R L, Kosik K S. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 41.Lee V M, Trojanowski J Q. The disordered neuronal cytoskeleton in Alzheimer's disease. Curr Opin Neurobiol. 1992;2:653–656. doi: 10.1016/0959-4388(92)90034-i. [DOI] [PubMed] [Google Scholar]
- 42.Lin C L, Bristol L A, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein J D. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/s0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- 43.Mandelkow E, Mandelkow E M. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 44.Mandelkow E M, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16:355–363. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- 45.Mayeda A, Krainer A R. Mammalian in vitro splicing assays. 1997. Methods Mol. Biol. [DOI] [PubMed] [Google Scholar]
- 46.Moore M J, Query C C, Sharp P A. Splicing of precursors to messenger RNAs by the spliceosome. In: Gesteland R, Atkins J, editors. RNA world. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1993. pp. 1–30. [Google Scholar]
- 47.Mount S M. A catalogue of splice junction sequences. Nucleic Acids Res. 1982;10:459–472. doi: 10.1093/nar/10.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene. 1994;141:171–177. doi: 10.1016/0378-1119(94)90567-3. [DOI] [PubMed] [Google Scholar]
- 49.Nandabalan K, Price L, Roeder G S. Mutations in U1 snRNA bypass the requirement for a cell type-specific RNA splicing factor. Cell. 1993;73:407–415. doi: 10.1016/0092-8674(93)90239-m. [DOI] [PubMed] [Google Scholar]
- 50.Poorkaj P, Bird T D, Wijsman E, Nemens E, Garruto R M, Anderson L, Andreadis A, Wiederholt W C, Raskind M, Schellenberg G D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. . (Erratum, 44:428.) [DOI] [PubMed] [Google Scholar]
- 51.Puig O, Gottschalk A, Fabrizio P, Seraphin B. Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes Dev. 1999;13:581–592. doi: 10.1101/gad.13.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reed R. Initial splice-site recognition and pairing during pre-mRNA splicing. Curr Opin Genet Dev. 1996;6:215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 53.Reyes J L, Gustafson E H, Luo H R, Moore M J, Konarska M M. The C-terminal region of hPrp8 interacts with the conserved GU dinucleotide at the 5′ splice site. RNA. 1999;5:206–220. doi: 10.1017/s1355838299981785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reyes J L, Kois P, Konforti B B, Konarska M M. The canonical GU dinucleotide at the 5′ splice site is recognized by p220 of the U5 snRNP within the spliceosome. RNA. 1996;2:213–225. [PMC free article] [PubMed] [Google Scholar]
- 55.Seiwert S D, Steitz J A. Uncoupling two functions of the U1 small nuclear ribonucleoprotein particle during in vitro splicing. Mol Cell Biol. 1993;13:3134–3145. doi: 10.1128/mcb.13.6.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Senapathy P, Shapiro M B, Harris N L. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 57.Sirand-Pugnet P, Durosay P, Brody E, Marie J. An intronic (A/U)GGG repeat enhances the splicing of an alternative intron of the chicken beta-tropomyosin pre-mRNA. Nucleic Acids Res. 1995;23:3501–3507. doi: 10.1093/nar/23.17.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sirand-Pugnet P, Durosay P, Clouet O, Brody E, Marie J. beta-Tropomyosin pre-mRNA folding around a muscle-specific exon interferes with several steps of spliceosome assembly. J Mol Biol. 1995;251:591–602. doi: 10.1006/jmbi.1995.0458. [DOI] [PubMed] [Google Scholar]
- 59.Solnick D. Alternative splicing caused by RNA secondary structure. Cell. 1985;43:667–676. doi: 10.1016/0092-8674(85)90239-9. [DOI] [PubMed] [Google Scholar]
- 60.Solnick D, Lee S I. Amount of RNA secondary structure required to induce an alternative splice. Mol Cell Biol. 1987;7:3194–3198. doi: 10.1128/mcb.7.9.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spillantini M G, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 62.Spillantini M G, Goedert M, Crowther R A, Murrell J R, Farlow M R, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spillantini M G, Murrell J R, Goedert M, Farlow M R, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teigelkamp S, Newman A J, Beggs J D. Extensive interactions of PRP8 protein with the 5′ and 3′ splice sites during splicing suggest a role in stabilization of exon alignment by U5 snRNA. EMBO J. 1995;14:2602–2612. doi: 10.1002/j.1460-2075.1995.tb07258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varani L, Hasegawa M, Spillantini M G, Smith M J, Murrell J R, Ghetti B, Klug A, Goedert M, Varani G. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci USA. 1999;96:8229–8234. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vilardell J, Warner J R. Regulation of splicing at an intermediate step in the formation of the spliceosome. Genes Dev. 1994;8:211–220. doi: 10.1101/gad.8.2.211. [DOI] [PubMed] [Google Scholar]
- 67.Wu J Y, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 68.Zhang W J, Wu J Y. Functional properties of p54, a novel SR protein active in constitutive and alternative splicing. Mol Cell Biol. 1996;16:5400–5408. doi: 10.1128/mcb.16.10.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zuo P, Manley J L. The human splicing factor ASF/SF2 can specifically recognize pre-mRNA 5′ splice sites. Proc Natl Acad Sci USA. 1994;91:3363–3367. doi: 10.1073/pnas.91.8.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]