

Abstract
While researchers know that tumor mutational burden (TMB) is low in hepatocellular carcinoma (HCC), prior studies have not investigated TMB in cirrhosis, small early HCC and progressed HCC. HCC (n = 18) and cirrhosis (n = 6) cases were identified. TMB was determined by a 1.7 megabase, 409-gene next-generation sequencing panel. TMB values were defined as the number of nonsynonymous variants per megabase of sequence. There was no significant difference between cirrhosis versus small early HCC or between cohorts when stratified by size, early versus progressed, differentiation or morphology. There was a significant difference between cirrhosis and small early HCC versus progressed HCC (p = 0.045), suggesting TMB may be related to HCC progression. TMB similarities in small early HCC and background cirrhosis suggest TMB is not a useful tool for diagnosing small early HCC. Additional study is needed to address TMB in histological and molecular subsets of HCC.
Keywords: : HCC, hepatocellular carcinoma, progressed hepatocellular carcinoma, small early hepatocellular carcinoma, TMB, tumor mutational burden
Liver cancer is the sixth-most common cancer and the fourth-most common cause of cancer-related deaths worldwide, with increasing incidence in the last several decades [1,2]. The most common primary liver cancer is hepatocellular carcinoma (HCC), comprising 75–85% of cases [1]. The risk factors for HCC vary from region to region, but HCC typically develops in patients with chronic liver disease associated with chronic viral hepatitis, alcohol consumption and metabolic syndrome, among others [3].
Treatment of cancer with immunotherapy is a rapidly evolving field in oncology, and one of the early biomarkers to predict response to immune checkpoint inhibitors has been the immunohistochemical expression of programmed death ligand 1 (PD-L1) on tumor cells [4]. Recent studies have shown that mismatch-repair deficiency can also predict objective clinical response to immune checkpoint inhibitors across many tumor types [5]. Tumor mutational burden (TMB) is an emerging biomarker that is also predictive of response to immune checkpoint inhibitors [6]. TMB is defined as the total number of somatic nonsynonymous mutations present in the coding region expressed as mutations per megabase (muts/Mb) in a tumor [6]. Currently, static pan-cancer cutoffs for low, intermediate, and high TMB cutoff are being utilized; however, the TMB landscape is changing and will most likely move toward values that are tissue specific and percentile based in the future [7]. Recently, the FDA has approved TMB (≥10 muts/Mb as detected by an FDA-approved assay) as a pan-tumor biomarker for treatment with pembroluzimab [8]. Finally, while both PD-L1 expression and high TMB predict response to immunotherapy across different tumor types, they are independent biomarkers in most cancer types with different rates of response predicted [9].
In HCC, many genomic alterations have been found, though there is no single oncogenic driver mutation in the majority of HCCs [10]. Studies have shown that HCCs have generally low (<5 muts/Mb) TMB, are very rarely hypermutated and are on the low median end of the TMB spectrum in carcinomas with values ranging from 0.42 to 65.6 muts/Mb and median ranging from 2.56 to 5 muts/Mb [11–14]. However, research on HCC with higher TMB has yielded interesting results [11,15–17]. In a recent study on SHR-1210 (anti-PD-1 antibody) combined with apatinib (VEGFR2 inhibitor) in advanced HCC, HCC patients with high TMB (>7.2 muts/Mb) responded favorably to treatment, with improved survival compared with those patients without high TMB [17]. In another study of 17 HCC cases, Ang et al. showed there were no significant differences in TMB between patients whose disease progressed, remained stable or responded to immune checkpoint inhibitors [11]. Other studies have shown that higher TMB is associated with tumor recurrence after radical hepatectomy, as well as significantly poorer overall and progression-free survival [15,16]. While much has been published on the correlation between high TMB and response to immunotherapy and prognosis, not much has been published on the mutational burden of cirrhotic liver adjacent to HCC and its precursor lesions [15,16]. Furthermore, no studies have evaluated whether TMB holds any diagnostic value as an ancillary test to histopathological examination.
In cirrhotic livers, high-grade dysplastic nodules (HGDNs) evolve to small early nonprogressed HCC, which may then develop into progressed and advanced HCC [18]. HGDNs are defined as vaguely nodular lesions with architectural and/or cytological atypia (such as small change cell) and the variable presence of unpaired arteries [19]. Early HCCs are defined as vaguely nodular malignant lesions with the following histological features: increased nuclear–cytoplasm ratio, variable numbers of intratumoral portal tracts, a pseudoglandular pattern, diffuse fatty change and varying numbers of unpaired arteries [19]. Although such morphological definitions are proposed, there are overlapping features between HGDNs and early HCC. According to the International Consensus Group for Hepatocellular Neoplasia, stromal invasion is the most helpful and distinguishing feature in differentiating small early HCC from HGDNs [19,20]. However, stromal invasion is not always present or may only be present focally and can therefore be missed on needle biopsy [20]. Progressed HCCs are defined as conspicuous tumors that are distinctly nodular with infiltrative and expansile growth patterns, and these HCCs commonly have a tumor capsule [21].
Based on morphology alone, it is often difficult to differentiate HGDNs from small early HCC. While studies have shown that TMB is generally lower in the majority of HCC compared with other carcinomas, no previous studies have investigated mutational burden in non-neoplastic cirrhotic liver compared with small early and progressed HCC [11–14]. The present study was undertaken to investigate mutational burden in background cirrhosis compared with small early HCC, small progressed HCC and large progressed HCC; to evaluate its utility in diagnosis of HCC; and to understand its role in early HCC development.
Materials & methods
After institutional review board approval, 18 cases of HCC from 18 patients were retrieved from departmental surgical pathology archives, including small early HCCs (n = 6), small progressed HCCs (n = 6), large well-differentiated HCCs (n = 3) and large moderately-differentiated HCCs (n = 3). Based on the definitions of the current edition of the WHO, small early HCCs are defined as vaguely nodular malignant lesions that are <2 cm in size with the following histological features: increased nuclear-cytoplasm ratio, variable numbers of intratumoral portal tracts, a pseudoglandular pattern, diffuse fatty change and varying numbers of unpaired arteries [19,21]. Progressed HCCs are defined as conspicuous tumors that are distinctly nodular with infiltrative and expansile growth patterns, and these HCCs commonly have a tumor capsule [21]. In the current study, small HCCs were defined as ≤1.5 cm, and large HCCs were defined as ≥2 cm. Each HCC was further classified into subtypes (e.g., steatohepatitic HCC, lymphocyte-rich HCC and HCC not otherwise specified [NOS]) defined by the WHO and by differentiation as defined by the WHO [21]. In addition, background cirrhosis of the six small early HCCs was also included for analysis. Relationships between TMB and morphological alterations (e.g., steatohepatitic features, cholestasis, inflammatory infiltrates and architectural pattern) were noted. Cases in which the distinction between small early HCC and high-grade dysplastic nodule was not certain were excluded. High-grade dysplastic nodules were not included as a separate category because it is not known whether there is a difference in TMB between small early HCC and background cirrhosis.
The areas with cirrhosis and HCC were microdissected in all cases for evaluation of TMB. Nonsynonymous somatic variants in select regions of 409 oncogenes and tumor-suppressor genes (1.7 megabases of genomic coverage) were detected by the Ion Oncomine™ Tumor Mutation Load Assay running on the Ion Chef™ and S5XL™ NGS instruments (Thermo-Fisher Scientific, MA, USA). Sequence data was captured by Ion Torrent Suite Software v5.10.2 and somatic prediction was conducted using the Ion Reporter Software (IRS) v5.10.5. TMB values were defined as the number of nonsynonymous variants (missense and nonsense single-nucleotide variants [SNVs], plus insertion and deletion variants [INDELs] and expressed as mutations per megabase [muts/Mb]).
Continuous variables were summarized as means and/or medians with ranges, and other variables as percentages. Data was analyzed using Mann–Whitney U test. All p-values were based on a 2-sided test of statistical significance. A p-value < 0.05 was considered statistically significant.
Results
Patient characteristics
Of the eighteen HCC patients, thirteen patients (72.2%) were male and five (27.7%) were female. Patient age ranged from 50 to 75 years (mean: 61 years; median: 60 years). The ethnicities of the patient cohort included Caucasian (n = 9), Hispanic (n = 8) and Asian (n = 1). The etiology of cirrhosis of the background liver included hepatitis B virus (n = 1), hepatitis C virus (n = 3), hepatitis B and C virus (n = 2), nonalcoholic steatohepatitis (n = 6), alcoholic steatohepatitis (n = 2), alpha-1 antitrypsin deficiency (n = 2), hemochromatosis (n = 1) and autoimmune hepatitis (n = 1). These characteristics are summarized in Table 1.
Table 1. . Patient characteristics.
| Age | n = 18 |
| Mean (years) | 61 |
| Median (years) | 60 |
| Sex | |
| Female | 5 |
| Male | 13 |
| Ethnicity | |
| Caucasian | 9 |
| Hispanics | 8 |
| Asian | 1 |
| Etiology of cirrhosis | |
| Hepatitis B | 1 |
| Hepatitis C | 3 |
| Hepatitis B and C | 2 |
| Non-alcoholic steatohepatitis | 6 |
| Alcoholic steatohepatitis | 2 |
| Alpha-1 antitrypsin deficiency | 2 |
| Hemochromatosis | 1 |
| Autoimmune hepatitis | 1 |
Pathologic characteristics
All cases of small early HCC (Figure 1A) with background cirrhosis (Figure 1B) and progressed HCC (Figure 1C) were untreated and did not have extrahepatic disease. According to the HCC subtypes defined by the WHO eighth edition, subtypes included the steatohepatitic variant (n = 4) and lymphocyte-rich HCC (n = 1) (Table 2) [21]. Cases with ≥90% steatohepatitic morphology were classified as the steatohepatitic subtype. Three cases also showed a smaller amount (≤50%) of steatohepatitic morphology and were characterized as HCC, NOS, with partial steatohepatitic morphology, for the purposes of the current study. The remaining ten cases did not fit any subtypes defined by the WHO and were categorized as HCC NOS. Three of the eighteen cases had intratumoral cholestasis and pseudoglandular architecture, including one small early HCC and two small progressed HCCs.
Figure 1. . Cirrhosis and hepatocellular carcinoma.

(A) Small early hepatocellular carcinoma (HCC) (400x, hematoxylin and eosin [H&E] stain). (B) Background cirrhosis of small early HCC (100x, H&E stain). (C) Small progressed HCC (400x, H&E stain).
Table 2. . HCC subtypes.
| HCC subtypes | Cases (n) |
|---|---|
| Steatohepatitic HCC | 4 |
| Lymphocyte-rich HCC | 1 |
| HCC NOS | 10 |
| HCC NOS with partial steatohepatitic morphology | 3 |
HCC: Hepatocellular carcinoma; NOS: Not otherwise specified.
Pathological features, including size, lymph–vascular invasion and pathological tumor stage, were evaluated in each HCC category and stratified by early versus progressed status (Table 3). All small and large HCCs were adequately sampled and evaluated based on the WHO criteria for differentiation [21].
Table 3. . Pathological characteristics of HCC.
| WHO criteria of HCC by size | Mean size, cm (range) | LVI, n (%) | pT stage |
|---|---|---|---|
| Small early HCC (n = 6) | 0.72 (0.5–1.0) | 0 (0%) | T1a (n = 1), T2 (n = 5) |
| Small progressed HCC (n = 6) | 0.98 (0.7–1.5) | 0 (0%) | T1a (n = 5), T2 (n = 1) |
| Large well-differentiated HCC (n = 3) | 9.4 (2.0–19.8) | 0 (0%) | T1b (n = 2), T2 (n = 1) |
| Large moderately-differentiated HCC (n = 3) | 3.9 (3.0–5.2) | 0 (0%) | T1b (n = 2), T3 (n = 1) |
HCC: Hepatocellular carcinoma; LVI: Lymph-vascular invasion; pT stage: Pathologic tumor stage.
Mutational burden
The mutational burden across all cases had low TMB based on pan-cancer definitions of low, intermediate, and high TMB, including background cirrhosis, with mutational burden ranging from 0 to 3.5 muts/Mb [11–14]. The distribution of TMB in the 18 HCC tumor samples is shown in Figure 2A, ranging from 0 to 3.5 muts/Mb. The mean TMB for the entire cohort of HCCs was 1.4 muts/Mb. The mean mutational burdens for cirrhosis, small early HCC, small progressed HCC, large well-differentiated HCC and large moderately-differentiated HCC were 0.9, 0.8, 1.7, 1.4 and 2.2, respectively (Figure 2B).
Figure 2. . Mutational burden in background cirrhosis and hepatocellular carcinoma tumor samples.
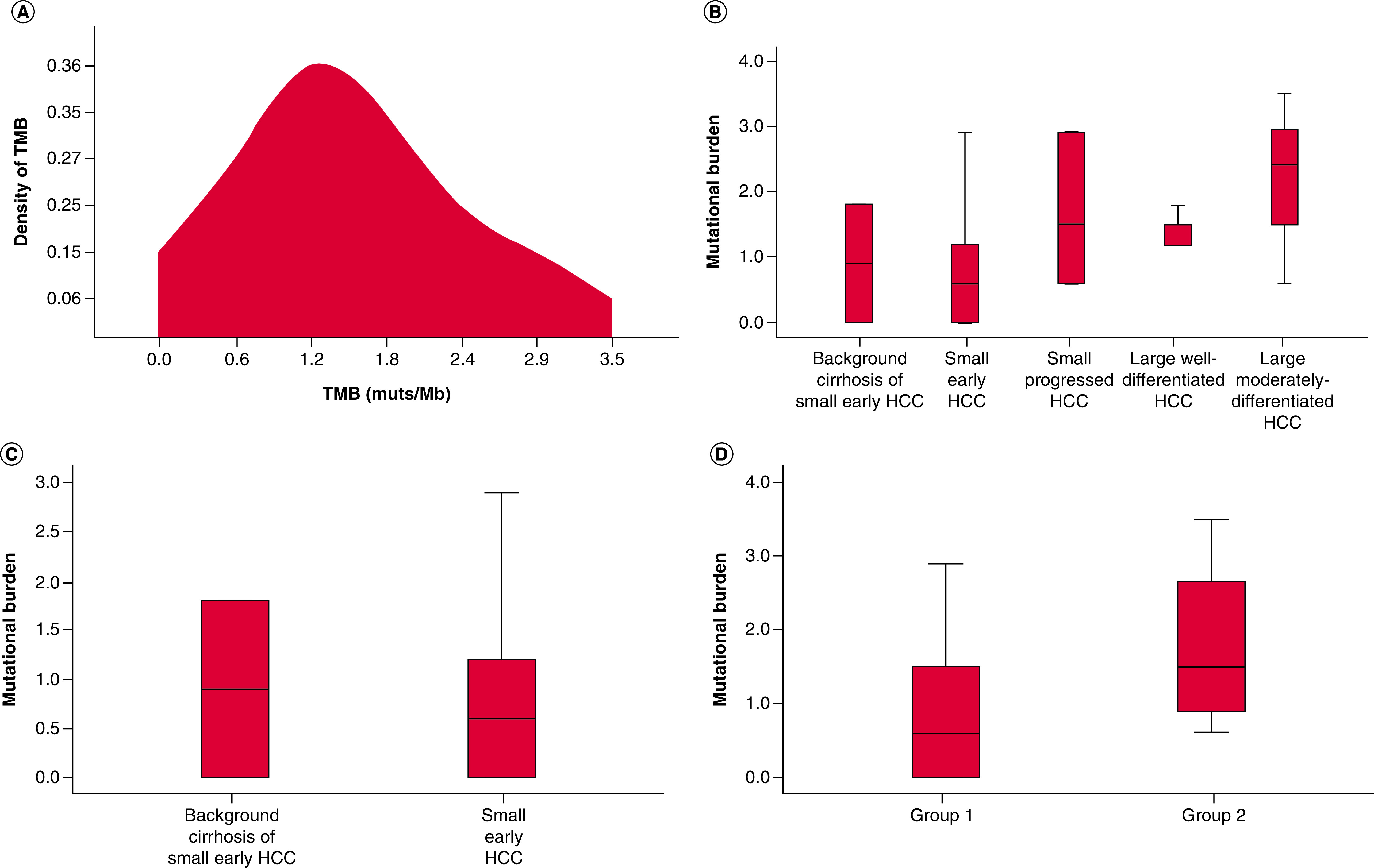
(A) TMB distribution in hepatocellular carcinoma (HCC) tumor samples and density of tumor mutational burden (TMB) represent the percentage of HCC at different TMB values. (B) Mutational burden in cirrhosis and early and progressed HCCs. (C) Mutational burden in the background cirrhosis (of small early HCC) vs small early HCC themselves. (D) Mutational burden in cirrhosis and small early HCC (group 1) versus small and large progressed HCC (group 2).
HCC: Hepatocellular carcinoma; TMB: Tumor mutational burden.
Background cirrhosis of small early HCC versus small early HCC
The mutational burden for background cirrhosis ranged from 0 to 1.8 muts/Mb, with a mean value of 0.9 muts/Mb. TMB for small early HCCs ranged from 0 to 2.9, with a mean value of 0.8 muts/Mb. There was no significant difference in TMB between background cirrhosis in small early HCCs and small early HCCs (p = 0.87, Figure 2C).
Small early HCC versus small progressed HCC
TMB for small progressed HCCs ranged from 0.6 to 2.9 muts/Mb, with a mean value of 1.7 muts/Mb. There was no significant difference in TMB between small early HCC and small progressed HCC (p = 0.19).
Small HCC versus large HCC
TMB for all small HCCs ranged from 0 to 2.9 muts/Mb, with a mean value of 1.3 muts/Mb. TMB for all large HCCs ranged from 0.6 to 3.5 muts/Mb, with a mean value of 1.8 muts/Mb. There was no significant difference between small HCC and large HCC (p = 0.29).
Large well-differentiated HCC versus large moderately-differentiated HCC
TMB for large well-differentiated HCCs ranged from 1.2 to 1.8 muts/Mb, with a mean value of 1.4 muts/Mb. TMB for large moderately-differentiated HCCs ranged from 0.6 to 3.5 muts/Mb, with a mean value of 2.2 muts/Mb. There was no significant difference between small HCC and large HCC (p = 0.66).
Small early HCC & background cirrhosis versus small & large progressed HCC
For additional comparison, small early HCC and its background cirrhosis were grouped together (group 1) for comparison to small progressed HCCs and large progressed HCCs (group 2). The mean TMB for group 1 was 0.9 muts/Mb, compared with 1.7 muts/Mb for group 2 (p = 0.045, Figure 2D).
Relationships between TMB & HCC for viral etiologies versus nonviral etiologies
TMB for HCCs with HBV and/or HCV etiologies (n = 6) ranged from 0.0 to 2.9 muts/Mb, with a mean value of 0.88 muts/Mb. TMB for HCCs with nonviral etiologies (n = 12) ranged from 0.6 to 3.5 muts/Mb, with a mean value of 1.7 muts/Mb. Though there was no significant difference between HCC for viral versus nonviral etiologies (p = 0.085), the samples are too small to draw any firm conclusions.
Relationships between TMB & morphologic findings in HCC
Three cases with cholestasis and pseudoglandular architecture had TMBs ranging from 0.6 to 2.9 muts/Mb, with a mean of 1.6 muts/Mb. The remaining fifteen cases without cholestasis had TMBs ranging from 0 to 3.5 muts/Mb, with a mean of 1.4 muts/Mb. There was no significant difference in TMB between HCC with and without cholestasis (p = 0.86). Seven cases with steatohepatitic features had TMBs ranging from 0 to 2.4 muts/Mb with a mean of 0.9 muts/Mb. The remaining eleven cases without steatohepatitic features had TMBs ranging from 0 to 3.5 muts/Mb, with a mean of 1.6 muts/Mb. There was no significant difference between TMB in HCC with steatohepatitic features versus TMB in HCC without steatohepatitic features (p = 0.12).
Discussion
Extensive research on TMB in HCC has demonstrated a correlation between higher TMB and poor prognosis. These HCCs exhibit an immune-rich microenvironment and tend to be inflammation-driven, which makes them candidates for immune checkpoint inhibitors. However, no studies have examined the differences in mutational burden in background cirrhosis versus HCC in varying stages of development. This pilot study addresses whether TMB values change during carcinogenesis of HCCs and if TMB values can be helpful in the diagnostic workup of HCCs.
In the current cohort, all cases of HCC and background cirrhosis were found to have relatively low mutational burden, consistent with previous literature [11–14]. There was no significant difference in TMB between the cohorts of HCC when stratified by size, early versus progressed status or differentiation (well vs moderately differentiated). However, when cirrhosis and small early HCCs were considered together, there was a significant difference versus small and large progressed HCCs. These findings suggest that while small early HCCs have a similar mutational burden to background cirrhosis, mutational burden may progress with histology irrespective of tumor size.
In clinical practice, differentiating between high-grade dysplastic nodules and well-differentiated HCC can be a diagnostic conundrum, as morphology alone may not suffice. Heat-shock protein 70 (HSP70), glutamine synthetase (GS) and glypican-3 (GPC3) immunostains, can be helpful in supporting the diagnosis of well-differentiated HCC with increased reliability and objectivity [22,23]. Sensitivity for detection of HCC is 58.7% with two positive immunostains and 25.0% with three positive immunostains while combined positivity for either two or three immunostains is 100% [22]. However, if only one of these three immunostains is positive, the sensitivity for detection of HCC is 93.5%, but specificity drops to 85.7%, as 14.3% of high-grade dysplastic nodules are positive for one immunostain [22]. Even with the aid of immunohistochemistry, differentiating a well-differentiated HCC from a high-grade dysplastic nodule remains challenging. Thus, additional molecular techniques may be useful in resolving this difficult histological differential diagnosis.
The current study found that there was no significant difference in mutational load between small early HCCs and the background cirrhotic liver (p = 0.87). While mutational burden was not directly evaluated in high-grade dysplastic nodules, the similarity of mutational burden between background cirrhotic liver and small early HCC suggests that mutational burden is not a useful diagnostic tool for distinguishing between high-grade dysplastic nodule and small early HCC.
The molecular subtyping of HCC has been expanding as increasing numbers of molecular alterations are identified in HCC [15,24,25]. Studies have shown that the phenotypes of HCC have been linked to specific genetic mutations, oncogenic pathways and/or gene expression profiles [25]. Though specific mutations were not evaluated in the current study, three HCC cases (one small early HCC and two small progressed HCCs) in the current cohort had cholestasis, pseudoglandular architecture and lack of inflammatory infiltrates, a morphological phenotype that has been associated with CTNNB1 mutations [25–27]. Previous studies of HCC mutation profiles showed a positive correlation between high TMB and CTNNB1 mutations [12,28]. However, TMB in these three cases of HCC in the current study were on the lower end of the TMB spectrum. Additional studies with identification of genetic alterations and molecular subtypes are needed, and may yield a deeper insight to the biology and pathogenesis between cirrhosis, precursor lesions and HCC.
Another example of phenotypes of HCC linked to genetic alterations is steatohepatitic HCC [25,27]. Investigators have found that this subtype of HCC falls into the nonproliferative category of the transcriptomic categorization of HCC, which is characterized by well-differentiated morphology, maintenance of hepatocytic markers and chromosomal stability [25,27]. In the current cohort, there were seven HCC cases with steatohepatitic features that had lower TMB with a mean of 0.9 muts/Mb. Steatohepatitic HCC falls into the G4 transcriptomic subgroup, which shares a similar gene expression profile to non-neoplastic mature hepatocytes [24,25,27]. This may explain the less aggressive histological phenotype of steatohepatitic HCC with a lack of satellite nodules and vascular invasion [27]. In the current study, steatohepatitic HCCs exhibited this less aggressive histological phenotype and showed a trend toward lower TMB than HCCs without steatohepatitic features. Although this trend was not statistically significant (p = 0.12), this finding further suggests that steatohepatitic HCC may represent a unique biological subtype of HCC and is in keeping with a low proliferation transcriptomic phenotype.
In other tumor types, such as colon and lung, high TMB has been associated with microsatellite instability, and lymphocyte-rich tumors have been associated with microsatellite instability and/or high TMB [29–35]. In contrast, lymphocyte-rich HCC (also known as lymphoepithelioma-like HCC) is not associated with microsatellite instability or a high number of somatic mutations [36,37]. In the current study, the case of lymphocyte-rich HCC also showed a lower TMB score of 1.0 muts/Mb, consistent with the previous literature.
The current study was undertaken to examine mutational burden in background cirrhosis, small early HCC and progressed HCC. The authors acknowledge there are limitations to this pilot study. One possible limitation was that TMB was calculated utilizing a targeted NGS panel. This raises the possibility of bias versus the gold standard of whole exome sequencing (WES). However, multiple prior studies have shown high concordance with the TMB value produced by the assay utilized in this study and those from both other targeted panels and WES [38,39]. A further limitation of the study is the small number of cases in the cohort, limiting the power of the study. With a small number of HCC cases, there was no control for variable histological subtypes of HCC, molecular subtypes of HCC or the etiology of chronic liver disease. Further studies with larger pure cohorts of cases are needed to evaluate and control for these factors.
Conclusion
There was no significant difference in TMB between the cohorts of HCC when stratified by size, progression (early vs progressed status), morphology or differentiation (well vs moderately differentiated) in this pilot study. However, there was a significant difference between cirrhosis/small early HCC versus small and large progressed HCC, suggesting that TMB may be associated with histological progression. While high-grade dysplastic nodules were not specifically studied, the lack of a difference in mutational burden between small early HCC and its background cirrhosis suggests that TMB is not useful as a diagnostic tool to distinguish between these two entities. Thus, the practical message of this study is that TMB does not appear to be a useful marker to differentiate cirrhotic and dysplastic nodules from early HCC. Future studies are needed to address TMB in specific histological and molecular subsets of HCC and in HCC arising from different etiologies of chronic liver disease.
Future perspective
Static pan-cancer cutoffs for low-, intermediate- and high-TMB cutoffs are currently being utilized in clinical practice. However, the landscape of TMB will likely change, as it moves toward values that are tissue specific and percentile based in the future [7]. While TMB is generally considered low compared with other carcinomas, as more research is done on TMB in HCCs, a subset of HCCs may be considered to have high TMB within the tissue-specific HCC cutoffs. Studying TMB in specific histological and molecular subsets of HCC and in HCC arising from different etiologies of chronic liver disease may also provide insight into the pathobiology of HCC and its response to immunotherapy.
Summary points.
While studies have shown that tumor mutational burden (TMB) is generally lower in the majority of hepatocellular carcinomas (HCCs) compared with other carcinomas, prior studies have not investigated TMB in non-neoplastic cirrhotic liver compared with small early HCC and progressed HCC.
This study investigated whether there were differences in mutational burden between background cirrhosis, small early HCC, small progressed HCC and large HCC.
There was no significant difference in TMB between the cohorts of HCC when stratified by size, progression (early vs progressed), morphology and differentiation (well vs moderately differentiated).
However, there was a significant difference between cirrhosis and small early HCC versus small and large progressed HCC, suggesting that TMB may be related to HCC progression.
The similarity in mutational burden of background cirrhosis and small early HCC suggests that TMB will not be a useful marker to differentiate cirrhotic and dysplastic nodules from early HCC.
Further studies are needed to address TMB in histological and molecular subsets of HCC and in HCC arising from different etiologies of chronic liver disease.
Footnotes
Author contributions
Each co-author listed participated sufficiently in the work to take responsibility for the content.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
Appropriate institutional review board approval was obtained.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68(6), 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Rawla P, Sunkara T, Muralidharan P, Raj JP. Update in global trends and aetiology of hepatocellular carcinoma. Contemp. Oncol. (Pozn) 22(3), 141–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hb E-S. Hepatocellular Carcinoma. N. Engl. J. Med. 365(2), 1118–1127 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 14(4), 847–856 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, N.Y.) 357(6349), 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9(1), 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Important article on the landscape of tumor mutational burden across different cancer types.
- 7.Fernandez EM, Eng K, Beg S et al. Cancer-specific thresholds adjust for whole exome sequencing-based tumor mutational burden distribution. JCO Precis. Oncol. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.U.S. Food and Drug Administration. Center for Drug Evaluation and Research. KEYNOTE-158 (NCT02628067) approval letter. June 17, 2020. Retrieved January 26, 2021, from https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors.
- 9.Yarchoan M, Albacker LA, Hopkins AC et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4(6), e126908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ocker M. Biomarkers for hepatocellular carcinoma: what's new on the horizon? World J. Gastroenterol. 24(35), 3974–3979 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ang C, Klempner SJ, Ali SM et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 10(40), 4018–4025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Rao X, Wen Z et al. Implications of driver genes associated with a high tumor mutation burden identified using next-generation sequencing on immunotherapy in hepatocellular carcinoma. Oncol. Lett. 19(4), 2739–2748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauriello A, Zeuli R, Cavalluzzo B et al. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in HCC patients not undergoing immunotherapy. Cancers 11(12), 1824 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Li M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 20(1), 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai H, Zhang Y, Zhang H, Cui C, Li C, Lu S. Prognostic role of tumor mutation burden in hepatocellular carcinoma after radical hepatectomy. J, Surg. Oncol. 121(6), 1007–1014 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Shrestha R, Prithviraj P, Anaka M et al. Monitoring immune checkpoint regulators as predictive biomarkers in hepatocellular carcinoma. Front. Oncol. 8, 269–269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Zhang Y, Jia R et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: an open-label, dose escalation and expansion study. Clin. Cancer Res. 25(2), 515–523 (2019). [DOI] [PubMed] [Google Scholar]; • Study showing favorable response to anti-PD-1 antibody and VEGFR2 inhibitor in patients with hepatocellular carcinoma (HCC) and higher tumor mutational burden.
- 18.Kobayashi M, Ikeda K, Hosaka T et al. Dysplastic nodules frequently develop into hepatocellular carcinoma in patients with chronic viral hepatitis and cirrhosis. Cancer 106(3), 636–647 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Kojiro M, Wanless IR, Alves V et al. Pathologic diagnosis of early hepatocellular carcinoma: a report of the international consensus group for hepatocellular neoplasia. Hepatology 49(2), 658–664 (2009). [DOI] [PubMed] [Google Scholar]; •• Key article on histologic differentiation between early and progressed HCC.
- 20.Desmet VJ. East–West pathology agreement on precancerous liver lesions and early hepatocellular carcinoma. Hepatology 49(2), 355–357 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Torbenson MS, Ig IOL, Park YN et al. WHO Classification of Tumours of the Digestive System. (5th), International Agency for Research on Cancer, Lyon: (2019). [Google Scholar]; • Article differentiating HCC from high-grade dysplastic nodules based on morphology and immunohistochemical stains.
- 22.Di Tommaso L, Destro A, Seok JY et al. The application of markers (HSP70 GPC3 and GS) in liver biopsies is useful for detection of hepatocellular carcinoma. J. Hepatol. 50(4), 746–754 (2009). [DOI] [PubMed] [Google Scholar]; • Article differentiating HCC from high-grade dysplastic nodules based on morphology and immunohistochemical stains.
- 23.Di Tommaso L, Franchi G, Park YN et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 45(3), 725–734 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Boyault S, Rickman DS, De Reyniès A et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45(1), 42–52 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Calderaro J, Ziol M, Paradis V, Zucman-Rossi J. Molecular and histological correlations in liver cancer. J. Hepatol. 71(3), 616–630 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Audard V, Grimber G, Elie C et al. Cholestasis is a marker for hepatocellular carcinomas displaying β-catenin mutations. J. Pathol. 212(3), 345–352 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Calderaro J, Couchy G, Imbeaud S et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 67(4), 727–738 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Wang S, Shi H, Liu T et al. Mutation profile and its correlation with clinicopathology in Chinese hepatocellular carcinoma patients. Hepatobiliary Surg Nutr. 10(2), 172–179 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC et al. Signatures of mutational processes in human cancer. Nature 500(7463), 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown SD, Warren RL, Gibb EA et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 24(5), 743–750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chae YK, Anker JF, Bais P, Namburi S, Giles FJ, Chuang JH. Mutations in DNA repair genes are associated with increased neo-antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 9(8), 7949–7960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le DT, Uram JN, Wang H et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372(26), 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mcgranahan N, Furness AJS, Rosenthal R et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351(6280), 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 348(6230), 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Song Z, Cheng G, Xu C, Wang W, Shao Y, Zhang Y. Clinicopathological characteristics of POLE mutation in patients with non-small-cell lung cancer. Lung Cancer 118, 57–61 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Chan AWH, Tong JHM, Pan Y et al. Lymphoepithelioma-like hepatocellular carcinoma: an uncommon variant of hepatocellular carcinoma with favorable outcome. Am. J. Surg. Pathol. 39(3), (2015). [DOI] [PubMed] [Google Scholar]
- 37.Sia D, Jiao Y, Martinez-Quetglas I et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 153(3), 812–826 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Chaudhary R, Quagliata L, Martin JP et al. A scalable solution for tumor mutational burden from formalin-fixed, paraffin-embedded samples using the Oncomine Tumor Mutation Load Assay. Transl. Lung Cancer Res. 7(6), 616–630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heydt C, Rehker J, Pappesch R et al. Analysis of tumor mutational burden: correlation of five large gene panels with whole exome sequencing. Sci. Rep. 10(1), 11387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]