

Abstract
Treatment with immune checkpoint blockade (ICB) has resulted in durable responses for a subset of patients with cancer, with predictive biomarkers for ICB response originally identified largely in the context of hypermutated cancers. Although recent clinical data have demonstrated clinical responses to ICB in certain patients with non-hypermutated cancers, previously established ICB response biomarkers have failed to accurately identify which of these patients may benefit from ICB. Here, we demonstrate that a replication stress response (RSR) defect gene expression signature, but not other proposed biomarkers, is associated with ICB response in 12 independent non-hypermutated cohorts of patients with cancer across 7 tumor types, including those of the breast, prostate, kidney, and brain. Induction or suppression of RSR deficiencies was sufficient to modulate response to ICB in pre-clinical models of breast and renal cancers. Mechanistically, we find that despite robust activation of checkpoint kinase 1 (Chk1) signaling in RSR-deficient cancer cells, aberrant replication origin firing causes exhaustion of replication protein A, resulting in accumulation of immunostimulatory cytosolic DNA. We further found that deficient RSR coincided with increased intratumoral dendritic cells in both mouse cancer models and human tumors. Together, this work demonstrates the RSR defect gene signature can accurately identify patients who may benefit from ICB across numerous non-hypermutated tumor types, and pharmacological induction of RSR defects may further expand the benefits of ICB to more patients.
One sentence summary:
Endogenous and induced replication stress response defects promote tumor immunogenicity via generation of cytosolic single-stranded DNA.
INTRODUCTION
Immune checkpoint blockade (ICB) has provided robust, durable benefit to a subset of patients. Many initial ICB trials were focused on highly mutated cancer types, such as melanoma and lung cancer, largely predicated on the idea that mutation-derived neoantigens would allow for generation of tumor-specific T cells (1). Subsequent analysis of patient responses in these highly mutated cancer types confirmed that increased tumor mutation burden (TMB) corresponded with improved patient outcomes (2, 3). The extreme hypermutator phenotype caused by deficiencies in DNA mismatch repair (MMR), which can be detected by microsatellite instability (MSI), has received FDA approval as the first tissue-agnostic biomarker for ICB therapy (4), with a second pan-cancer approval of high TMB (>10 mutations/megabase) as an ICB response biomarker (5). Further clinical studies in hypermutated cancers identified additional predictive biomarkers, such as programmed death-ligand 1 (PD-L1) protein expression (6), and various gene expression signatures (7–9).
We recently demonstrated distinct tumor immunobiology between hypermutated and non-hypermutated tumor types (10), finding that relative neoantigen load/tumor mutation burden was only a relevant factor for immune infiltration in hypermutated tumor types. Consistent with this finding, clinical data have failed to detect predictive value for TMB to identify ICB responders in many non-hypermutated tumor types, such as breast cancer (11), ccRCC (12) and GBM (13), and accumulating evidence suggests TMB does not universally predict sensitivity to ICB (14–16). Moreover, PD-L1 protein expression has shown mixed results for predicting response to ICB in TNBC (17, 18), and the utility of other transcriptional biomarkers remains poorly explored. Thus, there remains a critical gap in knowledge as to how to identify which patients with non-hypermutated cancer may benefit from ICB.
Defects in DNA damage response (DDR) pathways including MMR, homologous recombination repair, and nucleotide excision repair have been associated with ICB sensitivity (19). During deoxyribonucleic acid (DNA) synthesis, the replication machinery must overcome stalling due to obstacles such as DNA lesions, ribonucleic acid (RNA) transcriptional machinery, or insufficient nucleotides, collectively known as replication stress. To overcome these obstacles and maintain genomic integrity, cells activate replication stress response (RSR), which slows DNA replication to allow for repair, thereby preventing replication fork collapse (20). In normal cells, excessive replication-associated DNA damage, such as oncogene-induced hyperproliferation, will induce senescence or apoptosis, preventing tumorigenesis (21). Deficiencies in RSR facilitate growth of pre-malignant cells by supporting escape of this oncogene-induced senescence (22).
In this study, we demonstrated how RSR deficiencies from aberrant replication origin firing lead to accumulation of immunostimulatory cytosolic single-stranded DNA. Using pre-clinical murine breast cancer models, we found that RSR defects predicted sensitivity to ICB. Sensitivity or resistance to ICB could be caused by induction or suppression of RSR defects, respectively. Application of a transcriptional RSR defect (RSRD) score (23) predicted ICB response in 12 independent patient non-hypermutated tumor cohorts across 7 different tumor types. These findings may help identify which patients will benefit from ICB and also provide the biological rationale for clinical approaches to pharmacologically induce RSR defects that may expand the benefits of ICB to a greater number of patients.
RESULTS
RSRD gene expression score predicts functional replication stress response defects across breast cancer cell lines and primary organoid cultures.
We recently developed a gene expression signature indicative of RSR defects (RSRD) using isogenic mammary epithelial model cell lines (23). We applied this RSRD gene signature to a panel of 20 human breast cancer cell lines and found robust prediction of RSR function, as measured by ability to complete S-phase following release from hydroxyurea (HU)-induced replication stress (Fig. 1A and fig. S1, A to C), with an enrichment in RSR defects in TNBC cell lines. To ensure this result was not a cell culture artifact, we further validated it in primary organoid cultures that also showed robust prediction of RSR function (Fig. 1B). Inability to recover from HU-induced replication stress was confirmed at the level of single replication forks by DNA fiber analysis (Fig. 1C). RSRD-high cells also exhibited increased basal replication stress (24), as quantified by both decreased replication fork speed (fig. S1D) and increased phosphorylation of ataxia-telangiectasia and Rad3 related (ATR) (fig. S1, E and F). Increased endogenous replication stress was supported by accumulation of single-stranded DNA (ssDNA) detected by native 5-chloro-2’-deoxyuridine (CldU) staining, which was exacerbated by HU treatment (Fig. 1D).
Fig. 1. RSRD gene expression signature predicts functional RSR defects in breast cancer cell lines and organoids.
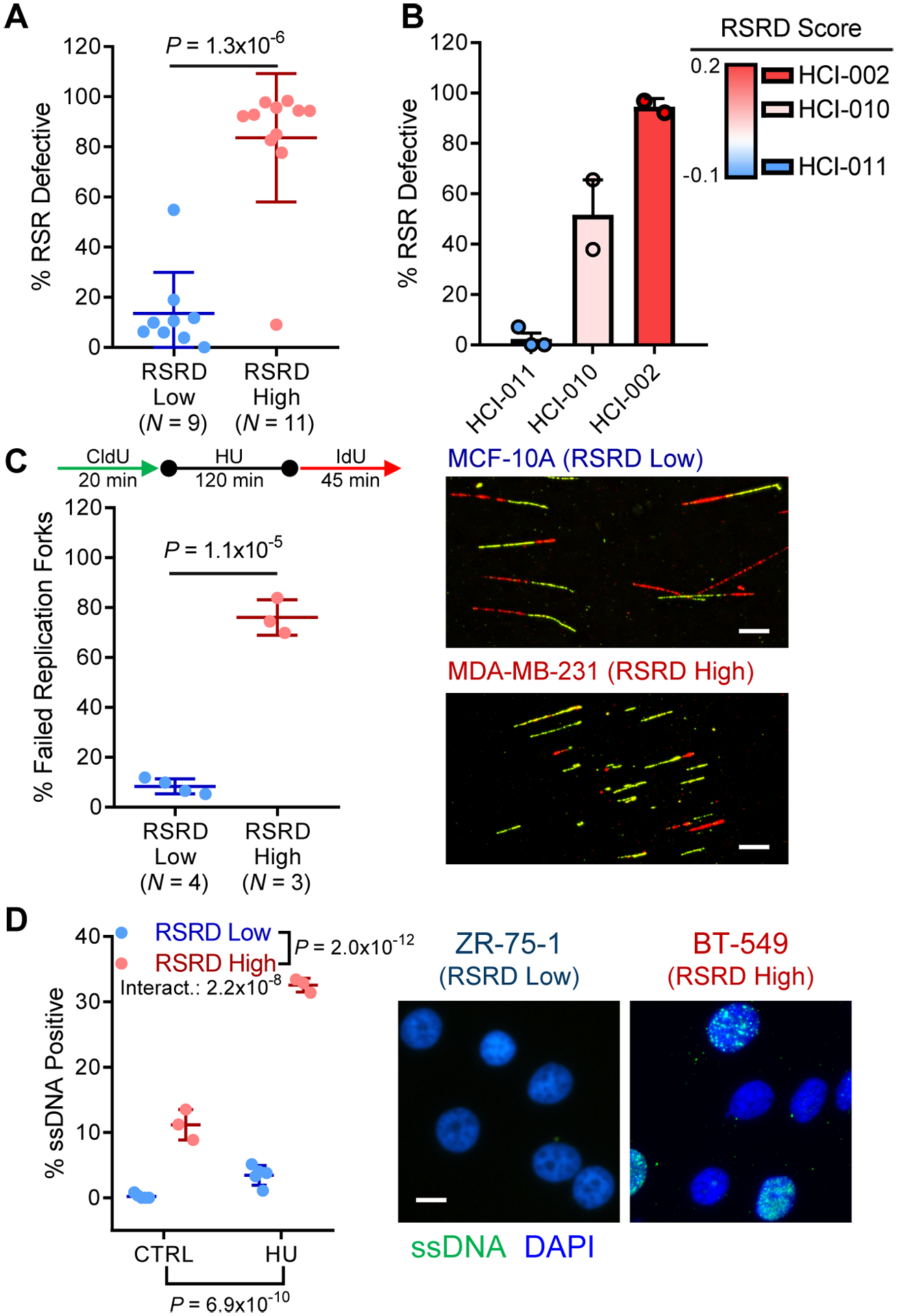
A, RSR function assessed by fraction of cells capable of completing cell cycle after hydroxyurea (HU)-induced replication stress (57). See fig. S1, A to C. Two-tailed t-test.
B, RSR deficiency as assessed by cell cycle assay in 3 organoid models derived from PDXs (58), sorted by RSRD score. Dots represent biological replicates.
C, Quantification of fork restart following HU-induced replication stress in a panel of RSRD-low and -high cell lines (left) and representative images (right). Replicating DNA was labeled with CldU (green) before stalling replication forks with HU. After HU washout, restarted replication forks were labeled with IdU (red). Percentage of failed replication forks was determined for at least 100 CldU-labeled fibers per cell line. Scale bar indicates 10 μm. Two-tailed t-test.
D, Native CldU staining to detect ssDNA (green) in control (CTRL) cells or following 6-hour incubation with HU along with nuclear counterstain (blue). Representative cell lines shown to the right. N = 3 cell lines per condition. Two-way ANOVA.
All data are mean ± s.d. with each dot representing an individual cell line.
Replication stress response defects are caused by aberrant origin firing driving RPA exhaustion from and lead to accumulation of immunostimulatory cytosolic DNA.
We next sought to mechanistically understand the observed RSR defects. Our original model system was based on defective checkpoint activation (25), though we detected no loss of DNA damage checkpoint activation in RSRD high cell lines as measured by checkpoint kinase 1 (CHK1) phosphorylation or by global phosphorylation of ataxia-telangiectasia mutated (ATM)/ATR substrates (Fig. 2, A and B). Nucleotide deficiency has also been implicated in oncogene-induced replication stress (26); however, we found no phenotypic changes following nucleoside supplementation (fig. S2, A and B). We next analyzed DNA synthesis following replication stress and found that whereas RSR-intact cells exhibited a gradual recovery of DNA synthesis, RSR-defective cells rapidly initiated DNA synthesis but ultimately failed to complete S-phase (Fig. 2C). Based on this observation, we hypothesized that RSRD cells may exhibit aberrant DNA replication origin firing despite activation of checkpoint kinases. Our hypothesis was supported by ultraviolet (UV)-resistant DNA synthesis in RSRD-high cells (Fig. 2D). We found that aberrant origin firing in RSRD high cells led to exhaustion of replication protein A (RPA) pools (Fig. 2E and fig. S2C) and rapid accumulation of DNA double-strand breaks (fig. S2D).
Fig. 2. RSRD-high cell lines undergo RPA exhaustion from aberrant replication origin firing causing accumulation of cytosolic DNA.
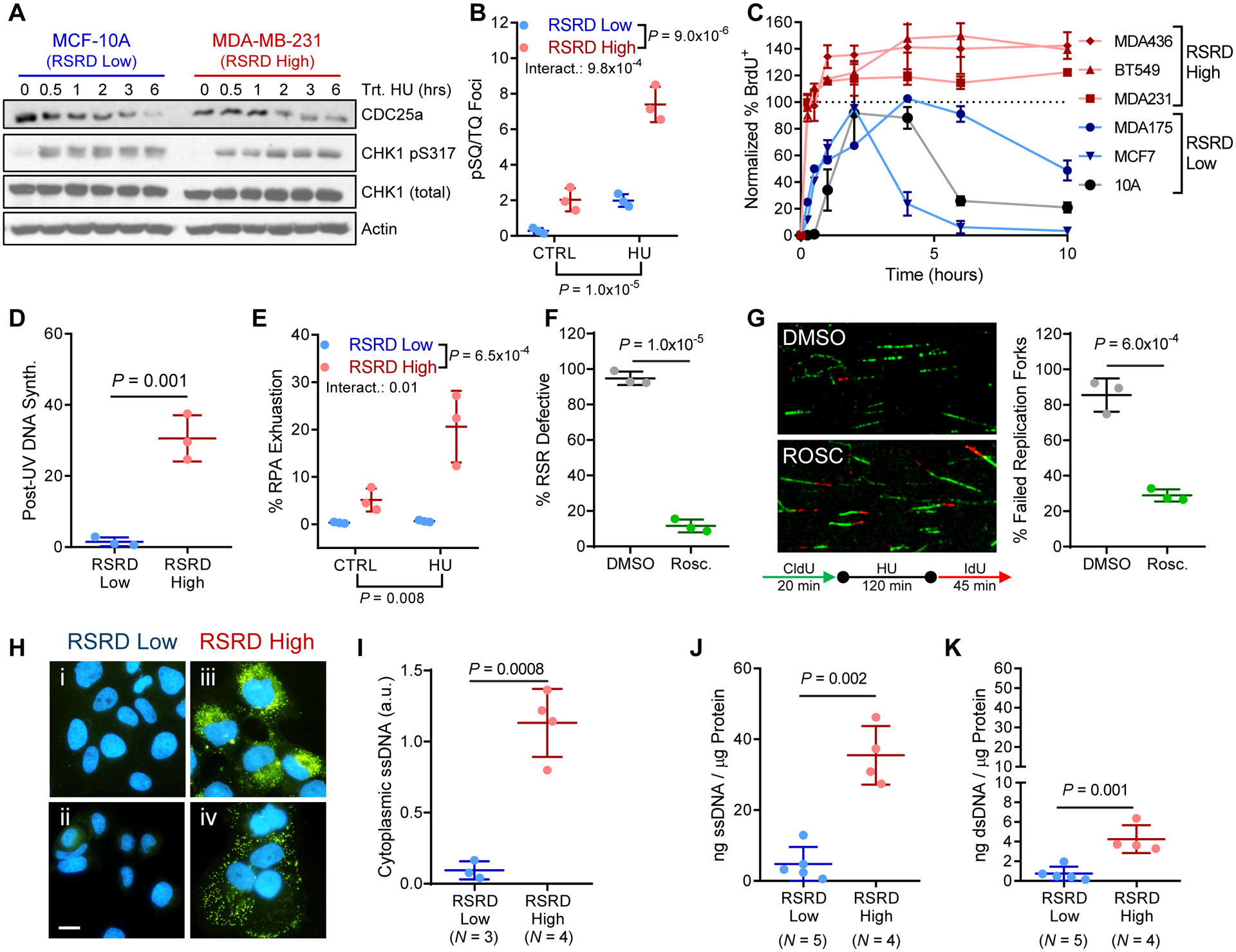
A, CHK1 activation following treatment with HU. RSRD-low MCF-10A and RSRD-high MDA-MB-231 show similar amounts of CHK1 phosphorylation and CDC25a degradation by Western blot.
B, Quantification of phosphorylation of chromatin-bound ATM/ATR substrates by staining with anti-pSQ/TQ (59). Cells were incubated in 2 mM HU or vehicle control (water) for 6 hours. Dots represent individual cell lines; mean ± s.d.; inset P values determined by two-way ANOVA.
C, Re-initiation of DNA synthesis following HU-induced replication stress. Cells were incubated with 2 mM HU for 16 hours and moved to fresh growth media for indicated times. BrdU was added 15 minutes prior to fixation to identify S-phase cells. Data reported as percentage of cells that are BrdU+ relative to vehicle-treated control cells.
D, UV-resistant DNA synthesis. S-phase cells were labeled with BrdU prior to UV-irradiation (5 J/m2) or mock-treated. Post-UV DNA synthesis was detected by EdU incorporation. Values reported as relative EdU intensity in BrdU positive cells in UV-irradiated compared to mock-treated cells. N = 3 cell lines per condition. Two-tailed t-test.
E, Percentage of cells with RPA exhaustion in control (CTRL) cells or following 6-hour incubation with HU. See fig. S2C. N = 3 cell lines per condition. Two-way ANOVA.
F, Percentage of cells that are RSR defective measured by ability to complete DNA synthesis after HU-induced replication stress per Fig. 1A, either following suppression of origin firing with 10 μM roscovitine or mock DMSO 2 hours prior to HU treatment and maintained throughout the experiment. N = 3 cell lines per condition. Two-tailed t-test.
G, Quantification of fork restart following HU-induced replication stress in RSRD-high cells either in presence or absence of suppression of origin firing with 10 μM roscovitine. N = 3 cell lines per condition. Two-tailed t-test.
H, Immunostaining for cytosolic ssDNA (green) and nuclei (blue) in RSRD-low (i, MCF-10A; ii, ZR-75–1) and RSRD-high cell lines (iii, BT549; iv, HCC38). Scale bar indicates 10 μm.
I, Quantification of cytoplasmic DNA immunostaining in (H) as integrated ssDNA intensity normalized to DAPI. Two-tailed t-test.
J and K, Quantification of cytosolic ssDNA (J) or dsDNA (K) concentrations, reported normalized to cytosolic protein concentration. Two-tailed t-test.
All data are mean ± s.d. with each dot representing the average value for an individual cell line.
To validate that aberrant origin firing was causing RSR defects, we used the cyclin-dependent kinase (CDK) inhibitor roscovitine to normalize origin firing. Treatment with roscovitine restored the ability of three independent RSRD-high cell lines to recover from HU-induced replication stress, as quantified both by bulk ability to complete the cell cycle (Fig. 2F) and at the level of single replication forks by DNA fiber analysis (Fig. 2G). Furthermore, in both presence and absence of HU, roscovitine treatment prevented accumulation of ssDNA (fig. S2E) and of double strand breaks in S-phase cells (fig. S2F).
Because roscovitine can inhibit origin firing/cell cycle through multiple CDKs (27), to better understand the mechanism of action we used siRNA to suppress expression of CDK1, CDK2, CDK5, CDK7, CDK9, CCNA1, and CCNA2. Although the percentage of cells in S-phase was slightly reduced by some siRNAs (fig. S2G), analysis of DNA damage specifically in cells actively synthesizing DNA revealed the largest suppression of DNA damage by suppression of CCNA2 (fig. S2H). Consistent with this, expression of CCNA2 was positively correlated with RSRD score in patients with breast cancer (fig. S2I). Evaluation of RSR function in TNBC cell lines with stable suppression of CCNA2 revealed that CCNA2 suppression could phenocopy the restored RSR function observed with treatment with roscovitine (fig. S2, J to L).
Based on the depletion of RPA pools necessary to protect ssDNA in RSRD-high cells, we hypothesized that the unprotected ssDNA may accumulate within the cytoplasm (28). Immunostaining for cytosolic ssDNA revealed accumulation in RSRD-high cell lines (Fig. 2, H and I), which was validated by direct quantification of cytosolic ssDNA following cellular fractionation (Fig. 2J). Increased accumulation of cytoplasmic double-stranded DNA (dsDNA) was also observed, albeit at an order of magnitude lower in quantity (Fig. 2K). Capillary electrophoresis of the precipitated cytosolic DNA further suggested the primary species was ssDNA, as most of the precipitated cytosolic DNA was sensitive to digestion by nucleases specific to ssDNA, but not by nucleases specific to dsDNA (fig. S3A). Further analysis of the size of precipitated cytosolic DNA fragments revealed a log-normal distribution of lengths centered around approximately 100 bases in length, with an average of 90.7% falling under the size limitation for passive diffusion out of the nucleus(29) (fig. S3, B and C). Retention of larger DNA fragments above the passive diffusion size limit in the nucleus may explain the observed relatively conserved size distribution of cytosolic DNA fragments. Similarly, suppression of nucleases RBBP8 (CtIP), DNA2, EXO1, and MRE11A indicated that although EXO1 may generate a portion of this ssDNA, the majority is likely generated by replication fork collapse as opposed to exonuclease activity (fig. S3, D to F).
RSRD score predicts response to immune checkpoint blockade in pre-clinical murine breast cancer models.
Healthy, unperturbed mammalian cells have minimal accumulation of cytosolic DNA, and cytosolic nucleic acid content derived from various sources such as viral infection or endogenous DNA damage binds to certain receptors, activating immunostimulatory pathways to restore homeostasis (30). Based on this immunostimulatory role for cytosolic DNA, we hypothesized that ICB may show increased efficacy in RSRD-high tumors. To test this, we analyzed RSRD score in panel of murine breast tumor models and selected five models with varying RSRD scores for further study: 2208L, 4T1, HRM1, T11, and E0771 (Fig. 3A). Using this panel, we first validated that the RSRD signature predicted functional RSR defects in murine breast cancer models, finding that RSRD score was also functionally predictive in murine breast cancer models (Fig. 3B). Furthermore, we found that RSR defective murine breast cancer models displayed increased cytosolic DNA (Fig. 3, C and D), consistent with our observation in human RSR defective breast cancer (fig. 2, J and K).
Fig. 3. RSRD score predicts response to immune checkpoint blockade in pre-clinical breast cancer models.
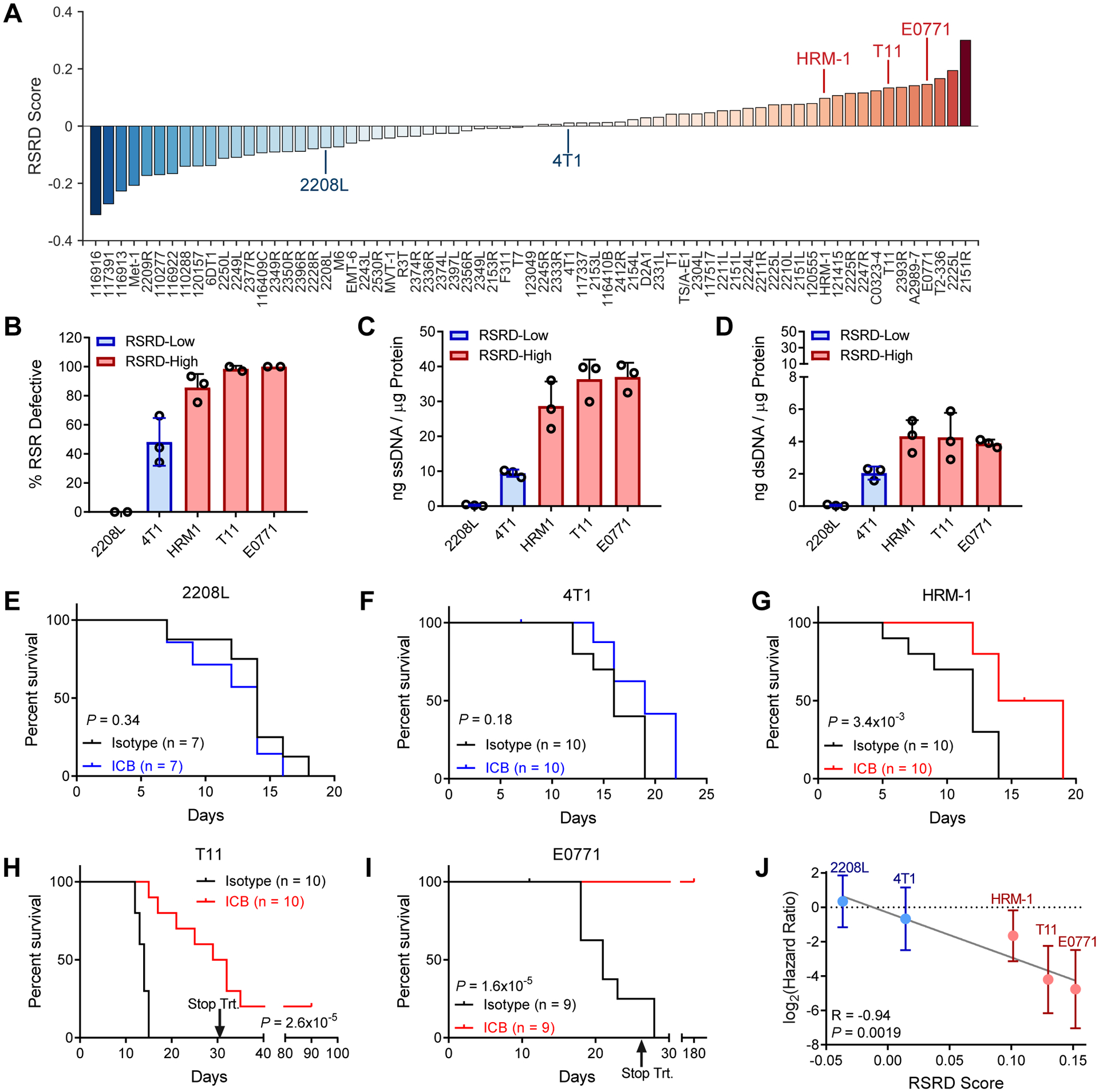
A, RSRD scores across mouse breast cancer models, with models selected for further analysis indicated.
B, Percentage of cells that are RSR-defective by HU cell cycle assay described in Fig. 1A, plotted as a function of RSRD score for selected models.
C and D, Quantification of cytosolic single-stranded (C) and double-stranded (D) DNA as described in Fig. 2, J and K for selected models. Each dot represents a biological replicate.
E to I, Kaplan Meier plots for selected RSRD-low (E, 2208L; F, 4T1) and RSRD-high (G, HRM-1; H, T11; I, E0771) in vivo models following treatment with ICB (10 mg/kg anti-PD1 and 5 mg/kg anti-CTLA4, thrice weekly) compared to isotype control. Significance determined by log-rank test.
J, Hazard ratio with 95% confidence interval determined by log-rank test for ICB treatment for models in (E to I) plotted as function of RSRD score. R, P values determined by Pearson correlation.
After confirming the RSRD signature was functionally active in murine breast cancer models, we next tested whether RSRD may be predictive of response to ICB in this system. ICB treatment failed to improve survival in either RSRD-low model (Fig. 3, E and F), but did improved survival in all three RSRD-high models (Fig. 3, G to I). Overall, there was a strong correlation between a higher RSRD score and improved ICB response, as indicated by a lower hazard ratio upon ICB treatment (P = 0.0019, Fig. 3J).
Modulating replication stress response function can induce sensitivity and resistance to immune checkpoint blockade.
To test if the RSRD phenotype is functionally associated with response to ICB, we first sought to evaluate if restoring RSR function would mitigate response to ICB. Consistent with inhibition of origin firing with roscovitine restoring RSR function in human cells, treatment of murine models with roscovitine was sufficient to mitigate cytosolic ssDNA (Fig. 4, A and B). For in vivo testing, we pre-treated RSRD-high T11 tumors with roscovitine for 3 days to restore RSR function, and then repeated the ICB treatment with continual roscovitine administration. While roscovitine monotherapy slightly improved overall survival, presumably by slowing cell proliferation, it abrogated any benefit achieved by treatment with ICB (Fig. 4C). To confirm that these effects were specific to restoration of RSR function, and not caused by roscovitine inhibiting immune cell function, we generated cell lines with stable suppression of Ccna2 in RSRD-high T11 cells (Fig. 4D). Stable suppression of Ccna2 restored RSR function (Fig. 4E) and reduced accumulation of cytosolic ssDNA (Fig. 4F), but did not alter the percentage of cells synthesizing DNA (Fig. 4G). In vivo treatment of shCcna2 or shCTRL cells with ICB confirmed that restoring RSR function by suppression of Ccna2 could induce resistance to ICB (Fig. 4H). Although depletion of STING moderately suppressed ICB response, it failed to do so to the same degree observed with restoration of RSR function (fig. S4, A and B), suggesting multiple cytosolic DNA sensors may be activate in RSRD-high tumors.
Fig. 4. ICB response can be modulated by induction or suppression of RSR defects.
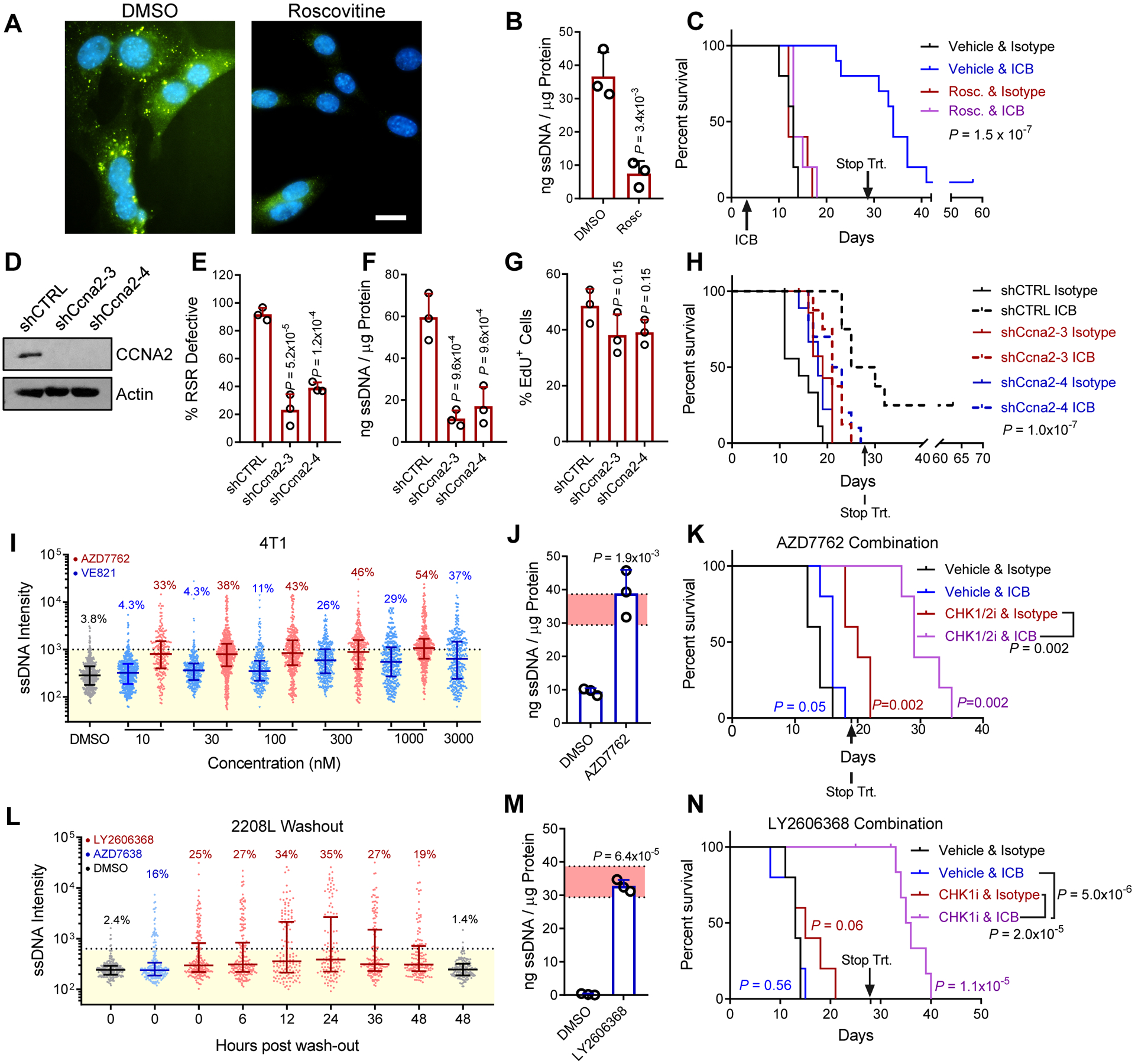
A, Immunostaining for ssDNA (green) and nuclei (blue) in RSRD-high T11 cells following treatment with 10 μM roscovitine (CDK inhibitor) for 36 hours. Scale bar indicates 10 μm.
B, RSRD-high T11 cells were treated with 10 μM roscovitine (CDK inhibitor) for 36 hours and then analyzed for cytoplasmic ssDNA as in 2J. Dots represent 3 biological replicates, values given as mean ± s.d. Two-tailed t-test.
C, Kaplan Meier plots for mice bearing RSRD-high/ICB-sensitive T11 tumor model treated with ICB in presence or absence of roscovitine. Roscovitine treatment started 3 days before initiation of ICB or IgG control antibodies. Treatment was ceased after 4 weeks. N = 10 mice per group. Significance determined by log-rank test.
D, Western blot confirming suppression of CCNA2 in T11 cells with stable expression of two shRNAs targeting Ccna2 (shCcna2-3, shCcna2-4) or non-targeting shRNA control (shCTRL).
E, Percentage of EdU positive T11 cells with stable expression of indicated shRNAs. One-way ANOVA and Holm-Sidak post-hoc test.
F, Percentage of RSR defective cells, determined as described in Fig. 1A, in T11 cells with stable expression of indicated shRNAs. One-way ANOVA and Holm-Sidak post-hoc test.
G, Quantification of cytosolic ssDNA, determined as described in Fig. 2J, in T11 cells with stable expression of indicated shRNAs. One-way ANOVA and Holm-Sidak post-hoc test.
H, Kaplan Meier plots for mice bearing RSRD-high T11 tumors with stable expression of indicated shRNAs treated with ICB or IgG control antibodies as described in Figure 3E, ceasing after 4 weeks. N = 8–10 per group. Log-rank test.
I, Staining for ssDNA after 12-hour treatment with varying concentrations of AZD7762 (CHKi), VE821 (ATRi), or DMSO vehicle control in RSRD-low 4T1 cells. Each dot represents and an individual cell.
J, Quantification of extracted cytosolic ssDNA relative to protein concentration in RSRD-low 4T1 cells following inhibition of CHK (100 nM AZD7762) for 24 hours and then analyzed for cytosolic ssDNA as in Fig. 2J. Shaded red area indicates mean ± std of RSRD-high cell lines. Dots represent 3 biological replicates, values given as mean ± std. Two-tailed t-test.
K, Kaplan Meier plots for mice bearing RSRD-low/ICB-resistant 4T1 tumors treated with either vehicle control, CHKi (25 mg/kg AZD7762, days 1/2), ICB (10 mg/kg anti-PD1 days 2/4/6), or combination of ICB and CHKi. Treatment ceased at day 19 (after three cycles) due to combination toxicity. N = 5 mice per arm, log-rank test.
L, Washout assay staining for ssDNA following 12-hour treatment with 100 nM LY2606368 (CHKi), 1 μM AZD6738 (ATRi) or DMSO vehicle control in RSRD-low 2208L cells. Each dot represents and an individual cell.
M, RSRD-low/ICB-resistant 2208L cells were treated with CHK1i (100 nM prexasertib) for 24 hours and then analyzed for cytoplasmic ssDNA as in Fig. 2J. Shaded red area indicates mean ± std of RSRD-high cell lines. Dots represent 3 biological replicates, values given as mean ± std. Two-tailed t-test.
N, Kaplan Meier plots for mice bearing RSRD-low/ICB-resistant 2208L tumors treated with either vehicle control, ICB, CHK1i (prexasertib), or combination of ICB and CHK1i. Treatment was ceased after 4 weeks. N = 10 mice per group. Significance determined by log-rank test.
After observing that restoration of RSR function could abrogate response to ICB, we next sought to test if induction of RSR defects could sensitize RSRD-low models to ICB, with the ultimate goal of identifying treatment strategies to expand the benefit of ICB to more patients. The top two clinically-relevant options for induction of RSR defects are inhibitors of ATR and CHK1, both critical regulators of RSR. Comparing the ATR inhibitor VE821 and CHK1/2 inhibitor AZD7762, we found that across all concentrations evaluated, CHK inhibition produced more ssDNA than ATR inhibition (Fig. 4I). Cytosolic fractionation validated that AZD7762 increased cytosolic ssDNA amounts compared to those observed in RSRD-high models (Fig. 4J). Combination of AZD7762 with ICB showed promising initial tumor growth responses, but ultimately the combination therapy proved too toxic. Despite stopping treatment after only 3 cycles, the combination still improved survival compared to either monotherapy alone (Fig. 4K).
To seek further improvement of this combination strategy, we next utilized the more CHK1-specific inhibitor prexasertib/LY2606368, which also induced more ssDNA than the ATR inhibitor AZD6738 at clinically achievable concentrations (fig. S4C). We recently found that Wee1 and ATR inhibitors exhibit persistent activity after drug removal, which could be leveraged to minimize toxicity observed in the context of combination with PARP inhibition (31). To evaluate if this concept was extendable to CHK inhibition, we analyzed ssDNA following LY2606368 washout for 48 hours and observed similar persistence (Fig. 4L). As observed with AZD7762, LY2606368 raised cytosolic ssDNA amounts to those observed in RSRD high cells (Fig. 4M). Based on the persistence of ssDNA for at least 48 hours following CHK1i washout, and to minimize combinatorial toxicity, CHK1i was administered only on days 1 and 4. Treatment of mice bearing highly ICB-resistant, RSRD-low 2208L tumors, with this dual ICB/CHK1i combinatorial strategy led to significantly improved survival compared to vehicle control (P = 1.1×10−5), CHKi alone (P = 2.0×10−5), and ICB alone (P = 5.0×10−6), whereas neither ICB (P = 0.56) nor CHKi (P = 0.06) alone significantly improved survival compared to vehicle control (Fig. 4N). No significant changes in mouse weights were observed using this therapeutic regimen (P = 0.99) (fig. S4D).
RSRD-high breast cancers have increased infiltration of dendritic cells and T cells in RSRD-high breast cancer.
To understand changes in the immune microenvironment between RSRD-high and RSRD-low tumors, we sought to understand how the RSRD score correlated with tumor cell expression of chemokines and cytokines. Altered chemokine/cytokine gene expression analysis in RSRD-high patient samples could be attributed to either modulation of expression in the tumor cells themselves, or cytokines secreted by RSRD-high tumor cells recruiting immune cells that in turn express additional chemokines/cytokines. To decouple alterations in chemokine/cytokine expression from tumor and immune cells, we looked for chemokines/cytokines correlated with RSRD score in both pure cell line cultures (Fig. 5A) and The Cancer Genome Atlas (TCGA) breast cancer samples (Fig. 5B). We found that RSRD score was significantly (FDR < 10%) positively correlated with expression of IL1A, IL1B, interferon alpha (IFNA1), GM-CSF (CSF2), IL11, and CXCL3 in both cell lines and primary breast cancer samples. Next, we used xCell (32) in the TCGA breast cancer cohort to interrogate how these increased cytokines/chemokines altered the immune cell microenvironment. After stratifying by subtype, numerous cell populations showed significant (FDR < 5%) Spearman correlation coefficients with RSRD score, including increases in multiple T-cell and dendritic cell populations (Fig. 5C).
Fig. 5. Immune microenvironment changes in RSRD-high breast cancer.
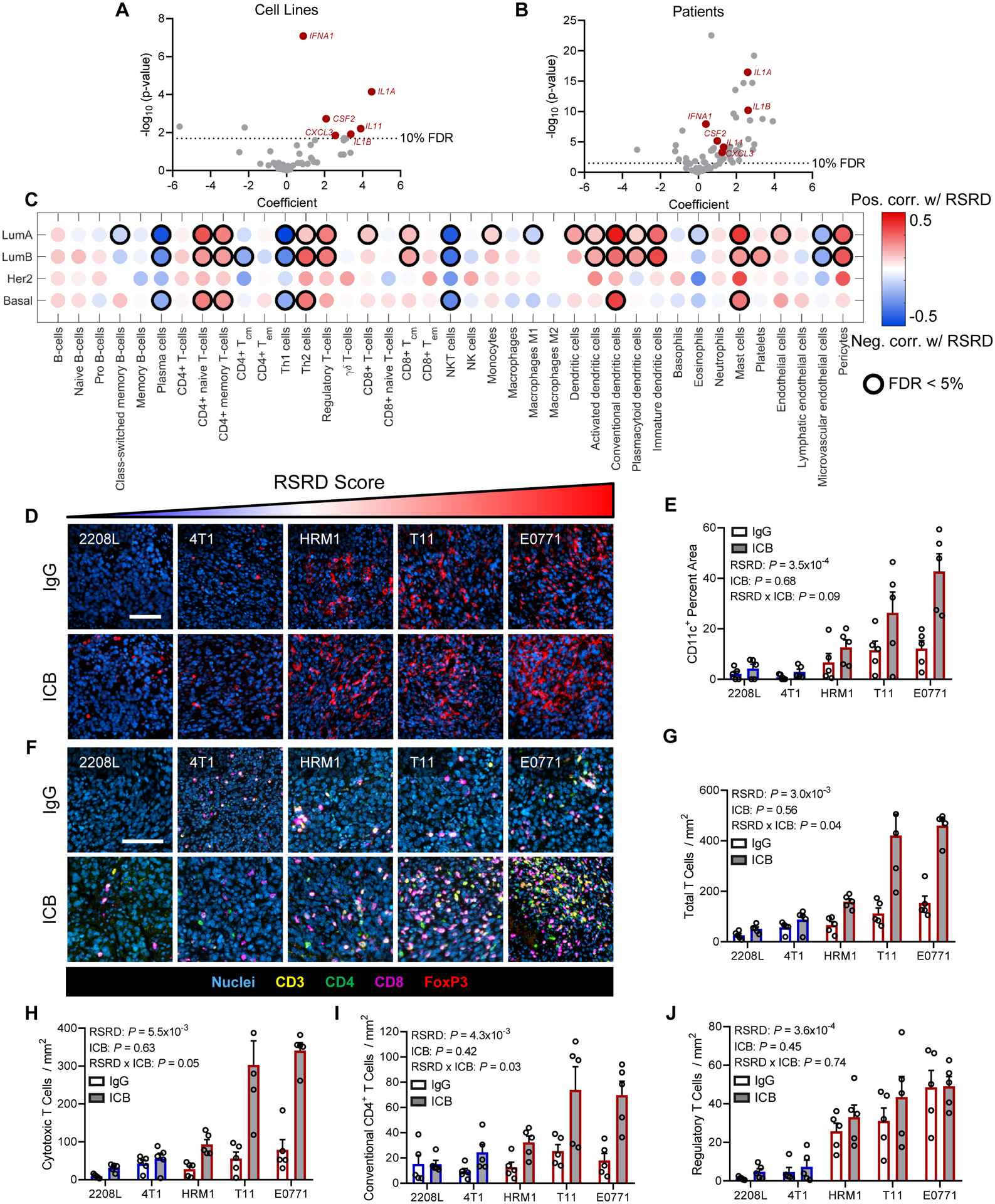
A and B, Relationship between RSRD score and expression of cytokines/chemokines in breast cancer cell lines (A) and samples from patients with breast cancer from TCGA (B). Highlighted cytokines indicate those significant in both cell lines and patients.
C, In silico immune populations from TCGA patients with breast cancer as determined by xCell. Dot color indicates magnitude of Spearman correlation coefficient. Relationships with an FDR<0.05 are indicated by black circles.
D, Representative images showing nuclei (blue) and dendritic cells (CD11c, red). Scale bar = 100 μm. Mice treated per Figure 3E–I for 10 days. IgG, IgG controls; ICB, immune checkpoint blockade.
E, Quantification of images from D. Dots represent tumors from independent mice (N = 5), values given as mean ± SEM. Significance determined by regression of average value for CD11c area per mouse with RSRD score and treatment status.
F, Representative multispectral images showing nuclei (blue), CD3 (all T cells, yellow), CD4 (CD4 T cells, green), CD8 (cytotoxic T cells, magenta), and FoxP3 (regulatory T cells, red). Scale bar indicates 100 μm. Mice treated per Fig. 3, E to I for 10 days.
G to J, Quantification of multispectral images from F, indicating total T cells (CD3+, G), cytotoxic T cells (CD3+CD8+, H), conventional CD4 T cells (CD3+CD4+FoxP3−, I), and regulatory T cells (CD3+CD4+FoxP3+, J). Dots represent tumors from independent mice (N = 5), values given as mean ± SEM. Significance determined by regression of average population value per mouse with RSRD score and treatment status.
We next sought to validate these computational predictions using our mouse pre-clinical models. For these experiments, tumors were immunostained following 10 days of treatment with either ICB or IgG controls. Quantification of dendritic cell infiltrates by staining for CD11c validated that RSRD high tumors were enriched in CD11c+ cells, which were further increased by treatment with ICB (Fig. 5, D and E). To analyze T-cell infiltrates, we developed a multispectral staining panel to probe CD3, CD4, CD8, and FoxP3 (Fig. 5F). Quantitative analysis of this panel indicated total T cells increased with RSRD score (P = 3×10−3), and that there was a significant positive interaction between RSRD score and ICB treatment (P = 0.04, Fig. 5G). Analysis of T cell subpopulations revealed a similar trend with cytotoxic CD8+ T cells (RSRD P = 5.5×10−3, RSRD*ICB P = 0.05, Fig. 5H) and conventional CD4+ T cells (RSRD P = 4.3×10−3, RSRD*ICB P = 0.03, Fig. 5I). Whereas Tregs were also increased with RSRD score as predicted (P = 3.6×10−4), there was no interaction effect observed with ICB treatment (Fig. 5J). Based on elevated amounts of dendritic cells present, we hypothesized that there may be increased immunological synapse activity leading to heightened T cell activation. Analyzing gene expression for components of the immune synapse, or supramolecular activation cluster (SMAC), indicated that RSRD-high tumors had increased expression of both central SMAC (c-SMAC) and peripheral SMAC (p-SMAC) components (Fig. S4E). Activation of c-SMAC was further supported by increased phosphorylation in RSRD-high tumors (fig. S4F) and may contribute to more active T cells in RSRD-high tumors. Consistent with these observed alterations in the immune microenvironment, we found that response to ICB in RSRD-high tumors was dependent on T cells because ICB treatment in nude mice lacking T cells no longer provided any survival benefit (fig. S4G), indicating that the observed response is not attributable to antibody-mediated cytotoxicity. To confirm this result, we re-challenged mice that had undergone complete tumor regression with ICB with tumor cells. Naïve immunocompetent mice implanted with RSRD-high E0771 and T11 tumors showed delayed tumor formation compared to nude mice, with a much longer delay in tumor formation in mice that had previously undergone a complete response to ICB (fig. S4, H and I).
High RSRD score predicts response to ICB in metastatic breast cancer patients.
In the TONIC breast cancer trial (11), a biopsy from a metastatic lesion was taken, and then patients were randomized to no treatment or four different low-dose induction treatment strategies for a two-week period to induce sensitivity to ICB. The four induction approaches utilized all induced DNA damage: (i) direct generation of DNA double-strand breaks with irradiation, (ii) generation of interstrand and intrastrand crosslinks with cyclophosphamide, (iii) inhibition of topoisomerase with the DNA intercalating agent doxorubicin, and (iv) generation of intrastand crosslinks with cisplatin. A second biopsy was taken prior to initiation of anti-PD1 therapy (Fig. 6A). Using RNA-seq data from biopsies collected immediately prior to anti-PD1 treatment (biopsy 2), we assessed the ability of RSRD score to predict response to anti-PD1 treatment and found that RSRD score predicted response, demonstrated by the receiver-operator characteristic (ROC) curve area under the curve (AUC) value of 0.78 (Fig. 6B). This predictive ability was moderately diminished by use of the pre-induction biopsies (fig. S5A). The original TONIC publication identified presence of stromal tumor-infiltrating lymphocytes (sTILs) and PD-L1 expression on immune cells by IHC as potential response biomarkers (11). Analyses of sTILs invasion and immune cell PD-L1 expression indicated that RSRD score is an independent biomarker of ICB response (fig. S5, B to D). Using in silico deconvolution of RNA-seq data, we also found a correlation between RSRD score and dendritic cells (Fig. 6C), matching prior in silico analysis of TCGA patient samples (Fig. 5C) and immunostaining of pre-clinical mouse models (Fig. 5, D and E).
Fig. 6. RSRD score is a dynamic biomarker to predict ICB response in metastatic breast cancer patients.
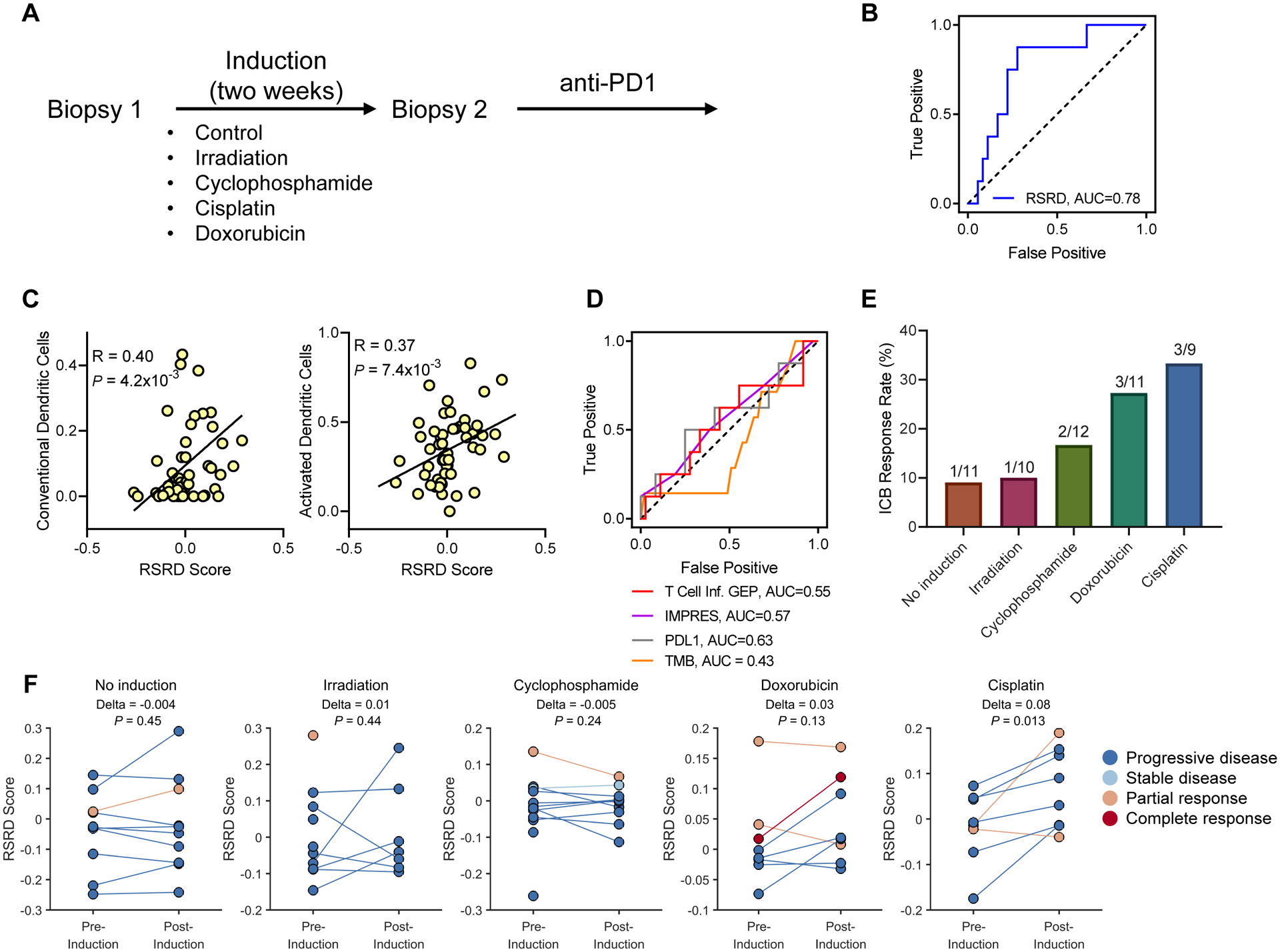
A, Schematic for metastatic breast cancer TONIC trial design (11).
B, ROC curves demonstrating ability of RSRD score to predict patients who achieved CR/PR/SD for at least 24 weeks following ICB treatment using RNA-seq from latest biopsy available.
C, Correlation of conventional/activated dendritic cells and RSRD score in the TONIC cohort. Spearman correlation coefficient.
D, ROC curves for alternative published ICB response biomarkers T Cell Inflamed (9, 33) gene expression signature, IMPRES (7), PD-L1 expression, and tumor mutation burden.
E, Response rate by induction treatment for patients in TONIC RNA-seq cohort.
F, Change in RSRD score following various induction strategies. Each pair of dots represents an individual patient (or single dots for patients lacking post-induction transcriptional data). Wilcoxon signed-rank test.
We recently demonstrated distinct tumor immunobiology between non-hypermutated tumor types, including breast cancer, and hypermutated tumors, such as melanoma and lung cancer, notably that relative neoantigen load/TMB was only a relevant factor in hypermutated types (10, 15). Consistent with this result, we found that TMB did not predict ICB response in the TONIC RNA-seq breast cancer cohort, consistent with prior results from the entire cohort (11) (Fig. 6D). We also compared the predictive ability of the RSRD score to PD-L1 expression and other published transcriptional biomarkers, including the T cell inflamed gene expression signature score, which was generated using multiple cancer types and has been applied across cancer types (9, 33) and the IMPRES gene expression score, which was generated from neuroblastoma data and validated in melanoma using the most independent cohorts published to date (N = 8) (7), but found that no other biomarker offered strong predictive ability (Fig. 6D).
When comparing patients within the RNA-seq TONIC cohort, we found that the various induction strategies induced varying response rates to anti-PD1 therapy (Fig. 6E), as was also described for the entire TONIC cohort in the original publication (11). The subset of samples with RNA-seq data generally mirrored response rates in the overall cohort, except for a slightly lower response rate for doxorubicin-induced (original = 35%, RNA-seq = 27%), and slightly higher response rate for cisplatin-induced (original = 23%, RNA-seq = 33.3%). To test if the RSRD gene expression signature could detect dynamic changes related to the induction treatments, we evaluated RSRD scores before and after various induction treatments (Fig. 6F). Patients who underwent no induction showed minimal change in RSRD score. Within the RNA-seq cohort, cisplatin-treated patients had the highest response rate, and we also detected a significant increase in RSRD score following induction with cisplatin (P = 0.01, Fig. 6F).
High RSRD score predicts ICB response in non-hypermutated tumor types.
We next sought to evaluate if prediction of ICB response by RSRD score could be generalized to other non-hypermutated cancer types treated with ICB. Analysis of an independent cohort of patients with cancer treated with ICB (3) confirmed the observation from the TONIC cohort that TMB does not offer prognostic value for ICB response in breast cancer (fig. S6, A and B). TMB also showed no prognostic value in ICB-treated patients with other non-hypermutated cancers, namely RCC and glioma (fig. S6, A to C). As was observed in breast cancer cell lines, RSRD-high ccRCC and GBM cells both exhibit increased cytosolic DNA, indicating that the RSRD signature is active in these other tumor types (fig. S7, A and B). To further evaluate the activity of the RSRD score in RCC, we next analyzed RSRD scores for two murine RCC tumor models, RENCA and RAG, compared to tumors derived from genetically engineered mouse models of kidney cancer driven by targeted deletion of either Vhl and Pbrm1 (34) or Vhl, Trp53, and Rb1 (35), as well as healthy kidney controls. RAG cells exhibited a high RSRD score, with a more intermediate score for RENCA cells (fig. S7C). As observed in murine breast cancer, high RSRD scores corresponded with functionally defective RSR (fig. S7D), increased accumulation of cytosolic ssDNA (fig. S7E), and increased sensitivity to treatment with ICB (fig. S7, F and G). Moreover, the combination of ICB with the CHK1 inhibitor prexasertib/LY2606368 in mice bearing RENCA tumors resulted in improved overall survival compared to either agent alone (fig. S7H), consistent with our observations in breast cancer models (Fig. 4, K and N).
Based on the ability of RSRD score to predict ICB response in breast cancer and observed activity in other pre-clinical models, we hypothesized that RSRD score may more broadly be predictive of clinical benefit in other non-hypermutated cancer types. To test if patients with RSRD-high ccRCC tumors exhibited increased response to ICB, we acquired samples from four cohorts of patients with ccRCC treated with ICB; a cohort treated with anti-PD1 from Ascierto et. al. (36); a cohort from NCT01358721 and an archival FFPE cohort treated with anti-PD1 from Miao et. al. (37); and patients treated with single agent anti-PD-L1 from the IMmotion150 trial (38). The RSRD score robustly predicted clinical responses to ICB across all 4 cohorts, as indicated by ROC curves with AUC values of 0.75 to 1.0 (Fig. 7A). Of other evaluated biomarkers, only the T cell inflamed gene expression signature score in the Miao archival cohort exceeded 0.7 (0.71), though it averaged an AUC of 0.48 in the other 3 cohorts analyzed. Furthermore, progression free survival (PFS) data from the Miao cohorts indicated that RSRD-high patients had improved outcomes by both univariate analysis (Fig. 7B) and multivariate analysis (Fig. 7C). Analysis of TCGA samples from patients with ccRCC who did not receive ICB demonstrated that high RSRD score was associated with worse prognosis, suggesting that improved survival in RSRD-high patients is ICB-specific (fig. S8, A and B). Previous studies have indicated that high amounts of endogenous retroviruses (39) and neoantigens (40) may promote ccRCC immunogenicity, but we did not find RSRD score to be associated with these factors (fig. S8, C to H).
Fig. 7. RSRD score predicts ICB response across multiple cancer types.
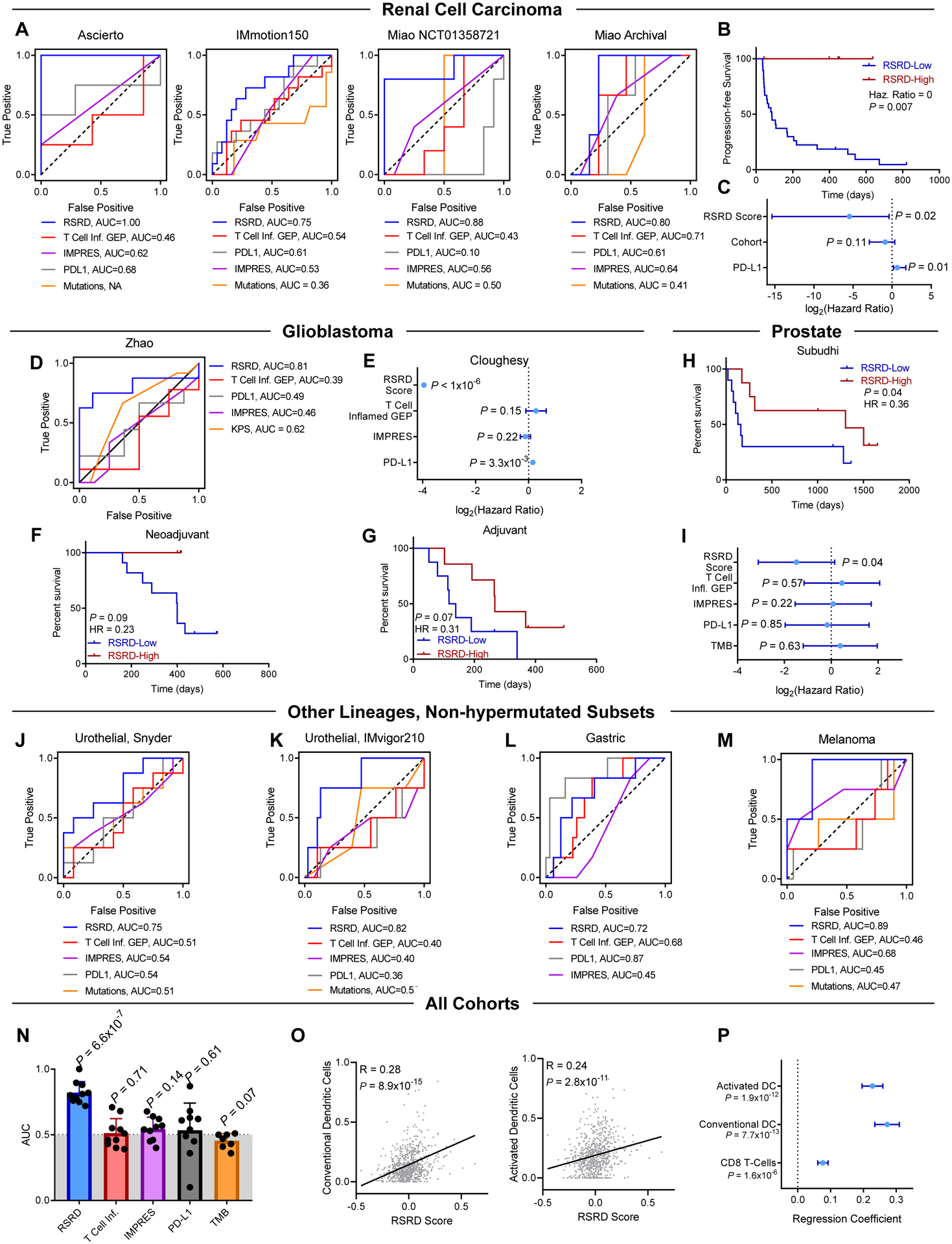
Prediction of response to ICB by published biomarkers T Cell Inflamed (9, 33) gene expression signature, IMPRES (7), PD-L1 expression, and mutation burden compared to RSRD score.
A, ROC curves demonstrating prediction of ICB response indicated biomarkers in cohorts of patients with ccRCC (36–38).
B, Progression free survival (PFS) for all in patients from Miao cohorts (37) based on RSRD score. Log-rank P-value.
C, Multivariate analysis of PFS in Miao cohorts (37) accounting for RSRD score, whether patients were treated in the first line, and PD-L1 expression, stratified by NCT1358721 vs archival cohort.
D, ROC curve showing ability of RSRD score to predict response in patients with GBM treated with anti-PD1 from the Zhao (41) cohort.
E, Overall survival analysis of anti-PD1-treated patients with GBM (13), considering treatment in either the neoadjuvant or adjuvant context as a clustering variable. All parameters were treated as continuous variables.
F and G, Kaplan-Meier curves showing overall survival from the Cloughesy (13) cohort treated either in the neoadjuvant (F) or adjuvant (G) context, and stratified by the RSRD score.
H, Overall survival of patients with metastatic prostate cancer treated with anti-CTLA4 stratified by RSRD score (53). Log-rank test.
I, Hazard ratio using overall survival data from (H) for indicated biomarkers. Error bars indicate 95% confidence interval. Log-rank test.
J to M, ROC curves showing ability of RSRD score and other indicated biomarkers to predict ICB response in low mutation subsets of patients with urothelial cancer treated with anti-PD-L1 from (J) Snyder (43) and (K) IMvigor210 (45), (L) patients with gastric cancer treated with anti-PD-L1 (54), and (M) patients with melanoma treated with anti-PD1 (55, 56).
N, Compilation of AUC values across all cohorts from Fig. 6 and 7 for evaluated biomarkers. P value assessed by one-sample t-test to determine significant difference from an AUC of 0.5 indicative of random assignment.
O, Correlation of conventional/activated dendritic cells and all patients treated with ICB. Spearman correlation coefficient.
P, Regression using a generalized linear mixed-effects model of RSRD score as a function of conventional dendritic cells, activated dendritic cells, or CD8 T-Cells. Patient cohorts were treated as a random effect. Regression coefficient with standard error.
Next, we sought to evaluate the predictive capability of the RSRD score in patients with GBM treated with ICB, which also exhibits the non-hypermutated phenotype of TMB/neoantigen-independent immunogenicity (10, 41). Analysis of RNA expression data from a patient cohort with GBM treated with anti-PD1(41) showed that the RSRD score alone robustly predicted clinical response (Fig. 7D). We confirmed this result in a second cohort of patients with GBM treated with anti-PD1 (13) in either the neoadjuvant or adjuvant setting. Although neoadjuvant treatment showed significantly better outcomes compared to adjuvant treatment (P = 0.01, fig. S8I), using a Cox proportional hazards model with treatment context as a clustering variable found that only a high RSRD score predicted improved overall survival (OS) (P <1×10−6, Fig. 7E). IMPRES score showed a trend towards improved prognosis, so we additionally performed multivariate analysis with both RSRD score and IMPRES score and found RSRD score still demonstrated robust prediction of outcomes following ICB treatment (fig. S8J). Despite small sample size in each arm, high RSRD score was still associated with improved OS regardless of whether anti-PD1 was administered in the neoadjuvant (HR = 0.23, P = 0.09, Fig. 7F) or adjuvant (HR = 0.31, P = 0.07, Fig. 7G) context. The RSRD score was not an independent prognostic biomarker in the TCGA cohort of patients with GBM who did not receive ICB (fig. S8K), suggesting the improved survival is specific to patients with GBM who received ICB.
We further validated the association between high RSRD score and improved response to ICB in a cohort of patients with metastatic prostate cancer treated with ipilimumab, a monoclonal antibody targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA4). Consistent with observations in ccRCC and GBM, we found that only RSRD score significantly predicted improved overall survival following treatment with anti-CTLA4 (HR = 0.36, P = 0.04, Fig. 7H–I). RSRD score was not prognostic in patients with prostate cancer in TCGA who would have predominately not received ICB (fig. S8L)
Our previous study also indicated that some tumor types may have both hypermutated and non-hypermutated subtypes which display distinct immunobiology, most strongly observed when comparing hypermutated and non-hypermutated urothelial cancers (10, 42). Although ICB responses tend to be enriched in patients with hypermutated urothelial cancer, a subset of patients with non-hypermutated tumors also exhibit clinical benefit (43–45). We hypothesized that the RSRD score may identify which patients with non-hypermutated urothelial cancer may benefit from ICB. To test this, we evaluated the ability of the RSRD score to predict response to anti-PD-L1 in two cohorts of patients with urothelial cancer (43, 45) stratified into hypermutated and non-hypermutated subsets. As hypothesized, in urothelial cancer the RSRD score showed good predictive accuracy in patients with non-hypermutated tumors (Fig. 7, J and K), but not in patients with hypermutated tumors or the unstratified bulk population (fig. S9, A to D). This phenomenon was similarly observed when analyzing cohorts of patients with non-hypermutated gastric cancer (Fig. 7L and fig. S9, E and F), and was observable in low-frequency non-hypermutated melanoma after pooling two cohorts for sufficient sample number (Fig. 7M and fig. S9, G to I). Consistent with observations in ccRCC and GBM, the RSRD score also outperformed other biomarkers in these non-hypermutated subsets (Fig. 7, J to M). Response rates between patients with hypermutated tumors and patients with RSRD-high/low-TMB tumors were comparable (63.1% vs 54.1%), and enriched over three-fold relative to biomarker-negative patients (16.8%) (Fig. S9, B, D, F, and I). Together, upon compiling all 11 cohorts with objective response rates, only the RSRD score showed substantial predictive accuracy (mean AUC = 0.82, P = 6.5×10−7), with the second highest average AUC observed using PD-L1 expression (mean AUC = 0.55) (Fig. 7N). Confidence intervals for individual cohort AUC values are shown in fig. S9J, along with plots showing the relationship between positive predictive value and negative predictive value in fig. S9, K to M. As observed in breast cancer (Fig. 5, c and E, and Fig. 6C), the RSRD score was also positively correlated with dendritic cell infiltration when analyzing all patient cohorts (Fig. 7O). This positive relationship between RSRD score and dendritic cells was conserved when using a generalized linear mixed effects model, with a weaker relationship between RSRD score and CD8 T-cells observed (Fig. 7P).
DISCUSSION
Despite observations of robust clinical responses to ICB in a subset of patients with tumors lacking extensive mutation burdens, we and others have observed a failure of biomarkers established in the context of hypermutated tumors to identify which patients with non-hypermutated cancer types may receive clinical benefit from ICB. This study identifies a gene expression signature reflective of defective replication stress response that is functionally linked with increased DNA damage, RPA exhaustion, aberrant replication fork firing, and accumulation of immunostimulatory cytosolic DNA. This RSRD signature accurately predicts response to ICB in patients with non-hypermutated cancers across 12 independent cohorts, whereas other proposed biomarkers including (PD-L1) protein expression (6), and various gene expression signatures (7–9) showed minimal predictive value. It is noteworthy that the RSRD score predicts ICB outcomes in non-hypermutated, yet immunologically distinct cancer types—highly T-cell-infiltrated ccRCC tumors and in marginally T cell-infiltrated GBM tumors for example. Future studies further detailing the immune microenvironment features that sensitize to ICB in RSR-deficient tumors will be necessary to understand the biology of ICB response.
A critical limitation the current study is the predictive capacity of our RSRD signature on retrospective data, requiring prospective validation. Moreover, technical details, including how recent a biopsy is required as well as tumor purity, will need to be considered when performing any prospective studies. Any combination trials with agents to induce RSR defects, including ATR or CHK1 inhibition, will need to carefully consider scheduling and/or dose de-escalation to avoid unintentional immunosuppression by these agents. It is probable that DDR inhibitor dosages required for inducing an immunologic response and sensitizing to ICB are lower than necessary for monotherapy. Thus, a focus in trial design on the minimal effective DDR inhibitor dose to achieve an immunologic effect will be critical to avoid immunosuppressive side effects that could counteract the benefit from activation of inflammatory signaling.
Here, we found that regardless of concentration or inhibitor used, the inhibition of CHK1/2 induced a more robust phenotype than that observed with ATR inhibition in RSRD-low cells. Although CHK1 is primarily activated by ATR in response to replication stress, if ATR is inhibited CHK1 can also be activated via DNA-dependent protein kinase (DNA-PK)(46). The activity of this DNA-PK compensatory pathway could explain the enhanced induction of the RSRD phenotype following CHK1/2 inhibition compared to ATR inhibition. Moreover, the CHK1/2 inhibitor AZD7762 can simultaneously inhibit both checkpoint kinases (cell-free CHK1 IC50 = cell-free CHK2 IC50 = 5 nM) (47), and although prexasertib/LY2606368 exhibits an order of magnitude higher affinity for CHK1 (cell-free IC50 = 0.9 nM) than CHK2 (cell-free IC50 = 8.0 nM), it would likely still inhibit both kinases to a degree (48). Our comparison of ATRi and CHK1/2i was also performed in the context of RSRD-low cells that generally have lower endogenous replication stress. Previous work has demonstrated that ATR inhibitors more specifically target cells with high replication stress compared to CHK1 inhibitors (46), so it is possible that repeating the comparison in cells with higher replication stress would produce a more equivalent phenotype.
Prospective studies to evaluate the predictive ability of the RSRD gene expression score, genotype-to-phenotype relationships in RSRD-high tumors, and effects of pharmacological induction of RSR defects, are underway. For example, NCT04266912 is a correlative-rich early clinical phase study of avelumab (anti-PD-L1) and ATR inhibitor M6620/VX970. Additional trials in progress include combination of ATR inhibitor BAY1895344 and pembrolizumab in solid tumors (NCT04095273), ATR inhibitor M6620/VX970 and pembrolizumab in squamous non-small cell lung cancer (NCT04216316), CHK1 inhibitor LY2606368 and LY3300054 anti-PD-L1 in solid tumors (NCT03495323), and ATR inhibitor AZD6738 and durvalumab in biliary tract cancers (NCT04298008).
Mutations in select DDR genes have previously been used as biomarkers for response to various therapeutic modalities, such as PARP inhibition. However, gene mutation information alone does not always fully inform functional DDR defects. Detected mutations do not always have a known functional relevance, and even with known mutations, DDR function can be restored through secondary events (49). Furthermore, DDR defects may arise by pathway modulation outside of mutational events (50). Thus, relying on DDR gene mutations alone to serve as predictive biomarkers for treatment selection may exclude patients who have functional deficiencies in DDR pathways. Utilization of gene expression-based biomarkers circumvents these problems and allows for dynamic monitoring of DDR function, including following therapeutic interventions as observed here for patients with breast cancer from the TONIC trial.
In conclusion, this work demonstrates that defects in replication stress response arise from aberrant replication origin firing, exhausting RPA pools and causing accumulation of immunostimulatory cytosolic ssDNA. The RSRD gene expression signature can dynamically detect defects in replication stress response and can predict clinical benefit to ICB in patients with non-hypermutated tumor types, who currently lack accurate predictive biomarkers for ICB treatment stratification. Use of ICB in patients with RSRD-high tumors, as well as combination therapy approaches using DDR inhibitors to induce RSRD-high phenotype with ICB, might expand the number of patients who may benefit from these treatment modalities, with prospective validation in multiple early phase clinical trials in development and underway.
MATERIALS AND METHODS.
Study design
The initial objective of this study was to understand causes of DNA replication stress response defects in breast cancer, as well as to identify any therapeutic vulnerabilities induced in RSR-defective cancer cells. After identifying that RSR-defective pre-clinical murine breast cancer models are sensitive to immune checkpoint blockade, results were validated in cohorts of patients with breast cancer. Association of RSR defect score with ICB sensitivity was then generalized to multiple cancer types with similar immunobiology using pre-clinical models and clinical patient cohorts. All in vivo mouse experiments were approved by the Institutional Animal Care and Use Committee. After randomization, tumor measurements were performed by a blinded investigator. Microscopy was performed semi-blinded, coupled with automated image quantification. Multiple independent biological replicates were performed for each experiment, with sample size given in the corresponding figure legend along with details on statistical analysis. Sample sizes for mouse studies were chosen based on previous studies; sample sizes for human cohorts were based on data availability. Detailed methods are provided in Supplementary Materials, including descriptions of cell lines (table S1), antibodies (table S2), and shRNAs/siRNAs (table S3).
Statistical analysis
Statistical analysis was performed in Matlab, R, and GraphPad Prism. Unless otherwise noted, comparisons between two groups were performed using a two-sided t-test. For samples not normally distributed, a Wilcoxon rank-sum test (unpaired data) or Wilcoxon signed-rank test (paired data) was used. For comparing the effects of a perturbation on two groups, two-way analysis of variance (ANOVA) was used. No meaningful differences in variance were noted between groups being compared statistically. Univariate survival was assessed by log-rank test; multivariate survival was assessed using the Cox proportional hazards model. Correlation between two parameters were assessed using the Spearman correlation coefficient unless otherwise specified. Multiple comparisons were accounted for by the method of Benjamini and Hochberg.
Supplementary Material
Acknowledgements:
We thank G. Mills for critical comments throughout the study. We are appreciative of many MD Anderson Cancer Center core facilities funded by grant CA016672: the Functional Genomics Core (shRNA and ORFeome Core) for reagents and technical assistance, the Characterized Cell Line Core for STR DNA fingerprinting and mycoplasma testing, the Research Histology Core Laboratory for tissue processing, and the Research Animal Support Facility.
Funding:
Support for this work was provided by Department of Defense Era of Hope Scholar Award grant W81XWH-10-1-0558, George and Barbara Bush Endowment for Innovative Cancer Research, and National Cancer Institute grant R01CA247862 to SYL. DJM was supported by Susan G. Komen grant PDF17483544 and National Cancer Institute grant K99CA240689. PGP was supported by Young Investigator Award from the Kidney Cancer Association. JMR was supported by National Cancer Institute grants CA148761 and CA016303. We acknowledge the joint participation of the Diana and Adrienne Helis Malvin Medical Research Foundation and Baylor College of Medicine to support XHFZ and JMR. CBP was partially supported by research funding from the Kidney Cancer Association (PI: Pilie), Conquer Cancer Foundation (PI: Pilie), Prostate Cancer Foundation (PI: Pilie), Department of Defense (DOD CA160728, PI: Jonasch), and National Institutes of Health (NIH/NCI CA016672, Biostatistics Shared Resource Group, PI: Pisters).
Competing interests:
D.J.M., P.G.P., E.J., and S-Y.L have a pending patent (“Replication stress response biomarkers for immunotherapy response”; WO-2019173456-A1; US Patent App. 16/978,758) on the RSRD gene expression signature. A.B.H. has stock options and is an advisory board member of Caris Life Sciences. A.B.H. also serves on the advisory board of WCG Oncology, has received licensing fees from Celldex Therapeutics and DNAtrix and received research funding from Merck. M.K. has advisory role for BMS, Roche, MSD and Daiichi Sankyo, and the institute receives research funding from AstraZeneca, BMS and Roche. E.J. has received research funding from Arrowhead Pharmaceuticals, research funding and consultation fees from Aravive, Merck, and Novartis, consultation fees from Aveo, Eisai, Ipsen, NiKang and Pfizer, and has received royalties from UpToDate and consultation fees from Elsevier (PracticeUpdate). All other authors declared no conflicts of interest.
Footnotes
List of Supplementary Materials.
Data and materials availability:
All data associated with this study are present in the paper or supplementary materials. The results here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. All TCGA data were downloaded using the TCGA data portal (https://portal.gdc.cancer.gov/) from the Pan-Cancer Atlas release (April 2018), except for phospho-proteomic data which was acquired from the corresponding manuscript’s supplementary information (51). Cell line gene expression data were acquired from CCLE (52). The TONIC breast cancer trial (11) data was acquired from EGAS0001003535, with associated patient outcome data acquired directly from the authors. All data from the Subudhi (53) cohort of metastatic prostate cancer treated with anti-CTLA4 were acquired from the paper’s supplementary materials. The Ascierto (36) anti-PD1 ccRCC cohort was downloaded from GEO (GSE67501). The Miao (37) anti-PD1 ccRCC data were downloaded from the publication’s supplemental information. For the ccRCC anti-PD-L1 IMmotion150 trial (38), data were downloaded from EGA (EGAS00001002928). The Zhao (41) GBM anti-PD1 cohort clinical data, including investigator-associated response, were downloaded from the publication’s supplemental data. Sequencing data (FASTQ files) were downloaded from SRA (PRJNA482620). The Cloughesy (13) GBM anti-PD1 clinical and transcriptional data were all downloaded from GEO (GSE121810). All data for urothelial cancer patients treated with anti-PD-L1 from Snyder et. al. (43) were downloaded from http://doi.org/10.5281/zenodo.546110. Data for the anti-PD-L1 IMvigor210 cohort(45) were downloaded from EGA (EGAS00001002556). Clinical data for metastatic gastric cancer treated with anti-PD-L1(54) were retrieved from the publication. RNA-seq data were downloaded from ENA (PRJEB25780). The Riaz (55) anti-PD1 melanoma cohort clinical data were downloaded from the publication’s supplemental information, and the RNA-seq data were downloaded from https://www.github.com/riazn/bms038_analysis. The Hugo (56) anti-PD1 melanoma cohort clinical data were downloaded from the publication’s supplemental information and the RNA-seq data were downloaded from GEO (GSE78220). Murine breast cancer data were acquired from GEO accessions GSE27101 and GSE69006. Newly generated murine RCC data are given in table S4 with additional data obtained from Harlander et al. supplemental data (35) and GSE83688 from Nargund et al. (34). Murine mouse breast cancer models T11 and 2208L are available from Dr. Jeffrey Rosen at Baylor College of Medicine following completion of a Material Transfer Agreement.
References and Notes
- 1.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD, Tumor neoantigens: building a framework for personalized cancer immunotherapy, J. Clin. Invest 125, 3413–3421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA, Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade., Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, Kaley TJ, Kendall SM, Motzer RJ, Hakimi AA, Voss MH, Russo P, Rosenberg J, Iyer G, Bochner BH, Bajorin DF, Al-Ahmadie HA, Chaft JE, Rudin CM, Riely GJ, Baxi S, Ho AL, Wong RJ, Pfister DG, Wolchok JD, Barker CA, Gutin PH, Brennan CW, Tabar V, Mellinghoff IK, DeAngelis LM, Ariyan CE, Lee N, Tap WD, Gounder MM, D’Angelo SP, Saltz L, Stadler ZK, Scher HI, Baselga J, Razavi P, Klebanoff CA, Yaeger R, Segal NH, Ku GY, DeMatteo RP, Ladanyi M, Rizvi NA, Berger MF, Riaz N, Solit DB, Chan TA, Morris LGT, Tumor mutational load predicts survival after immunotherapy across multiple cancer types, Nat. Genet 51, 202–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemery S, Keegan P, Pazdur R, First FDA Approval Agnostic of Cancer Site — When a Biomarker Defines the Indication, N. Engl. J. Med 377, 1409–1412 (2017). [DOI] [PubMed] [Google Scholar]
- 5.FDA approves pembrolizumab for adults and children with TMB-H solid tumors (2020) (available at https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors).
- 6.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn M-J, Felip E, Lee J-S, Hellmann MD, Hamid O, Goldman JW, Soria J-C, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L, Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer, N. Engl. J. Med 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, Boland G, Flaherty K, Herlyn M, Ruppin E, Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma., Nat. Med 24, 1545–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, Wucherpfennig KW, Liu XS, Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response, Nat. Med 24, 1550–1558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, Zhang C, Lunceford JK, Joe A, Cheng J, Webber AL, Ibrahim N, Plimack ER, Ott PA, Seiwert TY, Ribas A, McClanahan TK, Tomassini JE, Loboda A, Kaufman D, Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy, Science (80-.) 362, eaar3593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, Yi S, Lin S-Y, Sahni N, Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers, Nat. Commun 9, 1317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong SF, Wiersma TG, Russell NS, Lalezari F, Schouten PC, Bakker NAM, Ketelaars SLC, Peters D, Lange CAH, van Werkhoven E, van Tinteren H, Mandjes IAM, Kemper I, Onderwater S, Chalabi M, Wilgenhof S, Haanen JBAG, Salgado R, de Visser KE, Sonke GS, Wessels LFA, Linn SC, Schumacher TN, Blank CU, Kok M, Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial, Nat. Med 25 (2019), doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 12.McDermott DF, Lee J-L, Szczylik C, Donskov F, Malik J, Alekseev BY, Larkin JMG, Matveev VB, Gafanov RA, Tomczak P, Tykodi SS, Geertsen PF, Wiechno PJ, Shin SJ, Pouliot F, Alonso Gordoa T, Li W, Perini RF, Schloss C, Atkins MB, Pembrolizumab monotherapy as first-line therapy in advanced clear cell renal cell carcinoma (accRCC): Results from cohort A of KEYNOTE-427., J. Clin. Oncol 36, 4500 (2018). [Google Scholar]
- 13.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM, Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma, Nat. Med 25, 477–486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein O, Kee D, Markman B, Carlino MS, Underhill C, Palmer J, Power D, Cebon J, Behren A, Evaluation of TMB as a predictive biomarker in patients with solid cancers treated with anti-PD-1/CTLA-4 combination immunotherapy, Cancer Cell 39, 592–593 (2021). [DOI] [PubMed] [Google Scholar]
- 15.McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, Jonasch E, Khasraw M, Heimberger AB, Lim B, Ueno NT, Litton JK, Ferrarotto R, Chang JT, Moulder SL, Lin S-Y, High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types, Ann. Oncol 32, 661–672 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rousseau B, Foote MB, Maron SB, Diplas BH, Lu S, Argilés G, Cercek A, Diaz LA, The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors, N. Engl. J. Med 384, 1168–1170 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miles D, Gligorov J, André F, Cameron D, Schneeweiss A, Barrios C, Xu B, Wardley A, Kaen D, Andrade L, Semiglazov V, Reinisch M, Patel S, Patre M, Morales L, Patel SL, Kaul M, Barata T, O’Shaughnessy J, Zhang Q, Xu B, Shao Z, Wang X, Geng C, Yan X, Tong Z, Shen K, Yin Y, Sun T, Yang J, Feng J, Yan M, Wang Y, Liu Q, Zhang S, De Laurentiis M, Santoro A, Guarneri V, Colleoni M, Natoli C, Cortesi L, Placido S, Gianni L, Ferrau F, Livi L, Zambelli A, Del Mastro L, Tonini G, Montemurro F, Bianchi G, Pedersini R, Prete S, Allegrini G, Naso G, Vici P, Loirat D, Mailliez A, Priou F, Tredan O, Dalenc F, Perrin C, Gligorov J, Timar David M, Dohollou N, Teixeira L, Brocard F, Arnaud A, Delaloge S, Spano J-P, Mansi L, Andrade L, Damian F, Pedrini J, Aleixo S, Hegg R, Junior R, Reinisch M, Schmidt M, Wenzel C, Grischke E-M, Schneeweiss A, Just M, Harbeck N, Schumacher C, Peters U, Fischer D, Forstbauer H, Liersch R, Warner E, Bouganim N, Doyle C, Price Hiller J, Vandenberg T, Pavic M, Robinson A, Roldan Urgoiti G, Califaretti N, Alacacioglu A, Gumus M, Yalcin B, Cicin I, Kose F, Uygun K, Kaplan M, Cubukcu E, Wardley A, Harries M, Miles D, Doval D, Gupta S, Mohapatra P, Chatterjee S, Ghadyalpatil N, Singhal M, Nag S, Agarwal A, Wolf I, Gal Yam E, Yerushalmi R, Peretz T, Fried G, Ben Baruch N, Katz D, Hamilton E, Kayali F, Brufsky A, Telli M, Wright G, Oyola R, Rakowski T, Graff S, Tjulandin S, Semiglazov V, Aparicio A, Ruiz Borrego M, Merino L, Guerra Martinez J, Lopez E, Yamashita T, Ohtani S, Inoue K, Ito Y, Niikura N, Nakayama T, Sagara Y, Yanagita Y, Kamada Y, Kaneko K, Kaen D, Nervo A, Eniu A, Schenker M, Priester P, Melichar B, Zimovjanova M, Sormova P, Sufliarsky J, Kakalejcik M, Belbaraka R, Errihani H, Le Than D, Pham D, Aravantinos G, Papadimitriou C, Koumakis G, Papandreou C, Podolski P, Tabane K, Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer, Ann. Oncol (2021), doi: 10.1016/j.annonc.2021.05.801. [DOI] [PubMed] [Google Scholar]
- 18.Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, Iwata H, Masuda N, Otero MT, Gokmen E, Loi S, Guo Z, Zhao J, Aktan G, Karantza V, Schmid P, Luis F, Gonzalo GA, Diego K, Ruben K, Matias M, Mirta V, Sally BH, Stephen B, Philip C, Sherene L, Dhanusha S, Andrea G, Donatienne T, Carlos B, Leandro B, Fabiano C, de F. J. Ruffo Roberto H, Domicio Carvalho L, Fernando Cezar Toniazzi L, Roberto Odebrecht R, Antonio Orlando SN, Felipe S, David C, Danielle C, Cristiano F, Xinni S, Joanne Y, Alejandro A, Carlos G, Claudio S, Cesar S, Eduardo Y, Alvaro GD, Jesus S, Petra H, Zdenek K, Bohuslav M, Katarina P, Jana P, Vesna G, Erik J, Jeanette J, Soren L, Tamas L, Herve B, Isabelle D, Anthony G, Anne-Claire HB, Luis T, Jens-Uwe B, Peter F, Dirk F, Nadia H, Jens H, de KF Anna S Christian K, Sibylle L, Diana L, Tjoung-Won PS, Raquel Von S, Pauline W, Louis C, Ava K, Kai Cheong Roger N, Peter A, Tibor C, Zsuzsanna K, Laszlo L, Karoly M, Gabor R, John C, Catherine K, Seamus OR, Saverio C, Antonietta Da. A., Enrico R, Tomoyuki A, Takaaki F, Kenichi I, Takashi I, Yoshinori I, Tsutomu I, Hiroji I, Yoshimasa K, Koji M, Yasuo M, Hirofumi M, Seigo N, Naoki N, Shoichiro O, Akihiko O, Yasuaki S, Eiji S, Masato T, Yuko T, Kenji T, Koichiro T, Junichiro W, Naohito Y, Yutaka Y, Teruo Y, Anita B, Mastura MY, Angel GV, Alejandro JR, Jorge MR, Flavia MV, Jessica RC, Karin B, Vivianne TH, David P, Ewa C, Ewa NZ, Zbigniew N, Barbara R, Joanna S, Cezary S, Rafal T, Bogdan Z, Alexander A, Natalia F, Oleg L, Andrey M, Vladimir M, Guzel M, Jin Hee A, Seock-Ah I, Keun Seok L, Kwong Hwa P, Yeon Hee P, de B Begona las H., Javier C, Josefina CJ, Luis de la C. M., Jose GS, Maria G, Esther H, Esther ZA, Chien-Ting L, Mei-Ching L, Chiun-Sheng H, Chao-Jung T, Ling-Ming T, Cagatay A, Gul B, Irfan C, Erhan G, Seyda G, Nil MM, Mustafa O, Ozgur O, Sinan Y, Steve C, Janine G, Iain M, Peter S, Nicholas T, Mark T, Christopher T, Duncan W, Hryhoriy A, Oleksandr B, Igor B, Oleksii K, Olena K, Hanna K, Anna K, Iurii L, Alla N, Natalya O, Olga P, Andrii R, Sergii S, Yaroslav S, Dmytro T, Grygorii U, Ihor V, Sibel B, Madhu C, Michael C, Patrick C, Scott C, Jennifer D, Keerthi G, Jeffrey H, Kent H, William I, Randa L, Janice L, Raul M, Susan M, Rita N, Ira O, Coral O, Timothy P, Amit P, Brian P, Hope R, Irina R, Michael S, Robert S, Michael S, Laura S, Bradley S, Michaela T, Frances VA, Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial, Lancet 396, 1817–1828 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Shih DJH, Lin SY, Role of DNA repair defects in predicting immunotherapy response, Biomark. Res 8, 1–8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osborn AJ, Elledge SJ, Zou L, Checking on the fork: The DNA-replication stress-response pathway, Trends Cell Biol. 12, 509–516 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Halazonetis TD, Gorgoulis VG, Bartek J, An Oncogene-Induced DNA Damage Model for Cancer Development, Science (80-.) 319, 1352–1356 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F, Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication, Nature 444, 638–642 (2006). [DOI] [PubMed] [Google Scholar]
- 23.McGrail DJ, Lin CCJ, Dai H, Mo W, Li Y, Stephan C, Davies P, Lu Z, Mills GB, Lee JS, Lin SY, Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype, Cell Rep. 23, 2095–2106 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Flynn RL, Zou L, ATR: a master conductor of cellular responses to DNA replication stress, Trends Biochem. Sci 36, 133–140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGrail DJ, Lin CCJ, Dai H, Mo W, Li Y, Stephan C, Davies P, Lu Z, Mills GB, Lee JS, Lin SY, Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype, Cell Rep 23, 2095–2106 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B, Nucleotide deficiency promotes genomic instability in early stages of cancer development, Cell 145, 435–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petermann E, Woodcock M, Helleday T, Chk1 promotes replication fork progression by controlling replication initiation., Proc. Natl. Acad. Sci. U. S. A 107, 16090–16095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, König N, Kurth T, Wieczorek D, Kast K, Cardoso MC, Günther C, Lee-Kirsch MA, RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA, Nat. Commun 7 (2016), doi: 10.1038/ncomms11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP, Simple rules for passive diffusion through the nuclear pore complex, J. Cell Biol 215, 57–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L, Cytosolic DNA Sensing in Organismal Tumor Control, Cancer Cell, 1–18 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, Jeong KJ, Ding Z, Liang J, Wang SW, Dai H, Lee S, Sahni N, Mercado-Uribe I, Kim T, Chen K, Lin S-Y, Peng G, Westin SN, Liu J, O’Connor MJ, Yap TA, Mills GB, Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy, Cancer Cell 35, 851–867.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aran D, Hu Z, Butte AJ, xCell: digitally portraying the tissue cellular heterogeneity landscape., Genome Biol. 18, 220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK, IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade, J. Clin. Invest 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, Aras O, Han S, Oyama T, Takeda S, Ray CE, Dong Z, Berge M, Hakimi AA, Monette S, Lekaye CL, Koutcher JA, Leslie CS, Creighton CJ, Weinhold N, Lee W, Tickoo SK, Wang Z, Cheng EH, Hsieh JJ, The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma, Cell Rep. 18, 2893–2906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, Moch H, Wild PJ, Frew IJ, Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice, Nat. Med 23, 869–877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, Xu H, Pritchard TS, Fan J, Cheadle C, Cope L, Drake CG, Pardoll DM, Taube JM, Topalian SL, The Intratumoral Balance between Metabolic and Immunologic Gene Expression Is Associated with Anti-PD-1 Response in Patients with Renal Cell Carcinoma, Cancer Immunol. Res 4, 726–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, Horak C, Wind-Rotolo M, Tracy A, Giannakis M, Hodi FS, Drake CG, Ball MW, Allaf ME, Snyder A, Hellmann MD, Ho T, Motzer RJ, Signoretti S, Kaelin WG, Choueiri TK, Van Allen EM, Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma, Science (80-.) 359, 801–806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, Fong L, Joseph RW, Pal SK, Reeves JA, Sznol M, Hainsworth J, Rathmell WK, Stadler WM, Hutson T, Gore ME, Ravaud A, Bracarda S, Suárez C, Danielli R, Gruenwald V, Choueiri TK, Nickles D, Jhunjhunwala S, Piault-Louis E, Thobhani A, Qiu J, Chen DS, Hegde PS, Schiff C, Fine GD, Powles T, Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma, Nat. Med 24, 749–757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, Smith CC, Vincent BG, Serody JS, Beckermann KE, Ganesan S, Bhanot G, Rathmell WK, Endogenous retrovirus expression is associated with response to immune checkpoint pathway in clear cell renal cell carcinoma, JCI Insight 3 (2018), doi: 10.1172/jci.insight.121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M, Chambers T, Salgado R, Savas P, Loi S, Birkbak NJ, Sansregret L, Gore M, Larkin J, Quezada SA, Swanton C, Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis, Lancet Oncol. 18, 1009–1021 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, Filip I, Orenbuch R, Goetz M, Yamaguchi JT, Cloney M, Horbinski C, Lukas RV, Raizer J, Rae AI, Yuan J, Canoll P, Bruce JN, Saenger YM, Sims P, Iwamoto FM, Sonabend AM, Rabadan R, Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastomaNat. Med. 25, 462–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C, Emerging landscape of oncogenic signatures across human cancers., Nat. Genet 45, 1127–1133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD, Chang E, Aksoy BA, Al-Ahmadie H, Yusko E, Vignali M, Benzeno S, Boyd M, Moran M, Iyer G, Robins HS, Mardis ER, Merghoub T, Hammerbacher J, Rosenberg JE, Bajorin DF, Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis, PLoS Med. 14, 1–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenberg JE, Hoffman-Censits J, Powles T, Van Der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R, Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial, Lancet 387, 1909–1920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel III EE, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Şenbabaoğlu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Höglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, Powles T, TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells, Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buisson R, Boisvert JL, Benes CH, Zou L, Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase, Mol. Cell 59, 1011–1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM, AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies., Mol. Cancer Ther 7, 2955–66 (2008). [DOI] [PubMed] [Google Scholar]
- 48.King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS, LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms, Mol. Cancer Ther 14, 2004–2013 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, Jaspers JE, Watanabe K, Pieterse M, Kersbergen A, Sol W, Celie PHN, Schouten PC, van den Broek B, Salman A, Nieuwland M, de Rink I, de Ronde J, Jalink K, Boulton SJ, Chen J, van Gent DC, Bartek J, Jonkers J, Borst P, Rottenberg S, REV7 counteracts DNA double-strand break resection and affects PARP inhibition, Nature 521, 541–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pilié PG, Tang C, Mills GB, Yap TA, State-of-the-art strategies for targeting the DNA damage response in cancer, Nat. Rev. Clin. Oncol, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang K, Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyö D, Ellis MJ, Carr SA, Proteogenomics connects somatic mutations to signalling in breast cancer, Nature 534, 55–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, Sellers WR, Next-generation characterization of the Cancer Cell Line Encyclopedia, Nature (2019), doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subudhi SK, Vence L, Zhao H, Blando J, Yadav SS, Xiong Q, Reuben A, Aparicio A, Corn PG, Chapin BF, Pisters LL, Troncoso P, Tidwell RS, Thall P, Wu C, Zhang J, Logothetis CL, Futreal A, Allison JP, Sharma P, Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer, Sci. Transl. Med 12, eaaz3577 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, Liu XQ, Sher X, Jung H, Lee M, Lee S, Park SH, Park JO, Park YS, Lim HY, Lee H, Choi M, Talasaz A, Kang PS, Cheng J, Loboda A, Lee J, Kang WK, Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer, Nat. Med 24 (2018), doi: 10.1038/s41591-018-0101-z. [DOI] [PubMed] [Google Scholar]
- 55.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, Bhatia S, Hwu W-J, Gajewski TF, Slingluff CL, Chowell D, Kendall SM, Chang H, Shah R, Kuo F, Morris LGT, Sidhom J-W, Schneck JP, Horak CE, Weinhold N, Chan TA, Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab., Cell 171, 934–949.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS, Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma, Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D, Cyclin-dependent kinase 9-cyclin K functions in the replication stress response., EMBO Rep. 11, 876–882 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeRose YS, Wang G, Lin Y-C, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL, Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes., Nat. Med 17, 1514–20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuoka S, a Ballif B, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ, ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage, Science (80-.) 316, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 60.McGrail DJ, Lin CC-J, Garnett J, Liu Q, Mo W, Dai H, Lu Y, Yu Q, Ju Z, Yin J, Vellano CP, Hennessy B, Mills GB, Lin S-Y, Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm, npj Syst. Biol. Appl 3, 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, Korving J, van Boxtel R, Duarte AA, Lelieveld D, van Hoeck A, Ernst RF, Blokzijl F, Nijman IJ, Hoogstraat M, van de Ven M, Egan DA, Zinzalla V, Moll J, Boj SF, Voest EE, Wessels L, van Diest PJ, Rottenberg S, Vries RGJ, Cuppen E, Clevers H, A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity, Cell 172, 373–386.e10 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Barretina J, Caponigro G, Stransky N, Venkatesan K, a Margolin A, Kim S, Wilson CJ, Lehár J, V Kryukov G, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, a Mapa F, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, a Garraway L, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity Supp, Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Y, Bertram CC, Shi Q, Zinkel SS, Proapoptotic Bid mediates the Atr-directed DNA damage response to replicative stress, Cell Death Differ. 18, 841–852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mordes DA, Glick GG, Zhao R, Cortez D, TopBP1 activates ATR through ATRIP and a PIKK regulatory domain., Genes Dev. 22, 1478–89 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puddu F, Piergiovanni G, Plevani P, Muzi-Falconi M, Copenhaver GP, Ed. Sensing of Replication Stress and Mec1 Activation Act through Two Independent Pathways Involving the 9-1-1 Complex and DNA Polymerase ε, PLoS Genet. 7, e1002022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ercilla A, Llopis A, Feu S, Aranda S, Ernfors P, Freire R, Agell N, New origin firing is inhibited by APC/CCdh1 activation in S-phase after severe replication stress., Nucleic Acids Res. 44, 4745–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jackson DA, Pombo A, Replicon Clusters Are Stable Units of Chromosome Structure: Evidence That Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells, J. Cell Biol 140, 1285–1295 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, Feunteun J, Garber JE, Livingston DM, BRCA1 haploinsufficiency for replication stress suppression in primary cells, Nat. Commun 5, 5496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coquel F, Silva M-J, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A-L, Promonet A, Cribier A, Sarrazin A, Niedzwiedz W, Lopez B, Costanzo V, Krejci L, Chabes A, Benkirane M, Lin Y-L, Pasero P, SAMHD1 acts at stalled replication forks to prevent interferon induction, Nature 557, 57–61 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, Duran M, Pauli C, Shaw C, Chadalavada K, Rajasekhar VK, Genovese G, Venkatesan S, Birkbak NJ, McGranahan N, Lundquist M, LaPlant Q, Healey JH, Elemento O, Chung CH, Lee NY, Imielenski M, Nanjangud G, Pe’er D, Cleveland DW, Powell SN, Lammerding J, Swanton C, Cantley LC, Chromosomal instability drives metastasis through a cytosolic DNA response, Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification, Nat. Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or supplementary materials. The results here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. All TCGA data were downloaded using the TCGA data portal (https://portal.gdc.cancer.gov/) from the Pan-Cancer Atlas release (April 2018), except for phospho-proteomic data which was acquired from the corresponding manuscript’s supplementary information (51). Cell line gene expression data were acquired from CCLE (52). The TONIC breast cancer trial (11) data was acquired from EGAS0001003535, with associated patient outcome data acquired directly from the authors. All data from the Subudhi (53) cohort of metastatic prostate cancer treated with anti-CTLA4 were acquired from the paper’s supplementary materials. The Ascierto (36) anti-PD1 ccRCC cohort was downloaded from GEO (GSE67501). The Miao (37) anti-PD1 ccRCC data were downloaded from the publication’s supplemental information. For the ccRCC anti-PD-L1 IMmotion150 trial (38), data were downloaded from EGA (EGAS00001002928). The Zhao (41) GBM anti-PD1 cohort clinical data, including investigator-associated response, were downloaded from the publication’s supplemental data. Sequencing data (FASTQ files) were downloaded from SRA (PRJNA482620). The Cloughesy (13) GBM anti-PD1 clinical and transcriptional data were all downloaded from GEO (GSE121810). All data for urothelial cancer patients treated with anti-PD-L1 from Snyder et. al. (43) were downloaded from http://doi.org/10.5281/zenodo.546110. Data for the anti-PD-L1 IMvigor210 cohort(45) were downloaded from EGA (EGAS00001002556). Clinical data for metastatic gastric cancer treated with anti-PD-L1(54) were retrieved from the publication. RNA-seq data were downloaded from ENA (PRJEB25780). The Riaz (55) anti-PD1 melanoma cohort clinical data were downloaded from the publication’s supplemental information, and the RNA-seq data were downloaded from https://www.github.com/riazn/bms038_analysis. The Hugo (56) anti-PD1 melanoma cohort clinical data were downloaded from the publication’s supplemental information and the RNA-seq data were downloaded from GEO (GSE78220). Murine breast cancer data were acquired from GEO accessions GSE27101 and GSE69006. Newly generated murine RCC data are given in table S4 with additional data obtained from Harlander et al. supplemental data (35) and GSE83688 from Nargund et al. (34). Murine mouse breast cancer models T11 and 2208L are available from Dr. Jeffrey Rosen at Baylor College of Medicine following completion of a Material Transfer Agreement.