

Abstract
Objectives
Primary non-response (PNR) to anti-tumor necrosis factor-α (TNFα) biologics is a serious concern in patients with inflammatory bowel disease (IBD). We aimed to identify the genetic variants associated with PNR.
Patients and methods
Patients were recruited from outpatient GI clinics and PNR was determined using both clinical and endoscopic findings. A case-control genome-wide association study was performed in 589 IBD patients and associations were replicated in an independent cohort of 293 patients. Effect of the associated variant on gene expression and TNFα secretion was assessed by cell-based assays. Pleiotropic effects were investigated by Phenome-wide Association Study (PheWAS).
Results
We identified rs34767465 as associated with PNR to anti-TNFα therapy (OR:2.07, 95%CI:1.46–2.94, p=2.43×10−7, [Replication OR:1.8, 95%CI:1.04–3.16, p=0.03]). rs34767465 is a multiple-tissue expression quantitative trait loci for FAM114A2. Using RNA-sequencing and protein quantification from HapMap lymphoblastoid cell lines (LCLs), we found a significant decrease in FAM114A2 mRNA and protein expression in both heterozygous and homozygous genotypes when compared to wild type LCLs. TNFα secretion was significantly higher in THP-1 cells [differentiated into macrophages] with FAM114A2 knockdown versus controls. Immunoblotting experiments showed that depletion of FAM114A2 impaired autophagy related pathway genes suggesting autophagy mediated TNFα secretion as a potential mechanism. PheWAS showed rs34767465 was associated with comorbid conditions found in IBD patients (derangement of joints [p=3.7×10−4], pigmentary iris degeneration [p=5.9×10−4], diverticulum of esophagus [p=7×10−4]).
Conclusions
We identified a variant rs34767465 associated with PNR to anti-TNFα biologics, which increases TNFα secretion through mechanism related to autophagy. rs34767465 may also explain the comorbidities associated with IBD.
Keywords: Inflammatory bowel disease, genome-wide association study, anti-TNFα, single nucleotide polymorphism, primary non-response
Introduction
Despite substantial efficacy of anti-tumor necrosis factor-alpha (anti-TNFα) biologics in the treatment of inflammatory bowel disease (IBD), 8–42% of patients do not respond [primary non-responders] to therapy[1–5]. Primary non-response (PNR) to anti-TNFα has serious implications on disease course, including reduced likelihood of response to a second anti-TNF agent, and a greater need for surgery[6]. Moreover, these agents are expensive and are associated with significant risks of infections and autoimmune complications[7]. Therefore, identifying the factors that predict efficacy is crucial to allow clinicians to effectively use these biologics.
Biomarkers like C-reactive protein (CRP) level, TNFα level and Anti-Neutrophil Cytoplasmic Antibodies (ANCA) have been the subject of several investigations for their association with anti-TNF α response and have conflicting findings[8–14]. Investigation of the genetic profiles of PNR by targeting genes related to cytokines and their receptors or immunoglobulin receptors revealed associated genetic variants within IL-13 receptor (IL13Rα2), IL-23 receptor (IL23R), TNF-receptor I (TNFRI), neonatal Fc receptor (VNTR2/VNTR3), apoptosis-related genes (Fas ligands, caspase 9), IgG Fc receptor IIIa (FcYRIIIa), and MAP kinases [10, 12, 14–16]. None of the described genetic factors were reproduced in large studies. These targeted approaches may have missed genetic polymorphisms that can identify the patients unlikely to respond to the anti-TNFα biologics that more unbiased approach such as genome-wide association study (GWAS) captures. We conducted the first pharmacogenomics GWAS in IBD and identified and replicated a genetic locus associated with PNR to anti-TNFα therapy. Cell-based assay showed the variant increases TNFα secretion through mechanism related to autophagy. Phenome-wide association study (PheWAS) revealed its association with IBD-related comorbidities.
Methods
Study participants
All participants provided written informed consent as part of the University of Chicago institutional review board approved protocols. Participants were selected based on (i) established diagnosis of IBD by a gastroenterologist (Crohn’s Disease [CD], Indeterminate Colitis [IC] and Ulcerative Colitis [UC]) by clinical, radiological, and endoscopic examination via chart review (ii) treatment initiation with anti-TNFα biologics (Remacade® (infliximab), Humira® (adalimumab) and CIMZIA® (certolizumab)) (iii) European genetic ancestry. Patients were excluded if (i) Non-European ancestry (ii) 18 years of age or younger at treatment (iii) Prior therapy with anti-TNF agents (iv) duration of anti-TNF therapy could not be determined through chart review. This study was performed in accordance with the Declaration of Helsinki.
Determination of anti-TNF response phenotype in the discovery and replication cohort
The discovery and replication cohorts were recruited from the outpatient GI clinic from February 2009 to June 2012. Demographic and health information of the patients were collected through retrospective review of the electronic heath records. Endoscopic and clinical findings after induction therapy (8 weeks for UC or 12 weeks for CD) were used to determine response to therapy. Patients were categorized as primary non-responders when (i) there was no clinical remission (as noted by the gastroenterologist as marked reduction in diarrhea and abdominal pain, or in the case of patients with fistulae, a decrease in the drainage, size, or number of fistulae for CD, and marked reduction in the amount of diarrhea, hematochezia, and abdominal pain for UC) or (ii) active inflammation via colonoscopy (lack of endoscopic healing) after anti-TNFα induction. If the dose was increased after induction, we allowed an additional 8–12 week to determine response status. Patient had to be discontinued on therapy to be deemed primary non-responders. Responders were patients with favorable clinical and endoscopic response to anti-TNFα induction followed by a continued clinical response after 8–12 weeks of induction therapy in the absence of steroid therapy. Patients that stopped therapy prior to the end of induction due to anaphylactic reactions were excluded from the study. Secondary non-response was defined as loss of response (LOR) during maintenance after successful induction. If anti-drug antibodies were detected through routine clinical care after LOR, we categorized these patient as secondary non-responders. However, antibody levels were not available on all subjects. Because of the retrospective nature of our study, secondary non-responders were included as responders.
Genotyping
DNA was collected through the biobank of the Translational Research Initiative of the Department of Medicine (TRIDOM) of the University of Chicago Hospitals. The discovery cohort of 676 self-reported white patients was genotyped with the Illumina Infinium OmniExpressExome-8 (Illumina, San Diego, CA, USA) at the RIKEN Center for Genomic Medicine (Yokohama, Japan)[17]. Single nucleotide polymorphisms (SNPs) and sample-level quality control and imputation were performed as previously described[18]. Principal components (PC) 1 and 2 were used to determine genetic ancestry of all individuals with any outliers removed (Supplementary Figure 1). A total of 589 patients and 6,489,541 variants passed quality control filters and were used for analysis (Supplementary Figure 2). The discovery cohort comprised of 104 primary non-responders and 485 responders to anti-TNF therapy. Patients who were enrolled after genotyping of the discovery cohort, served as an independent replication cohort. The replication cohort was genotyped by pyrosequencing and comprised of 32 primary non-responders and 261 responders to anti-TNF therapy.
Cell Culture, transfection and RNAi
Commercially available lymphoblastoid cell lines (LCLs) from 1000 genome CEU population (purchased from Coriell Institute) were used to evaluate the effect of the SNP on the FAM114A2 mRNA and protein expression. Samples were selected based on their genotype at rs34767465 (GM07000, GM12004, GM12005, GM06985, GM11830, GM11831, GM12760, GM12044, GM12413). LCLs were grown in RPMI-1640 medium containing 10% heat inactivated fetal bovine serum (FBS) supplemented with 10 mM HEPES.
To investigate the functional effect of FAM114A2 on TNFα secretion, human monocyte THP-1 cells (ATCC:TIB-202) were grown in RPMI-1640 medium containing 10% heat inactivated FBS supplemented with 10 mM HEPES and seeded in 6-well plates at a density of 3.0×106/well before transfection. FAM114A2 siRNAs (OriGene:SR307384) and scrambled control were transfected using Lipofectamine RNAiMAX Reagent (ThermoFisher:13778030) according to the manufacturer’s instruction. Following knockdown, THP-1 monocytes were differentiated into macrophages by incubation with 150mM phorbol 12-myristate 13-acetate (PMA, Sigma:P8139) for 24h[19]. Concentration of TNFα in the cell culture supernatant of THP-1 cells-derived macrophages was determined by ELISA (ThermoFisher Scientific:KHC3011), according to the manufacturer’s protocol.
Immunoblotting
LCLs and THP-1 cells-derived macrophages were lysed in RIPA lysis buffer (Thermo Scientific, 11965092) supplemented with protease inhibitor cocktail (Roche, 11836170001). Whole cell lysates were centrifuged, and protein concentration was determined using Pierce Coomassie Protein Assay kit (Thermo Scientific, 23200). Equal amounts of proteins were separated by standard SDS/PAGE and transferred to PVDF membrane. The following antibodies were used to quantify protein levels: FAM114A2 (PA5–57441, ThermoFisher), β-tubulin (#86298, CST), GAPDH (MAB374, Millipore), LC3 (2775, Cell Signaling), and p62 (5114, Cell signaling). The protein quantification was performed by Gels Analyze tool of Image J. Representative results obtained from at least three independent experiments were presented.
Phenome-Wide Association Study (PheWAS)
rs34767465 was tested for its association across a wide range of diseases and traits defined by the phecode system[20] in eMERGE phase3 cohort[21]. Out of 83,717 eMERGE patients, 81,920 patients had both the genotype and phecodes, and were used for this study. Case-control status for each individual was defined for each phecode. Individuals with ≥ 3 instances of a phecode were considered as a case, and control status was assigned based on the absence of a given phecode. Samples with ≥1 to <3 phecodes were removed from analysis for any given phecode. A total of 1552 phecodes that occurred in a minimum of 25 cases were tested as previously described (Supplementary Figure 2)[22].
Statistical Analyses
Differences in baseline characteristics between cases and controls were assessed by χ2 test or Fisher exact test for categorical variables and t-test for continuous parameters. To identify variants associated with PNR, a case-control GWAS was conducted in the discovery cohort and significant SNPs were tested in the replication cohort. Summary statistics from the discovery and replication cohorts were analyzed in a fixed-effect meta-analysis using METAL (version-2011)[23], which assumes equal genetic effect between the two cohorts. A two-sided p-value of 5×10−8 was considered the Bonferroni corrected significance threshold for the discovery cohort[24] and meta-analysis. In the replication cohort, a two-sided p-value<0.05 was considered significant as only one SNP was tested in replication. Association analysis was conducted by logistic regression using an additive model. Heterogeneity of the associations across the two cohorts was assessed by Cochran’s Q statistics. Genetic analysis was conducted using SNPTESTv2.5.2 for the discovery cohort and PLINK(version-1.9) for the replication cohort[25, 26]. The gene region plot for the associated SNP was generated with LocusZoom (version-0.4.8)[27]. For PheWAS analysis, logistic regression was used to test the associations between the selected variant and each phecode after adjusting for sex, decade of birth, first three PCs and eMERGE recruitment site. eMERGE contains merged genotype and phenotype data from multiple recruitment sites. Therefore, we adjusted our regression model by site to account for any confounding biases. A two-sided p-value of p=3.2×10−5 was considered the Bonferroni corrected significance threshold for PheWAS. Missing data were handled by the listwise deletion method. Statistical analyses were performed using R (version-3.5.2) software.
Results
Demographic and clinical characteristics of patient and control groups are shown in Table 1. We found 17.7% and 10.9% PNR in our Discovery and Replication cohorts respectively. Age and sex were similar between cases and controls in both the discovery and replication cohorts. Antibiotic use (p<0.0001), methotrexate (p=0.018) and azathioprine (p=0.018) were significantly associated with PNR and were included as covariates in the GWAS analysis. PC1 (p=0.01) was associated with the PNR and used as a covariate to correct for population substructure. Genomic inflation factor (based on median χ2) was 0.9859035, indicating no population stratification.
Table 1.
Demographic and clinical characteristics of the study groups
| Discovery Cohort | Replication Cohort | ||||
|---|---|---|---|---|---|
| Variables | Cases (n = 104) | Controls (n = 485) | Cases (n = 32) | Controls (n = 261) | |
| Age, years (mean ±SD) | 36.62±14.39 | 35.88±14.23 | 36.53±16.88 | 30.29±15.14 | |
| Sex (%) | Female | 50 (48.1) | 232 (47.8) | 13 (40.6) | 137 (52.5) |
| Male | 54 (51.9) | 253 (52.2) | 19 (59.4) | 124 (47.5) | |
| Smoking n (%) | 40 (38.5) | 187 (38.1) | 15 (46.9) | 83 (31.8) | |
| GI dysplasia/ Cancer n (%) | 5 (4.8) | 34 (7) | 3 (9.4) | 14 (5.4) | |
| Calcineurin inhibitor n (%) | 2 (1.9) | 6 (1.2) | - | 3 (1.1) | |
| * Antibiotics n (%) | 26 (25) | 225 (46.4) | 13 (40.6) | 115 (45.1) | |
| * Methotrexate n (%) | 9 (8.7) | 88 (18.1) | 5 (16.1) | 65 (25) | |
| * Azathioprine n (%) | 46 (44.2) | 276 (56.9) | 11 (34.4) | 128 (49.8) | |
| Aminosalicylates n (%) | 33 (32) | 140 (29.2) | 11 (35.5) | 60 (23.6) | |
| Anti-TNFα therapy | |||||
| Infliximab n (%) | 66 (64.1) | 328 (67.9) | 14 (43.8) | 154 (59) | |
| Adalimumab n (%) | 29 (28.2) | 146 (30.2) | 17 (53.1) | 98 (37.5) | |
| Certolizumab n (%) | 9 (8.6) | 11 (2.3) | 1 (3.1) | 9 (3.4) | |
| IBD type | |||||
| Crohn’s Disease n (%) | 73 (70.2) | 387 (80) | 28 (87.5) | 255 (98) | |
| Ulcerative Colitis n (%) | 28 (26.9) | 85 (17.5) | - | - | |
| Indeterminate Colitis n (%) | 3 (2.8) | 13 (2.6) | 4 (12.5) | 6 (2.3) | |
p <0.05, statistically significant in the discovery cohort
GWAS findings
rs34767465 on chromosome 5, along with 9 SNPs in high linkage disequilibrium with this lead SNP, showed suggestive association with PNR to anti-TNFα therapy (OR:2.07, 95%CI:1.46–2.94, p=2.43×10−7) (Table 2, Figure 1A). While 7 of the 9 SNPs were imputed, two of them (rs13173354 and rs9324761) were present on the genotyping platform. rs201833877 on chromosome 11 also showed suggestive association with PNR (OR:3.17, 95%CI:1.87–5.36, p=7.18×10−7), however the variant is an insertion/deletion within a long repeat sequence and could not be replicated by pyrosequencing or TaqMan assay. Association of rs34767465 was confirmed in the replication cohort (OR:1.8, 95%CI:1.04–3.16, p=0.03). Near genome-wide significance was achieved when the cohorts were combined via meta-analysis (meta-analysis p=5.2×10−8), with no significant heterogeneity between cohorts detected (Cochran Q statistic P=0.21). rs34767465 is an intergenic variant located between FAM114A2 and GALNT10 (Figure 1B), and is a cis expressed quantitative locus (cis-eQTL) for FAM114A2 in skeletal muscle (Figure 1C) and whole blood[28–30].
Table 2:
GWAS loci for non-response to anti-TNF biologics
| Study Cohort | Chr | SNP | MA | MAF (%) | OR (95% CI) | p value |
|---|---|---|---|---|---|---|
| Discovery Cohort (N=589) # | 5 | rs34767465* | T | 17.5 | 2.07 (1.46–2.94) | 2.43×10−07 |
| 5 | rs34902560* | G | 17.5 | 2.06 (1.45–2.93) | 2.57×10−07 | |
| 5 | rs13173354$ | A | 17.5 | 2.06 (1.46–2.93) | 2.97×10−07 | |
| 5 | rs34826562* | T | 17.5 | 2.05 (1.44–2.90) | 3.13×10−07 | |
| 5 | rs6580051* | A | 17.5 | 2.05 (1.44–2.90) | 3.32×10−07 | |
| 5 | rs9324761$ | G | 17.5 | 2.05 (1.44–2.90) | 3.43×10−07 | |
| 5 | rs6882924* | G | 17.5 | 2.05 (1.44–2.90) | 3.69×10−07 | |
| 5 | rs2053309* | A | 17.5 | 2.05 (1.44–2.90) | 3.75×10−07 | |
| 5 | rs11415520* | AC | 17.5 | 2.05 (1.44–2.90) | 3.75×10−07 | |
| 5 | rs66483716* | G | 17.5 | 2.05 (1.44–2.91) | 3.79 ×10−07 | |
| 11 | rs201833877* | T | 5 | 3.17 (1.87–5.36) | 7.18 ×10−07 | |
| Replication Cohort (N = 293) | 5 | rs34767465 | T | 23 | 1.8 (1.04–3.16) | 0.03 |
Adjusted to azathioprine, methotrexate, antibiotics and principal component 1
Imputed variant
Variant present in the genotyping platform
Figure 1.
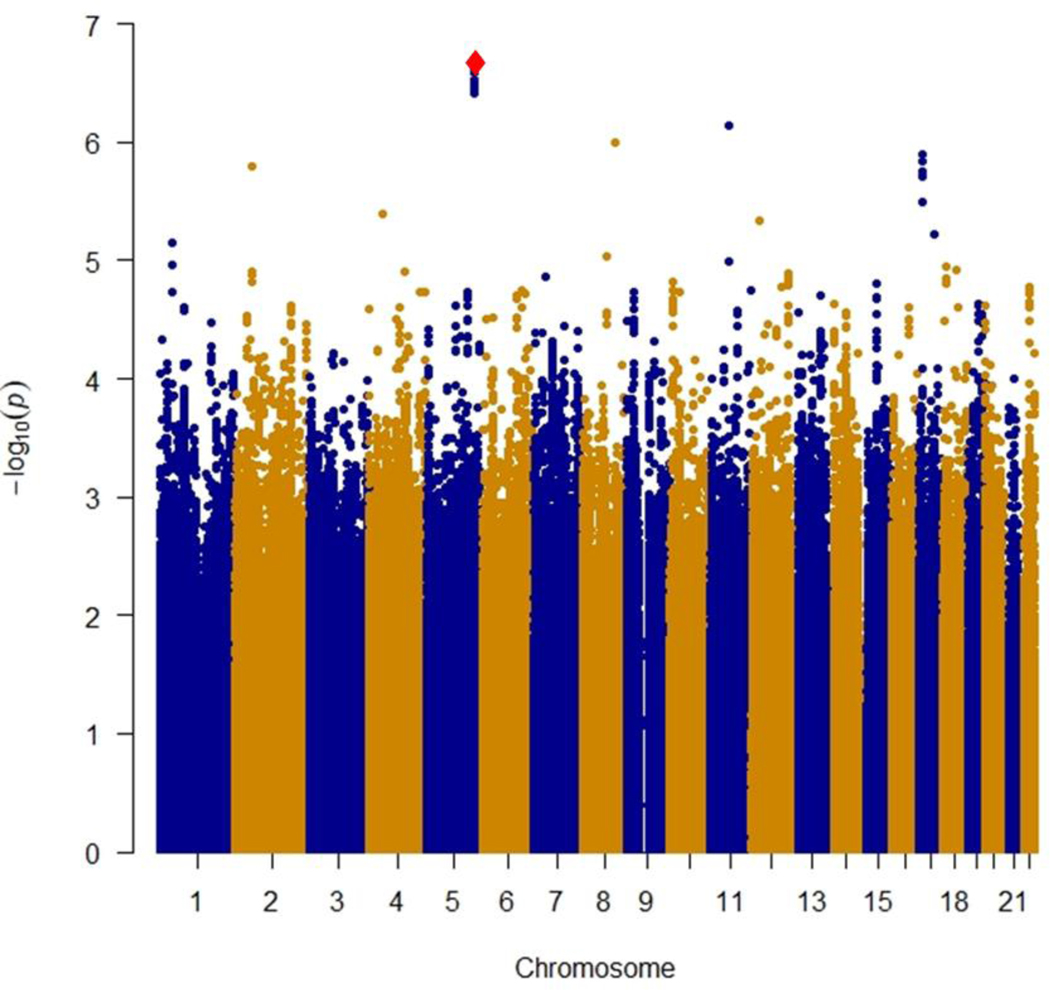


A): Manhattan plot of near-significant loci associated with primary non-response to anti-TNFα biologics in the discovery cohort
Single-nucleotide polymorphisms (SNPs) are plotted on the x-axis according to their positions on each chromosome against association with primary non-response to anti-TNFα biologics on the y-axis (−log10P value). Near-significant associations were observed in chromosome 5 [Genome-wide significant threshold: P = 5.0 × 10−8]. The diamond identifies our most significant SNP association (rs34767465).
B): Locus-specific plot of rs34767465 on chromosome 5
The x-axis represents the genomic position in chromosome 5 and the left y-axis represents the –log10P of association with primary non-response to anti TNF∝ biologics in the discovery cohort. The colors of the circles denote linkage disequilibrium (r2) between rs34767465 and nearby SNPs (based on pairwise r2 values from the 1000 Genomes Project European population). The right y-axis show the estimated recombination rate (obtained from HapMap). Genes at this locus are indicated in the lower panel of the plot. Chromosomal positions are based on hg19 genome build.
C): Violin plot of FAM114A2 expression for cis eQTL rs34767465 in Genotype-Tissue Expression (GTEx, release v7)
The allelic effect of rs34767465 on normalized FAM114A2 gene expression levels are shown by boxplots within violin plots. [Normalized effect size = −0.15, p = 0.0000027]. A and T alleles indicate the major and minor allele types, respectively with the number of subjects shown under each genotype. The plots indicate the density distribution of the samples in each genotype. The white line in the box plot (black) shows the median value of the gene expression at each genotype.
To determine if the association of rs34767465 with PNR was influenced by the IBD sub-types, GWAS restricted to only CD patients (Case:73, Controls:387) showed an association of 1.8-fold risk (OR:1.8, 95%CI:1.18–2.76, p=1.1×10−3), while 2.89-fold risk of PNR in only UC patients (Case:28, Controls:85) (OR:2.89, 95%CI:1.45–5.78, p=4.1×10−4).
Among the previously reported loci associated with PNR(Supplementary Table1), rs17200795 (OR:1.4, p=0.03) and rs7956809 (OR:0.31, p=0.03) showed nominal association in the discovery cohort.
Effect of rs34767465 on FAM114A2 gene expression
RNA sequencing data from 89 HapMap CEU LCLs (62 of wild type, 24 of heterozygous, 3 of homozygous) showed 14% decrease in the mRNA level of FAM114A2 in both rs34767465 heterozygous and homozygous LCLs, compared to wildtype (Figure 2A). However, the decrease was not statistically significant in homozygous LCLs due to small sample size. Immunoblotting showed 40% decrease in FAM114A2 protein level in heterozygotes and 68% decrease in homozygotes, compared to wildtypes, consistent with the gene expression data (Figure 2B and 2C).
Figure 2: The effect of rs34767465 on FAM114A2 gene and protein expression in LCLs.

A) Relative gene expression of FAM114A2 by rs34767465 genotypes in LCLs (number of subjects per genotype group - WT = 62, Het = 24, Hom = 3).
B) Immunoblot gel image showing decreased FAM114A2 protein expression by rs34767465 genotypes. Three different LCL lines with known genotypes were used.
C) Quantitative analysis of immunoblotting results using Image J. Protein values are normalized by β-tubulin expression and are shown relative to WT expression.
Wild type-WT, Heterozygous-Het, Homozygous-Hom, LCL-Lymphoblastoid Cell lines.
* p<0.05 ** p<0.01.
Effect of FAM114A2 knockdown on TNFα secretion
Significant increase of 24% and 41% in relative TNFα secretion was observed with siRNAs-mediated FAM114A2 knockdowns (Figure 3A). Immunoblotting showed that knockdown of FAM114A2 decreased the expression of both LC3 subtypes (89% of LC3-I, 24% of LC3-II, and 86% of the ratio of LC3-I/ LC3-II) and 100% increase in the relative expression of P62 protein expression, suggesting depletion of FAM114A2 impaired autophagy related pathways, thereby promoting TNFα secretion. (Figure 3B and 3C).
Fig. 3. Functional analysis of FAM114A2 in THP-1 cells.

A) FAM114A2 siRNAs and scramble were transfected into THP-1 cells and untreated control, following which THP-1 cells were treated with PMA (150mM) for 24 hours to transform the cells to macrophages. Cell culture supernatant was collected for TNFα ELISA analysis. All TNFα level values are shown as relative to PMA treated scramble control. All experiments were conducted in triplicate.
B) Immunoblot analysis showing FAM114A2 gene knockdown increased the autophagy related protein, P62, and decreased the LC3 protein level - Representative Immunoblotting image shown.
C) Quantitative analysis of Immunoblotting results using Image J. All protein expression values were normalized to GAPDH and are shown relative to scramble control in each group. All experiments were conducted in triplicate.
phorbol 12-myristate 13-acetate – PMA, * p<0.05 ** p<0.01.
PheWAS findings
The mean age of the PheWAS cohort was 49.2±24.9 years and 44165 (53.9%) were females. rs34767465 was strongly associated with derangement of joints (OR:1.3, p=2×10−5) and osteoarthritis (OR:1.13, p=3.6×10−5), when adjusted for decade of birth, gender and PC 1, 2 and 3 (Supplementary Figure 3A, Table 3). The other phenotypes that were close to Bonferroni-corrected significance were pigmentary iris degeneration (OR:1.86, p=7.5×10−5) and acquired diverticulum of the esophagus (OR:1.97, p=2.5×10−4). None of the phenotypes crossed Bonferroni-corrected significance level (p=3.2×10−5) after adjusting for eMERGE recruitment site along with decade of birth, gender, PC1, PC2 and PC3 (Supplementary Figure 3B).
Table 3:
PheWAS analyses for significant loci rs34767465
| Phecode | Description | Category | Case | Control | OR, p value* | OR, p value$ |
|---|---|---|---|---|---|---|
| 742.9 | Other derangement of joint | musculoskeletal | 833 | 78075 | 1.3, 2 × 10−5 | 1.23, 3.7 × 10−4 |
| 740.1 | Osteoarthritis; localized | musculoskeletal | 4205 | 71883 | 1.13, 3.6 × 10−5 | 1.01, 0.66 |
| 379.51 | Pigmentary iris degeneration | sense organs | 99 | 81738 | 1.86, 7.5 × 10−5 | 1.72, 5.9 × 10−4 |
| 530.6 | Diverticulum of esophagus, acquired | digestive | 70 | 81673 | 1.97, 2.5 × 10−4 | 1.9, 7.0 × 10−4 |
Adjusted to decade of birth, gender, PC1, PC2, PC3
Adjusted to decade of birth, gender, PC1, PC2, PC3, eMERGE recruitment site
Discussion
Most studies have investigated the genetic profiles of PNR by targeting single or a few variants in the immune-related genes. Few studies also used Illumina immune-chip to query most of the genetic loci in the immune system for their association with PNR[2, 31]. However, such studies exclusively targeted the variants of the immune system and might have missed other loci that can identify the primary non-responders that more unbiased approach such as GWAS captures. We therefore used a genome-wide approach by genotyping with Illumina Infinium OmniExpressExome-8 that covers the entire human genome, and our genotype data was imputed to further include SNPs that were not represented on the genotyping chip. Our study is the first pharmacogenomic GWAS to identify a novel variant rs34767465 associated with 2-fold increased risk of PNR to anti-TNFα therapy in patients with IBD.
rs34767465 is reported as a cis-eQTL for FAM114A2 in skeletal muscle and whole blood in GTEx and NESDA NTR conditional eQTL catalog. We confirmed the effect of the rs34767465T allele on reduced expression of FAM114A2, at both the mRNA and protein level. A phosphoproteomic study showed that Leucine-rich repeat kinase 2 (LRRK2) kinase inhibition led to ≥50% alteration in the phosphorylation of FAM114A2[32]. LRRK2 is a major susceptibility gene for CD and deficiency of the gene in mice confers enhanced susceptibility to experimental colitis[33]. We did not observe significant difference in the LRRK2 mRNA or protein levels by knockdown of FAM114A2 and vice versa, in LCLs (data not shown). Effect of LRRK2-mediated phosphorylation of FAM114A2 was not explored due to lack of available FAM114A2 phosphoprotein antibody. Our study showed that FAM114A2 knockdown in THP-1 cells-derived macrophages led to an impaired autophagy pathway, thereby increasing TNFα secretion. Dysfunctional autophagy plays an important role in IBD pathogenesis by altering processes like defense against infections, pro-inflammatory cytokine production by macrophages, antigen presentation by dendritic cells and the endoplasmic reticulum stress response in enterocytes[34]. We speculate that FAM114A2 regulates TNFα levels by inducing autophagy and patients with rs34767465T allele have reduced FAM114A2 expression leading to impaired autophagy and increased TNFα secretion. These patients therefore may not respond to the standard dose of anti-TNFα and tailored dosing might be helpful for an optimal response. Suboptimal drug concentrations are associated with LOR, leading to treatment failure and drug discontinuation. Kennedy et.al. reported that the only factor independently associated with PNR was low drug concentration and demonstrated that adequate drug concentrations at week 14 were associated with clinical remission, decreased risk of developing anti-drug antibodies and better long-term outcomes[35]. Therefore, ensuring adequate drug concentrations is of utmost importance during both the initiation phase to prevent PNR as well as the maintenance phase to avoid secondary LOR. Given that drug levels are not routinely taken in IBD patients we are not able to assess if non-response could be due to lower drug concentration.
We also observed rs201833877 associated with PNR in the discovery cohort. Based on GTEx data, this variant is a multi-tissue splice-QTL for MS4A7, a gene that is expressed in microglia and brain border macrophages and is associated with immune function[36, 37]. However, the variant is an insertion/deletion within a long repeat sequence and could not be reliably genotyped in the replication cohort.
Frequency of PNR may differ by IBD disease subtypes. We observed a 15%PNR in CD patients versus 32%PNR in UC patients in our discovery cohort. Previous studies on anti-TNFα non-response report 10–15% PNR in CD patients and 30–32% PNR in UC patients[2–5]. Therefore, even with a small sample size, the observed frequency of PNR in our GWAS cohort by disease subtypes are in line with previously reported frequency in CD and UC patients. Since majority of patients in the discovery cohort (78%) and all of the replication cohort patients had CD, we observed an overall frequency of 17.7% PNR in the discovery cohort and 10.9% in the replication cohort.
Previously reported PNR-related variants, rs17200795 and rs7956809 showed nominal association with PNR in our study. While rs17200795 showed increased odds of PNR rs7956809 was associated with decreased odds of PNR, i.e. opposite direction of effect to that reported by Barber et. al,[2] Therefore, further studies are warranted to explore the association of these variants with PNR. No other previously reported variants were associated with PNR in the discovery cohort. The lack of association could be due to the differences in the definition of PNR that often varies between studies. While some studies defined response based on the CRP levels or calprotectin levels, others looked at the endoscopic response or clinical response such as UC Disease Activity Index and CD Activity Index. However, these measures are not always assessed in routine clinical care of IBD and therefore were not available for most of our cohort. Additionally, we observed no association of previously identified IBD susceptibility loci with PNR in our cohort. This may stem from differences in study design in which all subjects in our study had IBD, while in previous susceptibility studies, control were subjects with no IBD.
PheWAS analysis of rs34767465 identified associations with comorbidities like derangement of joints, osteoarthritis, pigmentary iris degeneration and diverticulum of the esophagus. However, these phenotypes did not cross the Bonferroni corrected significance threshold after including the eMERGE recruitment site as a covariate, in addition to other covariates. The prevalence of IBD and PNR might be enriched relative to the general population at some eMERGE sites, and therefore eMERGE recruitment site was added to control for potential confounding.
rs34767465 associated conditions are known to co-occur in patients with IBD. CD and diverticulitis share clinical and radiologic features and studies suggest that even the characteristic pathology of CD can be a secondary reaction to diverticulitis[38]. Sultan et al., observed an increased frequency of sigmoid and rectal inflammation, extraintestinal manifestations (EIMs), and an older age of IBD onset in cases with diverticulosis. The study speculated that diverticula may lead to changes in the colonic microflora that can in turn promote IBD[39]. A variety of chronic comorbidities have been associated with IBD, as the disease might share pathogenic pathways with other conditions. The most common EIMs in IBD are musculoskeletal disorders, often associated with colonic involvement, and present as either articular (arthritis) or periarticular inflammation including enthesitis, myositis, or soft tissue rheumatism (fibromyalgia)[40]. A study showed that peripheral arthritis in IBD patients is influenced by major histocompatibility complex haplotypes HLA-B27, HLA-B35, HLA-DR, and HLA-B44, and therefore could be genetically driven[41]. Ophthalmologic problems can occur as an EIM of the disease or may be drug-related[42]. A study by Santeford et al, demonstrated that autophagy is essential for maintaining ocular immune privilege, and deletion of multiple autophagy genes in macrophages leads to an inflammation-mediated eye disease[43]. Hence rs34767465 not only influences IBD pathogenesis leading to non-response to anti-TNFα therapy but can also explain the associated comorbidities that are often observed in IBD patients. rs34767465 may guide physicians’ drug choice and therapeutic drug monitoring to prevent PNR to anti-TNFα therapy and IBD associated comorbidities.
This study has several limitations. First, the small sample size prevented genome-wide significance. However, the association replicated in an independent cohort and achieved near genome-wide significance threshold on meta-analysis of the combined discovery and replication cohorts. Second, although rs201833877 was also associated with PNR, it is an insertion/deletion of a long repeat sequence and could not be replicated. Third, we could not establish the link between LRRK2 and FAM114A2 due to lack of availability of a phosphoprotein antibody. Fourth, we did not cross the Bonferroni-corrected significance threshold for interesting phenotypes in the PheWAS analyses, which could be due to low number of cases, seen in the diverticulum of esophagus phenotype. Fifth, because of the retrospective nature of our study, we were unable to ascertain disease activity scores before therapy for all subjects, hence this measure could not be used to insure matching between our cases and control. Lastly, there was a lack of information on plasma biomarkers like anti-TNFα concentration, CRP levels or anti-drug antibody level for our patient cohorts. Although studies suggest the utility of these parameters in determining PNR[44, 45], these are not part of routine clinical care in IBD therapy and therefore were not available for most of the patients. Hence, we are not able to account for these variables in our analysis.
Conclusion
This is the first GWAS to identify a novel variant associated with PNR to anti-TNFα biologic therapy and may explain the comorbidities associated with IBD. The findings may help guide physicians’ drug choice and therapeutic drug monitoring to prevent PNR to anti-TNFα therapy and IBD-associated comorbidities, though outcomes studies are needed to determine the benefit of these findings to patients. Furthermore, our study did not address genomic association to secondary LOR and thus further discovery effort in these patients are needed.
Supplementary Material
Supplementary Figure 1: Principal component analysis of the Discovery Cohort and three HapMap populations
Principal components analysis was performed using the LD-pruned genome-wide SNP genotypes of discovery Cohort and the three HapMap populations, CEU (Utah Residents with Northern and Western European Ancestry, shown in blue), YRI (Yoruba in Ibadan, Nigeria, shown in red) and ASN (Asian, shown in green) to infer continuous axes of genetic variation. The analysis accounts for population-specific variations in allele distribution of the SNPs under investigation. The first two principal components of genetic ancestry (PC1 and PC2) shows that the HapMap populations formed three distinct clusters and the discovery cohort of European ancestry clustered with the CEU population as expected. Sample size: Discovery cohort 589, CEU= 112, YRI= 113, ASN= 170.
The figure outlines the genome-wide association study that identified and replicated a genetic locus rs34767465 associated with primary non-response to anti-TNFα therapy. rs34767465 was then tested for any association with a wide range of clinical conditions via a phenome-wide association study to investigate potential clinical relevance. rs34767465 was also studied for its effect on gene expression and TNFα secretion by cell-based assays.
Plots represent 1,552 phenotypes tested for association with rs34767465. Phenotypes grouped along the x axis by categorization within the PheWAS code hierarchy. The Y axis reflects the P value for each phenotype. The blue horizontal line represents nominal p-value threshold of 0.05, the red horizontal line represents Bonferroni corrected p-value threshold of 3.2 × 10−5, respectively.
A) PheWAS analysis adjusted to decade of birth, gender, PC1, PC2, PC3
B) PheWAS analysis adjusted to decade of birth, gender, PC1, PC2, PC3 and eMERGE recruitment site
Acknowledgement
The authors thank PGRN-CGM International Collaborative Studies for providing genotyping service.
Funding: MAP received pilot funding from the Digestive Disease Research Core Centers (NIDDK P30DK42086). JH received U01 and RO1 (HG011172 and AI130830). LP received funding from Michael J. Fox Foundation for Parkinson’s Research and NIH R01 NS097901.
Footnotes
Conflict of Interest: None declared
References:
- 1.Papamichael K, Gils A, Rutgeerts P, Levesque BG, Vermeire S, Sandborn WJ, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182–97. [DOI] [PubMed] [Google Scholar]
- 2.Barber GE, Yajnik V, Khalili H, Giallourakis C, Garber J, Xavier R, et al. Genetic Markers Predict Primary Non-Response and Durable Response To Anti-TNF Biologic Therapies in Crohn’s Disease. Am J Gastroenterol. 2016;111(12):1816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iijima H, Kobayashi T, Nagasaka M, Shinzaki S, Kitamura K, Suzuki Y, et al. Management of Primary Nonresponders and Partial Responders to Tumor Necrosis Factor-alpha Inhibitor Induction Therapy among Patients with Crohn’s Disease. Inflamm Intest Dis. 2020;5(2):78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SC, Jeen YT. Current and emerging biologics for ulcerative colitis. Gut Liver. 2015;9(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su C, Salzberg BA, Lewis JD, Deren JJ, Kornbluth A, Katzka DA, et al. Efficacy of anti-tumor necrosis factor therapy in patients with ulcerative colitis. Am J Gastroenterol. 2002;97(10):2577–84. [DOI] [PubMed] [Google Scholar]
- 6.Papamichael K, Rivals-Lerebours O, Billiet T, Vande Casteele N, Gils A, Ferrante M, et al. Long-Term Outcome of Patients with Ulcerative Colitis and Primary Non-response to Infliximab. J Crohns Colitis. 2016;10(9):1015–23. [DOI] [PubMed] [Google Scholar]
- 7.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. Jama. 2006;295(19):2275–85. [DOI] [PubMed] [Google Scholar]
- 8.Arnott ID, McNeill G, Satsangi J. An analysis of factors influencing short-term and sustained response to infliximab treatment for Crohn’s disease. Aliment Pharmacol Ther. 2003;17(12):1451–7. [DOI] [PubMed] [Google Scholar]
- 9.Bank S. A cohort of anti-TNF treated Danish patients with inflammatory bowel disease, used for identifying genetic markers associated with treatment response. Dan Med J. 2015;62(5). [PubMed] [Google Scholar]
- 10.Hlavaty T, Pierik M, Henckaerts L, Ferrante M, Joossens S, van Schuerbeek N, et al. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol Ther. 2005;22(7):613–26. [DOI] [PubMed] [Google Scholar]
- 11.Lacruz-Guzman D, Torres-Moreno D, Pedrero F, Romero-Cara P, Garcia-Tercero I, Trujillo-Santos J, et al. Influence of polymorphisms and TNF and IL1beta serum concentration on the infliximab response in Crohn’s disease and ulcerative colitis. Eur J Clin Pharmacol. 2013;69(3):431–8. [DOI] [PubMed] [Google Scholar]
- 12.Louis E, El Ghoul Z, Vermeire S, Dall’Ozzo S, Rutgeerts P, Paintaud G, et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment Pharmacol Ther. 2004;19(5):511–9. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Borra J, Lopez-Larrea C, Gonzalez S, Fuentes D, Dieguez A, Deschamps EM, et al. High serum tumor necrosis factor-alpha levels are associated with lack of response to infliximab in fistulizing Crohn’s disease. Am J Gastroenterol. 2002;97(9):2350–6. [DOI] [PubMed] [Google Scholar]
- 14.Pierik M, Vermeire S, Steen KV, Joossens S, Claessens G, Vlietinck R, et al. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20(3):303–10. [DOI] [PubMed] [Google Scholar]
- 15.Billiet T, Dreesen E, Cleynen I, Wollants WJ, Ferrante M, Van Assche G, et al. A Genetic Variation in the Neonatal Fc-Receptor Affects Anti-TNF Drug Concentrations in Inflammatory Bowel Disease. Am J Gastroenterol. 2016;111(10):1438–45. [DOI] [PubMed] [Google Scholar]
- 16.Jurgens M, Laubender RP, Hartl F, Weidinger M, Seiderer J, Wagner J, et al. Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am J Gastroenterol. 2010;105(8):1811–9. [DOI] [PubMed] [Google Scholar]
- 17.Perera MA, Cavallari LH, Limdi NA, Gamazon ER, Konkashbaev A, Daneshjou R, et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet. 2013;382(9894):790–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De T, Alarcon C, Hernandez W, Liko I, Cavallari LH, Duarte JD, et al. Association of Genetic Variants With Warfarin-Associated Bleeding Among Patients of African Descent. JAMA. 2018;320(16):1670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31(12):1102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanaway IB, Hall TO, Rosenthal EA, Palmer M, Naranbhai V, Knevel R, et al. The eMERGE genotype set of 83,717 subjects imputed to ~40 million variants genome wide and association with the herpes zoster medical record phenotype. Genet Epidemiol. 2019;43(1):63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26(9):1205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131(5):747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–13. [DOI] [PubMed] [Google Scholar]
- 27.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansen R, Hottenga JJ, Nivard MG, Abdellaoui A, Laport B, de Geus EJ, et al. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum Mol Genet. 2017;26(8):1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penninx BW, Beekman AT, Smit JH, Zitman FG, Nolen WA, Spinhoven P, et al. The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res. 2008;17(3):121–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boomsma DI, de Geus EJ, Vink JM, Stubbe JH, Distel MA, Hottenga JJ, et al. Netherlands Twin Register: from twins to twin families. Twin Res Hum Genet. 2006;9(6):849–57. [DOI] [PubMed] [Google Scholar]
- 31.Burke KE, Khalili H, Garber JJ, Haritunians T, McGovern DPB, Xavier RJ, et al. Genetic Markers Predict Primary Nonresponse and Durable Response to Anti-Tumor Necrosis Factor Therapy in Ulcerative Colitis. Inflamm Bowel Dis. 2018;24(8):1840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luerman GC, Nguyen C, Samaroo H, Loos P, Xi H, Hurtado-Lorenzo A, et al. Phosphoproteomic evaluation of pharmacological inhibition of leucine-rich repeat kinase 2 reveals significant off-target effects of LRRK-2-IN-1. J Neurochem. 2014;128(4):561–76. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z, Lenardo MJ. The role of LRRK2 in inflammatory bowel disease. Cell Res. 2012;22(7):1092–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iida T, Onodera K, Nakase H. Role of autophagy in the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2017;23(11):1944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kennedy NA, Heap GA, Green HD, Hamilton B, Bewshea C, Walker GJ, et al. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: a prospective, multicentre, cohort study. Lancet Gastroenterol Hepatol. 2019;4(5):341–53. [DOI] [PubMed] [Google Scholar]
- 36.DePaula-Silva AB, Gorbea C, Doty DJ, Libbey JE, Sanchez JMS, Hanak TJ, et al. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J Neuroinflammation. 2019;16(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Salum T, Villablanca A, Matthaus F, Tittarelli A, Baeza M, Pereda C, et al. Molecular signatures associated with tumor-specific immune response in melanoma patients treated with dendritic cell-based immunotherapy. Oncotarget. 2018;9(24):17014–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peppercorn MA. The overlap of inflammatory bowel disease and diverticular disease. J Clin Gastroenterol. 2004;38(5 Suppl 1):S8–10. [DOI] [PubMed] [Google Scholar]
- 39.Sultan K, Fields S, Panagopoulos G, Korelitz BI. The nature of inflammatory bowel disease in patients with coexistent colonic diverticulosis. J Clin Gastroenterol. 2006;40(4):317–21. [DOI] [PubMed] [Google Scholar]
- 40.Bourikas LA, Papadakis KA. Musculoskeletal manifestations of inflammatory bowel disease. Inflamm Bowel Dis. 2009;15(12):1915–24. [DOI] [PubMed] [Google Scholar]
- 41.Nunez C, Alecsandru DM, Mendoza JL, Urcelay E, Diaz-Rubio M, de la Concha EG, et al. Genetic markers linked to rheumatoid arthritis are also strongly associated with articular manifestations in ulcerative colitis patients. Hum Immunol. 2006;67(4–5):324–30. [DOI] [PubMed] [Google Scholar]
- 42.Troncoso LL, Biancardi AL, de Moraes HV Jr., , Zaltman C Ophthalmic manifestations in patients with inflammatory bowel disease: A review. World J Gastroenterol. 2017;23(32):5836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santeford A, Wiley LA, Park S, Bamba S, Nakamura R, Gdoura A, et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy. 2016;12(10):1876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hyams JS, Davis Thomas S, Gotman N, Haberman Y, Karns R, Schirmer M, et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): a multicentre inception cohort study. Lancet. 2019;393(10182):1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bar-Yoseph H, Levhar N, Selinger L, Manor U, Yavzori M, Picard O, et al. Early drug and anti-infliximab antibody levels for prediction of primary nonresponse to infliximab therapy. Aliment Pharmacol Ther. 2018;47(2):212–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Principal component analysis of the Discovery Cohort and three HapMap populations
Principal components analysis was performed using the LD-pruned genome-wide SNP genotypes of discovery Cohort and the three HapMap populations, CEU (Utah Residents with Northern and Western European Ancestry, shown in blue), YRI (Yoruba in Ibadan, Nigeria, shown in red) and ASN (Asian, shown in green) to infer continuous axes of genetic variation. The analysis accounts for population-specific variations in allele distribution of the SNPs under investigation. The first two principal components of genetic ancestry (PC1 and PC2) shows that the HapMap populations formed three distinct clusters and the discovery cohort of European ancestry clustered with the CEU population as expected. Sample size: Discovery cohort 589, CEU= 112, YRI= 113, ASN= 170.
The figure outlines the genome-wide association study that identified and replicated a genetic locus rs34767465 associated with primary non-response to anti-TNFα therapy. rs34767465 was then tested for any association with a wide range of clinical conditions via a phenome-wide association study to investigate potential clinical relevance. rs34767465 was also studied for its effect on gene expression and TNFα secretion by cell-based assays.
Plots represent 1,552 phenotypes tested for association with rs34767465. Phenotypes grouped along the x axis by categorization within the PheWAS code hierarchy. The Y axis reflects the P value for each phenotype. The blue horizontal line represents nominal p-value threshold of 0.05, the red horizontal line represents Bonferroni corrected p-value threshold of 3.2 × 10−5, respectively.
A) PheWAS analysis adjusted to decade of birth, gender, PC1, PC2, PC3
B) PheWAS analysis adjusted to decade of birth, gender, PC1, PC2, PC3 and eMERGE recruitment site