

Abstract
Interleukin-17C (IL-17C) is an understudied member of the IL-17 family of cytokines. Its synthesis is induced by both cytokines and pathogenic stimuli in a variety of cell types, most often expressed at mucosal and barrier surfaces. IL-17C expression is dysregulated in a variety of autoinflammatory and autoimmune diseases including inflammatory bowel disease, psoriasis, and atopic dermatitis, yet it is protective against bacterial infections of the gut, skin, and lungs. In this review we highlight studies on IL-17C regulation and its function at human mucosal surfaces. Understanding the relationship between IL-17C and autoinflammatory and autoimmune diseases of the mucosa and defining the beneficial and pathogenic functions of the cytokine in inflammatory responses are the first steps in determining the potential for IL-17C as a therapeutic target.
Keywords: IL-17C, barrier surface, inflammatory bowel disease, H. pylori, psoriasis, chronic obstructive pulmonary disease
1. BACKGROUND
The interleukin-17 (IL-17) family of cytokines consists of 6 members, IL-17A through IL-17F. IL-17A, the founding member, is expressed by T helper 17 (Th17) cells, T cytotoxic 17 (Tc17) cells, type 3 innate lymphoid cells, neutrophils, and NK cells [1]. IL-17F, despite being the alphabetically last member of the family, is derived from a lot of the same cellular sources as IL-17A, shares the closest amino acid sequence homology at 50% [2]. The other IL-17 family members are produced by intestinal epithelial cells and chondrocytes (IL-17B), epithelial cells (IL-17C), skeletal muscle, brain, heart, and lung tissue (IL-17D), and mononuclear leukocytes (IL-17E, alias IL-25) [1, 3]. Members of this cytokine family are secreted as homodimers or heterodimers in the case of IL-17A/IL-17F, and signal through heterodimeric receptors [1]. A shared receptor polypeptide chain, referred to as IL-17RA, forms a heterodimer with other receptor subunits to create the functional receptors for IL-17A, IL-17A/F, and IL-17F (IL-17RC), IL-17C (IL-17RE), and IL-17E (IL-17RB) [1].
IL-17B and IL-17C were discovered in 2000 when Li et al. used the Basic Local Alignment Search Tool (BLAST) to identify proteins with sequence homology to IL-17A [4]. Yet, serious research on IL-17C did not begin until a decade later, when two groups, Song et al. and Chang et al. determined the functional receptor for IL-17C was the heterodimer IL-17RA and IL-17RE [5, 6]. While the shared IL-17RA is reported to be expressed on a wide variety of cell types including epithelial cells, endothelial cells, fibroblasts, and macrophage [1], the expression of IL-17RE was originally limited to epithelial cells in the gut and Th17 cells [5–7]. Since this initial discovery IL-17RE expression has been reported at additional mucosal sites including the skin, lungs, and trachea [7]. Thus, IL-17C has been implicated in a number of autoimmune disorders and bacterial infections associated with mucosal sites throughout the body. Recently, Nies and Panzer reviewed IL-17C with an emphasis on mouse studies; this review will expand upon their efforts, focusing on IL-17C research in human mucosal tissue [8].
2. REGULATION OF IL-17C EXPRESSION
While the broadly studied cytokines IL-17A and IL-17F are expressed by a wide variety of immune cells, leukocytes are not a major source of IL-17C [7]. Rather, IL-17C is predominantly expressed by epithelial cells at mucosal surfaces of the gastrointestinal and respiratory tract and barrier surface of the skin [6, 7, 9–11]. Studies in the gastrointestinal tract demonstrated that IL-17C is produced by cells expressing Chromogranin A (enteroendocrine cells) and mucin (goblet cells) [9, 12], and studies of the skin found IL-17C expression in keratinocytes, mononuclear cells, and endothelial cells [13]. Aside from these reports, little has been done to further identify specific cells which produce this cytokine. In terms of regulation, IL-17C is induced by a number of bacterial insults and cytokine stimuli, which will be covered in more detail in the following sections.
2.1. TLR Stimulus
At mucosal surfaces immune surveillance is mediated by a large family of Pattern Recognition Receptors (PRRs) that recognize Pathogen-Associated Molecular Patterns (PAMPs), such as flagellin, lipopolysaccharides, foreign DNA and RNA molecules, or endogenous Damage-Associated Molecular Patterns (DAMPs), which are “self” molecules that under conditions of cellular stress or death, initiate inflammatory responses via PRRs [14]. A subset of PRRs known as toll-like receptors (TLRs) detect and initiate innate inflammatory responses triggered by common microbial motifs. TLRs found on the surface of epithelial cells (TLRs 1, 2, 4, 5, and 6) detect extracellular pathogens, while TLRs found in the lumen of endosomes (TLR 3, 7, 8, and 9) detect intracellular and viral pathogens [15].
One of the first investigations into TLR regulation of IL-17C studied the human colonic adenocarcinoma cell line, HCT-15, stimulated with peptidoglycan (PGN; TLR2 agonist), Poly I:C (TLR3 agonist), flagellin (TLR5 agonist), or CpG (TLR9 agonist). IL-17C protein secretion measured by ELISA showed that of the four TLRs tested, TLR2 and TLR5 stimulation induced 1.5 ng/mL of IL-17C, whereas TLR3 and TLR9 stimulation did not induce IL-17C expression above background (Table 1; Figure 1a) [7]. TLR5 stimulation of IL-17C expression was also reported in three other human colonic cell lines: NCM460, DLD-1, and HT-29 (Table 1) [10]. In that TLR2 and TLR5 are triggered by peptidoglycan and flagellin, respectively, both components of bacteria, these findings support a role for IL-17C in defense against bacteria in the gut. However, due to the HCT-15 cell line expressing only TLR2 and TLR5, it is likely that the observed differences in the ability of select TLRs to regulate IL-17C expression may reflect more on TLR expression levels in this cell line than on the inherent signal pathways that regulate IL-17C directly. Thus, the contribution of TLR regulation of IL-17C remains largely unresolved.
Table 1 |.
Molecules that regulate the expression of IL-17C in mucosal tissue
| Stimulant | Signaling Pathways | Cell Type | Reference | |
|---|---|---|---|---|
| Bacterial / TLR | E. coli | N/A* | Colonic epithelial cells, tracheal epithelial cells, keratinocytes | [7] |
| TLR2 (PGN) | MyD88 | HCT-15 | [7] | |
| TLR3 (Poly I:C) | TRIF, NF-κB (p65) | NHBE cells | [16] | |
| TLR5 (Flagellin) | MyD88 | HCT-15 | [7] | |
| MyD88, TRIF | NCM460 | [10] | ||
| N/A | DLD-1, HT-29 | [10] | ||
|
| ||||
| Cytokine | TNF-α | p38 MAPK, not STAT3 | Normal human keratinocytes | [13] |
| NF-κB p65 and p50 | Keratinocytes | [11] | ||
| N/A | HCT-15 | [7] | ||
| TNF-α and IL-17A | NF-κB, p38, ERK1/2, Akt, MCPIP | HT-29 | [9] | |
| N/A | Keratinocytes | [19] | ||
| IL-36γ | N/A | Keratinocytes | [37] | |
| IL-1β | NF-κB (p65) | NHBE | [27] | |
| N/A | HCT-15 | [7] | ||
| IL-1β and IL-13 Negative Regulation | JAK1/2 and STAT6 reduce NF-κB binding to IL-17C promotor | NHBE and HBE1 cells | [27] | |
N/A – signaling pathways not investigated in given study
Figure 1: Regulation of expression of IL-17C.
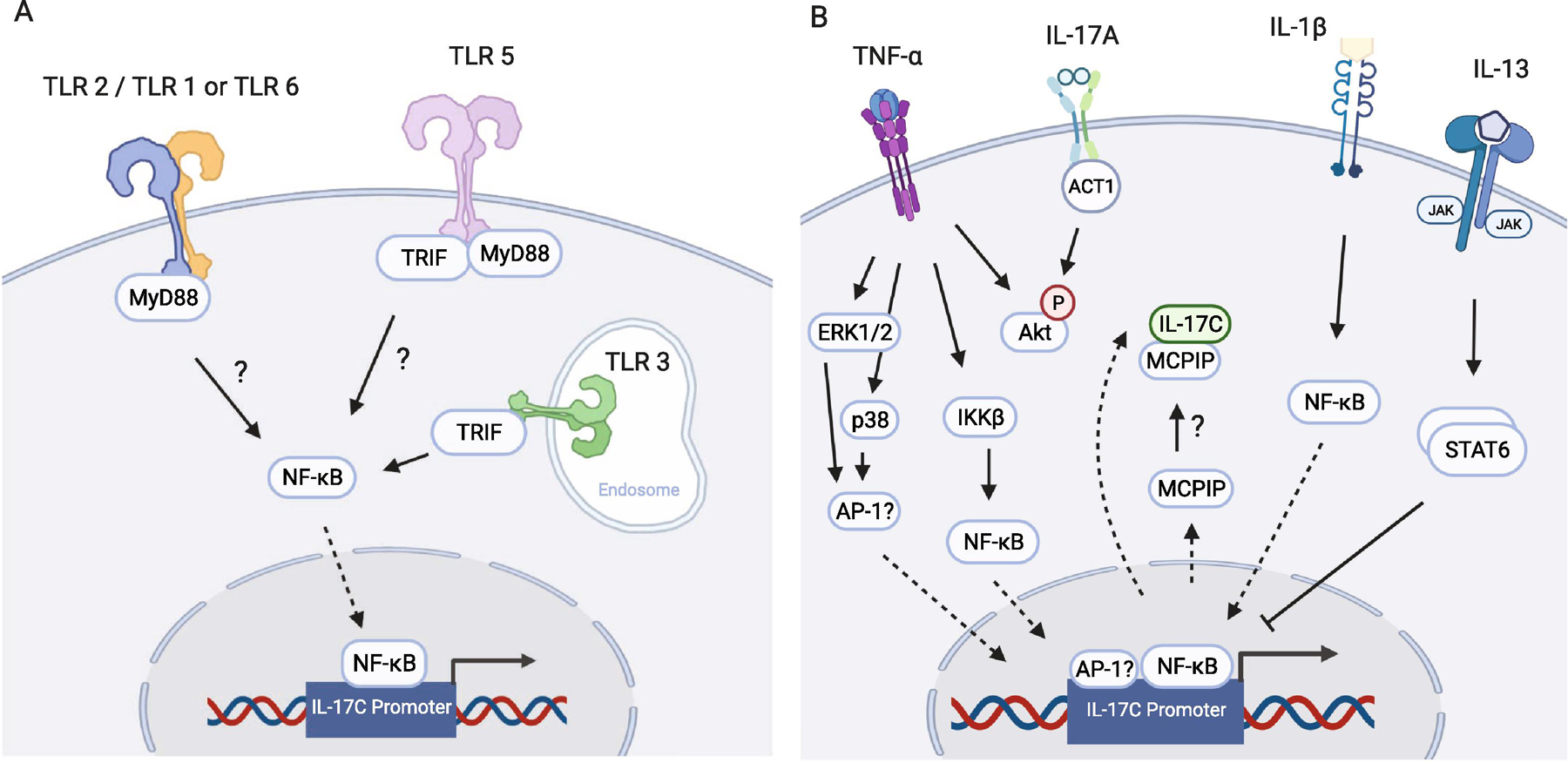
(A) TLR 2, TLR 3, and TLR 5 upregulate the expression of IL-17C at mucosal surfaces [see Table 2 for references]. (B) Expression of IL-17C downstream of TNF-α is mediated by ERK1/2, p38, and NF-κB. In the presence of IL-17A, phosphorylation levels of Akt may play a role in synergy between TNF-α and IL-17A stimulation of the expression of IL-17C. IL-17A is also known to increase the expression of MCPIP, which may sequester IL-17C in the cell, preventing release. IL-1β increases IL-17C expression through NF-κB. Co-treatment with IL-13 reduces IL-1β-mediated IL-17C expression via STAT6. Solid arrows represent activation pathways, whereas dashed arrows represent translocation through the nuclear membrane. Images created using BioRender.com
Focusing specifically on the contribution of IL-17C to mucosal immune protection, poly I:C stimulation of TLR3 in normal human bronchial epithelial (NHBE) cells in both submerged and air-liquid interface (ALI) culture conditions increased both IL-17C mRNA and protein expression (Table 1; Figure 1a) [16]. As TLR3 is classically considered a PRR for viral recognition, this study suggests a new role of IL-17C as important in response to viral pathogens at mucosal surfaces in addition to bacterial pathogens. To advance this field, a full characterization of all TLRs in primary culture epithelial spheroids, in contrast to transformed cell lines, would better inform our understanding of the spectrum of pathogenic insults that stimulate IL-17C expression.
2.2. Cytokine Stimulus
In addition to bacterial and viral-induced expression of IL-17C, cytokines produced by immune and non-immune cells at mucosal surfaces also regulate IL-17C. One of the most well studied cytokine inducers of IL-17C is tumor necrosis factor-alpha (TNF-α). This cytokine is multifunctional, regulating cellular survival and apoptosis and promoting the expression of other cytokines and chemokines [17, 18]. Normal human keratinocytes stimulated with TNF-α expressed IL-17C in both a MAPK- and NF-κB-dependent mechanism [11, 13, 19]. TNF-α stimulation of two colonic cell lines, HCT-15 and HT-29, also enhances the expression of IL-17C, suggesting that the relationship between TNF-α and IL-17C may extend into additional human tissues (Table 1; Figure 1B) [7, 9].
Functional cooperation and synergy between TNF-α and IL-17A are well established in the literature [20, 21]. Thus, it is not surprising that IL-17A has been shown to synergistically enhance TNF-α-mediated production of IL-17C. Stimulating HT-29 cells with IL-17A and TNF-α increases the expression of IL-17C mRNA and protein 14-fold, as contrasted to 2-fold for IL-17A and 6-fold for TNF-α alone [9]. Induction of IL-17C expression by TNF-α and IL-17A was also reported in keratinocytes (Table 1) [19, 22]. Using a combination of siRNA silencing and pharmacological inhibition, TNF-α- and IL-17A-mediated regulation of IL-17C was shown to be dependent on IKK2 and NF-κB in the NF-κB activation pathway, ERK1/2 and p38 in the MAPK pathway, and kinase Akt (Table 1; Figure 1B) [9]. IL-17A upregulates the expression of MCPIP, a molecule known to sequester and prevent the release of IL-6 [23]. When MCPIP was silenced, IL-17C release increased, suggesting that MCPIP may also play an inhibitory role in IL-17C regulation and release (Figure 1B) [9]. The interaction between TNF-α and IL-17A in regulating IL-17C expression in multiple tissue types suggests a ubiquitous role for IL-17C in inflammatory conditions.
In addition to the induction of IL-17C by the T cell-derived cytokines TNF-α and IL-17A, the innate immune cytokine IL-1β also regulates IL-17C expression. IL-1β is synthesized by a variety of cell lineages in the form of a proprotein, and in the presence of inflammatory stimuli, is cleaved by caspase-1 in the inflammasome and released from the cell, where it acts to determine cell fate (proliferation, differentiation, or apoptosis) and promotes the expression of other inflammatory cytokines [24–26]. IL-1β stimulation of IL-17C production was demonstrated in colonic (colonic adenocarcinoma cell line HCT-15) and bronchial cells (NHBE and immortalized bronchial epithelial cell line HBE1) [7, 27]. Studies performed in NHBE cells determined that in both submerged and ALI culture conditions, IL-1β induced IL-17C expression in a dose-dependent manner and was dependent on the NF-κB subunit p65 [27].
When NHBE cells are co-stimulated with IL-1β and the Th2-derived cytokine IL-13, IL-17C expression is inhibited by IL-13 in a dose-dependent fashion [27]. This inhibition requires JAK1/2 and STAT6 activation downstream of the IL-13 receptor, which, in turn, reduces the binding of p65 to the promoter of IL-17C (Table 1) [27]. Since IL-1β promotes the differentiation of naïve CD4+ T cells into Th17 cells, and IL-13 reduces the expression of IL-1β and inhibits its activity, this interplay between innate-derived and Th2-derived cytokines underscores the importance of homeostatic balance in immune cell function and cytokine expression and brings to light an important role that IL-17C may play at the intersection of complex inflammatory immune networks.
3. FUNCTION OF IL-17C
Research into the function of IL-17C at human mucosal surfaces is underdeveloped. In recognition that IL-17C is an early response cytokine in the defense against bacterial pathogens, such as E. coli and H. pylori, IL-17C upregulates the expression of a variety of antimicrobial peptides including human beta-defensin 2 (hBD2) expressed by keratinocytes [7, 28], Lipocalin 2 (LCN2) expressed in the gastric mucosa [12], and granzyme B expressed in the colonic epithelium (Table 2) [10]. These proteins function to sequester ions from bacteria limiting bacterial growth and to kill pathogens via cell lysis. The ubiquitous expression of IL-17C throughout barrier surfaces during infection and its ability to induce the expression of protective molecules suggest a critical role for this cytokine in the body’s first line of defense against invading pathogens.
Table 2 |.
Gene expression upregulated by IL-17C stimulation of human mucosal tissues
| Gene | Stimulant | Read-out | Cell Type | Reference | |
|---|---|---|---|---|---|
| Antimicrobial Agent | Granzyme-B (GZMB) | Flagellin | IPA analysis | NCM460 | [10] |
| Human Beta Defensin 2 (hBD2) | TNF-α & IL-17C* | mRNA | keratinocytes | [28] | |
| IL-17C^ | keratinocytes | [7] | |||
| IL-17C | mRNA | NHBE cells | [16] | ||
| Lipocalin-2 (LCN2) | H. pylori | IPA analysis | gastric mucosa | [12] | |
| TNF-α & IL-17C | mRNA | keratinocytes | [28] | ||
| Peptidase Inhibitor 3 (PI3) | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| S100A7 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| S100A8 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| H. pylori | IPA analysis | gastric mucosa | [12] | ||
| S100A9 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| H. pylori | IPA analysis | gastric mucosa | [12] | ||
| S100A12 | IL-17C | mRNA | NHBE cells | [16] | |
|
| |||||
| Cytokines and Chemokines | CCL20 | TNF-α & IL-17C | mRNA | keratinocytes | [28] |
| CSF3 | IL-17C | mRNA | NHBE cells | [16] | |
| CXCL8 | H. pylori | IPA analysis | gastric mucosa | [12] | |
| G-CSF | IL-17C | protein | keratinocytes | [7] | |
| IFN-γ | H. pylori | IPA analysis | gastric mucosa | [12] | |
| IL-1 β | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| Flagellin | IPA analysis | NCM460 | [10] | ||
| IL-17C.Fc (IgG1) fusion protein | protein | THP-1 cells | [4] | ||
| IL-1F5 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-1F9 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-6 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-17C | mRNA | human dermal microvascular endothelial (hDME) cells | [28] | ||
| H. pylori | IPA analysis | gastric mucosa | [12] | ||
| IL-8 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-17C | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-19 | TNF-α & IL-17C | mRNA | keratinocytes | [28] | |
| IL-23A | H. pylori | IPA analysis | gastric mucosa | [12] | |
| TNF-α | IL-17C | mRNA | hDME cells | [28] | |
| TNF-α & IL-17C | mRNA | keratinocytes | [28] | ||
| IL-17C.Fc (IgG1) fusion protein | protein | THP-1 cells | [4] | ||
| H. pylori | IPA analysis | gastric mucosa | [12] | ||
|
| |||||
| Other | BCL2 | H. pylori | IPA analysis | gastric mucosa | [12] |
Annotations for Table 2– following page
TNF-α (2ng/mL); IL-17A (200ng/mL)
IL-17C = concentrations ranged from 2ng/mL to 200ng/mL
IL-17C also stimulates the release of cytokines (Table 2) that enhance inflammatory responses in the skin [7, 28], the gastrointestinal tract [10, 12], and the lungs [16]. Among known IL-17C-regulated cytokines are IL-1β, TNF-α, and IL-6. As mentioned in the Cytokine Stimulus Section 2.2 of this review, both IL-1β and TNF-α have been well established as inducers of IL-17C expression. Thus, the discovery that IL-17C enhances expression of IL-1β and TNF-α signifies a complicated communication and regulatory network among these cytokines during an inflammatory event. Interestingly, IL-17C enhances the expression of IL-6, a crucial cytokine for the differentiation of naïve CD4+ T cells to Th17 cells [29]. In addition, the presence of IL-1β during Th17 differentiation yields a more pathogenic cell phenotype, thus indicating that IL-17C may indirectly contribute to the expansion of these inflammatory cells, which are often connected to autoinflammatory disease [30, 31]. The activation of inflammatory Th17 cells induces the expression of IL-17A, which will further increase the expression of IL-17C, thus establishing an interesting relationship between IL-17C and Th17 cells.
Although IL-17C is not always a potent inducer of antimicrobial agents, cytokines, or chemokines by itself, cooperative signaling with TNF-α has been shown. In keratinocytes, mRNA expression of 15 different molecules involved in immunity was shown to be significantly upregulated in the presence of IL-17C and TNF-α compared to TNF-α stimulation alone [28]. Considering TNF-α has been implicated in a variety of inflammatory and autoimmune diseases [32], the ability for IL-17C to enhance TNF-α-mediated gene expression suggests that, in some settings, IL-17C exacerbates disease. A comprehensive list of additional cytokines and chemokines regulated by IL-17C can be found in Table 2.
Very little research is available regarding the signaling cascades downstream of IL-17C ligation of its IL-17RE/IL-17RA heterodimeric receptor expressed on human epithelial cells and Th17 cells [5–7]. When HT-29 cells were stimulated with IL-17C, enhanced phosphorylation of p65/RelA, p38, ERK, and JNK was observed, suggesting that like IL-17A, IL-17C activates the NF-κB and MAPK pathways (Figure 2) [6]. Additional literature in mice has reported that IL-17C requires the adaptor protein Act1 to initiate downstream signaling, which will likely be observed in human cells as well (Figure 2) [5]. Altogether, IL-17C has been identified to regulate both innate and adaptive immunity, although the mechanism by which this occurs is incompletely understood.
Figure 2: Signaling cascade downstream of IL-17C.
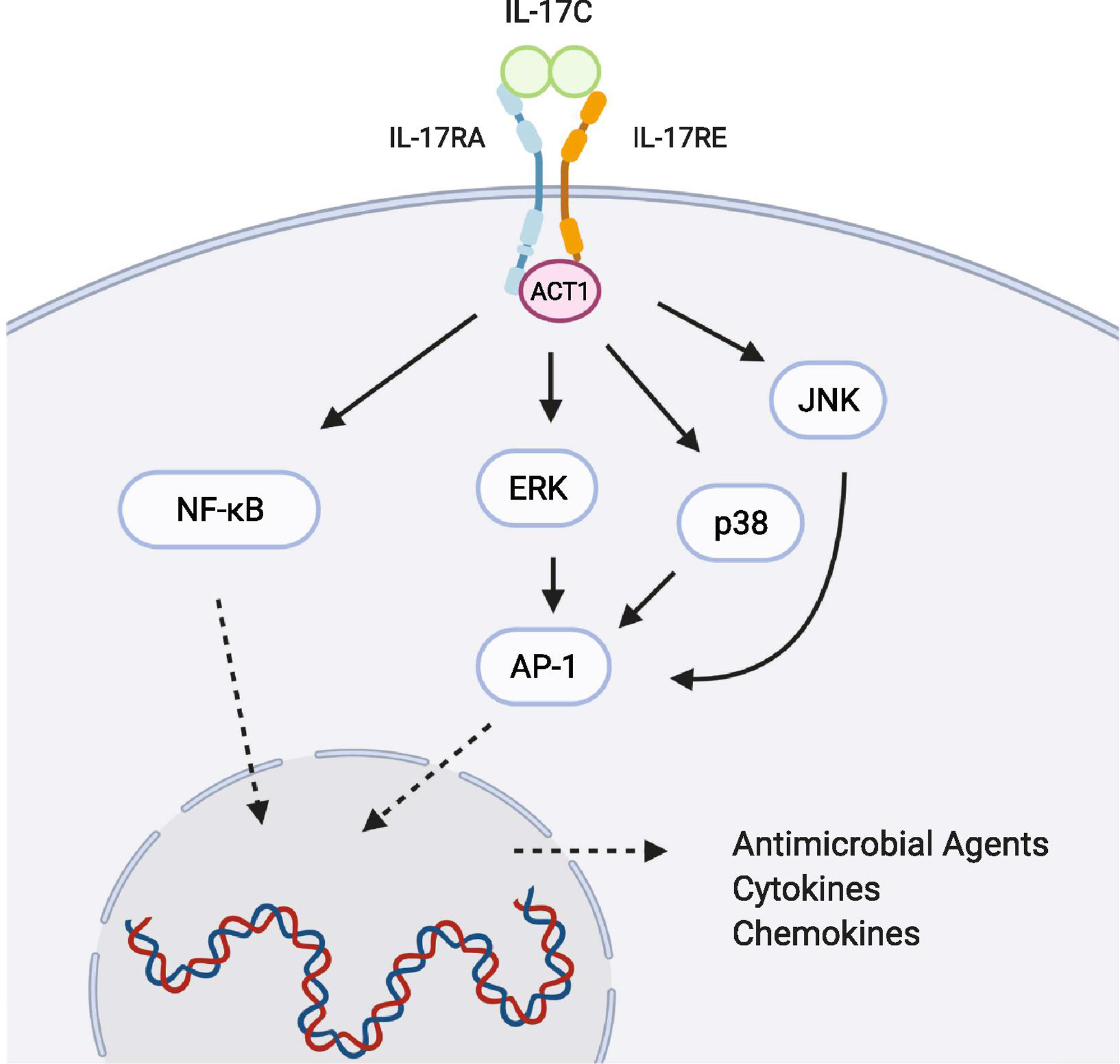
IL-17C activates the transcription factors NF-κB (RelA and p50) and AP-1. Phosphorylation of ERK, p38, and JNK has been demonstrated in colon epithelial cells. List of antimicrobial agents, cytokines, and chemokines are highlighted in Table 2. Solid arrows represent activation pathways, whereas dashed arrows represent translocation through the nuclear membrane. Image created using BioRender.com
4. IL-17C AND PATHOGENESIS AT MUCOSAL SURFACES
4.1. The Gut - IBD
IL-17C plays an integral role in regulation of the colonic inflammation associated with Crohn’s Disease (CD) and Ulcerative Colitis (UC) - the two forms of human Inflammatory Bowel Disease (IBD). Analysis of IL-17C levels in the intestinal tissue of IBD patients revealed that IL-17C mRNA expression is upregulated 5-fold in UC, but not CD, and directly correlates with increased inflammatory activity [10, 33]. Elevated IL-17C serum levels have also been seen in IBD patients when compared to healthy controls [9]. Upon further analysis of IBD patients divided into CD and UC affected individuals, IL-17C serum levels were significantly increased in UC and not CD, matching the intestinal findings. Moreover, within the CD group, IL-17C mRNA expression was significantly increased in inflamed tissue relative to uninvolved tissue - highlighting the role of IL-17C in both forms of IBD [9]. Within inflamed colonic tissue of both UC and CD patients, elevated IL-17C mRNA expression correlated with increased mRNA expression of the cytokines IL-17A, TNF-α, CCL20, IL-23, and IL-22 [9]. In light of the known increased activity of IL-17A and TNF-α in IBD [9, 34, 35], their ability to induce IL-17C expression in vitro, and the increased levels of IL-17C in inflamed lesions suggest a potential critical role for IL-17C in IBD pathogenesis.
During the treatment of IBD, psoriasiform skin lesions are a known side effect of anti-TNF-α therapy [36]. Within these lesions, increased IL-36γ mRNA and protein expression has been shown to subsequently increase the expression of IL-17C [37]. Although IL-17C has been shown to enhance both TNF-α- and IL-22-mediated expression of inflammatory mediators [7, 28], the addition of IL-17C in IL-36γ stimulated keratinocytes inhibited the expression of the antimicrobial peptide hBD2 [37]. This finding not only suggests a novel inflammatory signaling axis within psoriatic skin lesions but underscores the complexity of IL-17C function within different contexts.
Another study demonstrated that IL-17C mRNA and protein expression is upregulated in human colorectal cancer (CRC) samples and correlates with increases in IL-17A and IL-23 [38]. Since patients with IBD are at significantly increased risk of CRC [39, 40], these results further implicate IL-17C in many aspects of intestinal immunity. Overall, these findings underscore a relationship between IL-17C expression and various aspects of disease progression and, thus, highlight the need for defining the precise role of IL-17C in IBD pathogenesis or repair.
A contribution of IL-17C to disease status in IBD patients is consistent with mechanistic studies in the dextran sodium sulfate (DSS)-induced colitis model in mice. When mice are challenged with DSS, IL-17C mRNA and protein expression is increased in colonic tissue [7], and DSS exposure in IL-17RE−/− mice induced more severe disease, that included greater weight loss, persistent inflammation, and increased recruitment of macrophages and neutrophils to the colon when compared to IL-17RE+/+ mice [7]. The protective function of IL-17C in DSS-induced colitis was replicated in a similar study where DSS-treated mice lacking IL-17C exhibited greater weight loss, colonic shortening, and mRNA expression of the inflammatory cytokines and chemokines IL-17A, IL-6, RANTES, and CCL20 than DSS-treated wild type mice [41]. Taken together, it is critical to recognize that in human studies, increased expression of IL-17C or any other immune mediator during disease progression may reflect a failed protective function. Further human studies must be performed to support this hypothesis.
4.2. The Stomach - H. pylori
As previously discussed, IL-17C expression is upregulated in response to bacterial infection via stimulation of TLRs [7, 10]. Consistent with those reports, IL-17C mRNA levels are significantly upregulated in gastric mucosal biopsies of H. pylori-infected patients when compared to uninfected controls [12]. Notably, expression of the remaining IL-17 family members (IL-17A, B, D, E, and F) did not differ significantly from healthy controls [12]. Immunohistochemical analysis revealed that IL-17C is primarily derived from epithelial cells of the infected gastric mucosa rather than fibroblasts or localized immune cells [12]. Network mapping generated with Ingenuity Pathway Analysis (QIAGEN) predicted that IL-17C activates the expression of 8 genes (CXCL8, IFN, TNF-α, IL-6, IL-23A, S100A9, LCN2, and itself) during H. pylori infection, all of which are immune mediators involved in the migration of mononuclear leukocytes, recruitment of lymphocytes, or antimicrobial responses [12]. Thus, it is likely that IL-17C is an active participant in the host response to an H. pylori infection.
4.3. The Skin - Psoriasis & Atopic Dermatitis
Although not a classic mucosal tissue, skin is an important barrier surface where IL-17C has been implicated in autoimmune and inflammatory diseases. IL-17C mRNA expression is upregulated about 6000-fold in punch biopsies taken from human psoriatic skin lesions [13]. Interestingly, IL-17C protein expression is approximately 4-fold higher in psoriatic skin lesions and 125-fold higher than IL-17A protein in these lesions, demonstrating that IL-17C is the most predominant IL-17 family member in human involved psoriatic skin [28]. IL-17C mRNA levels are also increased in atopic dermatitis (AD) skin lesions, comparable to mRNA levels typically observed in psoriatic skin and greater than that found in uninvolved skin from the same patient [42]. Moreover, immunohistochemical analysis of inflamed skin from both AD and psoriatic patients showed increased levels of IL-17C in both keratinocytes and the immune cells infiltrating the dermis compared to healthy controls [42]. In a study intradermal injection of IL-17C in wild type mice lead to subsequent leukocyte infiltration and thickening of the epidermis were reported [7]. Conversely, an IL-17C−/− mouse treated with imiquimod showed significantly less neutrophilic infiltrate and lower expression of inflammatory cytokines (IL-17A, IL-17F, IL-22, CSF-3, CXCL1, IL-1β, and TNF) than their wild type counterparts [7]. Another study demonstrated IL-17C neutralization in a murine model of psoriasis, in which IL-23 is injected intradermally, resulted in decreased skin inflammation and lower levels of serum inflammatory cytokines (IL-17A, IL-22, IL-1β, S100A9/8, and LCN2) [42]. Note that these same inflammatory mediators are also predicted to be induced by IL-17C in the gastric mucosa of patients with H. pylori [12]. Similarly, in a flaky tail mouse model of spontaneous AD, IL-17C neutralization led to a less severe clinical score, as evidenced by decreases in both swelling and acanthosis [42]. Using the same neutralizing anti-IL17C antibody, another group showed that inhibition of IL-17C ex vivo leads to a significant downregulation of the inflammatory molecules HBD2 and IL-36γ in psoriatic skin lesions and LCE3A and IL-36γ in AD skin biopsies [43]. Due to the extensive characterization of IL-17C in mouse models of psoriasis and AD, and the complementary findings of increased IL-17C in human psoriatic and AD patients, we propose that the physiological role that IL-17C plays in perpetuating inflammation in mice will also be relevant in human disease. These reports demonstrate that IL-17C contributes to the inflammatory processes that characterize psoriasis and AD. Given that IL-17C is induced during an immune response to bacteria at other mucosal surfaces, future research should explore the potential role of IL-17C in bacterial skin infections.
4.4. Respiratory Tract - pulmonary, oral, and nasal mucosa
The respiratory tract represents an additional mucosal site in which IL-17C expression increases during an immune response against invading pathogens. Chronic Obstructive Pulmonary Disease (COPD) is exacerbated by bacterial pathogens that often cause pulmonary inflammation and lung cancer. In human lung cancer cells infected with Nontypeable Haemophilus influenzae (NTHi), IL-17C contributed to tumor growth through the recruitment of neutrophils to the tumor microenvironment [44]. Similarly, human bronchial epithelial (HBE) cells treated with IL-17C increased expression of CXCL1, a neutrophil chemoattractant [45], and when co-infected with human rhinovirus (HRV) and either NTHi or Pseudomonas aeruginosa, IL-17C mRNA and protein expression was synergistically upregulated [45]. In a follow up study, this group reported that HBE cells treated with HRV release IL-17C from their basolateral surface in a dose-dependent manner [46]. Released IL-17C subsequently binds to its receptor on the basolateral surface of HBE cells in a paracrine/autocrine manner and stimulates the release of basolateral CXCL1. In summary, IL-17C exacerbates microbial-induced COPD through neutrophil recruitment.
Recurrent aphthous ulcers (RAU) are an ulcerative disease of the oral mucosa linked to abnormalities in the immune system, although the precise cause of the disease is unknown. IL-17C mRNA expression is upregulated about 600-fold in human oral keratinocytes (HOKs) isolated from RAU lesions [47], and upon IL-17C stimulation, these HOKs increase TNF-α mRNA production, suggesting that IL-17C cooperates with additional cytokines to propagate inflammation in RAU [47]. In contrast to RAU, periodontitis is triggered by bacterial infection of the oral mucosa. In gingival biopsies taken from patients with aggressive periodontitis, CpG methylation of the IL-17C genomic region was significantly reduced compared to healthy tissues [48], suggesting a corresponding increase in gene expression, further emphasizing the function of IL-17C in an oral bacterial infection.
The nasal passage represents an additional mucosal site for pathogen invasion. When human nasal epithelial cells are treated with staphylococcal enterotoxin B, IL-17C expression is upregulated [49]. Similarly, IL-17C mRNA and protein expression is upregulated during Pseudomonas aeruginosa (PAO1 strain) infection of nasal epithelial cells. A key immunoregulatory activity is revealed in this model in that IL-17C directly inhibits PAO1 siderophore activity, therefore suppressing the infection [50], highlighting a novel mechanism for the anti-bacterial function of IL-17C.
5. Future Directions for the Field
IL-17C appears to propagate inflammation in autoimmune, autoinflammatory, and infectious diseases (Table 3) by teaming with Th17-derived cytokines, such as IL-17A and TNF-α. The literature clearly demonstrates that IL-17C plays a substantial role in the regulation of inflammatory pathways at mucosal surfaces. While current literature focuses on the gut, skin, and respiratory tract, it is likely that IL-17C also functions in the urogenital tract. Recent reports have implicated IL-17A and other Th17 cytokines in the response to bacteria in the urogenital tract [51, 52], therefore, we predict that future studies will reveal an involvement for IL-17C. In a murine model of intravaginal Chlamydia trachomatis infection, a Th1 and Th17 immune response drives the clearance of pathogens while contributing to immunopathological tissue damage [53]. Similarly, IL-17A is required for effective control and clearance of both uropathogenic Escherichia coli and Streptococcus pyogenes infection within the murine genital tract [51, 52]. Research is now needed to confirm and elucidate the role of IL-17C in human urinary tract infections and urogenital inflammatory diseases.
Table 3 |.
IL-17C in human disease
| Mucosal Site | Disease | Main Findings | Reference |
|---|---|---|---|
| Gut | Inflammatory Bowel Disease (IBD) | Increased in UC; correlates with inflammatory activity | [9, 10] |
| Helicobacter pylori | Upregulated in gastric mucosa; activates immune response | [12] | |
| Anti-TNF-α induced psoriaform skin lesion in CD | Elevated in lesions; propagates inflammation | [32] | |
| Colorectal Cancer (CRC) | Elevated in human CRC | [34] | |
|
| |||
| Skin | Psoriasis | Upregulated in lesions | [13, 28] |
| Atopic Dermatitis (AD) | Elevated in keratinocytes and infiltrating immune cells in lesions | [38] | |
| Inhibition of IL-17C results in downregulation of inflammatory molecules | [39] | ||
|
| |||
| Respiratory Tract | Chronic Obstructive Pulmonary Disorder (COPD) | Increased in viral-bacterial coinfection of human bronchial epithelial (HBE) cells | [41] |
| Released by Human Rhinovirusinfected HBE cells and contributes to neutrophil recruitment | [42] | ||
| Recurrent Aphthous Ulcers (RAU) | Upregulated in human oral keratinocytes (HOKs) in lesions | [43] | |
| IL-17C stimulation upregulates TNF-α mRNA production in HOKs | [43] | ||
| Periodontitis | CpG methylation of IL-17C DNA is reduced in aggressive periodontitis | [44] | |
| Staphylococcal enterotoxin B infection of the nasal passage | Upregulated in response to infection | [45] | |
| Pseudomonas aeruginosa infection of nasal passage | Controls infection by inhibiting PAO1 siderophore activity | [46] | |
Currently, the neutralization of Th17 derived cytokines, especially IL-17A and TNF-α, is widely used in the treatment of inflammatory diseases. Specific examples include antibodies that target IL-17A (Secukinumab and Ixekizumab) in the treatment of psoriasis [54] and anti-TNF-α therapies (Infliximab, Adalimumab, and Certolizumab) that have been extensively used to treat Crohn’s Disease [55] and rheumatoid arthritis [56]. In ongoing clinical trials of anti-IL-17A therapy for Hidradenitis Suppurativa, an inflammatory skin condition, downregulation of IL-17C expression is a primary outcome, further highlighting the role of IL-17C in inflammatory processes [57, 58].
IL-17A and TNF-α synergize to upregulate the production of IL-17C, and IL-17C promotes inflammation in autoimmune diseases, therefore, anti-IL17C therapy presents a potential therapeutic option for inflammatory mucosal diseases. This hypothesis is supported by numerous model studies in mice showing that antagonists to IL-17C or to IL-17RE are effective in the treatment of inflammatory diseases such as IBD, psoriasis, AD, asthma, and COPD [59–63]. A recent review of IL-17C [8] cited an early clinical trial of the first anti-human IL-17C antibody being used in the treatment of AD. Since the publication of their review, this clinical trial was terminated in Phase 2 after a futility analysis revealed a low probability of achieving the study’s target decrease in eczema area and severity index [64]. Although this cumulative data hint at IL-17C being a therapeutic target, this failure in the clinic suggests otherwise. As is widely observed in the cytokine literature, is overexpression of an immune mediator the cause of the ongoing inflammatory response or the consequence of that response? We postulate that since IL-17C is expressed earlier in infection than other IL-17 cytokine family members [6, 7] and that IL-17C upregulates the expression of a variety of other inflammatory mediators and chemokines, future investigations into the possibility that blocking IL-17C activity might limit chronic inflammation during autoimmune and autoinflammatory disorders are warranted.
6. CONCLUDING REMARKS
IL-17C is primarily expressed by epithelial cells and functions through the heterodimeric IL-17RE/A receptor complex at various mucosal sites. Unlike the well-characterized family members IL-17A and IL-17F, the complex immunological functions and mechanisms of IL-17C are not fully known. In their recent review, Nies and Panzer highlighted the kinetics of IL-17C expression, showing that IL-17C is an early responder in multiple mucosal disease models followed by a subsequent activation of IL-17A and IL-17F [6–8]. Our review furthered their efforts, focusing on the complicated contribution of IL-17C in the human host response to cytokines and pathogenic stimuli at mucosal surfaces, as well as during autoimmune, autoinflammatory, and bacterial diseases. In light of evidence connecting IL-17C to inflammation, further research would need to be pursued to gauge the feasibility of IL-17C as a therapeutic target.
HIGHLIGHTS.
Regulation of IL-17C by TLR and cytokine stimulation
Overview of the biological role of IL-17C at mucosal sites
Relationship between IL-17C and diseases at mucosal surfaces
Insight on the current and future use of IL-17C as a therapeutic target
ACKNOWLEDGMENTS
This work was supported by NIDA of the National Institutes of Health (NIH) under award DP1 DA037997. SMS was supported by NIGMS of the NIH under award number T32 GM008056. AM was supported as a summer student by HLBI of the NIH under award number 5R25 HL103152.
Abbreviations:
- IL
interleukin
- PGN
peptidylglycan
- Th17
helper T cells-17
- TLR
toll-like receptors
- ALI
air-liquid interface
- CD
Crohn’s Disease
- UC
Ulcerative Colitis
- IBD
Inflammatory Bowel Disease
- CRC
Colorectal Cancer
- DSS
Dextran Sodium Sulfate
- AD
Atopic Dermatitis
- COPD
Chronic Obstructive Pulmonary Disease
- NTHi
Nontypeable Haemophilus influenzae
- HBE
Human Bronchial Epithelial
- HRV
Human Rhinovirus
- RAU
Recurrent Aphthous Ulcer
- HOK
Human Oral Keratinocyte
- MCPIP
Monocyte chemotactic protein-induced protein
- IκB
Inhibitor of nuclear factor-κB
- IKK2
kinase
- NF-κB
Nuclear Factor-kappa B
- NF-κB
Nuclear factor-kappaB
- Act1
activator 1
- ERK
Extracellular Signal-Related Kinase
- JNK
JUN N-Terminal Kinase
- RelA
NF-κB p65
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION of INTERESTS
The authors have no conflicts of interest.
References
- [1].Song X, He X, Li X, Qian Y, The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cellular & molecular immunology, 2016. 13(4): p. 418–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iwakura Y, Ishigame H, Saijo S, Nakae S, Functional specialization of interleukin-17 family members. Immunity, 2011. 34(2): p. 149–62 [DOI] [PubMed] [Google Scholar]
- [3].Starnes T, Broxmeyer HE, Robertson MJ, Hromas R, Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol, 2002. 169(2): p. 642–6 [DOI] [PubMed] [Google Scholar]
- [4].Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI, Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proceedings of the National Academy of Sciences of the United States of America, 2000. 97(2): p. 773–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C, Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity, 2011. 35(4): p. 611–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y, IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol, 2011. 12(12): p. 1151–8 [DOI] [PubMed] [Google Scholar]
- [7].Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L Jr., Singh H, Ouyang W, Pappu R, IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol, 2011. 12(12): p. 1159–66 [DOI] [PubMed] [Google Scholar]
- [8].Nies JF, Panzer U, IL-17C/IL-17RE: Emergence of a Unique Axis in TH17 Biology. Front Immunol, 2020. 11: p. 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Friedrich M, Diegelmann J, Schauber J, Auernhammer CJ, Brand S, Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal immunology, 2015. 8(4): p. 943–58 [DOI] [PubMed] [Google Scholar]
- [10].Im E, Jung J, Rhee SH, Toll-like receptor 5 engagement induces interleukin-17C expression in intestinal epithelial cells. J Interferon Cytokine Res, 2012. 32(12): p. 583–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Johansen C, Riis JL, Gedebjerg A, Kragballe K, Iversen L, Tumor necrosis factor alpha-mediated induction of interleukin 17C in human keratinocytes is controlled by nuclear factor kappaB. J Biol Chem, 2011. 286(29): p. 25487–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tanaka S, Nagashima H, Cruz M, Uchida T, Uotani T, Jimenez Abreu JA, Mahachai V, Vilaichone RK, Ratanachu-Ek T, Tshering L, Graham DY, Yamaoka Y, Interleukin-17C in human Helicobacter pylori gastritis. Infect Immun, 2017. 85(10): p. 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Johansen C, Vinter H, Soegaard-Madsen L, Olsen LR, Steiniche T, Iversen L, Kragballe K, Preferential inhibition of the mRNA expression of p38 mitogen-activated protein kinase regulated cytokines in psoriatic skin by anti-TNFalpha therapy. Br J Dermatol, 2010. 163(6): p. 1194–204 [DOI] [PubMed] [Google Scholar]
- [14].Takeuchi O, Akira S, Pattern recognition receptors and inflammation. Cell, 2010. 140(6): p. 805–20 [DOI] [PubMed] [Google Scholar]
- [15].Kawai T, Akira S, TLR signaling. Cell Death Differ, 2006. 13(5): p. 816–25 [DOI] [PubMed] [Google Scholar]
- [16].Kusagaya H, Fujisawa T, Yamanaka K, Mori K, Hashimoto D, Enomoto N, Inui N, Nakamura Y, Wu R, Maekawa M, Suda T, Chida K, Toll-like receptor-mediated airway IL-17C enhances epithelial host defense in an autocrine/paracrine manner. Am J Respir Cell Mol Biol, 2014. 50(1): p. 30–9 [DOI] [PubMed] [Google Scholar]
- [17].Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M, Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev, 2008. 226: p. 87–102 [DOI] [PubMed] [Google Scholar]
- [18].van Horssen R, Ten Hagen TL, Eggermont AM, TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist, 2006. 11(4): p. 397–408 [DOI] [PubMed] [Google Scholar]
- [19].Johansen C, Bertelsen T, Ljungberg C, Mose M, Iversen L, Characterization of TNF-alpha- and IL-17A-Mediated Synergistic Induction of DEFB4 Gene Expression in Human Keratinocytes through IkappaBzeta. J Invest Dermatol, 2016. 136(8): p. 1608–16 [DOI] [PubMed] [Google Scholar]
- [20].Guilloteau K, Paris I, Pedretti N, Boniface K, Juchaux F, Huguier V, Guillet G, Bernard FX, Lecron JC, Morel F, Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1{alpha}, and TNF-{alpha} Recapitulates Some Features of Psoriasis. J Immunol, 2010. 184(9): p. 5263–70 [DOI] [PubMed] [Google Scholar]
- [21].Hartupee J, Liu C, Novotny M, Li X, Hamilton T, IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol, 2007. 179(6): p. 4135–41 [DOI] [PubMed] [Google Scholar]
- [22].Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG, Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol, 2011. 131(3): p. 677–87 [DOI] [PubMed] [Google Scholar]
- [23].Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S, Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature, 2009. 458(7242): p. 1185–90 [DOI] [PubMed] [Google Scholar]
- [24].Dinarello CA, Biology of interleukin 1. FASEB J, 1988. 2(2): p. 108–15. [PubMed] [Google Scholar]
- [25].Ekert PG, Silke J, Vaux DL, Caspase inhibitors. Cell Death Differ, 1999. 6(11): p. 1081–6 [DOI] [PubMed] [Google Scholar]
- [26].Lopez-Castejon G, Brough D, Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev, 2011. 22(4): p. 189–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yamanaka K, Fujisawa T, Kusagaya H, Mori K, Niwa M, Furuhashi K, Kono M, Hamada E, Suda T, Maekawa M, IL-13 regulates IL-17C expression by suppressing NF-kappaB-mediated transcriptional activation in airway epithelial cells. Biochem Biophys Res Commun, 2018. 495(1): p. 1534–40 [DOI] [PubMed] [Google Scholar]
- [28].Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, Chen CS, Fu W, Gudjonsson JE, McCormick TS, Ward NL, Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol, 2013. 190(5): p. 2252–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 2006. 441(7090): p. 235–8 [DOI] [PubMed] [Google Scholar]
- [30].Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ, Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature, 2010. 467(7318): p. 967–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y, Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol, 2014. 192(4): p. 1449–58 [DOI] [PubMed] [Google Scholar]
- [32].Bradley JR, TNF-mediated inflammatory disease. J Pathol, 2008. 214(2): p. 149–60 [DOI] [PubMed] [Google Scholar]
- [33].Moraes L, Magnusson MK, Mavroudis G, Polster A, Jonefjall B, Tornblom H, Sundin J, Simren M, Strid H, Ohman L, Systemic Inflammatory Protein Profiles Distinguish Irritable Bowel Syndrome (IBS) and Ulcerative Colitis, Irrespective of Inflammation or IBS-Like Symptoms. Inflamm Bowel Dis, 2020. 26(6): p. 874–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Breese EJ, Michie CA, Nicholls SW, Murch SH, Williams CB, Domizio P, Walker-Smith JA, MacDonald TT, Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology, 1994. 106(6): p. 1455–66 [DOI] [PubMed] [Google Scholar]
- [35].Busch MA, Grondahl B, Knoll RL, Pretsch L, Doganci A, Hoffmann I, Kullmer U, Bahner V, Zepp F, Meyer CU, Gehring S, Patterns of mucosal inflammation in pediatric inflammatory bowel disease: striking overexpression of IL-17A in children with ulcerative colitis. Pediatr Res, 2020. 87(5): p. 839–846 [DOI] [PubMed] [Google Scholar]
- [36].George LA, Gadani A, Cross RK, Jambaulikar G, Ghazi LJ, Psoriasiform Skin Lesions Are Caused by Anti-TNF Agents Used for the Treatment of Inflammatory Bowel Disease. Dig Dis Sci, 2015. 60(11): p. 3424–30 [DOI] [PubMed] [Google Scholar]
- [37].Friedrich M, Tillack C, Wollenberg A, Schauber J, Brand S, IL-36gamma sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn’s disease. Inflamm Bowel Dis, 2014. 20(11): p. 1891–901 [DOI] [PubMed] [Google Scholar]
- [38].Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, Shi Y, Iwakura Y, Qian Y, Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity, 2014. 40(1): p. 140–52 [DOI] [PubMed] [Google Scholar]
- [39].Bernstein CN, Blanchard JF, Kliewer E, Wajda A, Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer, 2001. 91(4): p. 854–62 [DOI] [PubMed] [Google Scholar]
- [40].Itzkowitz SH, Yio X, Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol, 2004. 287(1): p. G7–17 [DOI] [PubMed] [Google Scholar]
- [41].Reynolds JM, Martinez GJ, Nallaparaju KC, Chang SH, Wang YH, Dong C, Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. J Immunol, 2012. 189(9): p. 4226–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, Auberval M, Lauffer F, Moran T, Ongenaert M, Van Balen M, Dupont S, Lepescheux L, Garcia T, Hartle S, Eyerich K, Fallon PG, Brys R, Steidl S, Neutralization of IL-17C Reduces Skin Inflammation in Mouse Models of Psoriasis and Atopic Dermatitis. J Invest Dermatol, 2018. 138(7): p. 1555–63 [DOI] [PubMed] [Google Scholar]
- [43].Lauffer F, Jargosch M, Baghin V, Krause L, Kempf W, Absmaier-Kijak M, Morelli M, Madonna S, Marsais F, Lepescheux L, Albanesi C, Muller NS, Theis FJ, Schmidt-Weber C, Eyerich S, Biedermann T, Vandeghinste N, Steidl S, Eyerich K, IL-17C amplifies epithelial inflammation in human psoriasis and atopic eczema. J Eur Acad Dermatol Venereol, 2020. 34(4): p. 800–9 [DOI] [PubMed] [Google Scholar]
- [44].Jungnickel C, Schmidt LH, Bittigkoffer L, Wolf L, Wolf A, Ritzmann F, Kamyschnikow A, Herr C, Menger MD, Spieker T, Wiewrodt R, Bals R, Beisswenger C, IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene, 2017. 36(29): p. 4182–90 [DOI] [PubMed] [Google Scholar]
- [45].Jamieson KC, Traves SL, Kooi C, Wiehler S, Dumonceaux CJ, Maciejewski BA, Arnason JW, Michi AN, Leigh R, Proud D, Rhinovirus and Bacteria Synergistically Induce IL-17C Release from Human Airway Epithelial Cells To Promote Neutrophil Recruitment. J Immunol, 2019. 202(1): p. 160–70 [DOI] [PubMed] [Google Scholar]
- [46].Jamieson KC, Wiehler S, Michi AN, Proud D, Rhinovirus Induces Basolateral Release of IL-17C in Highly Differentiated Airway Epithelial Cells. Front Cell Infect Microbiol, 2020. 10: p. 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Al-Samadi A, Kouri VP, Salem A, Ainola M, Kaivosoja E, Barreto G, Konttinen YT, Hietanen J, Hayrinen-Immonen R, IL-17C and its receptor IL-17RA/IL-17RE identify human oral epithelial cell as an inflammatory cell in recurrent aphthous ulcer. J Oral Pathol Med, 2014. 43(2): p. 117–24 [DOI] [PubMed] [Google Scholar]
- [48].Schulz S, Immel UD, Just L, Schaller HG, Glaser C, Reichert S, Epigenetic characteristics in inflammatory candidate genes in aggressive periodontitis. Hum Immunol, 2016. 77(1): p. 71–5 [DOI] [PubMed] [Google Scholar]
- [49].Jin J, Rha KS, Kim DW, Kim YM, IL-17C expression in nasal epithelial cells of chronic rhinosinusitis with nasal polyposis. Eur Arch Otorhinolaryngol, 2014. 271(5): p. 1097–105 [DOI] [PubMed] [Google Scholar]
- [50].Jeon YJ, Jo A, Won J, Lee KM, Yoon SS, Choi JY, Kim HJ, IL-17C Protects Nasal Epithelium from Pseudomonas aeruginosa Infection. Am J Respir Cell Mol Biol, 2020. 62(1): p. 95–103 [DOI] [PubMed] [Google Scholar]
- [51].Carey AJ, Weinberg JB, Dawid SR, Venturini C, Lam AK, Nizet V, Caparon MG, Walker MJ, Watson ME, Ulett GC, Interleukin-17A Contributes to the Control of Streptococcus pyogenes Colonization and Inflammation of the Female Genital Tract. Sci Rep, 2016. 6: p. 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sivick KE, Schaller MA, Smith SN, Mobley HL, The innate immune response to uropathogenic Escherichia coli involves IL-17A in a murine model of urinary tract infection. J Immunol, 2010. 184(4): p. 2065–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vicetti QCN Miguel RD, Pavelko SD, Cherpes TL, Intravaginal Chlamydia trachomatis Challenge Infection Elicits TH1 and TH17 Immune Responses in Mice That Promote Pathogen Clearance and Genital Tract Damage. PLoS ONE, 2016. 11(9): p. 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jinna S, Strober B, Anti-interleukin-17 treatment of psoriasis. Journal of Dermatological Treatment, 2016. 27(4): p. 311–315 [DOI] [PubMed] [Google Scholar]
- [55].Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P, Anti-TNF Therapy in Crohn’s Disease. Int J Mol Sci, 2018. 19: p. 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ma X, Xu S, TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep, 2013. 1(2): p. 177–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rockefeller U, Pharmaceuticals V, Biomarkers In Hidradenitis Suppurativa Participants Receiving Brodalumab. 2019.
- [58].Rockefeller U, An Alternative Dose Interval Study in Participants With Hidradenitis Suppurativa Receiving Brodalumab, 2020.
- [59].Gao Z, Kuestner R, Appleby M, Lewis K, McKernan P, Okada S, Taft D, Kuijper J, Jaspers S, Levin S, Il-17c antagonists and methods of using the same. US20070129302A1, 2007.
- [60].Appleby MW, Rixon MW, Lewis KB, Gao Z, Kuestner RE, Birks CW, Il-17c antagonists and methods of using the same. WO2008049070A2, 2008.
- [61].Bültmann A, Mühlbacher R, Garcia T, Brys RCX, Nelles L, Conrath K, Antagonists of il17c for the treatment of inflammatory disorders. US20190062422A1, 2019.
- [62].Gonzales L, Ouyang W, Pappu R, Ramirez-Carrozi V, Treatment of gastrointestinal inflammation and psoriasis and asthmainflammation and psoriasis a. EP3214442A1, 2017.
- [63].Vandeghinste NER, Brys RCX, Antagonists of il-17c for the treatment and/or prevention of atopic dermatitis. WO2017060289A1, 2017.
- [64].NV G, MOR106 clinical development in atopic dermatitis stopped for futility, Galapagos NV, Intrado GlobeNewswire, 2019, pp. 1–3. [Google Scholar]