

Abstract
Down syndrome (DS) is characterized by a collection of clinical features including intellectual disability, congenital malformations, and susceptibility to infections and autoimmune diseases. While the presence of an extra chromosome 21 is known to cause DS, the precise genetic annotation linked to specific clinical features is largely missing. However, there is growing evidence that two genes located on chromosome 21, IFNAR1 and IFNAR2, play an important role in disease pathogenesis. These genes encode the two subunits of the receptor for type I interferons (IFN-I), a group of potent antiviral and pro-inflammatory cytokines. Human monogenic diseases caused by uncontrolled IFN-I production and response have been well characterized, and they clinically overlap with DS but also have notable differences. Herein, we review the literature characterizing the role of IFN-I in DS and compare and contrast DS to other IFN-mediated conditions. The existing IFN-I literature serves as a rich resource for testable hypotheses to elucidate disease mechanisms in DS and is likely to open novel therapeutic avenues.
Keywords: Down syndrome, type I interferon, innate immunity, inborn errors of immunity
Introduction
Down syndrome (DS) is the most common genetic cause of intellectual disability in the US, affecting 1 in 700 newborns [1]. In addition to having cognitive disabilities, DS individuals frequently present with cardiac and gastrointestinal anomalies and have increased incidence of Alzheimer’s disease and hematological disorders [2], [3]. With improving health care and a doubling of life expectancy of individuals with DS in the past few decades [4], it has become clear that immune abnormalities are prominent in DS. Clinically, these immune features appear somewhat paradoxical. On one hand, people with DS appear to suffer from immune over-activation resulting in autoinflammatory and autoimmune diseases such as hypothyroidism, celiac disease, type I diabetes, and autoimmune skin diseases [2], [5]. On the other hand, individuals with DS show signs of immune suppression resulting in increased rates of infections, in particular otitis media and respiratory tract infections as documented for respiratory syncytial virus, influenza virus, and SARS-CoV-2 [6]–[9]. They are also more likely to undergo complications from these infections: longer hospital stays, intubation, and even mortality [2], [10], [11]. While the increased infection rate in people with DS was long ascribed to anatomical airway abnormalities, it has become clear that there are prominent immune-intrinsic defects in DS [12], [13].
DS is caused by trisomy of chromosome 21 (HSA21) in 95% of cases, and chromosomal translocation in 5% of cases (mostly t(14;21) or t(21;21)) [3]. The current databases list 233 protein-coding genes on HSA21, as well as 423 non-protein-coding genes and 188 pseudogenes [3]. Two genes on HSA21 stand out from an immune perspective: IFNAR1 and IFNAR2, which encode the two subunits of the type I interferon (IFN-I) receptor (IFNAR).
IFN-Is are cytokines with potent inflammatory and antiviral functions. In humans, there are 17 IFN-Is (13 IFNα subtypes, IFNβ, IFNκ, IFNε, IFNω) [14]. Engagement with their receptor leads to the cross-phosphorylation of JAK1 and TYK2 kinases, ultimately resulting in a phosphorylated (p)ISGF3 complex, which consists of pSTAT1/pSTAT2/IRF9, in the nucleus. This complex initiates the transcription of hundreds of IFN-stimulated genes (ISGs), establishing an antiviral and antiproliferative state in infected and neighboring cells [15].
Human genetic defects resulting in loss of function of IFN-I system result in increased susceptibility to various viral agents [15]. Conversely, the hyperactivity of IFN-I causes Type I Interferonopathies (IFNopathies), a group of inflammatory disorders characterized by cognitive defects and skin inflammation [15], [16]. Investigation of IFN-I signaling in DS started in the 1970s [17], yet no cohesive theory about IFN dysregulation in DS has been accepted today. In recent years, multiple studies have interrogated this pathway in DS and its potential role in the pathogenesis of the syndrome. While the duplication of a full chromosome presents significant challenges in the evaluation of a single pathway, a wealth of data has been accumulated examining IFN signaling in DS. Herein, we review the literature on the effects of increased gene dosage of IFNAR1 and IFNAR2 in DS and consider the data supporting and refuting the theory of DS as an IFN-mediated disease.
I -. Triplication of Type I, II and III IFN receptor genes in Down syndrome
3 copies of 4 IFN receptor genes in Down syndrome.
There are currently three prevailing hypotheses aimed at explaining the genetic causality of DS: 1) a simple gene-dosage imbalance, in which syndrome features result from direct increased expression of genes on HSA21 and the downstream effects of this overexpression (for instance increased APP copy number causing increased accumulation of amyloid precursor protein in the brain which could explain the increased incidence of Alzheimer’s disease). We will refer to this as the “cis-acting” hypothesis. 2) A global dysregulation caused by specific genes on HSA21 that disrupt overall biological homeostasis via effects such as chromatin availability (HMGN1, BRWD1), splicing regulation (U2AF1L5, RBM1, U2AF1, DYRK1A), post-transcriptional regulation (ADARB1, micro-RNAs [miRNAs]), protein turnover (USP25), and metabolism (SOD1). We will refer to this as the “trans-acting” hypothesis. 3) The presence of an extra chromosome regardless of the genes it encodes, in which aneuploidy itself disturbs cellular homeostasis. We will refer to this as the “chromosome-intrinsic” hypothesis. All three processes are most likely at play in DS etiology [3], [18]. Specific effects of single genes, their interactions with the rest of the genome, and nonspecific disturbances caused by trisomy must be systematically studied to establish the pathogenesis of DS.
Four subunits of IFN receptors (IFN-Rs) are encoded on HSA21: IFNAR1, IFNAR2, IFNGR2, and IL10RB. IFNAR1 and IFNAR2 encode the 2 subunits of the IFN-I receptor. IFNGR2 encodes a subunit of type II IFN receptor, and IL10RB encodes a subunit of receptor for type III IFN, but also a subunit of receptors for three interleukins (IL-10, IL-22, and IL-26). Given that limited data exists on the effects of IFNGR2 and IL10RB triplication in DS, we will focus this review on IFNAR1 and IFNAR2. Besides, the presence of both IFNAR subunits on HSA21 make IFNAR1 and IFNAR2 particularly interesting candidates. In addition, these genes are known to be expressed in all nucleated cells in typical individuals and are expressed at low levels [19], [20]. Thus, small changes in expression of these genes may result in significant functional differences. Moreover, low and average-expressed genes are thought to be particularly affected by trisomy based on single-cell transcriptome analyses of T21 and other aneuploidies [21], making IFNAR1 and IFNAR2 likely to be differentially expressed.
Increased expression of IFN-R genes in Down syndrome.
As early as 1974, before the IFN-I genes or their receptors had even been cloned, Y.H. Tan observed that “cells that were trisomic for chromosome 21 were three to seven times more sensitive to protection [from vesicular stomatitis virus (VSV)] by human interferon than the normal diploid or trisomic 18 or 13 fibroblasts” [17]. A decade later, T21 fibroblasts were used to study biochemical changes triggered when IFN-I engages its receptor, and the group observed a linear relationship between the number of HSA21 copies present and the dose of IFNα or IFNβ required to induce a maximum response [22].
In recent years, studies have further delineated the expression of IFN-R genes in DS. Bulk RNA sequencing (RNA-seq) experiments in multiple cell lines and primary immune cells unveiled that the majority of genes mapped to HSA21 are expressed ~1.5 times more highly in DS than in controls, apart from a few variably expressed outliers that do not include the IFN-R genes [18], [23], [24]. At the protein level, IFNAR1, IFNAR2, and IFNGR2 but not IL10RB were found to be on average ~1.5x more highly expressed in T21 B-EBVs and monocytes, although the range of expression was wide and there was a large overlap with the controls [25] (Figure 1). Recently, the IFNAR1 expression was found to be increased in DS compared to controls in 85 of 100 white blood cell subsets surveyed, but the level of increase was variable depending on cell type [24]. Although more studies are warranted to systematically quantify IFNAR1 and IFNAR2 levels across cell types, these early data suggest that increased expression of IFNAR in T21 is not strictly uniform for reasons that are still unknown.
Figure 1.
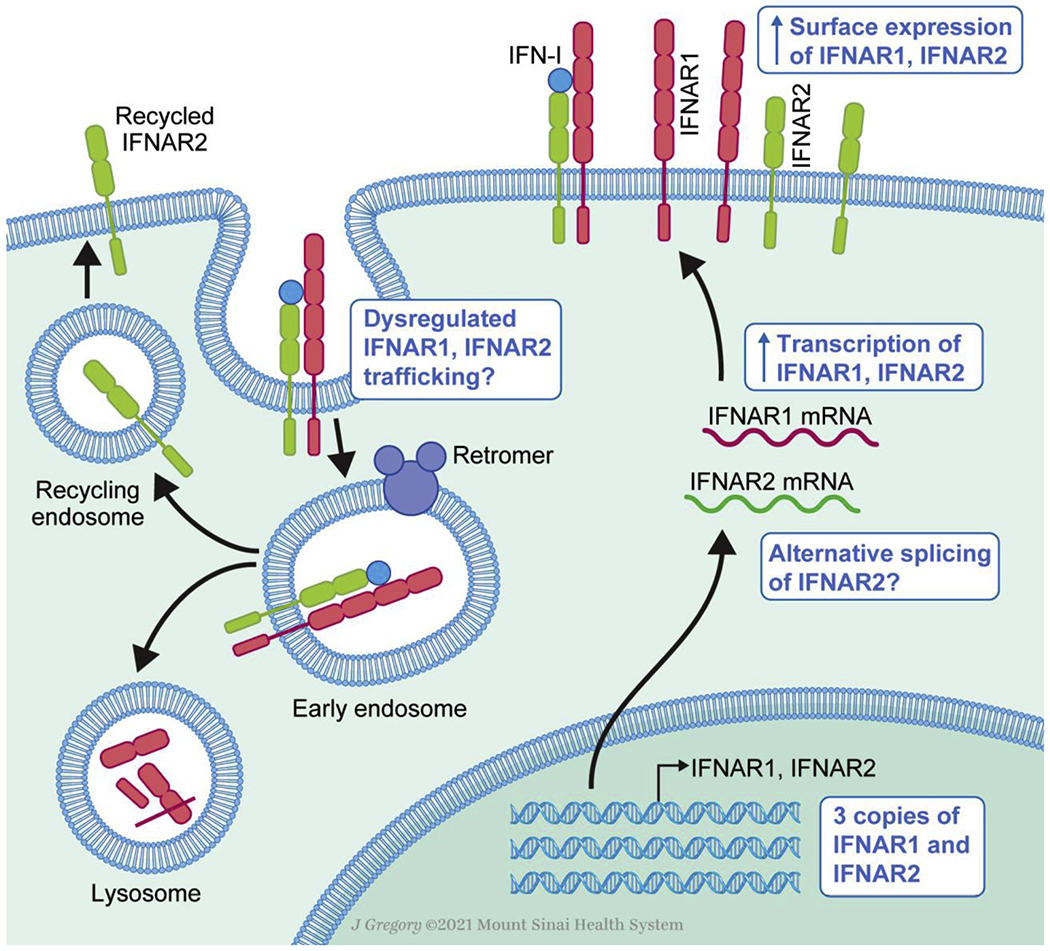
IFNAR1 and IFNAR2 expression and dynamics in health and Down syndrome. Diagram of transcription, translation and surface expression of the IFN-I receptor (right) and its trafficking after signaling resulting in lysosomal degradation of IFNAR1 and recycling of IFNAR2 (left). Blue boxes denote alterations characterized in Down syndrome.
Unknowns.
While in principle the increased gene dosage of IFNAR1 and IFNAR2 in DS has been established, many questions remain. Firstly, the effects of HSA21 “trans-acting” genes have not been studied. For example, HSA21 encodes post-transcriptional regulators such as ADARB1 and miRNAs. Proteomic studies in DS found that the global protein expression was slightly less than the mRNA expression (~1.3-1.4 fold over control vs ~1.5 fold), which most likely results from post-transcriptional regulation [18]. Further studies examining the impact of these elements on IFNAR1 and IFNAR2 expression are warranted and may likely be cell type specific.
Furthermore, important splicing regulators are located on HSA21, including U2AF1L5, RBM1, U2AF1, and DYRK1A (a dual specificity kinase that phosphorylates splicing factors). Their effects are poorly understood, but there is evidence that alternative splicing occurs in T21 iPSC-derived neurons [26]. This has yet to be explored in the context of the IFN-Rs. IFNAR2 is known to exist in three isoforms, IFNAR2a (a soluble truncated form), IFNAR2b (a transmembrane truncated form that lacks the intracellular domain), and IFNAR2c (the long transmembrane form that mediates IFN-I signaling) [27]. Alternative splicing of this gene or differential ratios of these isoforms could have profound consequences on IFN-I signaling.
Finally, IFNAR surface expression is dynamic and changes upon IFN-I signaling. After cytokine binding, the two subunits are internalized by the retromer complex and IFNAR2 is recycled to the plasma membrane while IFNAR1 is sorted to the lysosome and degraded [28] (Figure 1). If this process is inhibited so that IFNAR subunits remain at the plasma membrane, IFN-dependent signaling and downstream gene transcription are increased [28]. In DS, does increased expression of IFNAR result in more “naïve” subunits remaining at the surface after IFN signaling, resulting in heightened signaling? Moreover, retromer dysregulation has been documented in DS [29], [30], which could also lead to further upregulation of IFN signaling. Beyond gene production, the dynamics of IFNAR availability at the plasma membrane should be further explored in DS.
II -. Increased IFN signaling in Down syndrome
Increased response to IFN-I in Down syndrome.
T21 fibroblasts, B-EBVs, and primary monocytes and lymphocytes hyper-respond to IFNα and IFNβ in terms of proximal signaling, leading to augmented levels of pSTAT1 [23], [25], [31] (Figure 2). There is also evidence of increased ISG induction downstream of IFN-I stimulation in T21 fibroblasts compared to disomic controls [23] (Figure 2). Since all nucleated cells express IFNAR1 and IFNAR2, albeit to varying degrees, the impact of this hyper-response across cell types is of particular interest. Although the heterogeneity of this response has not been widely studied in DS, there is evidence that it is variable across immune cell types.
Figure 2.
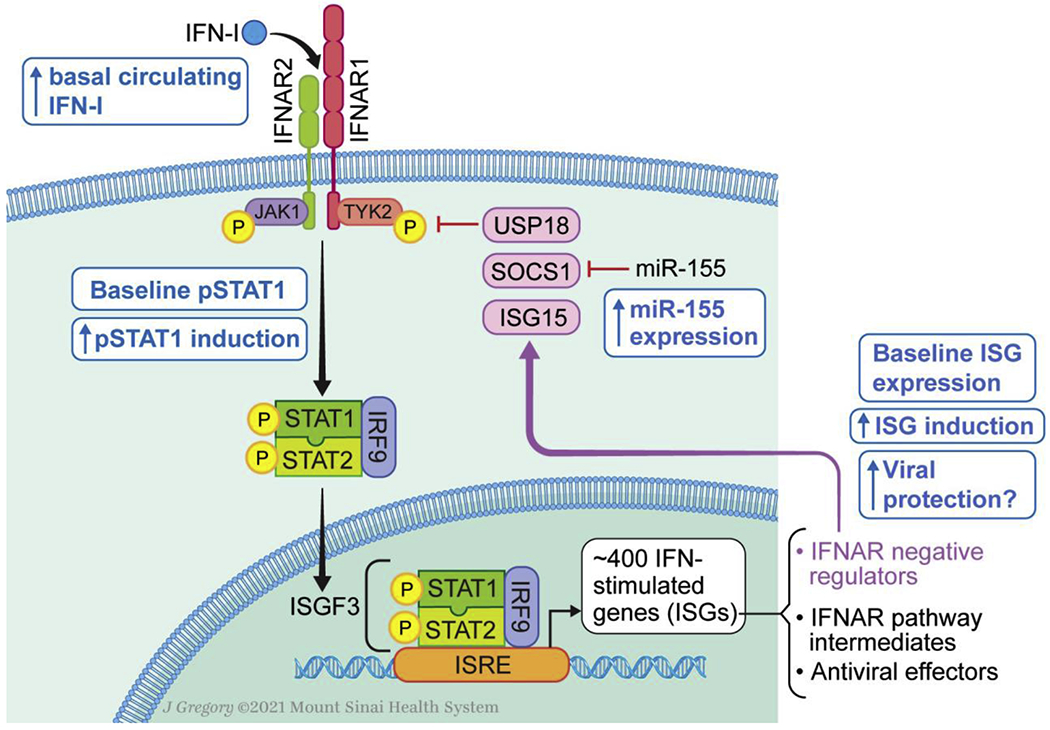
Mechanisms of Type I Interferon signaling dysregulation in Down syndrome. Diagram of JAK-STAT signaling upon IFN-I binding followed by induction of IFN-stimulated genes (ISGs). Downstream effects of ISGs including viral protection and negative regulation of the IFN-Ireceptor are outlined. Blue boxes denote alterations characterized in Down syndrome.
Contrary to other immune cells, T21 CD8+ TEMRAs do not hyper-respond to IFNα in one study [31] or even hypo-respond compared to controls in another study [25]. This correlates with the higher expression of activation and senescence markers in these T cells subsets [25], [31]. Whether this phenotype is due to initial IFN-I hyper-activity (cis-effect) or another HSA21-related mechanism (trans-effect) has yet to be elucidated. In T cells, over-activation could be caused by an autoimmunity-prone state conferred by cis-effects of genes on HSA21 (IFN-Rs, AIRE) and/or by dysregulation in metabolism and/or DNA repair pathways which are known to be features of aneuploidies (chromosome-intrinsic effect) [18].
The downstream effects of IFN dysregulation on DS phenotype remains largely unexplored. One study showed that upregulation of one ISG, IDO1, results in the increased production of neurotoxic metabolites kynurenine and quinolinic acid, which could explain some neurological defects in DS [32]. These results were replicated in mouse models of DS. Interestingly, IDO1 and kynurenine also mediate suppression of CD4 and CD8 cells and induction of T regulatory cells [33], [34]. In a DS mouse model, stimulation with a synthetic immune agonist, poly(I:C), was shown to induce higher levels of ISGs and resulted in increased morbidity and mortality compared to WT controls [35]. Combined, these results highlight the potential widespread impact of altered IFN-I signaling in DS.
Baseline IFN signaling in Down syndrome.
There is extensive evidence of IFN-I signaling in DS immune cells and patient-derived cell lines in the absence of exogenous IFN-I stimulation. The extent of this signaling and the pathogenic effects it might have remain largely unexplored, however. RNA-seq experiments have revealed that the majority of the differentially expressed genes in T21 cell lines and PBMCs were not located on HSA21 but were instead related to IFN-I signaling [23], [31]. Of note, re-analysis of these datasets by a different group did not come to the same conclusion [18]. Transcriptomic analysis in monocytes isolated from individuals with DS, non-DS controls, and patients with gain-of-function (GOF) mutations in STAT1 who constitutively activate IFN-I and IFN-II signaling revealed that people with DS have elevated expression of ISGs at baseline [25] (Figure 2). However, these levels are variable among individual with DS, with some clustering with the healthy controls while others clustered with the STAT1 GOF patients. These results highlight the heterogeneity of the DS population. This heterogenous feature is not unique to IFN signaling but is also true for all phenotypic traits of the syndrome [36]. Furthermore, mild STAT1 phosphorylation was also detected at baseline in T21 monocytes, and total STAT1, an ISG, was elevated in T21 monocytes, albeit to lower levels than those of STAT1 GOF patients [25] (Figure 2). This supports the notion that individuals with DS have constitutive IFN-I signaling, although it is not to the level of a known IFN-mediated disease and thus warrants further evaluation to understand its contribution to the syndrome and consider potential therapeutic avenues.
Unknowns
Previously described IFN-mediated diseases are caused either by defects in DNA and RNA-digesting enzymes (TREX1, SAMHD1, RNASEH2A, etc.) resulting in continual IFN-I production triggered by accumulated nucleic acids, by activating mutations in pathogen-sensing receptors (DDX58, TMEM173) also resulting in chronic IFN-I secretion, or finally by loss of IFN-I negative regulators (ISG15, USP18 and STAT2 ) [15], [16]. While there is evidence that DS cells have basal IFN-I signaling as discussed above, it remains unclear if an increased expression of IFN-I receptor in DS would result in spontaneous production of IFN-I. Elevated circulating levels of IFNα were found basally in 12% of tested individuals with DS using an ultra-sensitive assay [25], but no mechanism has been proposed for this spontaneous secretion. One interesting model is the constitutive IFN-I release caused by defects in the IFNAR negative regulators ISG15 and USP18 [37], [38]. With these genetic mutations, defective shutdown of IFN-I signaling is sufficient to cause spontaneous IFN-I production through mechanisms that are still not established. Another interesting avenue is the possibility of IFN-I siloes. Indeed, when IFNAR1 and IFNAR2 are internalized by the retromer, bound cytokine is engulfed with it and is “siloed” in the cell and may continue to signal [39]. Perhaps increased presence of IFNAR results in increased cytokine siloes in DS, which could explain the presence of IFNα in some individuals and the baseline ISG induction.
Furthermore, the negative regulation of IFNAR has also not been studied in DS. miR-155, which is located on HSA21, targets the IFNAR negative regulator SOCS1 [40]–[42], thus its increased expression could lead to IFN-I hyperactivity. The transcriptional repression mediated by the five miRNAs located in on HSA21 has been extensively studied in the context of Alzheimer’s disease in DS [43], but their effects on the immune system are largely unknown apart from a recent association between B cell dysfunction and increased miR-155 and miR-125b expression in DS [44]. On the other hand, negative regulators of IFNAR are induced by IFN-I signaling, including SOCS1, USP18, and ISG15 [14]. Therefore, increased expression of IFNAR could also result in increased negative regulation. The interplay of repression and induction of IFNAR negative regulators must be elucidated in DS.
Finally, the downstream effects of increased IFN-I signaling in DS remain poorly understood. Apart from a single study examining the effects of one ISG in DS [32], very little is known about how increased ISG induction contributes to DS pathology. The known anti-proliferative and pro-apoptotic properties of IFN-I in DS are also of interest. For instance, increased apoptosis has been noted in neutrophils, eosinophils, and B cells from individuals with DS, but the role of IFN-I in this process has not been studied [45], [46]. Examination of the downstream effects of increased IFN-I signaling in DS is still in its infancy and could potentially shed light on many observed phenotypic abnormalities of DS.
III -. Down syndrome: a single-chromosome interferonopathy?
Neurological manifestations: overlap between DS and Type I Interferonopathies
Type I IFNopathies are a group of diseases mediated by the chronic production and/or response to IFN-I. These result from single-gene defects at various points upstream of or in IFN-I signaling [15], [16]. The phenotypic spectrum of these diseases is wide, ranging from the highly penetrant and rare Aicardi-Goutières syndrome to the diffuse autoimmune disease systemic lupus erythematosus (SLE) [16]. Interestingly, however, there is some phenotypic overlap among all IFNopathies, mainly the involvement of the central nervous system (CNS) and the skin [16], [47].
The presence of basal ganglia calcifications (BGCs) is highly indicative of chronic IFN-I signaling and is found in the majority of these patients, although it is not always reported given that they are mostly found via head computed tomography (CT), a test that is not widely administered nowadays. Interestingly, studies from the 1980s report an increased incidence of BGCs in individuals with DS compared to controls, with up to 45% of patients showing signs of intracranial calcifications [48]–[50]. The presence of BGCs increases with age in DS [50], but they were also recently described in a young girl with DS which is exceedingly rare [51].
Symptomatically, CNS involvement in IFNopathies manifests as developmental delay, seizures, and dystonia, although these are not specific to this group of diseases [16], [47]. These manifestations all occur in DS [2] (Figure 3). Developmental delay is one of the syndromic features that John Langdon Down described when he first characterized the disease in 1866 [52]. Individuals with DS are also more likely to suffer from seizure disorders (incidence of 8%) than non-DS controls [53]. There is therefore a large overlap in the CNS phenotype of DS and type I IFNopathies.
Figure 3.
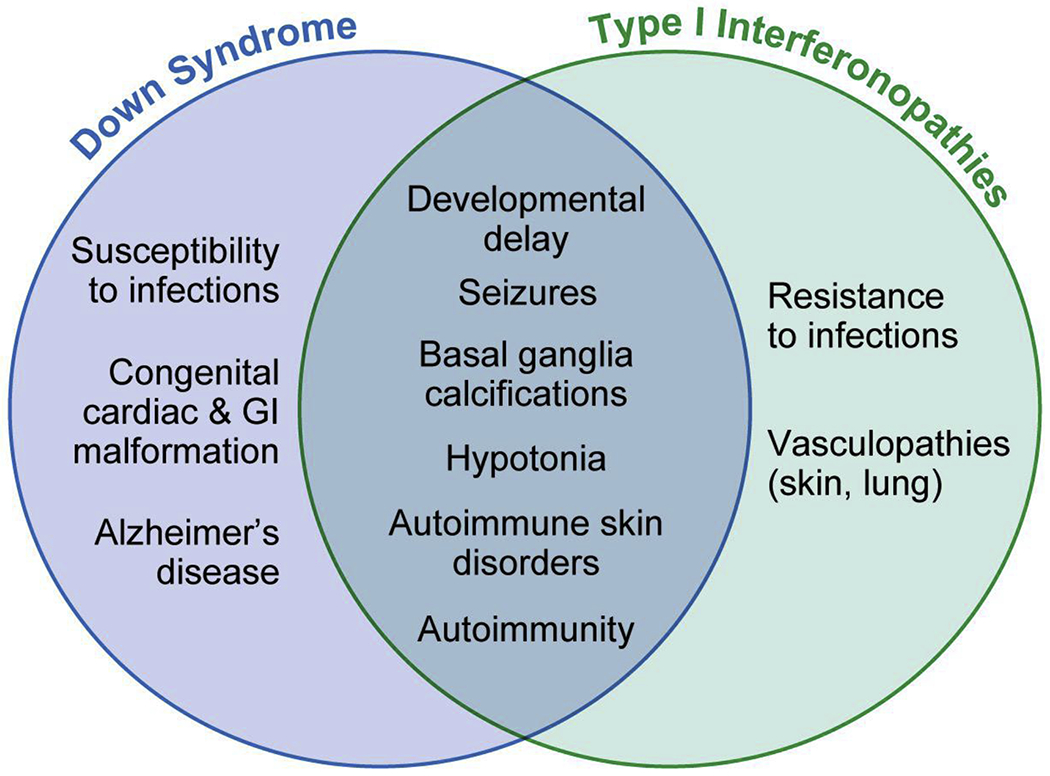
Specific and overlapping clinical features of Down syndrome monogenic Type I Interferonopathies.
Skin manifestations: DS and Type I Interferonopathies are distinct
The second most commonly seen manifestation across type I IFNopathies is skin involvement. These mainly present as skin vasculopathy with chilblains and livedo reticularis [54] (Figure 3). Livedo reticularis occurs in 9-13% of children with DS [5], but no chilblain lesions have been described in DS. On the other hand, Hidradenitis suppurativa (HS) is known to be more prevalent in DS [55], but there is no association between HS and type I IFNopathies [56]. Nonspecific autoimmune skin manifestations such as alopecia aerata and psoriasis which are known to occur in SLE [57] have an increased incidence in DS [5]. Interestingly, the Jak inhibitor Tofacitinib was recently administered to two patients with DS for alopecia aerata after corticosteroid injections had failed [58], and as a first line therapy in a patient with DS and psoriatic arthritis [59]. All three patients had significant improvement of symptoms. Tofacitinib is a pan-JAK inhibitor and therefore acts to block signaling downstream of a slew of cytokines, including IFN-I. While these results remain somewhat anecdotal given the small sample size, they support that IFN-I may be involved in the pathophysiology of this autoimmune condition in DS.
Unknowns
A prominent immune manifestation of DS is the presence of autoimmune disorders. Indeed, individuals with DS are more likely to develop autoimmune thyroid disease, type I diabetes, and celiac disease [2]. Although autoimmunity is not a classical feature of type I IFNopathies, most patients do present with autoantibodies [47]. Autoimmunity is also characteristic of SLE, which is thought to be largely IFN-mediated [16]. Although we do not purport that autoimmunity can be solely be explained by IFN-I dysregulation in DS, as B and T cell dysfunction (reviewed elsewhere [13]) certainly play a role, the link to SLE emphasizes the possibility of IFN-I involvement in the development of tolerance. The large body of knowledge accumulated to delineate the role of IFN-I in mediating autoimmunity in SLE provides precious clues to investigate this process in DS [60].
Another unexplained clinical presentation of immune dysfunction in DS is the increased susceptibility to infections. Indeed, as previously stated, individuals with DS have higher rates of morbidity and mortality due to viral and bacterial infections. It is estimated that infections account for 34-40% of deaths in DS [2]. This feature is in opposition to the concept of DS as a type I IFNopathy. Indeed, these patients do not suffer from increased incidence of infections and in some cases are even thought to be more resistant to viral infections than controls [15], [61], which is what is expected given the antiviral effect of IFN-I.
So far, no mechanistic links between immune defects in DS and increased susceptibility to infection have been made. In their recent study, Kong et al. hypothesized that increased susceptibility to cutaneous candidiasis in DS could be due to increased IFNβ repression of the Th17 signaling axis, but their results refuted this hypothesis [25]. Nonetheless, the IFN-I literature provides possible explanations for the seemingly paradoxical susceptibility to infection in the presence of increased IFN-I signaling. Studies have highlighted the importance of the levels and timing of IFN, as the cytokine is protective early in disease but later becomes pathologic [62], [63]. Recent findings of autoantibodies mediating increased disease severity in COVID-19 provide another potential mechanism by which people with DS, who are known to be prone to autoimmunity, may develop worse viral disease [64].
Finally, the interplay between IFN-I signaling and other cytokines is of particular interest in DS. Indeed, multiple studies have shown that several cytokines are increased in children and adults with DS, including IL-1, IL-6, IL-22, IFNγ, and TNFα [65], [66]. Increase in the clinical marker of inflammation C-reactive protein and decrease in components of the complement cascade (indicative of complement activation) have also been reported [66], [67]. Combined, these data paint a picture of global immune dysregulation in DS. IFN-Is can themselves directly act on cytokine and chemokine release [68]. Altered JAK and STAT availability due to signaling downstream of IFNAR could also dysregulate responses to a slew of cytokines that themselves act via these JAKs and STATs, as has been demonstrated in other genetic diseases of altered cytokine signaling [69]. Finally, although the triplication of IFNGR2 and IL10RB are understudied, there is evidence that T21 cell lines express higher levels of these receptor subunits and that they hyper-respond to stimulation with IFNγ [25]. IFNγ is a potent checkpoint for cytokine signaling and could also play a role in soluble immune dysregulation [70]. The role and interplay between the receptors to type I, type II, and type III IFNs, and their impact on the rest of the secretome is another exciting area of DS immunology that deserves further exploration.
Conclusion:
Taken together, the literature shows that IFN-I signaling is profoundly altered in DS. Although there is strong biochemical and clinical evidence that DS is, at least in part, an IFN-mediated disease, it remains distinct from previously described IFN-mediated diseases that stem from single-pathway defects. While these monogenic diseases can provide useful clues to understand DS pathology, they have strong limitations which must be considered when studying a “mono-chromosomic” IFN-mediated disease. The 1.5-fold increase in IFNAR1 and IFNAR2 copy number has intrinsic consequences, but these must be evaluated together with other genes located on HSA21 and in the context of aneuploidy to fully understand the genetic mechanisms underlying DS. Perhaps, IFN-I mediated activity truly combines cis-acting, trans-acting and chromosome-intrinsic origins of pathophysiology.
Today, the field of immunology in DS has been dominated by observational studies cataloguing major differences in immune subsets between DS individuals and the general population. Studies with a strong genetic hypothesis that provide mechanistic clues to understand observed phenotypes have been limited. The evaluation of IFNAR1 and IFNAR2 gene dosage effects is an emerging field that promises to fill gaps in our understanding of why people with DS suffer from severe immune dysfunction and may even extend to uncovering mechanisms of other DS features like neurologic defects and developmental delay.
For instance, IFN-I is known to play an important role in the CNS, which is of particular interest in DS, a disease whose most prominent features include intellectual disability, dementia, and seizures. Early mouse experiments revealed that excessive IFN-I signaling in the brain led to neurotoxicity [71], [72]. Conversely, a recent study found that constitutive IFNβ production in the brain is necessary for protection from the neurotropic virus HSV-1 [73]. These studies, in addition to the prominent CNS manifestations of IFNopathies discussed above, highlight the privileged role of IFN-I signaling in the brain. DS mouse models show improvement in neuron survival in the presence of IFN-neutralizing antibodies or when the mouse is genetically manipulated to partially restore IFN-R genes to WT, disomic, numbers [74], [75]. While these studies show promise, much remains to be unveiled about the role of IFN-I in the CNS.
Another promising field worth investigating is IFN-I signaling in utero. DS is in many ways a developmental condition, and there is a wealth of evidence that IFN-I can be nefarious at every step in pregnancy, from implantation to placental formation to fetal brain development [76]. IFN-induced transmembrane proteins (IFITMs), ISGs that are beneficial when acting to block viral entry, can become pathogenic when interfering with cell fusion necessary for placenta formation [77]. It is estimated that 30-40% of T21 pregnancies result in spontaneous miscarriage [78]. Abnormal placental development has also been documented in T21 humans and mouse models of DS [79]. IFN-I signaling in-utero in T21 has not been studied and could provide key insights into early developmental defects in DS.
Finally, the growing field of IFN-I signaling in DS also opens novel therapeutic avenues. Indeed, many drugs targeting the IFN-I pathway with increasing precision have been developed in recent decades [80]. The majority of them block JAKs downstream of IFNAR and other cytokine receptors. They have been successfully used to treat IFN-mediated diseases and are well tolerated, resulting in their FDA approval for a plethora of inflammatory diseases [80]. Their use in DS have only been reported in three cases so far, all of them with successful outcomes [58], [59], and there is currently a clinical trial underway to test these drugs in more patients [81]. This provides an exciting new therapeutic approach to address the immune features in DS, and perhaps even other manifestations of the syndrome as the role of IFN becomes better defined.
Highlights.
Both subunits of the Type I Interferon (IFN-I) receptor, IFNAR1 and IFNAR2, are located on chromosome 21, which is triplicated in people with Down syndrome (DS).
IFNAR1 and IFNAR2 are more highly expressed in cells from individuals with DS.
IFN-I signaling is present at baseline in individuals with DS, and cells derived from these individuals hyper-respond to IFN-I stimulation.
There is overlap in the clinical presentations of DS and Type I Interferonopathies, a group of monogenic diseases caused by excessive IFN-I signaling.
Acknowledgments
We would like to thank Jill K. Gregory for the illustrations. Used with permission of Mount Sinai Health System.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors whose names are listed below certify that they have no affiliations with or involvement in any organization or entity with any financial interest, or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
References
- [1].Mai CT et al. , “National population-based estimates for major birth defects, 2010–2014,” Birth Defects Res, vol. 111, no. 18, pp. 1420–1435, November. 2019, doi: 10.1002/bdr2.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bull MJ, “Down syndrome,” N. Engl. J. Med, vol. 382, no. 24, pp. 2344–2352, 2020, doi: 10.1056/NEJMra1706537. [DOI] [PubMed] [Google Scholar]; *Comprehensive review of the clinical characteristics of Down syndrome.
- [3].Antonarakis SE et al. , “Down syndrome,” Nat. Rev. Dis. Prim, vol. 6, no. 1, pp. 1–20, January. 2020, doi: 10.1038/s41572-019-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Comprehensive review of the known genetic disturbances in Down syndrome.
- [4].De Graaf G, Buckley F, and Skotko BG, “Estimation of the number of people with Down syndrome in the United States,” Genet. Med, vol. 19, no. 4, pp. 439–447, April. 2017, doi: 10.1038/gim.2016.127. [DOI] [PubMed] [Google Scholar]
- [5].Madan V, Williams J, and Lear JT, “Dermatological manifestations of Down’s syndrome,” 2006, doi: 10.1111/j.1365-2230.2006.02164.x. [DOI] [PubMed] [Google Scholar]
- [6].Löwensteyn YN et al. , “Respiratory syncytial virus-related death in children with down syndrome: The RSV GOLD Study,” Pediatr. Infect. Dis. J, vol. 39, no. 8, pp. 665–670, 2020, doi: 10.1097/INF.0000000000002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pérez-Padilla R et al. , “Pandemic (H1N1) 2009 Virus and Down syndrome patients,” Emerg. Infect. Dis, vol. 16, no. 8, pp. 1312–1314, 2010, doi: 10.3201/eid1608.091931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Clift AK, Coupland CAC, Keogh RH, Hemingway H, and Hippisley-Cox J, “COVID-19 Mortality Risk in Down Syndrome: Results From a Cohort Study Of 8 Million Adults,” Ann. Intern. Med, pp. M20–4986, October. 2020, doi: 10.7326/M20-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chan M et al. , “The burden of respiratory syncytial virus (RSV) associated acute lower respiratory infections in children with Down syndrome: A systematic review and meta-analysis,” J. Glob. Health, vol. 7, no. 2, Dec. 2017, doi: 10.7189/jogh.07.020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ram G and Chinen J, “Infections and immunodeficiency in Down syndrome,” Clin. Exp. Immunol, vol. 164, no. 1, pp. 9–16, Apr. 2011, doi: 10.1111/j.1365-2249.2011.04335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Malle L et al. , “Individuals with Down syndrome hospitalized with COVID-19 have more severe disease,” Genet. Med, 2020, doi: 10.1038/s41436-020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dieudonné Y et al. , “Immune Defect in Adults With Down Syndrome: Insights Into a Complex Issue,” Front. Immunol, vol. 11, p. 840, May 2020, doi: 10.3389/fimmu.2020.00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Verstegen RHJ and Kusters MAA, “Inborn Errors of Adaptive Immunity in Down Syndrome,” Journal of Clinical Immunology, vol. 40, no. 6. Springer, pp. 791–806, 01-August-2020, doi: 10.1007/s10875-020-00805-7. [DOI] [PubMed] [Google Scholar]; *Most recent review of defects of adaptive immune system in Down syndrome.
- [14].Schreiber G, “The molecular basis for differential type i interferon signaling,” Journal of Biological Chemistry, vol. 292, no. 18. American Society for Biochemistry and Molecular Biology Inc., pp. 7285–7294, 05-May-2017, doi: 10.1074/jbc.R116.774562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Taft J and Bogunovic D, “The Goldilocks Zone of Type I IFNs: Lessons from Human Genetics,” J. Immunol, vol. 201, no. 12, pp. 3479–3485, December. 2018, doi: 10.4049/jimmunol.1800764. [DOI] [PubMed] [Google Scholar]
- [16].Rodero MP and Crow YJ, “Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview,” J. Exp. Med, vol. 213, no. 12, pp. 2527–2538, November. 2016, doi: 10.1084/jem.20161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tan YH, Schneider EL, Tischfield J, Epstein CJ, and Ruddle FH, “Human chromosome 21 dosage: Effect on the expression of the Interferon induced antiviral state,” Science, vol. 186, no. 4158, pp. 61–63, October. 1974, doi: 10.1126/science.186.4158.61. [DOI] [PubMed] [Google Scholar]
- [18].Hwang S, Cavaliere P, Li R, Zhu LJ, Dephoure N, and Torres EM, “Consequences of aneuploidy in human fibroblasts with trisomy 21,” Proc. Natl. Acad. Sci, vol. 118, no. 6, p. e2014723118, February. 2021, doi: 10.1073/pnas.2014723118. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Evidence of global distrubances caused by the presence of an extra chromosome.
- [19].Piehler J, Thomas C, Christopher Garcia K, and Schreiber G, “Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation,” Immunol. Rev, vol. 250, no. 1, pp. 317–334, November. 2012, doi: 10.1111/imr.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moraga I, Harari D, Schreiber G, Uzé G, and Pellegrini S, “Receptor Density Is Key to the Alpha2/Beta Interferon Differential Activities,” Mol. Cell. Biol, vol. 29, no. 17, pp. 4778–4787, September. 2009, doi: 10.1128/mcb.01808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stamoulis G et al. , “Single cell transcriptome in aneuploidies reveals mechanisms of gene dosage imbalance,” Nat. Commun, vol. 10, no. 1, pp. 1–11, December. 2019, doi: 10.1038/s41467-019-12273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yap WH, Teo TS, and Tan YH, “An early event in the interferon-induced transmembrane signaling process,” Science, vol. 234, no. 4774, pp. 355–358, October. 1986, doi: 10.1126/science.2429366. [DOI] [PubMed] [Google Scholar]
- [23].Sullivan KD et al. , “Trisomy 21 consistently activates the interferon response,” Elife, 2016, doi: 10.7554/eLife.16220.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Waugh KA, Araya P, Pandey A, Sullivan KD, Hsieh EW, and Espinosa JM Correspondence, “Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome,” CellReports, vol. 29, pp. 1893–1908.e4, 2019, doi: 10.1016/j.celrep.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kong X-F et al. , “Three Copies of Four Interferon Receptor Genes Underlie a Mild Type I Interferonopathy in Down Syndrome,” J. Clin. Immunol, 2020, doi: 10.1007/s10875-020-00803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Robust characterization of IFN-I signaling at protein and RNA levels in T21 cell lines and PBMCs from individuals with Down syndrome.
- [26].Gonzales PK, Roberts CM, Fonte V, Jacobsen C, Stein GH, and Link CD, “Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21,” PLoS One, vol. 13, no. 3, March. 2018, doi: 10.1371/journal,pone.0194581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Weerd NA and Nguyen T, “The interferons and their receptors—distribution and regulation,” Immunol. Cell Biol, vol. 90, no. 5, pp. 483–491, May 2012, doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chmiest D et al. , “Spatiotemporal control of interferon-induced JAK/STAT signalling and gene transcription by the retromer complex,” Nat. Commun, vol. 7, no. 1, pp. 1–15, December. 2016, doi: 10.1038/ncomms13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Curtis ME, Yu D, and Praticò D, “Dysregulation of the Retromer Complex System in Down Syndrome,” Ann. Neurol, vol. 88, no. 1, pp. 137–147, July. 2020, doi: 10.1002/ana.25752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Filippone A and Praticò D, “Endosome Dysregulation in Down Syndrome: A Potential Contributor to Alzheimer Disease Pathology,” Ann. Neurol, p. ana.26042, February. 2021, doi: 10.1002/ana.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Araya P et al. , “Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity,” Proc. Natl. Acad. Sci. U. S. A, vol. 116, no. 48, pp. 24231–24241, November. 2019, doi: 10.1073/pnas.1908129116. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Characterization of disturbances in T cell compartment of individuals with Down syndrome, not limited to IFN-I signaling.
- [32].Powers RK et al. , “Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors,” Nat. Commun, vol. 10, no. 1, pp. 1–11, December. 2019, doi: 10.1038/s41467-019-12739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maria NI et al. , “Association of Increased Treg Cell Levels With Elevated Indoleamine 2,3-Dioxygenase Activity and an Imbalanced Kynurenine Pathway in Interferon-Positive Primary Sjögren’s Syndrome,” Arthritis Rheumatol, vol. 68, no. 7, pp. 1688–1699, July. 2016, doi: 10.1002/art.39629. [DOI] [PubMed] [Google Scholar]
- [34].Mándi Y and Vécsei L, “The kynurenine system and immunoregulation,” Journal of Neural Transmission, vol. 119, no. 2. Springer, pp. 197–209, 09-February-2012, doi: 10.1007/s00702-011-0681-y. [DOI] [PubMed] [Google Scholar]
- [35].Tuttle KD, Waugh KA, Araya P, Baxter J, Espinosa JM, and S. KD Correspondence, “Article JAK1 Inhibition Blocks Lethal Immune Hypersensitivity in a Mouse Model of Down Syndrome,” 2020, doi: 10.1016/j.celrep.2020.108407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Antonarakis SE, “Down syndrome and the complexity of genome dosage imbalance,” Nature Reviews Genetics, vol. 18, no. 3. Nature Publishing Group, pp. 147–163, 01-March-2017, doi: 10.1038/nrg.2016.154. [DOI] [PubMed] [Google Scholar]
- [37].Bogunovic D et al. , “Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency,” Science, vol. 337, no. 6102, pp. 1684–1688, September. 2012, doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Meuwissen MEC et al. , “Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome.,” J. Exp. Med, vol. 213, no. 7, pp. 1163–74, June. 2016. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Altman JB et al. , “Type I IFN is siloed in endosomes,” Proc. Natl. Acad. Sci. U. S. A, vol. 117, no. 30, pp. 17510–17512, July. 2020, doi: 10.1073/pnas.1921324117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Su C, Hou Z, Zhang C, Tian Z, and Zhang J, “Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells,” Virol. J, vol. 8, 2011, doi: 10.1186/1743-422X-8-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yao R et al. , “MicroRNA-155 Modulates Treg and Th17 Cells Differentiation and Th17 Cell Function by Targeting SOCS1,” PLoS One, vol. 7, no. 10, October. 2012, doi: 10.1371/journal.pone.0046082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang D et al. , “MiRNA-155 Regulates the Th17/Treg Ratio by Targeting SOCS1 in Severe Acute Pancreatitis,” Front. Physiol, vol. 9, no. JUN, p. 686, June. 2018, doi: 10.3389/fphys.2018.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Alexandrov PN, Percy ME, and Lukiw WJ, “Chromosome 21-Encoded microRNAs (mRNAs): Impact on Down’s Syndrome and Trisomy-21 Linked Disease,” Cell. Mol. Neurobiol, vol. 38, no. 3, pp. 769–774, April. 2018, doi: 10.1007/s10571-017-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Farroni C et al. , “Dysregulated miR-155 and miR-125b are related to impaired B-cell responses in down syndrome,” Front. Immunol, vol. 9, no. NOV, p. 2683, November. 2018, doi: 10.3389/fimmu.2018.02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gemen EFA, Verstegen RHJ, Leuvenink J, and De Vries E, “Increased circulating apoptotic lymphocytes in children with Down syndrome,” Pediatr. Blood Cancer, vol. 59, no. 7, pp. 1310–1312, December. 2012, doi: 10.1002/pbc.24246. [DOI] [PubMed] [Google Scholar]
- [46].K. & Yasui KA, Shinozaki K, Nakazawa K, Agematsu T, “Presenility of granulocytes in Down syndrome individuals.,” Am. J. Med. Genet, vol. 84, pp. 406–412,1999. [PubMed] [Google Scholar]
- [47].Lee-Kirsch MA, “The Type I Interferonopathies,” Annu Rev Med, no. 68, pp. 297–315,2017, doi: 10.1146/annurev-med-050715-104506. [DOI] [PubMed] [Google Scholar]
- [48].Wisniewski KE, French JH, Rosen JF, Kozlowski PB, Tenner M, and Wisniewski HM, “Basal Ganglia Calcification (BGC) in Down’s Syndrome (DS)—Another Manifestation of Premature Aging,” Ann. N. Y. Acad. Sci, vol. 396, no. 1, pp. 179–189, 1982, doi: 10.1111/j.1749-6632.1982.tb26852.x. [DOI] [PubMed] [Google Scholar]
- [49].Ieshima A, Kisa T, Yoshino K, Takashima S, and Takeshita K, “A morphometric CT study of Down’s syndrome showing small posterior fossa and calcification of basal ganglia,” Neuroradiology, vol. 26, no. 6, pp. 493–498, November. 1984, doi: 10.1007/BF00342687. [DOI] [PubMed] [Google Scholar]
- [50].Takashima S and Becker LE, “Basal ganglia calcification in Down’s syndrome,” 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Panda PK, Elwadhi A, and Sharawat IK, “Intracranial calcification and seizures in down syndrome,” BMJ Case Reports, vol. 14, no. 4. BMJ Publishing Group, p. e243180, 23-April-2021, doi: 10.1136/bcr-2021-243180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Down J, “Observations on an ethnic classification of idiots.,” Ment. Retard, vol. 33, pp. 54–6, 1866. [PubMed] [Google Scholar]
- [53].Stafstrom CE, Patxot OF, Gilmore KE, and Wisniewski KE, “Seizures in children with Down syndrome: etiology, characteristics and outcome,” Dev. Med. Child Neurol, vol. 33, no. 3, pp. 191–200, 1991, doi: 10.1111/j.1469-8749.1991.tb05108.x. [DOI] [PubMed] [Google Scholar]
- [54].Volpi S, Picco P, Caorsi R, Candotti F, and Gattorno M, “Type I interferonopathies in pediatric rheumatology,” Pediatr. Rheumatol, vol. 14, no. 1, p. 35, December. 2016, doi: 10.1186/s12969-016-0094-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lam M, Lai C, Almuhanna N, and Alhusayen R, “Hidradenitis suppurativa and Down syndrome: A systematic review and meta-analysis,” Pediatr. Dermatol, vol. 37, no. 6, pp. 1044–1050, November. 2020, doi: 10.1111/pde.14326. [DOI] [PubMed] [Google Scholar]
- [56].Kanazawa N, “Designation of Autoinflammatory Skin Manifestations With Specific Genetic Backgrounds,” Frontiers in Immunology, vol. 11. Frontiers Media S.A., p. 475, 18-March-2020, doi: 10.3389/fimmu.2020.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].“Overview of cutaneous lupus erythematosus - UpToDate.” [Online]. Available: https://www-uptodate-com.eresources.mssm.edu/contents/overview-of-cutaneous-lupus-erythematosus?search=sle&topicRef=4668&source=see_link. [Accessed: 01-Apr-2021].
- [58].Rachubinski AL et al. , “CASE REPORT Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata,” JAAD Case Reports, vol. 5, pp. 365–367, 2019, doi: 10.1016/j.jdcr.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Evidence of efficacy of Jak inhibitor therapy in two children with DS.
- [59].Pham AT, Rachubinski AL, Enriquez-Estrada B, Worek K, Griffith M, and Espinosa JM, “JAK inhibition for treatment of psoriatic arthritis in Down syndrome,” Rheumatology, February. 2021, doi: 10.1093/rheumatology/keab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crow MK, Olferiev M, and Kirou KA, “Type I Interferons in Autoimmune Disease,” Annual Review of Pathology: Mechanisms of Disease, vol. 14. Annual Reviews Inc., pp. 369–393, 24-January-2019, doi: 10.1146/annurev-pathol-020117-043952. [DOI] [PubMed] [Google Scholar]
- [61].Speer SD et al. , “ISG15 deficiency and increased viral resistance in humans but not mice,” Nat. Commun, vol. 7, no. May, p. 11496, 2016, doi: 10.1038/ncomms11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Channappanavar R et al. , “Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice,” Cell Host Microbe, vol. 19, no. 2, pp. 181–193, February. 2016, doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Channappanavar R et al. , “IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes,” J. Clin. Invest, vol. 129, no. 9, pp. 3625–3639, September. 2019, doi: 10.1172/JCI126363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bastard P et al. , “Auto-antibodies against type I IFNs in patients with life-threatening COVID-19 Downloaded from,” Science, 2020, doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang Y et al. , “Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: A meta-analysis,” Oncotarget, vol. 8, no. 48, pp. 84489–84496, 2017, doi: 10.18632/oncotarget.21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sullivan KD et al. , “Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation,” Sci. Rep, 2017, doi: 10.1038/s41598-017-13858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Licastro F, Chiappelli M, Ruscica M, Carnelli V, and Corsi MM, “Altered cytokine and acute phase response protein levels in the blood of children with Downs syndrome: Relationship with dementia of Alzheimer’s type,” Int. J. Immunopathol. Pharmacol, vol. 18, no. 1, pp. 165–172, January. 2005, doi: 10.1177/039463200501800117. [DOI] [PubMed] [Google Scholar]
- [68].González-Navajas JM, Lee J, David M, and Raz E, “Immunomodulatory functions of type I interferons,” Nat. Rev. Immunol, vol. 12, 2012, doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Milner JD et al. , “Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations Key Points,” Blood, vol. 21, p. 25, 2015, doi: 10.1182/blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ivashkiv LB, “IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy,” Nature Reviews Immunology, vol. 18, no. 9. Nature Publishing Group, pp. 545–558, 01-September-2018, doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gresser I et al. , “Interferon-induced disease in mice and rats,” Ann. N. Y. Acad. Sci, vol. 350, no. 1, pp. 12–20, 1980, doi: 10.1111/j.1749-6632.1980.tb20602.x. [DOI] [PubMed] [Google Scholar]
- [72].Campbell IL et al. , “Structural and functional neuropathology in transgenic mice with CNS expression of IFN-α,” in Brain Research, 1999, vol. 835, no. 1, pp. 46–61, doi: 10.1016/S0006-8993(99)01328-1. [DOI] [PubMed] [Google Scholar]
- [73].Gao D et al. , “TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons,” J. Clin. Invest, vol. 131, no. 1, January. 2021, doi: 10.1172/JCI134529. [DOI] [PMC free article] [PubMed] [Google Scholar]; *IFN-I has a priviledged role in cortical neurons and is constitutively secreted.
- [74].Maroun LE, Heffernan TN, and Hallam DM, “Partial IFN-a /b and IFN-g Receptor Knockout Trisomy 16 Mouse Fetuses Show Improved Growth and Cultured Neuron Viability,” Mary Ann Liebert, Inc, 2000. [DOI] [PubMed] [Google Scholar]
- [75].Maroun LE, “Anti-interferon immunoglobulins can improve the trisomy 16 mouse phenotype,” Teratology, vol. 51, no. 5, pp. 329–335, 1995, doi: 10.1002/tera.1420510509. [DOI] [PubMed] [Google Scholar]
- [76].Yockey LJ and Iwasaki A, “Interferons and Proinflammatory Cytokines in Pregnancy and Fetal Development,” Immunity. 2018, doi: 10.1016/j.immuni.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Buchrieser J et al. , “IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise,” Science, vol. 365, no. 6449, pp. 176–180, July. 2019, doi: 10.1126/science.aaw7733. [DOI] [PubMed] [Google Scholar]; *Evidence of pathogenic role of IFN-I signaling in pregnancy.
- [78].Savva GM, Morris JK, Mutton DE, and Alberman E, “Maternal age-specific fetal loss rates in Down syndrome pregnancies,” Prenat. Diagn, vol. 26, no. 6, pp. 499–504, June. 2006, doi: 10.1002/pd.1443. [DOI] [PubMed] [Google Scholar]
- [79].Adams AD, Guedj F, and Bianchi DW, “Placental development and function in trisomy 21 and mouse models of Down syndrome: Clues for studying mechanisms underlying atypical development,” Placenta, vol. 89, pp. 58–66, January. 2020, doi: 10.1016/j.placenta.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gadina M et al. , “Janus kinases to jakinibs: From basic insights to clinical practice,” Rheumatol. (United Kingdom), vol. 58, no. Suppl 1, pp. i4–i16, February. 2019, doi: 10.1093/rheumatology/key432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].“Tofacitinib for Immune Skin Conditions in Down Syndrome - Full Text View - ClinicalTrials.gov..” [Online]. Available: https://clinicaltrials.gov/ct2/show/NCT04246372. [Accessed: 17-May-2021].