

Acute kidney injury (AKI) is manifested by a rapid decline of renal function that is associated with high morbidity and mortality (1). However, the underlying mechanisms of AKI remain poorly understood, and effective treatment for AKI is unavailable. Both hypoxia-inducible factor (HIF) and NF-κB have been implicated in gene regulation in AKI, but very little is known about their cross talk.
Renal tubular cells are high in oxygen consumption. Thus, a disturbance of oxygen homeostasis, especially ischemia or hypoxia, is a main cause of AKI. The HIF family is a family of “master” regulators of gene expression in response to hypoxia or ischemia, conditions of decreased availability of oxygen. When activated, HIF mounts adaptive responses to hypoxia at the molecular, cellular, and tissue levels (2). HIF consists of a hypoxia-inducible α-subunit and a constitutively expressed β-subunit. In response to hypoxia, HIF-α is induced and moves to the nucleus to dimerize with the β-subunit to form functional HIF leading to target gene expression. There are three α-subunits, i.e., HIF-1α, HIF-2α, and HIF-3α. In the kidney, HIF-1α is expressed by most renal tubular epithelial cells, whereas HIF-2α is mainly expressed in renal interstitial fibroblasts and endothelial cells (2). In general, both HIF-1α and HIF-2α are activated by hypoxia in AKI but in different phases: HIF-1α takes part in the initial adaptation process of hypoxia as it is rapidly induced and then falls to a low level within 72 h, whereas HIF-2α accumulation begins under prolonged hypoxic conditions (2). Inflammation is another important pathological factor in AKI. It has been previously reported that renal tubular cells are not only a mere victim of inflammation but also the propagator of intrarenal inflammation. NF-κB, consisting of five subunits [RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100)], is the main proinflammatory transcription factor in AKI (3, 4).
In an article recently published in the American Journal of Physiology-Renal Physiology, Li et al. (5) provide the evidence for a cross talk between NF-κB and HIF in AKI. Specifically, they demonstrated the regulation of HIF-1 by NF-κB. This finding not only sheds new light on understanding the pathophysiology of AKI but also suggests rational strategies for therapeutic interventions. In their study, Li et al. (5) verified a significant increase of HIF-1α in renal tubular cells in AKI induced by ischemia-reperfusion (I/R), unilateral ureteral obstruction (UUO), or sepsis. They then showed that tubular Hif1a mRNA expression was strongly associated with inflammation, highlighting the possible connection between tubular HIF-1α and inflammation in AKI. They further conducted a series of in vivo and in vitro experiments, leading to the finding that NF-κB may transcriptionally regulate HIF-1α in renal tubule cells. Notably, their chromatin immunoprecipitation assay showed that NF-κB p65 could directly bind to the Hif1a gene promoter. Interestingly, HIF-1α could be induced by IL-1β in the presence of oxygen via NF-κB. Thus, NF-κB may induce HIF-1 in the absence of hypoxia. Together, these results indicate an intriguing interaction between hypoxia and inflammation in renal pathophysiology. At the molecular level, this interaction may be mediated at least partly by NF-κB regulation of Hif1a transcription (Fig. 1).
Figure 1.
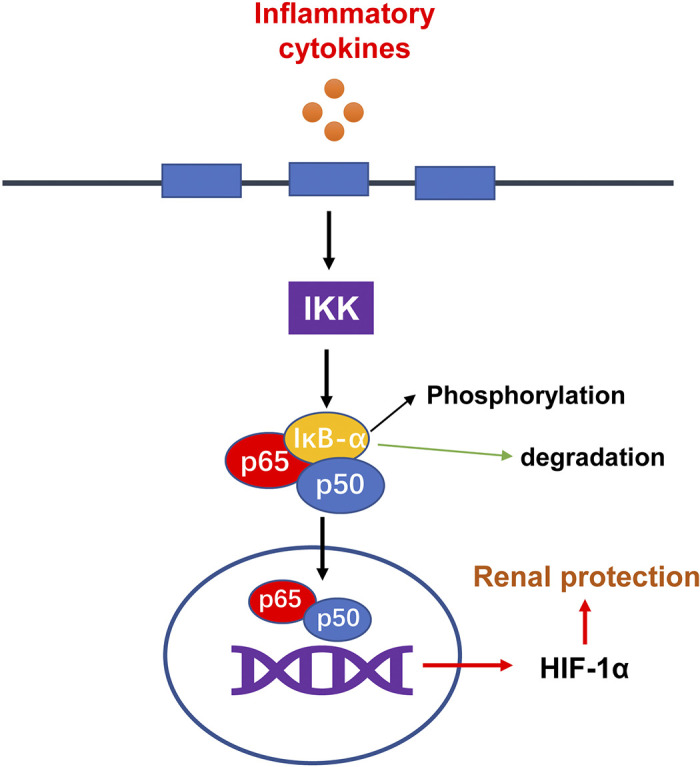
The NF-κB-hypoxia-inducible factor (HIF)-1 pathway in acute kidney injury. Inflammatory cytokines activate IκB kinase (IKK), leading to the degradation and dissociation of IκB from NF-κB. NF-κB then moves to into the nucleus to directly promote the transcription of Hif1a, which may protect renal tubular cells in acute kidney injury.
Despite these interesting findings and ideas, a number of questions remain open. First, in addition to HIF-1, HIF-2α is also activated in AKI, albeit in different cells. Does NF-κB regulate HIF-2α as well? In this study, Li et al. (5) just detected HIF-1 in AKI, and whether HIF-2 is subjected to NF-κB regulation is unknown. In this regard, He et al. (6) suggested that HIF-2α was indirectly regulated by NF-κB in AKI. They found that low-dose LPS pretreatment protected mice from subsequent ischemic or toxic AKI. Mechanistically, they demonstrated that low-dose LPS pretreatment activated NF-κB, which induced HIF-2α expression in renal tubular cells for renal protection (6). Second, the interaction between HIF and NF-κB in renal tubular cells was explored at only one time point in AKI models. It remains an open question as to how long this cross talk persists and whether there is a signal to diminish this cross talk. Third, this study mainly demonstrated that NF-κB can regulate HIF. Can HIF regulate NF-κB? In 2019, Pan et al. (7) reported that HIF-1 could regulate NF-κB in AKI. They showed that HIF-1α activated by delayed remote ischemic preconditioning conferred renoprotection against septic AKI in mice through inhibiting NF-κB activation. However, this is an indirect regulation, and whether HIF can directly regulate NF-κB in AKI remains unclear. Regardless, these studies suggest a possible interesting interaction between NF-κB and HIF in AKI, shedding new light on pathogenesis and suggesting novel strategies for AKI prevention and treatment.
GRANTS
Z.D. is a recipient of the Senior Research Career Scientist award from the United States Department of Veterans Affairs and is supported by grants from the United States Department of Veterans Affairs (BX000319 and BX005236) and the National Institutes of Health (DK058831 and DK087843).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.L., drafted manuscript; Z.D., edited and revised manuscript; Z.L. and Z.D., approved final version of manuscript.
REFERENCES
- 1.Tang C, Dong Z. Epigenetic regulation in acute kidney injury: new light in a dark area. Kidney Int 88: 665–668, 2015. doi: 10.1038/ki.2015.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shu S, Wang Y, Zheng M, Liu Z, Cai J, Tang C, Dong Z. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells 8: 207, 2019. doi: 10.3390/cells8030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, Tang C, He L, Yang D, Cai J, Zhu J, Shu S, Liu Y, Yin L, Chen G, Liu Y, Zhang D, Dong Z. The negative feedback loop of NF-kappaB/miR-376b/NFKBIZ in septic acute kidney injury. JCI Insight 5: e142272, 2020. doi: 10.1172/jci.insight.142272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12: 695–708, 2011. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Ji J, Wen Y, Cao J, Kharbuja N, Ni W, Yin D, Feng S, Liu H, Lv L, Liu B, Wang B. HIF-1α is transcriptionally regulated by NF-κB in acute kidney injury. Am J Physiol Renal Physiol 321: F225–F235, 2021. doi: 10.1152/ajprenal.00119.2021. [DOI] [PubMed] [Google Scholar]
- 6.He K, Chen X, Han C, Xu L, Zhang J, Zhang M, Xia Q. Lipopolysaccharide-induced cross-tolerance against renal ischemia-reperfusion injury is mediated by hypoxia-inducible factor-2alpha-regulated nitric oxide production. Kidney Int 85: 276–288, 2014. doi: 10.1038/ki.2013.342. [DOI] [PubMed] [Google Scholar]
- 7.Pan T, Jia P, Chen N, Fang Y, Liang Y, Guo M, Ding X. Delayed remote ischemic preconditioning confers renoprotection against septic acute kidney injury via exosomal miR-21. Theranostics 9: 405–423, 2019. doi: 10.7150/thno.29832. [DOI] [PMC free article] [PubMed] [Google Scholar]