

Abstract
Cutaneous signs and symptoms may facilitate the diagnosis or can help in identifying complications or side effects of overtreatment of inherited metabolic diseases. The principal manifestations can be grouped into vascular lesions, ichthyosis, papular and nodular skin lesions, abnormal pigmentation, photosensitivity, skin laxity, hair shaft involvement, and nail abnormalities. We have summarized associations of these cutaneous signs and symptoms in 252 inherited metabolic diseases. This represents the sixth of a series of articles attempting to create and maintain a comprehensive list of clinical and metabolic differential diagnoses according to system involvement.
Keywords: Skin disorders, abnormal pigmentation, ichthyosis, photosensitivity, hair shaft involvement, cutis laxa, vascular lesions, skin eruptions
1. Introduction
This is the sixth in a series of articles that intends to provide a comprehensive list of inherited metabolic diseases associated with specific signs and symptoms. The first five issues of the footprints were dedicated to inherited metabolic diseases (IMDs) associated with movement disorders [1], metabolic liver disorders [2], those with psychiatric presentations [3], metabolic cardiovascular disease [4] and those with cerebral palsy phenotypes [5]. The list follows the classification as included in the knowledge base of inborn errors of metabolism (IEMbase) [6], the proposed nosology of inborn errors of metabolism [7], and the recently-published international classification of inherited metabolic diseases (ICIMD) [8]. The sixth issue of our series will be dedicated to metabolic dermatological diseases. Just as genodermatoses represent hereditary disorders of genetic origin with skin manifestations, we can speak of “metabodermatoses” as inherited metabolic diseases with skin manifestations. The main manifestations are grouped into vascular lesions, ichthyosis, papular and nodular skin lesions, abnormal pigmentation, photosensitivity, skin laxity, hair disorders, and nail abnormalities [9].
1.1. Vascular Lesions
These affect vascular components within the dermis.
1.1.1. Angiokeratoma
These flat or raised vascular skin lesions develop classically as clusters, punctate, and reddish or blue-black angiectases. They do not blanch with pressure and the largest lesions may appear hyperkeratotic. Angiokeratomata may occur in GM1 gangliosidosis (GLB1), fucosidosis (FUCA1), aspartylglucosaminuria (AGA), and galactosialidosis (CTSA), but the classical symptoms of these disorders usually prompt the diagnosis well before the appearance of angiokeratoma. Fabry disease (GLA), β-mannosidosis (MANBA), and Schindler disease type II (NAGA) may also present with angiokeratomata.
1.1.2. Acrocyanosis, Angiomas, and Telangiectasia
Ten percent of patients with primary mitochondrial disorders present with skin or hair abnormalities, and others involve secondary mitochondrial pathology [10]. Orthostatic acrocyanosis is a prototypical sign of ethylmalonic encephalopathy, but may be observed in other multisystem mitochondrial disorders with onset in early infancy [11]. Acrocyanosis appears as a bilateral mottled discoloration of the entire foot or hand due to vasospasm of small arterioles and venules with secondary dilatation of capillaries. The skin becomes bright red, without trophic changes or pain. Acrocyanosis is a striking recognizable feature of ethylmalonic encephalopathy (mitochondrial sulfur dioxygenase deficiency, ETHE1). Acrocyanosis and livedo reticularis may be observed in hyperoxaluria type I (AGXT).
Capillary malformations appearing as light pink to deep-red angiomas or as telangiectasia occur in inherited metabolic diseases, including neuraminidase deficiency (NEU1), Smith-Lemli-Opitz syndrome (DHCR7), transaldolase deficiency (TALDO1), and prolidase deficiency (PEPD). Visible veins are frequently observed in pyrroline-5-carboxylate synthetase (ALDH18A1) and pyrroline-5-carboxylate reductase 1 (PYCR1) deficiencies (see ‘Skin laxity’, below). More rarely, vascular signs have been observed in patients with a number of congenital disorders of glycosylation (CDG), along with neurologic, facial, and other multisystem abnormalities. These include DPM1-CDG, MGAT2-CDG, and SLC35A1-CDG [12].
1.1.3. Ulcers secondary to thromboembolic disease
Ulcers secondary to thromboembolic disease may also develop in classical homocystinuria (cystathionine β-synthase deficiency; CBS) during adolescence and adulthood. Inflammatory skin lesions of various degrees are clinical features of hyperzincemia and hypercalprotectinemia.
1.2. Cutaneous lesions
For the purpose of this review, we include here lesions involving the epidermis (excluding melanocytes) or dermis (excluding vessels and connective tissue).
1.2.1. Ichthyosis and Erythroderma
Lipids in the stratum corneum have the important function of preventing water evaporation through the epidermis. These lipids in the extracellular space of the stratum corneum are composed of 50% acylceramides, 25% cholesterol, and 15% free fatty acids [13]. Specifically, corneocytes in the stratum corneum are surrounded by a monolayer of omega-hydroxylated-acylceramides that form the corneocyte lipid envelope. These acylceramides are composed of a sphingoid base attached to an omega-hydroxylated ultra-long chain fatty acid (usually 30-34 carbons long); the omega-hydroxyl group of this ultra-long fatty acid is further esterified by linoleic acid, culminating in an acylceramide about 70 carbons in length (Fig. 1) [14]. The vast majority of steps along the acylceramide biosynthetic pathway have been associated with ichthyosis (Fig. 1) [15]. Pertinent references describing epidermal acylceramide disruption due to involvement of different steps along this pathway are provided in Supplemental Table 1.
Figure 1.
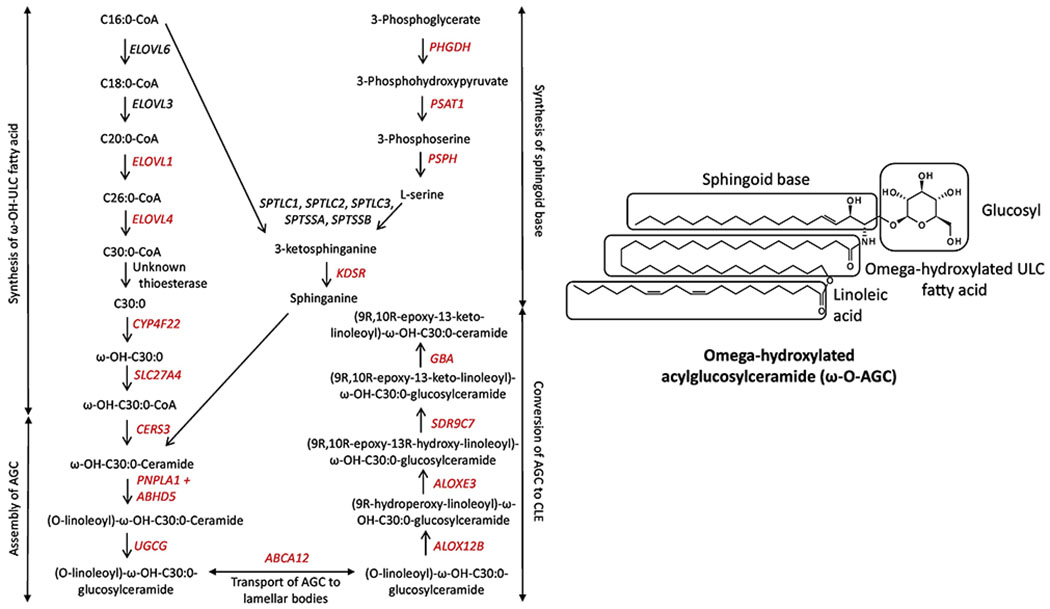
Acylceramide biosynthetic pathway. Genes in red have been associated with ichthyosis/erythrokeratoderma in humans. AGC = acylglucosylceramide; CLE = corneocyte lipid envelope; ULC = ultra-long-chain. For the chemical structures of all molecules in this figure, please refer to Xiao et al. [15].
Epidermal cholesterol also plays an important role in the lipid barrier. SULT2B1 catalyzes the transfer of the 5’-sulfate from the donor 3’-phosphoadenosine 5’-phosphosulfate (PAPS) to the hydroxyl group (OH) of cholesterol; consequently, cholesterol is converted to cholesterol sulfate by sulfonation. Deficiency of SULT2B1 leads to an autosomal recessive form ichthyosis [16]. This cholesterol sulfate is later desulfated in the outer epidermis by STS (steroid sulfatase). Deficiency of this sulfatase leads to X-linked ichthyosis, characterized by accumulation of cholesterol sulfate in the stratum corneum (where it makes up to 10-12% of lipids in affected patients, compared to 1% in normal controls) [17]. Patients may have no other symptoms than dark, scaly skin. Mild corneal opacities, without vision loss, are found in about 25%.
The skin may be the key to the diagnosis of the multisystemic Refsum disease (PHYH) and Sjogren–Larsson syndrome (ALDH3A2), which manifest with cataracts, ichthyosis and retinal involvement. Ichthyotic lesions are also prominent in infants with CHILD syndrome (NSDHL). MEDNIK syndrome or Martinelli disease (AP1S1), is a severe neurocutaneous adaptinopathy with multisystemic involvement [18]. MEDNIK-like syndrome (Keratitis-Ichthyosis-Deafness Syndrome, Autosomal Recessive or KIDAR syndrome) is a recently described disorder due to biallelic loss-of-function variants in AP1B1 [19]. MEDNIK-like syndrome largely phenocopies MEDNIK syndrome, as both disorders are characterized by a neurocutaneous phenotype with intellectual disability, developmental delay, enteropathy, deafness, hepatopathy, ichthyosis, erythroderma, palmoplantar keratoderma, and sparse hair. However, neuropathy, a clinical symptom characteristic of MEDNIK syndrome, has not been described so far in MEDNIK-like disease, probably due to the young age of the patients reported. In addition, the skin phenotype shows some differences, as MEDNIK syndrome shows a picture of erythrokeratodermia variabilis, with fixed and migratory plaques of affected skin upon a normal background. This phenotype stands in contrast to that of patients with MEDNIK-like syndrome, who have more generalized erythroderma. Moreover, the clinical features of the AP1B1 defect seem to vary among patients according to the specific variant, as some patients show a milder phenotype characterized by ichthyosis, failure to thrive, thrombocytopenia, photophobia, and progressive hearing loss without intellectual impairment [20]. Another syndromic ichthyosis, CEDNIK syndrome, resembles MEDNIK syndrome clinically [21].
Ichthyosis is also seen in arthrogryposis-renal dysfunction-cholestasis syndrome, a rare, fatal autosomal recessive disorder caused by variants in genes involved in intracellular trafficking (VPS33B or VIPAR), as these are essential in lamellar body biogenesis [22]. Ichthyosis has been described within a growing group of congenital disorders of glycosylation (CDG), including MPDU1-CDG, dolichol kinase deficiency (DOLK-CDG), SRD5A3-CDG and PIGL-CDG [23]. MPDU1-CDG is a defect in the N-glycan assembly in the endoplasmic reticulum (ER) characterized by ichthyosis and/or erythroderma, psychomotor retardation, seizures, hypotonia, gastrointestinal problems, visual impairment, dwarfism and transient growth hormone deficiency. DOLK-CDG is a defect in dolichol kinase, the last step of the dolichol phosphate biosynthesis. It shows a clinical spectrum with non-syndromic dilated cardiomyopathy and in some patients a severe phenotype with ichthyosis, epilepsy, microcephaly, visual impairment, hypoglycemia and death within the first 6 months. SRD5A3-CDG is a defect in polyprenol reductase, involved in the biosynthesis of dolichol. This disorder causes a cerebello-oculo-cutaneous syndrome, its skin component consists of ichthyosis, erythroderma and/or dry skin. PIGL-CDG or CHIME syndrome is characterized by colobomas, congenital heart defects, early-onset migratory ichthyosiform dermatosis, intellectual disability and ear anomalies, besides other clinical manifestations. This is a defect in an ER-localized enzyme that catalyzes the second step of GPI-anchor biosynthesis, the de-N-acetylation of N-acetylglucosaminylphosphatidylinositol that occurs on the cytoplasmic side of the ER [23].
1.2.2. Papular and Nodular Skin Lesions
Papules are small (<1 cm) elevated lesions of variable shape. Nodules are circumscribed, palpable, solid, round or ellipsoidal lesions, and are located deeper in the dermis or subcutaneous fat. The presentation of ceramidase deficiency is striking from early infancy, with subcutaneous nodules around joints (notably interphalangeal and metacarpal regions, ankle, wrist, knee, and elbow) and pressure points, with painful joint swelling. Hunter disease (IDS), uniquely among the mucopolysaccharidoses, has localized nodular accumulations of glycosaminoglycans in scapular skin.
Multiple and symmetrical lipomas are the most common skin disorders with mitochondrial encephalomyopathies [24], usually in adults bearing mitochondrial DNA variants.
Diffuse, large, flat, and sometimes yellowish tuberous xanthomata are seen along with tendinous xanthomata, xanthelasma, and corneal arcus in familial hypercholesterolemia. A similar picture can occur in sitosterolemia, but interdigital webspace xanthomas are typically considered pathognomonic of familial hypercholesterolemia. Distinctive xanthomata occur in severe hypertriglyceridemia in lipoprotein lipase deficiency or other chylomicronemias, with eruptive yellow papules on a slightly erythematous base, typically on the buttocks, shoulders, and extensor surfaces of the extremities. In cerebrotendinous xanthomatosis, accumulation of abnormal sterol derivates induces xanthomata in the brain and tendons.
1.3. Abnormal Pigmentation
In most cases, this is due to the abnormal production of melanin and thus involve the melanocytes, predominantly located in the epidermis, but occasionally also located in the dermis. Sometimes, an abnormal pigment other than melanin can also give rise to hyperpigmentation, such as a p-quinone polymer in alkaptonuria, or hemosiderin in hemochromatosis.
1.3.1. Hypopigmentation
Skin hypopigmentation, usually results from impaired production, metabolism, or distribution of melanin. Oculocutaneous albinism, characterized by a generalized reduction in pigmentation, is most commonly caused by a deficiency in tyrosinase, an enzyme in the biosynthetic pathway of melanin.
Patients with phenylketonuria untreated from birth are usually fair skinned and blue-eyed; early treatment has proved that it is not the variant but the abnormal chemical environment in the untreated state that interferes with normal pigmentation, as tyrosine (a precursor of melanin) is decreased if phenylalanine can’t be hydroxylated. Also, patients with cystinosis and some with homocystinuria have thin hypopigmented skin and fine brittle hair. Moreover, skin hypopigmentation is a characteristic sign of infantile sialic acid storage disease.
Hypopigmented skin and hair, twisted hairs or pili torti, and pale and lax skin in Menkes syndrome (ATP7A) (“kinky hair disease”) are due to decreased keratin fiber strength, impaired tyrosinase (a copper-dependent enzyme) activity, and consequent reduced melanin synthesis.
Several disorders of intracellular trafficking interfere with the biogenesis of melanosomes and are associated with cutaneous hypopigmentation and in most cases are associated with immune and/or hematological dysfunction, e.g., Hermansky–Pudlak syndrome (various genes), Chediak–Higashi syndrome (LYST), and Griscelli syndrome types 1 and 2 (MYO5A; RAB27A). Cutaneous (and ocular) hypopigmentation also occurs in Vici syndrome (EPG5), a severe disorder involving dysregulation of autophagy.
Some CDGs may present with pigmentary alterations (e.g., ST3GAL5-CDG or POFUT1-CDG). Hypomelanosis has also been found in a few patients with Kearns–Sayre syndrome [10].
1.3.2. Hyperpigmentation
Defective activity of homogentisate dioxygenase (HGD) in alkaptonuria causes accumulation of homogentisic acid, which is oxidized by a polyphenol oxidase into the p-quinone molecule benzoquinone acetic acid that undergoes polymerization [25]. This quinone polymer is a melanin-like pigment that binds to connective tissue, resulting in ochronosis. Pigmentations is prominent in the ears and may also occur early in the sclerae and nose as salt-and-pepper spots which may become confluent. It may be widely distributed in older patients, especially distally on the fingers. Patients who develop chronic kidney disease have decreased capacity to excrete homogentisic acid, and thus develop diffuse and rapidly-progressive ochronosis [26].
Extensive dermal melanocytosis has been described in several lysosomal disorders, most prominently GM1 gangliosidosis and Hurler disease. Nerve growth factor (NGF) is a ligand for the Trk tyrosine kinase receptor; the accumulated GM1 ganglioside or heparan sulfate binds to the Trk tyrosine kinase receptor, and is thought to increase its signal activity in the presence of NGF. This disruption of the Trk/NGF signaling pathway has been hypothesized to result in failure of complete migration of melanocytes from the neural crest to the epidermis, with an early arrest in the dermis [27]. Melanin absorbs light, resulting in black color; however, when melanin is present in the dermis, light travels through connective tissue, and collagen bundles in the dermis scatter shorter wavelengths of light (Tyndall effect) corresponding to the blue end of the spectrum (Fig. 2). This is akin to the reason why an iris without melanin looks blue (despite lacking any pigment), since collagen bundles in the iris stroma scatter blue light.
Figure 2.
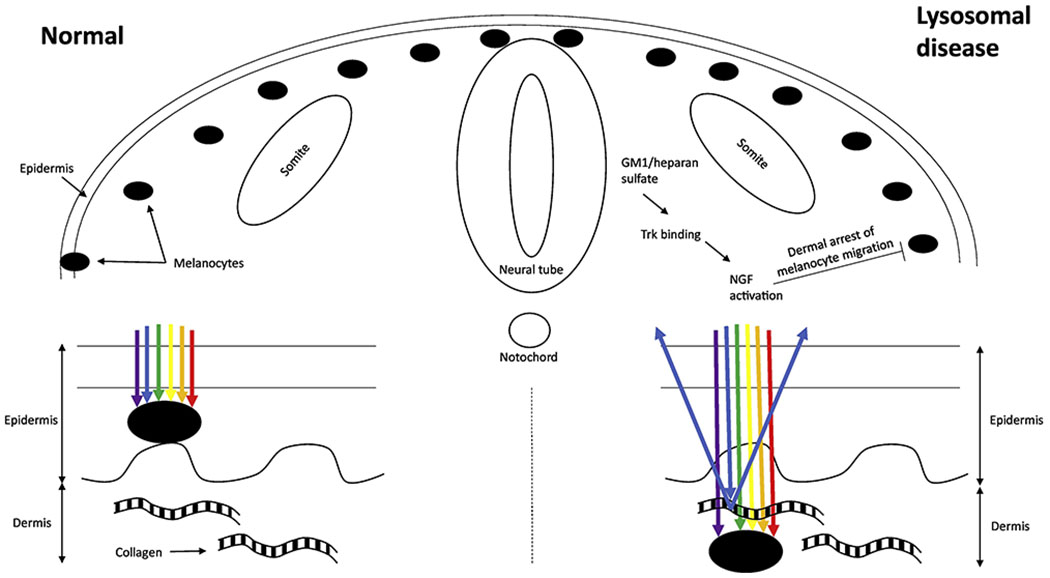
Proposed mechanism for extensive dermal melanocytosis in lysosomal diseases.
Skin hyperpigmentation is characteristic of adrenal insufficiency and may provide the first substantial diagnostic clue in late childhood for adrenoleukodystrophy, or later, of adrenomyeloneuropathy. Adrenal insufficiency can cause skin hyperpigmentation in Kearns-Sayre syndrome. In hereditary hemochromatosis, bluish-gray or bronze hyperpigmentation can be seen. Patients with Wilson disease (ATP7B) have reticulated brownish hyperpigmentation of the lower legs and blue lunulae. There is additionally premature graying of the hair. Cutaneous hyperpigmentation can be observed in Gaucher type 1 (GBA) [28].
1.4. Photosensitivity
The skin “sees” light by using opsin photopigments, akin to the photoreceptor mechanism found in the retina. In fact, humans express a wide variety of opsins in different skin cells, including melanocytes, keratinocytes, dermal fibroblasts, and hair follicle cells. The detection of light by these opsins triggers an array of physiological processes in the skin, including melanogenesis and photoaging [29].
Photosensitive skin lesions characterize many of the porphyrias. In erythropoietic protoporphyria (FECH), photosensitivity begins early in life, with painful burning, stinging, and pruritus in sun-exposed areas followed by erythema, edema, erosions, and scarring. Congenital erythropoietic porphyria (UROS) is the most dramatic cutaneous porphyria, with pink urine and red teeth, vesicular and bullous lesions and fragile skin prone to ulcers or erosions. Residual scarring, alternating hyperpigmentation/ depigmentation, and eventual mutilation of fingers, nasal tips, and ears may occur. Patients with porphyria cutanea tarda type II (due to heterozygous UROD variants) present as adults with fragile skin (erosions due to minor trauma). The onset of hepatoerythropoietic porphyria (due to biallelic UROD variants) is neonatal or shortly thereafter; the skin is fragile, with vesicles and blisters after sun exposure. Facial hypertrichosis is common. Variegate porphyria (PPOX), diagnosed from puberty onwards, may feature pseudosclerodermatous changes as a late skin manifestation.
Fluorescent porphyrins include protoporphyrin IX, uroporphyrin, and coproporphyrin. These porphyrins can accumulate in the skin, where they strongly absorb electromagnetic radiation in the UV region, resulting in photoexcited porphyrins with electrons in high-energy states. The first excited state is a singlet porphyrin with very short half-life (<0.01 microseconds), the excess energy of which can be dissipated either via ionization or by fluorescence (returning to the ground state) or by transitioning to a triplet, a still excited state but with lower energy and longer half-life (microseconds to miliseconds). This longer half-life increases the probability that the photoexcited triplet porphyrin will react with other molecules. The triplet porphyrin can return to the ground state via phosphorescence, or by transferring energy to oxygen to form singlet oxygen or other reactive oxygen species (ROS). These ROS are the primary mediators of porphyrin-induced photosensitivity (Fig. 3). This porphyrin-induced oxidative stress is also the basis of photodynamic therapy, a treatment option for cancer relying on the preferential accumulation of porphyrins by cancer cells, which can then be killed by exposure to electromagnetic radiation that excites the photosensitizing porphyrins. The acute pain upon exposure to light appears to be mediated by the activation of TRPA1 and TRPV1 channels, which play an integral role in pain [30].
Figure 3.
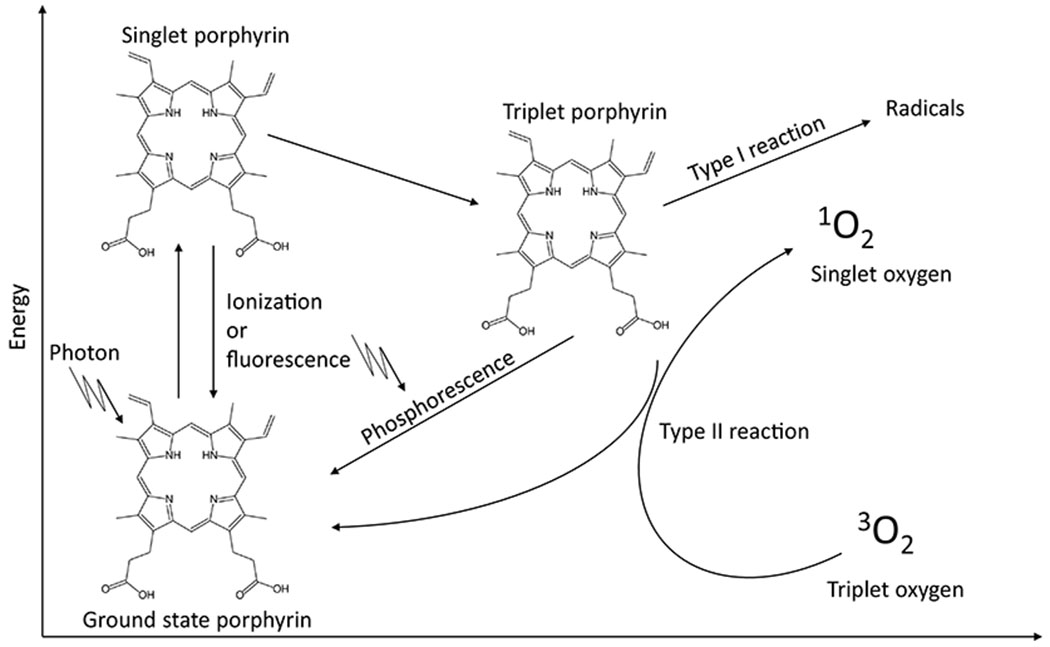
Mechanism of photosensitivity in porphyrias. The absorption of electromagnetic radiation results in an excited singlet porphyrin, which can return to the ground state by ionization, or by emission of electromagnetic radiation (resulting in fluorescence). The singlet porphyrin can also result in a lower energy (but still excited) triplet porphyrin, that can in turn return to the ground state by emission of electromagnetic radiation (resulting in phosphorescence), by directly transferring energy to biological substrates resulting in the formation of free radicals (type I reaction), or by transferring energy to molecular oxygen, resulting in the formation of an excited singlet oxygen and other ROS (type II reaction). These ROS initiate a cascade of epidermal inflammation.
Hartnup disease (SLC6A19) is characterized by a pellagra-like light-sensitive rash, arising from deficient renal and intestinal absorption of tryptophan, an important precursor in NAD biosynthesis. Previously proposed theories for the photosensitivity of pellagra include cutaneous deficiency of urocanic acid (believed to protect against UV radiation), accumulation of kynurenic acid (a photosensitizer), or a reduction in the epidermal repair process due to NAD and NADP deficiency [31]; however, solid evidence for any of these hypotheses was lacking. More recently, a mouse model of pellagra revealed that the photosensitivity is related to upregulation of Ptgs2 (encoding cyclooxygenase-2 or COX-2, an enzyme important for the synthesis of prostaglandins), leading to increased activation of EP4 (prostaglandin receptor) signaling. Specifically, UV induces skin inflammation via production of ROS, which in turn upregulates prostaglandin E2 (PGE2) production. Treatment with a COX-2 inhibitor (indomethacin) led to attenuation of photosensitivity in niacin deficiency [32].
1.5. Skin Laxity
This manifestation results from the involvement of connective tissue in the dermis. Cutis laxa represents a highly heterogeneous group of disorders characterized by loose and/or wrinkled skin. The lack of elasticity of skin contrasts with the hyperelasticity of classical Ehlers–Danlos syndrome. A variety of defects in subunits of a V-ATPase pump important for Golgi Ph maintenance have been associated with cutis laxa; these include ATP6V0A2, ATP6V1A, ATP6V1E1, ATP6AP1, ATP6AP2 deficiencies, which belongs to the group of CDG type II. ALG8-CDG may also present with cutis laxa. Disorders of vesicular trafficking (COG7 deficiency, geroderma osteodysplasticum) have also been associated with cutis laxa, likely from Golgi involvement. Disorders of phosphatidylinositol metabolism can also lead to cutis laxa, such as phosphatidylinositol 4-kinase type 2-alpha deficiency (PI4K2A), phosphatidylinositol 4,5-bisphosphate phospholipase C γ2 deficiency (PLCG2), and Lenz-Majewski syndrome (PTDSS1), the latter affecting phosphatidylinositol 4-phosphate metabolism [33,34].
Skin and/or joint laxity, are among the characteristic findings of delta-1-pyrroline-5-carboxylate synthetase (ALDH18A1) and pyrroline-5-carboxylate reductase 1 (PYCR1) deficiencies, due to defective proline biosynthesis. Skin laxity can also be observed in transaldolase deficiency (TALDO1) and in arterial tortuosity syndrome caused by homozygous or compound heterozygous pathogenic variants in the gene encoding glucose transporter GLUT10 (SLC2A10).
1.6. Hair Disorders
1.6.1. Hair shaft abnormalities
These range from changes in color, density, length, and structure of the hair to loss of hair (alopecia). Trichorrhexis nodosa is a characteristic sign in some patients with argininosuccinic aciduria (ASL). The presentation resembles alopecia visually, but close examination reveals abundant very short, fragile hairs, with characteristic nodules on microscopy. Abnormal hair appearance also occurs in Menkes disease (ATP7A), typically with pili torti, possibly with trichorrhexis nodosa or monilethrix (segmental narrowing of the hair shaft). In addition, patients show skin laxity. In occipital horn syndrome (ATP7A), hair appears coarse, and the skin is soft, mildly extensible, and bruises easily.
Pili torti are found in Bjornstad syndrome, an autosomal recessive disorder due to variants in the BCS1L gene. Pathogenic variants in the RMRP gene cartilage-hair hypoplasia, characterized by fine, sparse, light-colored hair, with reduced hair shaft diameter.
Universal alopecia totalis (including absent lanugo hair) can be seen in patients with holocarboxylase synthetase (HLCS) deficiency. Patients with biotinidase (BTD) deficiency have patchy alopecia reminiscent of acrodermatitis enteropathica.
1.6.2. Hypertrichosis
Hypertrichosis is common in several lysosomal storage diseases. Diffuse hypertrichosis is typical of SURF1 deficiency [35,36].
1.7. Other
Skin and hair abnormalities are prominent in multiple carboxylase deficiency due to holocarboxylase synthetase (HLCS) [37] or biotinidase deficiency (BTD) [38]. Patients surviving the initial episode of metabolic decompensation often develop a bright red patchy or generalized body eruption, often desquamative and typically periorificial, associated with alopecia.
Patients with methylmalonic aciduria and propionic aciduria can have skin lesions reminiscent of acrodermatitis enteropathica (superficial scalded skin, desquamation, periorificial dermatitis, psoriasiform lesions, and alopecia), sometimes known as acrodermatitis dysmetabolica, or acrodermatitis acidemica [39]. Similar lesions have been described in maple syrup urine disease (BCKDHA, BCKDHB, DBT), glutaric aciduria type 1 (GCDH), and urea cycle disorders [39].
Patients with glutamine synthetase deficiency manifest necrolytic migratory erythema (NME) [40], similar to that seen in patients with glucagonomas. The pathomechanism of NME in glucagonoma is thought to involve hypoaminoacidemia, as a decreased plasma concentration of most amino acids (essential and non-essential, ketogenic or glucogenic) is seen [41], likely from increased amino acid clearance [42,43], and the rash improves with amino acid infusion [44]. However, NME has not been described in patients with other defects in amino acid biosynthesis (such as primary serine deficiencies, or asparagine synthetase deficiency); it is unknown why a selective deficiency of glutamine leads to NME. Interestingly, glutamine is known to regulate the proliferation of alpha-cells that synthesize glucagon [45]. In addition, glucagon receptor inhibition leads to increased expression of glutamine synthetase in a mouse model of ornithine transcarbamylase deficiency [46]; conversely, it is possible that glucagon excess (as seen in patients with glucagonomas) might downregulate glutamine synthetase expression. Thus, a close interrelationship exists between glucagon and glutamine.
Patients with NAXD and NAXE deficiencies have skin blisters, with light microscopy of skin biopsies revealing separation between the dermis and epidermis [47]. The accumulated toxic metabolites NADHX and NADPHX inhibit NAD-dependent and NADP-dependent dehydrogenases, respectively [48–50]. In particular, 3-phosphoglycerate dehydrogenase, necessary for serine biosynthesis, is an NAD-dependent enzyme known to be inhibited by NADHX [48]; however, the skin manifestations of NAXD and NAXE deficiencies (skin blisters) are not at all similar to those of serine deficiency (ichthyosis). The manifestations are more similar to pemphigus or pemphigoid; interestingly, patients with pemphigus vulgaris develop antimitochondrial antibodies that penetrate keratinocytes to react with various mitochondrial proteins, activating a pathway of acantholysis [51]. Autoantibody profiling by proteomics revealed that antibodies against NDUFS1, an NADH-dependent mitochondrial dehydrogenase, were over 10 times more frequent in patients with pemphigus vulgaris than in controls [52]. A polymorphism in MT-ND4, encoding another NADH-dependent mitochondrial dehydrogenase, is positively associated with bullous pemphigoid [53]. Since the specific location of dermoepidermal separation is different according to the disease (suprabasal in pemphigus vulgaris, subepidermal in bullous pemphigoid, and either intraepidermal, between the epidermal basal cells and basement membrane, or between the basal membrane and dermis in different types of epidermolysis bullosa), it would be important to report immunofluorescence and electron microscopy of skin biopsies from patients with NAXD and NAXE deficiencies, as better characterization of the exact location of involvement might provide clues about the pathomechanism of skin blisters in these diseases.
Recurrent morbilliform rashes and erythematous macules and papules can be frequently observed in mevalonic aciduria (MVK), a disorder of cholesterol biosynthesis.
Cutaneous ulcers, mainly severe progressive ulceration of lower extremities, perhaps complicated by secondary infection, are a hallmark finding of prolidase deficiency (PEPD). Skin changes also include telangiectasias and premature graying of hair.
Hyperkeratotic lesions on the palms and soles are a characteristic sign of oculocutaneous tyrosinemia or tyrosinemia type II (TAT). Skin lesions usually begin in early infancy and are painful, nonpruritic, and frequently associated with hyperhidrosis.
Punctate palmoplantar keratoderma type I (AAGAB) represents a disorder of vesicular trafficking characterized by multiple hyperkeratotic centrally indented papules from early adolescence or later that are distributed irregularly on the palms and soles. Aplasia cutis congenita (Adams–Oliver syndrome) is a genetically heterogeneous disorder defined by aplasia cutis congenita of the scalp vertex and terminal transverse limb defects.
Recurrent skin infections may occur in SLC35C1-CDG. Hypoplastic nails can be found in ALG12-CDG and in PIGV-CDG.
Mitochondrial disorders rarely cause linear skin defects, or more specifically, linear areas of erythematous skin hypoplasia in the head and neck. This syndrome is genetically heterogeneous and has been associated with variants in HCCS, COX7B or NDUFB11. What these three gene deficiencies have in common is that they are inherited in an X-linked fashion, and only female patients (or male individuals with more than one X chromosome) manifest disease. Naturally, women are mosaic for the X chromosome, and this explains why the linear skin defects follow the lines of Blaschko [54].
2. Diagnosis
We categorized the signs and symptoms of metabolic disease presenting with dermatological abnormalities as: ‘Vascular’, ‘Ichthyosis’, ‘Papulonodular’, ‘Abnormal Pigmentation’, ‘Photosensitivity’, ‘Cutis laxa’, ‘Hair shaft involvement’, and ‘Nail abnormalities’ (Supplemental Table 2). Supplemental Table 3 (also available at www.iembase.org/gamuts) summarizes in detail the specific type of cardiovascular involvement associated with various IMDs. We found 252 relevant IMDs associated with skin, hair or nails involvement.
Hair shaft involvement and abnormal pigmentation are the most common cardiovascular abnormalities reported in 64/252 (25%) and 59/252 (23%) of IMDs with integumentary involvement, respectively, followed by papular and nodular skin lesions in 15%, ichthyosis in 12%, cutis laxa in 10%, vascular lesions in 9%, nail abnormalities in 8% and photosensitivity in 4% (Figure 4).
Figure 4.
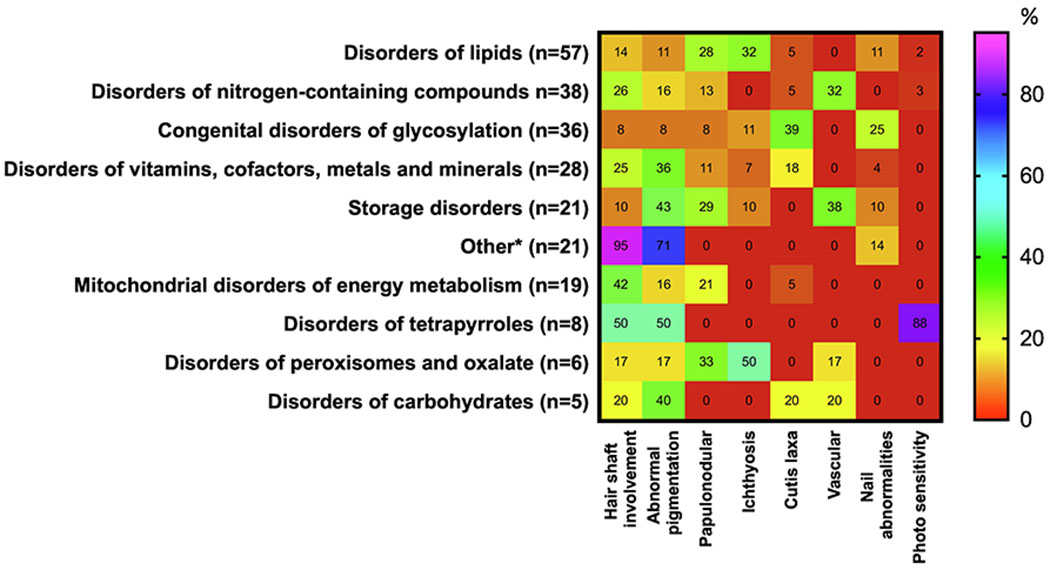
Occurrence (%) of symptoms associated with disorders affecting skin, hair and nails in 10 categories of IMDs. The percentages for cardiovascular involvement were calculated using as the denominator the total number of IMDs in each category presenting with any skin, hair or nails involvement. Heat scale ranges from red (0%) for diseases with no particular symptoms reported to violet (100%) for diseases with particular symptoms occurring with highly frequency. For definition of 10 categories of disorders affecting skin, hair and nails see Supplemental Table 2. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
A list of key laboratory investigations to aid in the diagnosis of the various listed IMDs is summarized in Table 1. For more details see Supplemental Table 3. The diagnostic approach, as with all IMDs, should focus on treatable disorders.
Table 1.
Biochemical investigations in metabolic diseases affecting skin, hair and nails.
| Basic tests | Profiles | Special tests |
|---|---|---|
|
| ||
| Blood count | Amino acids (P, U) | Copper (S, U) |
| ASAT/ALAT (P) | Organic acids (U) | Transferrin (S) |
| CK (P) | Acylcarnitines (DBS, P) | Iron (S) |
| ALP (P) | Sialotransferins (S) | Ferritin (S) |
| Lactate (P) | Sterols (P) | Carnitine (P) |
| Glucose (P) | VLCFA (P) | Glycogen (L) |
| Ammonia (B) | Lipid panel (S) | Lysosomal Enzymes (S) |
| Coagulation factors | Monogenic (CSF) | Neopterin (CSF) |
| Porphyrins (U) | Vitamins (S) | |
| Interferon-alpha (CSF) | ||
3. Prognosis and treatment
Biotin treatment is spectacularly effective in biotinidase deficiency, with rapid and complete remission of skin, neurological, and metabolic abnormalities, but usually not of other impairments. Skin lesions in patients with branched-chain amino acid metabolism disorders often rapidly resolve after dietary correction of isoleucine deficiency. Niacin supplementation is recommended to treat the pellagra-like dermatitis of Hartnup disease. Chenodeoxycholic acid can lead to reduction in the size of xanthomas in cerebrotendinous xanthomatosis [55]. Lipid-lowering medications such as statins or anti-PCSK9 antibodies might similarly address xanthomas in genetic etiologies of hypercholesterolemia. The skin lesions of porphyria cutanea tarda typically improve as plasma porphyrin levels normalize with phlebotomy. Afamelanotide, an alpha-melanocyte-stimulating hormone analog administered via subcutaneous implant, increases pigmentation and duration of sun exposure without pain, leading to improved quality of life in patients with erythropoietic protoporphyria [56].
Supplemental table 3 includes information on primary treatment options for the mentioned IMDs, i.e., treatment that addresses at least one aspect of the pathophysiology of the disease, when available.
4. Conclusions
We provide a comprehensive list of metabolic diseases associated with skin, hair or nail involvement, as well as a proposed battery of standard laboratory and biochemical tests to aid in diagnosis based on the many possible IMDs in the aforementioned list. This represents the sixth issue in a series of educational summaries providing a comprehensive and updated list of metabolic differential diagnosis according to system involvement. The full list can be accessed for free at www.iembase.org/gamuts, and will be curated and updated on a regular basis.
Supplementary Material
Supplemental Table 1. References demonstrating disrupted epidermal ceramide metabolism for disorders along the acylceramide biosynthetic pathway.
Supplemental Table 2. Integumentary abnormalities (terms reported for skin, hair and nails) in inherited metabolic disorders.
Supplemental Table 3. Integumentary involvement in inherited metabolic disorders (IMDs). IMDs with integumentary involvement as a primary or prominent feature are in bold. For IMDs associated with multiple types of integumentary involvement, the most prevalent one is also marked in bold.
We found 252 IMDs associated with various types of dermatologic involvement, and provide a list of tests to aid in their diagnosis.
This is the sixth issue in a series of educational summaries providing a comprehensive list of metabolic differential diagnosis.
We will curate and update this list on a continual basis in the IEMbase website.
Acknowledgements:
This work was supported, in part, by the Intramural Research Program of the National Human Genome Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Ferreira CR, Hoffmann GF, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. I. Movement disorders, Mol Genet Metab. 127 (2019) 28–30. 10.1016/j.ymgme.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferreira CR, Cassiman D, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases, Mol Genet Metab. 127 (2019) 117–121. 10.1016/j.ymgme.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Horvath GA, Stowe RM, Ferreira CR, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. III. Psychiatric presentations, Mol Genet Metab. 130 (2020) 1–6. 10.1016/j.ymgme.2020.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ferreira CR, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. IV. Metabolic cardiovascular disease, Mol Genet Metab. 132 (2021) 112–118. 10.1016/j.ymgme.2020.12.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Horvath GA, Blau N, Ferreira CR, Clinical and biochemical footprints of inherited metabolic disease. V. Cerebral palsy phenotypes, Mol Genet Metab. (2021). 10.1016/j.ymgme.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N, Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism, Genet Med. 20 (2018) 151–158. 10.1038/gim.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ferreira CR, van Karnebeek CDM, Vockley J, Blau N, A proposed nosology of inborn errors of metabolism, Genet Med. 21 (2019) 102–106. 10.1038/s41436-018-0022-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ferreira CR, Rahman S, Keller M, Zschocke J, ICIMD Advisory Group, An international classification of inherited metabolic disorders (ICIMD), J Inherit Metab Dis. 44 (2021) 164–177. 10.1002/jimd.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Martinelli D, El Hachem M, Bertini E, Dionisi-Vici C, Skin and Hair Disorders, in: Hoffmann GFF, Zschocke J, Nyhan WL (Eds.), Inherited Metabolic Diseases: A Clinical Approach, 2nd ed., Springer-Verlag, Berlin Heidelberg, 2017: pp. 341–370. 10.1007/978-3-662-49410-3. [DOI] [Google Scholar]
- [10].Feichtinger RG, Sperl W, Bauer JW, Kofler B, Mitochondrial dysfunction: a neglected component of skin diseases, Exp Dermatol. 23 (2014) 607–614. 10.1111/exd.12484. [DOI] [PubMed] [Google Scholar]
- [11].Bodemer C, Rötig A, Rustin P, Cormier V, Niaudet P, Saudubray JM, Rabier D, Munnich A, de Prost Y, Hair and skin disorders as signs of mitochondrial disease, Pediatrics. 103 (1999) 428–433. 10.1542/peds.103.2.428. [DOI] [PubMed] [Google Scholar]
- [12].Dyer JA, Winters CJ, Chamlin SL, Cutaneous findings in congenital disorders of glycosylation: the hanging fat sign, Pediatr Dermatol. 22 (2005) 457–460. 10.1111/j.1525-1470.2005.00117.x. [DOI] [PubMed] [Google Scholar]
- [13].Feingold KR, Thematic review series: skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis, J Lipid Res. 48 (2007) 2531–2546. 10.1194/jlr.R700013-JLR200. [DOI] [PubMed] [Google Scholar]
- [14].Opálka L, Kováčik A, Pullmannová P, Maixner J, Vávrová K, Effects of omega-O-acylceramide structures and concentrations in healthy and diseased skin barrier lipid membrane models, J Lipid Res. 61 (2020) 219–228. 10.1194/jlr.RA119000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao C, Rossignol F, Vaz FM, Ferreira CR, Inherited disorders of complex lipid metabolism: A clinical review, J Inherit Metab Dis. (2021). 10.1002/jimd.12369. [DOI] [PubMed] [Google Scholar]
- [16].Heinz L, Kim G-J, Marrakchi S, Christiansen J, Turki H, Rauschendorf M-A, Lathrop M, Hausser I, Zimmer AD, Fischer J, Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans, Am J Hum Genet. 100 (2017) 926–939. 10.1016/j.ajhg.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Elias PM, Williams ML, Choi E-H, Feingold KR, Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis, Biochim Biophys Acta. 1841 (2014) 353–361. 10.1016/j.bbalip.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martinelli D, Dionisi-Vici C, AP1S1 defect causing MEDNIK syndrome: a new adaptinopathy associated with defective copper metabolism, Ann N Y Acad Sci. 1314 (2014) 55–63. 10.1111/nyas.12426. [DOI] [PubMed] [Google Scholar]
- [19].Alsaif HS, Al-Owain M, Barrios-Llerena ME, Gosadi G, Binamer Y, Devadason D, Ravenscroft J, Suri M, Alkuraya FS, Homozygous Loss-of-Function Mutations in AP1B1, Encoding Beta-1 Subunit of Adaptor-Related Protein Complex 1, Cause MEDNIK-like Syndrome, Am J Hum Genet. 105 (2019) 1016–1022. 10.1016/j.ajhg.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boyden LM, Atzmony L, Hamilton C, Zhou J, Lim YH, Hu R, Pappas J, Rabin R, Ekstien J, Hirsch Y, Prendiville J, Lifton RP, Ferguson S, Choate KA, Recessive Mutations in AP1B1 Cause Ichthyosis, Deafness, and Photophobia, Am J Hum Genet. 105 (2019) 1023–1029. 10.1016/j.ajhg.2019.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O’brien TJ, Bercovich D, Shalev S, Geiger D, Bergman R, Horowitz M, Mandel H, A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma, Am J Hum Genet. 77 (2005) 242–251. 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rogerson C, Gissen P, VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function, Biochim Biophys Acta Mol Basis Dis. 1864 (2018) 1609–1621. 10.1016/j.bbadis.2018.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jaeken J, Rymen D, Matthijs G, Congenital disorders of glycosylation: other causes of ichthyosis, Eur J Hum Genet. 22 (2014) 444. 10.1038/ejhg.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Musumeci O, Barca E, Lamperti C, Servidei S, Comi GP, Moggio M, Mongini T, Siciliano G, Filosto M, Pegoraro E, Primiano G, Ronchi D, Vercelli L, Orsucci D, Bello L, Zeviani M, Mancuso M, Toscano A, Lipomatosis Incidence and Characteristics in an Italian Cohort of Mitochondrial Patients, Front Neurol. 10 (2019) 160. 10.3389/fneur.2019.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roberts NB, Curtis SA, Milan AM, Ranganath LR, The Pigment in Alkaptonuria Relationship to Melanin and Other Coloured Substances: A Review of Metabolism, Composition and Chemical Analysis, JIMD Rep. 24 (2015) 51–66. 10.1007/8904_2015_453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA, Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation, Mol Genet Metab. 77 (2002) 136–142. 10.1016/s1096-7192(02)00121-x. [DOI] [PubMed] [Google Scholar]
- [27].Hanson M, Lupski JR, Hicks J, Metry D, Association of dermal melanocytosis with lysosomal storage disease: clinical features and hypotheses regarding pathogenesis, Arch Dermatol. 139 (2003) 916–920. 10.1001/archderm.139.7.916. [DOI] [PubMed] [Google Scholar]
- [28].Goldblatt J, Beighton P, Cutaneous manifestations of Gaucher disease, Br J Dermatol. 111 (1984) 331–334. 10.1111/j.1365-2133.1984.tb04731.x. [DOI] [PubMed] [Google Scholar]
- [29].de Assis LVM, Tonolli PN, Moraes MN, Baptista MS, de Lauro Castrucci AM, How does the skin sense sun light? An integrative view of light sensing molecules, Journal of Photochemistry and Photobiology C: Photochemistry Reviews. 47 (2021) 100403. 10.1016/j.jphotochemrev.2021.100403. [DOI] [Google Scholar]
- [30].Babes A, Sauer SK, Moparthi L, Kichko TI, Neacsu C, Namer B, Filipovic M, Zygmunt PM, Reeh PW, Fischer MJM, Photosensitization in Porphyrias and Photodynamic Therapy Involves TRPA1 and TRPV1, J Neurosci. 36 (2016) 5264–5278. 10.1523/JNEUROSCI.4268-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wan P, Moat S, Anstey A, Pellagra: a review with emphasis on photosensitivity, Br J Dermatol. 164 (2011) 1188–1200. 10.1111/j.1365-2133.2010.10163.x. [DOI] [PubMed] [Google Scholar]
- [32].Sugita K, Ikenouchi-Sugita A, Nakayama Y, Yoshioka H, Nomura T, Sakabe J-I, Nakahigashi K, Kuroda E, Uematsu S, Nakamura J, Akira S, Nakamura M, Narumiya S, Miyachi Y, Tokura Y, Kabashima K, Prostaglandin E2 is critical for the development of niacin-deficiency-induced photosensitivity via ROS production, Sci Rep. 3 (2013) 2973. 10.1038/srep02973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sohn M, Ivanova P, Brown HA, Toth DJ, Varnai P, Kim YJ, Balla T, Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions, Proc Natl Acad Sci U S A. 113 (2016) 4314–4319. 10.1073/pnas.1525719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sohn M, Balla T, Lenz-Majewski syndrome: How a single mutation leads to complex changes in lipid metabolism, J Rare Dis Res Treat. 2 (2016) 47–51. 10.29245/2572-9411/2017/1.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].E O, I B, Sh R, Fj H, E S, E C, F W, M S, Hypertrichosis in patients with SURF1 mutations, American Journal of Medical Genetics. Part A 138 (2005). 10.1002/ajmg.a.30972. [DOI] [PubMed] [Google Scholar]
- [36].Baertling F, Mayatepek E, Distelmaier F, Hypertrichosis in presymptomatic mitochondrial disease, J Inherit Metab Dis. 36 (2013) 1081–1082. 10.1007/s10545-013-9593-3. [DOI] [PubMed] [Google Scholar]
- [37].Seymons K, De Moor A, De Raeve H, Lambert J, Dermatologic signs of biotin deficiency leading to the diagnosis of multiple carboxylase deficiency, Pediatr Dermatol. 21 (2004) 231–235. 10.1111/j.0736-8046.2004.21308.x. [DOI] [PubMed] [Google Scholar]
- [38].Yang Y, Yang J-Y, Chen X-J, Biotinidase deficiency characterized by skin and hair findings, Clin Dermatol. 38 (2020) 477–483. 10.1016/j.clindermatol.2020.03.004. [DOI] [PubMed] [Google Scholar]
- [39].Tabanlioğlu D, Ersoy-Evans S, Karaduman A, Acrodermatitis enteropathica-like eruption in metabolic disorders: acrodermatitis dysmetabolica is proposed as a better term, Pediatr Dermatol. 26 (2009) 150–154. 10.1111/j.1525-1470.2008.00803.x. [DOI] [PubMed] [Google Scholar]
- [40].Häberle J, Görg B, Toutain A, Rutsch F, Benoist J-F, Gelot A, Suc A-L, Koch HG, Schliess F, Häussinger D, Inborn error of amino acid synthesis: human glutamine synthetase deficiency, J Inherit Metab Dis. 29 (2006) 352–358. 10.1007/s10545-006-0256-5. [DOI] [PubMed] [Google Scholar]
- [41].Tamura A, Ogasawara T, Fujii Y, Kaneko H, Nakayama A, Higuchi S, Hashimoto N, Miyabayashi Y, Fujimoto M, Komai E, Kono T, Sakuma I, Nagano H, Suzuki S, Koide H, Yokote K, Iseki K, Oguma R, Matsue H, Nojima H, Sugiura K, Yoshitomi H, Ohtsuka M, Rahmutulla B, Kaneda A, Inoshita N, Ogawa S, Tanaka T, Glucagonoma With Necrolytic Migratory Erythema: Metabolic Profile and Detection of Biallelic Inactivation of DAXX Gene, J Clin Endocrinol Metab. 103 (2018) 2417–2423. 10.1210/jc.2017-02646. [DOI] [PubMed] [Google Scholar]
- [42].Almdal TP, Heindorff H, Bardram L, Vilstrup H, Increased amino acid clearance and urea synthesis in a patient with glucagonoma, Gut. 31 (1990) 946–948. 10.1136/gut.31.8.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barazzoni R, Zanetti M, Tiengo A, Tessari P, Protein metabolism in glucagonoma, Diabetologia. 42 (1999) 326–329. 10.1007/s001250051158. [DOI] [PubMed] [Google Scholar]
- [44].Thomaidou E, Nahmias A, Gilead L, Zlotogorski A, Ramot Y, Rapid Clearance of Necrolytic Migratory Erythema Following Intravenous Administration of Amino Acids, JAMA Dermatol 152 (2016) 345–346. 10.1001/jamadermatol.2015.3538. [DOI] [PubMed] [Google Scholar]
- [45].Dean ED, Li M, Prasad N, Wisniewski SN, Von Deylen A, Spaeth J, Maddison L, Botros A, Sedgeman LR, Bozadjieva N, Ilkayeva O, Coldren A, Poffenberger G, Shostak A, Semich MC, Aamodt KI, Phillips N, Yan H, Bernal-Mizrachi E, Corbin JD, Vickers KC, Levy SE, Dai C, Newgard C, Gu W, Stein R, Chen W, Powers AC, Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation, Cell Metab. 25 (2017) 1362–1373.e5. 10.1016/j.cmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cavino K, Sung B, Su Q, Na E, Kim J, Cheng X, Gromada J, Okamoto H, Glucagon Receptor Inhibition Reduces Hyperammonemia and Lethality in Male Mice with Urea Cycle Disorder, Endocrinology. 162 (2021). 10.1210/endocr/bqaa211. [DOI] [PubMed] [Google Scholar]
- [47].Zhou J, Li J, Stenton SL, Ren X, Gong S, Fang F, Prokisch H, NAD(P)HX dehydratase (NAXD) deficiency: a novel neurodegenerative disorder exacerbated by febrile illnesses, Brain. 143 (2020) e8. 10.1093/brain/awz375. [DOI] [PubMed] [Google Scholar]
- [48].Becker-Kettern J, Paczia N, Conrotte J-F, Zhu C, Fiehn O, Jung PP, Steinmetz LM, Linster CL, NAD(P)HX repair deficiency causes central metabolic perturbations in yeast and human cells, FEBS J. 285 (2018) 3376–3401. 10.1111/febs.14631. [DOI] [PubMed] [Google Scholar]
- [49].Prabhakar P, Laboy JI, Wang J, Budker T, Din ZZ, Chobanian M, Fahien LA, Effect of NADH-X on cytosolic glycerol-3-phosphate dehydrogenase, Arch Biochem Biophys. 360 (1998) 195–205. 10.1006/abbi.1998.0939. [DOI] [PubMed] [Google Scholar]
- [50].Yoshida A, Dave V, Inhibition of NADP-dependent dehydrogenases by modified products of NADPH, Arch Biochem Biophys. 169 (1975) 298–303. 10.1016/0003-9861(75)90344-6. [DOI] [PubMed] [Google Scholar]
- [51].Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA, Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology, J Biol Chem. 285 (2010) 3695–3704. 10.1074/jbc.M109.081570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, Jasinskas A, Leiferman KM, Liang L, Marchenko S, Nakajima-Sasaki R, Pittelkow MR, Zone JJ, Grando SA, Pemphigus vulgaris autoantibody profiling by proteomic technique, PLoS One. 8 (2013) e57587. 10.1371/journal.pone.0057587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Russlies J, Fähnrich A, Witte M, Yin J, Benoit S, Gläser R, Günter C, Eming R, Erdmann J, Gola D, Gupta Y, Holtsche MM, Kern JS, König IR, Kiritsi D, Lieb W, Sadik CD, Sárdy M, Schauer F, van Beek N, Weidinger A, Worm M, Zillikens D, Schmidt E, Busch H, Ibrahim SM, Hirose M, Polymorphisms in the Mitochondrial Genome Are Associated With Bullous Pemphigoid in Germans, Front Immunol. 10 (2019) 2200. 10.3389/fimmu.2019.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Indrieri A, Franco B, Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder, Genes (Basel). 12 (2021). 10.3390/genes12020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Duell PB, Salen G, Eichler FS, DeBarber AE, Connor SL, Casaday L, Jayadev S, Kisanuki Y, Lekprasert P, Malloy MJ, Ramdhani RA, Ziajka PE, Quinn JF, Su KG, Geller AS, Diffenderfer MR, Schaefer EJ, Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis, J Clin Lipidol. 12 (2018) 1169–1178. 10.1016/j.jacl.2018.06.008. [DOI] [PubMed] [Google Scholar]
- [56].Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach J-C, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ, Afamelanotide for Erythropoietic Protoporphyria, N Engl J Med. 373 (2015) 48–59. 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. References demonstrating disrupted epidermal ceramide metabolism for disorders along the acylceramide biosynthetic pathway.
Supplemental Table 2. Integumentary abnormalities (terms reported for skin, hair and nails) in inherited metabolic disorders.
Supplemental Table 3. Integumentary involvement in inherited metabolic disorders (IMDs). IMDs with integumentary involvement as a primary or prominent feature are in bold. For IMDs associated with multiple types of integumentary involvement, the most prevalent one is also marked in bold.