

Abstract
We have previously shown in a model of claudin-low breast cancer that regulatory T cells (Tregs) are increased in the tumor microenvironment (TME) and express high levels of PD-1. In mouse models and patients with triple negative breast cancer (TNBC), it is postulated that one cause for the lack of activity of α-PD-1 therapy is the activation of PD-1 expressing Tregs in the TME. We hypothesized that the expression of PD-1 on Tregs would lead to enhanced suppressive function of Tregs and worsen anti-tumor immunity during PD-1 blockade. To evaluate this, Tregs were isolated from claudin-low tumors and functionally evaluated ex vivo. We compared transcriptional profiles of Tregs isolated from tumor bearing mice with or without α-PD-1 therapy using RNA-Seq. We found several genes associated with survival and proliferation pathways, for example Jun, Fos, and Bcl2, were significantly upregulated in Tregs exposed to α-PD-1 treatment. Based on these data, we hypothesized that α-PD-1 treatment on Tregs results in a pro-survival phenotype. Indeed, Tregs exposed to PD-1 blockade had significantly higher levels of Bcl-2 expression, and this led to increased protection from glucocorticoid-induced apoptosis. Additionally, we found in vitro and in vivo that Tregs in the presence of α-PD-1 proliferated more than control Tregs. PD-1 blockade significantly increased the suppressive activity of Tregs at biologically relevant Treg: Tnaive cell ratios. Altogether, we show that this immunotherapy blockade increases proliferation, protection from apoptosis, and suppressive capabilities of Tregs, thus leading to enhanced immunosuppression in the TME.
Introduction
Breast cancer is the most prevalent malignancy in women, accounting for 30% of newly diagnosed cancer cases1. In 2021, more than 44,000 women and men in the U.S. are expected to die from breast cancer2. The clinical prognosis of patients with breast cancer is dependent on tumor grade, involvement of lymph nodes, and expression of the hormone and growth factor receptors estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)3. The triple negative breast cancer (TNBC) subtype is characterized by the lack of expression of ER, PR, and HER2. This clinical subtype can be further divided into molecular groups including the basal-like and claudin-low subtypes, which include the majority of TNBC tumors4,5. The basal-like and claudin-low subtypes are defined by increased expression of tumor proliferative genes and high infiltration of immune cells6. TNBC has the worst prognosis of the breast cancer subtypes due to the lack of targeted therapies that define the other breast cancer subtypes. Because of this, surgery, radiation, and chemotherapy are first-line treatments for TNBC, but due to immune involvement, immunotherapeutic strategies to treat these types of tumors hold great promise.
Immunotherapy has been a promising new approach to cancer treatment in the last decade. Immunotherapy involves enhancing the patient’s immune cells to kill tumor cells. PD-1/PD-L1 signaling is an important adaptive immune response pathway to ensure the immune system is only activated at the appropriate times to minimize inflammation in the setting of persistent antigen. PD-1 expression on T cells from cancer patients is critical to the progressive dysfunction of these cells in the TME7 . Tumors can utilize this immune suppression mechanism by overexpressing PD-L18, the ligand to the PD-1 receptor, thus dampening anti-tumor immune activity in the TME. Most of the previous studies evaluating the function of PD-1 have been focused on cytotoxic CD8+ T cell function in the context of both chronic viral infections and cancer9. CD8+ T cell exhaustion is characterized by the loss of proliferation, reduction in pro-inflammatory cytokine production, and diminished cytotoxic activity10. This loss of function can be reversed by blocking the PD-1/PD-L1 signaling axis, restoring cytokine production, proliferation, and leading to an enhanced immune response11. The role of PD-1 on other types of immune cells in the TME is much less well understood.
Our group has previously shown that TNBC is typically heavily infiltrated with both adaptive and innate immune cells12. Most recently, the IMPassion130 Study demonstrated a significant improvement in progression-free survival in patients treated with the anti-PD-L1 monoclonal antibody (mAb) atezolizumab plus nab-paclitaxel, a microtubule disrupting chemotherapy agent, compared to those receiving the chemotherapy alone13. Despite this, the clinical response for patients with TNBC treated with anti-PD-1 or PD-L1 mAb therapy alone was modest, with 6–19% of patients responding to therapy and with none of these patients responding persistently. Additionally, in many cancers refractory to PD-1 blocking therapy, it has been reported that a subset of these patients can experience hyperprogression of cancer14–16 from anti-PD-1 immunotherapy. The reason for this hyperprogression is not well understood, although it is noteworthy that Tregs are increased after PD-1 blockade in these patients14,17,18. Thus, while PD-1 expression on cytotoxic CD8+ T cells may be the primary target of immune checkpoint inhibition, it is becoming evident that PD-1 expressed by other immune cell subsets could contribute significantly to the effectiveness of checkpoint blockade19–22.
A major limitation to characterizing the function of PD-1 on non-CD8+ T cells has been the lack of tumor models with substantial expression of PD-1 on immune cells other than CD8+ tumor infiltrating lymphocytes. Although it has been demonstrated in various tumor settings that Tregs often express high levels of PD-1, until now a suitable model for studying checkpoint blockade in tumors highly infiltrated with PD-1+ Tregs was not feasible. Work from our group has shown that in a mouse model of claudin-low breast cancer the frequency of CD4+Foxp3+ Tregs expressing PD-1 was greater than the frequency of PD-1+ CD4+ Tconv and CD8+ T cells subsets23. As Tregs provide an important mechanism of immune suppression and evasion in cancer progression24, we used our previous model and two additional models to evaluate the hypothesis that PD-1+ Tregs could be enhancing immune suppression in the TME after PD-1 blockade and potential mechanisms for this finding.
Methods and Materials
Mice and cell lines
BALB/cJ, BALB/c Foxp3-GFP, BALB/c Thy1.1, and C57BL/6J (B6) females and were purchased from The Jackson Laboratory (Bar Harbor, ME). Female mice (8–14 weeks) were used for all experiments. T11 and T12 (claudin-low) tumor models have been described23,25. T12 cells were prepared by harvesting a T12 tumor from a tumor bearing mouse, followed by manual and chemical digestion for form a single cell suspension. E0771 cells were obtained from American Type Culture Collection (ATCC). All tumor cells lines found to be free of mycoplasma as determined by PCR testing. BALB/c mice were injected with 1 × 104 T11 (claudin-low) cells in PBS or 1×105 T12 (claudin-low) cells in Matrigel HC low-growth factor. B6 mice injected with 2.5×105 E0771 (luminal) cells in PBS. Tumors were orthotopically transplanted by intradermal injection into a mammary fat pad and measured twice per week using calipers. Tumor width × height was recorded, and mice were sacrificed at the specified tumor size or at the IACUC-approved end point of 2cm2.
Study Approval
All animal experiments were conducted in accordance with protocols approved by the University of North Carolina Institutional Animal Care and Use Committee (IACUC).
Isolation of murine tumor-infiltrating lymphocytes (TILs)
Murine tumors were resected and digested in Liberase TL (Roche #5401020001), DNase I (Sigma #D4527), Hyaluronidase (Sigma), and Collagenase XI (Sigma #C9697), as previously described26. Single cell suspensions were enriched for lymphocytes by isolating cells at the interface of a 44% Percoll (Sigma #P1644) in media and Lympholyte-M (Cedarlane #CL5031) gradient.
Antibodies and flow cytometry reagents
Flow cytometry monoclonal antibodies against murine CD45 (30-F11 #11-0451-82), Foxp3 (FJK-16S #45-5773-82), PD-1 (J43 #48-9981-82), Ki67 (SolA15 #17-5698-80), Thy1.1 (HIS51 #45-0900-80), CTLA-4 (UC10-4B9 # 12-1522-82), and GITR (DTA-1 #25-5874-82) were purchased from Invitrogen. Monoclonal antibodies against murine CD4 (GK1.5 #100414), CD8 (53-6.7 #100722), PD1 (RMP1-30 #109103), LAP-TGFβ (TW7-16B4 #141405), CD25 (PC61 #102051), and BrdU (Bu20a #339808) were purchased from BioLegend. Monoclonal antibodies against murine Bcl-2 (3F11 #556537) were purchased from BD Biosciences, and monoclonal antibodies against murine Bim (C34C5 #948055) were purchased from Cell Signaling Technology. Cell viability was determined using Aqua Fluorescence Reactive Dye (Life Technologies #L34965). For flow cytometry, cells were surface stained, fixed/permeabilized overnight using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience #00-5523-00), and intracellular staining performed the following day according to manufacturer’s instructions. Apoptosis was measured using PE Annexin V Apoptosis Detection Kit (BD Pharmingen #559763). Data were acquired using the BD FACSCanto or BD LSRFortessa (BD Biosciences, San Jose, CA). Acquired data were analyzed using FlowJo Flow Cytometry Analysis Software (FlowJo LLC, Ashland, OR).
Proliferation assays using BrdU incorporation
Tumor bearing BALB/c mice were injected with 2mg BrdU intraperitoneally in 200μl DPBS 24 hours before TIL isolation. Isolated TILs were stained using APC BrdU Flow Kit (BD Biosciences #51-9000019AK) adapting the manufacturer’s protocol. Briefly, cells were stained for surface antigens, then resuspended in BD Cytofix/Cytoperm buffer for 30 min on ice. Cells were washed with Perm/Wash and resuspended in BD Cytoperm Permeabilization Buffer Plus for 10 min on ice. Cells were then re-fixed/permeabilized overnight using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience #00-5523-00). Cells were then treated with 30μg DNase for 1 hour at 37°C. Cells were then stained for intracellular proteins including BrdU for 30 min at room temperature. Data were acquired using the BD FACSCanto (BD Biosciences, San Jose, CA). Acquired data were analyzed using FlowJo Flow Cytometry Analysis Software (FlowJo LLC, Ashland, OR).
In vivo antibodies
Monoclonal antibodies used for in vivo antibody inhibition were purchased from BioXCell (#BE0033-2). Mice undergoing immune checkpoint inhibition received intraperitoneal injection of 200μg anti-PD-1 (J43) or 200μg anti-PD-1 (J43) antigen binding fragments (Fabs) created using Pierce Fab Preparation Kit (ThermoFisher #44985) on day +7 post-tumor implantation when the tumor was palpable and then every 3–4 days throughout the experiment.
RNA-Seq
Foxp3+GFP+ Tregs isolated from tumors were sorted using a MoFlo XDP (Backman Coulter, Pasadena, CA) to greater than 90% purity. RNA was isolated from sorted Tregs using RNeasy Micro Kit (Qiagen, Germantown, MD). RNA-Seq libraries constructed with NuGEN Ovation SoLo (NuGEN Technologies, Redwood City, CA). Samples were sequenced using Illumina HiSeq 2500 Rapid Run (Illumina, San Diego, CA). Differential gene-expression analysis was performed using DESeq227. Ingenuity pathway analysis was performed in web portal (https://www.quiagenbioinformatics.com/products/ingenuity-pathway-analysis/).
Treg suppression and proliferation assays
For the Treg suppression assays we evaluated tumor infiltrating Tregs. Foxp3+GFP+ cells were sorted from tumors of T11 (claudin-low) bearing mice using a MoFlo XDP (Beckman Coulter, Pasadena, CA) or FACSAria II (BD Biosciences, San Jose, CA) cell sorter to greater than 90% purity. APCs were isolated from WT BALB/cJ splenocytes following CD90 microbead-depletion (Miltenyi #130-049-101) and irradiation at 30 Gy. Responder cells were isolated from BALB/c Thy1.1 mice using a T recovery column kit (Cedarlane #CL101). Isolated cells were then B220 and CD25 depleted using phycoerythrin (PE) conjugated antibodies and anti-PE magnetic bead sorting (Miltenyi #130-048-801). Responder cells were stained with the Cell Proliferation Dye eFluor 670 (eBioscience #65-0840) and plated at varying Treg:TEffector cell ratios with soluble α-CD3 (eBioscience #16-0031-85). Cells were co-cultured for 3 days, stained, and FACS analyzed.
For the assays measuring proliferation of Tregs ex vivo, we evaluated tumor infiltrating Tregs. Foxp3+GFP+ cells were sorted on a cell sorter similar to above to greater than 90% purity. The sorted Tregs were then stained with the Cell Proliferation Dye eFluor670 (eBioscience #65-0840) and plated with irradiated APCs and soluble α-CD3 with or without α-PD-1 Fabs in the cell culture. Fabs of PD-1 made from antibody clone J43 were used in vitro cultures to eliminate effects from Fc mediated activity of the antibodies. Cells were cultured for 3 days, stained, and FACS analyzed.
Treg apoptosis assays
For the assays measuring ex vivo Treg apoptosis, we evaluated tumor infiltrating Tregs. After isolation of TILs, the isolated lymphocytes were enriched for total T cells using a T recovery column kit (Cedarlane #CL101). T cells were then cultured with 10uM Dexamethasone (Sigma D4902) for 24 hours with or without α-PD-1 Fabs (BioXCell #BE0033-2) in the cell culture. Cells were then harvested, stained with PE Annexin V Apoptosis Detection Kit (BD Pharmingen #559763), and FACS analyzed.
Bcl-2 inhibition in vivo
Bcl-2 inhibition was accomplished using Venetoclax (ABT-199). ABT-199 was purchased from MedChemExpress (MedChemExpress Cat. No. HY-15531). ABT-199 was formulated in a mixture of 60% Phosal 50 PG (Fisher #NC0130871), 30% PEG 400 (Sigma #202398-5G), and 10% Ethanol (Fisher BP2818-500). Mice were dosed with ABT-199 or Vehicle alone in 0.2mL at 100mg/kg/day by oral gavage. Mice were treated starting at day 3 after tumor injection and daily for the duration of tumor growth.
MTT assay with ABT-199
T11 cells were plated in 96 well plate in complete media and incubated overnight. Venetoclax (ABT-199) was dissolved in DMSO, diluted in complete media, and added to the T11 cells at a starting concentration of 20μM. T11 cells with ABT-199 were incubated at 37°C, 5% CO2 for 48 hours. Cells were then harvested and cell death was determined using MTT Cell Growth Assay (Sigma CGD1) following manufacture’s protocols. ABT-199 dose response curve and IC50 was calculated using Prism (GraphPad, San Diego, CA).
Results
In our model of claudin-low breast cancer, a large number of Tregs infiltrating the tumor expressed PD-1. The level of PD-1 expression on Tregs was not uniform (Figure 1A) with the majority of PD-1+ Tregs expressing low levels of the protein while some Tregs expressed higher levels of PD-1 (Figure 1B). While fewer in proportion, the PD-1hi Treg population had a significant increase in suppressive molecules such as CTLA4 (p=0.05) and proteins critical to Treg function such as the high affinity IL-2 receptor alpha subunit, CD25 (p=0.028) (Figure 1C).
Figure 1: Infiltrating Tregs increase in the tumor after PD-1 blockade.
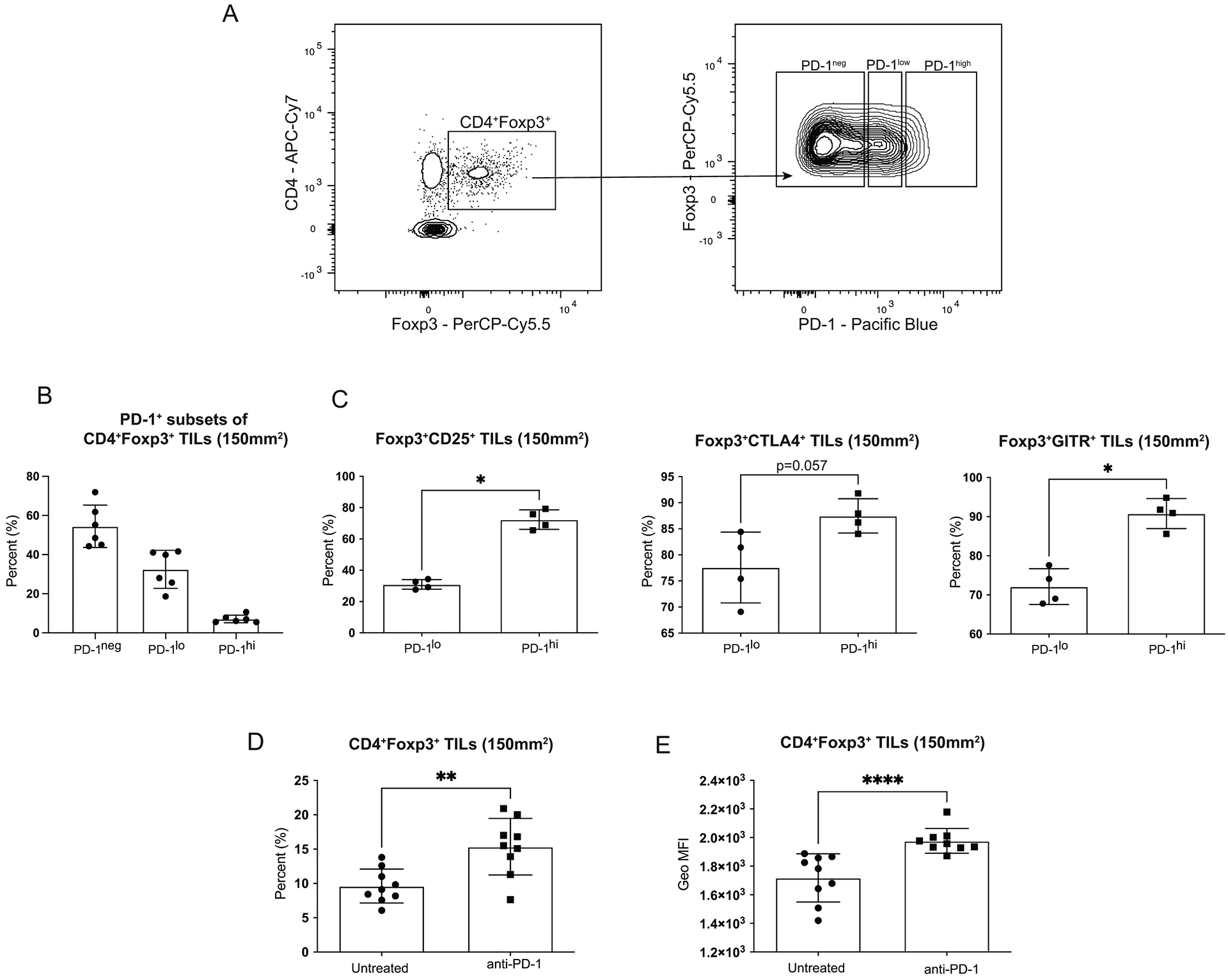
Mice were injected with 1 × 104 T11 (claudin-low) tumor cells. Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and analyzed by FACS. Cells were gated on Lymphocytes/ Single Cells/ Live/ CD3+/ CD4+Foxp3+ then analyzed for Treg markers. (A) Representative flow plots gated on CD4+Foxp3+ Tregs showing PD-1 expression levels. (B) Percent PD-1neg, PD-1lo, and PD-1hi CD4+Foxp3+ Tregs (n=6). (C) Percent CD4+Foxp3+ Tregs expressing CD25 or CTLA4 in PD-1lo versus PD-1hi populations (n=4). (D-E) Mice were untreated or treated with 200μg α-PD-1 antibody (J43) injected IP twice a week for the duration of the experiment. (D) Percent CD4+Foxp3+ Tregs from CD45+ gated population (n=9). (E) Geometric Mean Fluorescence Intensity of Foxp3 in CD4+Foxp3+ cells (n=9). Statistical significance determined by Mann-Whitney test. * denotes p < 0.05. ** denotes p < 0.01. **** denotes p < 0.0001.
While the functional differences between these PD-1+ Treg populations is unknown, it has been shown only intermediate PD-1-expressing CD8+ T cells can be rescued by PD-1 blockade, while PD-1-high T cells are committed to exhaustion28. Since we observed a low percentage of PD-1-high expressing cells, we assessed the outcome of PD-1 blockade on the PD-1+ Tregs infiltrating the claudin-low tumors. We compared CD4+Foxp3+ tumor infiltrating lymphocytes (TILs) from untreated mice to mice treated with α-PD-1 antibody and saw a significant increase in the frequency of Tregs in mice treated with PD-1 blockade (p=0.004) (Figure 1D). We also observed a significant increase in Foxp3 levels measured by the geometric mean fluorescence intensity (MFI) of Foxp3 in Tregs treated with PD-1 blockade (p<0.001) (Figure 1E). Higher Foxp3 levels has been directly associated with increased suppressive capabilities in Tregs29, thereby suggesting that Tregs treated with PD-1 blockade could lead to increased immunosuppression in the TME in claudin-low tumors.
To determine if there were transcriptional differences between Tregs isolated from untreated claudin-low tumors versus Tregs from tumors treated with PD-1 blockade, we sorted GFP+ Tregs from Foxp3GFP reporter mice and performed RNA-Seq. This demonstrated transcriptional changes in the Tregs from tumors treated with PD-1 blockade (Figure 2A). We found 27 significantly differentially regulated genes in Tregs isolated from mice treated with PD-1 blockade when compared to untreated controls (padj<0.05) (Table I). We used Ingenuity Pathway Analysis (IPA) to determine if any biological pathways were affected by PD-1 blockade in our RNA-Seq data. IPA predicted that the apoptosis pathway was inhibited when Tregs were treated with PD-1 blockade. In addition, Jun and Fos (p=0.001), genes responsible for T cell proliferation,30 were significantly upregulated in Tregs from tumors treated with PD-1 blockade (Figure 2B). Bcl-2, an anti-apoptotic protein, was also significantly upregulated (p=0.028) in Tregs isolated from tumors treated with PD-1 blockade (Figure 2B). Based on these data, we hypothesized that PD-1 blockade in claudin-low tumors was promoting a pro-survival phenotype in Tregs.
Figure 2: Treg transcriptional profile changes with PD-1 blockade compared to untreated.
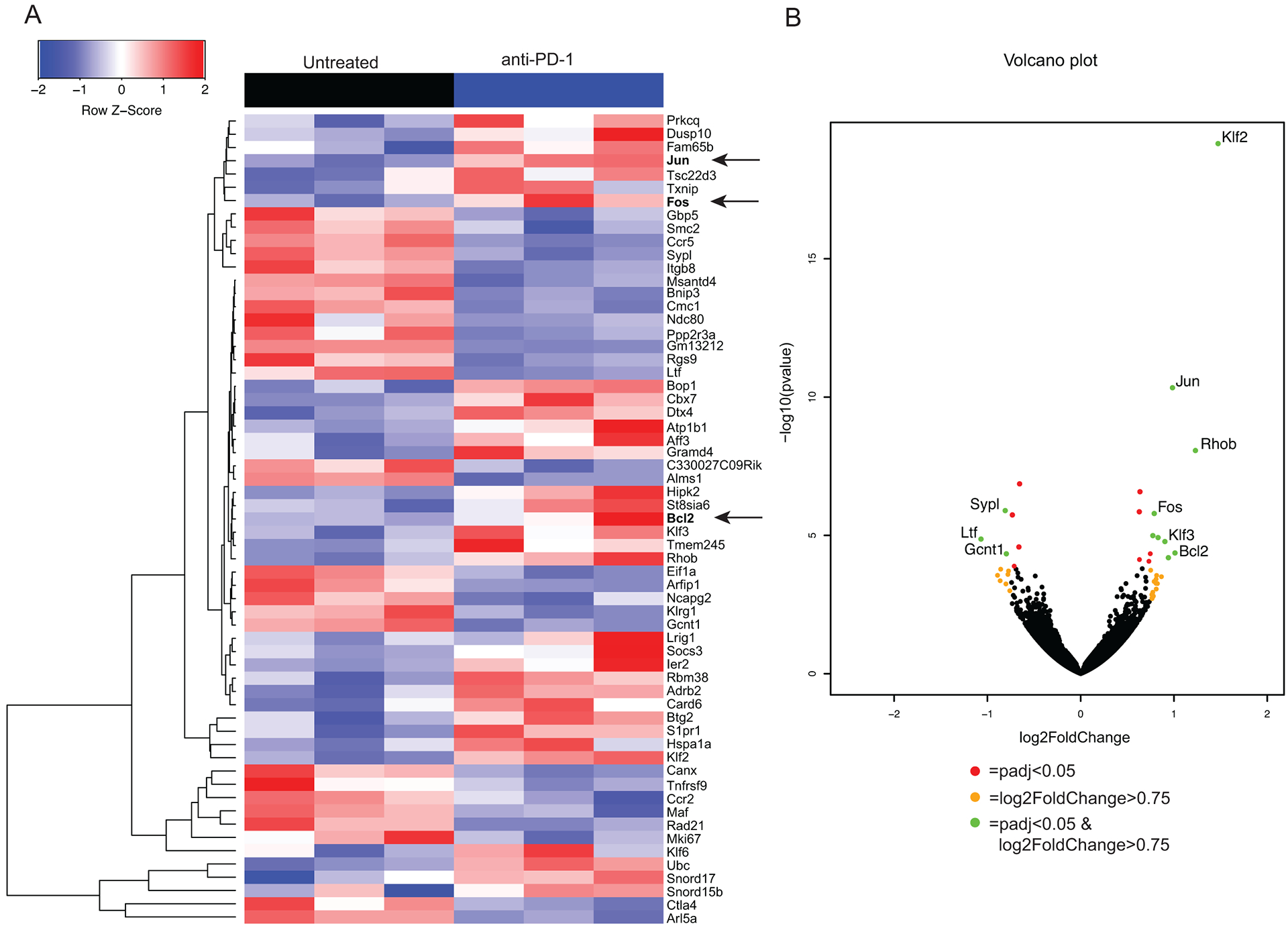
Foxp3-GFP mice were injected with 1 × 104 T11 (claudin-low) cells and were untreated or treated with 200 μg α-PD1 antibody (J43) injected IP twice a week. Tumors were harvested at 150 mm2, digested, enriched for lymphocytes, and GFP+ Tregs were sorted to greater than 90% purity using MoFlo XDP cell sorter. RNA was isolated from sorted cells and RNA-Seq was performed on the HiSeq 2500 Rapid Run platform. (n=6) (A) Samples were clustered using hierarchal clustering. Z-score of raw counts normalized among samples within each group. (B) Volcano plot showing significantly differentially regulated genes with a p adjusted value <0.05 and a log2 Fold Change >0.75.
Table I.
Genes significantly regulated in Tregs treated with PD-1 blockade versus untreated.i
| Upregulated | |||
|---|---|---|---|
| Gene | baseMean | Log2FoldChange | padj |
| Klf2 | 1351.561 | 1.473 | 5.55E-16 |
| Jun | 1034.796 | 0.984 | 1.86E-07 |
| Rhob | 300.810 | 1.230 | 2.31E-05 |
| Ubc | 5773.062 | 0.635 | 0.0004 |
| S1pr1 | 1512.735 | 0.629 | 0.0016 |
| Fos | 809.493 | 0.788 | 0.0016 |
| Ier2 | 646.815 | 0.774 | 0.0083 |
| Adrb2 | 328.347 | 0.830 | 0.0088 |
| Klf3 | 254.169 | 0.903 | 0.0104 |
| Atp1b1 | 72.096 | 1.011 | 0.0221 |
| Tsc22d3 | 942.382 | 0.746 | 0.0221 |
| Bcl2 | 203.146 | 0.940 | 0.0288 |
| Snord17 | 5426.186 | 0.630 | 0.0319 |
| Card6 | 481.202 | 0.731 | 0.0350 |
| Downregulated | |||
| Gene | baseMean | Log2FoldChange | padj |
| Arl5a | 5260.143 | −0.655 | 0.0003 |
| Sypl | 553.684 | −0.809 | 0.0016 |
| Ccr5 | 652.223 | −0.732 | 0.0016 |
| Ltf | 48.023 | −1.069 | 0.0092 |
| Itgb8 | 972.937 | −0.662 | 0.0152 |
| Gcnt1 | 300.171 | −0.797 | 0.0220 |
| Klrg1 | 370.393 | −0.712 | 0.0499 |
Foxp3-GFP mice were injected with 1 × 104 T11 (claudin-low) cells and were untreated or treated with 200μg α-PD1 antibody (J43) injected IP twice a week. Tumors were harvested at 150 mm2, digested, enriched for lymphocytes, and GFP+ Tregs were sorted to greater than 90% purity using MoFlo XDP cell sorter. RNA was isolated from sorted cells and RNA-Seq was performed on the HiSeq 2500 Rapid Run platform. (n=6) Differential gene-expression analysis was performed using DESeq2. Genes listed are significantly upregulated or downregulated with an adjusted p value < 0.5.
To test this hypothesis, we first evaluated the proliferative potential of Tregs in vitro. Tregs cultured with α-PD-1 Fabs proliferated significantly more than Tregs without α-PD-1 in the culture (p<0.0001) (Figure 3A–B). To confirm that the significant proliferation of Tregs resulted from activation through CD3/CD28 engagement rather than an artifact of the α-PD-1 Fabs, Tregs were cultured with α-PD-1 Fabs alone without α-CD3. PD-1 blockade alone did not lead to Treg proliferation (Supplementary Figure S1), suggesting that the increase in proliferation is due to the release of the inhibitory signal from PD-1, thus allowing the Tregs to proliferate. We next investigated if the increase in Treg proliferation was also present in vivo in the TME. To address this question, we evaluated cellular proliferation by BrdU incorporation. When immune cells were isolated early during tumor growth (tumor size of 50mm2), the Tregs proliferated significantly more than CD8+ or CD4+Foxp3− T cells (p=0.029) (Figure 3C). We could not detect a difference in proliferation between Tregs from mice treated with α-PD-1 versus untreated (data not shown) on day 15 post tumor injection (50mm2). We then evaluated proliferation at day 23 after tumor injection, by measuring expression of proliferation marker Ki-6731. We saw a non-significant increase in the frequency and total number of proliferating Tregs from mice treated with α-PD-1 Fabs compared to untreated mice (Figure 3D–E). We have previously published in this model of claudin-low breast cancer that Tregs infiltrating into the tumors have significantly higher levels of PD-1 expression than CD8+ T cells23. We predicted that PD-1 blockade would have a reduced impact on CD8+ T cells that it does on Tregs because of the reduced PD-1 expression. Indeed, PD-1 blockade did not increase the frequency of CD8+Ki67+ T cells compared to untreated mice (Figure 3F). Tregs not only have increased proliferation when exposed to α-PD-1, but in our model of claudin-low breast cancer, the Tregs proliferate at a higher rate than other T cell subsets (Figure 3C) suggesting an increased potential for Treg-mediated suppression in the TME.
Figure 3: PD-1 blockade increases Treg proliferation.
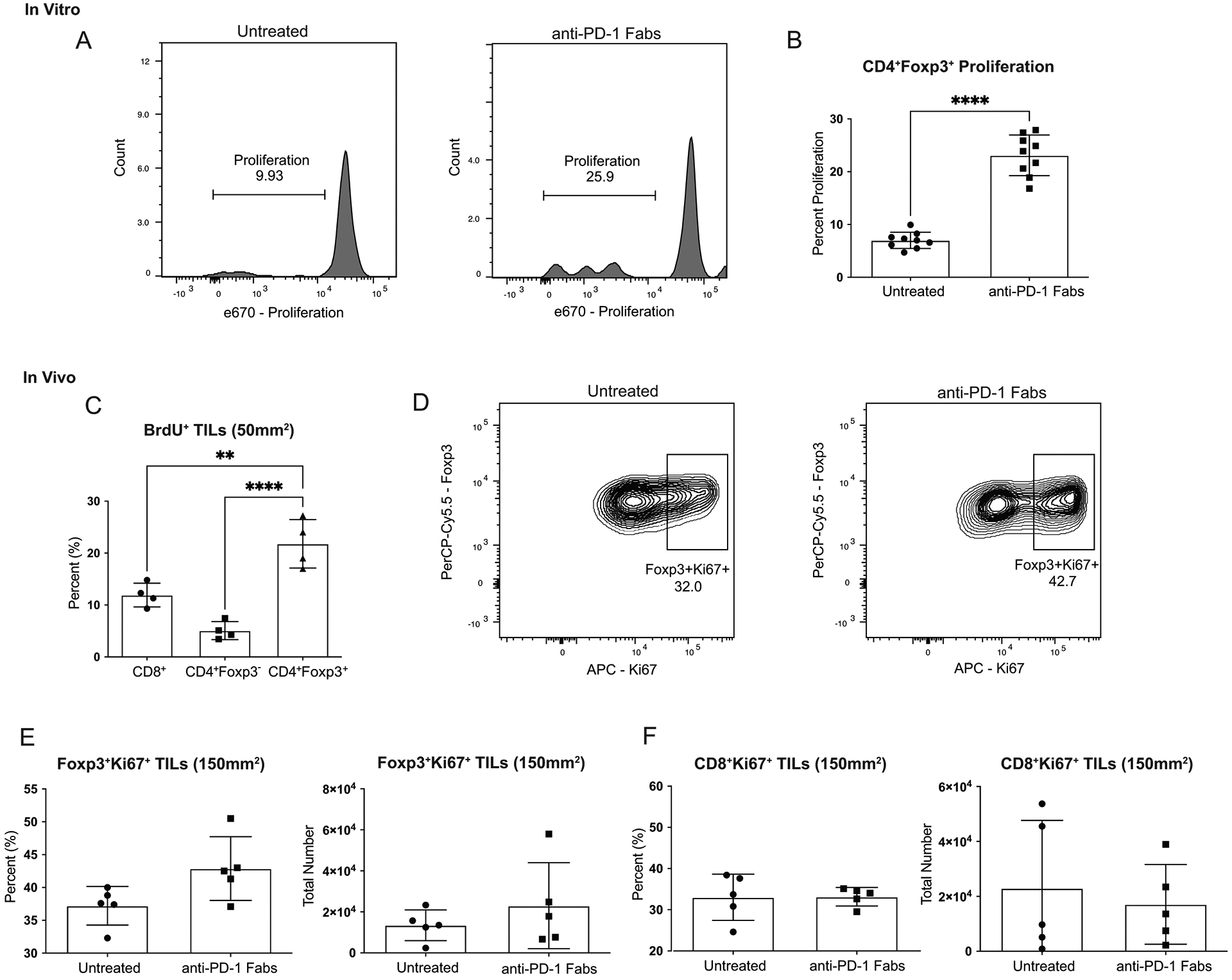
Mice were injected with 1 × 104 T11 (claudin-low) tumor cells. (A-B) Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and GFP+ Tregs were sorted using MoFlo-XDP cell sorter. Tregs stained with proliferation dye were incubated with or without α-PD-1 Fabs, irradiated APCs, and soluble α-CD3 in culture for 72 hours. Cells were gated on Lymphocytes/ Single Cells/ Live/ Thy1.1-/ Foxp3+ and then analyzed for proliferation using e670 proliferation dye. (A) Representative flow plots gated on proliferation of Tregs cultured without or with α-PD-1 Fabs. (n=9) (B) Percent proliferating CD4+Foxp3+ Tregs from in vitro culture. (C) Mice were injected with α-PD-1 Ab and 2mg BrdU. Tumors were harvested at 50mm2, digested, enriched for lymphocytes, and measured for BrdU incorporation by flow cytometry (n=4). (D-F) Tumors were harvested at 150mm2, enriched for lymphocytes, and Ki67 expression in CD4+Foxp3+ Tregs or CD8+ T cells analyzed by FACS (n=5). Cells were gated on Lymphocytes/ Single Cells/ Live/ CD45+/ CD4+Foxp3+ or CD8+ where indicated. Statistical significance determined by Mann-Whitney test. * denotes p < 0.05. **** denotes p < 0.0001.
From our RNA-Seq data, we found that Bcl-2 was significantly upregulated in Tregs during treatment with α-PD-1 (Figure 2B). As an anti-apoptotic protein, Bcl-2 expression can protect cells from apoptosis induced by various stimuli32. To validate our RNA-Seq data, we confirmed that Bcl-2 protein was upregulated in Tregs isolated from tumors treated with α-PD-1. Mice were injected with claudin-low tumors, treated with α-PD-1 or left untreated, and then tumors were harvested at 150mm2 to analyze protein expression by flow cytometry. The frequency of Bcl-2+CD4+Foxp3+ cells was approximately 8 times higher in Tregs from α-PD-1 treated mice when compared to untreated (Figure 4A–B). Mice treated with α-PD-1 also had a significant increase in the levels of Bcl-2 in Tregs when compared to Tregs from untreated mice (p=0.018) (Figure 4D). We also measured the pro-apoptotic protein Bim and saw no difference in Bim frequency or expression levels between the two treatment groups of Tregs (Figure 4C and E). Bcl-2/Bim ratios are often used as a measure for survival potential in cells. Tregs exposed to α-PD-1 had significantly higher Bcl-2/Bim ratios than untreated Tregs (p=0.028) (Figure 4F), suggesting a potential for increased protection of Tregs from apoptosis in the TME. Thus, both the increased frequency of Bcl-2+ Tregs and the increased expression of Bcl-2 in Tregs could enhance resistance of Tregs to apoptosis upon treatment with α-PD-1 mAb therapy.
Figure 4: Tregs exposed to PD-1 blockade have increased Bcl-2 expression.
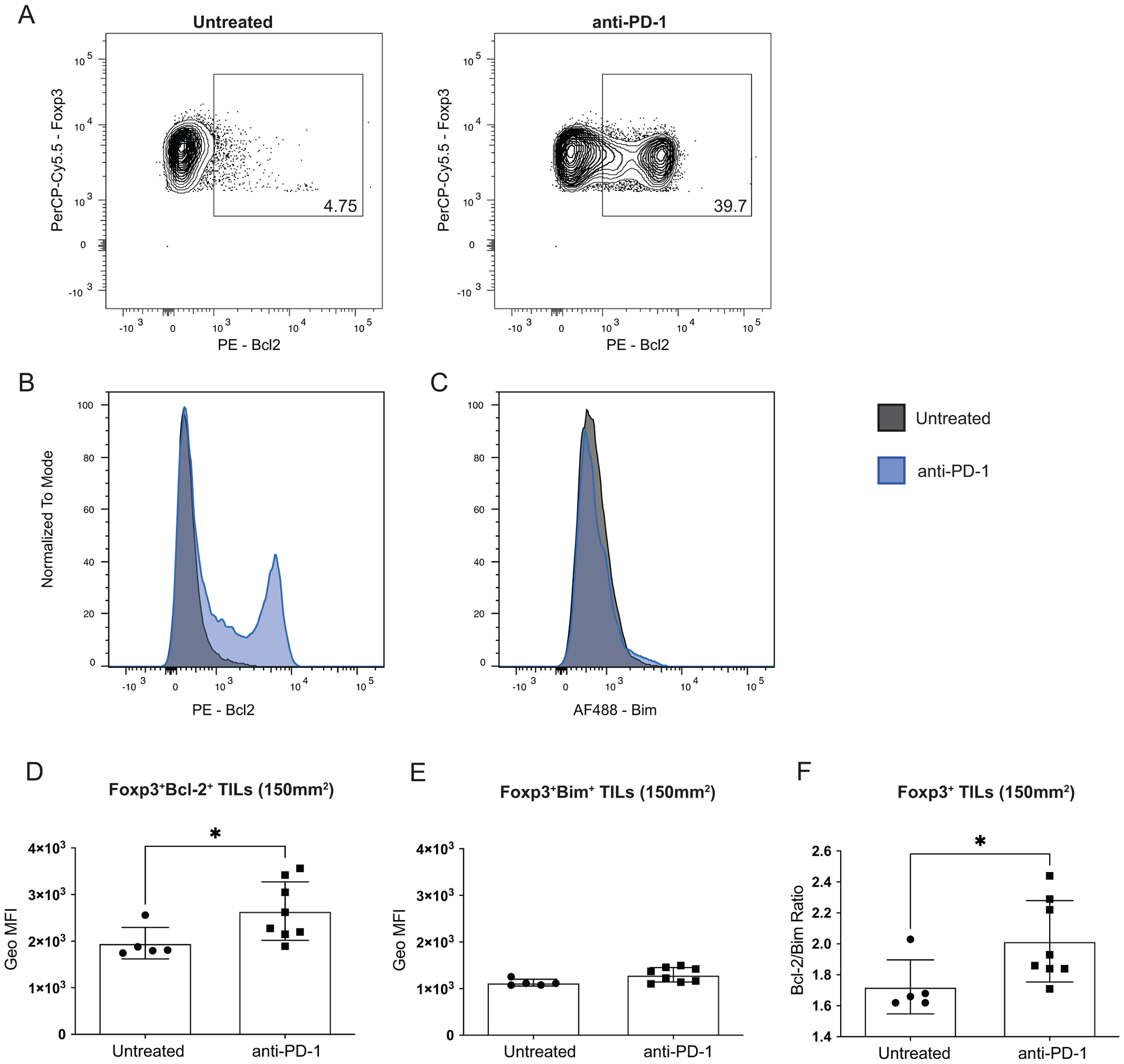
BALB/c mice were injected with 1 × 104 T11 (claudin-low) tumor cells. Mice were untreated or treated with 200 μg α-PD-1 antibody (J43) injected IP twice a week for the duration of the experiment. Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and analyzed by FACS. Cells were gated on Lymphocytes /Single Cells /Live /CD45+ /Foxp3+ from tumors of mice without or with α-PD-1 treatment. (A) Representative flow plots showing frequency of Bcl2+ cells of CD4+Foxp3+ (n=9). (B-C) Histogram overlays of Bcl2 and Bim expression in CD4+Foxp3+ (n=9). (D) Geometric Mean Fluorescence Intensity (MFI) of Bcl-2 in CD4+Foxp3+ cells in untreated compared to mice treated with α-PD-1 (n=5 Untreated n=8 α-PD-1). (E) MFI of Bim in CD4+Foxp3+ cells in untreated compared to mice treated with α-PD-1 (n=5 Untreated n=8 α-PD-1). (F) Ratio of Bcl-2 to Bim MFIs from (D-E) in CD4+Foxp3+ cells (n=5 Untreated n=8 α-PD-1). Statistical significance determined by Mann-Whitney test. * denotes p < 0.05.
Because we found a significant increase in levels of Bcl-2 in the Tregs from T11 (claudin-low) tumors (Figure 4A), we sought to test if these Tregs were protected from apoptosis ex vivo. Anti-apoptotic Bcl-2 expression has been shown to inhibit glucocorticoid (GC)-induced apoptosis, so we tested if Bcl-2 expressed in Tregs could protect them from Dexamethasone (Dex)-induced apoptosis. Tumor infiltrating T cells were isolated from control Foxp3-GFP T11 (claudin-low) tumor bearing mice as well as from mice treated with α-PD-1 twice weekly and then cultured ex-vivo with or without Dex. Apoptosis in Tregs was assessed using Annexin V/7-AAD staining. There was greater protection from apoptosis in Tregs from mice treated with α-PD-1 and cultured in Dex than Tregs from untreated mice (p < 0.0001) (Figure 5A–B). Interestingly, we did not see this significant decrease in cell death in CD8+ T cells from mice treated with α-PD-1 (Fig 5C), suggesting that this protection from apoptosis may be specific to Tregs in the TME. We confirmed our findings in an additional model of claudin-low breast cancer (T12). There was a decrease in Tregs undergoing apoptosis when treated with PD-1 blockade and this decrease was sustained with the addition of Dex (Supplementary Figure S2A). To determine if this protection from apoptosis could be attributed to Bcl-2, we added Venetoclax (ABT-199), a potent and selective Bcl-2 inhibitor. When Bcl-2 was inhibited in vitro, there was no longer a reduction in Tregs undergoing apoptosis with PD-1 blockade (Supplementary Figure S2A). To determine if this protection from apoptosis after PD-1 blockade was specific to claudin-low breast cancer, we employed a model of luminal breast cancer (E0771). We determined that unlike the T11 and T12 claudin-low models of breast cancer where there is a greater frequency of Tregs than CD8+ T cells expressing PD-1, the E0771 model of breast cancer had a higher frequency of CD8+ T cells that were PD-1+ (Supplementary Figure S2B). When we assessed apoptosis in Tregs in mice with E0771 tumors, these cells were not protected from apoptosis induced by Dex (Supplementary Figure S2C–D). Interestingly, CD8+ T cells from mice treated with PD-1 blockade were protected from apoptosis in the E0771 luminal breast cancer model (Supplementary Figure S2E). Thus, protection from apoptosis was directly correlated with the difference in the expression of PD-1 by Tregs and CD8+ T cells.
Figure 5: Tregs are protected from apoptosis after PD-1 blockade.
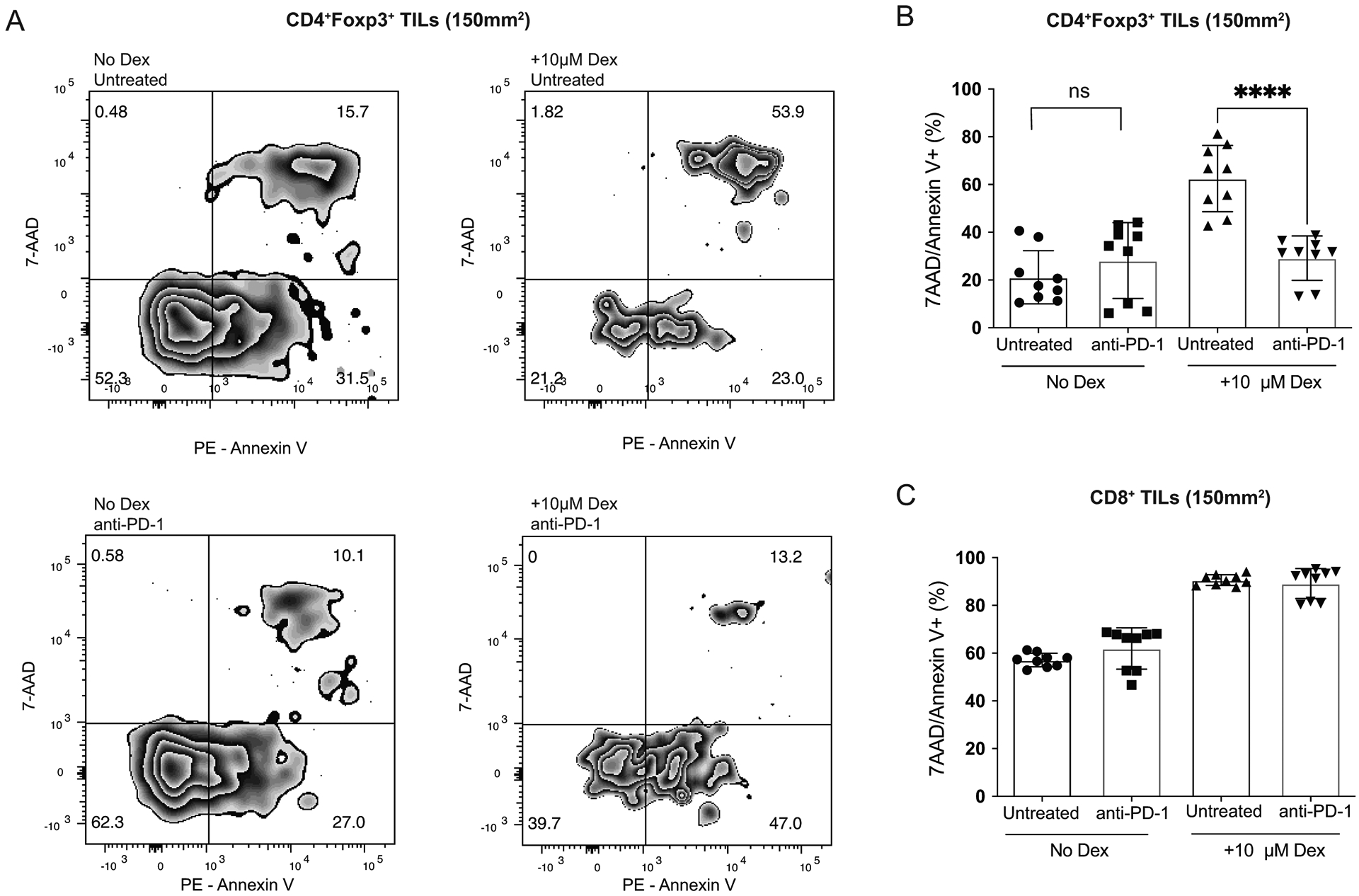
BALB/c Foxp3-GFP mice were injected with 1 × 104 T11 (claudin-low) tumor cells. Mice were untreated or treated with 200 μg α-PD-1 antibody (J43) injected IP twice a week for the duration of the experiment. Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and total T cells were isolated using cell isolation column (n=9). Isolated total T cells were cultured in 96 well plate in complete media or complete media + 10μM Dexamethasone. Apoptosis was measured using Annexin V and 7-AAD staining. (A) Representative flow plots gated on GFP+ Tregs isolated from the tumor of mice either untreated or treated with α-PD-1 cultured with or without Dex. (B) Percent CD4+Foxp3+7-AAD/Annexin V+ Tregs from CD45+ parent population. (C) Percent CD8+/7-AAD/Annexin V+ T cells from CD45+ parent population. Statistical significance determined by Mann-Whitney test. **** denotes p < 0.0001.
We also treated mice with a Bcl-2 inhibitor to determine if there would be increased apoptosis in Tregs. Mice treated with Bcl-2 inhibitor ABT-199 had delayed tumor growth and increased survival irrespective of α-PD-1 treatment (Supplementary Figure S3A–B). While it is possible that ABT-199 had a direct effect on the T11 tumor cells themselves, the EC50 against T11 cells in vitro was 2 μM (Supplementary Figure S3D) while the IC50 of ABT-199 on Bcl-2 expressing hematopoietic cells is 4nM33, suggesting that in our system ABT-199 does not have potent activity against T11 (claudin-low) tumor cells and is likely acting by inhibiting Treg function. However, ABT-199 therapy did not enhance the efficacy of anti-PD-1 mAb in this model. While the total number of Tregs infiltrating into the tumor after treatment with ABT-199 was similar, the number of CD8+ T cells was significantly decreased (Supplementary Figure S3C), indicating that the effect of Bcl-2 inhibition on the presence of T cells in the TME is non-specific, and may contribute to the lack of synergy using ABT-199 with checkpoint inhibitors.
Higher levels of Foxp3 expression have been associated with increased suppressive capabilities in Tregs29. Elevated levels of Foxp3 in Tregs treated with PD-1 blockade described earlier (Figure 1E) prompted us to examine whether Tregs treated with PD-1 blockade had increased suppressive capabilities. We interrogated several pathways that could be used by Tregs to suppress anti-tumor immune responses in the TME from T11 (claudin-low) bearing mice with or without PD-1 blockade. Expression of the inhibitory receptor CTLA-4, the high affinity Il-2 receptor chain CD25, secretion of the suppressive cytokine TGF-β, and expression of Glucocorticoid induced TNF receptor (GITR) are well characterized either as mechanisms of suppression utilized by Tregs34,35 or characteristic of Treg function (CD25) in limiting the availability of IL-2. All of these are known to contribute to their suppressive capabilities36. After PD-1 blockade, there was an increase in the mean frequency of Tregs expressing suppressive markers CTLA-4, GITR, and TGF-β (p=0.09, p=0.05, p=0.07, respectively) with only the difference in GITR meeting the pre-defined definition of statistical significance for these studies (Figure 6A). There were no significant differences in the level of expression of these molecules in Tregs as determined by the MFI (Figure 6B). We wanted to confirm our findings in an additional model of claudin-low breast cancer (T12) that we have previously demonstrated to be enriched in Tregs and refractory to PD-1 blockade therapy23. After PD-1 blockade in the T12 (claudin-low) model, there was an increase in the mean frequency of Tregs expressing GITR and TGF-β (Supplementary Figure S4A). There was no difference in CD25 expression on Tregs from mice treated with PD-1 blockade, but the MFI of CD25 was increased on Tregs after PD-1 blockade (Supplementary Figure S4B). Based on these findings, we then sought to test if Tregs exposed to PD-1 blockade had increased suppressive capabilities. To address this, Foxp3-GFP T11 (claudin-low) tumor bearing mice were treated with α-PD-1 or left untreated and the tumors were harvested around 150mm2 for isolation of TILs. Tregs that had been exposed in the TME to PD-1 blockade were significantly better at suppressing naïve CD8+ T cell proliferation in an ex vivo setting than Tregs from mice that were untreated (Figure 6C). These differences in suppression were significant at a 2:1 (p=0.005) and a 1:1 (p=0.02) ratio of Tregs to CD8+ T cells (Figure 6C). Based on our previous work, the ratio of Tregs to CD8+ T cells in the T11 (claudin-low) TME is approximately 1.5:123; thus, the suppressive effect observed in our experiment is biologically relevant to Treg-dependent inhibition of conventional T cell activation at the ratios used in vitro.
Figure 6: PD-1 blockade results in increased suppressive capabilities in Tregs.
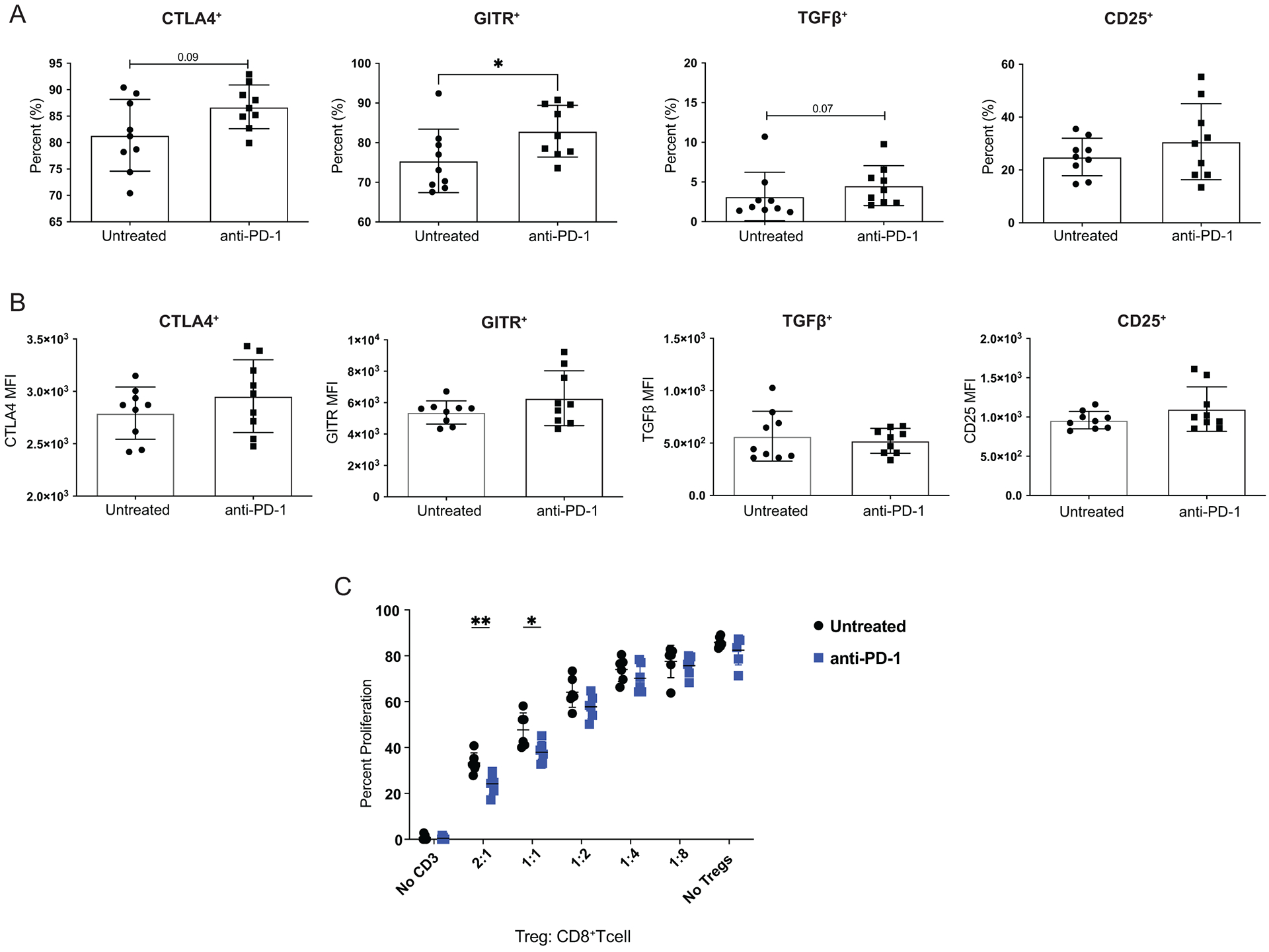
Mice were injected with 1 × 104 T11 (claudin-low) tumor cells. Mice were untreated or treated with 200 μg α-PD-1 antibody (J43) injected IP twice a week for the duration of the experiment. (A-B) Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and analyzed by FACS. Cells were gated on Lymphocytes/ Single Cells/ Live/ CD3+/ CD4+Foxp3+ then analyzed for Treg markers. (A) Percent CD4+Foxp3+ Tregs expressing suppressive molecules; CTLA4, GITR, TGF-β, and CD25 from mice treated with α-PD-1 versus untreated (n=9). (B) Geometric Mean Fluorescence Intensity of suppressive molecules in CD4+Foxp3+ cells (n=9). Statistical significance determined by Mann-Whitney test. (C) Tumors were harvested at 150mm2, digested, enriched for lymphocytes, and GFP+ Tregs were sorted using MoFlo-XDP cell sorter. Naive T cells were stained with proliferation dye and were incubated with sorted Tregs, irradiated APCs, and soluble α-CD3 in culture for 72 hours. Statistical significance determined by multiple t-tests. * denotes p < 0.05. ** denotes p < 0.01.
Discussion
Triple negative breast cancer (TNBC) has the worst prognosis of the breast cancer subtypes despite being heavily immune infiltrated37. The standard dogma in cancer immunotherapy is that tumors with immune infiltration have the capacity to mount a productive anti-tumor immune response and are therefore good candidates for immune checkpoint blockade. However, PD-1 is not only expressed on CD8+ cytotoxic T lymphocytes but also on different populations of CD4+ T and NK cells. Here, we show in a murine model that faithfully reproduces tumors found in patients with claudin-low breast cancer that PD-1 is most frequently expressed on Foxp3+ Tregs. Blockade of PD-1 was associated with enhanced suppression, increased proliferation and diminished apoptosis of Tregs in vitro, which was also reproduced in the TME. These data suggest that the activity of checkpoint inhibitors is more complicated than currently evaluated. The presence of a substantial immune infiltrate may not predict a response to immune checkpoint therapy if a significant number of the immune cells that express PD-1 are Tregs, which behave differently than conventional T cells upon checkpoint inhibition.
The mechanism(s) for the enhanced function of Tregs in the presence of α-PD-1 mAb therapy is not currently clear. Our data indicate that α-PD-1 therapy affects at least three different pathways for Treg activity. First, we found increased proliferation of Tregs in the presence of α-PD-1 mAb therapy. This is consistent with findings evaluating the effects of α-PD-1 mAb on the proliferation of CD8+ T cells11, and could be related to the increased expression of Jun and Fos in Tregs from α-PD-1 treated animals. The second pathway is increased resistance to apoptosis. Previous work has demonstrated a critical role for the expression of Bcl family member proteins and decreased expression of Bim in the maintenance of Tregs38. We found that α-PD-1 therapy enhanced Bcl-2 expression and diminished glucocorticoid-induced apoptosis in Tregs. Interestingly, we found that the Bcl-2 inhibitor ABT-199 could improve the median time for tumor growth in mice receiving T11 tumors, which was independent of co-administration with anti-PD-1 therapy. Given the extremely modest activity of ABT-199 in vitro against T11 tumor cells, these data suggest that inhibition of Bcl-2 in T11 tumors may also be due to diminished function of Tregs. Finally, Tregs exposed to α-PD-1 therapy had enhanced suppressive function, which correlates with the increased expression of Foxp3 by those cells.
There are currently multiple ongoing clinical trials in TNBC where Pembrolizumab (humanized α-PD-1 antibody) is being given as a monotherapy39. In all reported trials to date the overall response rate to PD-1 inhibition in TNBC is reported to be between 4–20%, with only a small fraction of patients seeing any benefit from therapy40. Our previous work has suggested that immune infiltration alone is not a reliable biomarker to predict overall response rate to immune checkpoint therapy, but instead the complete microenvironment including immunosuppression in the TME should be considered23. While the expected outcome of PD-1 therapy is that the inhibitory signal on cytotoxic T cells will be blocked, thereby allowing them to remain functional and lead to tumor killing, it is unknown if PD-1 blockade functions similarly on other immune cell subsets that express PD-1. It has been hypothesized that therapeutic benefit from immune checkpoint blockade could be masked due to enhanced immunosuppression in the TME, leading to hyperprogression of cancer14–16. Our study supports this hypothesis by demonstrating that PD-1 blockade promoted a pro-survival phenotype and enhanced suppression from PD-1+ regulatory T cells in the TME.
Most of the previous studies looking at the role of PD-1 on Tregs have been in vitro studies from peripheral Tregs. These studies broadly demonstrate that Tregs cultured in vitro with PD-1 blocking antibody enhance proliferation of Tregs14,17,41,42, although these studies are limited by the fact that Treg function and proliferation were measured from peripheral Tregs rather than tissue-infiltrating Tregs. Our study is novel in that we directly measure the proliferative capacity and suppressive function of tumor infiltrating Tregs treated with PD-1 blockade in vivo.
It was somewhat unexpected that the number of significantly expressed genes on Tregs in mice treated with/without anti-PD-1 mAb was quite modest. The evaluation of persistent expression of PD-1 on T cell exhaustion in vitro is difficult, and as a consequence, we chose to perform our screen using Tregs isolated from mice after in vivo treatment with anti-PD-1 mAb or control. One limitation to this approach was the performance of bulk RNA-Seq on Tregs sorted from tumors, only 50% of which express PD-1 (Figure 1B). Inclusion of PD-1 negative Tregs in our gene expression data may have minimized any changes to transcript regulation of Tregs from anti-PD-1 therapy. Furthermore, we could not assume that all PD-1-expressing Tregs would be exposed to saturating amounts of the antibody. Additional factors that might limit changes in gene expression could be due to the timing of the administration of the antibody in relationship to the time of the RNA-seq evaluation. Nonetheless, we confirmed our findings by measuring protein expression of the relevant genes, thus allowing us to evaluate pathways that could mediate changes in Treg function in the presence of anti-PD-1 mAb therapy.
In summary, we have shown in claudin-low tumors that Tregs express significant levels of PD-1. Blockade of PD-1 on these cells by α-PD-1 therapy leads to enhanced Treg proliferation, suppressive function, and resistance to apoptosis. The increased proliferation we observe is accompanied by increased expression of Jun and Fos, while the resistance to apoptosis is associated with increased expression of Bcl-2. These studies suggest that the activity and toxicity of checkpoint inhibitor therapy may be correlated with differences in expression of PD-1 on CD8+ versus Treg cells. We demonstrate in this study a model of breast cancer refractory to checkpoint inhibition that can be used to determine mechanistically how PD-1high Tregs in the TME alter outcomes to immunotherapy. This hypothesis should be tested clinically and specifically evaluated in the treatment of patients with triple negative breast cancer, especially those of the claudin-low/mesenchymal subtype.
Supplementary Material
Key Points.
Anti-PD-1 promotes the function of Tregs in a claudin-low model of breast cancer
PD-1 blockade increases proliferation, survival, and suppressive function of Tregs
Acknowledgments
The authors would like to acknowledge the UNC Flow Cytometry Core Facility for their assistance in sorting and the high throughput sequencing facility (HTSF) who sequenced samples.
Financial Support
This work is supported by UNC Basic Immune Mechanisms (T32AI007273-33) Breast Cancer SPORE (P50 CA058223, CMP and JSS) and an ROI Grant from the State of North Carolina. The UNC Flow Cytometry Core Facility and HTSF are supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. Research reported in this publication was supported in part by the National Cancer Institute (P50 CA058223), the North Carolina Biotech Center Institutional Support Grant 2012-IDG-1006 and 2005-IDG-1016, and the University Cancer Research Fund. This work as also supported by R01 HL139730.
References
- 1.American Cancer Society. Cancer Facts & Figures, <https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf> (2019).
- 2.SusanG Komen. Breast Cancer Fact Sheet, <https://ww5.komen.org/uploadedFiles/_Komen/Content/About_Breast_Cancer/Facts_and_Statistics/Breast_Cancer_in_Women/BCFactSheetOct2017.pdf> (2019, July).
- 3.Bianchini G, Balko JM, Mayer IA, Sanders ME & Gianni L. 2016. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 13: 674–690, doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purrington KS, Visscher DW, Wang C, Yannoukakos D, Hamann U, Nevanlinna H, Cox A, Giles GG, Eckel-Passow JE, Lakis S, Kotoula V, Fountzilas G, Kabisch M, Rudiger T, Heikkila P, Blomqvist C, Cross SS, Southey MC, Olson JE, Gilbert J, Deming-Halverson S, Kosma VM, Clarke C, Scott R, Jones JL, Zheng W, Mannermaa A, Jane Carpenter for AI, Eccles DM, Vachon CM & Couch FJ. 2016. Genes associated with histopathologic features of triple negative breast tumors predict molecular subtypes. Breast Cancer Res Treat. 157: 117–131, doi: 10.1007/s10549-016-3775-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prat A, Adamo B, Cheang MC, Anders CK, Carey LA & Perou CM. 2013. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 18: 123–133, doi: 10.1634/theoncologist.2012-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM & Vincent BG. 2016. Genomic Analysis of Immune Cell Infiltrates Across 11 Tumor Types. J Natl Cancer Inst. 108, doi: 10.1093/jnci/djw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M & Davis MM. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 5: 677–685, doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 8.Pardoll DM 2012. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 12: 252–264, doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ 2011. T cell exhaustion. Nat Immunol. 12: 492–499. [DOI] [PubMed] [Google Scholar]
- 10.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R & Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 77: 4911–4927, doi: 10.1128/jvi.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ & Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 439: 682–687, doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 12.Iglesia MD, Vincent BG, Parker JS, Hoadley KA, Carey LA, Perou CM & Serody JS. 2014. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 20: 3818–3829, doi: 10.1158/1078-0432.CCR-13-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA & Investigators IMT. 2018. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 379: 2108–2121, doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 14.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, Morikawa H, Kawazoe A, Kinoshita T, Shitara K, Sakaguchi S & Nishikawa H. 2019. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 116: 9999–10008, doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, Mazieres J, Zalcman G, Brosseau S, Le Moulec S, Leroy L, Duchemann B, Lefebvre C, Veillon R, Westeel V, Koscielny S, Champiat S, Ferte C, Planchard D, Remon J, Boucher ME, Gazzah A, Adam J, Bria E, Tortora G, Soria JC, Besse B & Caramella C. 2018. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 4: 1543–1552, doi: 10.1001/jamaoncol.2018.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim CG, Kim KH, Pyo KH, Xin CF, Hong MH, Ahn BC, Kim Y, Choi SJ, Yoon HI, Lee JG, Lee CY, Park SY, Park SH, Cho BC, Shim HS, Shin EC & Kim HR. 2019. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann Oncol. 30: 1104–1113, doi: 10.1093/annonc/mdz123. [DOI] [PubMed] [Google Scholar]
- 17.Woods DM, Ramakrishnan R, Laino AS, Berglund A, Walton K, Betts BC & Weber JS. 2018. Decreased Suppression and Increased Phosphorylated STAT3 in Regulatory T Cells are Associated with Benefit from Adjuvant PD-1 Blockade in Resected Metastatic Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 24: 6236–6247, doi: 10.1158/1078-0432.CCR-18-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodagatta-Marri E, Meyer DS, Reeves MQ, Paniagua R, To MD, Binnewies M, Broz ML, Mori H, Wu D, Adoumie M, Del Rosario R, Li O, Buchmann T, Liang B, Malato J, Arce Vargus F, Sheppard D, Hann BC, Mirza A, Quezada SA, Rosenblum MD, Krummel MF, Balmain A & Akhurst RJ. 2019. alpha-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by alpha-TGFbeta antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer. 7: 62, doi: 10.1186/s40425-018-0493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW, Zhang L, Iannello A, Mathur N, Jardine KE, Kirn GA, Bell JC, McBurney MW, Raulet DH & Ardolino M. 2018. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. 128: 4654–4668, doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quatrini L, Wieduwild E, Escaliere B, Filtjens J, Chasson L, Laprie C, Vivier E & Ugolini S. 2018. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat Immunol. 19: 954–962, doi: 10.1038/s41590-018-0185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, Brandt C, Kane L, Campos LS, Lu L, Belz GT, McKenzie AN, Teichmann SA, Dougan G & Liu P. 2016. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature. 539: 102–106, doi: 10.1038/nature20105. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Hou S, Fang Q, Liu X, Liu X & Qi H. 2018. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity. 49: 264–274 e264, doi: 10.1016/j.immuni.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, Reisdorf S, Anders CK, Carey LA, Parker JS, Perou CM, Vincent BG & Serody JS. 2017. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. 127: 3472–3483, doi: 10.1172/JCI90499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallimore A, Quezada SA & Roychoudhuri R. 2019. Regulatory T cells in cancer: where are we now? Immunology. 157: 187–189, doi: 10.1111/imm.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Usary J, Darr DB, Pfefferle AD & Perou CM. 2016. Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes. Curr Protoc Pharmacol. 72: 14 38 11–11, doi: 10.1002/0471141755.ph1438s72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgents JE, Moran TP, West ML, Davis NL, Johnston RE & Serody JS. 2010. The immunosuppressive tumor environment is the major impediment to successful therapeutic vaccination in Neu transgenic mice. J Immunother. 33: 482–491, doi: 10.1097/CJI.0b013e3181d756bb. [DOI] [PubMed] [Google Scholar]
- 27.Love MI, Huber W & Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550, doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackburn SD, Shin H, Freeman GJ & Wherry EJ. 2008. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 105: 15016–15021, doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chauhan SK, Saban DR, Lee HK & Dana R. 2009. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 182: 148–153, doi: 10.4049/jimmunol.182.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huen NY, Pang AL, Tucker JA, Lee TL, Vergati M, Jochems C, Intrivici C, Cereda V, Chan WY, Rennert OM, Madan RA, Gulley JL, Schlom J & Tsang KY. 2013. Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. Int J Cancer. 133: 373–382, doi: 10.1002/ijc.28026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerdes J, Schwab U, Lemke H & Stein H. 1983. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 31: 13–20, doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- 32.Vaux DL, Cory S & Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 335: 440–442, doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 33.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH & Elmore SW. 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 19: 202–208, doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt A, Oberle N & Krammer PH. 2012. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 3: 51, doi: 10.3389/fimmu.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu J, Yamazaki S, Takahashi T, Ishida Y & Sakaguchi S. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 3: 135–142, doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 36.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M & Byrne MC. 2002. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16: 311–323, doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 37.Waks AG & Winer EP. 2019. Breast Cancer Treatment. JAMA. 321: 316, doi: 10.1001/jama.2018.20751. [DOI] [PubMed] [Google Scholar]
- 38.Tischner D, Gaggl I, Peschel I, Kaufmann M, Tuzlak S, Drach M, Thuille N, Villunger A & Jan Wiegers G. 2012. Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and limits their functionality in mice. J Autoimmun. 38: 59–69, doi: 10.1016/j.jaut.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, Hui R, Curigliano G, Toppmeyer D, O’Shaughnessy J, Loi S, Paluch-Shimon S, Card D, Zhao J, Karantza V & Cortes J. 2017. Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple-negative breast cancer (mTNBC): KEYNOTE-086 cohort A. Journal of Clinical Oncology. 35: 1008–1008, doi: 10.1200/JCO.2017.35.15_suppl.1008. [DOI] [Google Scholar]
- 40.Wein L, Luen SJ, Savas P, Salgado R & Loi S. 2018. Checkpoint blockade in the treatment of breast cancer: current status and future directions. Br J Cancer. 119: 4–11, doi: 10.1038/s41416-018-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franceschini D, Paroli M, Francavilla V, Videtta M, Morrone S, Labbadia G, Cerino A, Mondelli MU & Barnaba V. 2009. PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J Clin Invest. 119: 551–564, doi: 10.1172/JCI36604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong M, La Cava A & Hahn BH. 2013. Blockade of programmed death-1 in young (New Zealand Black x New Zealand White)F1 mice promotes the suppressive capacity of CD4+ regulatory T cells protecting from lupus-like disease. J Immunol. 190: 5402–5410, doi: 10.4049/jimmunol.1202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.