

Summary
Positive-strand RNA viruses replicate in close association with rearranged intracellular membranes. For hepatitis C virus (HCV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), these rearrangements comprise endoplasmic reticulum (ER)-derived double membrane vesicles (DMVs) serving as RNA replication sites. Cellular factors involved in DMV biogenesis are poorly defined. Here, we show that despite structural similarity of viral DMVs with autophagosomes, conventional macroautophagy is dispensable for HCV and SARS-CoV-2 replication. However, both viruses exploit factors involved in autophagosome formation, most notably class III phosphatidylinositol 3-kinase (PI3K). As revealed with a biosensor, PI3K is activated in cells infected with either virus to produce phosphatidylinositol 3-phosphate (PI3P) while kinase complex inhibition or depletion profoundly reduces replication and viral DMV formation. The PI3P-binding protein DFCP1, recruited to omegasomes in early steps of autophagosome formation, participates in replication and DMV formation of both viruses. These results indicate that phylogenetically unrelated HCV and SARS-CoV-2 exploit similar components of the autophagy machinery to create their replication organelles.
Keywords: autophagy, PI3P, class III PI3K, DFCP1, Beclin 1, hepatitis C virus, coronavirus, DMV, replication organelle, daclatasvir
Graphical abstract
Highlights
-
•
Conventional macroautophagy is dispensable for HCV and SARS-CoV-2 replication
-
•
Components of the class III PI3K complex promote HCV and SARS-CoV-2 replication
-
•
Class III PI3K and DFCP1 contribute to membranous replication organelle formation
Twu et al. investigate involvement of autophagy machinery components in HCV and SARS-CoV-2 replication. Conventional macroautophagy is dispensable for replication of either virus. However, factors involved in autophagosome biogenesis, including components of the class III PI3K complex, contribute to viral replication. Most likely they promote membranous replication organelle formation.
Introduction
Positive-strand RNA viruses replicate in host cells in close association with altered intracellular membranes (Belov et al., 2012; Knoops et al., 2008; Limpens et al., 2011; Romero-Brey et al., 2012). The transformed host membranes, often referred to as replication organelles, not only serve as viral replication platforms, but they may also provide shielding of the replication machinery against host innate immune-sensing systems (Knoops et al., 2008; Scutigliani and Kikkert, 2017). The replication organelles can be grouped into two types: the invaginated vesicle/spherule type, and the double-membrane vesicle (DMV) type (Paul and Bartenschlager, 2013). Virus-induced DMVs, ranging from ∼100 to 300 nm in diameter, depending on the virus, possess structures resembling those of autophagosomes (reviewed in Blanchard and Roingeard [2015], Paul and Bartenschlager [2015], and Wolff et al. [2020]).
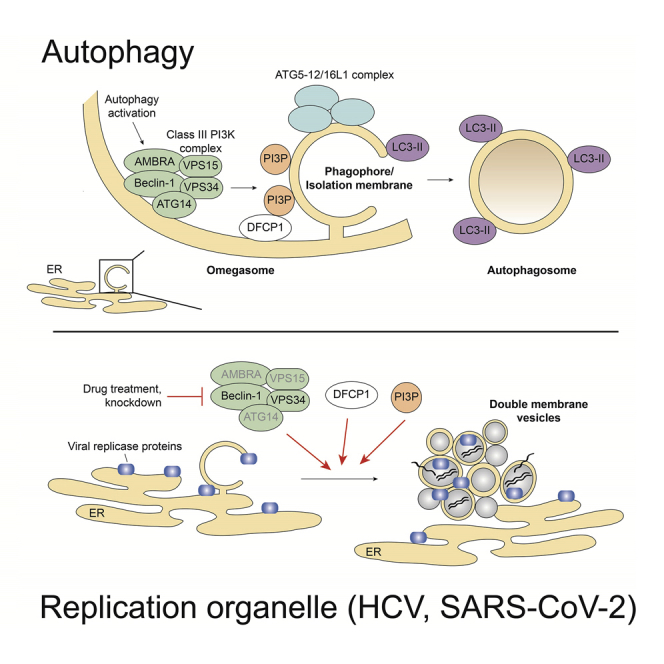
Macroautophagy (hereafter autophagy) is an intracellular lysosomal degradative pathway that plays an important role in the maintenance of cellular homeostasis through the degradation of proteins or damaged organelles. Cellular stresses, such as starvation or viral infection, induce autophagy by activation of the Unc-51-like kinase 1 (ULK1) complex that in turn activates the class III phosphatidylinositol (PI) 3-kinase (PI3K) complex (Figure 1A). This complex consists of several components, including the regulatory component Beclin 1 and the catalytic lipid kinase subunit VPS34 that converts PI into PI3-phosphate (PI3P) (Figure 1A). PI3P recruits its effector proteins, such as the double FYVE-containing protein 1 (DFCP1) and the WD repeat domain phosphoinositide-interacting proteins (WIPIs) to the isolation membrane that is derived from endoplasmic reticulum (ER), mitochondria, Golgi, or other membrane sources. The ATG5-12/16L1 complex, together with proteolytically cleaved and phosphatidylethanolamine (PE)-conjugated LC3 (designated LC3-II), coordinate the elongation and closure of the autophagosome. Finally, autophagosomes fuse with lysosomes creating autolysosomes that degrade and recycle engulfed components (Figure 1A) (reviewed in Lamb et al. [2013], Mizushima et al. [2011], and Vescovo et al. [2014]).
Figure 1.

Autophagy ATG5-12/16L1 complex is not required for HCV and SARS-CoV-2 replication
(A) Schematic depiction of the early steps of autophagosome formation and involved factors. When cells encounter stress, such as starvation or viral infection, activity of the mTOR complex is inhibited, resulting in activation of the ULK1 complex and the class III-PI3K complex. The latter synthesizes PI3P, which leads to the recruitment of proteins such as DFCP1 and WIPIs to the isolation membrane. The ATG5-12/16L1 complex and LC3-II (LC3-I conjugated to phosphatidylethanolamine) coordinate the elongation and closure of the autophagosome. Factors labeled with red letters are analyzed in this study.
(B) Tomographic slices and 3D renderings of DMVs induced in HCV- and SARS-CoV-2-infected cells. Figure modified according to Romero-Brey et al. (2012) and Cortese et al. (2020), respectively, with permission of the publishers.
(C) CRISPR-Cas-9-based KO of ATG5 and ATG16L1 in Huh7-Lunet/T7 cells was confirmed using western blotting with ATG5 and ATG16L1 antibodies, respectively. GAPDH was used as loading control.
(D and E) Huh7-Lunet/T7 cells with ATG5 or ATG16L1 KO were subjected to starvation or cultured under regular conditions and treated or not with 100 nM bafilomycin A1 (BafA1) for 2 h prior to cell lysis or fixation.
(D) Effect of ATG5 or ATG16L1 KO on LC3 lipidation was determined by western blotting. Tubulin served as loading control.
(E) Fixed cells were stained for LC3, and the number of LC3 puncta in at least 50 cells were analyzed for each condition using the “Analyzed Particles” function in the ImageJ software package. Example confocal microscopy images are shown in Figure S1A.
(F) Effect of ATG5 and ATG16L1 KO on cell viability was determined at 24, 48, and 72 h post-seeding by measuring ATP content using a CellTiter-Glo assay.
(G) Huh7-Lunet/T7 cells with given ATG KOs were transfected with a subgenomic HCV reporter replicon (sgJFH1). At given time points after transfection, luciferase activity, reflecting HCV replication, was measured. Values were normalized to transfection efficiency using the 4 h post-transfection value.
(H) Huh7-Lunet/T7/ACE2 cells with given ATG KOs were infected with SARS-CoV-2 at the indicated MOI. Virus replication was determined by qRT-PCR using virus-specific primers after 24 h. Values were normalized to ypoxanthine-guanine phosphoribosyltransferase (HPRT) RNA levels that were quantified in parallel.
Data in (E)–(H) represent the mean ± SEM from two independent experiments. ∗p < 0.05; ns, not significant. See also Figure S1.
Hepatitis C virus (HCV), a major causative agent of chronic liver disease, including liver cirrhosis and hepatocellular carcinoma, is a positive-strand RNA virus belonging to the Flaviviridae family. HCV replicates its RNA genome most likely within DMVs that are derived from the ER (Romero-Brey et al., 2012) (Figure 1B). These DMVs accumulate in the cytoplasm of infected cells, often in close proximity to lipid droplets (Lee et al., 2019b; Paul et al., 2013; Romero-Brey et al., 2012). Another DMV-forming virus is severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a member of the Coronaviridae family. By the end of July 2021, SARS-CoV-2 had caused around 205 million infections worldwide with more than 4 million deaths as a result of coronavirus disease 2019 (COVID-19) (World Health Organization, 2021). As in the case of HCV, DMVs induced by SARS-CoV-2 are derived from the ER (Cortese et al., 2020; Snijder et al., 2020) (Figure 1B). Moreover, DMV-like structures can be induced by the sole expression of viral polyprotein fragments, which comprise nonstructural protein 3-4A-4B-5A-5B (NS3-5B) in the case of HCV and nsp3-4 in the case of SARS-CoV and Middle Eastern respiratory syndrome (MERS)-CoV (Oudshoorn et al., 2017; Romero-Brey et al., 2012, 2015; Wolff et al., 2020).
The structural similarities between virally induced DMVs and autophagosomes suggest that viruses might utilize autophagy or components involved in the formation of autophagosomes for the biogenesis of the viral replication organelle. In HCV, previous studies show that infection increases the PE-conjugated LC3-II form, indicating autophagy activation (Ait-Goughoulte et al., 2008; Dreux et al., 2009; Sir et al., 2008). In addition, it has been reported that perturbation of the ATG5-12/16L1 complex impairs HCV replication and decreases DMV number and size (Dreux et al., 2009; Fahmy and Labonté, 2017; Guévin et al., 2010; Tanida et al., 2009). In the case of coronaviruses, autophagy appears to contribute to DMV formation in murine hepatitis virus (MHV), infectious bronchitis virus (IBV), and SARS-CoV (Cottam et al., 2011; Prentice et al., 2004a; Prentice et al., 2004b). Results of these studies suggest that autophagy might be important for virus-induced DMV formation in HCV and SARS-CoV-2 (reviewed in Miller et al. [2020]). However, it is unclear whether autophagy per se is required for DMV formation or whether distinct components of the autophagy machinery are hijacked by these viruses to induce their replication organelles. Moreover, the underlying mechanism of DMV biogenesis remains to be determined.
To address these questions, we studied the role of different components of the autophagy machinery for their role in HCV and SARS-CoV-2 replication and formation of virus-induced DMVs. We found that autophagy per se is not required for the replication of either virus. However, pharmacological inhibition of class III PI3K activity or knock down of VPS34 suppressed replication and DMV formation in HCV and SARS-CoV-2 replicating cells. Consistently, PI3P production was increased in cells infected with either virus. Finally, we report that the PI3P effector protein DFCP1 promotes viral replication and membrane alterations induced by HCV and SARS-CoV-2. These data suggest that the phylogenetically unrelated HCV and SARS-CoV-2 exploit same components of the autophagosome formation machinery to build up their membranous replication organelles.
Results
Autophagy ATG5-12/16L1 complex is not required for efficient replication of HCV and SARS-CoV-2
To determine whether autophagy per se is required for HCV and SARS-CoV-2 replication, we generated ATG5 and ATG16L1 knockout (KO) cell pools using CRISPR-Cas-9 technology and Huh7-Lunet/T7 cells (Figure 1C). First, we characterized these cells for impact on autophagy. LC3 lipidation and autophagy flux were significantly impaired in those KO cells (Figure 1D). LC3 puncta formation induced upon starvation treatment was also decreased (Figures 1E and S1A). These results indicated that starvation-induced autophagy was inhibited in KO cells without impact on cell viability (Figure 1F). For HCV, ATG5 KO had no effect on viral replication, while ATG16L1 reduced replication very moderately, but not to a statistically significant extent (Figure 1G).
In the case of SARS-CoV-2, we first compared replication kinetics between Huh7-Lunet/T7 and A549 cells, each stably expressing ACE2 (Huh7-Lunet/T7/ACE2 and A549/ACE2, respectively) (Cortese et al., 2020). As shown in Figure S1B, both cell pools supported robust infection and replication as revealed by production of infectious progeny viruses. Although Huh7-derived cells were more susceptible to cell death as compared to A549/ACE2 cells (Figure S1B), Huh7 cell numbers were comparable between infected control (Ctrl) KO, ATG5 KO, and ATG16L1 KO cells (Figure S1C). By using these conditions, we observed no significant effect of ATG5 and ATG16L1 depletion in our Huh7-derived cells on SARS-CoV-2 replication as determined by qRT-PCR (Figure 1H). The same result was found when we measured nucleocapsid protein by using two different approaches (Figures S1D and S1E). These results suggest that ATG5-12/16L1-dependent autophagy does not contribute to the replication of these viruses.
PI3P production is important for efficient replication of HCV and SARS-CoV-2
Autophagosome formation is highly dependent on PI3P production by the class III PI3K complex consisting of AMBRA (activating molecule in Beclin 1-regulated autophagy protein 1), VPS34, VPS15, Beclin 1, and ATG14L (Matsunaga et al., 2009) (Figure 1A). In our previous study, we showed that HCV NS5A interacts with the class III PI3K complex via the receptor for activated protein C kinase 1 (RACK1) (Lee et al., 2019a). Therefore, we further investigated the role of the class III PI3K complex and PI3P production in RNA replication and replication organelle formation by HCV and, given the analogous DMV structures, SARS-CoV-2.
A previous study suggests that inhibition of PI3K activity by wortmannin impairs HCV replication (Mohl et al., 2016). However, wortmannin has a rather broad spectrum targeting all classes of PI3K as well as PI4K (Balla and Balla, 2006; Balla et al., 1997; Liu et al., 2005), with the latter being essential for HCV replication and formation of DMVs, where high amounts of PI4P accumulate (Reiss et al., 2011). Therefore, we employed the VPS34-specific compound PIK-III (Dowdle et al., 2014). To exclude that PIK-III affects PI4K activation by HCV and PI4P production, we expressed the HCV NS3-5B polyprotein in Huh7-Lunet/T7 cells and determined PI4P distribution after immunostaining with a PI4P-specific antibody. This expression system was used because VPS34 inhibition might affect viral replication, which in turn would impair replication organelle formation, whereas in the replication-independent expression system this is not the case. We found that PIK-III treatment had no effect on the elevation of PI4P in HCV polyprotein-expressing cells (Figures S2A and S2B).
Having excluded an effect of PIK-III on PI4K, we investigated the role of PI3K for HCV and SARS-CoV-2 replication. For HCV studies, highly permissive Huh7-derived cells stably expressing the viral receptor CD81 were infected with the HCV reporter virus JcR2a (Poenisch et al., 2015) and treated with increasing concentrations of PIK-III, or the PI4K inhibitor PI4KA-G1 (Bojjireddy et al., 2014), or the highly potent HCV NS5A inhibitor daclatasvir (Gao et al., 2010), with the latter two drugs serving as positive controls. In all cases, we observed profound inhibition of HCV replication, with the VPS34 inhibitor reaching a half-maximal effective concentration (EC50) of around 320 nM and a selectivity index (ratio of EC50 and half-maximal cytotoxic concentration [CC50]) >10 (Figure 2A; Table 1).
Figure 2.

PI3P production is required for efficient HCV and SARS-CoV-2 replication
(A) Huh7-Lunet/CD81H cells were infected with the HCV reporter virus JcR2a and treated with given drugs 4 h after infection. Luciferase activity reflecting viral RNA replication (red lines) and cell viability (ATP content; black lines) was measured at 24 h post-infection. DCV, daclatasvir.
(B) (Upper) Schematic depiction of the experimental setup. Huh7-Lunet/T7 cells were transfected twice with two different VPS34-targeting siRNAs using a 24-h interval, prior to electroporation with a subgenomic HCV reporter replicon 48 h after the second siRNA transfection. (Lower) knockdown (KD) efficiency was determined by western blotting. GAPDH served as loading control.
(C) Luciferase activity reflecting HCV RNA replication was measured 24, 48, and 72 h after electroporation. For each replicon RNA transfection, values were normalized to the 4-h value reflecting transfection efficiency, and these values were normalized to those obtained with NT siRNA-transfected cells. Data in (A) and (C) represent the mean ± SEM from three independent experiments.
(D) A549/ACE2 cells were treated with serial dilutions of given drugs 30 min before infection with SARS-CoV-2 (MOI of 1). Infected cells were fixed 24 h post-infection and the percentage of inhibition was determined using N protein-specific immunostaining. Cell viability (ATP content) was measured at the same time point.
(E) Calu-3 cells were treated with PIK-III 2 h after infection with SARS-CoV-2 (MOI of 5). Total RNA of infected cells was extracted at 6 h post-infection and analyzed using qRT-PCR to determine replication efficiency.
Data in (D) and (E) represent the mean ± SEM from two independent experiments.
(F) (Upper) Schematic of the experimental setup. A549/ACE2 cells were transfected twice with siRNAs using a 24-h interval, prior to SARS-CoV-2 infection 48 h after the second siRNA transfection. (Lower) Confirmation of KD efficiency by western blotting. GAPDH served as loading control.
(G) Percentage of SARS-CoV-2 replication was determined using N protein-specific immunostaining at 24 h post-infection. Data represent the mean ± SEM from three independent experiments. See also Figure S2.
Table 1.
Inhibition of HCV and SARS-CoV-2 replication by selected drugs
| (nM) | HCV |
SARS-CoV-2 |
||||
|---|---|---|---|---|---|---|
| Infection |
Replicon RNA transfection |
|||||
| EC50 | CC50 | EC50 | CC50 | EC50 | CC50 | |
| Daclatasvir | <0.006 | >25 | <0.006 | >25 | – | – |
| PI4KA-G1 | 23.8 | >4,000 | 19.8 | >4,000 | – | – |
| PIK-III | 318.2 | >4,000 | 309.5 | >4,000 | 172.5 | ∼50,000 |
| Remdesivir | – | – | – | – | 79.2 | >10,000 |
To determine whether PIK-III affected HCV entry or RNA replication, we transfected Huh7-derived cells with subgenomic replicons encoding for firefly luciferase, enabling us to monitor post-entry steps. Transfected cells were treated with any of the three drugs starting 4 h after transfection until cell harvest 20 h later. We observed a dose-dependent inhibition of HCV replication by PIK-III with an EC50 very similar to the one determined with the infection system (∼310 nM; Figure S2C; Table 1). Although this compound was ∼15-fold less active in suppressing HCV replication as compared to the PI4K inhibitor (Table 1), these results suggest that VPS34 activity is required for viral RNA replication.
We corroborated these data by using small interfering RNA (siRNA)-mediated VPS34 depletion (Figure 2B). Cells were twice transfected with either siRNA prior to transfection of a subgenomic HCV reporter replicon encoding the firefly luciferase. Viral replication was analyzed 24, 48, and 72 h after HCV RNA transfection by using luciferase assay (Figure 2C), and cell viability was determined by quantification of the ATP content (Figure S2E, left panel). VPS34 depletion significantly reduced HCV replication, without impacting cell viability. For independent validation, we also depleted Beclin 1, a key regulator of VPS34 activity, and determined the impact on HCV replication. We observed a consistent reduction of HCV replication in Beclin 1-depleted cells (Figures S2D and S2E), corroborating the important role of the class III PI3K complex for HCV RNA replication.
To determine the role of PI3K for SARS-CoV-2 replication, we infected A549/ACE2 cells with SARS-CoV-2 and treated the cells with PIK-III or the nucleoside analog remdesivir, which served as a positive control (Figure 2D; Table 1). In the case of PIK-III, we observed a dose-dependent reduction of viral replication with an EC50 of ∼170 nM, which is at least ∼250-fold below the CC50 value measured in this cell system (Table 1). We also observed inhibition of SARS-CoV-2 replication by PIK-III using Calu-3 cells, a commonly used human lung cancer cell line (Zhu et al., 2010) (Figure 2E). These results suggest that PI3P production by PI3K is required also for SARS-CoV-2 replication, consistent with earlier studies (Williams et al., 2021; Yuen et al., 2021).
For corroboration of these results, we determined the impact of VPS34 and Beclin 1 knockdown (KD) in A549/ACE2 cells on SARS-CoV-2 replication (Figures 2F and S2D, respectively). In both cases, KD diminished SARS-CoV-2 replication without overt impact on cell viability (Figures 2G and S2F, respectively). This result suggests that the class III PI3K complex contributes to HCV and SARS-CoV-2 replication.
HCV and SARS-CoV-2 enhance PI3P production in infected cells
To monitor PI3P production and subcellular distribution in HCV- and SARS-CoV-2-infected cells, we generated two GFP-tagged DFCP1 constructs as reported earlier by Axe et al. (2008): first, a GFP-tagged DFCP1 wild-type, which binds to PI3P via FYVE-type zinc finger domains, and second, a DFCP1 mutant deficient in PI3P binding (GFP-DFCP1∗) (Figure S3A). Each protein was stably expressed in Huh7-Lunet/T7 cells, and functionality of the sensor was validated using serum-starved cells. As shown previously, the abundance of GFP-DFCP1 puncta was profoundly enhanced in starved cells, reflecting increased PI3P production during autophagosome formation (Axe et al., 2008) (Figures S3B and S3C). No such increase was observed in starved cells expressing GFP-DFCP1∗, showing specificity of the sensor for PI3P (Figures S3D and S3E). Of note, upon treatment of the cells with the VPS34 inhibitor PIK-III, abundance of GFP-DFCP1 puncta in starved cells was reduced to background levels, consistent with impaired induction of autophagosome formation (Figures S3B and S3C). These results validate GFP-DFCP1 for the monitoring of PI3P production in cells.
Next, we analyzed how HCV affects PI3P production. Huh7-Lunet/T7 cells expressing GFP-DFCP1 were transfected with a full-length HCV genome, cultured for 48 h, and then treated with DMSO or PIK-III for 2 h prior to cell fixation. We found that the number of GFP-DFCP1 puncta was strongly increased in HCV-replicating cells and diminished upon PIK-III treatment (Figures 3A and 3B).
Figure 3.

HCV and SARS-CoV-2 increase PI3P production in infected cells
(A and B) Huh7-Lunet/T7 cells stably expressing the PI3P sensor protein GFP-DFCP1 were electroporated with genomic HCV RNA (strain Jc1). After 48 h, cells were treated with 1 μM PIK-III for 2 h and fixed for immunofluorescence (IF).
(A) Representative confocal microscopy images showing GFP-DFCP1 and HCV NS5A. Nuclear DNA was stained with DAPI. Upper left inserts show magnifications of boxed areas in the overviews.
(B) Quantification of GFP-DFCP1 puncta.
(C and D) Huh7-Lunet/T7/ACE2 cells stably expressing GFP-DFCP1 were infected with SARS-CoV-2 (MOI of 1). After 1 h, culture medium was replaced with DMEM containing 1 μM PIK-III for 20 h and fixed for IF.
(C) Representative confocal microscopy images showing GFP-DFCP1, SARS-CoV-2 nsp3, and nucleocapsid. Nuclear DNA was stained with DAPI. Upper left inserts show magnifications of boxed areas in the overviews. Scale bars, 10 μm (overview images) and 2 μm (magnified images).
(D) Quantification of GFP-DFCP1 puncta.
In (B) and (D), at least 60 cells were analyzed for each condition, and GFP-DFCP1 puncta were counted by using the “Analyzed Particles” function in ImageJ. All data represent the mean ± SEM from two independent experiments. ∗∗∗p < 0.001. See also Figures S3–S5.
We further investigated the role of NS5A in HCV-induced PI3P production using the NS5A inhibitor daclatasvir, which we had earlier shown to block DMV formation (Berger et al., 2014). Huh7-Lunet/T7 cells stably expressing GFP-DFCP1 were transfected with the HCV NS3-5B encoding expression plasmid, treated with daclatasvir, and analyzed by confocal microscopy. Consistent with the data obtained with HCV replicating cells (Figures 3A and 3B), expression of NS3-5B increased the number of GFP-DFCP1 puncta, indicating elevated PI3P production, which was blunted when cells were treated with daclatasvir (Figure S4). This effect was specific because the reduction of GFP-DFCP1 abundance by daclatasvir was not observed in cells expressing NS3-5B with NS5A containing the daclatasvir-resistance mutation Y93H (Figure S4).
With the aim to monitor PI3P production and subcellular distribution in the context of SARS-CoV-2 replication, we infected Huh7-Lunet/T7/ACE2 cells and cultured them in medium containing DMSO or 1 μM PIK-III. After 20 h, cells were fixed and nucleocapsid and nsp3 were detected by immunofluorescence, along with the PI3P sensor. Very similar to HCV, abundance of GFP-DFCP1 puncta was increased in SARS-CoV-2-infected cells and reduced upon treatment with the VPS34 inhibitor PIK-III (Figures 3C and 3D). The analogous result was obtained with cells in which DMV formation was induced by expression of hemagglutinin (HA)-nsp3-4-V5, arguing that PI3P accumulation does not require viral replication as we found for HCV (Figure S5; compare with Figure S4).
In summary, these data suggest that both HCV and SARS-CoV-2 induce PI3P production. In both cases the sole expression of proteins inducing replication organelle-like structures (DMVs by HCV NS3-5B and SARS-CoV-2 nsp3-4) suffices to induce PI3P accumulation.
PI3P production is important for HCV and SARS-CoV-2 expression-induced DMVs
The results described so far indicate that inhibition of VPS34 by PIK-III blocks PI3P accumulation in HCV NS3-5B and SARS-CoV-2 nsp3-4-expressing or -infected cells, and hampers viral replication, arguing that formation of DMVs, the sites of viral RNA replication (Klein et al., 2020; Paul et al., 2013; Romero-Brey et al., 2012; Snijder et al., 2020; Wolff et al., 2020), requires PI3P. To put this assumption to the test, we determined the impact of PIK-III on the formation of DMV-like structures induced by viral protein expression. With respect to HCV, we transfected Huh7-Lunet/T7 cells with the NS3-5B expression construct and treated the cells with 1 μM PIK-III (corresponding to ∼3-fold EC50) or DMSO 5 h after transfection until cell harvest. Drug treatment neither affected cell viability in transfected cells (Figure S6A) nor the expression level of viral proteins as determined by NS5A-specific western blotting (Figure 4A). In addition, transfection efficiency quantified by counting the number of NS5A-expressing cells (Figure 4B) and abundance of NS5A determined by fluorescence microscopy were comparable between cells treated with DMSO or PIK-III (Figure S6B).
Figure 4.

PI3K inhibition reduces DMV formation induced by HCV NS3-5B and SARS-CoV-2 nsp3-4 expression
(A–D) Huh7-Lunet/T7 cells were transfected with the HCV NS3-5B expression plasmid. Five hours after transfection 1 μM PIK-III was added to the cells that were lysed or fixed 24 h after transfection.
(A) Cell lysates were analyzed by western blotting. GAPDH served as loading control. Numbers below the NS5A gel lanes indicate relative protein levels as determined by image analysis. Values were normalized to the loading control; the DMSO-treated sample was set to 1.
(B) Fixed cells were stained for NS5A, and transfection efficiency was determined by counting the number of NS5A-positive cells from five randomly selected areas.
(C and D) Fixed cells were processed for TEM analysis and the number of DMVs was determined.
(C) Representative TEM images. Yellow boxed areas in overview images are enlarged in the right panels. Red asterisks indicate DMVs. Scale bars, 1,000 nm (overview image) and 500 nm (magnified image).
(D) Profiles of DMV-positive cells were analyzed using TEM images taken at ×4,000 original magnification. For systematic random sampling, two to seven square areas of 100 μm2 were placed on a whole-cell image and the number of DMVs was determined. Displayed analyses are based on 13 DMSO-treated cells and 46 PIK-III-treated cells.
(E–H) Huh7-Lunet/T7 cells were transfected with the SARS-CoV-2 HA-nsp3-4-V5 expression plasmid, and 24 h later, 1 μM PIK-III was added to the cells. After 48 h, cells were lysed or fixed for further analysis.
(E) Cell lysates were evaluated by western blotting. GAPDH was used as loading control. Numbers below the HA-nsp3 lanes were determined as described in (A).
(F) Fixed cells were stained with an HA-specific antibody and transfection efficiency was determined by counting the number of HA-nsp3-positive cells from five randomly selected areas.
(G and H) Fixed cells were processed for TEM analysis and number of DMVs was determined. Cells were analyzed and data are displayed as described for (C) and (D). (H) Analyses are based on 15 DMSO treated cells and 49 PIK-III treated cells.
All data represent the mean ± SEM. ∗∗∗p < 0.001; ns, not significant; according to two-tailed, unpaired Student’s t test. See also Figure S6.
Next, we analyzed the cells by transmission electron microscopy (TEM) to determine the impact of PIK-III treatment on DMV formation. For this analysis we took into account the transfection efficiency (around 35%; Figure 4B) and examined 46 PIK-III-treated cells, which on a statistical basis includes at least 16 transfected cells. We found that PIK-III treatment significantly reduced the number of DMVs compared to DMSO-treated control cells (Figures 4C and 4D), suggesting that PI3P production is required for HCV NS3-5B-induced DMV formation.
For SARS-CoV-2, we analyzed DMV formation in an analogous manner using the HA-nsp3-4-V5 expression construct and 1 μM PIK-III, corresponding to ∼10-fold EC50. Also, in this case, drug treatment did not affect cell viability, viral protein expression level, and transfection efficiency (Figures S6C, 4E, 4F, and S6D, respectively). Of note, PIK-III treatment significantly reduced the number of HA-nsp3-4-V5-induced DMVs (Figures 4G and 4H), arguing that PI3P is also required for DMV formation in the case of SARS-CoV-2.
Previous studies suggest that MHV-induced DMVs are degraded by ER-associated degradation (ERAD) tuning, involving vesicle trafficking to lysosomes (Mihelc et al., 2021). To study whether the reduced DMV number observed in PIK-III-treated cells resulted from impaired DMV formation or increased DMV degradation, we investigated the colocalization of HA-nsp3 with LC3 and lysosomal-associated membrane protein 1 (LAMP-1), markers for ERAD tuning and lysosomes, respectively. We did not observe colocalization between HA-nsp3 and LC3 or LAMP1 (Figures S6E and S6F, respectively), arguing that PI3K inhibition interferes with DMV formation rather than accelerating DMV degradation.
The PI3P effector protein DFCP1 is required for HCV and SARS-CoV-2 replication and DMV formation
DFCP1 is a PI3P effector protein that associates with omegasomes, the sites from which phagophores form (Figure 1A). However, the exact function of the protein remains to be determined (Axe et al., 2008; Lamb et al., 2013). A previous study suggests that inhibition of DFCP1 impairs HCV replication (Mohl et al., 2016), but the underlying mechanism has not been addressed. It is also unclear whether DFCP1 contributes to SARS-CoV-2 replication. Therefore, we investigated the role of DFCP1 in HCV and SARS-CoV-2 replication using a KD approach. Huh7-derived cells were transfected with two different siRNAs, and cell lysates prepared 48 h after transfection were analyzed by western blotting. Both siRNAs reduced DFCP1 amounts to background levels without affecting cell viability (Figures 5A and 5B). Consistent with the earlier report, DFCP1 KD significantly reduced HCV replication (Figure 5C). To study the effect of DFCP1 KD on SARS-CoV-2 replication, we used A549/ACE2 cells. Also, in these cells, KD efficiency was high and did not affect cell viability (Figures 5D and 5E, respectively). Notably, DFCP1 KD also drastically reduced SARS-CoV-2 replication (Figure 5F).
Figure 5.

DFCP1 is critically involved in HCV and SARS-CoV-2 replication
(A) DFCP1 KD in Huh7-Lunet/T7 cells was achieved by siRNA transfection (two different siRNAs) and confirmed by western blotting. GAPDH served as loading control.
(B and C) Huh7-Lunet/T7 cells were transfected twice with siRNAs using a 24-h interval, prior to electroporation with a subgenomic HCV reporter replicon 48 h after the second siRNA transfection.
(B) Cell viability (ATP content) was measured 24, 48, and 72 h after the last siRNA transfection.
(C) Luciferase activity reflecting HCV RNA replication was measured 24, 48, and 72 h after electroporation.
(D) DFCP1 KD in A549/ACE2 cells was evaluated by using western blotting as described for (A).
(E and F) A549/ACE2 cells were transfected with siRNAs 48 h prior to infection with SARS-CoV-2 (MOI of 1).
(E) Cell viability (ATP content) was measured 24 h after siRNA transfection.
(F) Relative SARS-CoV-2 replication was determined by N protein-specific immunostaining at 24 h post-infection and normalization of the values to those obtained with NT siRNA treated cells.
All data represent the mean ± SEM from three independent experiments.
To examine the role of DFCP1 in DMV formation, we employed our expression-based systems to avoid confounding effects caused by replication inhibition resulting from DFCP1 KD. For HCV, Huh7-Lunet/T7 cells were transfected with either DFCP1 siRNA or the non-targeting control, followed by transfection with the NS3-5B expression construct. In this case we used a variant of this construct encoding a NS5A with an EGFP insertion (NS5AEGFP) that does not affect NS5A function (Figure 6B) (Schaller et al., 2007). This variant was employed because we analyzed the cells by correlative light and electron microscopy (CLEM) in order to make sure that analyzed cells indeed had been transfected. Forty-eight hours after transfection, cells were harvested and analyzed by western blotting and CLEM. KD with either siRNA reduced DFCP1 to below our western blotting detection limit, while it did not affect expression level of NS5AEGFP (note that the double band corresponds to basal and hyperphosphorylated NS5A) (Figure 6A). Analysis of fixed cells by CLEM revealed significant reduction of DMV abundance by KD with either siRNA as compared to cells transfected with the control siRNA (Figures 6B and 6C).
Figure 6.

DFCP1 is required for HCV NS3-5B and SARS-CoV-2 nsp3-4-induced DMV formation
(A–C) Huh7-Lunet/T7 cells were transfected twice with siRNAs in a 24-h interval, prior to transfection with the HCV NS3-5B/5AEGFPexpression plasmid 48 h after the second siRNA transfection. Cells were lysed or fixed 24 h after transfection of the expression construct.
(A) Cell lysates were evaluated by western blotting. GAPDH was used as loading control.
(B and C) Transfected cells were first identified by GFP signal, then fixed and further processed for CLEM.
(B) Representative CLEM images. Upper panels in each column show bright-field image (left), fluorescent image (middle), and TEM cell overview (right). The middle panel in each column shows a magnified overview of a cell section. Yellow boxes indicate areas that are shown as enlargements in the bottom panel of each column. Red asterisks indicate DMVs. The design of the HCV expression construct is shown on the top.
(C) Cell profiles were analyzed using TEM images taken at ×4,000 original magnification as described for Figure 4D. At least 10 GFP-positive cells were counted for each condition.
(D–F) Huh7-Lunet/T7 cells were consecutively transfected twice with siRNAs and the SARS-CoV-2 NeonGreen (NG)-HA-nsp3-4-V5 expression plasmid as described for (A).
(D) Confirmation of DFCP1 KD and NG/HA-nsp3-4-V5 expression by western blotting. GAPDH was used as loading control.
(E and F) Transfected cells were identified by confocal microscopy using the NG signal, fixed, and further processed for CLEM analysis. Cells were analyzed and data are displayed as described for (B) and (C). At least 10 NG-positive cells were analyzed for each condition. The design of the SARS-CoV-2 expression construct is shown on the top of panel (E).
Data in (C) and (F) represent mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001.
In the case of SARS-CoV-2, we conducted the analogous experiments but using a monocistronic expression construct encoding NeonGreen followed by HA-nsp3-4-V5 (Figure 6E), with NeonGreen allowing the identification of transfected cells and correlation with EM images and the HA-tag allowing the detection of processed HA-nsp3. Consistent with the replication data, KD of DFCP1 also impaired DMV formation induced by SARS-CoV-2 HA-nsp3-4-V5, which was not due to effects of DFCP1 depletion on HA-nsp3-4-V5 abundance (Figures 6D–6F). Taken together, our data suggest that DFCP1 is a critical host factor for efficient replication of HCV and SARS-CoV-2. The most likely contribution of DFCP1 to replication is to support the biogenesis of DMVs, which are the predominant elements of the replication organelles and the sites of HCV and SARS-CoV-2 RNA replication.
Discussion
Here, we show that components involved in autophagosome formation, but not conventional macroautophagy itself, are required for HCV and SARS-CoV-2 replication, most likely by contributing to the biogenesis of the replication organelles, composed primarily of DMVs. Earlier studies suggest that the ATG5-12/16L1 complex is required for HCV replication (Dreux et al., 2009; Fahmy et al., 2018; Fahmy and Labonté, 2017; Guévin et al., 2010; Tanida et al., 2009), which is at variance with our results showing that KO of ATG5 and ATG16L1 does not affect HCV replication. This inconsistency might result from the different approaches used to inhibit ATG proteins. While earlier studies employed siRNA KD, which might have caused insufficient depletion of ATG proteins, we used CRISPR-Cas9-based KO cells unable to express targeted genes. Although during selection for KO cells, we might have enriched for cells compensating the defect, such as upregulation of ATG16L2 replacing ATG16L1 in the complex with ATG5-12 (Ishibashi et al., 2011), we note that two functional assays demonstrated a block of ATG5-12/16L1-dependent autophagy in our cell pools, yet virus replication was not significantly affected. In the case of coronaviruses, the role of ATG5 for viral replication is discussed controversially. Prentice et al., 2004a, Prentice et al., 2004b reported that MHV replication is strongly reduced by ATG5. In contrast, two other groups report that ATG5 is not required for MHV and SARS-CoV replication (Schneider et al., 2012; Zhao et al., 2007), consistent with our data. These discrepancies might originate from different cell lines and virus strains used in these studies, but further studies are required to clarify these inconsistencies.
By using a biosensor, we found that PI3P levels are upregulated in HCV- and SARS-CoV-2-infected cells. This elevation appears to be important for viral replication, because treatment of the cells with the class III PI3K-specific inhibitor PIK-III reduced HCV and SARS-CoV-2 replication, consistent with previous studies employing other PI3K inhibitors (Mohl et al., 2016; Silvas et al., 2020; Williams et al., 2021; Yuen et al., 2021). However, VPS34 is involved in autophagy and endocytic pathways (Jaber et al., 2016; Juhász et al., 2008), raising the question whether the PI3P we detected resides on autophagy-related membranes directly involved in DMV formation or on endosomal membranes involved in cargo transport to DMV formation sites. Although the role of PI3P in endosomal transport processes such as the delivery of cholesterol to HCV-induced DMVs cannot be excluded (Stoeck et al., 2017), our data suggest that PI3P produced by PI3K is directly involved in HCV and SARS-CoV-2 replication. Consistent with this assumption, KD or KO of ATG14L (Lee et al., 2019a) and depletion of Beclin 1 (Figure S2) (Gassen et al., 2021), which are both components of the PI3K complex (Figure 1A), inhibit HCV replication. Consistently, in the case of SARS-CoV-2, we observed reduced viral replication in Beclin 1 KD cells (Figure S2F), arguing that the class III PI3K complex, or components thereof, is involved in HCV and SARS-CoV-2 replication.
While PI3K inhibition decreased HCV and SARS-CoV-2 replication, even at the highest non-cytotoxic concentration of PIK-III, residual viral replication was detectable. One possible explanation for this observation is the use of alternative sources for PI3P. In addition to PI3K, PI3P can be generated by phosphatases such as phosphatidylinositol 4-phosphatase or 5-phosphatase from other phosphoinositides, thus compensating for loss of VPS34 function. Alternatively, even at highest PIK-III concentrations, we still observed few GFP-DFCP1 puncta, indicating that very low amounts of PI3P might suffice to promote replication of these two viruses. In any case, we note that suppression of viral replication by VPS34 inhibition has also been reported for tombusviruses, including cucumber leaf spot virus, turnip crinkle virus, and red clover necrotic mosaic virus that create replication organelles corresponding to vesicle-like invaginations of intracellular membranes. In cells infected with these viruses, VPS34 translocates to the replication compartment through interaction with the viral replicase to facilitate replication (Feng et al., 2019). Thus, the host cell pathways studied here might be exploited by multiple positive-strand RNA viruses to create their replication organelles.
Previous studies demonstrated that HCV and coronavirus infection increases the abundance of LC3-II in infected cells, although it has not been confirmed whether autophagy flux was enhanced or reduced (Cottam et al., 2011; Dreux et al., 2009; Prentice et al., 2004b; Sir et al., 2008; Su et al., 2011; Taguwa et al., 2011). PI3P production and LC3 lipidation are linked to conventional (macro)autophagy, because PI3P production precedes the recruitment of autophagy-related proteins and autophagosome formation. We confirm elevated PI3P production in HCV- and SARS-CoV-2-infected cells and demonstrate upregulation of PI3P as determined with the PI3P sensor. Sensitivity of this probe for PI3P was validated by the lack of its recruitment when using a PI3P non-binding DFCP1 mutant, arguing against the possibility that the probe was recruited by the DFCP1 moiety independent from PI3P. With respect to HCV, our results are supported by a previous study showing that the PI3K inhibitor 3-methyladenine (3-MA) or VPS34 KD reduces the abundance of LC3-II in HCV-infected cells and HCV replication, respectively (Su et al., 2011). In contrast, two other studies report that inhibition of PI3P production neither affects HCV-induced LC3 lipidation nor DFCP1 puncta formation (Mohl et al., 2016; Sir et al., 2012). One possible explanation for these discrepancies might be the use of different 3-MA treatment durations, ranging from 3 to 40 h between the different studies. Another explanation could be the different assay used to determine HCV replication.
In the case of coronaviruses, lipidation or redistribution of LC3 has been described for cells infected with transmissible gastroenteritis virus and infectious bronchitis virus (Cottam et al., 2011; Guo et al., 2016). In contrast, Reggiori et al. (2010) reported an association between MHV-induced DMVs and non-lipidated LC3 and the importance of non-lipidated LC3 for replication and DMV formation. Since the presence of LC3-I is a marker for ERAD vesicles (Calì et al., 2008a, 2008b), this study suggests that the ERAD pathway, rather than autophagy, is hijacked by MHV for DMV formation. Notably, we did not observe colocalization between nsp3 and LC3 (Figure S6E), indicating that different strategies might be used by different coronaviruses for DMV formation.
While we provide evidence that the class III PI3K is required for HCV- and SARS-CoV-2-induced DMV formation, the turnover of these structures remains unclear. As shown in a recent publication, DMVs induced by MHV are subjected to lysosomal degradation late in infection when the ER becomes the main budding compartment for new virions (Mihelc et al., 2021). At least in the expression-based systems, we neither observed enhanced degradation of HA-nsp3 upon PI3K inhibition (Figures 4E and S6D) nor colocalization with LAMP1 (Figure S6F), arguing against DMV degradation. Further studies are required to dissect DMV turnover in the case of SARS-CoV-2.
We have recently shown that NS5A, in cooperation with RACK1, facilitates the formation of the class III PI3K complex (Lee et al., 2019a). Here, we further investigated how NS5A affects PI3P production and, thus, DMV formation. Inhibition of NS5A by daclatasvir strongly reduced NS3-5B-induced GFP-DFCP1 puncta formation. This effect was specific as PI3P levels were unaffected by daclatasvir in cells expressing a drug-resistant replicase, indicating that NS5A is involved in HCV-induced PI3P production, consistent with our earlier report showing that daclatasvir blocks HCV-induced DMV formation.
DFCP1 is a PI3P effector protein that, upon starvation, associates with omegasomes (Axe et al., 2008; Nanao et al., 2015). However, the exact function of this protein in autophagy remains unclear. Recent evidence suggests that DFCP1 contributes to the growth of lipid droplets and the interaction between lipid droplets and the ER (Gao et al., 2019; Li et al., 2019). In the present study, we show that DFCP1 positively regulates HCV and SARS-CoV-2 replication, possibly by promoting formation of DMVs, the replication sites of these viruses. We previously observed DMV-decorated ER wrapping around lipid droplets in HCV-infected cells (Lee et al., 2019b), arguing that DFCP1 might affect DMV formation through regulating the contact between ER and lipid droplets, at least in HCV. Although it is unclear whether the same applies to SARS-CoV-2, we note that lipid droplets have been observed in close proximity of virus particles, and it has been proposed that lipid droplets might serve as an assembly platform (Dias et al., 2020). However, we did not observe such proximity of lipid droplets and SARS-CoV-2 particles or DMVs by using various high-resolution imaging techniques (Cortese et al., 2020), although recent reports suggest that inhibition of lipid droplet formation interferes with the SARS-CoV-2 replication cycle (Dias et al., 2020; Williams et al., 2021). Further studies are required to clarify whether lipid droplets contribute to some specific steps of the SARS-CoV-2 life cycle or are more generally required as lipid or energy source.
In conclusion, we show that although ATG5-12/16L1-dependent autophagy is dispensable for HCV and SARS-CoV-2 replication, components of the autophagosome biogenesis machinery such as elements of the PI3K complex and DFCP1 are critically involved in viral replication, most likely by contributing to the formation of DMVs, the viral sites of RNA replication. These results underscore how evolutionarily distinct viruses hijack cellular pathways and factors in a convergent manner. Identification of commonly used host cell factors might pave the way for host-targeting broad-spectrum antivirals that are urgently needed for better preparedness against future pandemics.
Limitations of study
Our study is based on the use of cell lines that, although widely used to study HCV and SARS-CoV-2 replication, do not necessarily reflect the in vivo situation. While Huh7 cells employed to characterize HCV-induced DMV formation reflect the tropism of this virus, for technical reasons and to enable direct comparison, for SARS-CoV-2-induced DMVs we used the same cell line, which does not correspond to the primary tropism of this virus, even though many studies have used this cell line to characterize the SARS-CoV-2 replication cycle. Another limitation might be our focus on the KO of ATG5 and ATG16L1 to study the possible dependency of HCV and SARS-CoV-2 replication on macroautophagy. However, at least ATG5 is not required for macroautophagy, raising the possibility that alternative autophagy pathways might still contribute to the replication of these viruses. Finally, although the sole expression of nsp3-4 of SARS-CoV-2 suffices to induce DMVs, we cannot exclude the contribution of other viral proteins to this process. Further studies are needed to verify these possibilities.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Atg5 (D5F5U) Rabbit | Cell Signaling Technology | Cat#12994 |
| Atg16L1 (D6D5) Rabbit | Cell Signaling Technology | Cat#8089 |
| Anti-LC3 pAb (Polyclonal Antibody) | MBL International | Cat#PM036 |
| Purified Anti-PtdIns(4)P (PI4P) IgM | Echelon Biosciences | Cat#Z-P004 |
| PI3 Kinase Class III antibody (VPS34) | Cell Signaling Technology | Cat#3811 |
| Beclin-1 Antibody | Cell Signaling Technology | Cat#3738 |
| SARS-CoV-2 N protein | Sino Biological | Cat#40143-MM05 |
| Anti-SARS Nsp3 antibody | Abcam | Cat#ab181620 |
| mouse monoclonal anti-NS5A (9E10) | Gift from C. M. Rice | N/A |
| Monoclonal Anti-HA antibody | Sigma Aldrich | Cat#H3663 |
| ZFYVE1 (DFCP1) polyclonal antibody | Thermo Fisher Scientific | Cat#PA5-96710 |
| GAPDH (D16H11) XP® Rabbit mAb | Cell Signaling Technology | Cat#5174S |
| goat polyclonal anti-mouse HRP | Sigma Aldrich | Cat#A0168 |
| goat polyclonal anti-rabbit HRP | Sigma Aldrich | Cat#SAB3700852 |
| Alexa Fluor® 488, donkey anti rabbit | Thermo Fisher Scientific | Cat#A21206 |
| Alexa Fluor® 488, donkey anti mouse | Thermo Fisher Scientific | Cat#A21202 |
| Alexa Fluor® 568, donkey anti mouse | Thermo Fisher Scientific | Cat#AA10037 |
| Alexa Fluor® 488, Goat anti mouse IgM | Thermo Fisher Scientific | Cat#A21042 |
| DAPI (4’,6-Diamidino-2- Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat#D1306 |
| Chemicals, peptides, and recombinant proteins | ||
| Daclatasvir | MedChemExpress | Cat#HY-10466 |
| Bafilomycin A1 | Sigma Aldrich | Cat#B1793 |
| PI4KA-G1 | Gift from V. Lohmann (Bojjireddy et al., 2014) | N/A |
| PIK-III | Cayman Chemical | Cat#17002 |
| Remdesivir | MedChemExpress | Cat#HY-104077 |
| EBSS | Sigma Aldrich | Cat#E2888 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| TransIT®-LT1 Transfection Reagent | Mirus Biology | Cat#MIR2306 |
| Lipofectamine® RNAiMAX Reagent | Thermo Fisher Scientific | Cat#13778 |
| Gateway LR Clonase II Enzyme mix | Thermo Fisher Scientific | Cat#11791100 |
| In-Fusion® HD Cloning Kit | Clontech Laboratories | Cat#639650 |
| NucleoSpin RNA extraction kit | Machery-Nagel | Cat#740955 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat#4368814 |
| iTaq Universal SYBR green supermix | BioRad | Cat#1725121 |
| Experimental models: Cell lines | ||
| HEK293T | Graham et al., 1977 | N/A |
| Huh7.5 | Blight et al., 2002 | N/A |
| Huh7-Lunet/T7 | Backes et al., 2010 | N/A |
| Huh7-Lunet/CD81 high (H) | Koutsoudakis et al., 2006 | N/A |
| A549/ACE2 | Klein et al., 2020 | N/A |
| Calu-3 | Cortese et al., 2020 | N/A |
| Oligonucleotides | ||
| A full list of oligos is provided in Table S1 | N/A | N/A |
| Recombinant DNA | ||
| GFP-DFCP1/pMRX-puro | Hamasaki et al., 2013 | N/A |
| pWPI_GFP-DFCP1 | This paper | N/A |
| pWPI_GFP-DFCP1∗ | This paper | N/A |
| pWPI_ACE2 | Klein et al., 2020 | N/A |
| lentiCRISPR v2_Puro_Ctrl | Addgene | N/A |
| lentiCRISPR v2_Puro_ATG5 | This paper | N/A |
| lentiCRISPR v2_Puro_ATG16L1 | This paper | N/A |
| pCMV-dR8.91 | Stoeck et al., 2017 | N/A |
| pMD2.G | Stoeck et al., 2017 | N/A |
| pFK_Jc1 | Pietschmann et al., 2006 | N/A |
| pFK_JcR2a | Reiss et al., 2011 | N/A |
| pFK_i389LucNS3-3′_JFH_δg | (Wakita et al., 2005) | N/A |
| pFK_Con1ET | (Lohmann et al., 2003) | N/A |
| pTM_NS3-5B_JFH/NS5AEGFP | Schaller et al., 2007 | N/A |
| pTM_NS3-5B_JFH | Backes et al., 2010 | N/A |
| pTM_NS3-5B_NS5A Y93H | Berger et al., 2014 | N/A |
| SARS2_HA-3-4-V5 | Tabata et al., 2021 | N/A |
| piRO_Z-NG | Goellner et al., 2020 | N/A |
| SARS2_NG-HA-3-4-V5 | Tabata et al., 2021 | N/A |
| Software and algorithms | ||
| FIJI | N/A | https://imagej.nih.gov/ij/ |
| GraphPad Prism 5.0 | LaJolla, CA, USA | N/A |
| CellProfiler | Broad Institute, USA | https://cellprofiler.org |
| CellProfiler Analyst | Broad Institute, USA | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ralf Bartenschlager (ralf.bartenschlager@med.uni-heidelberg.de).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement that can be retrieved via the following link: https://www.klinikum.uni-heidelberg.de/zentrum-fuer-infektiologie/molecular-virology/requests-for-reagents
Experimental models and subject details
Cell lines and culture conditions
HEK293T (Graham et al., 1977), Huh7.5 (Blight et al., 2002), Huh7-Lunet/T7 (Backes et al., 2010), Huh7-Lunet/CD81 high (H) (Koutsoudakis et al., 2006) and A549/ACE2 cells (Klein et al., 2020) were maintained in Dulbecco’s modified Eagle medium (DMEM, Thermo Fisher Scientific), supplemented with 2 mM L-glutamine, nonessential amino acids, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 10% fetal calf serum (DMEM cplt). Huh7-Lunet/T7 cells stably expressing the bacteriophage T7 RNA polymerase were cultured in the presence of 5 μg/ml zeocin, while Huh7-Lunet SGR, Huh7-Lunet/CD81H, and A549/ACE2 cells were maintained in DMEM cplt containing 250 μg/ml G418. Starvation treatment was achieved by culturing cells in Earle’s Balanced Salts (EBSS, Sigma) for 2 h.
Virus stock production
For HCV production, in vitro transcripts of full-length HCV constructs Jc1 or JcR2a (Pietschmann et al., 2006; Poenisch et al., 2015) were transfected into Huh7.5 cells by electroporation as described below. Virus was harvested at 48 or 72 h post-transfection, filtered through a 0.45 μm-pore size filter and stored at −70°C until use. Tissue culture infectious dose 50 (TCID50) was determined as previously described (Kärber, 1931; Spearman, 1908). Unless stated otherwise, cells were infected with HCV using a MOI of 0.1.
The SARS-CoV-2 isolate Bavpat1/2020 was kindly provided by Prof. Christian Drosten (Charité Berlin, Germany) through the European Virology Archive at passage 2. Working virus stocks were generated by passaging the virus two times in VeroE6 cells. Virus titers were measured using plaque assay performed as previously described (Cortese et al., 2020).
Method details
Antibodies and immunofluorescence reagents
All antibodies and immunofluorescence reagents used in this study are listed in Key Resources Table.
DNA plasmid constructs
To generate the lentiviral construct pWPI_GFP-DFCP1, the GFP-DFCP1 sequence was first inserted into the pENTR1A vector using plasmid GFP-DFCP1/pMRX-puro (Hamasaki et al., 2013) as template and primers specified in Key Resources Table. Thereafter, the GFP-DFCP1 sequence was inserted into the pWPI vector using the LR recombinase of the Gateway system (Thermo Fisher Scientific) according to the instructions of the manufacturer. The construct encoding the inactive PI3P sensor (pWPI_GFP-DFCP1∗) was generated by PCR-based mutagenesis using pWPI_GFP-DFCP1 as template and primers specified in Key Resources Table. The amplicon was inserted into pWPI_GFP-DFCP1 using the InFusion system as recommended by the manufacturer (Clontech Laboratories).
The SARS CoV-2 HA-nsp3-4-V5 expression construct (Tabata et al., 2021) was generated by inserting a synthetic DNA fragment (provided by Biocat, Heidelberg) into the pcDNA3.1 vector. In this construct, codon-optimized SARS-CoV-2 nsp3-4 is tagged with HA and V5 at the N- and C terminus, respectively, and transcribed under control of the T7 RNA polymerase promoter. This T7 based system allows cytoplasmic transcription, thus avoiding possible splicing of the mRNA in the case of nuclear transcription as would happen e.g., in commonly used CMV promoter based expression constructs. The SARS2_NG-HA-3-4-V5 expression construct (Tabata et al., 2021) was obtained by using the expression vector piRO_Z-NG (Goellner et al., 2020) and replacing the Zika virus sequence by the HA-nsp3-4-V5 SARS-CoV2 coding region using the InFusion system (Clontech Laboratories) and primers listed in Key Resources Table. Other plasmids used in this study are listed in Key Resources Table.
Plasmid DNA transfection
For plasmid transfection, 1x105 /ml target cells were seeded in 6- or 24-well plates or glass-bottom culture dishes containing gridded coverslips (MatTek Corporation). After overnight culture, pTM_NS3-5B_JFH, pTM_NS3-5B_JFH/NS5AEGFP, SARS2_HA-3-4-V5, or SARS2_NG-HA-3-4-V5 plasmids were transfected using the TransIT-LT1 Transfection Reagent (Mirus Bio) according to the manufacturer’s protocol. Cells were harvested at time points specified in the result section for further analyses.
Lentivirus production and establishment of stable cell lines
Cells stably expressing GFP-DFCP1 and GFP-DFCP1∗ as well as Huh7-Lunet/T7/ACE2 cells were obtained by lentiviral transduction. Lentiviral vectors pWPI_GFP-DFCP1 or pWPI_GFP-DFCP1∗ were transfected into HEK293T cells along with packaging (pCMV-dR8.91) and envelope (pMD2.G) plasmids using polyethylenimine as reported earlier (Klages et al., 2000; Stoeck et al., 2017). Supernatants were harvested at 48 h and 72 h post-transfection, filtered and used to transduce target cells. Two days after transduction, cells were cultured in medium containing 10 μg/ml Blasticidin (Sigma). To generate ATG5 and ATG16L1 KO cells, annealed oligonucleotides were inserted into a lentiCRIPSRv2 plasmid (Addgene), in which single guide (sg) RNAs are transcribed under control of an U6 promoter. sgRNA sequences targeting ATG5 and ATG16L1 are 5′-AACTTGTTTCACGCTATATC-3′ and 5′-GCTGCAGAGACAGGCGTTCG-3′, respectively. Huh7-Lunet/T7 cells were infected with lentiviruses generated as described above and subjected to selection using DMEM cplt containing 3 μg/ml puromycin (Sigma Aldrich) for 1 week prior to use. For ectopic ACE2 expression, Huh7-Lunet/T7 cells with ATG5 or ATG16L1 KO, or with GFP-DFCP1 or mutant expression, were additionally transduced with a lentivirus encoding full length human ACE2 followed by selection with G418 (Thermo Fisher Scientific).
HCV replication assay
HCV replicons or genomes encoding firefly or Renilla luciferase, respectively, were transfected into Huh7-derived cells and viral replication was measured by luciferase assay as reported earlier (Reiss et al., 2011; Stoeck et al., 2017). In brief, cells were lysed in luciferase lysis buffer (25 mM glycylglycin, 15 mM MgSO4, 4 mM EGTA, 1% Triton X-100, 10% glycerol, pH 7.8) and frozen at −20°C. Luminescence was measured after adding the respective substrate (coelenterazine for Renilla luciferase and luciferin, glycyl-glycine, ATP and dithiothreitol for firefly luciferase) by using a Mithras LB940 plate luminometer (Berthold Technologies).
SARS-CoV-2-replication assay (immunostaining)
To determine SARS-CoV-2 replication, an immunostaining detecting the nucleocapsid (N) protein was used as reported recently (Steuten et al., 2021). In brief, cells were seeded at a density of 1.5 × 104 cells per well of a flat bottom 96-well plate (Corning). On the next day, SARS-CoV-2 (MOI = 1.0) was added to the compound containing medium. At 16 or 24 h post-infection, plates were fixed with 10% formaldehyde and inactivated by incubation of the culture plates in 6% formaldehyde for 30 min. Cells were washed with PBS prior to permeabilization with 0.2% Triton X-100 in PBS for 15 min. After blocking of the cells with 2% skim milk, dissolved in PBS containing 0.02% Tween 20, N protein-specific antibody was added for 1 h. Cells were washed with PBS prior to addition of the secondary antibody conjugated with horseradish peroxidase. After 1 h at 37°C, cells were washed and the signal was developed using TMB substrate. Reaction was quenched by addition of 0.5 M H2SO4 and absorbance was measured at 450 nm using a Tecan XFluor4 reader (Wiesbaden, Germany).
qRT-PCR-based SARS-CoV-2-replication assay
Total RNA was extracted from SARS-CoV-2-infected cells using NucleoSpin RNA extraction kit (Machery-Nagel) following manufacturer’s protocol. Reverse transcription (RT) reaction for cDNA synthesis was performed using the high capacity cDNA RT kit (ThermoScientific). Each cDNA was diluted 1:5 in nuclease free H2O and qPCR was performed using iTaq Universal SYBR green mastermix (Bio-Rad). Primers for qPCR were designed using Primer3 for SARS-CoV-2-ORF1 (Forward 5′- GAGAGCCTTGTCCCTGGTTT-3′, Reverse 5′-AGTCTCCAAAGCCACGTACG-3′) and HPRT (Forward 5′-CCTGGCGTCGTGATTAGTG-3′, Reverse 5′-ACACCCTTTCCAAATCCTCAG-3′). Relative abundance for SARS-CoV-2 Orf1 mRNA was corrected for PCR efficiency and normalized to HPRT transcript level.
Image-based SARS-CoV-2-replication assay
Huh7-Lunet-derived cells expressing human ACE2 were seeded into 96-well imaging plates (Greiner) and infected with SARS-CoV-2. At 24 h post infection, cells were fixed with paraformaldehyde followed by inactivation with 6% formaldehyde for 30 min. After inactivation, cells were immunostained using antibodies specific for SARS-CoV-2 nucleocapsid (N) protein, and imaging was performed with a Zeiss Cell Discoverer 7 microscope, followed by image analysis as described earlier (Pahmeier et al., 2021). In short, nuclei were segmented using the CellProfiler Analyst software package. The segmented nuclear mask was expanded by 5 pixels to obtain cytoplasmic region and N intensity was measured in this area. Next, a semi-supervised machine learning classification of cells into N-positive or -negative classes was performed using CellProfiler Analyst. Percentage infected cells and total number of cells analyzed (minimum 5,000 cells per sample) for ATG5 or ATG16L1 knockout cells was calculated by normalization to their respective values obtained with control KO cells.
Cell viability assays
Cytotoxicity was measured by using two different assays as specified in the results section. First, we used the CellTiter-Glo Luminescent Assay kit (Promega), quantifying intracellular ATP content, according to the manufacturer’s instructions. In brief, cells were lysed by adding an amount of CellTiter-Glo Reagent equal to the cell culture medium volume. After gentle mixing and 10 min incubation at room temperature, luminescence was measured using a Mithras LB940 plate luminometer (Berthold Technologies). Second, cell cytotoxicity was measured by WST-1 assay (Roche). Cells were seeded into 96 well-plates in triplicates and cultured for duration indicated in the result section. Culture medium was removed and cells were incubated in 100 μl/well of WST-reagent at 37°C for 30 min. Cell viability was analyzed by measuring absorbance at 450 nm using a Tecan XFluor4 reader (Wiesbaden, Germany).
Plaque assay
Plaque assay was performed as described previously (Klein et al., 2020). In short, VeroE6 cells were seeded in duplicate prior to inoculation with serial 10-fold dilutions of SARS-CoV-2 containing supernatants obtained from the time-course of infection of Huh7-Lunet/ACE2 and A549/ACE2 cells. After 1 h at 37°C, inoculum was replaced by serum-free MEM (GIBCO, Life Technologies), supplemented with 0.8% carboxymethylcellulose (Sigma-Aldrich). Cells were fixed three days post infection with formaldehyde for 30 min. Plates were plunged in 6% formaldehyde for inactivation and transported outside the BSL3 area. After washing the plates with water, the wells were stained with 1% crystal violet prepared in 10% ethanol for 30 min. After rinsing the plates with water, the number of plaques was counted and virus titers were calculated.
siRNA-based knockdown (KD)
For siRNA transfection, Huh7-Lunet/T7 cells were seeded at a density of 1x106 cells per 6-cm diameter dish or 5x105 cells per well of a 6-well plate. Cells were transfected twice with 50 pmole or 25 pmole siRNAs, respectively, at 24-h intervals using the Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific) according to the instructions of the manufacturer. For reverse transfection, 3 pmole siRNA were mixed with 0.25 μl RNAiMAX Reagent and spotted into a well of a 96-well plate. Target A549/ACE2 cells were seeded at a density of 2x104 cells per well of a 96-well plate and 48-72 h after transfection, cells were further processed as described in the results section.
Preparation of in vitro transcripts and RNA transfection by electroporation
RNA transcripts were generated from linearized plasmid DNAs by in vitro transcription using 80 mM HEPES (pH 7.5), 12 mM MgCl2, 2 mM spermidine, 40 mM dithiothreitol, 3.125 mM of each nucleotide, 1 U/μl RNasin (Promega), 0.6 U/μl T7 RNA polymerase and 10 μg DNA template. Reaction mixture was incubated at 37°C for 2 h in a total volume of 100 μl. Another 0.3 U/μl T7 RNA polymerase was added after 2 h incubation and reaction was continued for another 2 h. Reaction was stopped by adding 2 U of RNase-free DNase (Promega) per μg DNA and 1 h incubation at 37°C. RNA was purified with acidic phenol-chloroform, precipitated with isopropanol, and dissolved in RNase-free water.
For RNA transfection, target cells were prepared by trypsinization and resuspended in Cytomix (van den Hoff et al., 1992) supplemented with 5 mM glutathione and 2 mM ATP at a concentration of 107 cells/ml. For electroporation, 5 μg (subgenomic) or 10 μg (genomic) transcripts were mixed with 400 μl cell suspension, cells were transferred into a 0.4-cm cuvette (Bio-Rad) and subjected to an electrical pulse of 975 μF and 270 V in a Gene Pulser system (Bio-Rad).
Immunofluorescence analysis
Immunofluorescence (IF) was conducted as previously described (Zayas et al., 2016). In brief, cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 30 min, or, in the case of SARS-CoV-2 infection, with paraformaldehyde followed by inactivation with 6% formaldehyde for 30 min. Fixed cells were permeabilized in 0.5% Triton X-100/PBS and blocked with 10% FCS/PBS for 10 min and 1 h respectively. Cells were incubated with primary antibodies (see Key Resources Table for detail) and Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific) (diluted 1:1,000 in PBS with 10% FCS) for 1 h each. DAPI (1:500 diluted) was added to the secondary antibody solution for staining of nuclear DNA. IF images were generated with a Leica TCS SP8 (Leica Microsystems) and analyzed using the ImageJ software package. GFP-DFCP1 or LC3 puncta were counted using custom-ImageJ pipeline containing the “Analyzed Particles” function.
Western blot analysis
Cell lysates were mixed with 2x sample buffer (120 mM Tris-HCl [pH 6.8], 60 mM SDS, 100 mM DTT, 1.75% glycerol, 0.1% bromophenol blue) and incubated for 15 min at 95°C. Proteins were separated by electrophoresis into SDS - 10% polyacrylamide gels and transferred to a PVDF membrane. Membranes were blocked with PBST supplemented with 5% skim milk. Primary antibodies (see Key Resources Table for detail) were diluted in PBST containing 0.5% skim milk and membranes were incubated with the antibodies overnight at 4°C. After several washings, membranes were incubated for 1 h at room temperature with the corresponding secondary antibody. Signals were detected using the Western lightning plus-ECL reagent (PerkinElmer) and an Intas ChemoCam Imager 3.2 (Intas).
Transmission electron microscopy (TEM) and image analysis
Sample preparation was performed as described previously (Paul et al., 2011; Stoeck et al., 2017). Briefly, 24 h after transfection cells were washed with PBS, fixed for 30 min with CaCo buffer (2.5% glutaraldehyde and 2% sucrose in 50 mM sodium cacodylate, pH 7.4), supplemented with 50 mM KCl, 2.6 mM MgCl2 and 2.6 mM CaCl2, post-fixed with 2% OsO4 and then treated with 0.5% uranyl acetate. Cells were dehydrated and embedded into Epon resin. After polymerization of the embedded samples, 70 nm thick slices were generated using a microtome. Images were obtained with a JEOL JEM1400 (JEOL) microscope and processed using the ImageJ software package. DMV number and size were quantified by manually mapping the DMVs using ImageJ.
Correlative light electron microscopy (CLEM)
CLEM was conducted as reported earlier (Cortese et al., 2020). In brief, cells were seeded onto dishes containing gridded coverslips (MatTek Corporation). Twenty-four h after transfection with plasmids pTM_NS3-5B_JFH/NS5AEGFP or SARS2_NG-HA-3-4-V5, samples were first analyzed using a confocal microscope (Nikon) with a 10x objective lens to locate GFP-NS5A- or NeonGreen-positive cells, and the differential interference contrast (DIC) signal of these cells were acquired for correlation to electron microscopy images. Cells were then fixed and processed for TEM as described above. GFP-positive cells were located in electron microscopy using the grid coordinates and DIC signals, and their images were acquired and analyzed as described above for TEM.
Quantification and statistical analysis
Statistical analyses were performed using the Graphpad Prism software package (V5.03). All statistical details of experiments can be found in the figure legends. Significance values were calculated by applying 1-way ANOVA, unless stated otherwise in the figure legends. Statistical significances are depicted by asterisks in the figures as follows: (∗) for p < 0.05, (∗∗) for p < 0.01, and (∗∗∗) for p < 0.001.
Acknowledgments
We thank Ulrike Herian for excellent technical assistance and Sarah Göllner and Volker Lohmann for providing experimental resources. We also acknowledge the excellent support provided by the Infectious Diseases Imaging Platform (IDIP) headed by Vibor Laketa and the University of Heidelberg Electron Microscopy Core Facility (EMCF Heidelberg) headed by Stefan Hillmer. The SARS-CoV-2 isolate Bavpat1/2020 was kindly provided by Prof. Christian Drosten (Charité Berlin, Berlin, Germany) through the European Virology Archive (Ref-SKU: 026V-03883) at passage 2. We are grateful to all members of the Molecular Virology unit for continuous stimulating discussions. R.B. was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; project nos. 112927078–TRR 83, 240245660–SFB1129, and 272983813–TRR 179) and by the project “Virological and immunological determinants of COVID-19 pathogenesis – lessons to get prepared for future pandemics (KA1-Co-02 “COVIPA”)”, a grant from the Helmholtz Association's Initiative and Networking Fund. V.P. is supported by a European Molecular Biology Organization (EMBO) long-term fellowship (ALTF 454-2020).
Author contributions
Conceptualization, W.-I.T., K.T., and R.B.; methodology, W.-I.T. and J.-Y.L.; investigation, W.-I.T., J.-Y.L., K.T., H.K., V.P., B.C., and U.H.; computing resources, V.P. and J.-Y.L.; writing – original draft, W.-I.T. and R.B.; editing and final approval, W.-I.T., J.-Y.L., K.T., H.K., V.P., B.C., U.H., and R.B.; supervision, R.B.; funding acquisition, R.B.
Declaration of interests
The authors declare no competing interests.
Published: November 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110049.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ait-Goughoulte M., Kanda T., Meyer K., Ryerse J.S., Ray R.B., Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes P., Quinkert D., Reiss S., Binder M., Zayas M., Rescher U., Gerke V., Bartenschlager R., Lohmann V. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 2010;84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A., Balla T. Phosphatidylinositol 4-kinases: Old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–361. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Balla T., Downing G.J., Jaffe H., Kim S., Zólyomi A., Catt K.J. Isolation and molecular cloning of wortmannin-sensitive bovine type III phosphatidylinositol 4-kinases. J. Biol. Chem. 1997;272:18358–18366. doi: 10.1074/jbc.272.29.18358. [DOI] [PubMed] [Google Scholar]
- Belov G.A., Nair V., Hansen B.T., Hoyt F.H., Fischer E.R., Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C., Romero-Brey I., Radujkovic D., Terreux R., Zayas M., Paul D., Harak C., Hoppe S., Gao M., Penin F., et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology. 2014;147 doi: 10.1053/j.gastro.2014.07.019. 1094–105.e25. [DOI] [PubMed] [Google Scholar]
- Blanchard E., Roingeard P. Virus-induced double-membrane vesicles. Cell. Microbiol. 2015;17:45–50. doi: 10.1111/cmi.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight K.J., McKeating J.A., Rice C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojjireddy N., Botyanszki J., Hammond G., Creech D., Peterson R., Kemp D.C., Snead M., Brown R., Morrison A., Wilson S., et al. Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J. Biol. Chem. 2014;289:6120–6132. doi: 10.1074/jbc.M113.531426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calì T., Galli C., Olivari S., Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008;371:405–410. doi: 10.1016/j.bbrc.2008.04.098. [DOI] [PubMed] [Google Scholar]
- Calì T., Vanoni O., Molinari M. The endoplasmic reticulum crossroads for newly synthesized polypeptide chains. Prog. Mol. Biol. Transl. Sci. 2008;83:135–179. doi: 10.1016/S0079-6603(08)00604-1. [DOI] [PubMed] [Google Scholar]
- Cortese M., Lee J.Y., Cerikan B., Neufeldt C.J., Oorschot V.M.J., Köhrer S., Hennies J., Schieber N.L., Ronchi P., Mizzon G., et al. Integrative imaging reveals SARS-CoV-2-induced reshaping of subcellular morphologies. Cell Host Microbe. 2020;28:853–866.e5. doi: 10.1016/j.chom.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottam E.M., Maier H.J., Manifava M., Vaux L.C., Chandra-Schoenfelder P., Gerner W., Britton P., Ktistakis N.T., Wileman T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias S.S.G., Soares V.C., Ferreira A.C., Sacramento C.Q., Fintelman-Rodrigues N., Temerozo J.R., Teixeira L., Nunes da Silva M.A., Barreto E., Mattos M., et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020;16:e1009127. doi: 10.1371/journal.ppat.1009127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle W.E., Nyfeler B., Nagel J., Elling R.A., Liu S., Triantafellow E., Menon S., Wang Z., Honda A., Pardee G., et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- Dreux M., Gastaminza P., Wieland S.F., Chisari F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmy A.M., Labonté P. The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci. Rep. 2017;7:40351. doi: 10.1038/srep40351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmy A.M., Khabir M., Blanchet M., Labonté P. LC3B is not recruited along with the autophagy elongation complex (ATG5-12/16L1) at HCV replication site and is dispensable for viral replication. PLoS ONE. 2018;13:e0205189. doi: 10.1371/journal.pone.0205189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Xu K., Kovalev N., Nagy P.D. Recruitment of Vps34 PI3K and enrichment of PI3P phosphoinositide in the viral replication compartment is crucial for replication of a positive-strand RNA virus. PLoS Pathog. 2019;15:e1007530. doi: 10.1371/journal.ppat.1007530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Nettles R.E., Belema M., Snyder L.B., Nguyen V.N., Fridell R.A., Serrano-Wu M.H., Langley D.R., Sun J.H., O’Boyle D.R., 2nd, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G., Sheng Y., Yang H., Chua B.T., Xu L. DFCP1 associates with lipid droplets. Cell Biol. Int. 2019;43:1492–1504. doi: 10.1002/cbin.11199. [DOI] [PubMed] [Google Scholar]
- Gassen N.C., Papies J., Bajaj T., Emanuel J., Dethloff F., Chua R.L., Trimpert J., Heinemann N., Niemeyer C., Weege F., et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021;12:3818. doi: 10.1038/s41467-021-24007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goellner S., Cerikan B., Cortese M., Neufeldt C.J., Haselmann U., Bartenschlager R. Replication-independent generation and morphological analysis of flavivirus replication organelles. STAR Protoc. 2020;1:100173. doi: 10.1016/j.xpro.2020.100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Guévin C., Manna D., Bélanger C., Konan K.V., Mak P., Labonté P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology. 2010;405:1–7. doi: 10.1016/j.virol.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Yu H., Gu W., Luo X., Li R., Zhang J., Xu Y., Yang L., Shen N., Feng L., Wang Y. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci. Rep. 2016;6:23864. doi: 10.1038/srep23864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y., et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Ishibashi K., Fujita N., Kanno E., Omori H., Yoshimori T., Itoh T., Fukuda M. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12–5-16L2 complex. Autophagy. 2011;7:1500–1513. doi: 10.4161/auto.7.12.18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N., Mohd-Naim N., Wang Z., DeLeon J.L., Kim S., Zhong H., Sheshadri N., Dou Z., Edinger A.L., Du G., et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein Armus. J. Cell Sci. 2016;129:4424–4435. doi: 10.1242/jcs.192260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász G., Hill J.H., Yan Y., Sass M., Baehrecke E.H., Backer J.M., Neufeld T.P. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J. Cell Biol. 2008;181:655–666. doi: 10.1083/jcb.200712051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931;162:480–483. [Google Scholar]
- Klages N., Zufferey R., Trono D. A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol. Ther. 2000;2:170–176. doi: 10.1006/mthe.2000.0103. [DOI] [PubMed] [Google Scholar]
- Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B., Stanifer M.L., Boulant S., Bartenschlager R., Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020;11:5885. doi: 10.1038/s41467-020-19619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsoudakis G., Kaul A., Steinmann E., Kallis S., Lohmann V., Pietschmann T., Bartenschlager R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006;80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb C.A., Yoshimori T., Tooze S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Tabata K., Twu W.I., Rahman M.S., Kim H.S., Yu J.B., Jee M.H., Bartenschlager R., Jang S.K. RACK1 mediates rewiring of intracellular networks induced by hepatitis C virus infection. PLoS Pathog. 2019;15:e1008021. doi: 10.1371/journal.ppat.1008021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.Y., Cortese M., Haselmann U., Tabata K., Romero-Brey I., Funaya C., Schieber N.L., Qiang Y., Bartenschlager M., Kallis S., et al. Spatiotemporal coupling of the hepatitis C virus replication cycle by creating a lipid droplet-proximal membranous replication compartment. Cell Rep. 2019;27:3602–3617.e5. doi: 10.1016/j.celrep.2019.05.063. [DOI] [PubMed] [Google Scholar]
- Li D., Zhao Y.G., Li D., Zhao H., Huang J., Miao G., Feng D., Liu P., Li D., Zhang H. The ER-localized protein DFCP1 modulates ER-lipid droplet contact formation. Cell Rep. 2019;27:343–358.e5. doi: 10.1016/j.celrep.2019.03.025. [DOI] [PubMed] [Google Scholar]
- Limpens R.W., van der Schaar H.M., Kumar D., Koster A.J., Snijder E.J., van Kuppeveld F.J., Bárcena M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio. 2011;2 doi: 10.1128/mBio.00166-11. e00166–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Shreder K.R., Gai W., Corral S., Ferris D.K., Rosenblum J.S. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Lohmann V., Hoffmann S., Herian U., Penin F., Bartenschlager R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. Journal of Virology. 2003;77(5):3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K., Saitoh T., Tabata K., Omori H., Satoh T., Kurotori N., Maejima I., Shirahama-Noda K., Ichimura T., Isobe T., et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Mihelc E.M., Baker S.C., Lanman J.K. Coronavirus infection induces progressive restructuring of the endoplasmic reticulum involving the formation and degradation of double membrane vesicles. Virology. 2021;556:9–22. doi: 10.1016/j.virol.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K., McGrath M.E., Hu Z., Ariannejad S., Weston S., Frieman M., Jackson W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy. 2020;16:2131–2139. doi: 10.1080/15548627.2020.1817280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Mohl B.P., Bartlett C., Mankouri J., Harris M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016;97:680–693. doi: 10.1099/jgv.0.000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanao T., Koike M., Yamaguchi J., Sasaki M., Uchiyama Y. Cellular localization and tissue distribution of endogenous DFCP1 protein. Biomed. Res. 2015;36:121–133. doi: 10.2220/biomedres.36.121. [DOI] [PubMed] [Google Scholar]
- Oudshoorn D., Rijs K., Limpens R.W.A.L., Groen K., Koster A.J., Snijder E.J., Kikkert M., Bárcena M. Expression and cleavage of Middle East respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. MBio. 2017;8 doi: 10.1128/mBio.01658-17. e01658-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahmeier F., Neufeldt C.J., Cerikan B., Prasad V., Pape C., Laketa V., Ruggieri A., Bartenschlager R., Cortese M. A versatile reporter system to monitor virus-infected cells and its application to dengue virus and SARS-CoV-2. J. Virol. 2021;95:e01715–e01720. doi: 10.1128/JVI.01715-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Bartenschlager R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013;2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Bartenschlager R. Flaviviridae replication organelles: Oh, what a tangled web we weave. Annu. Rev. Virol. 2015;2:289–310. doi: 10.1146/annurev-virology-100114-055007. [DOI] [PubMed] [Google Scholar]
- Paul D., Romero-Brey I., Gouttenoire J., Stoitsova S., Krijnse-Locker J., Moradpour D., Bartenschlager R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011;85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Hoppe S., Saher G., Krijnse-Locker J., Bartenschlager R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013;87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T., Kaul A., Koutsoudakis G., Shavinskaya A., Kallis S., Steinmann E., Abid K., Negro F., Dreux M., Cosset F.L., Bartenschlager R. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poenisch M., Metz P., Blankenburg H., Ruggieri A., Lee J.Y., Rupp D., Rebhan I., Diederich K., Kaderali L., Domingues F.S., et al. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog. 2015;11:e1004573. doi: 10.1371/journal.ppat.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., McAuliffe J., Lu X., Subbarao K., Denison M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004;78:9977–9986. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F., Monastyrska I., Verheije M.H., Calì T., Ulasli M., Bianchi S., Bernasconi R., de Haan C.A., Molinari M. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.S., et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Merz A., Chiramel A., Lee J.Y., Chlanda P., Haselman U., Santarella-Mellwig R., Habermann A., Hoppe S., Kallis S., et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Berger C., Kallis S., Kolovou A., Paul D., Lohmann V., Bartenschlager R. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. MBio. 2015;6:e00759. doi: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller T., Appel N., Koutsoudakis G., Kallis S., Lohmann V., Pietschmann T., Bartenschlager R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 2007;81:4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M., Ackermann K., Stuart M., Wex C., Protzer U., Schätzl H.M., Gilch S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012;86:10112–10122. doi: 10.1128/JVI.01001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scutigliani E.M., Kikkert M. Interaction of the innate immune system with positive-strand RNA virus replication organelles. Cytokine Growth Factor Rev. 2017;37:17–27. doi: 10.1016/j.cytogfr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvas J.A., Jureka A.S., Nicolini A.M., Chvatal S.A., Basler C.F. Inhibitors of VPS34 and lipid metabolism suppress SARS-CoV-2 replication. bioRxiv. 2020 doi: 10.1101/2020.07.18.210211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir D., Chen W.L., Choi J., Wakita T., Yen T.S., Ou J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir D., Kuo C.F., Tian Y., Liu H.M., Huang E.J., Jung J.U., Machida K., Ou J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012;287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Limpens R.W.A.L., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., Faas F.F.G.A., Koster A.J., Bárcena M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18:e3000715. doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman C. The method of “right and wrong cases” (“constant stimuli”) without Gauss’s formulae. Br. J. Psychol. 1908;2:227–242. [Google Scholar]
- Steuten K., Kim H., Widen J.C., Babin B.M., Onguka O., Lovell S., Bolgi O., Cerikan B., Neufeldt C.J., Cortese M., et al. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis. 2021;7:1457–1468. doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck I.K., Lee J.Y., Tabata K., Romero-Brey I., Paul D., Schult P., Lohmann V., Kaderali L., Bartenschlager R. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. J. Virol. 2017;92 doi: 10.1128/JVI.01196-17. e01196-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W.C., Chao T.C., Huang Y.L., Weng S.C., Jeng K.S., Lai M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011;85:10561–10571. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata K., Prasad V., Paul D., Lee J.-Y., Pham M.-T., Twu W.-I., Neufeldt C.J., Cortese M., Cerikan B., Si Tran C., et al. Convergent use of phosphatidic acid for hepatitis C virus and SARS-CoV-2 replication organelle formation. bioRxiv. 2021 doi: 10.1101/2021.05.10.443480. 2021.2005.2010.443480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguwa S., Kambara H., Fujita N., Noda T., Yoshimori T., Koike K., Moriishi K., Matsuura Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011;85:13185–13194. doi: 10.1128/JVI.06099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Fukasawa M., Ueno T., Kominami E., Wakita T., Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5:937–945. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- van den Hoff M.J., Moorman A.F., Lamers W.H. Electroporation in “intracellular” buffer increases cell survival. Nucleic Acids Res. 1992;20:2902. doi: 10.1093/nar/20.11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovo T., Refolo G., Romagnoli A., Ciccosanti F., Corazzari M., Alonzi T., Fimia G.M. Autophagy in HCV infection: keeping fat and inflammation at bay. BioMed Res. Int. 2014;2014:265353. doi: 10.1155/2014/265353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . 2021. WHO coronavirus disease (COVID-19) dashboard.https://covid19.who.int/?gclid=EAIaIQobChMItd3v2JH_8wIVFCM4Ch2KxAl1EAAYASABEgKRe_D_BwE [PubMed] [Google Scholar]
- Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H.G., Mizokami M., Bartenschlager R., Liang T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Medicine. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.G., Jureka A.S., Silvas J.A., Nicolini A.M., Chvatal S.A., Carlson-Stevermer J., Oki J., Holden K., Basler C.F. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell Rep. 2021;36:109479. doi: 10.1016/j.celrep.2021.109479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff G., Melia C.E., Snijder E.J., Bárcena M. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 2020;28:1022–1033. doi: 10.1016/j.tim.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen C.K., Wong W.M., Mak L.F., Wang X., Chu H., Yuen K.Y., Kok K.H. Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J. Med. Virol. 2021;93:2076–2083. doi: 10.1002/jmv.26583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayas M., Long G., Madan V., Bartenschlager R. Coordination of hepatitis C virus assembly by distinct regulatory regions in nonstructural protein 5A. PLoS Pathog. 2016;12:e1005376. doi: 10.1371/journal.ppat.1005376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Thackray L.B., Miller B.C., Lynn T.M., Becker M.M., Ward E., Mizushima N.N., Denison M.R., Virgin H.W., 4th Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Chidekel A., Shaffer T.H. Cultured human airway epithelial cells (calu-3): A model of human respiratory function, structure, and inflammatory responses. Crit. Care Res. Pract. 2010;2010:394578. doi: 10.1155/2010/394578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.