

Abstract
Immunity to pulmonary infection typically requires elicitation of lung-resident T cells that subsequently confer protection against secondary infection. The presence of tissue resident T cells in SARS-CoV-2 convalescent patients is unknown. Using a sublethal mouse model of COVID-19, we determined if SARS-CoV-2 infection potentiated antigen-specific pulmonary resident CD4+ and CD8+ T cells responses and if these cells mediated protection against secondary infection. S protein specific T cells were present in resident and circulating populations. However, M and N protein specific T cells were only detected in the resident T cell pool. Using an adoptive transfer strategy, we found that T cells from SARS-CoV-2 immune animals did not protect naïve mice. These data indicate resident T cells are elicited by SARS-CoV-2 infection but are not sufficient for protective immunity.
INTRODUCTION
Establishing correlates of protection is a critical step in vaccine development. The focus of current vaccine strategies to prevent SARS-CoV-2 infection, the causative agent of COVID-19, has centered on driving high neutralizing antibody titers to block viral entry into host cells [1]. While the protective nature of antibodies in COVID-19 is well established, the role of T cells in immunity, particularly those resident in the lung tissue, remains a lingering question [2]. Antigen-specific CD4+ and CD8+ T cells have been detected in the peripheral blood of COVID-19 convalescent patients [3, 4]. However, it is unclear whether SARS-CoV-2 infection elicits a pulmonary resident T cell response and if T cells alone are sufficient to prevent lethal disease.
Understanding the contributions of specific immune subsets to protection requires the use of animal models of disease where antibodies or cells can be added or eliminated prior to infection. Convalescent human plasma protects mice expressing the human angiotensin converting enzyme 2 (hACE2) from lethal SARS-CoV-2 infection while purified convalescent IgG from rhesus macaques decreased viral loads using both prophylactic and therapeutic treatment strategies [5, 6]. These passive transfer studies confirmed the protective nature of antibodies in COVID-19. Recently, the role of CD8+ T cells in convalescent non-human primates has also been addressed. Convalescent rhesus macaques depleted of their CD8+ T cells had partially impaired control of SARS-CoV-2 in the upper respiratory track indicating CD8+ T cells play a role in restricting viral replication [5]. Similarly, depletion of CD4+ and CD8+ T cells during primary SARS-CoV-2 infection in rhesus macaques delayed viral clearance, but the animals ultimately cleared the infection and the response to secondary challenge was not impacted [7]. These data suggest T cells play a role in viral clearance and may be important as antibody responses wane over time, but notably, did not address whether T cells alone are sufficient for protective immunity.
When evaluating the role T cells are playing in the immune response, it is essential to consider the anatomical location of the response. Pulmonary resident T cells are retained in the airway and/or lung parenchyma after a variety of viral and bacterial infections and are positioned to swiftly respond to secondary respiratory exposure to pathogens [8–13]. Underscoring the critical role tissue resident T cells play in defense against infection, it has been demonstrated by several groups that these cells alone are sufficient for survival of multiple acute viral and bacterial infections [11–13]. In contrast to the tissue resident T cells which rapidly respond to localized infection, circulating T cells replenish the resident pool to either maintain immunity or work in concert with resident T cells to control disseminated infection [8, 10]. Together, these examples highlight the importance of identifying the location of T cells elicited by respiratory infection and their contribution to protective immunity as expansion of appropriate pools of T cells will dictate both vaccine design and delivery. Herein, we established a sublethal SARS-CoV-2 primary infection model in hACE2 mice to determine if natural infection promoted establishment of SARS-CoV-2 specific resident T cells and, if present, what contribution these cells made to survival of secondary lethal infection.
MATERIAL AND METHODS
SARS-CoV-2
SARS-CoV-2 USA/WA1/2020 was obtained from BEI Resources. Viral stocks were generated in Vero cells and frozen at −80°C until use.
Mice and inoculations
Specific-pathogen free, 6-10 week old, male and female B6.Cg-Tg(K18-ACE2)2Prlmn/J (K18-hACE2; stock #034860) mice were purchased from The Jackson Laboratory and/or bred in-house. For SARS-CoV-2 infections, mice were intranasally inoculated with 101-104 PFU in 25 μl PBS to a single nare after anesthetizing with ketamine/xylazine. Prior to euthanizing animals for tissue harvest, mice were anesthetized with ketamine/xylazine and then retro-orbitally injected with 2.5 μg CD45.2 FITC (Biolegend) in 100 μl PBS to label circulating cells. All animal studies were approved by and conducted in accordance with Rocky Mountain Laboratories’ Animal Care and Use Committee under animal study protocol 2020-075-E.
Tissue harvest and processing
Lungs were aseptically removed after intravascular staining of circulating cells and subsequent euthanasia. Single cell suspensions were generated as previously described [8]. Briefly, lungs were minced and then digested with Liberase TM (Sigma-Aldrich). Red blood cells were lysed with ACK lysis buffer (Life Technologies) and the total number of viable cells was determined using trypan blue exclusion using a TC20 automated cell counter (Bio-Rad).
Flow cytometry analysis
Single cell suspensions were stained with Zombie Near-IR (Biolegend) to distinguish live and dead cells. Cells were stained with the following antibodies (Biolegend or BD Biosciences): CD3 BUV395, CD4 AF700, CD8 BV605, CD44 PE-Cy7, CD62L Pacific Blue, CD69 PE, CD103 APC, IFN-γ PE, TNF-α APC, and/or IL-2 Pacific Blue. Prior to intracellular cytokine staining, 1.5E6 lung cells were stimulated for 5 hours with Influenza A NP or SCV2 M, N, or S+ Peptivator pools (Miltenyi Biotec) or for 4 hours with PMA/ionomycin (Biolegend). During the last 4 hours of culture, brefeldin A (Biolegend) was added to inhibit cytokine secretion. Data acquisition was performed on a Symphony Flow Cytometer (BD Biosciences) and data analyzed using FlowJo 10 (BD Biosciences). The gating scheme utilized for analysis, including assessment of cytokine production among cells stimulated with peptide pools is shown in Supplemental Figs. 1 and 2.
Adoptive transfer of T cells
Total pulmonary T cells were isolated from the naïve hACE2 animals or total pulmonary Teff from SARS-CoV-2 immune hACE2 mice via cell sorting using the MACSQuant Tyto (Miltenyi Biotec). Where indicated, 3.5E5 cells were intratracheally transferred into naïve hACE2 mice in 50 μl of sterile saline or 1.4E6 cells were intravenously transferred into naïve hACE2 mice in 100 μl of sterile saline. Twenty-four hours later, mice were intranasally challenged with either 103 or 102 PFU of SARS-CoV-2 and survival determined.
Statistical analysis
Statistically significant differences between two groups were determined using an unpaired two-tailed t test. For T cell cytokine production, a paired two-tailed t test was utilized to compare the mock and peptide stimulated samples. In all cases, statistical significance was set at p < 0.05. Significant differences in survival between groups was determined using a log-rank (Mantel-Cox) test.
RESULTS AND DISCUSSION
Dose-dependent survival of K18-hACE2 mice after SARS-CoV-2 infection
We first determined the LD50 of SARS-CoV-2 in hACE2 mice to identify doses of infection that resulted in either sub-lethal or lethal intranasal infection. Animals inoculated with 103 or 104 PFU required euthanasia by day 6 post-challenge (Fig. 1A). Approximately 50% and 20% of animals infected with 102 or 101 PFU, respectively, succumbed to infection (Fig. 1A). Thus, the calculated LD50 was 92 PFU. The susceptibility of hACE2 mice to higher doses of SARS-CoV-2 in our hands is consistent with a previous report [14]. We next determined if sublethal SARS-CoV-2 infection induced protective immunity against lethal secondary challenge. Twenty-eight days after primary infection, animals were re-infected with 103 PFU. As expected, all naïve controls succumbed by day 7 post-challenge (Fig. 1B). In contrast, 100% of animals previously infected with 102 PFU survived secondary lethal challenge (Fig. 1B). Given the presence of complete protective immunity at a primary infection dose of 102, we utilized this as an immunizing dose for all subsequent studies.
Figure 1. Dose-dependent survival of hACE2 mice after SARS-CoV-2 infection.
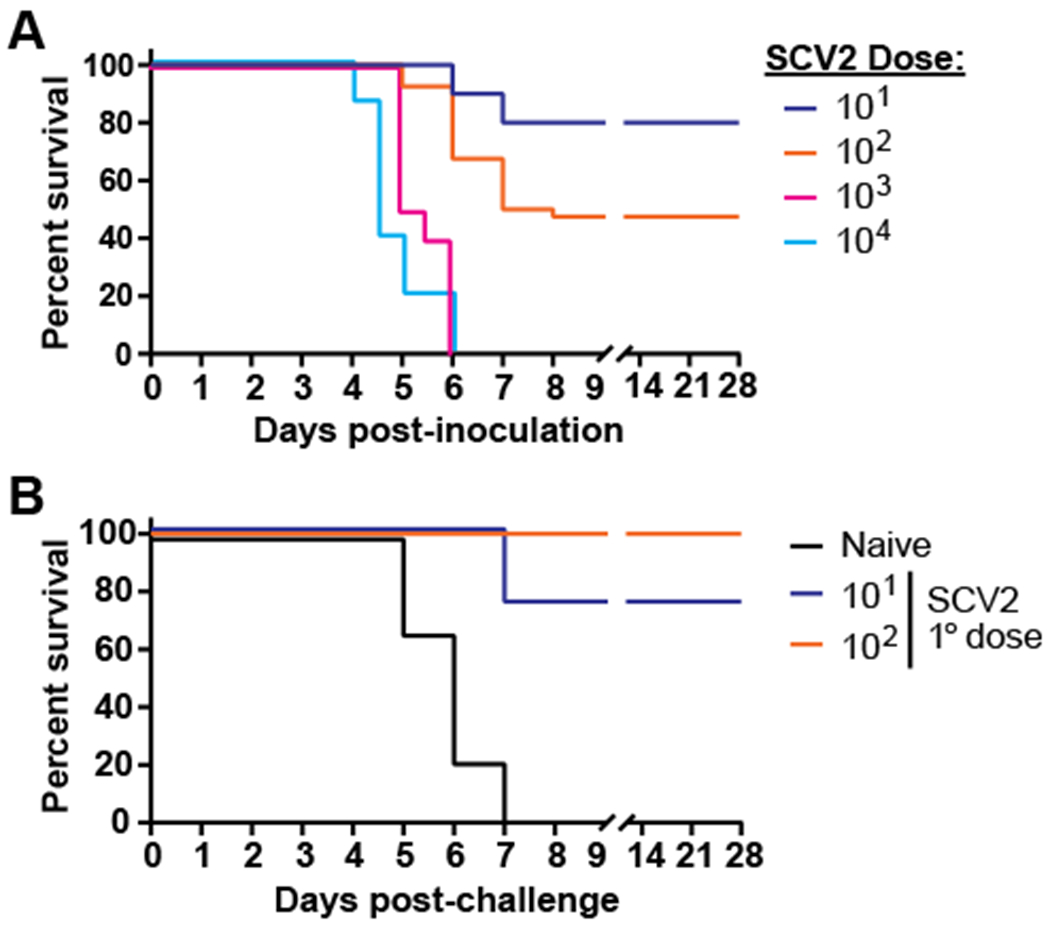
A) hACE2 mice were intranasally infected with increasing doses of SARS-CoV-2 and were monitored for signs of illness. n=10-40 mice/group with data combined from 6 independent experiments. B) Naïve animals or mice that survived 101 or 102 PFU primary infection with of SARS-CoV-2 were intranasally challenged with 103 PFU 28 days after the initial infection and were monitored for signs of illness. n=4-9 mice/group with data combined from 2 independent experiments.
Infection with SARS-CoV-2 elicits tissue resident T cell responses
Pulmonary infections often lead to the recruitment and retention of T cells into the lung establishing a pool of tissue resident effector T cells. These cells are critical for defense against a variety of pathogens whereas T cells circulating through the lung via the vasculature are dispensable [11–13, 15]. However, infection in the pulmonary compartment is no guarantee that pools of tissue resident effector T cells will develop. For example, we have observed that animals which survived intranasal Klebsiella. pneumoniae infection and are 100% protected against lethal secondary challenge do not have detectable tissue resident T cells in their lungs (unpublished observation). It is unknown if SARS-CoV-2 infection triggers expansion of resident T cells. Moreover, if these cells are present, it is also unknown if antigen specificity among resident effector T cells is similar to those in circulation. Therefore, we next determined if sublethal SARS-CoV-2 infection resulted in generation of pulmonary resident effector T cells followed by identification of their antigen specificity. In contrast to naïve controls, both resident CD4+ and CD8+ effector T cells (CD44hi; Teff) were detected in SARS-CoV-2 lungs 28 days after infection (Fig. 2A–D). The resident T cells from SARS-CoV-2 infected mice expressed markers associated with tissue-resident memory including CD69 and CD103 (Fig. 2E, F). These immune animals also had significantly greater numbers of circulating CD4+ and CD8+ Teff (Fig. 2G, H). Interestingly, the magnitude of the response to SARS-CoV-2 (10-25% resident of CD4+ or CD8+ T cells) was lower than that observed for other pulmonary viral and bacterial infections. For example, 50-80% of CD4+ and CD8+ T cells after Influenza A and >45% of CD4+ following B. pertussis infection are tissue resident T cells [11, 12]. Notably, Influenza A and B. pertussis persist in the lung for much longer than SARS-CoV-2. Thus, one possibility for the paucity of resident T cells present after SARS-CoV-2 infection could be a consequence of its rapid clearance from the host which negates the requirement for a strong T cell response to ultimately control and clear the infection.
Figure 2. Infection with SARS-CoV-2 elicits tissue resident T cell responses.
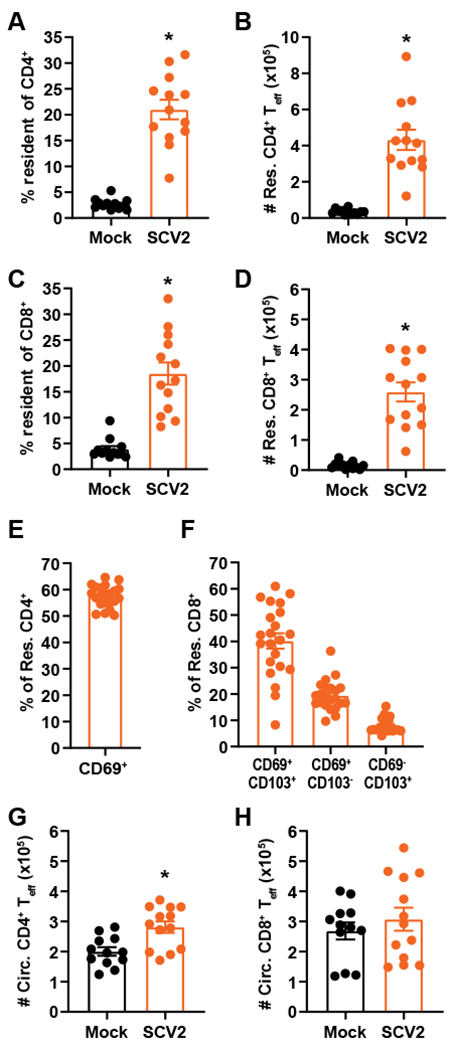
hACE2 mice were intranasally infected with 102 PFU of SARS-CoV-2. Twenty-eight days later, the A) percent resident of CD4+, B) number of resident CD4+ Teff, C) percent resident of CD8+, D) number of resident CD8+ Teff, E) percent CD69+ of resident CD4+ from SCV2 infected mice, F) percent expressing CD69 and/or CD103 of resident CD8+ from SCV2 infected mice, G) number of circulating CD4+ Teff, and H) number of circulating CD8+ Teff was determined using flow cytometry. Uninfected mice served as negative controls. n=12-13 mice/group and are combined from 3 independent experiments for A-D, G, H; n=22 mice combined from 2 independent experiments for E, F. Each point represents an individual mouse; error bars indicate SEM. Statistical significance was determined using an unpaired t-test; * indicates p < 0.05.
Resident and circulating T cells are specific for M, N, and S antigens
The presence of increased numbers of resident and circulating CD4+ and CD8+ Teff in SARS-CoV-2 immune animals led us to ask which antigen(s) these cells were responding to. Both CD4+ and CD8+ T cells isolated from the peripheral blood of COVID-19 patients produce Th1 cytokines IFN-γ and TNF-α in response to antigen stimulation [3, 4]. Therefore, we compared the ability of pulmonary resident and circulating T cells from SARS-CoV-2 immune mice to produce these cytokines in response to non-specific and antigen-specific stimuli. All T cell subsets readily produced both IFN-γ and TNF-α in response to non-specific stimulation with PMA and ionomycin demonstrating SARS-CoV-2 infection in mice also drives a strong Th1 response (Supplemental Fig. 3). To identify antigen-specific responses, immune lung cells were stimulated with overlapping peptide pools from either Influenza A NP (negative control) or SARS-CoV-2 M, N, or S. The response to each peptide was directly compared within each immune mouse to the corresponding mock stimulated sample. Immune circulating CD4+ T cells produced IFN-γ in response to S (Fig. 3A). Within the resident CD4+ pool, responses specific for M, N, and S were detected (Fig. 3B, C). Resident CD8+ T cells responded to S and there was a trend towards a response to N, but the difference did not reach statistical significance (Fig. 3E). Notably, only CD4+ T cells within the resident pool produced cytokine in response to M and N (Fig. 3B, C). Our data extend the studies using human peripheral blood by examining antigen specificity within pulmonary resident T cells and revealed circulating T cells shared specificity for some, but not all, antigens. Thus, while admittedly challenging, the ability to examine tissue resident T cell responses in human lungs at autopsy or airway cells obtained by bronchoalveolar lavage among convalescent patients provides an important opportunity to understand the pulmonary immune response not only to SARS-CoV-2, but other infections as well [16]. Together these data suggest anatomical antigen-specificity for T cell responses to SARS-CoV-2 and underscores the utility of including multiple antigens in future vaccine iterations. Further, the use of several antigens could lessen the chance that viral mutations in a single antigen would render a vaccine ineffective.
Figure 3. Resident and circulating T cells are specific for M, N, and S antigens.
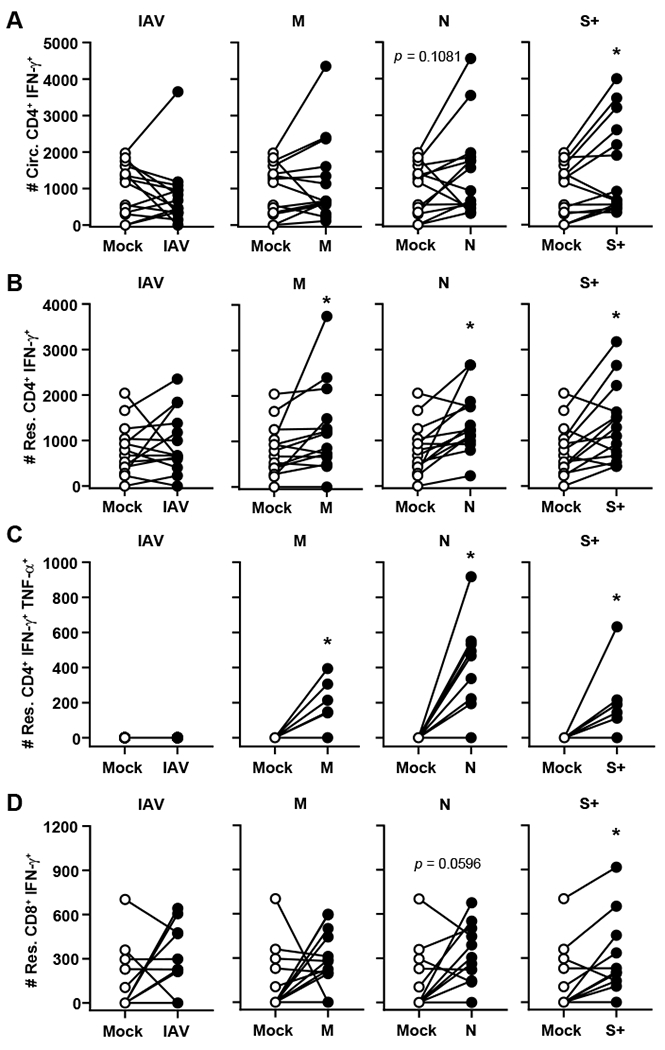
hACE2 mice were intranasally infected with 102 PFU of SARS-CoV-2. Twenty-eight days later, lung cells were mock stimulated, stimulated with PMA/Ionomycin, or IAV NP, SCV2 M, SCV2 N, or SCV2 S+ peptide pools. The production of A) IFN-γ by circulating CD4+ T cells, B) IFN-γ by resident CD4+ T cells, C) IFN-γ and TNF-α by resident CD4+ T cells, and D) IFN-γ by resident CD8+ T cells was determined by flow cytometry. n=14 mice/group and are combined from 3 independent experiments. Each point represents an individual mouse; error bars indicate SEM. Statistical significance was determined using a paired two-tailed t test to compare the mock and peptide stimulated samples; * indicates p < 0.05.
Adoptive transfer of pulmonary T cells does not protect against SARS-CoV-2 challenge
Since we observed the presence of tissue resident effector T cells in lungs of SARS-CoV-2 mice we next addressed whether these cells could independently protect the naïve host against lethal SARS-CoV-2 infection. Therefore, we adoptively transferred immune T cells into a naïve host prior to SARS-CoV-2 infection. Due to the limited number of Teff in naïve animals, mice that received total pulmonary T cells purified from naïve lungs served as negative controls. We purified total Teff from SARS-CoV-2 immune animals because we reasoned SARS-CoV-2-specific T cells would be enriched within the Teff pool giving us optimal opportunity to assess whether immune T cells were sufficient for protective immunity. Initially, we challenged mice with 103 PFU following adoptive transfer of either naïve T cells or immune Teff into naïve hosts. We observed equivalent lethality in both groups indicating T cells were not sufficient for protection against this dose of SARS-CoV-2 (data not shown). It was possible that T cells may make a minor contribution to control of secondary SARS-CoV-2 infection that was overwhelmed by a high challenge dose. Therefore, we examined the ability of adoptively transferred Teff could mediate protection against an approximate LD50 challenge. One day after transfer of naïve T cells or immune Teff cells mice were intranasally challenged with 102 PFU SARS-CoV-2. Following infection, 20% of animals receiving naïve donor T cells succumbed to SARS-CoV-2 infection (Fig. 4A). In contrast to control animals, there was increased mortality in the immune Teff group (40%) compared to those receiving naïve T cells, however this difference was not statistically significant (Fig. 4A). Due to the small volume necessary for intratracheal adoptive transfer, we limited the number of cells given to prevent aggregates that could block airways. Ultimately, this led to fewer cells transferred than are present in an immune lung (Fig. 2). Thus, we transferred almost five times as many immune Teff intravenously to reflect the number of resident and circulating Teff present in the immune lung and then infected with 102 PFU of SARS-CoV-2 one day later. As observed with intratracheal transfer, immune Teff were unable to significantly improve survival compared to naïve animals (Fig. 4B). Together, these data indicate immune effector T cells were not sufficient for protective immunity and at lower challenge doses, may be pathogenic when cells are delivered directly into the airspace.
Figure 4. Adoptive transfer of pulmonary T cells does not protect against SARS-CoV-2 challenge.
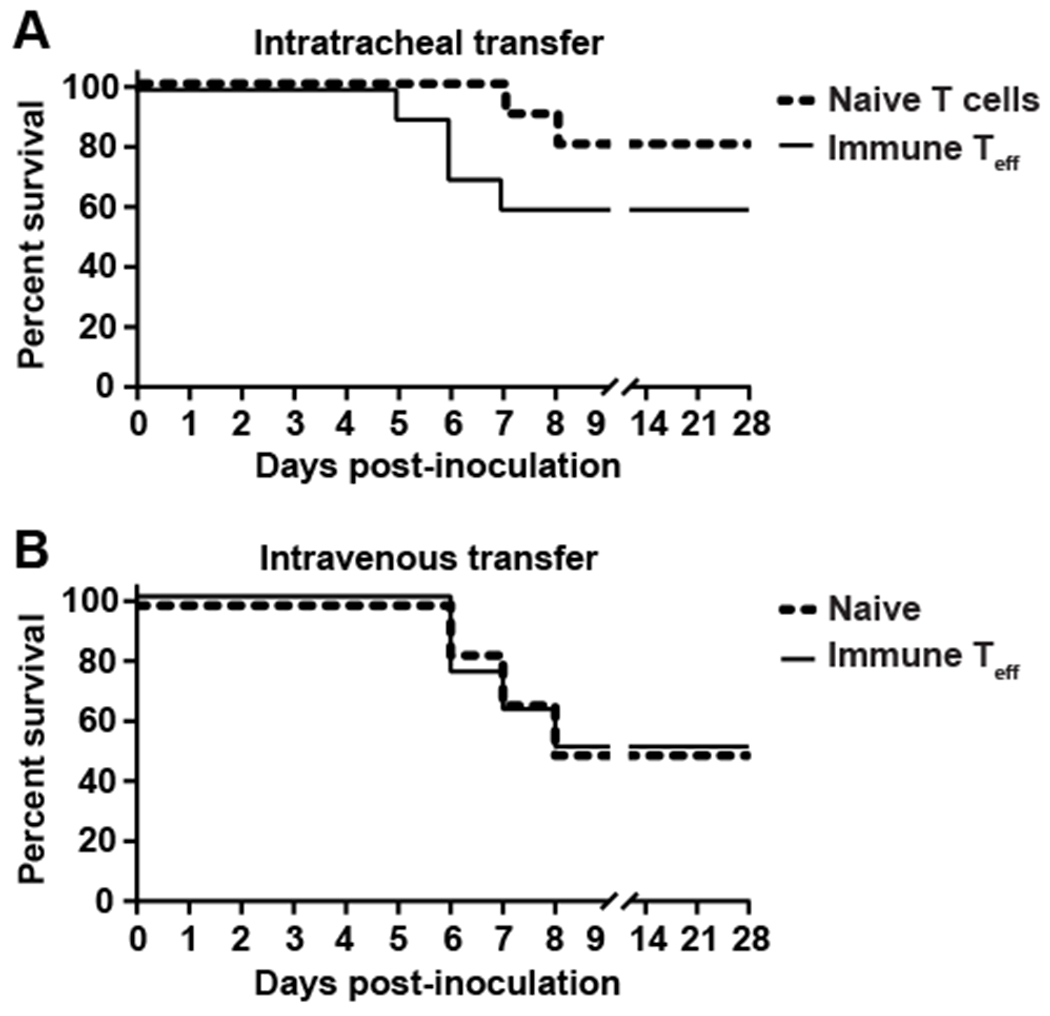
A) Total T cells from naïve hACE2 mice or total Teff from hACE2 immune animals were purified and 3.5x105 cells were intratracheally transferred into naïve hACE2 animals or B) 1.4x106 total Teff from hACE2 immune animals were intravenously transferred into naïve hACE2 animals. One day later, mice were intranasally challenged with 102 PFU of SARS-CoV-2 and monitored for signs of illness. n=10 mice/group (A) or n=6-8 mice/group (B) and are combined from 2 independent experiments. Statistically significant differences in survival between groups was determined using a log-rank (Mantel-Cox) test and found not to be statistically significant.
Our finding that T cells did not independently confer protective immunity against to SARS-CoV-2 is in contrast to previous reports examining immune requirements following infection with SARS. Specifically, airway-resident CD4+ T cells were critical for vaccine-mediated immunity to mouse adapted SARS in Balb/c mice [13]. However, depletion of CD4+ T cells in that study did not result in uniform lethality. This suggested that while T cells contribute to protection other immune cell subset(s) also actively participate in protective immunity [13]. The disconnect between our data and this earlier SARS work could be a consequence of the strategy used to generate immune T cells. In the study cited above immune animals were generated by using a vaccine directed against SARS. It is possible that unlike a natural infection the vaccine promoted an immune response in which T cells gained the capability to more actively participate in defense against infection. Another possibility is that there were inherent difference in the development of immunity based on the strain of mouse used in the study (hACE2 B6 versus Balb/c) and/or differences in the underlying protective mechanisms required for SARS versus SARS-CoV-2. Another possibility for the inability of adoptively transferred immune Teff to confer protective immunity in our model of SARS-CoV-2 infection could be their restriction to the lung. While we reasoned adoptively transferring total Teff from immune animals into the airways of naïve hACE2 mice would allow these cells optimal interaction with infected epithelial cells immediately after SARS-CoV-2 infection, SARS-CoV-2 can infect organs outside of the lung [14]. The consequences of extra-pulmonary SARS-CoV-2 infection to disease lethality is still under debate but limiting the T cell response to the airway could be detrimental for controlling viral replication in other locations. In addition to the possibility that extra- pulmonary infection contributed to the lack of function for pulmonary immune T cells in protection against SARS-CoV-2, the function of adoptively transferred T cells could also have been negatively impacted by alveolar macrophages (AMs). AMs negatively regulate dendritic cell function and in the context of SARS infection, suppress activation and migration of dendritic cells which are responsible for initiating anti-SARS T cell responses [17–19]. Finally, transferred T cells may not receive optimal cytokine signaling due to the acute nature of SARS-CoV-2 infection in mice coupled with dampened inflammatory cytokine responses that are necessary to potentiate existing T cell responses [14].
Although T cells were not solely responsible for the protective immune response, they likely remain an important component of the response to SARS-CoV-2. Indeed, CD8+ T cell depletion in convalescent rhesus macaques revealed this cellular subset is required for optimal viral control [5]. These data raise the possibility that resident T cells could coordinate the overall immune response in the tissue as demonstrated in other infectious models [20–22]. For example, a newly identified population of lung resident CD4+ T cells termed resident helpers given their shared characteristics of T follicular helper cells and resident memory T cells, coordinates the response of B cells and CD8+ T cells in Influenza A [22]. Likewise, skin resident CD4+ T cells coordinate the recruitment of circulating T cells to control Leishmania infection [20]. Resident T cells may act similarly as cellular conductors within the lung to efficiently coordinate the diverse response that eliminates SARS-CoV-2. While it remains to be seen whether T cells are long-lived after SARS-CoV-2, anti-SARS T cells were found in convalescent humans after antibody responses were no longer detectable [15, 23–25]. Thus, eliciting long-lived, antigen-diverse T cell responses by future vaccine candidates may be a critical component for provoking durable immunity against COVID-19.
Supplementary Material
Key points.
SARS-CoV-2 infection elicits pulmonary resident CD4+ and CD8+ T cells
Pulmonary T cells are not sufficient for protection against secondary SARS-CoV-2
ACKNOWLEDGEMENTS
Funding for this work was provided by the Division of Intramural Research, NIAID, NIH. We thank Dr. Benjamin Schwarz for his thoughtful comments as well as Dr. Brian Smith and RML’s animal care staff for managing and maintaining the hACE2 breeding colony.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Disease.
Footnotes
The authors have no conflicts of interest.
REFERENCES
- 1.Kyriakidis NC, Lopez-Cortes A, Gonzalez EV, Grimaldos AB, and Prado EO, SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines, 2021. 6(1): p. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarjour NN, Masopust D, and Jameson SC, T Cell Memory: Understanding COVID-19. Immunity, 2021. 54(1): p. 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, Marrama D, de Silva AM, Frazier A, Carlin AF, Greenbaum JA, Peters B, Krammer F, Smith DM, Crotty S, and Sette A, Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell, 2020. 181(7): p. 1489–1501 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekine T, Perez-Potti A, Rivera-Ballesteros O, Stralin K, Gorin JB, Olsson A, Llewellyn-Lacey S, Kamal H, Bogdanovic G, Muschiol S, Wullimann DJ, Kammann T, Emgard J, Parrot T, Folkesson E, Karolinska C-SG, Rooyackers O, Eriksson LI, Henter JI, Sonnerborg A, Allander T, Albert J, Nielsen M, Klingstrom J, Gredmark-Russ S, Bjorkstrom NK, Sandberg JK, Price DA, Ljunggren HG, Aleman S, and Buggert M, Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell, 2020. 183(1): p. 158–168 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, Liu J, Peter L, Atyeo C, Zhu A, Bondzie EA, Dagotto G, Gebre MS, Jacob-Dolan C, Li Z, Nampanya F, Patel S, Pessaint L, Van Ry A, Blade K, Yalley-Ogunro J, Cabus M, Brown R, Cook A, Teow E, Andersen H, Lewis MG, Lauffenburger DA, Alter G, and Barouch DH, Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature, 2021. 590(7847): p. 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng J, Wong LR, Li K, Verma AK, Ortiz ME, Wohlford-Lenane C, Leidinger MR, Knudson CM, Meyerholz DK, McCray PB Jr., and Perlman S, COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature, 2021. 589(7843): p. 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasenkrug KJ, Feldmann F, Myers L, Santiago ML, Guo K, Barrett BS, Mickens KL, Carmody A, Okumura A, Rao D, Collins MM, Messer RJ, Lovaglio J, Shaia C, Rosenke R, van Doremalen N, Clancy C, Saturday G, Hanley P, Smith B, Meade-White K, Shupert WL, Hawman DW, and Feldmann H, Recovery from acute SARS-CoV-2 infection and development of anamnestic immune responses in T cell-depleted rhesus macaques. bioRxiv, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts LM, Wehrly TD, Ireland RM, Crane DD, Scott DP, and Bosio CM, Temporal Requirement for Pulmonary Resident and Circulating T Cells during Virulent Francisella tularensis Infection. J Immunol, 2018. 201(4): p. 1186–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, and Barber DL, Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol, 2014. 192(7): p. 2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, and Harty JT, Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol, 2017. 2(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, and Mills KHG, Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J Immunol, 2017. 199(1): p. 233–243. [DOI] [PubMed] [Google Scholar]
- 12.Zens KD, Chen JK, and Farber DL, Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight, 2016. 1(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Zhao J, Mangalam AK, Channappanavar R, Fett C, Meyerholz DK, Agnihothram S, Baric RS, David CS, and Perlman S, Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity, 2016. 44(6): p. 1379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, Fox JM, Chen RE, Earnest JT, Keeler SP, Ritter JH, Kang LI, Dort S, Robichaud A, Head R, Holtzman MJ, and Diamond MS, SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol, 2020. 21(11): p. 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson RV, Crane DD, and Bosio CM, Long lived protection against pneumonic tularemia is correlated with cellular immunity in peripheral, not pulmonary, organs. Vaccine, 2010. 28(40): p. 6562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farber DL, Tissues, not blood, are where immune cells function. Nature, 2021. 593(7860): p. 506–509. [DOI] [PubMed] [Google Scholar]
- 17.Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G, and Thepen T, Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med, 1993. 177(2): p. 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strickland DH, Thepen T, Kees UR, Kraal G, and Holt PG, Regulation of T-cell function in lung tissue by pulmonary alveolar macrophages. Immunology, 1993. 80(2): p. 266–72. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Zhao J, Van Rooijen N, and Perlman S, Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog, 2009. 5(10): p. e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, and Scott P, Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med, 2015. 212(9): p. 1405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, and Masopust D, T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science, 2014. 346(6205): p. 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Son YM, Cheon IS, Wu Y, Li C, Wang Z, Gao X, Chen Y, Takahashi Y, Fu YX, Dent AL, Kaplan MH, Taylor JJ, Cui W, and Sun J, Tissue-resident CD4(+) T helper cells assist the development of protective respiratory B and CD8(+) T cell memory responses. Sci Immunol, 2021. 6(55). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng H, Yang LT, Wang LY, Li J, Huang J, Lu ZQ, Koup RA, Bailer RT, and Wu CY, Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology, 2006. 351(2): p. 466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang F, Quan Y, Xin ZT, Wrammert J, Ma MJ, Lv H, Wang TB, Yang H, Richardus JH, Liu W, and Cao WC, Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J Immunol, 2011. 186(12): p. 7264–8. [DOI] [PubMed] [Google Scholar]
- 25.Yang LT, Peng H, Zhu ZL, Li G, Huang ZT, Zhao ZX, Koup RA, Bailer RT, and Wu CY, Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin Immunol, 2006. 120(2): p. 171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.