

Abstract
We describe here for the first time a lipid‐binding‐domain (LBD) in p38γ mitogen‐activated protein kinase (MAPK) involved in the response of T cells to a newly identified inhibitor, CSH71. We describe how CSH71, which binds to both the LBD and the ATP‐binding pocket of p38γ, is selectively cytotoxic to CTCL Hut78 cells but spares normal healthy peripheral blood mononuclear (PBMC) cells, and propose possible molecular mechanisms for its action. p38γ is a key player in CTCL development, and we expect that the ability to regulate its expression by specifically targeting the lipid‐binding domain will have important clinical relevance. Our findings characterize novel mechanisms of gene regulation in T lymphoma cells and validate the use of computational screening techniques to identify inhibitors for therapeutic development.
Keywords: ATP‐binding pocket, computational virtual screening of chemicals, cutaneous T‐cell lymphoma, cytotoxicity, lipid‐binding domain, NMR spectroscopy, TCR signaling pathway, the MAP kinase p38γ, TNF alpha

Abbreviations
AA, amino acid (aa)
BMK1, ERK/big MAP kinase 1
CRE, CAMP‐responsive element
CSP, chemical shift perturbation
CTCL, cutaneous T‐cell lymphoma
DEF, docking site for ERK, FxF
DLGH1, Drosophila disc large tumor suppressor human
EAE, experimental autoimmune encephalomyelitis
eIF‐4E/MNK, eukaryotic translation initiation factor 4E/MAPK‐interacting kinase
ERK, extracellular signal‐regulated kinase
HD, healthy donors
HRD, His–Arg–Asp
HTVS, high‐throughput virtual screening
IP, immunoprecipitation
JNK/SAPK, c‐jun N‐terminal or stress‐activated protein kinases
LBD, lipid‐binding‐domain
LiVS, ligand virtual screening
MAPK, mitogen‐activated protein kinase
MKI, MAPK insert
MKK, MAP kinase kinases
NCI DTP, National Cancer Institute Developmental Therapeutics Program
NFAT, nuclear factor of activated T cells
PBMC, peripheral blood mononuclear cells
PDZ domain, refers to a shared domain (80‐90 aa) of three proteins PSD95, DLGH1, and ZO‐1 that were first discovered
PDZ‐binding motif, a motif that recognizes and interacts with PDZ domain
PSD95, postsynaptic density protein
STD, saturation transfer difference
TCR, T‐cell receptor
TFs, transcription factors
TLR5, Toll‐like receptor 5
UDScore, universal diversity score
ZO‐1, zonula occludens‐1 protein
The human p38 mitogen‐activated protein kinase (MAPK) family consists of p38α, p38β, p38γ, and p38δ. p38α and p38β are ubiquitously expressed, but p38γ and p38δ are expressed only in specific tissue types. The expression of these isoforms, and the phosphorylation of their diverse protein targets, activate many biological pathways. Sequence comparisons indicate that the p38 isoforms are about 60% similar to one another; they are also about 40%–45% similar to three other members of the MAPK family: extracellular signal‐regulated kinases (ERKs), c‐jun N‐terminal or stress‐activated protein kinases (JNK/SAPK), and ERK/big MAP kinase 1 (BMK1) [1].
p38 kinases are directly activated by dual kinase MAP kinase kinases (MKK), in particular MKK3 and MKK6, and they play key roles in many tissues under normal or stressful physiological conditions. Half of the substrates of p38 kinases are transcription factors (TFs), emphasizing their role in gene regulation at the transcriptional level.
A defining characteristic of each member of the p38 family is a conserved activation loop with a unique TGY (Thr180 and Tyr 182) [2, 3] dual‐phosphorylation motif that promotes the canonical p38 pathway. However, a T‐cell‐specific alternate p38 activation pathway exists via the phosphorylation of Y323 (p‐Y323) by the tyrosine kinase ZAP70. This pathway is essential for T‐cell activation [4] and differentiation [5].
p38γ is considered a promising therapeutic target for treating cutaneous T‐cell lymphoma (CTCL) [6] because its expression, which is typically undetectable in normal T cells, is increased in CTCL [6]. p38γ expression has been shown to support the proliferation of diverse cancers, including colon [7], prostate [8], esophageal [9], breast [10], and liver [11]. Because p38γ shares an ATP‐binding site with other MAP kinases, it is necessary to target its non‐ATP‐binding, or allosteric site, to specifically inhibit p38γ activity. Recently, the non‐ATP‐binding site of p38α, also described as a lipid‐binding domain, or lipid‐binding‐domain (LBD), has been thoroughly studied by several research groups [12, 13, 14].
A connection between the alternate activation pathway on Tyr 323 and the lipid‐binding domain was first made by studies of the autophosphorylation of p38 by the MAPK insert (MKI), which undergoes significant conformational changes upon ligand binding on the LBD [12, 15, 16]. Several molecular mechanisms of p38 autophosphorylation at T180 have been proposed, including dimerization [16, 17]. In this model, the phosphorylation of Y323 opens up a rigid region of peptides in the highly conserved HRD, followed by the auto‐monophosphorylation of T180 via the proximal p‐Y323 [17] of the other p38 monomer.
Most studies of alternative p38 activation have relied on cell‐free in vitro assays. One study [17] hypothesized that the rigid HRD, which is highly conserved among kinases but has an unclear function [16, 17, 18], becomes pliable before autophosphorylation occurs. This possibility is supported by the fact that a single‐point mutation that mimics the phosphorylation of p38α at Y323 (Y323T) causes inter‐lobe orientation changes. X‐ray crystallography further revealed p‐Y323‐directed inter‐lobe orientation changes as well as conformation changes on the phosphorylation lip (TGY180‐182 of p38α) that result in the formation of a docking site for ERK, FxF (DEF) pocket that serves as an additional docking site for other important substrates [16, 17].
p38β is also elevated in CTCL [6]. It is the only isoform capable of spontaneous auto‐monophosphorylation of the TGY motif in HEK cells overexpressing MKK3/6 [19]. The autophosphorylation of p38β at Thr180 is generated by a short region composed of α ‐G Helix and MKI, the latter of which is a major motif of the lipid‐binding domain (LBD) of p38α/β [15, 20, 21]. The finding is intriguing; however, the study was conducted in the HEK cell line, which lacks ZAP70 expression and has no TCR signaling pathway. Therefore, the spontaneous autophosphorylation observed must be due to allosteric effects of the classical p38 activation pathway, rather than the alternative activation pathway triggered by the phosphorylation of Y323 by ZAP70.
Direct protein–protein interactions between p38γ and other proteins, such as the cytoskeletal protein tyrosine phosphatase PTPN3 [22] (a negative regulator of early T‐cell receptor (TCR) signal transduction and T‐cell activation [23]) and nuclear factor of activated T cells 4 (NFATC4) [3], have been previously reported. We have proposed that p38γ interferes with TCR signaling by interacting with Drosophila disc large tumor suppressor human (DLGH1) and subsequently up‐regulating NFATC4 via a unique PDZ‐binding domain in p38γ [24]. Although we have previously reported nuclear p38 [25], its function remains elusive.
The NFAT family of proteins consists of five distinct members in humans: NFAT1 (NFATC2), NFAT2 (NFATC1), NFAT3 (NFATC4), NFAT4 (NFATC3), and NFAT5 [26]. They are TFs that modulate important functions in diverse tissues with cell‐type specificity. Several studies have concluded that NFATs mediate the activation‐induced cell death (AICD) of T and B cells and therefore serve as tumor suppressors in lymphoid cells [27]. NFATs modulate TNF expression through several NFAT‐binding motifs in the promoter region of TNF gene [28]. NFATs also interact with ERK though their FxF motif, which docks in the ERK DEF site [29].
To better study the effect of ligand binding to the LBD/MKI of p38 on its auto‐monophosphorylation activity and biological function, we devised a cell‐based system using a CTCL cell line with a fully functional ZAP70 kinase capable of phosphorylating either Y323 (in p38α/β) or Y326 (in p38γ) and in which TCR signaling is intensively activated. We confirm the presence in p38γ of a lipid‐binding domain similar to that in p38α and study the effect of ligand binding to this region. Finally, we conducted virtual ligand screening of 270 000 proteins to identify a candidate molecule that is selectively cytotoxic to CTCL cells.
Our study reveals the molecular mechanism of the conformational change that p38 undergoes in response to drugs targeting its lipid‐binding domain, that is, the opening of the DEF pocket to accommodate substrates such as the TFs that modulate TCR signaling. It also suggests pathways of novel drug development for the treatment of CTCL.
Experimental procedures
NMR titration and chemical shift perturbation methods (2D NMR) to uncover the LBS on p38γ
2D; Ileδ1‐[13CH3]; Leu, Val‐[13CH3, 12CD3]‐labeled p38γ sample was prepared, and the NMR spectra were collected and analyzed as described [6]. In the complex sample, 100 μm p38γ was added stepwise with β‐OG or CSH71 to a molar ratio of 1 : 10. NMR chemical shift perturbation (CSP)s were calculated as.
where Δδ H, Δδ N, and Δδ C are the chemical shift differences between the free and bound states in the proton, nitrogen, and carbon dimensions, respectively.
CSP normalization method
The CSP for each compound was normalized separately. The values in each dataset (different compounds) were between 0 and 1. CSPs were calculated and further normalized using the min‐max method using the following equation: normalized CSP = (CSP − CSPmin)/(CSPmax − CSPmin); where CSPmax and CSPmin are the largest and smallest CSPs in each titration of the compound.
Virtual ligand screening targeting the LBD
The LiVS pipeline was performed as described previously [30] using Glide software (Schrödinger). Briefly, the Glide high‐throughput virtual screening (HTVS) mode, which is fast but less accurate, was first implemented to dock the entire NCI DTP library (270 000 compounds) onto the LBD of p38γ. The 10 000 top‐ranked compounds were further docked and scored using Glide standard precision (SP) mode. Next, the 1000 top‐ranked compounds from SP docking were redocked and rescored using the Glide extra precision (XP) mode, and were further analyzed, filtered, and narrowed down to 100 compounds using Lipinski's rule of five [31] for drug‐likeness, high‐throughput screen frequent hitters (PAINS) [32], and protein reactive chemicals such as oxidizers or alkylators (ALARM) [33]. Molecular diversity was maximized by using UDScore (Universal Diversity Score), our in‐house‐developed compound library diversity score to measure library diversity, a universal score independent of library size.
Cell culture and screening NCI compounds to determine cytotoxicity in Hut78
Hut78 cells cultured as previously described [6], were used to validate selected compounds. Eighty candidate compounds were obtained from the NCI DTP Open Chemical Repository. To determine the cytotoxic effect of each compound on cell viability in Hut78 cells, CellTiter‐Glo assays were used (Promega, Madison, WI, USA). We seeded 7000 cells/well when using a 96‐well plate and cells were collected at 2 time points, 48 and 96h treatments, respectively. IC50 values were determined using CalcuSyn software (Biosoft, Cambridge, UK), as described previously [6]. Each experiment was performed in triplicate.
Kinase assay with p38 isoforms in vitro
To characterize the selected small molecules that inhibited p38γ, we performed kinase assays in vitro using an ADP‐Glo kit (Promega), as described previously [6]. In detail, we followed the protocol according to the manusfacture production. the active kinase forms of p38 isoforms (α, β, γ, and δ) were obtained from SignalChem and incubated with increasing doses of selected small molecules. A synthetic peptide substrate (IPTTPITTTYFFFKKK) was added to the mixture at a final concentration of 0.2 µg·µL−1, followed by 10 µm ATP. The luminescent ADP‐Glo kinase assay measures ADP produced from ATP consumption in the reaction. Staurosporine was used as a positive control. Luminescence was monitored at an integration time of 0.5 s using an automated BMG PHERAstar plate reader (BMG Labtech, Cary, NC, USA). Each experiment was performed in triplicate.
NMR and competition saturation transfer difference experiment
Inactive p38γ protein was expressed in E. coli as described previously [6]. The compounds identified using LiVS were prepared as stock solutions in DMSO‐d6 and stored at −20 °C. Stock solution concentrations were determined using NMR with a known concentration of the reference compound sodium trimethylsilylpropanesulfonate (DSS). The buffer for NMR experiments was composed of 15 mm Tris‐d11 dissolved in D2O, TCEP 1 mm, MgCl2 2 mm, and TSP‐d4 50 μm; buffer pH was 7.0. NMR saturation transfer difference (STD) [19] experiments were carried out at 25 °C on a 700 MHz Ascend magnet (Bruker) equipped with a 5‐mm triple resonance cryogenic probe (spectral width 14 ppm, with 32k data points). The saturation frequency was set at −0.2 ppm, and the reference experiment frequency was set at −30 ppm. The 50 ms gauss pulse saturation train was 3.8 s long with field strength of 86 Hz. The T2 filter spin‐lock was 80 ms long with field strength of 4960 Hz. The total number of scans was 5120, and the saturation and reference experiment were acquired in an interleaved manner using Bruker stddiffgp19.3 pulse sequence. The data were analyzed using Bruker TopSpin 3.1 software. The STD was calculated using (Iref – Isat)/Iref, where the Iref and Isat are the peak intensity in reference and saturated spectrum, respectively. The STD error was estimated from the noise intensity of the spectrum in the range between 9.5 and 11 ppm. The sample prepared for STD measurement of CSH71 was composed of 1 µm p38γ, 50 µm CSH71, 2% DMSO‐d6. For the competition STD study, an additional 100 µm ATP was added to the sample. We also noticed that there were some residual STD values from free CSH71 under the conditions of the STD experiments used for the protein ligand complex. We obtained our final reported STD values for CSH71 in complex with protein by subtraction of the residual STD values of the free CSH71 sample obtained under the same conditions.
Microarray analysis and RNA sequencing analysis
The Affymetrix GeneChip Human Gene 1.0‐ST array (Affymetrix, Santa Clara, CA, USA) was used to define gene expression profiles from the samples. Synthesis and labeling of cDNA targets, hybridization, and scanning of GeneChips were carried out by the Integrative Genomics Core Facility at City of Hope. Briefly, cRNA was generated according to the manufacturer's protocol using Affymetrix's GeneChip Whole Transcript Sense Target Labeling Assay Hybridization cocktails containing 5.5 µg of fragmented, end‐labeled cDNA were prepared and applied to GeneChip Human Gene 1.0 ST arrays. Hybridization was performed for 16 h, and the arrays were washed and stained with a GeneChip Fluidics Station 450 using FS450_0007 script. Arrays were scanned at 5 μm resolution using Affymetrix GCS 3000 7G.
Raw intensity measurements of all probe sets were background‐corrected, normalized, and converted into expression measurements using Affymetrix’s Expression Console v1.1.1. The Bioconductor “ArrayTools” package was then used to identify the genes differentially expressed between the treated and untreated samples. Significant genes were selected with a cutoff of adjusted P < 0.05 and fold change of 2. The DAVID functional annotation tool https://david.ncifcrf.gov/content.jsp?file=citation.htm ) was then used to identify modulated KEGG signaling pathways.
Immunoprecipitation and western blot analysis
Immunoprecipitation with beads
Antibodies were added to protein lysates at recommended dilutions and incubated at 4 °C overnight with rotation. Magnetic beads (Dynabeads protein G, #10004D Life Technologies, Waltham, MA, USA) were washed with lysis buffer then added to the lysate‐antibody mix. The bead‐antibody‐lysate mixtures were left to incubate at room temperature for 60 min with rotation. Beads were washed with cold PBST 4 times and subsequently resuspended in loading buffer and boiled for 5 min. The samples in loading buffer were separated from beads and moved to a fresh clean tube. Western blot (WB) analysis was performed as described previously [6]. Primary antibodies purchased from Cell Signaling Technology (Danvers, MA, USA) were used at the following dilutions: p38α (#9218, 1 : 1000), p38β (#2339, 1 : 1000), p38δ (#2308, 1 : 1000), p38γ (#2307, 1 : 1000), p‐p38 T180/Y182 (#4511, 1 : 1000), NFACT C1 (#8032, 1 : 1000), Lamin A/C (#4777, 1 : 5000), and GAPDH (#2118, 1 : 5000). Primary anti‐SAPK3 (ab205926) was purchased from abcam (Cambridge, MA, USA) and used at 1 : 1000 dilution. Primary p‐p38 Y323 antibody (#12322‐1) was purchased from Signalway Antibody and used at 1 µg·mL−1. Primary anti‐PTPN3 (#NBP2‐78797, 1 : 1000) was purchased from Novus, Goat anti‐rabbit HRP‐linked antibody (GE #NA934, 1 : 2500) was used as secondary antibody.
Nuclear and cytosolic protein extraction
Nuclear and cytosolic proteins were extracted from cells using the NE‐PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher 78833, Waltham, MA, USA). Briefly, fresh cells were pelleted and washed with cold 1x PBS, followed by resuspension in CER I buffer by vortexing. After a 10‐min incubation on ice, CER II buffer was added to the mixture, which was then pelleted via centrifugation. After centrifugation, the supernatant (containing cytoplasmic proteins) and pellet (containing nuclei) were separated by pipetting. Similarly, nuclear proteins were extracted from pellets by adding NER buffer and performing vigorous vortexing and subsequent centrifugation. Supernatants were collected as nuclear protein lysates. Protein lysates were quantified using the BCA assay, mixed with loading dye containing β‐mercaptoethanol, and boiled for 5 min on a hotplate.
Confocal fluorescence microscopy
Materials and methods of Immunofluorescence staining and microscopy with Hut 78 cells followed instructions as previously described [34]. For DLGH1 staining, we used antibody against Goat‐DLGH1, (Invitrogen, Waltham, MA, USA, Cat. #PA5‐18790) and detected using Texas red conjugated donkey anti‐goat IgG (red) (ThermoFisher, Cat. #PA1‐28662); for alternative p38 activation detection, we stained cells with Rabbit antibody against phosphor‐p38 at Y323 (ThermoFisher, Cat. # PA5‐40258), Donkey anti‐Rabbit 650 Dylight (abcam, ab96894) was then used as secondary antibody. For IL17RA staining, we chose mouse antibody IL17RA (R & D, Minneapolis, MN, USA, MAB4481) as primary antibody, and FITC conjugated Goat anti‐mouse as secondary antibody. Stained cells were washed and mounted with the mounting medium Vectashield (Vector Laboratories, Burlingame, CA, USA). The cells were counterstained with 4‐6‐diamidino‐2‐phenylindole (DAPI) (blue) and analyzed by fluorescence microscopy with a confocal scanning microscope (Zeiss LSM 510 META or Zeiss LSM 700, Oberkochen, Germany) with a × 63 or ×100 objective. Confocal laser scanning microscopy was performed at Light Microscopy Shared Resource Facility of City of Hope.
Statistical analysis
Data are expressed as the mean ± SD. The statistical significance of differences was analyzed using Student's t‐test, where P < 0.05 was considered significant.
Results
Conservation of the lipid‐binding domain among p38 isoforms
It is well known that p38α harbors a lipid‐binding domain (LBD) that exerts allosteric functions on its ATP‐binding pocket [12, 17]. We compared amino acid sequences in the lipid‐binding domain among all four p38 isoforms and found the site to be highly conserved in the p38 family (Fig. 1A). The sequence and structural similarities between the p38γ and p38α isoforms suggest that p38γ may also contain a lipid‐binding domain.
Fig. 1.
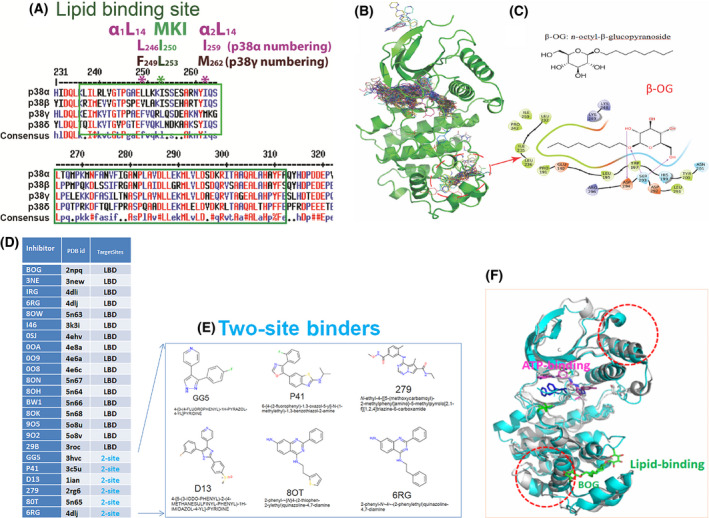
Lessons we learnt from p38α on LBD. (A) Consensus amino acids sequences alignment diagram in the lipid‐binding domain among four p38 isoforms by clustalW2. MARP insert harbors LBD which is between two αL14 helixes, α1L14 and α2L14. L246 and I259 are two conserved residues of p38α in two L14 loops that indicated in *. Likewise, the counterparts of above two residues in p38 γ are F249 and M262 that are conserved. (B) structures of ligands bound to p38α from 253 PBD entries that have X‐ray Crystallography structures. Most of the ligands are single‐site binders, either on the ATP‐binding site or the lipid‐binding site, while some ligands bound to two sites simultaneously. Circled is the area β‐OG bound at the lipid‐binding pocket. (C) Chemical structure of β‐OG (n‐octyl‐β‐glucopyranoside, top) and 2‐dimensional (2D) interaction diagram of β‐OG in complex with p38α. The hydrophobic tail of β‐OG is surrounded by hydrophobic amino acids of p38α (bottom). (D) 17 small ligands of the p38α solely target lipid‐binding domain (LBD) and 6 small molecules targets both LBD and ATP binding pocket (data summarized from X‐ray crystallography). The column of TargetSites shows the number of sites for a ligand; 2‐site (teal color) means that the inhibitor binds at both the lipid‐binding site and ATP‐binding site simultaneously, and LBD indicates the inhibitor can only bind LBS site of p38α. Only one PDB id is listed for each small molecule. (E) Two site binders of p38α with X‐ray crystallography structure consist of 6 molecules: GG5 (PBD: 3hvc); p41 (PBD: 3c5u); D13 (PBD: 1ian); 279 (PBD: 2rg6); 80T (PBD: 5n65); and 6RG (PBD: 4dlj) are ligands for two sites, that is, the ATP binding and the lipid‐binding of p38α. (F) β‐OG lipid binding of p38α may cause allosteric effects on ATP binding. We superimposed two public PBD datasets from RSCB, the DFG‐out conformation of p38α alone (PDB 2baj) is displayed as cyan cartoons; and the inactive p38α bound β‐OG as grey cartoons (PDB 3gcv). When β‐OG bound, the structure is changed significantly near both the lipid‐binding and ATP‐binding sites (depicted as dotted red circles), which suggests that the binding of β‐OG at the lipid‐binding site causes conformational changes near the ATP‐binding site and thereby affect its kinase activity allosterically.
The lipid‐binding domain in p38α was so named because the first small molecule discovered to bind to this site was the lipid‐like detergent n‐octyl‐beta‐glucopyranoside (β‐OG, Fig. 1B,C). We confirmed that β‐OG was among 17 small molecules (Fig. 1D) that exclusively bind the LBD of p38α by comparing 253 publicly available X‐ray crystallographic structures of p38α‐compounds deposited in the RCSB Protein Data Bank (PDB). The chemical structure of β‐OG (Fig. 1C, top) and its 2D interaction with p38α (Fig. 1C, bottom) depict the hydrophobic tail of the chemical surrounded by hydrophobic residues of p38α. In addition to the 17 molecules that bind only the LBD, six molecules—GG5 (PBD: 3hvc), p41 (PBD: 3c5u), D13 (PBD: 1ian), 279 (PBD: 2rg6), 80T (PBD: 5n65), and 6RG (PBD: 4dlj)—bind to two sites on p38α: the ATP‐binding site and the lipid‐binding domain (Fig. 1D–E). The biological functions of these two site‐binding molecules are still unclear [11, 12, 13, 14, 35, 36, 37, 38].
p38α also contains a Docking site for ERK via motif of ERK – FXFP, or DEF site near the LBD [16]. We hypothesized that ligand binding to the LBD allosterically triggers the opening of the nearby DEF pocket to accommodate the binding of diverse substrates and render a spectrum of biological effects. To test this hypothesis, we showed that molecules that bind LBD allosterically alter the structure of the ATP‐binding site, as evidenced by the crystal structures of p38α in complex with β‐OG (Fig. 1F, PDB: 2npq).
Demonstration of an LBD on p38γ using NMR
To determine whether p38γ also has an LBD, we used two‐dimensional NMR CSP, or chemical shift perturbation, to investigate the interaction between β‐OG and p38γ. Two‐dimensional NMR spectroscopy allows the study of site‐specific interactions with mm binding affinities under near‐physiological conditions, and it reveals structural information that may otherwise be difficult to obtain by X‐ray diffraction. It can rapidly identify binding sites on target proteins, as well as conformational changes triggered by compound binding.
We monitored the chemical shift perturbation of p38γ in both 1H‐15N HSQC and 1H‐13C HMQC spectra alone, and in the presence of β‐OG (Fig. S1A,B). We found that residues with significant NMR CSPs (magenta) were localized primarily around the MKI motif (CSP cutoff for amides at 0.02 ppm (Fig. S1C) and methyls at 0.03 ppm (Fig. S1D)). Figure 2A summarizes both spectra NMR CSP and indicates those residues that are greatly perturbed by β‐OG (orange). Figure S1E shows that all our NMR CSP procedures have titrated molar ratios, β‐OG (the variable) to p38γ (the constant). Ratios (p38γ/ β‐OG) are 1 : 0 (red, no β‐OG as control), 1 : 2.5 (yellow), 1 : 5 (green), 1 : 7.5 (cyan), and 1 : 10 (blue). Based on these results, we generated a three‐dimensional structure of β‐OG bound to the LBD of p38γ (Fig. 2B) using p38γ crystallography structure coordinate datasets downloaded from RCSB PDB [PDB id 1cm8 [39]]. The NMR titration data indicate specific binding of β‐OG to p38γ in the LBD.
Fig. 2.
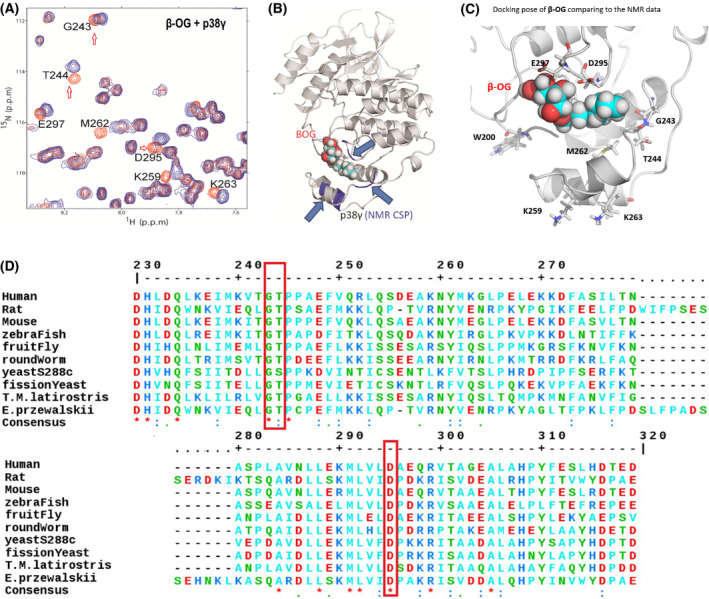
NMR demonstrates a lipid‐binding site on p38γ for β‐OG. (A) 2D NMR CSP experiment locates the p38γ residues that significantly shifted due to addition of β‐OG, indicated in orange. NMR CSPs data by monitoring 1H‐15N HSQC and 1H‐13C HMQC spectra upon the addition of β‐OG alone and p38γ to β‐OG with a molar ratio of 1 : 10. The red hollow arrows indicate three residues, G243, T244, and D295 that are conserved among species from human to yeast (See also Figure 2D, red bracket). (B) The structures of p38γ in complex with β‐OG. A 3D structural model presenting where β‐OG bound to LBD of p38γ, based on NMR CSP titration data. The p38γ crystallography structure coordinate datasets were downloaded from RCSB PDB [PDB id 1cm8 [39]]. (C) β‐OG binds p38 γ in the MKI region of in the lipid‐binding domain and residues which greatly perturbed residues in NMR CSP assay are W200, M262, G243, D295, and E 297. K259 and K263 are consistent with that of alpha p38 which are perturbed by B‐OG in Figure 1F. (D) ClustalW2 consensus alignment diagram of amino acid sequences in the lipid‐binding domain of p38γ among indicated species. β‐OG NMR CSP perturbed residues of human p38γ: G243, T244, and D295 (in red brackets) are conserved among species including yeast. Three residues E297, K259, and M262 share homology with mouse.
Our results indicate that LBD‐bound β‐OG perturbs several nearby residues, including W200, M262, G243, D295, and E297 (Fig. 2A,C). Two of these residues are among three (indicated in Fig. 2A by red arrows) that are highly conserved from yeast to humans (Fig. 2D), suggesting an important function for the LBD in p38γ. Taken together, our results demonstrate for the first time that p38γ contains an LBD for the ligand β‐OG.
Identification of compounds that bind p38γ LBD using virtual ligand screening
To identify additional compounds that bind to the LBD of p38γ and thus could serve as potential inhibitors of the enzyme, we implemented an in‐house‐developed Ligand Virtual Screening (LiVS) pipeline [30] to screen the NCI DTP library (270 000 compounds) in silico. LiVS has been demonstrated to be a powerful tool for novel drug screening. We used LiVS to dock small molecule compounds from the National Cancer Institute Developmental Therapeutics Program (NCI DTP) compound library to the LBD and assess their biological effects.
Computational Druggable Site Prediction [30] identified the p38γ LBD predicted by NMR as a top druggable site. We superimposed the LBD of p38γ onto the known crystal structure of inactive, non‐phosphorylated p38α (PDB 3new) and tested a selection of FDA‐approved drug probes using Druggable Site Prediction (DSP) [30]. We found that approximately 23% of probes bound the LBD as one of their top preferential docking sites (Fig. 3A, Site2, MaxGScore = −8.9 kcal·mol−1), whereas approximately 41% preferentially bound the ATP‐binding site (Fig. 3A, Site1, MaxGScore = −9.82 kcal·mol−1). Note that MaxGScore is a measure of a ligand’s binding affinity to the target protein: the lower the score, the higher the binding affinity. Approximately 7% of the probes preferentially bound to Site3 (MaxGScore = −7.76 kcal·mol−1), which connects to Site2 as one site that appears as a tunnel (Fig. 3A).
Fig. 3.
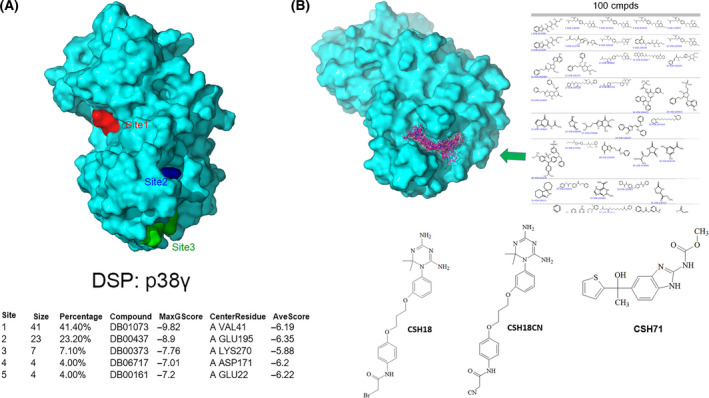
Development of inhibitors targeting the lipid‐binding site of p38γ. (A) The three best druggable sites on p38γ are located using Druggable Site Prediction (DSP) method where FDA‐approved drug probes prefer to bind (Site 1 in red, Site 2 in blue, and Site 3 in green). The table at the bottom shows that 41% of FDA drug probes prefer to bind at the ATP‐binding site (Site 1, MaxGScore of −9.82 kcal·mol−1), with 23% of FDA drug probes bound to Site 2, (MaxGScore of −8.9), the second‐preferentially druggable site on p38γ which is the lipid‐binding domain. Both Site 2 and Site 3 cover the lipid‐binding domain of p38γ. (B) An illustration of multiple steps to develop inhibitors bound to the lipid‐binding site of p38γ, which includes Docking of the NCI DTP compound library (270,000 compounds) to the lipid‐binding site of p38γ (PBD:1cm8, top left); 100 compounds were selected using virtual ligand screening (top right); 80 available compounds were ordered from NCI DTP for cytotoxicity assays; two compounds, CSH18 (NSC109833) and CSH71 (NSC381863) were selected due to their cytotoxicity against Hut78 CTCL cells, with IC50 = 3.4 µm and 33 nm, respectively. As CSH18 is a speculated alkylator, we synthesized CSH18CN, which structure is depicted in the middle (Bottom).
Next, we performed virtual ligand screening using Glide docking of the compounds in the NCI DTP library onto p38γ. We used LiVS to screen small molecules with high affinity for p38γ, followed by comprehensive score measurement for the diversity of the library. Using virtual screening computation techniques, we narrowed down the 270 000 compounds in the full NCI DTP library to 10 000, then 1000, and finally 100 compounds with the highest affinity for p38γ. Figure 3B (top right) illustrates a portion of the top 100 matches docked with the LBD of p38γ. Finally, we compared docking scores and performed structural diversity analysis of the 100 compounds to identify 80 candidates, which we obtained from NCI DTP for further experimental validation.
Biological validation of CSH71 and CSH18CN using cytotoxicity measurements in CTCL cells
We treated Hut78 CTCL cells with increasing doses of each of the candidate compounds for 72 h to determine whether any were cytotoxic against CTCL. Of the 80 compounds computationally predicted to bind to the p38γ LBD, two—CSH18 (NSC109833) and CSH71 (NSC381863)—exhibited drastically different structures (Fig. 3B, bottom). Both were cytotoxic to Hut78 cells, with IC50 = 3.4 µm and 33 nm, respectively (Table 1). Mass spectrometry indicates that CSH71 from NCI DTP is a mix of three fractions (Fig. S2A). Here, we focused on Fraction 3 which is pure CSH71 (Fig. S2B). We also synthesized CSH71 commercially. CSH18 (NSC109833) is predicted to be an alkylator, therefore, we replaced the ‐Br group with –CN (CSH18CN, Fig. 3B, bottom), which we also had commercially synthesized. The purity of both commercially synthesized compounds is over 95% (data provided upon request).
Table 1.
IC50 (µm) of Hematopoietic cell lines that are sensitive to CSH18 and CSH71.
| CSH18 | CSH71 | ||
|---|---|---|---|
| Hut78 | 3.4 | 0.033 | CTCL |
| HL60 | 3 | 0.73 | AML |
| MOLT4 | 0.24 | 0.29 | ALL |
| K562 | 0.54 | 0.27 | CML |
| CCRF‐CEM | 0.1 | 0.05 | AML |
| SR | 0.11 | 0.27 | Lymphoma |
| RPMI‐8226 | 7.8 | 4.8 | MM |
Pure CSH71 showed cytotoxic effects in Hut78 cells, with IC50 = 2.2 µm at 72 h (Fig. S3A). In comparison, β‐OG is cytotoxic to Hut78 cells at IC50 = 1.76 mm. Although CSH71 and CSH18CN are each cytotoxic to Hut78 cells (Fig. 4A. 48 h; and B. 96 h, top), peripheral blood mononuclear cells (PBMC) from healthy donors (HD)—in which p38γ expression is undetectable—were unaffected even at high doses.
Fig. 4.
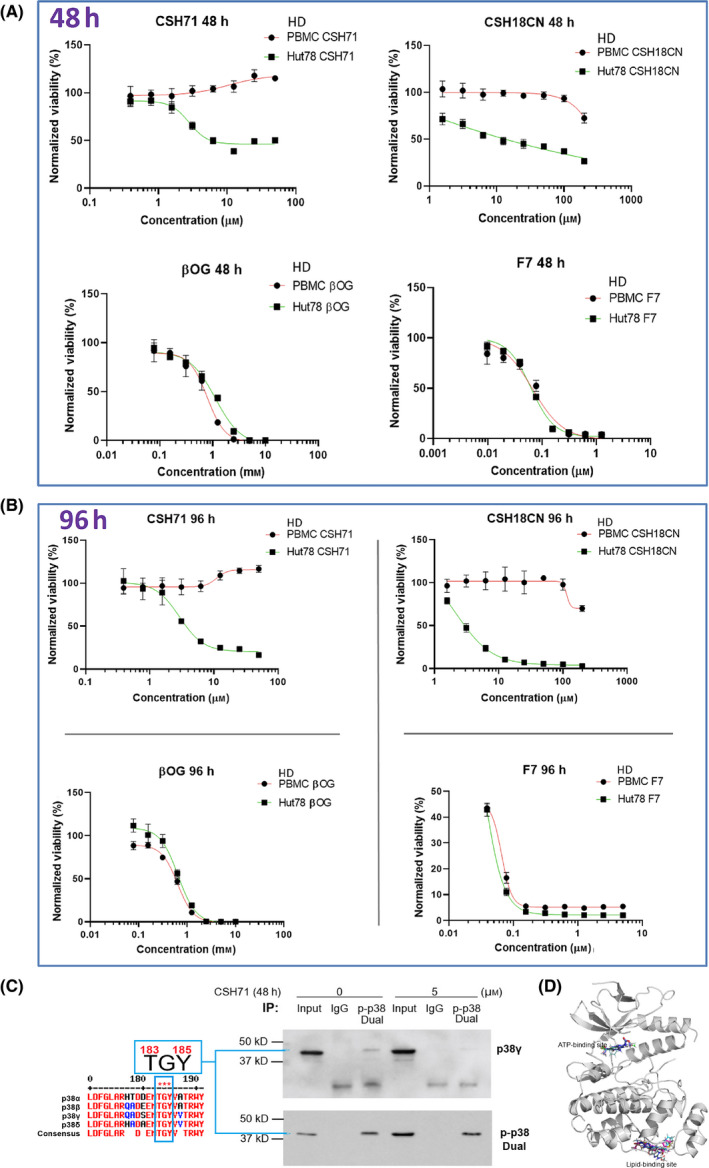
Cytotoxicity measurements of pure CSH71 and CSH18CN in CTCL cells. (A and B) Cytotoxicity assays and IC50s determination of newly synthesized compounds which are considered as two site binders CSH71 and CSH18CN Hut78 CTCL cells treated with increasing doses, respectively, for 48 h (A.) and 96 h. (B). F7 and β‐OG, served as one‐site binder, ATP binding and lipid‐binding of p38γ, respectively. PBMC from healthy donors (HD) used as a control as its p38γ level is at undetected level. Rest of text we focus on CSH71 for further analysis of its biological activity due to its interaction with the p38γ LBS. (C) Enrichment of dual‐phosphorylated p38 by Immunoprecipitation (IP). Sequence of p38 isoforms that all contain TGY motif dual activation site T180/183 GY182/185 (for p38γ the motif is T183‐G‐Y185, all other isoforms are T180 and Y182, Left). Right: IP results, first pulling down with dual‐p‐p38 of p38 antibody, then detect with p38γ and dual‐p p38 antibody, respectively, in cytosol of Hut78 cells both untreated (o µm) and CSH71‐treated (5 µm). The blue arrow points to p38γ that contains dual phosphorylation p‐p38 at T183‐G‐Y185. (D) CSH71 has only two predicted binding sites of p38γ by all around docking pose commutating analysis; and the MaxGScores of ATP‐binding and lipid‐binding, are −6.82, and −6.18 kcal·mol−1, respectively, which also indicates the binding affinity of CSH71 in LBD is much weaker than that of β‐OG with a MaxGScore of −8.9 kcal·mol−1.
As described above, β‐OG binds only to the LBD of p38γ. To investigate the effects of molecules that bind solely to the ATP‐binding site of p38γ, we used the multi‐kinase inhibitor F7/PIK75, which we have previously shown binds to the ATP‐binding site in p38γ. Interestingly, unlike CSH18 and CSH71, which bind two sites on p38γ, F7 (which targets only the ATP‐binding site) and β‐OG (which targets only the LBD), are cytotoxic to both healthy PBMC and Hut78 cells (Fig. 4A 48 h and B 96 h, bottom).
In vitro kinase activity assays in the presence of increasing concentrations of CSH71 up to 200 µM indicate it has no effect on p38γ kinase activity (Fig. S3D)— further suggesting that the kinetics of two‐site‐binding ligands differ from those that recognize only the ATP‐binding site. Further investigation on other p38 isoforms confirmed it also does not inhibit their kinase activity (Fig. S3C).
There are four isoforms of human p38, all of which contain a conserved motif (“TGY motif”) in each activation loop, regardless of its position—that is, whether it is positioned as dual phosphorylation of Thr180‐G‐Tyr182 (in p38 α, β, and δ) or Thr183‐G‐Tyr185 (in p38γ)—as the length of polypeptides of p38 isoforms are slightly different (See Fig. 4C left). We reasoned that the antibody against dual phosphorylation p‐p38 (CST # 4511) can recognize all dual phosphorylation combinations of this motif in all four p38 isoforms.
To validate the concept that p38γ contains a p‐p38 dual phosphorylation site positioned at Thr183‐G‐Tyr185, we first performed an immunoprecipitation (IP) study by pulling down all dual‐phosphorylated p38 with antibodies against p‐p38 dual phosphorylation (CST #4511) and then detected p38γ among the immunoprecipitated dual‐phosphorylated p38 protein using a total p38γ antibody (CST #2307). By comparing that with isotope IgG control (Fig. 4C right, immunoprecipitation, IP experiment), we concluded using this indirect approach (i.e. IP) that the dual phosphorylation‐enriched p38 contains p38γ, in which the dual phosphorylation sites are positioned at Thr183 and Tyr185 based the above background information.
Computational analysis of the interaction of CSH71 with p38γ indicates that it binds to the ATP‐binding site as well as the LBD, with MaxGScores of −6.82, and −6.18 kcal·mol−1, respectively. Therefore, the binding affinity of CSH71 to p38γ is much weaker than that of β‐OG to the LBD (MaxGScore = −8.9 kcal·mol−1, Fig. 4D).
For decades, kinase inhibitor drug development has focused on blocking the molecule’s ATP‐binding site. Our data deliver an important novel message that simultaneously targeting both the lipid‐binding domain and ATP‐binding site may be a more effective and specific approach.
Demonstration of CSH71 binding to p38γ by 1D NMR STD
Saturation transfer difference is an NMR method based on the phenomenon that proton saturation on a protein can be transferred to an interacting ligand. The STD monitors the ligand proton peak intensity difference in the presence and absence of saturation on protein protons. STD is suitable to detect interaction with ligands with dissociation constants KD ranging from 10‐3 to 10‐8 M, and, because the region of the ligand that interacts most closely with the target protein shows the largest peak intensity changes, it is also a useful way to identify and map ligand binding epitopes. The extent of variation of STD values within the ligand is used to determine the specificity of the interaction [40].
To further understand the nature of CSH71 binding to p38γ, we performed STD NMR on the CSH71‐p38γ complex alone and in the presence of increasing concentrations of ATP (Fig. S4A,B) to determine whether ATP competes with CSH71 for binding to the p38γ ATP‐binding pocket (Fig. 4D). STD values varied from approximately 56% to 59% among the aromatic peaks of CSH71 in the absence or presence of ATP, respectively. These observations suggest that the interaction is not due to random binding [40, 41] of CSH71 with p38γ. The docking pose at the ATP‐binding site closely resembles the NMR STD result—with the five‐ring group inside the pocket, while the—COOCH3 tail of CSH71 remains outside the ATP pocket (Fig. S4A, top and Fig. S4C, left). CSH71 binding to the LBD pocket is more flexible, however (Fig. S4A, bottom and Fig. S4C, right). These results are consistent with a model in which CSH71 binds first to the ATP‐binding site, as predicted both computationally and by our NMR 1D result (in the absence of ATP).
To mimic conditions in living cells, we performed ATP replacement experiments using STD NMR on the CSH71‐p38γ complex in the presence of increasing concentrations of ATP. Although the high‐affinity binding of ATP displaces CSH71 from the ATP‐binding site, its addition does not reverse the changes of aromatic peaks of CSH71 on p38γ observed as the stable STD value (Fig. S4B), again indicating that CSH71 binds more than one site on p38γ.
Demonstration of CSH71 binding to p38γ at LBD by 2D NMR CSP
To more precisely locate the p38γ residues bound by CSH71, we performed 2D NMR chemical shift perturbations (CSP) titration experiments using CSH71. In 1H‐15N correlation spectra (amides), CSH71 resulted in significant CSPs on the residues V33, G39, A40, V41, Y326 (colored red, CSP > 0.02 ppm); disappeared residues F111 and G117 are shown as magenta sticks (CSP > 0.02 ppm; Fig. S5A,C). The disappearance of certain residues suggests tight binding of CSH71 to p38γ.
To compare independent NMR CSP results between two different chemicals—CSH71 (which binds to two sites on p38γ, blue round dots) and β‐OG (which binds only to the LBD, green triangles)—we normalized the 1H‐15N NMR CSP results for CSH71, and overlaid the results with those of β‐OG (Fig. 5A). The shifted residues in the ATP‐binding region induced by CSH71, with a cutoff value of 0.5 of normalized CSP value, are V33, G39, A40, V41, and K69. The shifted residues in the LBD induced by CSH71 are T244, I276, and E312 (Fig. 5A, blue round dot). β‐OG, in contrast, shifts residues solely in the region of the LBD: G264, T244, M262, D295, I259, and D294 (Fig. 5A, green triangles).
Fig. 5.
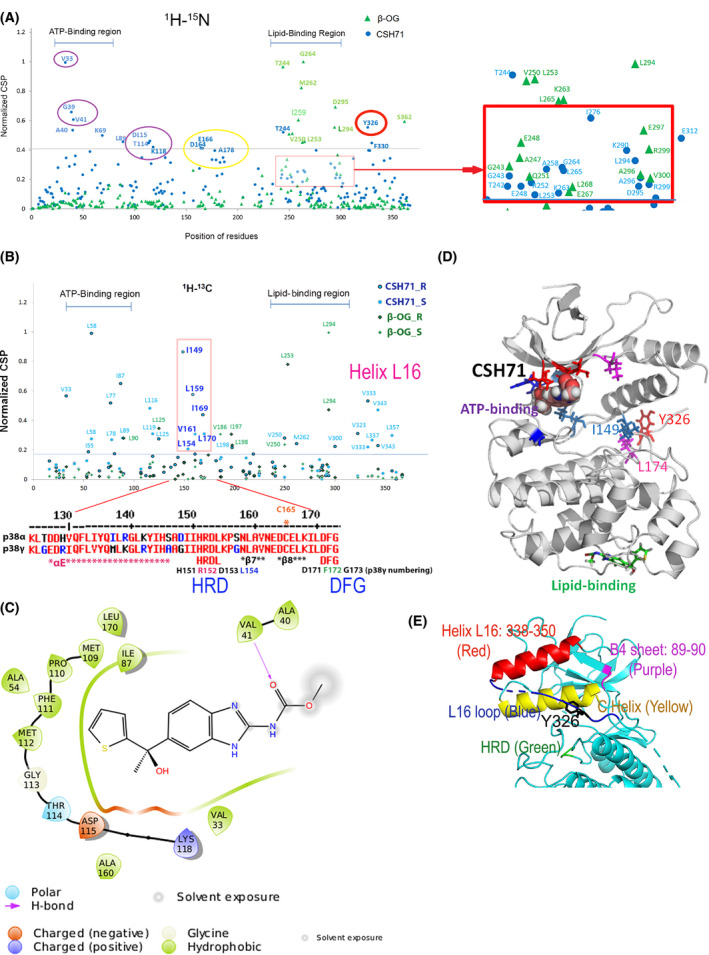
Compound CSH71 binds to two sites on p38γ in cell‐free based system. (A) NMR CSP 1H‐15N data for p38γ ‐ CSH71 (blue round dots) which prefers to bind to the ATP‐binding site, whereas β‐OG only binds to the lipid‐binding site (green triangles). The shifted residues V33, G39, V41, A40, and K69 (indicated by round blue dots, CSH71) are located at the ATP‐binding site of p38γ; red square showed the residues that CSH71 binds in the lipid‐binding domain significantly shifted are T244 (blue), I276 and E312. (B) NMR CSP the 1H‐13C HMQC spectra in the methyl region for p38γ showed HRD motif (151–153) and its adjacent residues (I149, L154, L159, V161, I169, and L170) highly disturbed in CSH71. It is consistent of others’ finding of p38a that pliable HRD folding of Helix L16 and L16 loop occurs upon compound binding of the LBD, which promotes trans‐auto phosphorylation via dimerization. (C) 2D interaction diagram of CSH71 in the ATP‐binding site of p38γ. V41 (Hydrogen bond); D115 (negative charged); and K118 (positive charged). Many hydrophobic are surrounding CSH71: V33, G39, and V41 on one side of the pocket; T114, D115, and K118 on the other side of the same pocket of p38γ. (D) Docking pose summarized NMR CSP 2D data of p38γ with CSH71, which shows how shifted C‐terminal residues that in Y326‐V348 segment (L16 loop and L16 helix) posit with those of are located in the ATP‐binding site. It is worth to note that along with two residues I149 and L174, Y326 of p38γ, a counterpart of the alternative phosphorylation site Y323 of p38α, in the interlobe region, may greatly shifted, making possible for opening up DEF pocket based on other’s studies (R. A. Engh [14] and O. Livnah [20]). (E) 3D structure of N‐terminal segments/domain of p38γ that contains residues that greatly shifted by CSH71. It illustrates a unique structure in the N‐lobe p38γ which contains an N‐terminal C Helix (yellow) that is sandwiched by C‐terminal sequences Helix L16 (red) and L16 loop (blue) where Y326 resides, the site for alternative phosphorylation by ZAP70. The conserved His‐Arg‐Asp (H151R152D153 motif, colored green), also greatly shifted and likely be protruding toward C Helix as binding of CSH71 to p38γ.
When we adjusted the basal line of normalized CSP to 0.18 (1H‐15N NMR), based on that of β‐OG, we can see more residues in the LBD shifted by CSH71—L198; M219; M262, and V300—as well as others near the alternative phosphorylation site—Y326 (amides shifted residues) and V323 and V333 (methyl shifted residues). In summary, both CSH71 and β‐OG shift residues G243, E248, L253, K263, L294, D295, A296, and R299, which confirms experimentally that CSH71 binds the LBD of p38γ (Fig. 5A).
Additionally, we also observed another motif Helix L16 in the C‐terminal that is greatly shifted in both 1H‐15N and 1H‐13C spectra (Fig. 5A,B). L16 loops covers residues L337, V343, and L357 (the methyl shifted residues), their sequence is at C‐terminal but 3D structure is at N‐lobe. In Fig. 5E, we showed that red Helix L16 flips back to the p38γ at N‐lobe, suggesting any shifts in the region affect the ATP‐binding pocket via allosteric effects.
We also observed HRD and the DFG site being shifted. In 1H‐13C spectra (methyls), L58 and I149 are shifted, and the disappeared residues are M109CE, L170CD1, and L174CD1 (Fig. S5B, D), in the region depicted in the overlaid NMR CSP spectra as covering the HRG and the DFG site (D171‐F172‐G173) (Fig. 5B). Our NMR CSP (methyl group) data support this notion and indicate that the HRD motif of p38γ and its adjacent residues I149, L154, L159, V161, I169, and L170 in 1H‐13C (Methyl, Fig. 5B, in the pink bracket) were highly perturbed by CSH71. 1H‐15N spectra residues D164‐ A178 that covers β8 and DFG motif also were shifted (Fig. 5A, yellow circle). Together with the computational 2D interaction diagram (Fig. 5C), this result suggests that the ligand‐LBD binding results in conformational changes in HRD by CSH71 (Fig. 5D).
In addition, L89 of β‐sheet 4 of N‐lobe p38γ (Fig. 5E) is also greatly shifted. It suggests the N‐lobe of the p38γ‐CSH71 complex experienced significant structural changes that can only be deciphered by X‐ray crystallography, which is out of the scope of this study. It is worth noting that residue Y326 of p38γ—the counterpart of the alternative phosphorylation site Y323 in the p38α and β isoforms—is significantly shifted by CSH71 (Fig. 5A, red circle). The biological significance of this shift is discussed in the following sections.
DEF site formation of p38γ
The DEF site of p38 is a functional domain that can accommodate various ligands, each of which triggers a specific biological outcome [3, 16, 42, 43]. Portions of the DEF are conserved across all four p38 isoforms (Fig. 6A, top). These segments also contain key residues of the lipid‐binding domain, as the MKI is one of its critical motifs. The X‐ray crystallographic structure of p38α indicates that W197 serves as an important anchor in the lipid‐binding pocket for lipid‐like ligands [13, 35, 37, 38]. Our NMR 2D CSP results suggest that W200 of p38γ (the counterpart of W197 in p38α) anchors β‐OG on the top in the lipid‐binding domain with α1L14 and α2L14 helixes at the bottom (Fig. 2C).
Fig. 6.
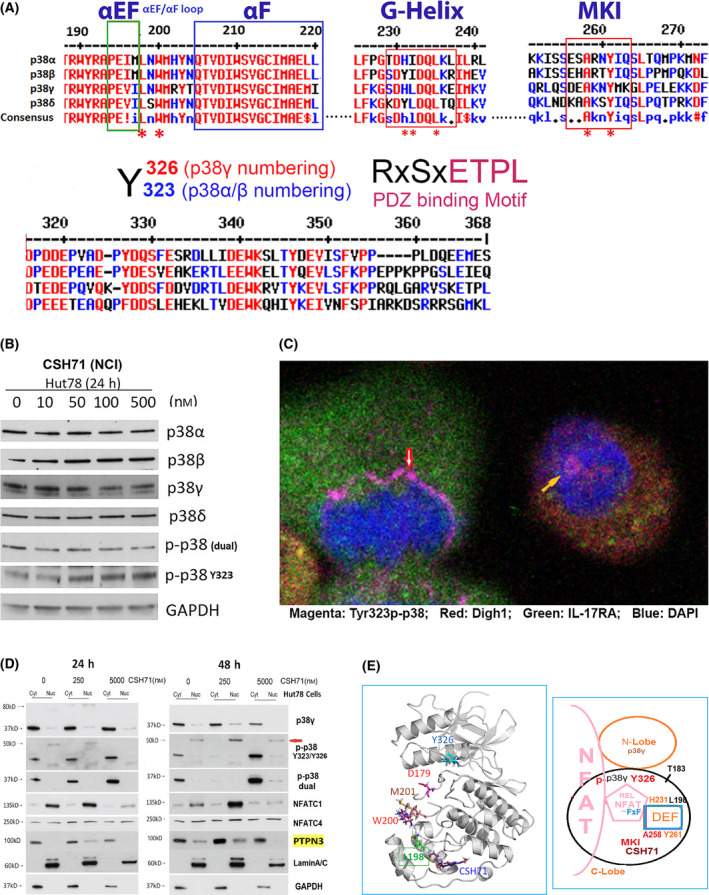
CSH71 inducing alternative p38 and NFAT activation. (A) Illustration of conserved segments of the DEF site interaction pocket (top), including α EF and α F loop, G‐Helix and MARP insert (MKI) which are structurally conserved among four p38 isoforms, which participates the formation of DEF pocket, many residues are perturbed by CSH71 binding underscored with *. The segments at bottom are p38s alternative activation site Y323/Y326 by ClustalW2 Consensus alignment diagram analysis with p38γ residue numbering. Residues ETPL is colored pink as PDZ binding Motif that solely existed in p38γ. (B) Western blot analysis to detect protein levels changes upon treatments of CSH71 (NCI) of four isoforms of p38 and two phosphorylated forms dual and alternative p38 together with GAPDH as protein internal loading control. (C) Confocal fluorescence microscopy analysis of untreated Hut78 cells indicates the subcellular location of alternative p38 activation by immunofluorescent staining using the specific antibody Y323 p‐p38. Co‐staining with antibodies against DLGH1 (red) and IL17RA (green) was used to identify downstream targets. DAPI was used to stain the nucleus. Alternative p38 activation is seen on the newly synthesized nuclear envelopes that surround anaphase chromatins (red arrow) and also within the nucleus (yellow arrow). (D) Hut 78 cell cytosolic and nuclear fractions lysates treated with indicated doses of CSH71 (250 nm and 5 µm) for 24 or 48 h are subject to WB analysis, using anti‐p38γ, anti‐p‐p38 at Y323 (alternative), anti‐p‐p38 (dual), anti‐PTPN3 and anti‐NFATC1, anti NFATC4 antibodies. Anti‐Lamin A/C antibody for the Lamin protein as the loading control for nuclear proteins and GAPDH for cytosol proteins. (E) Proposed model of interaction between NFAT with the DEF site of p38γ when the alternative site at Y326 is activated (phosphorylated). Left, 3D p38γ with residues reflect allosteric network from LBD to ATP binding site. Right, NFAT Interaction interact with p38γ via two docking motifs—FxF and LxL sequences; p38γ is also known as ERK6, which implicates similarity with the ERK family; in the REL domain of NFATc4, which contains an FxF motif (F681, F683) at the COOH‐terminal that allows interaction with the DEF pocket. Other NFAT members (NFATC1, NFATC2, and NFATC3) have FxY motifs in which possibly allows interaction with ERK. The LxL sequence is specific for other MAP kinases, such as c‐Jun NH2‐terminal protein kinase (JNK).
Further analysis of the NMR CSP data on the DEF pocket of p38γ indicated that residues I197 and L198 (the counterpart of L195 of p38α) were greatly shifted and that H231 (the counterpart of H228 of p38α) could be very dynamic and pliable. Residue H231 was not observable from a 2D spectrum of the apo form of p38γ that assumed even greater perturbation by CSH71. Residues L220, T221, and T224, which surround H231, are significantly shifted, as well as residues T244, A258, M262, K263, G264, and L265 in the MKI region (Fig. 5A,B and 2A,D). Three of these residues are conserved among species: H231, G243, and T244 (Fig. 2C,D indicated by *). Our data suggest that the DEF interaction pocket may open due to shifted segments HRD‐DFG, inter‐lobe (Y326) and β‐sheet 4 (L89) in the N‐lobe, which lead to significant structural changes on inter‐lobe of p38γ upon CSH71 binding. In theory, this would further promote the accommodation of TFs such as NFATs [29].
Residues predicted to be involved in the DEF pocket of p38γ are L198, H231 and A258, M262, and L265 (Fig. 2C; 5A, yellow circle; Fig. 5B, pink bracket), most of which are perturbed or disappeared in our CSP NMR assay in Fig. 5A,B. Figure 6E (bottom right) summarizes our data indicating that a DEF pocket is likely formed in p38γ upon LBD binding. This pocket then is able to accommodate NFAT via its FXF motif in the REL domain.
Alternative activation of p38 co‐related to TNFα mRNA expression upon treatment of CSH71 at lower dosage
p38 isoforms harbor both a phosphorylation lip (the dual phosphorylation site TGY [T183–Y185 in p38γ]) and a C‐terminal tail segment with the alternative phosphorylation site Y323 as well as the L16 loop and Helix L16. p38γ is unique in its alternative activation site at Y326, and the PDZ‐binding motif in the C‐terminal domain (RxSxETPL, Fig. 6A, bottom right). The PDZ‐binding motif modulates NFAT transcriptional activation [44] through an interaction with the PDZ domain of DLGH1, which is a p38γ kinase substrate (via its ATP site). DLGH1 activation is known to drive the TCR pathway to NFAT rather than NFkB [44].
To determine the efficacy of alternative p38 activation in Hut78 cells following treatment with CSH71, we monitored by western Blot the protein levels of p38 isoforms and p38 dual (p‐p38 TGY) and alternative phosphorylation forms (p‐p38 at Y323) of cells treated with increasing dosages of CSH71. Our data showed increasing phosphorylation of p38 at Y323 in response to increasing doses of CSH71 up to 500nM and a decrease in dual p38 phosphorylation at this dosage after 24 h of exposure (Fig. 6B).
Based our cytotoxicity studies of CSH71 in Hut78 cells (IC50 ~ 2 µm), we chose 500 nm as a low dose to study the effect on nuclear NFATs because CSH71 induces alternative p38 activation at concentrations between 50 and 500 nm (Fig. 6B). We treated Hut78 cells with a moderate dosage of 250 nm of CSH71, followed by total RNA isolation and microarray analysis, which we compared to untreated cells. The treatment induced the expression of the pro‐inflammatory cytokine TNFα in many top pathways (Table 2). It also caused the up‐regulation of the olfactory factor pathway and the down‐regulation of Toll‐like receptor 5 (TLR5) responses to the chemical stimulation of T cells.
Table 2.
Pathway analysis of DE Genes CSH71(NCI) 250 nm vs. Control.
| Pathway | Source | P‐value | q‐value | Top Hits_overlap |
|---|---|---|---|---|
| Upregulated | ||||
| Olfactory transduction ‐ Homo sapiens (human) | KEGG | 8.47E‐10 | 2.05E‐07 | OR5B21; OR52N1; OR52J3; OR56A3; OR4N5; OR1S1; OR11H1; OR2L2; OR10K2; OR6N1; OR8K3; OR2B3; OR6Y1; OR5W2; OR2L8; OR5A1 |
| HTLV‐I infection ‐ Homo sapiens (human) | KEGG | 0.00072468 | 0.01288891 | JUN; EGR1; ADCY8; ZFP36; TNF; CREM; ATF3 |
| MAPK signaling pathway ‐ Homo sapiens (human) | KEGG | 0.00353542 | 0.03619249 | GADD45A; JUN; TNF; DUSP5; DUSP4; FLNC |
| TNF signaling pathway ‐ Homo sapiens (human) | KEGG | 0.00376954 | 0.03619249 | PTGS2; JAG1; JUN; TNF |
| Inflammatory bowel disease (IBD) ‐ Homo sapiens (human) | KEGG | 0.00631366 | 0.05071297 | STAT4; TNF; JUN |
| Osteoclast differentiation ‐ Homo sapiens (human) | KEGG | 0.00698855 | 0.0527318 | CALCR; TNF; JUN; FOSL2 |
| Leishmaniasis ‐ Homo sapiens (human) | KEGG | 0.00837645 | 0.05637127 | PTGS2; TNF; JUN |
| Downregulated | ||||
| Pathway | source | p‐value | q‐value | Top Hits_overlap |
| Pathogenic Escherichia coli infection ‐ Homo sapiens (human) | KEGG | 3.10E‐05 | 0.00018592 | TUBA3E; TUBA3D; TLR5 |
| Gap junction ‐ Homo sapiens (human) | KEGG | 0.00460401 | 0.01381204 | TUBA3E; TUBA3D |
Microarray RNA analysis and pathway analysis of Hut78 CTCL cells with 100 nM CSH71 at 48 h treatment. Several significantly up‐regulated pathways are in bold, such as the Olfactory transduction pathway, MAPK signaling pathway and TNF pathway. Likewise, down‐regulated pathway, Pathogenic Escherichia coli infection, etc. Two representative signaling molecules TNF and TRL5 are highlighted in bold. p‐values for two most significantly pathways are Olfactory transduction (unregulated, P = 8.47E‐10, bold) and (down‐regulated, Pathogenic Escherichia coli infection, P = 3.10E‐05, bold), respectively.
A connection between p38 activation, inflammation, and TNFα expression was first established decades ago. p38 activation promotes the expression of pro‐inflammatory cytokines such as TNFα [1, 45], and the pathway has been implicated in experimental autoimmune encephalomyelitis (EAE), rheumatoid arthritis [46], multiple sclerosis [47], Alzheimer’s disease [48], and inflammatory bowel disease [49]. We previously showed that the knockdown of either p38γ or NFATC4 significantly reduces IL‐17A mRNA levels in Hut78 cells [24]. To further understand the role of IL‐17A in CTCL, we also monitored the expression of the IL‐17A receptor IL17RA—a ubiquitous type I transmembrane glycoprotein that plays a pathogenic role in many inflammatory and autoimmune diseases such as rheumatoid arthritis [50, 51] and CTCL [52]—and found that it is constitutively expressed in Hut78 cells (Fig. 6C).
Previous research has indicated that p38 controls IL‐17 production in a model of EAE by activating the eukaryotic translation initiation factor 4E/MAPK‐interacting kinase (eIF‐4E/MNK) pathway [53]. Studies on alternative p38 activation in T cells in EAE have concluded that p38 regulates the production of IL‐17 at a transcriptional level through the CAMP‐responsive element (CRE), which is the binding site for activation transcriptional factor ATF2 [54]. However, another study concluded that p38 activation in EAE affects Th17 differentiation rather than IL‐17 production [55]. Very likely, IL‐17RA positions itself for the upcoming abrupt changes in cytokine IL‐17 levels in inflammation, autoimmune disorders, and cancer.
Interestingly, confocal fluorescence microscopy of p‐Y323 in p38 and its substrates DLGH1 and IL17RA identified alternatively activated p38 on the newly synthesized nuclear envelopes that surround anaphase chromatin (Fig. 6C, red arrow) and within the interphase nucleus (yellow arrow) of Hut78 cells. This is an intriguing observation, because ZAP70 activity at the TCR complex occurs in the plasma membrane of T cells, and the mechanisms by which phosphorylated Y323/326 p38 migrates to, and remains in, the nucleus are unclear. Here, our data support the notion that Hut78, a malignant T‐cell line derived from a CTCL patient, is one of the best cell‐based systems in which to study alternative p38 activation by ZAP70.
Molecular mechanism of biological effects of CSH71 as a two site‐binding ligand on p38γ in CTCL cells
Given that many essential TFs in TCR signaling, including NFATs, are substrates of p38 [56], our results demonstrate not only that alternative p38 phosphorylation is activated in Hut78 cells, but also suggest that the role of p38 in the nucleus involves the regulation of NFAT transcriptional activity.
To determine the nature of nuclear p38γ and its actions on NFATs, we extracted cytosolic and nuclear fractions of Hut78 cells treated with 250 nm or 5 µm CSH71 for 24 or 48 h. The molecular weight of phosphorylated p38γ after 48 h treatment with 250 nm of CSH71 is approximately 40 kD in the cytosol and 50 kD in the nucleus (red arrow, Fig. 6D, 250 nm, 48 h). This suggests the occurrence of additional post‐translational modifications of p‐Y326 p38γ in the nucleus of live cells—most likely representing an intermediate/transient form of phosphorylation at pT180 –pY323/Y326.
Surprisingly, neither the dual‐phosphorylated form or the alternatively phosphorylated form of p38 are detectable in the cytosol after treatment with 250 nm CSH71 for 48 h, suggesting that certain tyrosine phosphatases are activated in cytosol at low doses of CSH71. (The treatment also causes the disappearance of the dual‐phosphorylated form from the nucleus.) We propose that two PDZ domain‐containing tyrosine phosphatases reported to be substrates of p38γ [57]—PTPN3 and PTPN4—are responsible. We further speculate that PTPN3 dephosphorylates p‐Y323/326 of p38α/γ in the cytosol (Fig. 6D, right). Our western blot analysis confirms these speculations in that 250 nm 48 h treatment increases amounts of tyrosine phosphatases PTPN3 (highlight with yellow), which is reportedly interacts with p38γ via its PDZ‐binding motif, may also contribute to the dephosphorylation of Y185 in dually phosphorylated p38γ (disappearance of the band p‐p38 in cytosol upon the treatment) that we observed in Fig. 6D (48 h, 250 nm treated). One study has indicated that PTPN3 activation enhances oncogenesis through the dephosphorylation of p38γ, which activates Ras [58]. We hypothesize that PTPN4, which, according to GeneCard, is found mainly in the nucleus, may keep NFATs in a non‐phosphorylated, transcriptionally active form in Hut 78 cells.
NFATC1/C4 are nuclear proteins with essential activity in T cells. Consistent with changes in p38γ expression, nuclear NFATC1 is significantly increased in cells treated with 250 nm CSH71, but its expression drastically decreases at higher (5 µm) dosages. Interestingly, we observed that the phosphorylated form of NFATC4 (MW = 250 kD) is equally expressed in both cytosol and nucleus of Hut78 cells at both the 24 h and 48 h time points (Fig. 6D). In cells treated with 5 µm CSH71, both dual‐ and alternative phosphorylation is dramatically increased in the cytosol lysates. This suggests that at this higher dosage CSH71 occupies both the ATP‐binding site and the LBD of p38γ, resulting in allosteric conformation changes that lead to stress‐related p38 activation, drastic reduction of NFAT affinity, and reduced nuclear translocation, subsequently lowering transcriptional activity and eventually causing cell death.
Discussion
The Engh group [17] has proposed a model of transient dimerization to explain the molecular mechanism of auto‐monophosphorylation at T180 (also known as alternative p38 phosphorylation) triggered by Y323 phosphorylation by ZAP70. In this model, the phosphorylation of Y323 in one p38 monomer causes a structural disruption at the L16 loop region, bringing it near T180 of another monomer and facilitating auto‐monophosphorylation. A rigid His–Arg–Asp (HRD) motif (H151‐D153 in p38γ) blocks this auto‐monophosphorylation when Y323 is unphosphorylated [17].
The HRD motif is conserved throughout all protein kinase families [18]. It supports the configuration of the activation segment [59], and its deformation prevents the auto‐monophosphorylation triggered by the phosphorylation of Y323 by ZAP70 [17]. When Y323 is mutated to T323, which bears conformational resemblance to p‐Y323, the C‐alpha atoms of the TGY motifs are shifted by more than 17Ao, promoting conformational changes in the active loop and changing inter‐lobe orientation to create a DEF site interaction pocket for substrates such as the TFs ATF2 and ELK1 [16]. (Note that in p38γ the alternative phosphorylation site is Y326.) In this study, we cannot tell upon which isoform of p38 the alternative phosphorylation has taken place, and however, our NMR, IP, and western blot results in Hut 78 cells strongly suggest that it occurs on p38γ (Figs 4C, 5A, D and 6D).
Along with identifying conservative segments defining the DEF site interaction pocket among p38 isoforms (Fig. 6A), our NMR CSP results pinpointed several residues—H231, G243, T244, A258, M262, K263, G264, and L265—in the region that are significantly shifted by CSH71 (Fig. 5A,B and 2D). We predict that the DEF interaction pocket opens due to shifted p‐Y326 in p38γ. The subsequent auto‐monophosphorylation at T180, followed by a shift of the entire inter‐lobe, would in theory further promote the accommodation of TFs such as ATF2 or NFATs [29]. The 50 kD band of p‐Y323 observed in our western blots (Fig. 6D) suggests an intermediate stage of auto‐monophosphorylation on T180/T183 that co‐exists with phosphorylation at Y323/Y326.
p38γ is also known as ERK6, and it shares similarities with the ERK protein kinase family. NFATC4 contains an FxF motif (F681, F683) at the COOH‐terminal end of the NFATC4 REL domain that allows interaction with the DEF pocket [3]. A similar motif is also found in other NFAT members (FxY motifs in NFATC1, NFATC2, and NFATC3), which possibly allows interaction with ERK. Two docking motifs—FxF and LxL—appear to facilitate interaction with ERK via its DEF site. The LxL sequence is specific for other MAP kinases, such as c‐Jun NH2‐terminal protein kinase (JNK) [60].
In Fig. 6D, we showed that NFATC1 is increasingly expressed in the nucleus of Hut78 cells in the presence of 250nM CSH71, which correlates with alternative p38 activation (p‐Y323) in the nucleus. This implies that NFATs interact with phosphorylated p38 via the DEF pocket and activate the TCR signaling pathway after their translocation into the nucleus. Coupled with our data in Fig. 5B,D, we now have novel insight as to how the DEF pocket of p38 opens when alternatively activated to allow NFAT docking, which leads to its entry into the nucleus and the transcription of downstream target mRNA upon CSH71 treatment at low dose (250nM).
CSH71 binds weakly to the ATP‐pocket of p38γ, based on its docking pose score (−6.82 kcal·mol−1), which is much lower than that of F7 (−9.81 kcal·mol−1, an ATP‐pocket binder) [6]. The STD value of CSH71 further supports this notion (Fig. S4B). Therefore, we conclude that CSH71 preferentially binds the LBD of p38γ in Hut78 cells, as it would be displaced from the ATP‐binding site by the abundant presence of ATP in live cells. This binding promotes Y326 phosphorylation, which results in opening of the DEF pocket as described. The folding of the pliable HRD motif with the L16 loop and Helix L16 occurs upon CSH71 binding to p38γ (Fig. 5B, pink bracket of shifted residues; Fig. 5E, HRD in green loop), which leading to auto‐monophosphorylation of T183. The increased levels of NFATC1 we observed upon CSH71 treatment (250 nm, Figs 6D and 7), further confirms other reports indicating that alternative p38 activation induces NFATC1 mRNA expression [2]. It is also consistent with previous studies that concluded that alternative p38 activation directs T cells toward the NFAT pathway via DLGH1 rather than the NF‐κB pathway [44]. Our data suggest a novel pathway in which, at low concentrations (250 nm), CSH71 binds to the LBD of p38γ (not the ATP‐binding site) and actives alternative p38 phosphorylation, resulting in the opening of the DEF pocket to accommodate its substrates based on molecular mechanisms proposed by the Engh [15] and Livnah [14] groups.
Fig. 7.
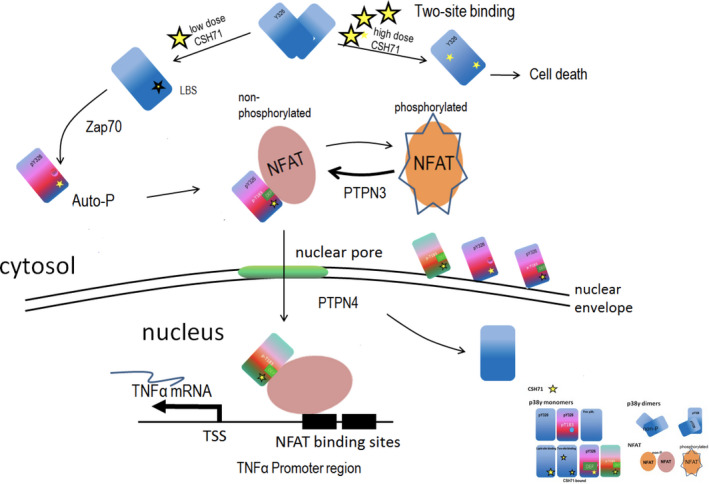
CSH71 as a LBD binder and its biological functionality in CTCL cells. Proposed schematic molecular mechanism by which CSH71 (250 nm) induces TNFα, first to increase cytosolic Y326 phosphorylation of p38γ at the TCR complex proximal to the plasma membrane of T cells and then migrate into the nucleus. Alternative p38 phosphorylation is activated in CTCL cells upon lipid binding, which extends conformational changes to the active loop of p38, followed by interlobe orientation changes, evidenced at Y326 and regions surrounding DEF pocket perturbation, which suggest formation of a DEF site interaction pocket and its availability to other substrates/factors, such as NFATs. Nuclear NFATs then subsequently transcriptionally activate TNFα mRNA in CTCL cells, evidenced by RNA sequencing and pathway analysis at CSH71 treatment (250 nm). At a higher dosage, it is very likely that CSH71 binds to two sites (ATP binding and LBS binding) of p38γ and prevents its dimerization, which further blocks the interaction of p38γ and NFAT thereby preventing its subsequent translocation into the nucleus and thereby cell death.
To further delineate the downstream signaling induced by CSH71 in Hut78 cells, we performed DE analysis after microarray RNA analysis. The pathway analysis (mRNA) of Hut78 cells treated with 100 nm CSH71 for 30 h are listed in Table 2. Our model best explains the increase in TNFα, a critical mediator of immune and inflammatory responses, observed in microarray analysis after CSH71 treatment because there are several NFAT‐binding sites in the promoter region of TNFα [28]. We posit that, at a low dose, CSH71 binding to p38γ causes opening of the DEF pocket to accommodate more transcription factor substrates, as indicated by increasing levels of NFAT in the nucleus, and this activates TNFα mRNA expression. In addition, we hypothesize that at this lower concentration, CSH71 activates tyrosine phosphatases PTPN3, as evidenced by the increased amount of PTPN3 in cytosol and the disappearance of the p‐Y323 band in western blots of the cytosol and nuclear fractions from treated cells (Fig. 6D, 48 h, 250 nm WB results). We predict that these phosphatases are PTPN3 (we confirmed in cytosol by WB) and PTPN4 (nucleus), which become active 48 h after the initial activation of alternative p38 phosphorylation (Fig. 6D, 24 h). Both tyrosine phosphatases have PDZ domains [61, 62, 63, 64], and, interestingly, both are substrates of p38γ, perhaps facilitated by binding via the PDZ‐binding motif [57, 58].
Taken together, Fig. 7 summarizes our experimental data as a molecular model of the proposed mechanism by which CSH71 promotes cytotoxicity in CTCL cells only at high doses. When present at low doses, CSH71 preferentially binds the LBD of p38γ, promoting Y326 phosphorylation and the resulting conformational changes in the DEF pocket to accommodate substrates such as TFs and/or co‐factors for transcriptional machinery. This complex subsequently enters the nucleus and activates the expression of TNFα, which promotes pro‐apoptotic activity in CTCL cells. In contrast, when present at high levels (5 µm), CSH71 occupies both the ATP‐binding site and the LBD. This causes phosphorylation of Y326 and a decrease in tyrosine phosphatase levels that leads to an overall increase in the inactive, phosphorylated form of NFAT in the cytosol. The dramatic reduction of NFAT levels in the nucleus causes cell death.
In summary, we have confirmed the presence of an LBD in p38γ and identified a molecule, CSH71, that binds to both the LBD and the ATP‐binding pocket. For the first time, we describe the biological impact of molecules that bind to two sites in p38γ, and we present a model to explain how targeting the LBD with novel drugs could selectively kill CTCL cells.
Conflict of interest
There are no conflicts to declare.
Author contributions
XHZ contributed to conceptualization, design and investigation, writing—original draft, and writing—review, editing and corresponding, initializing several key experiments such as screened CSH71 and CSH18, designing CSH18CN, and contributed to immunofluorescence staining and confocal microscopy experiments; CHC contributed to NMR CSP 2D experiments and data analysis and manuscript revisions; HL contributed to conceptualization of docking/virtual screening and generating compound list; JH contributed to cell viability assays, IP and WB experiments; XW contributed to microarray analysis; WH and JS contributed to NMR 1D and STD experiments and data analysis; DH contributed to compound purification; SN contributed to kinase activity assays; STR contributed to overseeing the lab.
Supporting information
Fig S1A–D NMR spectra and CSP plots indicate residues in LBD of p38γ that have significantly chemically shifted in response to addition of β‐OG. The ratio of β‐OG to p38γ is 10:1. (E) The titration effects of β‐OG to the LBD residues of p38γ by 1H‐15N correlation NMR spectra. (A) NMR spectra. Overlay of the 1H‐15N HMQC spectra in the amide region for p38γ, free (red) and in complex with β‐OG (blue). (B) NMR spectra. Overlay of the 1H‐13C HMQC spectra in the methyl region for p38γ, free (red) and in complex with β‐OG (blue). (C) CSP plots. The cutoff value in NMR spectra for amide (ppm>0.02) is indicated by red arrows. (D) CSP plots. The cutoff value in NMR spectra for methyl (ppm>0.03) is indicated by red arrows.
Fig S1E A zoomed‐in view of the partial region in 1H‐15N correlation spectra of β‐OG with p38γ. Three residues, T244, E297 and M262 in LBD of p38γ are significantly chemically shifted by ligand β‐OG. β‐OG was titrated into isotope‐labeled p38γ. The different color (Red, Yellow, Green, Cyan, Blue) indicted by arrows on each side of three residues refer to a progressive chemically‐shift in respond to increasing concentration of β‐OG in the presence of p38γ (p38γ : β‐OG = 1:0 red, 1:2.5 yellow, 1:5 green, 1:7.5 cyan and 1:10 blue, respectively).
Fig S2A Pure CSH71 are isolated from CSH71 (NCI) by HPLC. A. Mass spectrum of CSH71 (Fraction 3 indicated by arrow.) CSH71 (NCI) is from NCI DTP and we showed it contains 3 fractions including nocodazole (Fraction 2), a well‐known cell cycle arrest agent that arrests cells at G2 /M phase.
Fig S2B Purified 3 (pure CSH71) by MASS and is ready for the further experiments such as NMR assays etc.
Fig S3A Biological assessments of CSH71 and β‐OG. A. Increasing concentrations of CSH71 (pure) were applied to Hut78 CTCL cells for 72 h.
Fig S3B Hut78 CTCL cells were treated with indicated concentrations of β‐OG for 72h.
Fig S3C–D (C) Increasing concentrations of CSH71 (pure) were applied to Hut78 CTCL cells for 72 h. (D) To determine p38γ kinase activity of CSH71 by in vitro kinase activity measured by ADP‐glo assays (blue).
Fig S4A 1D NMR STD spectra p38 in complex with CSH71 indicate CSH71 bound p38γ. STD NMR 1D 1H spectra overlay of compound CSH71 aromatic region in the presence of p38γ.
Fig S4B 1D NMR aromatic proton chemical shifts by CSH71 and STD values in complex with p38γ in the absence and presence of ATP.
Fig S4C The docking pose in ATP‐binding site more agrees with NMR STD result as five‐ring group is located inside the pocket, while –COOCH3 tail is outside (left). In lipid‐binding, more flexible binding of CSH71 in the LBD pocket (right).
Fig S5 (A) NMR 2D CSP with CSH71 spectra indicate CSH71 bound p38γ. A. 2D NMR CSP titration experiment with CSH71 spectra: overlay of the 1H‐15N HSQC spectra in the amide region for p38γ, free (red) and in complex with CSH71 (blue). (B) 2D NMR CSP titration experiment 1H‐13C HMQC spectra in the methyl region overlay for p38γ, free (red) and in complex β‐OG (blue). (C) Cut‐off value for Amide CSP is >0.02 ppm for selected shifted residues V33, G39, A40, V41, and Y326. (D) Cut‐off value for Methyl CSP is >0.02 ppm for selected shifted residues L58 and I149.
Acknowledgements
Research reported in this publication included work performed in City of Hope Cores (Integrative Genomics and Bioinformatics Core, Drug Discovery & Structural Biology Core, and Mass Spectrometry and Proteomics Core) supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572 and the Light Microscopy Core at City of Hope. Other support included 1R01CA233922‐01 (ROSEN) and LLS Grant ID: 6576‐19 (ROSEN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Edited by Christian Griesinger
Contributor Information
Xu Hannah Zhang, Email: xuzhang@coh.org.
Steven T. Rosen, Email: srosen@coh.org.
Data accessibility
The data that support the findings of this study are openly available in NCBI's Gene Expression Omnibus and are accessible through https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE182484, GEO Series accession number GSE182484.
References
- 1. Zarubin T and Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15, 11–18. [DOI] [PubMed] [Google Scholar]
- 2. Alam MS, Gaida MM, Ogawa Y, Kolios AG, Lasitschka F and Ashwell JD (2014) Counter‐regulation of T cell effector function by differentially activated p38. J Exp Med 211, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang TT, Xiong Q, Enslen H, Davis RJ and Chow CW (2002) Phosphorylation of NFATc4 by p38 mitogen‐activated protein kinases. Mol Cell Biol 22, 3892–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, Fornace AJ Jr and Ashwell JD (2005) Alternative p38 activation pathway mediated by T cell receptor‐proximal tyrosine kinases. Nat Immunol 6, 390–395. [DOI] [PubMed] [Google Scholar]
- 5. Jun JE, Kulhanek KR, Chen H, Chakraborty A and Roose JP (2019) Alternative ZAP70‐p38 signals prime a classical p38 pathway through LAT and SOS to support regulatory T cell differentiation. Sci Signal 12, eaao0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang XH, Nam S, Wu J, Chen CH, Liu X, Li H, McKeithan T, Gong Q, Chan WC, Yin HH et al. (2018) Multi‐kinase inhibitor with anti‐p38gamma activity in cutaneous T‐cell lymphoma. J Invest Dermatol 138, 2377–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin N, Qi X, Tsai S, Lu Y, Basir Z, Oshima K, Thomas JP, Myers CR, Stoner G and Chen G (2016) p38γ MAPK is required for inflammation‐associated colon tumorigenesis. Oncogene 35, 1039–1048. [DOI] [PubMed] [Google Scholar]
- 8. Browne AJ, Gobel A, Thiele S, Hofbauer LC, Rauner M and Rachner TD (2016) p38 MAPK regulates the Wnt inhibitor Dickkopf‐1 in osteotropic prostate cancer cells. Cell Death Dis 7, e2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zheng S, Yang C, Liu T, Liu Q, Dai F, Sheyhidin I and Lu X (2016) Clinicopathological significance of p38beta, p38gamma, and p38delta and its biological roles in esophageal squamous cell carcinoma. Tumour Biol 37, 7255–7266. [DOI] [PubMed] [Google Scholar]
- 10. Qi X, Yin N, Ma S, Lepp A, Tang J, Jing W, Johnson B, Dwinell MB, Chitambar CR and Chen G (2015) p38γ MAPK is a therapeutic target for triple‐negative breast cancer by stimulation of cancer stem‐like cell expansion. Stem Cells 33, 2738–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomas‐Loba A, Manieri E, Gonzalez‐Teran B, Mora A, Leiva‐Vega L, Santamans AM, Romero‐Becerra R, Rodriguez E, Pintor‐Chocano A, Feixas F et al. (2019) p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 568, 557–560. [DOI] [PubMed] [Google Scholar]
- 12. Diskin R, Engelberg D and Livnah O (2008) A novel lipid binding site formed by the MAP kinase insert in p38 alpha. J Mol Biol 375, 70–79. [DOI] [PubMed] [Google Scholar]
- 13. Perry JJ, Harris RM, Moiani D, Olson AJ and Tainer JA (2009) p38alpha MAP kinase C‐terminal domain binding pocket characterized by crystallographic and computational analyses. J Mol Biol 391, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Comess KM, Sun C, Abad‐Zapatero C, Goedken ER, Gum RJ, Borhani DW, Argiriadi M, Groebe DR, Jia Y, Clampit JE et al. (2011) Discovery and characterization of non‐ATP site inhibitors of the mitogen activated protein (MAP) kinases. ACS Chem Biol 6, 234–244. [DOI] [PubMed] [Google Scholar]
- 15. Diskin R, Lebendiker M, Engelberg D and Livnah O (2007) Structures of p38alpha active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J Mol Biol 365, 66–76. [DOI] [PubMed] [Google Scholar]
- 16. Tzarum N, Diskin R, Engelberg D and Livnah O (2011) Active mutants of the TCR‐mediated p38alpha alternative activation site show changes in the phosphorylation lip and DEF site formation. J Mol Biol 405, 1154–1169. [DOI] [PubMed] [Google Scholar]
- 17. Rothweiler U, Aberg E, Johnson KA, Hansen TE, Jorgensen JB and Engh RA (2011) p38alpha MAP kinase dimers with swapped activation segments and a novel catalytic loop conformation. J Mol Biol 411, 474–485. [DOI] [PubMed] [Google Scholar]
- 18. Kornev AP, Haste NM, Taylor SS and Eyck LF (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beenstock J, Ben‐Yehuda S, Melamed D, Admon A, Livnah O, Ahn NG and Engelberg D (2014) The p38beta mitogen‐activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the alpha‐G helix and MAPK insert. J Biol Chem 289, 23546–23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bell M, Capone R, Pashtan I, Levitzki A and Engelberg D (2001) Isolation of hyperactive mutants of the MAPK p38/Hog1 that are independent of MAPK kinase activation. J Biol Chem 276, 25351–25358. [DOI] [PubMed] [Google Scholar]
- 21. Askari N, Diskin R, Avitzour M, Capone R, Livnah O and Engelberg D (2007) Hyperactive variants of p38alpha induce, whereas hyperactive variants of p38gamma suppress, activating protein 1‐mediated transcription. J Biol Chem 282, 91–99. [DOI] [PubMed] [Google Scholar]
- 22. Tian L, Kim MS, Li H, Wang J and Yang W (2018) Structure of HIV‐1 reverse transcriptase cleaving RNA in an RNA/DNA hybrid. Proc Natl Acad Sci USA 115, 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bauler TJ, Hughes ED, Arimura Y, Mustelin T, Saunders TL and King PD (2007) Normal TCR signal transduction in mice that lack catalytically active PTPN3 protein tyrosine phosphatase. J Immunol 178, 3680–3687. [DOI] [PubMed] [Google Scholar]
- 24. Zhang XH, Ngo VN, Sandoval N, Cui Q, Shi Y, Zain JM, Querfeld C, Guo C, Wu X and Rosen ST (2016) Role of p38γ ‐ NFATc4 ‐ IL17A pathway as a potential therapeutic target in cutaneous T cell lymphoma. Blood 128, 2725. 10.1182/blood.V128.22.2725.2725 [DOI] [Google Scholar]
- 25. Zhang HX, Yin Z, Zhang A, Pillai R, Armstrong R and Rosen ST (2020) DNMT1 and p38γ are inversely expressed in reactive non‐metastatic lymph nodes burdened with colorectal adenocarcinoma. eJHaem 1, 300–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheung CY and Ko BC (2013) NFAT5 in cellular adaptation to hypertonic stress ‐ regulations and functional significance. J Mol Signal 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serfling E, Berberich‐Siebelt F, Avots A, Chuvpilo S, Klein‐Hessling S, Jha MK, Kondo E, Pagel P, Schulze‐Luehrmann J and Palmetshofer A (2004) NFAT and NF‐kappaB factors‐the distant relatives. Int J Biochem Cell Biol 36, 1166–1170. [DOI] [PubMed] [Google Scholar]
- 28. Falvo JV, Tsytsykova AV and Goldfeld AE (2010) Transcriptional control of the TNF gene. Curr Dir Autoimmun 11, 27–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang TT, Xiong Q, Graef IA, Crabtree GR and Chow CW (2005) Recruitment of the extracellular signal‐regulated kinase/ribosomal S6 kinase signaling pathway to the NFATc4 transcription activation complex. Mol Cell Biol 25, 907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu W, Zhou M, Li Z, Li H, Polaczek P, Dai H, Wu Q, Liu C, Karanja KK, Popuri V et al. (2016) A selective small molecule DNA2 inhibitor for sensitization of human cancer cells to chemotherapy. EBioMedicine 6, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lipinski CA (2004) Lead‐ and drug‐like compounds: the rule‐of‐five revolution. Drug Discov Today Technol 1, 337–341. [DOI] [PubMed] [Google Scholar]
- 32. Baell JB and Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- 33. Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J and Hajduk PJ (2005) ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J Am Chem Soc 127, 217–224. [DOI] [PubMed] [Google Scholar]
- 34. Zhang XH, Zhao C, Seleznev K, Song K, Manfredi JJ and Ma ZA (2006) Disruption of G1‐phase phospholipid turnover by inhibition of Ca2+‐independent phospholipase A2 induces a p53‐dependent cell‐cycle arrest in G1 phase. J Cell Sci 119, 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buhrmann M, Hardick J, Weisner J, Quambusch L and Rauh D (2017) Covalent lipid pocket ligands targeting p38alpha MAPK mutants. Angew Chem Int Ed Engl 56, 13232–13236. [DOI] [PubMed] [Google Scholar]
- 36. Bührmann M, Wiedemann BM, Müller MP, Hardick J, Ecke M and Rauh D (2017) Structure‐based design, synthesis and crystallization of 2‐arylquinazolines as lipid pocket ligands of p38α MAPK. PLoS One 12, e0184627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tong L, Pav S, White DM, Rogers S, Crane KM, Cywin CL, Brown ML and Pargellis CA (1997) A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat Struct Biol 4, 311–316. [DOI] [PubMed] [Google Scholar]
- 38. Getlik M, Simard JR, Termathe M, Grutter C, Rabiller M, van Otterlo WA and Rauh D (2012) Fluorophore labeled kinase detects ligands that bind within the MAPK insert of p38alpha kinase. PLoS ONE 7, e39713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellon S, Fitzgibbon MJ, Fox T, Hsiao H‐M and Wilson KP (1999) The structure of phosphorylated P38γ is monomeric and reveals a conserved activation‐loop conformation. Structure 7, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 40. Mayer M and Meyer B (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- 41. Cala O and Krimm I (2015) Ligand‐orientation based fragment selection in STD NMR screening. J Med Chem 58, 8739–8742. [DOI] [PubMed] [Google Scholar]
- 42. Shaul YD and Seger R (2007) The MEK/ERK cascade: from signaling specificity to diverse functions. Biochem Biophys Acta 1773, 1213–1226. [DOI] [PubMed] [Google Scholar]
- 43. Tanoue T, Maeda R, Adachi M and Nishida E (2001) Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J 20, 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Round JL, Humphries LA, Tomassian T, Mittelstadt P, Zhang M and Miceli MC (2007) Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF‐kappaB transcription factors. Nat Immunol 8, 154–161. [DOI] [PubMed] [Google Scholar]
- 45. Feng YJ and Li YY (2011) The role of p38 mitogen‐activated protein kinase in the pathogenesis of inflammatory bowel disease. J Dig Dis 12, 327–332. [DOI] [PubMed] [Google Scholar]
- 46. Han ZS, Enslen H, Hu X, Meng X, Wu IH, Barrett T, Davis RJ and Ip YT (1998) A conserved p38 mitogen‐activated protein kinase pathway regulates Drosophila immunity gene expression. Mol Cell Biol 18, 3527–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sweeney SE and Firestein GS (2004) Signal transduction in rheumatoid arthritis. Curr Opin Rheumatol 16, 231–237. [DOI] [PubMed] [Google Scholar]
- 48. Krementsov DN, Thornton TM, Teuscher C and Rincon M (2013) The emerging role of p38 mitogen‐activated protein kinase in multiple sclerosis and its models. Mol Cell Biol 33, 3728–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson GV and Bailey CD (2003) The p38 MAP kinase signaling pathway in Alzheimer's disease. Exp Neurol 183, 263–268. [DOI] [PubMed] [Google Scholar]
- 50. Batalla A, Coto E, Gómez J, Eirís N, González‐Fernández D, Gómez‐De Castro C, Daudén E, Llamas‐Velasco M, Prieto‐Perez R, Abad‐Santos F et al. (2018) IL17RA gene variants and anti‐TNF response among psoriasis patients. Pharmacogenomics J 18, 76–80. 10.1038/tpj.2016.70 [DOI] [PubMed] [Google Scholar]
- 51. Munoz‐Aceituno E, Martos‐Cabrera L, Ovejero‐Benito MC, Reolid A, Abad‐Santos F and Dauden E (2020) Pharmacogenetics update on biologic therapy in psoriasis. Medicina (Kaunas) 56, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krejsgaard T, Ralfkiaer U, Clasen‐Linde E, Eriksen KW, Kopp KL, Bonefeld CM, Geisler C, Dabelsteen S, Wasik MA, Ralfkiaer E et al. (2011) Malignant cutaneous T‐cell lymphoma cells express IL‐17 utilizing the Jak3/Stat3 signaling pathway. J Invest Dermatol 131, 1331–1338. [DOI] [PubMed] [Google Scholar]
- 53. Noubade R, Krementsov DN, Del Rio R, Thornton T, Nagaleekar V, Saligrama N, Spitzack A, Spach K, Sabio G, Davis RJ et al. (2011) Activation of p38 MAPK in CD4 T cells controls IL‐17 production and autoimmune encephalomyelitis. Blood 118, 3290–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Namiki K, Matsunaga H, Yoshioka K, Tanaka K, Murata K, Ishida J, Sakairi A, Kim J, Tokuhara N, Shibakawa N et al. (2012) Mechanism for p38α‐mediated experimental autoimmune encephalomyelitis. J Biol Chem 287, 24228–24238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR and Ashwell JD (2011) Lack of the T cell–specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood 118, 3280–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murphy LO, MacKeigan JP and Blenis J (2004) A network of immediate early gene products propagates subtle differences in mitogen‐activated protein kinase signal amplitude and duration. Mol Cell Biol 24, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen KE, Lin SY, Wu MJ, Ho MR, Santhanam A, Chou CC, Meng TC and Wang AH (2014) Reciprocal allosteric regulation of p38gamma and PTPN3 involves a PDZ domain‐modulated complex formation. Sci Signal 7, ra98. [DOI] [PubMed] [Google Scholar]
- 58. Hou SW, Zhi HY, Pohl N, Loesch M, Qi XM, Li RS, Basir Z and Chen G (2010) PTPH1 dephosphorylates and cooperates with p38gamma MAPK to increase ras oncogenesis through PDZ‐mediated interaction. Can Res 70, 2901–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krupa A, Preethi G and Srinivasan N (2004) Structural modes of stabilization of permissive phosphorylation sites in protein kinases: distinct strategies in Ser/Thr and Tyr kinases. J Mol Biol 339, 1025–1039. [DOI] [PubMed] [Google Scholar]
- 60. Jacobs D, Glossip D, Xing H, Muslin AJ and Kornfeld K (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13, 163–175. [PMC free article] [PubMed] [Google Scholar]
- 61. Babault N, Cordier F, Lafage M, Cockburn J, Haouz A, Prehaud C, Rey FA, Delepierre M, Buc H, Lafon M et al. (2011) Peptides targeting the PDZ domain of PTPN4 are efficient inducers of glioblastoma cell death. Structure 19, 1518–1524. [DOI] [PubMed] [Google Scholar]
- 62. Genera M, Samson D, Raynal B, Haouz A, Baron B, Simenel C, Guerois R, Wolff N and Caillet‐Saguy C (2019) Structural and functional characterization of the PDZ domain of the human phosphatase PTPN3 and its interaction with the human papillomavirus E6 oncoprotein. Sci Rep 9, 7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCoy AJ (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prehaud C, Wolff N, Terrien E, Lafage M, Megret F, Babault N, Cordier F, Tan GS, Maitrepierre E, Menager P et al. (2010) Attenuation of rabies virulence: takeover by the cytoplasmic domain of its envelope protein. Sci Signal 3, ra5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1A–D NMR spectra and CSP plots indicate residues in LBD of p38γ that have significantly chemically shifted in response to addition of β‐OG. The ratio of β‐OG to p38γ is 10:1. (E) The titration effects of β‐OG to the LBD residues of p38γ by 1H‐15N correlation NMR spectra. (A) NMR spectra. Overlay of the 1H‐15N HMQC spectra in the amide region for p38γ, free (red) and in complex with β‐OG (blue). (B) NMR spectra. Overlay of the 1H‐13C HMQC spectra in the methyl region for p38γ, free (red) and in complex with β‐OG (blue). (C) CSP plots. The cutoff value in NMR spectra for amide (ppm>0.02) is indicated by red arrows. (D) CSP plots. The cutoff value in NMR spectra for methyl (ppm>0.03) is indicated by red arrows.
Fig S1E A zoomed‐in view of the partial region in 1H‐15N correlation spectra of β‐OG with p38γ. Three residues, T244, E297 and M262 in LBD of p38γ are significantly chemically shifted by ligand β‐OG. β‐OG was titrated into isotope‐labeled p38γ. The different color (Red, Yellow, Green, Cyan, Blue) indicted by arrows on each side of three residues refer to a progressive chemically‐shift in respond to increasing concentration of β‐OG in the presence of p38γ (p38γ : β‐OG = 1:0 red, 1:2.5 yellow, 1:5 green, 1:7.5 cyan and 1:10 blue, respectively).
Fig S2A Pure CSH71 are isolated from CSH71 (NCI) by HPLC. A. Mass spectrum of CSH71 (Fraction 3 indicated by arrow.) CSH71 (NCI) is from NCI DTP and we showed it contains 3 fractions including nocodazole (Fraction 2), a well‐known cell cycle arrest agent that arrests cells at G2 /M phase.
Fig S2B Purified 3 (pure CSH71) by MASS and is ready for the further experiments such as NMR assays etc.
Fig S3A Biological assessments of CSH71 and β‐OG. A. Increasing concentrations of CSH71 (pure) were applied to Hut78 CTCL cells for 72 h.
Fig S3B Hut78 CTCL cells were treated with indicated concentrations of β‐OG for 72h.
Fig S3C–D (C) Increasing concentrations of CSH71 (pure) were applied to Hut78 CTCL cells for 72 h. (D) To determine p38γ kinase activity of CSH71 by in vitro kinase activity measured by ADP‐glo assays (blue).
Fig S4A 1D NMR STD spectra p38 in complex with CSH71 indicate CSH71 bound p38γ. STD NMR 1D 1H spectra overlay of compound CSH71 aromatic region in the presence of p38γ.
Fig S4B 1D NMR aromatic proton chemical shifts by CSH71 and STD values in complex with p38γ in the absence and presence of ATP.
Fig S4C The docking pose in ATP‐binding site more agrees with NMR STD result as five‐ring group is located inside the pocket, while –COOCH3 tail is outside (left). In lipid‐binding, more flexible binding of CSH71 in the LBD pocket (right).
Fig S5 (A) NMR 2D CSP with CSH71 spectra indicate CSH71 bound p38γ. A. 2D NMR CSP titration experiment with CSH71 spectra: overlay of the 1H‐15N HSQC spectra in the amide region for p38γ, free (red) and in complex with CSH71 (blue). (B) 2D NMR CSP titration experiment 1H‐13C HMQC spectra in the methyl region overlay for p38γ, free (red) and in complex β‐OG (blue). (C) Cut‐off value for Amide CSP is >0.02 ppm for selected shifted residues V33, G39, A40, V41, and Y326. (D) Cut‐off value for Methyl CSP is >0.02 ppm for selected shifted residues L58 and I149.
Data Availability Statement
The data that support the findings of this study are openly available in NCBI's Gene Expression Omnibus and are accessible through https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE182484, GEO Series accession number GSE182484.