

Abstract
Directed evolution of heme proteins has opened access to new-to-nature enzymatic activity that can be harnessed to tackle synthetic challenges. Among these, reactions resulting from active site iron-nitrenoid intermediates present a powerful strategy to forge C–N bonds with high site- and stereoselectivity. Here we report a biocatalytic, intermolecular benzylic C–H amidation reaction operating at mild and scalable conditions. With hydroxamate esters as nitrene precursors, feedstock aromatic compounds can be converted to chiral amides with excellent enantioselectivity (up to >99% ee) and high yields (up to 87%). Kinetic and computational analysis of the enzymatic reaction reveals rate-determining nitrenoid formation followed by stepwise hydrogen atom transfer-mediated C–H functionalization.
Keywords: biocatalysis, nitrene transfer, P411 enzymes, asymmetric C-H functionalization
Graphical Abstact
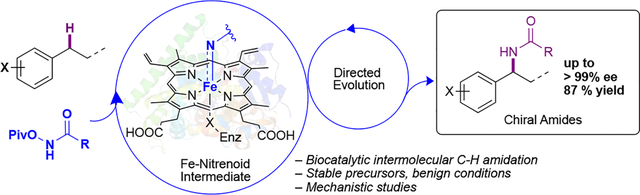
Directed evolution of P411 heme enzymes enables intermolecular C-H amidation with high yields and exquisite enantioselectivity. The biocatalytic process utilizes stable hydroxamate esters as nitrenoid precursors and is amenable to scaleup. Mechanistic studies reveal rate-determining nitrenoid formation followed by a stepwise, hydrogen atom transfer-mediated C-H functionalization.
From constituting the building blocks of life to appearing ubiquitously in natural products, therapeutics, polymers and synthetic materials, amides are arguably the most privileged among the nitrogen-containing functional groups. Nearly one-fourth of marketed drugs and about two-thirds of drug candidates bear at least one amide bond.[1,2] Amine acylation, the principal method for amide synthesis, is the most common reaction performed in the pharmaceutical industry.[2] However, the approach is fundamentally limited by its reliance on the availability of oxidized precursors (enantioenriched amines) as reactants. Likewise, since acylation with amide coupling reagents produce stoichiometric byproducts and possess poor atom economy, alternative methods to synthesize amides have been recognized as a pressing challenge in green chemistry.[3] Alternatively, access to amides can be envisaged through a direct C–H amidation reaction (Figure 1A). Selective C–H functionalization – an enduring frontier for organic synthesis – offers strategies to promote inert starting materials to useful intermediates, streamline synthesis by using abundant synthons, and at its pinnacle allow precise alteration of complex molecules.[4,5] Here we report that enzymatic C(sp3)–H functionalization of benzylic positions can provide highly enantioenriched amides from unfunctionalized starting materials. (Figure 1C).
Figure 1:

Objectives, state of the art and new findings. (A) intermolecular, enantioselective C-H amidation. (B) Previous examples. (C) New findings from this study.
The paradigm to achieve nitrene-transfer mediated, asymmetric C–H amidation was recently pioneered by Chang and coworkers with the introduction of dioxozolone derivatives as nitrene precursors.[6–8] In the presence of an appropriate organometallic catalyst (typically Rh, Ru, Ir, or Co), the resulting acyl(metal)nitrenoid species can undergo enantioselective C–H insertion reactions to form amides.[9] This strategy has largely been confined to intramolecular lactam formation[10–16] and some cases of C(sp2)-H functionalization.[17,18] Only three enantioselective, intermolecular versions have been reported for C(sp3)–H amidation. Two, by Matsunaga and coworkers, are restricted to thioamides and quinoline substrates as directing groups.[19,20] The third, more general report by Blakey and coworkers utilizes chiral Rh-indenyl catalysts to amidate allylic positions (Figure 1B).[21]
Over the last decade, heme enzymes housing the Fe-porphyrin cofactor have emerged as efficient biocatalysts for carbene and nitrene transfer reactions.[22,23] The active site of P411 enzymes (which bear a Ser in place of Cys for axial coordination to the heme iron atom) can be evolved to stabilize and direct the reactivity of Fe-nitrenoid species to C–H insertion and olefin functionalization reactions.[24–26] On this basis, the hypothesis that an active-site acyl Fe-nitrenoid species can be generated and harnessed for an intermolecular C–H amidation reaction seemed compelling. Unlike previously reported Fe-sulfonyl-nitrenoid and unprotected Fe-nitrenoid species, acyl(metal)nitrenes are vulnerable to deleterious, Curtius-type rearrangement processes to make unwanted isocyanate products that add a further challenge to reaction development.[16,27]
Hydroxylamine derivatives have been used successfully to generate metal[28–30] and active site nitrenoid species.[25,26] Thus, we hypothesized that pivalate esters of hydroxamic acids, through the loss of pivalic acid, may generate an active site Fe-acyl-nitrenoid intermediate that can engage in a formal intermolecular C-H insertion reaction (Figure 2A). Two transformations were selected as models (Figure 2B): (i) reaction I: benzylic amidation of 4-ethylanisole (1) with 3-phenyl-N-(pivaloyloxy)propanamide (2) and, (ii) reaction II: benzylic amidation of ethylbenzene (3) with N-(pivaloyloxy)acetamide (4). The reactions were screened with a panel of engineered P411 enzymes in whole-cell reactions. Only one variant, henceforth called iAMD0, provided the amidation product, detectable by mass spectroscopy at trace yields (<1 %) and trace total turnover numbers (<10 TTN). Directed evolution has allowed development of efficient enzymes even from such low activities.[31] Reaction I was chosen for the evolution campaign with the hypothesis that active site modification to accept these larger substrates may simultaneously predispose the evolved enzymes toward smaller reactants (like 3 and 4) rather than vice versa.
Figure 2:
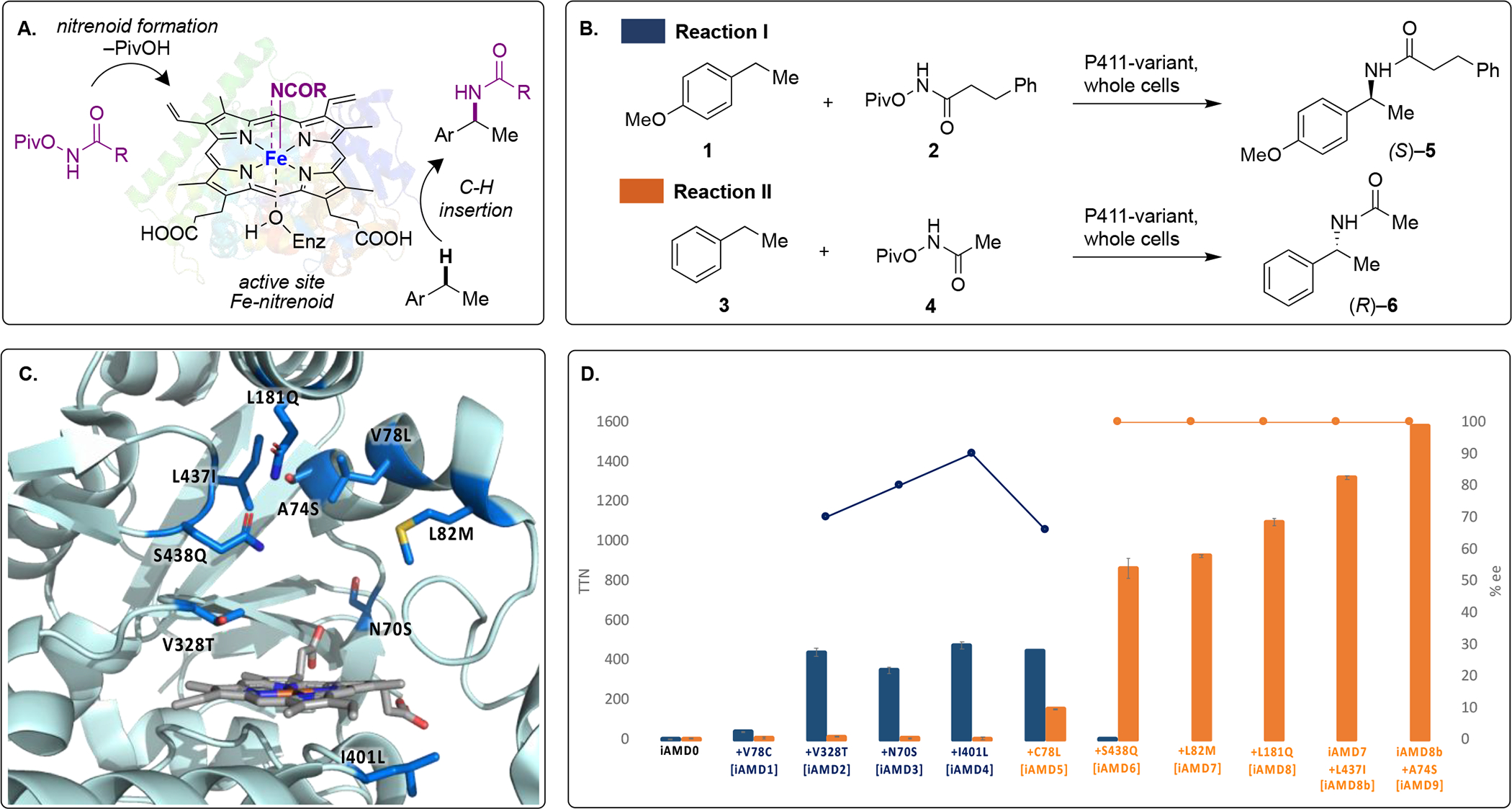
Directed evolution for enzymatic, intermolecular amidation. (A) Hypothesis of the putative reaction pathway (Piv = pivoloyl). (B) Model amidation reactions I and II. (C) Active site model of iAMD9 (based on crystal structure from PDB ID: 5ucw[24]) with mutated residues highlighted. (D) Directed evolution of amidation activity for reaction I (blue) and reaction II (orange). For each variant, bars indicate total turnover numbers (TTNs) for the whole-cell reactions, while circles indicate enantiomeric excess (ee) of product (S)-5 (blue) and (R)-6 (orange). Refer to Supplementary Information for experimental details.
Directed evolution involved screening sequential rounds of site-saturation mutagenesis (SSM, one site at a time) at residues close to the heme cofactor in the active site as well as in flexible loop regions of the protein (Figure 2C). SSM mutant libraries were screened for improved activity in whole-cell, 96-well reactions under anaerobic conditions. The crystal structure of a closely related P411 enzyme (PDB ID: 5ucw) was inspected to identify prospective target sites.[24] Four rounds of SSM and screening yielded variant iAMD4 incorporating mutations V78C, V328T, N70S, and I401L, which catalyzed the amidation reaction I with ~475 TTN and provided (S)-configured amide (5) with an enantiomeric enrichment (ee) of 90% (Figure 2D). The boost in activity was almost entirely a result of the mutation at position 328, which lies on a loop close to the distal heme pocket (Figure 2C). Mutations at sites 70 and 401 contributed to improving enantioselectivity. Note that previous reports of intermolecular enzymatic nitrene C-H functionalization are restricted to delivering only the (R)-configured product.[24,25] 2-Phenylethanamine, the expected Curtius-type rearrangement product of the acyl nitrenoid derived from 2, was not detected. However, this lineage showed no improvement in catalyzing reaction II. Poor activity was also noted in the reaction between 1 and 4. The hypothesis that these enzymes will be predisposed toward amidation with smaller substrates thus proved to be too simplistic. Hence, reaction II was chosen for subsequent rounds of directed evolution. The C78L mutant that confers moderate activity with reaction II maintains activity with reaction I, although a significant drop in enantioselectivity (66% ee) is seen for the formation of 5.
Mutation S438Q in variant iAMD6 amplified activity for reaction II by ~10 fold, but at the cost of nearly complete abolishment of activity for reaction I. With variant iAMD6, amide (R)-6 is obtained with exceptional enantioselectivity (>99% ee). This divergence in activities for the two model reactions as well as the opposite configurations of the respective product enantiomers highlights the incompatibility of 2 and 4 to function in the same active site in this lineage. Further rounds of SSM provided improvements in TTNs while maintaining high enantioselectivity. The evolution campaign concluded with variant iAMD9, which provided (R)-6 with 1580 TTN, an analytical yield of 57% and with >99% ee. This value compares favorably with previous C-H nitrene insertion reactions with P411 enzymes (~300–700 TTN for benzylic sulfonamidation and primary amination),[24,25] but is lower than that seen for carbene C-H insertion (upto 3750 TTN).[31] Constitutional isomers arising from competing functionalization of the primary carbon or the aromatic ring were not detected.
To gauge substrate scope, the evolved lineage was tested with various substituted ethylbenzene and aromatic derivatives (Figure 3). Methyl substitution at para-, ortho-, and meta-position (7-9) was tolerated. Naphthyl derivative (10), five- and six-membered rings (11, 21), and propylbenzenes were competent substrates (12, 13). Halogenated substrates (14–17) gave moderate to low TTNs (215–1000), consistent with the trend of electron-rich substrates performing better than electron-poor ones. The use of N-(pivaloyloxy)propionamide and methyl (pivaloyloxy)carbamate as nitrene precursors allowed the enzymatic synthesis of ethylamide (18) and methylcarbamate (19) derivatives, respectively. With the exception of the indenyl product (11), exquisite enantioselectivity (>99% ee, minor enantiomer not detected) was seen in all other cases. Variant iAMD4-L82M afforded (S)-5 with TTNs comparable to iAMD4 (470 and 475, respectively) but with near-perfect enantioselectivity (>99% ee).
Figure 3:

Substrate scope of benzylic amidation. (A) Analytical scale reactions performed with 5.0 mM substrate and 10.0 mM hydroxamate ester under whole-cell conditions. aWith iAMD9, bwith iAMD8, cwith iAMD8b-A74T, dwith iAMD4-L82M. (B) Scaleup reactions performed with iAMD8-expressing E. coli cells. Yields refer to isolated product. (C) Relative analytical yield of 20 in iAMD8 catalyzed amidation with related nitrene precursors. Refer to Supplementary Information for experimental details.
Gratifyingly, these conditions could be applied readily to millimole-scale reactions. High isolated yields were obtained at 1–4 mmol scales after a simple extraction of the reaction mixture with organic solvent followed by chromatographic purification (Figure 3B). Leftover starting material could be detected, whereas the nitrenoid precursor was fully consumed. We believe that complete substrate conversion is limited by catalyst deactivation during the course of the reaction.
For the iAMD8-catalyzed synthesis of 20, related nitrenoid precursors like 23 and 24 were also tolerated, with 2–4 fold reduced analytical yield but excellent product enantioselectivity (>99% ee); 25 yielded only trace product (Figure 3C).
The mechanistic landscape of enzymatic amidation was investigated using a combination of kinetic isotope effects (KIEs) and density functional theory (DFT) calculations. Non-competitive kH/kD initial rate measurements for the amidation of ethylbenzene (3) and d10-ethylbenzene (3-d10) were carried out in side-by-side reactions using whole-cell transformations with the iAMD8 variant. A KIE of near unity (1.06 ± 0.02) is indicative that C–H bond cleavage is not involved in the turnover-limiting step of the reaction (Figure 4). This value, and an observed first-order dependence of initial rates on the concentration of 4, is consistent with nitrenoid formation as the turnover-limiting step in the enzymatic reaction. Nitrenoid formation being rate determining is a likely reason why the same enzyme variant displays divergent reactivities with different nitrene precursors (see above). Competitive KIE experiments were utilized to investigate the C-H functionalization step for amide formation with substrate 3, since selectivity and substrate scope depend on the nature of this step.[32, 33] A competitive KIE of 8.2 ± 0.4 indicates that this step involves C-H bond cleavage. In previous heme-enzyme catalyzed nitrene C-H insertion studies, large competitive KIE values have also been noted (kH/kD = 4.7, 5.3)[34], with the observation of a non-competitive KIE greater than unity (kH/kD = 2.7[34], 1.6[35]), indicating contribution of C-H cleavage to the rate-determining step. These reports utilized azides as nitrenoid sources, which perhaps leads to facile nitrenoid formation.
Figure 4:

Kinetic studies of enzymatic amidation. Non-competitive KIE measurements performed in triplicate from independently determined rate-constants for 3 and 3-d10 carried out in side-by-side reactions. Competitive KIE measurements were performed from two batches of cells with 16 individual measurements of the ratio of products (6 and 6-d9) resulting from reaction of 3 and 3-d10 in the ‘same pot’.
For a quantitative interpretation of experimental KIEs, both concerted and stepwise pathways for C-H bond functionalization were studied using DFT. Nitrenoid insertion was modeled with a truncated porphyrin system chelated to a methoxide group to simulate the serinate residue of iAMD8. Potential energy surface scans on all reactants were performed to map the conformational and configurational space for this reaction. A UB3LYP-D3BJ/6-31+G(d)/LANL2DZ(Fe) functional and basis set were utilized for geometry and transition-state optimizations. Single point energy corrections were calculated at the UB3LYP-D3BJ/6-311+G(d,p)/LANL2TZ(f) level of theory and an implicit solvent model was applied (SMD-chlorobenzene) to simulate the dielectric of the enzyme pocket based on the benchmarks reported by Liu and Arnold.[35] Attempts to locate transition states (TS) for concerted nitrenoid insertion on any of the spin-state surfaces were unsuccessful, suggesting that this is likely not a viable pathway for C-H amidation.[35,36] Stepwise nitrenoid insertion involving hydrogen atom transfer (HAT) followed by carbon-nitrogen bond formation via a radical rebound mechanism was then examined (Figure 5). An exploration of different spin states indicated that the lowest energy TS for HAT proceeds on the triplet surface (TSHAT-Triplet) while the radical rebound TS proceeds on the quintet surface (TSCN-Quintet).
Figure 5:

Potential energy surface of the porphyrin model system for stepwise HAT followed by radical rebound. An IBO analysis was used to depict the spin density of TSHAT-Triplet and TSC-N-Quintet. The predicted KIE for TSHAT-Triplet is consistent with experiment. Energies and bond distances are in kcal/mol and angstroms respectively.
Upon closer inspection of TSHAT-Triplet, hydrogen atom transfer is colinear and the hydrogen is equidistant from the nitrogen and benzylic carbon atoms. An iron-nitrogen distance of 1.94 Å in combination with the electron density analysis of the nitrenoid connotes double bond-like character in TSHAT-Triplet. However, at the C-N bond-forming step the iron-nitrogen bond elongates to 2.38 Å, indicating a switch to single-bond character in TSCN-Quintet. The HAT TS has a higher activation barrier (ΔG‡ = 22.0 kcal/mol) than the radical rebound TS (ΔG‡ = 17.6 kcal/mol), thereby making HAT the first irreversible step for product formation for ethyl benzene (Figure 5, see the supporting information for energetics of all electronic states). A theoretical KIE for HAT was calculated from TSHAT-triplet using the formalism of Bigeleisen and Mayer[37] and including a Wigner tunneling correction; a theoretical kH/kD value of 8.5 is in excellent agreement with the experimental value of 8.2 ± 0.4 (Figure 5).
Spin density computations along the reaction coordinate for HAT further point to the radical nature of the transferring hydrogen. In TSHAT-Triplet, the spin density on the nitrogen and benzylic carbon are each 0.5, indicative of a concerted transfer of proton and electron as a hydrogen atom. Visualization of the spin density indicates this symmetric nature in the bonding orbitals; C-H bond breaking is displayed as a purple orbital and N-H bond formation is shown as an orange orbital (Figure 5, see the Supporting Information for the intrinsic bond orbital (IBO) analysis for the entire reaction coordinate). Following TSHAT-Triplet, the spin density on the benzylic carbon drops to 0.25 in IntHAT-Triplet indicating the delocalization of the radical – a feature that is preserved in TSC-N-Quintet as evident from the IBOs shown as green orbitals. These results obtained from the porphyrin model system in conjunction with the deuterium KIEs provide insights into the chemical underpinnings of the enzymatic C-H amidation reaction.
In conclusion, directed evolution of P411 enzymes has now enabled the intermolecular benzylic C–H amidation of feedstock aromatic compounds, a transformation unreported with small molecule catalysts. Mechanistic studies reveal that nitrene formation from the hydroxamate precursors is the rate- determining step in this enzymatic amidation process, with subsequent C-H functionalization occuring via a stepwise HAT–radical rebound pathway. Diversifying the repertoire of biocatalytic chemistry requires accessing novel enzymes that can provide starting points for further evolution. This finding positions us to expand enzymatic amidation to more challenging substrates and to explore other reactions of acyl-nitrene species.
Supplementary Material
Acknowledgements
This research was supported in part by the NIH National Institute of General Medical Sciences (R01GM125887), the ACS GCI Pharmaceutical Roundtable Research Grant (F.H.A., S.V.A.), and Binghamton University startup funds (J.S.H.). J.S.H. and M.S.C. acknowledge support from the XSEDE Science Gateways Program (CHE180061 and CHE210031), which is supported by NSF grant number ACI-1548562. We thank S. Brinkmann-Chen for helpful discussion and comments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Lundberg H, Tinnis F, Selander N, Adolfsson H, Chem. Soc. Rev 2014, 43, 2714–2742. [DOI] [PubMed] [Google Scholar]
- [2].Roughley SD, Jordan AM, J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- [3].Sabatini MT, Boulton Lee. T., Sneddon HF, Sheppard TD, Nat Catal 2019, 2, 10–17. [Google Scholar]
- [4].Davies HML, Morton D, J. Org. Chem 2016, 81, 343–350. [DOI] [PubMed] [Google Scholar]
- [5].Abrams DJ, Provencher PA, Sorensen EJ, Chem. Soc. Rev 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- [6].Park Y, Park KT, Kim JG, Chang S, J. Am. Chem. Soc 2015, 137, 4534–4542. [DOI] [PubMed] [Google Scholar]
- [7].Hong SY, Park Y, Hwang Y, Kim YB, Baik M-H, Chang S, Science 2018, 359, 1016–1021. [DOI] [PubMed] [Google Scholar]
- [8].Park Y, Kim Y, Chang S, Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- [9].Tosi E, de Figueiredo RM, Campagne J-M, Catalysts 2021, 11, 471. [Google Scholar]
- [10].Park Y, Chang S, Nat. Catal 2019, 2, 219–227. [Google Scholar]
- [11].Hong SY, Kim D, Chang S, Nat Catal 2021, 4, 79–88. [Google Scholar]
- [12].Lee E, Hwang Y, Kim YB, Kim D, Chang S, J. Am. Chem. Soc 2021, jacs.1c02550. [DOI] [PubMed] [Google Scholar]
- [13].Ju M, Zerull EE, Roberts JM, Huang M, Guzei IA, Schomaker JM, J. Am. Chem. Soc 2020, 142, 12930–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang H, Park Y, Bai Z, Chang S, He G, Chen G, J. Am. Chem. Soc 2019, 141, 7194–7201. [DOI] [PubMed] [Google Scholar]
- [15].Xing Q, Chan C-M, Yeung Y-W, Yu W-Y, J. Am. Chem. Soc 2019, 141, 3849–3853. [DOI] [PubMed] [Google Scholar]
- [16].Zhou Z, Chen S, Hong Y, Winterling E, Tan Y, Hemming M, Harms K, Houk KN, Meggers E, J. Am. Chem. Soc 2019, 141, 19048–19057. [DOI] [PubMed] [Google Scholar]
- [17].Liu L, Song H, Liu Y-H, Wu L-S, Shi B-F, ACS Catal 2020, 10, 7117–7122. [Google Scholar]
- [18].Liu W, Yang W, Zhu J, Guo Y, Wang N, Ke J, Yu P, He C, ACS Catal 2020, 10, 7207–7215. [Google Scholar]
- [19].Fukagawa S, Kato Y, Tanaka R, Kojima M, Yoshino T, Matsunaga S, Angew. Chem. Int. Ed 2019, 58, 1153–1157. [DOI] [PubMed] [Google Scholar]
- [20].Fukagawa S, Kojima M, Yoshino T, Matsunaga S, Angew. Chem. Int. Ed 2019, 58, 18154–18158. [DOI] [PubMed] [Google Scholar]
- [21].Farr CMB, Kazerouni AM, Park B, Poff CD, Won J, Sharp KR, Baik M-H, Blakey SB, J. Am. Chem. Soc 2020, 142, 13996–14004. [DOI] [PubMed] [Google Scholar]
- [22].Brandenberg OF, Fasan R, Arnold FH, Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang Y, Arnold FH, Acc. Chem. Res 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH, Nat. Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jia Z-J, Gao S, Arnold FH, J. Am. Chem. Soc 2020, 142, 10279–10283. [DOI] [PubMed] [Google Scholar]
- [26].Cho I, Prier CK, Jia Z, Zhang RK, Görbe T, Arnold FH, Angew. Chem. Int. Ed 2019, 58, 3138–3142. [DOI] [PubMed] [Google Scholar]
- [27].Li D, Wu T, Liang K, Xia C, Org. Lett 2016, 18, 2228–2231. [DOI] [PubMed] [Google Scholar]
- [28].Jat JL, Paudyal MP, Gao H, Xu Q-L, Yousufuddin M, Devarajan D, Ess DH, Kürti L, Falck JR, Science 2014, 343, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ma Z, Zhou Z, Kürti L, Angew. Chem. Int. Ed 2017, 56, 9886–9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Legnani L, Prina-Cerai G, Delcaillau T, Willems S, Morandi B, Science 2018, 362, 434–439. [DOI] [PubMed] [Google Scholar]
- [31].Zhang RK, Chen K, Huang X, Wohlschlager L, Renata H, Arnold FH, Nature 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simmons EM, Hartwig JF, Angew. Chem. Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- [33].Chan J, Lewis A, Gilbert M, Karwaski M-F, Bennet AJ, Nat. Chem. Biol 2010, 6, 405–407. [DOI] [PubMed] [Google Scholar]
- [34].Singh R, Kolev JN, Sutera PA, Fasan R, ACS Catalysis 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang Y, Cho I, Qi X, Liu P, Arnold FH, Nat. Chem 2019, 11, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang J, Gao H, Yang L, Gao YQ, ACS Catalysis 2020, 10, 5318–5327. [Google Scholar]; Recent report by Gao and coworkers on intermolecular C-H amination showing that the concerted nitrene insertion has a prohibitively high barrier on the singlet surface further supports our assertion that nitrene insertion is a stepwise pathway.
- [37].Bigeleisen J, Mayer MG, J. Chem. Phys 1947, 15, 261–267. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.