

Summary
Viral encephalitis is a major neglected medical problem. Host defense mechanisms against viral infection of the central nervous system (CNS) have long remained unclear. The few previous studies of CNS-specific immunity to viruses in mice in vivo and humans in vitro have focused on the contributions of circulating leukocytes, resident microglial cells and astrocytes, with neurons long considered passive victims of viral infection requiring protection from extrinsic antiviral mechanisms. The last decade has witnessed the gradual emergence of the notion that neurons also combat viruses through cell-intrinsic mechanisms. Forward genetic approaches in humans have shown that monogenic inborn errors of TLR3, IFN-α/β, or snoRNA31 immunity confer susceptibility to herpes simplex virus 1 (HSV-1) infection of the forebrain, whereas inborn errors of DBR1 underlie brainstem infections due to various viruses, including HSV-1. The study of human pluripotent stem cell (hPSC)-derived CNS-resident cells has unraveled known (i.e. TLR3-dependent IFN-α/β immunity) and new (i.e. snoRNA31- or DBRl-dependent immunity) cell-intrinsic antiviral mechanisms operating in neurons. Reverse genetic approaches in mice have confirmed that some known antiviral mechanisms also operate in mouse neurons (e.g. TLR3 and IFN-α/β immunity). The search for human inborn errors of immunity (IEIs) underlying various forms of viral encephalitis, coupled with mouse models in vivo, and hPSC-based culture models of CNS and peripheral nervous system cells and organoids in vitro, should shed further light on the cell- and tissue-specific mechanisms of host defense against viruses in the human brain.
Keywords: neurons, cell-intrinsic immunity, viral infection, inborn errors of immunity, mouse models, Human pluripotent stem cells
Introduction
Many viruses can underlie severe infection of the central nervous system (CNS) in humans. The incidence of viral encephalitis is at least 1-2 cases per 100,000 individuals per year, corresponding to a prevalence of ~1/1,000 births [1]. Viral encephalitis is life-threatening and most frequently affects children and the elderly [2,3]. Its clinical course is typically acute, but some forms can be chronic. There are at least 20 causal viruses, some of which are common, such as herpes simplex virus 1 (HSV-1), varicella zoster virus (VZV), enteroviruses (EV), influenza viruses (IV), measles virus (MV), JC virus (JCV) and noroviruses (NV), and some of which are rare, such as rabies virus, the Nipah and Hendra viruses, and West Nile virus (WNV) [4]. Most viruses are thought to reach the CNS by crossing the blood-brain barrier, although some viruses, including HSV-1, HSV-2, VZV and rabies virus, are known to invade the CNS via the peripheral nervous system (PNS). It is becoming increasingly clear that many viruses, including IV, EV, and WNV, can use both routes [5]. Regardless of the route taken to reach the brain, the invasion of the CNS by a neurotropic virus can result in CNS inflammation and neurodegeneration, with significant mortality and neurological sequelae in survivors. Many of the causal viruses have been identified. Patients with known cellular immunodeficiencies, such as those with acquired immunodeficiency related to human immunodeficiency virus (HIV) or organ transplantation, display higher vulnerability to viral infection of the CNS by some viruses including Epstein-barr virus (EBV), cytomegalovirus (CMV) and human herpes virus 6 (HHV6) [6]. However, viral encephalitis typically affects otherwise healthy people, and the molecular and cellular mechanisms underlying the pathogenesis of viral encephalitis remain unclear.
The CNS is widely considered an immunological sanctuary, although it is increasingly recognized that ‘immune responses’ mediated by circulating leukocytes and CNS-resident cells can take place in the CNS, either to protect the brain against damage due to autoimmune, infectious, neurodegenerative, and oncogenic processes, or to contribute to the autoimmune responses in this organ [7]. The mechanisms of host defense against viral infection of the CNS have long remained unknown. Most previous studies on CNS immunity have focused on the contribution of leukocytes and two populations of glial cells — microglia and astrocytes —with neurons generally considered to be passive victims of viral infections [8–13]. However, the last decade has witnessed the gradual emergence of the notion that neurons make use of intrinsic mechanisms to fight viral attacks. This notion emerged from initial studies testing the hypothesis that CNS-specific cell-intrinsic immunity, rather than leukocyte-mediated immunity, is crucial for host defense against HSV-1 in humans [14–16]. We review here recent progress towards understanding cell-intrinsic antiviral immunity in specific types of neurons, in both humans and mice.
Antiviral immunity in rat and mouse neurons: type I IFN-related mechanisms
Since the discovery of type I interferons (IFNs) in 1957 [17], tremendous efforts have been made to determine the role of these cytokines in defenses against diverse RNA and DNA viruses, in various mouse or rat models of infection in vivo and in vitro. Perhaps the first clue to the antiviral activity of type I IFNs in neurons came from the observation that the in vitro treatment of cultured isolated embryonic rat dorsal root ganglion neurons with recombinant IFN-α or IFN-β inhibited virus replication in these neurons in a dose-dependent manner [18]. Various studies have since shown that the protective effect of type I IFNs differs between types of neurons and viruses. IFN-α treatment in rat hippocampal neurons induces no protective response against Borna disease virus [19]. Similarly, IFN-β does not protect embryonic mouse hippocampal neurons against infection with Theiler’s murine encephalomyelitis virus (TMEV) or vesicular stomatitis virus (VSV), possibly due to the lack of induction of certain antiviral IFN-inducible genes (ISGs), such as the ApoL9b gene, by IFN-β in these neurons [20]. Nevertheless, mouse hippocampal neurons seem to have high basal levels of the type I IFNs thought to be crucial for the early control of MV infection [21]. Major differences in IFN responsiveness have also been observed between the granule cell neurons of the cerebellum and the cortical neurons in mice, with granule cell neurons having higher basal ISG expression levels and responding more strongly to IFN-β treatment, consistent with the lower susceptibility of granule cell neurons and the cerebellum to WNV infection [22]. IFN-β treatment has also been shown to inhibit VSV replication strongly in mouse primary olfactory neurons [23]. All these observations suggested a neuron cell type-specific protective effect of type I IFNs against viral infections, for at least some of the viruses tested.
Type I IFNs are produced by CNS-resident cells, including neurons, during viral infections [24,25]. Mouse models with specific gene knockouts have therefore been used for in vivo and in vitro studies investigating the requirement for cellular IFN pathways in host defense against viral infections of the CNS [22,26]. The outcome of experimental viral infections in mouse models may ultimately be influenced by viral load, virus strain, route of infection and animal strain. However, studies performed over the last decade have provided evidence of type I IFN-mediated cell-intrinsic antiviral mechanisms in mouse neurons in vitro and in vivo. In cultured trigeminal neurons isolated from mice lacking Stat1, a key transcription factor of the cellular type I IFN response pathway, defective neuron axonal IFN-β signaling resulted in a failure to restrict HSV-1 infection, as shown by comparisons with wild-type (WT) neurons. These findings were consistent with the uncontrolled viral infection observed in the trigeminal ganglia and brain stem of Stat1-deficient mice in vivo [27]. Hippocampal neurons from mice lacking Irf1, an ISG-inducing transcription factor with an unclear role in type I IFN responses, were found to display higher levels of VSV replication than WT mouse neurons, consistent with the impaired control of a late-phase viral infection of the mouse brain, which was not restricted by type I IFNs and affected the cerebrum, cerebellum and brain stem [28]. In mice devoid of type I IFN signaling (due to disruption of the Ifnar1 locus) specifically in calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα)-expressing neurons, infected with mouse hepatitis virus A59 (MHV-A-59), viral spread in Ifnar1-deficient mice was enhanced in the neurons of the forebrain, mid- and hindbrain, and the spinal cord, but not in the cerebellum [29]. Finally, type I IFN signaling in neurons seems to be essential for microglial activation and protection against lethal VSV encephalitis in mice, as shown by studies based on the conditional knockout of Ifnar1 (and therefore of type I IFN signaling) in neurons or microglia, which demonstrated the importance of type I IFN-dependent crosstalk between neurons, astrocytes and microglia [30]. Thus, studies of gene-targeted mouse models have provided clear evidence that neuron-intrinsic type I IFN-related immunity to viruses may be crucial for the brain region-specific control of viral infections.
Antiviral immunity in mouse neurons: other antiviral mechanisms
A role for apoptosis in antiviral immunity in neurons was first suggested by early investigations searching for mechanisms underlying the age-dependent morbidity and mortality of viral encephalitis [31]. In mouse in vivo and in vitro studies, an alphavirus, Sindbis virus (SV), caused acute encephalomyelitis in an age-dependent manner in mice, and this age-dependent susceptibility to fatal infection appeared to be due to a greater intrinsic susceptibility to the induction of apoptosis in the neurons of the dorsal root ganglion and spinal cord in young mice. The contributions of apoptosis and other programmed cell death pathways to host antiviral defense mechanisms have been studied in depth in many other organs and cell types [32], but relevant data for neurons and other CNS-specific cells remained limited. In one study published in 2017, cortical neurons isolated from mice lacking RIPK3, a key component of the cellular apoptosis and necroptosis pathways, produced abnormally low levels of chemokines upon infection with WNV [33]. This led the authors to suggest that the enhanced susceptibility to WNV of RIPK3-deficient mice in vivo was due to the suppression of neuronal chemokine expression, leading to lower levels of recruitment of T lymphocytes and myeloid cells to the CNS for the restriction of viral infection [33]. By contrast, in another study published in 2019 [34], the cell type-specific targeted expression or deletion of RIPK3 in forebrain cortical neurons decreased and increased, respectively, the viral load in the mouse brain following infection with Zika virus (ZIKV). This study also showed, in cultured mouse cortical neurons, that ZIKV infection activated the nucleotide sensor ZBP1 and downstream RIPK1 and RIPK3 signaling for the restriction of viral replication, by altering cellular metabolism via the upregulation of IRG1, an enzyme, and the production of the metabolite, itaconate [34]. These two studies indicated that key molecular components of the cell apoptosis and necroptosis pathways could control mouse cortical neuron-intrinsic immunity to viruses through their canonical and non-canonical functions.
Autophagy is a highly conserved catabolic pathway that is crucial for maintaining the basal turnover of cellular components. Interestingly, the in vivo evidence for an antiviral role of autophagy in mammalian animal hosts is mostly restricted to viruses specifically targeting neurons, including SIV and HSV-1 [35]. DRG neurons from Atg5 knockout mice, which have a defective autophagy pathway, displayed uncontrolled HSV-1 replication [36]. This observation was further confirmed in DRG neurons from mice with conditional deletions of Atg5 limited to neurons, suggesting that autophagy governs a peripheral nervous system cell-intrinsic antiviral mechanism against HSV-1 infection, at least in mouse DRG neurons. The formation of autophagic clusters, in a type I IFN signaling-dependent manner, has also been reported in cultured mouse trigeminal ganglion (TG) neurons, following infection with HSV-1, HSV-2, pseudorabies virus (PrV) and, to a lesser extent, yellow fever virus, but not after infection with MHV68, VACV, TMEV or VSV, although the physiological relevance of this observation remains unclear [37]. Thus, in parallel to type I IFN responses, a number of alternative mechanisms for governing neuron-intrinsic antiviral immunity in mice have been proposed, principally involving host cell death pathways, including apoptosis, necroptosis and autophagy, although key cell death pathway molecules may also be involved, through their non-canonical functions [33].
Neuron-intrinsic antiviral immunity in humans: TLR3-dependent type I IFN responses
Cell-intrinsic antiviral immunity had never been studied in human neurons before we used human pluripotent stem cell (hPSC)-derived CNS cells to dissect the cellular basis of the pathogenesis of HSV-1 encephalitis (HSE). Over the last 15 years, we have tested the hypothesis that childhood HSE results from single-gene inborn errors of immunity to HSV-1 in the human CNS [38,39]. Our human genetic studies have progressively led to the dissection of neuron-intrinsic antiviral immunity in humans, through genetically defined, patient-specific hPSC-based disease modeling [40]. HSV-1 enters the human body via the oral or nasal epithelium and infects neurons, subsequently establishing latency in the TG sensory ganglia [41]. More than 85% of adults worldwide have antibodies against HSV-1, which causes asymptomatic infection or benign, self-healing disease in most individuals. In about 1~2/10,000 infected individuals of all ages, HSV-1 invades the CNS via the olfactory bulb, causing forebrain HSE (~95% of cases), or via the TG nerves, causing brainstem HSE (~5% of cases) [3,42]. HSE is fatal in more than 70% of patients if left untreated, and most acyclovir-treated survivors develop mild to severe neurological sequelae [43]. Typically, HSE is isolated, and affects otherwise healthy individuals who are not particularly susceptible to other clinical forms of HSV-1 infection [3,44]. Remarkably, HSE has not been reported in children with AIDS or conventional primary immunodeficiencies of hematopoietic cells, suggesting that inborn errors of leukocytes are unlikely to be causal [6]. A definitive link between HSV and HSE was established in 1941 [45]. The pathogenesis of HSE has long remained unclear, despite identification of the causal virus. However, very rare cases of “syndromic” HSE combined with mycobacterial diseases have been observed: in a child with autosomal recessive (AR) STAT1 deficiency with impaired cellular responses to IFN-α/β, IFN-λ and IFN-γ [46–48], and a child with a IKBKG (NEMO) mutation whose cells displayed poor IFN-α/β and IFN-λ production [49]. Both disorders involved an impairment of cell-intrinsic immunity in leukocytes and, probably, in all other cell types. These observations suggested that human genetic defects affecting intrinsic immunity in the CNS, probably related to IFN-mediated antiviral defense, may underlie HSE, in at least some children.
Genetic studies of isolated forebrain HSE led to the discovery of single-gene inborn errors of the Toll-like receptor 3 (TLR3)-dependent pathway of interferon (IFN)-α/β and -λ production, with mono- or biallelic mutations of six TLR3 pathway genes (TLR3, UNC93B1, TICAM1 (TRIF), TRAF3, TBK1 or IRF3) [14,50–57] and one gene of the type I IFN response pathway (IFNAR1) [58] (Figure 1). Toll-like receptor 3 (TLR3) is an endosomal receptor of dsRNA [59], governing a pathway of IFN-α/β and IFN-λ induction. IFNAR1 is one of the two subunits of the receptor of type I IFNs. It governs activation of the downstream pathway by all type I IFNs. These findings, together with the previous observation of syndromic HSE in patients with X-linked recessive (XR) NEMO deficiency [49] or AR complete STAT1 deficiency [46], suggested a crucial role for TLR3-dependent IFN-α/β immunity in host defense against HSV-1 in the CNS. It has also been suggested that other mutations of these and otherTLR3 or IFN pathway genes may underlie HSE in children or adults [60–64]. Interestingly, a minority of patients with mutations of TLR3 have developed pneumonia due to influenza A virus or SARS-CoV-2, or ophthalmic zoster [65,66]. Nevertheless, TLR3-mediated responses to dsRNA and antiviral immunity seem to be redundant in most TLR3-expressing cell types, including leukocytes in particular, probably accounting for the lack of viral dissemination during the course of HSE [51,55]. TLR3- and type I IFN-mediated cell-intrinsic immunity was initially studied with dermal fibroblasts as surrogate cells, and then with hPSC-derived CNS- and PNS-resident cells from patients with forebrain HSE and mutations of TLR3 pathway genes. Consistent with the findings of studies showing that mouse Tlr3 is required for responses to HSV in neurons and astrocytes [67,68], TLR3 pathway-deficient human fibroblasts [14,51–55,57] and iPSC-derived cortical neurons and oligodendrocytes [69] were found to be much more susceptible to HSV-1 infection than control cells. More recently, the molecular basis of TLR3-dependent cell-intrinsic antiviral immunity was attributed to a role as the rheostat in controlling basal cellular levels of IFN-β immunity in fibroblasts and cortical neurons [70]. By contrast, in vitro-differentiated human UNC-93B-deficient astrocytes or neural stem cells, and TLR3-deficient peripheral TG neurons are as susceptible to infection as control cells [71], probably due to insufficient basal levels of IFN immunity, as pretreatment with IFN-α or poly(I:C) protected TG neurons from healthy controls, but not those from TLR3-deficient patients, against HSV-1 infection [71]. These data provided a plausible cellular basis for the pathogenesis of genetically driven forebrain HSE, suggesting that TLR3-dependent, type I IFN-mediated cortical neuron-intrinsic anti-HSV-1 immunity, as opposed to the innate and adaptive immunity mediated by leukocytes and related cells, was crucial for host defense against HSV-1 in the human forebrain [14,55].
Figure 1. Illustration of neuron-intrinsic type I IFN immunity or new antiviral mechanisms crucial for defense against viral infection of the central nervous system in humans.
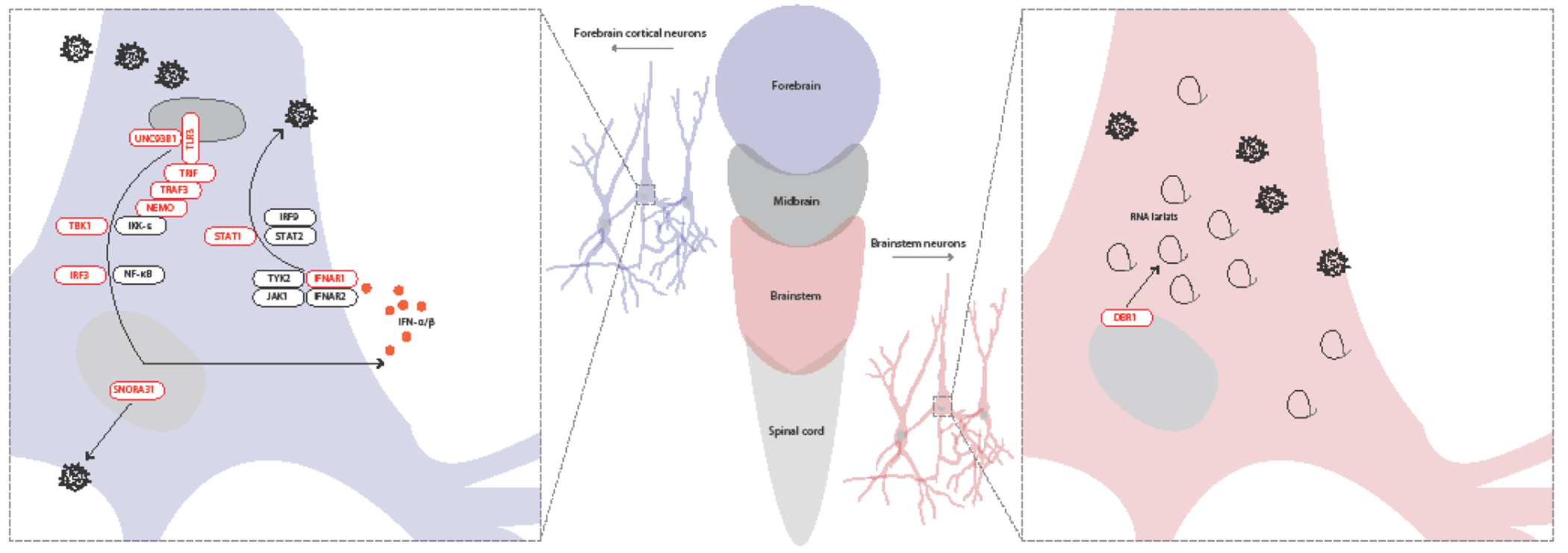
TLR3-type I IFN circuit and snoRNA31 immunity in forebrain HSV-1 infection (left), and DBR1-mediated RNA lariat metabolism in brainstem viral infection (right). TLR3 controls basal levels of IFN-β-mediated anti-HSV-1 immunity in cortical neurons and oligodendrocytes. DBR1 controls brainstem-specific immunity to viruses (HSV-1, influenza virus, norovirus), presumably in neurons and other cells of the brainstem, which is composed of the midbrain and hindbrain. In red: molecules shown to be involved in TLR3-IFN-α/β-mediated or snoRNA31-mediated immunity to HSV-1 in cortical neurons (left), or involved in DBR1-mediated RNA lariat metabolism controls brainstem viral encephalitis due to HSV-1, influenza virus and norovirus (right). In dark gray: other key molecules of the TLR3-type I IFN circuit that have not been shown to be involved in central nervous system antiviral immunity in humans.
Neuron-intrinsic immunity in humans: new antiviral mechanisms
An unbiased genome-wide search of new HSE-causing genes via whole-exome sequencing led to the discovery of rare heterozygous variants of SNORA31, which encodes small nucleolar RNA 31 (snoRNA31), in five unrelated patients with forebrain HSE [72]. SnoRNA31 is a 130-nucleotide snoRNA of the H/ACA box class. SNORA31 is highly conserved in the general population, and ubiquitously expressed in various human cell types [73]. SnoRNA31 had only one predicted function: directing the isomerization of uridine residues to generate pseudouridines in position 218 of the 18S ribosomal RNA (rRNA) and position 3,713 of the 28S rRNA [74]. Studies with hPSC-derived cortical neurons showed that SnoRNA31 was produced and functional in human cortical neurons, as a CRISPR/Cas9-introduced biallelic deletion within SNORA31 impaired the pseudouridylation of the uridine residue in position 218 of the ribosomal RNA 18S in isogenic hPSC-derived CNS neurons. Moreover, snoRNA31 is a CNS neuron-intrinsic HSV-1 restriction factor, as CRISPR/Cas9-introduced biallelic and monoallelic SNORA31 deletions render neurons highly susceptible to HSV-1. Accordingly, CNS cortical neurons derived from the iPSCs of patients with SNORA31 mutations are highly susceptible to HSV-1, like those from TLR3- or STAT1-deficient patients. Exogenous IFN-β renders neurons with SNORA31 and TLR3 mutations, but not those with STAT1 mutations, resistant to HSV-1 infection. Finally, transcriptomic analyses of SNORA31-mutated hPSC-derived cortical neurons have shown these cells to have normal responses to stimulation with TLR3 and IFN-α/β, but abnormal responses to HSV-1, suggesting that AD snoRNA31 deficiency impairs intrinsic immunity by a distinctive mechanism in vitro, and, by inference, underlies HSE in vivo by a distinctive mechanism. The modified transcriptome-level response to HSV-1 associated with snoRNA31 deficiency may affect the expression of one or more of the effectors induced by TLR3 or IFN-α/β, thereby impairing anti-HSV-1 immunity in these cells. Alternatively, snoRNA31 may interfere with HSV-1 propagation directly, by interacting with viral transcripts. Future studies should address the fine molecular mechanism by which snoRNA31 contributes to the control of HSV-1 in CNS cortical neurons and, potentially, in other CNS-resident cell types. The discovery of AD snoRNA31 deficiency as a genetic etiology of forebrain HSE demonstrated that snoRNA31 is a new CNS neuron-intrinsic HSV-1 restriction factor. It also provided evidence that snoRNAs can be essential for host defense.
The search for single-gene inborn errors of immunity underlying brainstem viral encephalitis (BVE) led to the discovery of another new cell-intrinsic antiviral mechanism, mediated by debranching enzyme 1 (DBR1). AR partial DBR1 deficiency was reported in 2018, in otherwise healthy children with BVE caused by various viruses, including HSV-1, influenza B virus (IBV), and norovirus [75]. DBR1 is the only known RNA lariat-debranching enzyme in humans. As inferred from studies performed in yeast, DBR1 hydrolyzes 2’5’-phosphodiester linkages at the branch points of intron lariat RNAs, facilitating their rapid turnover [76]. No connection between DBR1 and host immunity to infection was previously known. In most patients with devastating BVE, the brainstem is the only region of the CNS affected, suggesting that, if there is an inborn error of immunity underlying BVE, it may affect brainstem-specific immunity. Two of the five DBRl-deficient children developed brainstem HSE. DBR1 protein levels are highest in the brainstem and spinal cord, suggesting that DBR1 deficiency disrupts immunity in brainstem-resident cells [75]. DBRl-deficient fibroblasts from the patients, whose TLR3 and IFN-α/β response pathways were intact, were found to contain higher RNA lariat levels than control cells, this difference becoming even more marked during HSV-1 infection. Moreover, DBR1-deficient fibroblasts were highly susceptible to HSV-1 and VSV, like TLR3- and STAT1-deficient fibroblasts [75]. The underlying molecular mechanism remains elusive. The accumulation of RNA lariats may impair virus recognition by host cells, thereby damaging cell-intrinsic defenses against viral invasion. DBR1 may also regulate the processing of some host protein-coding RNAs, non-coding RNAs (ncRNAs) [77–79], or viral RNA lariats [80–83], thereby controlling cell-intrinsic defense against intracellular virus replication. It could be speculated that inherited DBR1 deficiency probably underlies viral infection of the brainstem through the disruption of brainstem-specific and cell-intrinsic immunity to viruses, including HSV-1. Future investigations should test these hypotheses in hPSC-derived brainstem neurons and other CNS cell types.
Concluding remarks
The human brain contains ~1011 neurons, of diverse subtypes, with only a limited regeneration capacity. Based on the large body of studies on cultured rat and mouse primary neurons and reverse genetic studies in vivo and in vitro, it has been possible to establish the roles of antiviral pathways (i.e. type I IFNs, cell death pathways) in various types of neurons, against multiple neurotropic viruses. Studies of the human inborn errors of immunity underlying viral encephalitis led to the discovery of HSE-causing gene mutations disrupting known (TLR3-dependent IFN-α/β immunity) and new (dependent on DBR1 or snoRNA31) antiviral mechanisms, providing opportunities for the use of hPSC-based disease modeling for experimental testing of the hypothesis that CNS-specific cell-intrinsic immunity, rather than leukocyte-mediated immunity, is crucial for host defense against HSV-1. It is now clear that neurons use direct mechanisms to fight viral attacks, with different molecular pathways used by different types of neurons. The added value of human forward genetic studies is that such studies provide information about host defense genes under natural conditions of infection, through an unbiased, genome-wide approach. Future studies making use of next-generation sequencing technologies and genome-wide computational tools [84–89] to search for inborn errors of immunity conferring predisposition to various types of isolated viral encephalitis, such as BVE due to IBV or norovirus [75], are likely to discover additional, key molecules controlling CNS-specific immunity to viruses. Such discoveries will, in turn, pave the way for further studies of CNS tissue-specific, cell-intrinsic, as opposed to leukocyte-mediated, innate or adaptive immunity to viruses in humans [16]. Known and new antiviral mechanisms are likely to be discovered. Each of the various types of neurons may be crucial for antiviral immunity in a specific region of the brain, for the control of one specific virus or a small subset of viruses, either as the master ‘protector’ or in coordination with other cells resident in or infiltrating into the brain during infection [30,69,90–95]. Both mouse in vivo models and human hPSC-mediated CNS- and PNS-specific cell and organoid models will be powerful tools for unraveling cell-intrinsic immunity to viruses in neurons and other cell types, in great breadth and depth.
Acknowledgments
We thank our patients and their families for participating in this study. We apologize to our colleagues whose works are not cited in this paper due to space limitation. We thank the members of both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. Our work is funded in part by the National Center for Advancing Translational Sciences/NIH/Clinical and Translational Science Award program (grant UL1TR001866); NIH grants R01AI088364, R01NS072381, and R21AI151663; grants from the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01); ANR grants SEAeHostFactors (ANR-18-CE15-0020-02) and CNSVIRGEN (ANR-19-CE15-0009-01); the Robertson Therapeutic Development Fund; the Tri-Institutional stem cell initiative fund (Tri-SCI); the Rockefeller University; INSERM; University of Paris; and the St. Giles Foundation. OH was supported by a NYSTEM training award (grant # C32559GG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Jmor F, Emsley HC, Fischer M, Solomon T, Lewthwaite P: The incidence of acute encephalitis syndrome in Western industrialised and tropical countries. Virol J 2008, 5:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Giraldo D, Wilcox DR, Longnecker R: The Innate Immune Response to Herpes Simplex Virus 1 Infection Is Dampened in the Newborn Brain and Can Be Modulated by Exogenous Interferon Beta To Improve Survival. mBio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Abel L, Plancoulaine S, Jouanguy E, Zhang SY, Mahfoufi N, Nicolas N, Sancho-Shimizu V, Alcais A, Guo Y, Cardon A, et al. : Age-Dependent Mendelian Predisposition to Herpes Simplex Virus Type 1 Encephalitis in Childhood. J Pediatr 2010, 157: 623–629. [DOI] [PubMed] [Google Scholar]
- [4].Stahl JP, Mailles A, Dacheux L, Morand P: Epidemiology of viral encephalitis in 2011. Med Mal Infect 2011, 41:453–464. [DOI] [PubMed] [Google Scholar]
- [5].Swanson PA 2nd, McGavern DB: Viral diseases of the central nervous system. Curr Opin Virol 2015, 11:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jouanguy E, Beziat V, Mogensen TH, Casanova JL, Tangye SG, Zhang SY: Human inborn errors of immunity to herpes viruses. Curr Opin Immunol 2020, 62:106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Romo-Gonzalez T, Chavarria A, Perez HJ: Central nervous system: a modified immune surveillance circuit? Brain Behav Immun 2012, 26:823–829. [DOI] [PubMed] [Google Scholar]
- [8].Ransohoff RM, Engelhardt B: The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 2012, 12:623–635. [DOI] [PubMed] [Google Scholar]
- [9].Wraith DC, Nicholson LB: The adaptive immune system in diseases of the central nervous system. J Clin Invest 2012, 122:1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ransohoff RM, Brown MA: Innate immunity in the central nervous system. J Clin Invest 2012, 122:1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chakraborty S, Nazmi A, Dutta K, Basu A: Neurons under viral attack: victims or warriors? Neurochem Int 2010, 56:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Patterson CE, Daley JK, Echols LA, Lane TE, Rall GF: Measles virus infection induces chemokine synthesis by neurons. J Immunol 2003, 171:3102–3109. [DOI] [PubMed] [Google Scholar]
- [13].O’Donnell LA, Conway S, Rose RW, Nicolas E, Slifker M, Balachandran S, Rall GF: STAT1-independent control of a neurotropic measles virus challenge in primary neurons and infected mice. J Immunol 2012, 188:1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, et al. : Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314:308–312. [DOI] [PubMed] [Google Scholar]
- [15].Zhang SY, Jouanguy E, Sancho-Shimizu V, von Bernuth H, Yang K, Abel L, Picard C, Puel A, Casanova JL: Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev 2007, 220:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang SY, Jouanguy E, Zhang Q, Abel L, Puel A, Casanova JL: Human inborn errors of immunity to infection affecting cells other than leukocytes: from the immune system to the whole organism. Curr Opin Immunol 2019, 59:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]; * With a review of cell-intrinsic immunity of both hematopoietic cells and non-hematopoietic cells, the authors suggest that studies of inborn errors of non-hematopoietic cell-intrinsic immunity are reshaping immunology by scaling immunity to infection up from the immune system to the whole organism.
- [17].Isaacs A, Lindenmann J: Virus interference. I. The interferon. Proc. R. Soc. London Ser. B 1957, 147:258–267. [PubMed] [Google Scholar]
- [18].Svennerholm B, Ziegler R, Lycke E: Herpes simplex virus infection of the rat sensory neuron. Effects of interferon on cultured cells. Arch Virol 1989, 104:153–156. [DOI] [PubMed] [Google Scholar]
- [19].Lin CC, Wu YJ, Heimrich B, Schwemmle M: Absence of a robust innate immune response in rat neurons facilitates persistent infection of Borna disease virus in neuronal tissue. Cell Mol Life Sci 2013, 70:4399–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kreit M, Paul S, Knoops L, De Cock A, Sorgeloos F, Michiels T: Inefficient type I interferon-mediated antiviral protection of primary mouse neurons is associated with the lack of apolipoprotein l9 expression. J Virol 2014, 88:3874–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cavanaugh SE, Holmgren AM, Rail GF: Homeostatic interferon expression in neurons is sufficient for early control of viral infection. J Neuroimmunol 2015, 279:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr., Diamond MS: Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 2013, 19:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Trottier MD Jr., Palian BM, Reiss CS: VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology 2005, 333:215–225. [DOI] [PubMed] [Google Scholar]
- [24].Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T: Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 2006, 103:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sorgeloos F, Kreit M, Hermant P, Lardinois C, Michiels T: Antiviral type I and type III interferon responses in the central nervous system. Viruses 2013, 5:834–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thaney VE, O’Neill AM, Hoefer MM, Maung R, Sanchez AB, Kaul M: IFNbeta Protects Neurons from Damage in a Murine Model of HIV-1 Associated Brain Injury. Sci Rep 2017, 7:46514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rosato PC, Leib DA: Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog 2015, 11:e1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S, Bruder D, Grashoff M, Korte M, Koster M, Kalinke U, Hauser H, et al. : Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog 2014, 10:e1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hwang M, Bergmann CC: Neuronal Ablation of Alpha/Beta Interferon (IFN-alpha/beta) Signaling Exacerbates Central Nervous System Viral Dissemination and Impairs IFN-gamma Responsiveness in Microglia/Macrophages. J Virol 2020, 94:e00422–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chhatbar C, Detje CN, Grabski E, Borst K, Spanier J, Ghita L, Elliott DA, Jordao MJC, Mueller N, Sutton J, et al. : Type I Interferon Receptor Signaling of Neurons and Astrocytes Regulates Microglia Activation during Viral Encephalitis. Cell Rep 2018, 25:118–129 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Using a mouse model of VSV central nervous system infection, this study showed that the activation, proliferation, and accumulation of microglia are regulated by type I IFN receptor signaling by neurons and astrocytes, but not microglia. Crosstalk between neurons, astrocytes, and microglia in the infected nervous system is, therefore, crucial for full microglial activation and protection against lethal encephalitis.
- [31].Griffin DE: The Gordon Wilson lecture: unique interactions between viruses, neurons and the immune system. Trans Am Clin Climatol Assoc 1996, 107:89–98. [PMC free article] [PubMed] [Google Scholar]
- [32].Bedoui S, Herold MJ, Strasser A: Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol 2020, 21:678–695. [DOI] [PubMed] [Google Scholar]
- [33].Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, Martinez J, Gale M Jr., Loo YM, Oberst A: RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169:301–313 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]; * In a mouse model of West Nile virus encephalitis, the authors showed that the enhanced susceptibility of Ripk3−/− mice was due to a suppression of neuronal chemokine expression and low levels of T-lymphocyte and inflammatory myeloid cell recruitment to the central nervous system.
- [34].Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, Gao X, Locasale JW, Martinez J, Gale M Jr., et al. : The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 2019, 50:64–76 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This paper shows that neuronal ZIKV infection activates the nucleotide sensor ZBP1 and the kinases RIPK1 and RIPK3, core components of virus-induced necroptotic cell death signaling. However, RIPK signaling restricts viral replication not by altering cell death, but by altering cellular metabolism through upregulation of the enzyme IRG1 and production of the metabolite itaconate.
- [35].Orvedahl A, Levine B: Autophagy and viral neurovirulence. Cell Microbiol 2008, 10:1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A: A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12:334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Katzenell S, Leib DA: Herpes Simplex Virus and Interferon Signaling Induce Novel Autophagic Clusters in Sensory Neurons. J Virol 2016, 90:4706–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Casanova JL: Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A 2015, 112:E7128–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Casanova JL: Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A 2015, 112:E7118–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Harschnitz O, Studer L: Human stem cell models to study host-virus interactions in the central nervous system. Nat Rev Immunol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A comprehensive review of recent progress in the application of human pluripotent stem cell mediated cellular models to the study of host antiviral immunity in the central nervous system.
- [41].Smith G: Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 2012, 66:153–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jubelt B, Mihai C, Li TM, Veerapaneni P: Rhombencephalitis / brainstem encephalitis. Curr Neurol Neurosci Rep 2011, 11:543–552. [DOI] [PubMed] [Google Scholar]
- [43].Stahl JP, Mailles A: Herpes simplex virus encephalitis update. Curr Opin Infect Dis 2019, 32:239–243. [DOI] [PubMed] [Google Scholar]
- [44].Gnann JW Jr., Whitley RJ: Herpes Simplex Encephalitis: an Update. Curr Infect Dis Rep 2017, 19:13. [DOI] [PubMed] [Google Scholar]
- [45].Smith MG, Lennette EH, Reames HR: Isolation of the virus of herpes simplex and the demonstration of intranuclear inclusions in a case of acute encephalitis. Am J Pathol 1941, 17:55–68. [PMC free article] [PubMed] [Google Scholar]
- [46].Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. : Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003, 33:388–391. [DOI] [PubMed] [Google Scholar]
- [47].Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, Hawkins K, Casanova JL, Arkwright PD: Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol 2006, 176:5078–5083. [DOI] [PubMed] [Google Scholar]
- [48].Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J, Zhang SY, Bustamante J, Vogt G, Lejeune J, et al. : A partial form of recessive STAT1 deficiency in humans. J Clin Invest 2009, 119:1502–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Audry M, Ciancanelli M, Yang K, Cobat A, Chang HH, Sancho-Shimizu V, Lorenzo L, Niehues T, Reichenbach J, Li XX, et al. : NEMO is a key component of NF-kappaB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J Allergy Clin Immunol 128:610–617 e611-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L, et al. : Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 2015, 212:1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, et al. : Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med 208:2083–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lim HK, Seppanen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, Dell W, Lorenzo L, Byun M, Pauwels E, et al. : TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk. Neurology 2014, 83:1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, et al. : Human TRAF3 Adaptor Molecule Deficiency Leads to Impaired Toll-like Receptor 3 Response and Susceptibility to Herpes Simplex Encephalitis. Immunity 33:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, et al. : Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121:4889–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. : TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317:1522–1527. [DOI] [PubMed] [Google Scholar]
- [56].Zhang SY, Casanova JL: Inborn errors underlying herpes simplex encephalitis: From TLR3 to IRF3. J Exp Med 2015, 212:1342–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, et al. : Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 2012, 209: 1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, AbuZaitun O, Lorenzo L, Khan T, Hasek M, Hernandez N, et al. : Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest 2021, 131:e139980. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This paper describes the identification of complete autosomal recessive IFNAR1 deficiency as an underlying cause of childhood herpes simples encephalitis. It confirms the crucial role of type I IFNs in the control of HSV-1 infection in the human brain.
- [59].Alexopoulou L, Holt AC, Medzhitov R, Flavell RA: Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413:732–738. [DOI] [PubMed] [Google Scholar]
- [60].Garcia-Morato B, Apalategi CA, Bravo-Galego L, Blaquez Moreno A, Soimon-Fuentes M, Garmendia J, Mendez Echevarria A, Del Rosal Rabes T, Dominguez-Solo A, Lopez-Granados E, et al. : Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J Allergy Clin Immunol 2019, 144:309–312. [DOI] [PubMed] [Google Scholar]
- [61].Mork N, Kofod-Olsen E, Sorensen KB, Bach E, Orntoft TF, Ostergaard L, Paludan SR, Christiansen M, Mogensen TH: Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun 2015, 16:552–566. [DOI] [PubMed] [Google Scholar]
- [62].Vitturi BK, Rosemberg S, Arita FN, da Rocha AJ, Forte WCN, Tilbery CP: Multiphasic disseminated encephalomyelitis associated with herpes virus infection in a patient with TLR3 deficiency. Mult Scler Relat Disord 2019, 36:101379. [DOI] [PubMed] [Google Scholar]
- [63].Sironi M, Peri AM, Cagliani R, Forni D, Riva S, Biasin M, Clerici M, Gori A: TLR3 Mutations in Adult Patients With Herpes Simplex Virus and Varicella-Zoster Virus Encephalitis. J Infect Dis 2017, 215:1430–1434. [DOI] [PubMed] [Google Scholar]
- [64].Armangue T, Baucells BJ, Vlagea A, Petit-Pedrol M, Esteve-Sole A, Deya-Martinez A, Ruiz-Garcia R, Juan M, Perez de Diego R, Dalmau J, et al. : Toll-like receptor 3 deficiency in autoimmune encephalitis post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm 2019, 6:e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, Dobbs K, Hernandez N, Goudin N, Hasek ML, et al. : Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med 2019, 216:2038–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liang F, Glans H, Enoksson SL, Kolios AGA, Lore K, Nilsson J: Recurrent Herpes Zoster Ophthalmicus in a Patient With a Novel Toll-Like Receptor 3 Variant Linked to Compromised Activation Capacity in Fibroblasts. J Infect Dis 2020, 221:1295–1303. [DOI] [PubMed] [Google Scholar]
- [67].Sato R, Kato A, Chimura T, Saitoh SI, Shibata T, Murakami Y, Fukui R, Liu K, Zhang Y, Arii J, et al. : Combating herpesvirus encephalitis by potentiating a TLR3-mTORC2 axis. Nat Immunol 2018, 19:1071–1082. [DOI] [PubMed] [Google Scholar]; ** This paper neatly shows, in a mouse model of HSV-1 infection of the central nervous system, that the TLR3-mTORC2 axis mediates the activation of molecules (including mTORC1) required for the induction of type I interferons, providing feasible mechanisms of TLR3 contribution to innate immune responses to HSV-1 in neurons and astrocytes.
- [68].Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, Ulhoi BP, Holm TH, Mogensen TH, Owens T, et al. : TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 122:1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, et al. : Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491: 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gao D, Ciancanelli MJ, Zhang P, Harschnitz O, Bondet V, Hasek M, Chen J, Mu X, Itan Y, Cobat A, et al. : TLR3 controls constitutive IFN-beta antiviral immunity in human fibroblasts and cortical neurons. J Clin Invest 2021, 131:e134529. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study shows that human TLR3 controls constitutive levels of IFNB mRNA and secreted bioactive IFN-β protein, thereby also controlling constitutive mRNA levels for IFN-stimulated genes (ISGs) in fibroblasts and cortical neurons. The mechanism by which TLR3 restricts viral growth in human fibroblasts and cortical neurons in vitro and, by inference, by which the human CNS prevents infection with HSV-1 in vivo, is, therefore, based on the control of early viral infection by basal IFN-β immunity.
- [71].Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, Liu B, Tchieu J, Steinbeck JA, Lafaille F, et al. : Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci USA 2018, 115:E8775–E8782. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Unlike human hPSC-derived cortical neurons, which possess a TLR3-dependent constitutive resistance sufficient to block incoming HSV-1 in the absence of prior antiviral signals, human hPSC-derived TG neurons are susceptible to HSV-1 because they require preemptive stimulation of the TLR3 or IFN-α/β receptors to induce antiviral immunity.
- [72].Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, Itan Y, Ewaleifoh O, Rapaport F, Carlile TM, et al. : Human SN0RA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nat Med 2019, 25:1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Five unrelated patients with forebrain HSE were each found to be heterozygous for one of four rare variants of SN0RA31, which encodes snoRNA31, an RNA molecule controlling central nervous system cortical neuron-intrinsic immunity to HSV-1 via a mechanism independent of the TLR3-type I IFN circuit.
- [73].Jorjani H, Kehr S, Jedlinski DJ, Gumienny R, Hertel J, Stadler PF, Zavolan M, Gruber AR: An updated human snoRNAome. Nucleic Acids Res 2016, 44:5068–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kiss AM: Human Box H/ACA Pseudouridylation Guide RNA Machinery. Molecular and Cellular Biology 2004, 24:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, et al. : Inborn Errors of RNA Lariat Metabolism in Humans with Brainstem Viral Infection. Cell 2018, 172:952–965 e918. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Biallelic DBR1 mutations were identified in unrelated patients with brainstem infection due to HSV-1, influenza virus, or norovirus. Autosomal recessive, partial DBR1 deficiency impairs RNA lariat metabolism and underlies viral infection of the brainstem in humans through the disruption of tissue-specific and cell-intrinsic immunity to viruses.
- [76].Nam K, Hudson RH, Chapman KB, Ganeshan K, Damha MJ, Boeke JD: Yeast lariat debranching enzyme. Substrate and sequence specificity. J Biol Chem 1994, 269:20613–20621. [PubMed] [Google Scholar]
- [77].Murray JL, Sheng J, Rubin DH: A role for H/ACA and C/D small nucleolar RNAs in viral replication. Mol Biotechnol 2014, 56:429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sedger LM: microRNA control of interferons and interferon induced anti-viral activity. Mol Immunol 2013, 56:781–793. [DOI] [PubMed] [Google Scholar]
- [79].Han B, Park HK, Ching T, Panneerselvam J, Wang H, Shen Y, Zhang J, Li L, Che R, Garmire L, et al. : Human DBR1 modulates the recycling of snRNPs to affect alternative RNA splicing and contributes to the suppression of cancer development. Oncogene 2017, 36:5382–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ulfendahl PJ, Kreivi JP, Akusjarvi G: Role of the branch site/3′-splice site region in adenovirus-2 E1A pre-mRNA alternative splicing: evidence for 5′- and 3′-splice site co-operation. Nucleic Acids Res 1989, 17:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Plotch SJ, Krug RM: In vitro splicing of influenza viral NS1 mRNA and NS1-beta-globin chimeras: possible mechanisms for the control of viral mRNA splicing. Proc Natl Acad Sci U S A 1986, 83:5444–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Perng GC, Jones C: Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Galvis AE, Fisher HE, Fan H, Camerini D: Conformational Changes in the 5′ End of the HIV-1 Genome Dependent on the Debranching Enzyme DBR1 During Early Stages of Infection. J Virol 2017, 91:e01377–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, Shang L, Boisson B, Casanova JL, Abel L: Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 2015, 112:5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, et al. : The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods 2016, 13:109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Velez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, et al. : The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A 2015, 112:13615–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Itan Y, Zhang SY, Vogt G, Abhyankar A, Herman M, Nitschke P, Fried D, Quintana-Murci L, Abel L, Casanova JL: The human gene connectome as a map of short cuts for morbid allele discovery. Proc Natl Acad Sci U S A 2013, 110:5558–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rapaport F, Boisson B, Gregor A, Beziat V, Boisson-Dupuis S, Bustamante J, Jouanguy E, Puel A, Rosain J, Zhang Q, et al. : Negative selection on human genes underlying inborn errors depends on disease outcome and both the mode and mechanism of inheritance. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89]. Zhang P, Cobat A, Lee YS, Wu Y, Bayrak CS, Boccon-Gibod C, Matuozzo D, Lorenzo L, Jain A, Boucherit S, et al. : A computational approach for detecting physiological homogeneity in the midst of genetic heterogeneity. Am J Hum Genet 2021, 118(3):e2001248118. * Development and validation of a novel effective and unbiased computational approach for unraveling genetic heterogeneity by detecting physiological homogeneity in next-generation sequencing data, at the level of patient cohorts.
- [90].Sepahi A, Kraus A, Casadei E, Johnston CA, Galindo-Villegas J, Kelly C, Garcia-Moreno D, Munoz P, Mulero V, Huertas M, et al. : Olfactory sensory neurons mediate ultrarapid antiviral immune responses in a TrkA-dependent manner. Proc Natl Acad Sci U S A 2019, 116:12428–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Fares M, Cochet-Bernoin M, Gonzalez G, Montero-Menei CN, Blanchet O, Benchoua A, Boissart C, Lecollinet S, Richardson J, Haddad N, et al. : Pathological modeling of TBEV infection reveals differential innate immune responses in human neurons and astrocytes that correlate with their susceptibility to infection. J Neuroinflammation 2020, 17:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, Tang X, Zhu J, Zhao Z, Jaffre F, et al. : A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27:125–136 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jacob F, Pather SR, Huang WK, Zhang F, Wong SZH, Zhou H, Cubitt B, Fan W, Chen CZ, Xu M, et al. : Human Pluripotent Stem Cell-Derived Neural Cells and Brain Organoids Reveal SARS-CoV-2 Neurotropism Predominates in Choroid Plexus Epithelium. Cell Stem Cell 2020, 27:937–950 e939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vaegter C, Nyengaard JR, et al. : Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 2016, 7:13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, Bateup HS, Szpara ML, Lee AY, Cox JS, et al. : Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun 2020, 11:3382. [DOI] [PMC free article] [PubMed] [Google Scholar]