

Abstract
Loss of photoreceptors due to retinal degeneration is a major cause of untreatable visual impairment and blindness. Cell replacement therapy, using retinal stem cell (RSC)-derived photoreceptors, holds promise for reconstituting damaged cell populations in the retina. One major obstacle preventing translation to the clinic is the lack of validated markers or strategies to prospectively identify these rare cells in the retina and subsequently enrich them. Here, we introduce a microfluidic platform that combines nickel micromagnets, herringbone structures, and a design enabling varying flow velocities among three compartments to facilitate a highly efficient enrichment of RSCs. In addition, we developed an affinity enrichment strategy based on cell-surface markers that was utilized to isolate RSCs from the adult ciliary epithelium. We showed that targeting a panel of three cell surface markers simultaneously facilitates the enrichment of RSCs to 1:3, relative to unsorted cells. Combining the microfluidic platform with single-cell whole-transcriptome profiling, we successfully identified four differentially expressed cell surface markers that can be targeted simultaneously to yield an unprecedented 1:2 enrichment of RSCs relative to unsorted cells. We also identified transcription factors (TFs) that play functional roles in maintenance, quiescence, and proliferation of RSCs. This level of analysis for the first time identified a spectrum of molecular and functional properties of RSCs as well as some distinguishing features.
Graphical Abstract
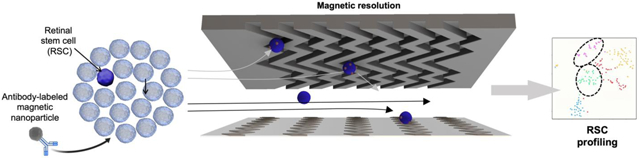
A microfluidic approach combined with single-cell RNA sequencing enabled prospective identification of retinal stem cells and discovery of novel cell-surface markers as well as transcription factors with functional roles in their maintenance, quiescence, and proliferation.
Introduction
Many retinopathies, while variable in their etiology, involve the degeneration of photoreceptors, leading to visual impairment and blindness. Photoreceptor cells, like most neurons, do not spontaneously regenerate once lost. Previous success with transplantation of autologous retinal pigmented epithelium (RPE) grafts has provided a critical proof of concept that warrants further development of stem cell-based therapies for retinal degenerative diseases.1 Adult retinal stem cells (RSCs) are rare quiescent cells located at the retinal periphery within the pigmented P-cadherin+ outer layer of the ciliary epithelium (CE) that is contiguous with the RPE, as depicted in Fig. 1a. These rare cells proliferate in vitro to form clonally derived spheres that are self-renewing and multipotential for all retinal fates, suggesting that they are true stem cells2-4 and not cells derived via trans-differentiation of any CE cell.5 Indeed, there is evidence that CE can give rise to photoreceptors during postnatal development.6 From a therapeutic viewpoint, expansion of RSCs in vitro or reactivation in situ in the eyes, would have a significant impact on autologous transplantation or endogenous stem cell activation, respectively. This can overcome immune rejection, tumorigenic effects, and ethical concerns associated with using embryonic stem cells.7
Figure 1.

RSC purification approach & magnetic resolution fluidic device design. (a) A mouse eye is enucleated, and primary cells are dissected from the ciliary epithelium (CE) of the mouse eye. (b) The CE is dissociated and incubated with antibodies specific to RSC cell-surface proteins. The antibodies are labeled with magnetic nanoparticles (MNPs) via biotin-streptavidin coupling. (c) i. The cells tagged with MNPs are loaded into a three-zone microfluidic device featuring surface ridges and nickel micromagnets. Cells with high magnetic loading are captured in the high capture zone, whereas cells with medium to low magnetic loading are captured in medium and low capture zones, respectively. (c) ii. Simulation of fluid flow within the device. High linear velocity regions generated by the grooved surface (red circle) bring the MNPs tagged cells in contact with the nickel micromagnets where they become captured. (c) iii. Average linear velocities calculated for each capture zone within the cell capture device. (d) Experimental workflow: The cells are first labeled with antibodies, sorted in the device to enrich the cells. The individual enriched cells are then subjected to single-cell RNA sequencing which allows for the identification of two clusters that contained RSCs with differentially expressed cell-surface markers and intracellular markers. The novel cell-surface markers were then used to further purify the RSCs from the CE from 1:500 to 1:2. To test if any of the intracellular marker-encoding genes and transcription factors have any functional roles in RSCs, they were assayed either in a mouse model or using siRNA knockdown in the clonal stem cell sphere assays.
A key enabling advance needed for next-generation therapeutic applications of RSCs is effective methods for their isolation and characterization. To isolate these cells, fluorescence-activated cell sorting (FACS) based on pigmentation and P-cadherin expression has been used to achieve an enrichment of 1 in 500 cells (0.2%). However, because of the physical and/or chemical stress induced by FACS, 90% of the cells typically die during sorting.4. We previously developed a deterministic lateral displacement (DLD)-based microfluidic device to isolate RSCs based on size. This device promoted cell tumbling to limit cell deformation through channels and thereby enhanced the size-sorting resolution. This method permitted an enrichment of ~0.5% and high cell recovery upward of 80%.8 However, this relatively low enrichment of RSCs precluded the ability to carry out transcriptomic profiling of RSCs, which is essential for identifying the gene regulatory networks that regulate stem cell self-renewal and progenitor cell fate specification. For this reason, past single-cell expression profiling studies in the retina either profiled mature cells9 or analysed large retinal cell populations during development.10
Single-cell profiling of rare RSCs is particularly challenging since it requires isolation of pure RSCs in the presence of high background of other retinal cells to enable further downstream analysis. In addition, the method should allow a high recovery of RSCs to minimize processing time and avoid cell death. Microfluidics provides an attractive tool for capturing rare stem cells within small volumes ranging from tens to hundreds of picolitres.11-14 In general, microfluidic isolation of rare cells relies on physical15-19 and/or affinity-based approaches20-22 and leverages electrical,23 optical,24 mechanical,25 acoustic,26 and magnetic forces for cell capture.27 Magnetic cell sorting approaches typically have a minimal effect on cell viability and integrity and is therefore particularly attractive for use with fragile cell types. Furthermore, magnetic sorting methods do not require any complex instrumentation and are not affected by experimental conditions, such as pH, temperature, or ionic strength.28
Based on the unmet needs in the area of single-cell RSC profiling and the potential therapeutic applications of these rare cells, we introduce a microfluidic platform that enables an unprecedented enrichment of RSCs along with off-chip single-cell RNA sequencing to generate a transcriptomic profile. This transcriptomic profile enabled us to identify novel cell-surface markers for RSCs and transcription factors (TFs) involved in RSC functions that are operative in different subpopulations of these cells.
Results and Discussion
A microfluidic approach for enrichment of RSCs
We envisioned being able to enrich RSCs using a gentle isolation procedure by loading cells with magnetic nanoparticles attached to cell-surface markers and introducing the cells into a microfluidic platform that contains surface ridges and nickel micromagnets to create a strong magnetic field and enable cell capture.29 In this approach, an antibody specific to a cell-surface marker is modified with magnetic nanoparticles (MNPs) via biotin-streptavidin coupling. The modified antibodies are incubated with dissociated CE cells dissected from the mouse eye (Fig. 1b). The cells labeled with MNPs are then loaded into a microfluidic device sandwiched between two arrays of magnets. The device features three main design considerations (Fig. 1ci). First, the linear velocity varies along the length of the device. The device features three distinct capture zones that contain regions of low, medium, and high linear velocity where cells with varied levels of labeling would be captured (Fig. 1cii, ciii). This design facilitates the isolation of heterogeneous cell subpopulations with distinct phenotypic features. The capture zones are connected via tubing to facilitate an independent retrieval of cells from each zone subsequent to cell capture. Second, we incorporate nickel micromagnets within each capture zone to enhance the external magnetic force, where the low capture zone is covered with more nickel structures to facilitate capturing cells with low cell-surface marker expression (Fig. S1). Although several ferromagnetic materials (e.g., cobalt)30 can be used, we opted to use nickel as it can be easily deposited with evaporation techniques. Third, we utilize surface ridges that are patterned on the upper side of the device. In comparison to traditional flat-walled microfluidic devices, the ridged surface creates microvortices that disrupt the laminar flow streamlines, thus enhancing the interaction between the MNPs-coated cells and internal micromagnets. This design is based on previously reported fluidic microvortices29 and was adopted to design a microvortex-generating herringbone chip for isolation of rare circulating tumor cells.31 The typical workflow involves sorting the cells labeled with MNPs within the device and the enriched RSCs are subjected to single-cell RNA sequencing to identify differentially expressed (DE) cell-surface markers that can prospectively be utilized to further enrich RSCs. In addition, loss-of-function assays are carried out to test if any of the differential TFs have any functional roles in adult RSCs or retinal progenitors using siRNA knockdowns of the respective genes (Fig. 1d).
Characterization of RSCs
Identifying new cell-surface markers for nanoparticle-based targeting was the first step required to enable this approach. CE cell populations were tested against a panel of 8 antibodies potentially marking the CE and RSCs. The Frizzled receptors are components of the Wnt signaling pathway, a large family of membrane receptors that are involved in retinal neurogenesis.32 ABCG2, a downstream target for Notch signaling, belongs to the ATP-binding cassette superfamily of transmembrane proteins and participates in the maintenance of RSCs or progenitors, under the regulation of notch signaling,33 and is expressed on other adult stem cell populations.32 Notch1 plays a selective role in retinal stem versus progenitor cells in both developing and adult eye,34-37 and overexpression of Notch1 in RSCs increases their symmetric divisions.35 Bone morphogenetic proteins such as BMP2 and BMP4 were identified as principal effectors of the antiproliferative effects on RSCs35. The fibroblast growth factor (FGF) was previously shown to control RSC renewal and differentiation. In addition, exposure to FGF favors RSC differentiation into photoreceptor-like cells38. P-cadherin was used for prospective enrichment of mouse RSCs using FACS4.
This screen identified 3 cell-surface markers, including Frizzled1 (Fz1), ABCG2, and Notch1 that permitted high enrichment of RSCs (Fig. 2a). The cells captured within the device were immunostained with antibodies specific for the three cell-surface markers and counted. Captured cells were also assessed using clonal sphere-forming assays as well as viability assays. The number of immunostained cells correlated positively with the number of formed spheres, which allowed us to identify a population of RSCs with a diameter of ~10 um,8 as shown by the fluorescence images provided in Fig. 2b. The results echoes our previous finding in which we demonstrated that a subset of RSCs (~10 um) can be isolated using a deterministic lateral displacement microfluidic device.8 Cell sorting using a combination of antibodies specific to the three markers, henceforth referred to as FAN, demonstrated that it is possible to isolate the RSC population with up to 54-fold enrichment relative to unsorted cells, giving a frequency of sphere formation averaging 26% (Fig. 2c). Conversely, sorted FAN− cells did not give rise to any RSC spheres, suggesting that the FAN biomarker panel can effectively capture the entire RSC population. In addition, the FAN biomarker panel permitted higher enrichment compared to each of the individual markers alone, revealing that no single marker was sufficient to isolate RSCs. The in vivo expression of FAN was confirmed by immunostaining of the mouse eye (Fig. 2d) and flow cytometric analysis of the dissociated CE cells subsequent to immunostaining (Fig. S2a). In addition, quantitative PCR revealed the expression of the FAN markers within sorted FAN+ versus FAN− cells, as well as in the primary RSC spheres (Fig. S2b). The sorted FAN− cell population exhibited lower gene expression of the FAN markers. The performance of the new approach was benchmarked against FACS. We found that a FAN+ cell population sorted by FACS had ~6% viability, whereas the microfluidics platform yielded ~74% cell viability (Fig. 2e). In addition, the progeny in the spheres that arose from FAN+ sorted single mouse and human RSCs were still able to show pan-retinal differentiation (Fig. S2c-f).
Figure 2.

Isolation and characterization of RSCs. (a) Preliminary screen of candidate antibodies using the magnetic resolution approach. Frequency of clonal sphere formation represents the ratio of formed spheres to the total number of isolated live cells normalized to unsorted cells. (b) Fluorescence images of single cells isolated in the device and immunostained with FITC-labeled ABCG2, Cy5-labeled Notch1, and AF594-labeled Fz1 antibodies. (c) Dissociated CE cells incubated with MNPs-labeled antibodies specific to Fz1, ABCG2, and Notch1 (FAN), exhibited the highest capture efficiency of RSCs. (d) Immunostaining of the CE in mouse eyes with ABCG2, Notch1 and Fz1 antibodies. (e) Percentage of viable cells sorted using the microfluidic device benchmarked against FACS. *** p<0.005 (Student’s t-test).
Deep sequencing of enriched primary cells reveals two distinct RSC clusters
Single-cell SMARTseq4 RNA deep sequencing39 was used to analyze 47,750 single-nucleus transcriptomes from 177 usable cells collected from a high-enrichment FAN+ fraction subsequent to cell sorting. Initial analyses of differentially expressed (DE) genes indicated that there were distinct cell populations (Fig. S3) with varying expression patterns of genes corresponding to the sorting antibodies (Fig. S4a, b). Using scClustVis unsupervised cluster visualization of the scRNA seq data,40 and 2D visualization of the cells using t-distributed stochastic neighbor embedding (t-SNE) grouped the transcriptomes into 5 distinct clusters (Fig. 3a), and heatmaps of the DE genes were generated (Fig. S5). Each of these 5 clusters were assigned to a likely specific cell type based on the expression of known marker genes (Fig. S3c). The DE genes in each cluster suggested that cluster 2 (18.6% of profiled cells) may contain RSCs. In addition, cluster 4 (13%) also expressed genes that were indicative of RSCs. The other three clusters expressed markers specific for trabecular meshwork (cluster 0, 29.4%), ciliary epithelium (cluster 1, 21.5%), and cornea/limbal (cluster 3, 17.5%) cells, resulting from the primary tissue dissection.
Figure 3.

SMART deep sequencing analyses of sorted FAN+ mouse CE cells. Single FAN+ cells sorted using the microfluidic device were sequenced using SMART-seq v4 after a cohort of cells were tested for sphere-forming ability (1:2 enrichment of RSCs). A total of 177 usable cells were analyzed. (a) t-SNE plot identifying 5 clusters. Uniquely DE genes from each cluster were used to annotate them as trabecular meshwork cells (0), ciliary epithelial cells (1), corneal/limbal cells (3), and retinal stem cells in both clusters 2 and 4, confirmed by RSC sphere-forming ability. (b) Heat map showing the cell-surface markers used to isolate the RSCs, the new cell-surface markers and transcription factors that are DE in each RSC cluster. Dot size indicates the proportion of cells in each cluster, while the colour shows mean normalized gene expression in the cells in which the gene was detected. (c) Cell sorting using antibodies specific to the identified cell-surface markers. The cells isolated from each capture zone were plated at clonal density (10 cells/μL) for sphere-forming assays. (d) CE cells were incubated with a combination of antibodies specific to the identified cell-surface markers, sorted using the microfluidic device and plated at clonal density. Error bars = s.e.m., N=3 for each.
Microfluidic enrichment of RSCs based on identified cell-surface markers
The scRNA seq data revealed four DE cell-surface marker-encoding genes which were expressed by the two proposed RSC clusters, including Cnr1 by cluster 2, Grm7 by cluster 4, and Nptxr and Il15ra found in both clusters (Fig. 3b, Fig. S6). To determine whether the two proposed RSC clusters comprised RSCs, we sorted these clusters using our microfluidic device by targeting the novel DE cell-surface markers and performed clonal sphere assays. The highest enrichment of RSCs was achieved when we targeted CE cells with an antibody specific to Grm7, suggesting that it is a DE marker for cluster 4 RSCs that may have greater propensity to proliferate. In addition, high enrichment of clonal sphere forming cells was attained when we targeted either Nptxr or Il15ra, which are expressed by both RSC populations. Conversely, targeting Cnr1, led to lower enrichment of clonal sphere-forming cells compared to Grm7, suggesting it may be a DE marker for a more quiescent RSC population (cluster 2). Overall, targeting a single cell-surface marker was not sufficient to capture all RSCs, as evident by the presence of some RSC sphere-forming cells in the effluents (Fig. 3c). As expected, targeting the 4 new markers simultaneously, hereafter referred to as CING, resulted in 138-fold enrichment of sphere-forming cells relative to unsorted cells (Fig. 3d). In addition, targeting CING enabled capturing all the RSC sphere-forming cells with no sphere formation detected in the effluent. This indicates that all the stem cells were located within clusters 2 and 4 (and not in clusters 0, 1 or 3). The expression of the CING biomarker panel was confirmed by immunohistochemistry (Fig. S6a) and by flow cytometric analysis of the dissociated CE cells (Fig. S6b). The higher enrichment achieved by targeting CING compared to FAN can be ascribed to the relative expression of FAN by some cells in all the cell clusters (Fig. 3b, Fig. S4).
Validation of TFs identified in RSCs using RNA interference
The transcriptional identity of RSCs determined using Enrichr,41 included distinct TFs that are expressed either in cluster 2 or in cluster 4 cells (Fig. 3b, Fig. S3b). We found that Creb1 is highly expressed by both cluster 2 and cluster 4 cells but has very low expression in the other three cell clusters (Fig. 3b, Fig. S7a). We also found that Hdac10 is expressed specifically by cluster 2 cells comprising the relatively quiescent RSC population. Loss-of-function studies were performed to assess the effects of these genes in adult RSCs or their progenitor cells. In the following clonal sphere assays, the failure to form RSC spheres may be a result of defects in either adult RSCs, their downstream progenitors or both. The effect on RSC proliferation was monitored by evaluating the total number of primary spheres that reflects the number of endogenous stem cells, or secondary spheres, which is indicative of the self-renewal ability of stem cells.2 The average diameter of clonally expanded spheres can be used as an estimate for the proliferation of retinal progenitors, given that more than 99% of the cells in each sphere are progenitor cells.2
First, we explored the role of Creb1 expression in mutant CE obtained from homozygous hypomorphic littermates, which have an approximately 80% depletion of Creb1.42 Both wild-type and heterozygous hypomorphs were used as a control. It is worth noting that Creb1 codes for a phosphorylation-dependant TF that contributes to the innate protection against photoreceptor degeneration and promotes neuronal survival in response to retinal injury.43 It also has been implicated in mouse brain neural progenitor survival and proliferation.44 We observed a large reduction in the number of primary spheres in the Creb1 homozygous hypomorphs relative to the wild-type and heterozygotes, thus indicating alterations in adult RSC proliferation and/or survival (Fig. 4a). In addition, the spheres were marginally smaller in diameter, thus indicating a role of Creb1 in the proliferation of retinal progenitors (Fig. 4b). We also tested the self-renewal capacity of Creb1 homozygous hypomorphic RSCs and found that they had a diminished capacity to form new secondary spheres (Fig. 4c). Knocking down Creb1 in adult RSCs in vitro using small interfering RNAs (siRNAs) reproduced the genetic hypomorph effect on stem cell number with a 50% loss of sphere formation (Fig. 4d). Most interesting, the differentiation of the RSC spheres derived from the Creb1 genotypes toward RPE, rod photoreceptors, and Müller glia was not affected (Fig. S7b). All together, these data demonstrate the involvement of Creb1 in the proliferation of RSCs and retinal progenitors, but not their differentiation.
Figure 4.

Validation of TFs in RSCs using RNA interference. (a) Frequency of sphere formation from Creb1 mutant CE (wild type: WT, heterozygous hypomorphs: Het, and homozygous hypomorphs: Homo). Homo had a 45-50% decrease in stem cell frequency, and the total number of stem cells per eye was also decreased by 56-65% compared to WT and Het (WT=165 +/− 9.4, Het=207.4 +/−16.9, Homo=71.9 +/−16.7; F2,21=28.47, p<0.0001). (b) Creb1 RSC sphere diameter also decreased slightly in the Homo showing an effect on progenitor cells (WT=93.75 +/−1.72, Het: 86.19 +/−1.76, Homo: 77.92 +/−1.04; F2,74=26.35, p<0.0001). (c) Self-renewal of RSCs was tested by dissociating the primary spheres into single cells and plating them at 1000 cells/well. The Homo Creb1 RSCs had a diminished capacity to form new secondary spheres (WT: 13.2 +/−1.63, Het: 12.2 +/−1.10, Homo: 7.2 +/− 1.66, F2,12=5.516, p=0.02). (d) siRNA knockdown of Creb1 in adult RSCs in vitro reproduced the genetic homozygous hypomorph effect on RSC number with a 50% loss of sphere formation compared to siRNA controls (t5=5.217; p=0.0034). (e) Primary CE cells were kept undifferentiated for 5 days in the presence of Hdac10 siRNA or NT siRNA, dissociated and then sorted using either Cnr1 (cluster 2) or Grm7 (cluster 4) populations. Hdac10 significantly increased the number of sphere-forming cells in both the Cnr1 and the Grm7 populations as well as the negative populations compared to controls (F1,24=13.56, p=0.0012). Separate qPCR experiments demonstrated that the siRNAs caused 75%-90% decrease in the expression of the target genes (data not shown). Error bars=s.e.m. * indicates where a column is significantly different from its control: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Second, we investigated the role of Hdac10, which codes for a transcriptional repressor that has implications in stem cell identity through epigenetic modulation.45 To investigate the possibility that loss of Hdac10 may shift the quiescent RSC population to a more active state, we pre-treated primary CE cells on a monolayer with Hdac10 or control non-targeting siRNAs for 5 days. We sorted the cells using our microfluidic approach by targeting either Cnr1(specific to cluster 2 containing the more quiescent RSCs) or Grm7 (specific to cluster 4 containing the more proliferative RSCs) with the specific MNPs-labeled antibody and replated the cells to perform sphere colony forming assay. We found that knocking down Hdac10 greatly increased the number of clonal RSC spheres in both the Cnr1+ (4-fold increase) and the Cnr1− (3.25-fold increase) populations as well as the Grm7+ (1.5-fold increase) population as compared to the non-targeting siRNA control population (Fig 4e). This suggests that the stem cells in the Hdac10 treated monolayers are shifting to a more activated state and/or there is an increase in the symmetric division of the stem cells before the sphere assay. It also appears that the Cnr1 marker retains its expression after 5 days of Hdac10 siRNA treatment since the greatest increase in sphere number was in the Cnr1+ fraction. Application of the Hdac10 knockdown only throughout the differentiation period of already formed primary RSC spheres had no effect on the differentiation of the clonal spheres to postmitotic retinal cell types. These data suggest functional roles for the DE TFs in RSC proliferation and maintenance, without having any effects on the differentiation profiles of their progeny (Fig. S7c).
Identification of a transitional state between quiescent and proliferative RSCs
A re-analysis of the presumed RSC clusters (2 & 4) using scClustVis software revealed a third significantly separate sub-cluster of RSCs, which may be a transitional population between the more quiescent (cluster 2) and the primed-to-proliferate RSCs (cluster 4) (Fig. S8). This may indicate that all three RSC clusters may be a single population of RSCs, but in three different states – one being quiescent, a second transitional state, and a third primed-to-proliferate state, similar to the cells populations previously described for muscle stem cells46 and neural stem cells.47 Indeed, the relative quiescence of adult retinal stem cells in vivo is induced by the postnatal release of bone morphogenetic proteins and serum frizzled-related protein 2 from the lens and cornea.32 The siRNA experiments suggest that Hdac10 might epigenetically retain the RSCs in a quiescent state, but once Hdac10 is inhibited – the RSCs move to a transitional or a primed-to-proliferate state, and then are more likely to actively proliferate and form a clonal RSC sphere in vitro.
Conclusions
Enabled by a microfluidic approach that allowed high-fidelity, high-yield isolation of retinal stem cells, we used whole-transcriptome profiling to obtain a global overview of RSCs at the single-cell level and identify their transcriptional identity for the first time. We have identified and tested a set of four cell-surface markers that are useful for the enrichment of RSCs from the adult ciliary epithelium. This finding defines an approach that we anticipate would be broadly applicable in basic developmental studies as well as clinical applications. We believe that the comprehensive gene expression profiling provided by this study will be broadly useful for better understanding of the genetic program of RSCs and the molecular mechanisms controlling their quiescence, self-renewal and multipotentiality. This may allow targeted therapies triggering the in-situ reactivation of quiescent endogenous RSCs and differentiation of their progeny into appropriate retinal cells to replace damaged cells.
Methods
Computational simulations
Magnetic and flow field simulations (S1) were carried out in COMSOL Multiphysics, with the goal of comparing the magnitude of magnetic force acting on each zone within the device with the magnitude of drag force opposing cell capture.
Cell capture occurs when the magnetic force acting on the cell is large enough to balance the drag force generated by the flow. The magnetic force acting on the cells labeled with magnetic nano-beads is given by (I):
| (I) |
Where Nb is the number of beads per cell, Vm is the bead volume, Δχbead [unitless] is the difference between the magnetic susceptibility of the bead and the medium, μ0 [H/m] is the permeability of free space (4π×10−7 H/m), and B [T] is the applied magnetic field. Here we used magnetic nanoparticles with diameter of 100 nm.
The transverse drag force acting on a cell, neglecting wall effects, at low Reynolds numbers is given by Stokes’ law (II):
| (II) |
Where r [m] is the cell radius (10 μm), η [Pa×s] is the dynamic viscosity of the medium (0.001 Pa×s), and v [m/s] is the velocity of the cell.
Primary mouse cell isolation and culture
Adult CD1 (RRID:IMSR_CRL:22) and C57Bl/6 (RRID:IMSR_CRL:27) mice were obtained from Charles River. B6.129S2Creb1tm1Gsc/J (RRID:IMSR_JAX:004445) breeding pairs were obtained from Dr. Alcino Silva’s repository at Jackson Labs via Dr. Sheena Joselyn (University of Toronto, Canada). Both sexes of animals were used for these experiments. Experimental procedures and husbandry were performed in accordance with the CCAC and approved by the Animal Care Committee at the University of Toronto. Cells were dissected as described and grown in a 5% CO2 incubator at 37°C.
Cells were isolated from the ciliary epithelium (CE) of the eye. Adult mice were killed by cervical dislocation, and their eyes were harvested in oxygenated artificial cerebrospinal fluid (aCSF: 124 mM NaCl, 5 mM KCl, 1.3 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM D-glucose). The neural retina was first dissected free of the retinal pigmented epithelium (RPE). The cornea and iris were cut away on one edge of the CE and the RPE was cut away of the other edge of CE, leaving the sclera overlaid with the ciliary muscle and the bilayered CE. Each tissue was treated with Dispase (Corning) for 10 min at 37°C to facilitate the removal of the RPE from the underlying basement membrane. Tissue was dissected and placed in a trypsin solution at 37°C for another 10 min [artificial CSF modified to contain high Mg2+ (3.2 mM MgCl2) and low Ca2+ (108μM CaCl2), 1.33 mg/ml trypsin (Sigma-Aldrich), 0.67 mg/ml hyaluronidase (Sigma-Aldrich), and 0.2 mg/ml kynurenic acid (Sigma-Aldrich)]. The CE was removed from the muscle and sclera, dissociated and then the cells were centrifuged at 150g for 5 min, and the enzyme solution was removed and replaced with serum-free media,2 containing trypsin inhibitor (1 mg/ml ovamucoid; Worthington). Cells were triturated until a single cell suspension was achieved and then centrifuged again as above. The cells were centrifuged again and resuspended in serum-free media, containing 20 ng/mL FGF2 (F0291, Sigma-Aldrich) and 2 ng/mL heparin (H3149, Sigma-Aldrich) and either plated directly in 24-well Nunc plates or run through the microfluidics device. The cells were either counted and plated at clonal density or sorted as described below and then plated. The procedure for the isolation of mouse retinal stem cells can be accessed online at JoVE.48
Primary human cell isolation and culture
Human eyes that have been consented to be used for research purposes and were procured from the Eye Bank of Canada (Toronto, ON) within 24 h post-mortem. No age or sex restriction was used in this study. All procedures using human tissue were approved by the University of Toronto Research Ethics Board. Cells were dissected as described and grown in a 5% CO2 incubator at 37°C. Single cells from the Pars Plana and Pars Plicata were isolated as previously described.48 The eyes were placed in oxygenated artificial cerebrospinal fluid (aCSF: 124 mM NaCl, 5 mM KCl, 1.3 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM D-glucose). The neural retina was first dissected free of the RPE. The cornea and iris were cut away on one edge of the CE and the RPE was cut away of the other edge of CE leaving the sclera over-laid with the ciliary muscle and the bilayered CE. The CE, muscle and Bruch’s membrane were peeled away from the sclera and the underlying Bruch’s membrane was mostly removed with a combination of peeling and microdissection scissors. Each tissue strip was treated with Dispase (Corning) for 20 min at 37°C to facilitate the removal of the RPE from the underlying basement membrane. Tissue was dissected and placed in a trypsin solution at 37°C for another 20 min [artificial CSF modified to contain high Mg2+ (3.2 mM MgCl2) and low Ca2+ (108μM CaCl2), 1.33 mg/ml trypsin (Sigma-Aldrich), 0.67 mg/ml hyaluronidase (Sigma-Aldrich), and 0.2 mg/ml kynurenic acid (Sigma-Aldrich)]. The CE was scraped away from the underlying muscle, which was removed from the dish. The cells were then transferred to a 15-mL conical Falcon tube, dissociated with a cotton-plugged glass pipette, and then centrifuged at 150g for 5 min, and the enzyme solution was removed and replaced with serum-free media, containing trypsin inhibitor (1mg/ml ovamucoid; Worthington). The cells were further dissociated with a small borehole glass pipette and then placed back in the centrifuge for another 5 minutes. The supernatant was then removed and replaced with growth factor media. The cells were then counted and plated at 10 cells/μl in serum-free media (SFM) containing FGF2 and heparin. Cells proliferated to form floating clonal sphere colonies in cell suspension, and these retinal spheres were counted after 7 days in culture and plated on laminin for differentiation using the same protocol as the mouse cells.
Device fabrication
Glass slides coated with a Ni layer (1.5 μM) (EMF Corp.) were used to fabricate the microfluidic device. A layer of positive photoresist (AZ1600) was spin-coated to generate the zigzag micromagnets using contact lithography. After exposure for 10 s, the photoresist was developed, then the spare Ni layer was wet-etched, and the top resist was removed. Polydimethoxysilane (PDMS, Dow Chemical) soft lithography was used to fabricate the microfluidic channel. First, silicon masters were fabricated using two different masks. Two layers of SU8 (Microchem) with different heights were patterned to create the surface ridges and the microfluidic channel. SU8 with thickness of 50 μm was first spin-coated to produce the channel and then the second layer of SU8 (thickness: 50 μm) was used to create the surface ridges. PDMS and curing agent at the ratio of 10:1 were poured on the chip master. This was followed by baking the PDMS at 67°C for 1 hour. Afterward, the replicas were peeled, and holes were punched for the inlet and outlet in the PDMS layer. PDMS replicas were attached to the glass slides with the zigzag Ni micromagnet using a 30s plasma treatment and left to bond overnight. Finally, silicon tubing was attached to the inlet and outlet of the device. Prior to use, the devices were conditioned with 1% Pluronic F68 (Sigma-Aldrich) in phosphate-buffered saline (PBS) for 1 hour to reduce the nonspecific adsorption.
Labeling antibodies with magnetic nanoparticles
Streptavidin-coated magnetic nanoparticles (10 μg, 100 nm, 4204-5, Chemicell) were incubated with 1 μg of either biotin-labeled ABCG2 antibody (144108-biotin, USBiological), biotin-labeled Notch1 antibody (Bbs-1335R-biotin, Bioss, RRID:AB_11060483), biotin-labeled Frizzled-1 antibody (BAM11201, R&D Systems, RRID:AB_356852), biotin-labeled P-cadherin antibody (bs-1159R-biotin, Bioss, RRID:AB_11066484), biotin-labeled FGFR2 antibody (bs-0675R-biotin, Bioss, RRID:AB_11043719), biotin-labeled BMPR1A antibody (bs-1509R-biotin, Bioss, RRID:AB_11099605), biotin-labeled BMPR1B antibody (bs-6639R-biotin, Bioss, RRID:AB_11103392), biotin-labeled BMPR2 antibody (bs-4237R-biotin, Bioss, RRID:AB_11063902), biotin-labeled Cnr1 antibody (bs-1683R-biotin, Bioss, RRID:AB_11057646), biotin-labeled Grm7 antibody (LS-C527894, LifeSpan BioSciences), biotin-labeled Il15ra antibody (LS-C471046, LifeSpan BioSciences), or biotin-labeled Nptxr antibody (ORB451807, Biorbyt Ltd.) in 100 μl of phosphate buffered saline (PBS) for 30 min at room temperature. Subsequently, the magnetic nanoparticles-labeled antibodies were pelleted using a magnetic-ring stand (Thermo Fisher Scientific) and washed three times with PBS solution.
Isolation of RSCs
Primary CE cells were incubated with either single or mixture of antibodies modified with magnetic nanoparticles (prepared in the previous step), for 45 min at room temperature. The cells were loaded into the microfluidic device at a flow rate of 2 ml/h. After washing three times with PBS, the magnets were removed, and the isolated cells were aspirated from each capture zone.
Post-sorting cell culture
After cell sorting using the microfluidic device, the cells were collected in the media, counted using a hemocytometer, and plated at clonal densities in Nunc 24-well plates. In experiments where noted, the cells were visually counted after 12-16 hours to assess initial cell survival. Spheres that were greater than 80 μm on day 7 were counted. We use this cut-off because it is difficult to dissociate any sphere below this cut-off, which would subsequently confound the assessment of its self-renewal ability.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cells using RNA isolation kit (35300, Norgen Biotek). The genomic DNA was removed using RNAse-free DNAse I kit (25710, Norgen Biotek), according to the manufacturer’s protocol. RNA was converted to cDNA using a SuperScript VILO cDNA Synthesis Kit (11754050, Thermo Fisher Scientific), according to the manufacturer’s protocol. A comparative Ct experiment was performed on a QuantStudio 6 real-time PCR (Applied Biosystems). The following Taqman assays (Thermo Fisher Scientific) were used: Pax6 (Mm00443081_m1), Rax (Mm01258704_m1), Vsx2/Chx10 (Mm00432549_m1), Abcg2 (Mm00496364_m1), Frizzled1 (Mm00445405_s1), Notch1 (Mm00627185_m1), NCadherin/Cdh2 (Mm01162497), PCadherin/Cdh3 (Mm01249209_m1), Ppib (Mm00478295_m1), Hdac10 (Mm01308119_g1), Zfp3 (Mm03053166_s1), Hey2 (Mm01180513_m1), Nrip3 (Mm00508049_m1). Quantification was performed using the delta Ct method with Actb (Mm02619580_g1) or Gapdh (Mm99999915_g1) as endogenous controls and dissociated primary ciliary epithelium as a calibrator. The assays were carried out in triplicates using 10 ng cDNA for each sample in a 384-well plate. The 20 μl reaction mixture consisted of 10 μl 2X TaqMan gene expression Master mix (4304437, Thermo Fisher Scientific), 1 μl of 20X assay, and 9 μl of cDNA (final concentration of 10 ng/μl). Cycling conditions for the qPCR were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Fluorescence-activated cell sorting (FACS)
Dissociated primary CE cells (1,000,000 cells) were incubated with a blocking buffer (1% BSA in PBS) for 30 min at room temperature, followed by another incubation for 30 min with 10 μl of 100 μg/ml of either FITC-labeled ABCG2 antibody (orb401130, Biorbyt Ltd), Cy5-labeled Notch1 antibody (bs-1335R-Cy5, Bioss; RRID:AB_11060483), or APC-labeled Frizzled1 antibody (FAB11201A, R&D Systems; RRID:AB_2108935), each-at-a-time, for 1 h at room temperature. Gating criteria for cell analysis were set according to the level of autofluorescence of non-fluorescent primary CE cells using a FACSAria system (BD Biosciences). Sorted cells were then counted on a hemacytometer and plated in FGF2+heparin in serum-free media at clonal density (<10 cells/μl).
Flow cytometry
Flow cytometry was used to confirm the expression of cell-surface markers by RSCs. Briefly, CE cells (100,000 cells) were incubated with a blocking buffer (2% BSA in PBS) for 30 min on ice. Afterward, the cells were fixed 4% paraformaldehyde solution (Sigma-Aldrich). The cells were incubated with 10 μl of biotin-labeled Cnr1 antibody (bs-1683R-biotin, Bioss, RRID:AB_11057646), biotin-labeled Grm7 antibody (LS-C527894, LifeSpan BioSciences), biotin-labeled Il15ra antibody (LS-C471046, LifeSpan BioSciences), or biotin-labeled Nptxr antibody (ORB451807, Biorbyt Ltd) in 100 μl of phosphate buffered saline (PBS), for 1 h at room temperature. The cells were then incubated with 2 μl APC-streptavidin for 30 min at room temperature. Control experiments were carried out in which the cells were incubated with 10 μL of 100 μg mL−1 biotin-labeled rabbit isotype control (Cat.# ab200208, Abcam) for 1 h at room temperature followed by incubation with APC-streptavidin. Subsequently, samples were injected into FACSCanto flow cytometer (BD Biosciences) and measurements were plotted as histograms. Absorbance values were normalized to an isotype control. A total of 10,000 cells were analyzed per cell sample.
Sphere differentiation and immunostaining
Individual clonal RSC spheres (mouse or human) were selected after 7 days of primary culture. RSC spheres that arose from single cells either plated directly or after isolation of the FAN+ fraction collected from the microfluidic device, were differentiated under pan-retinal conditions (1% FBS+FGF2+heparin) in Nunc 48-well plates coated with 50 ng/ml laminin (L2020, Sigma-Aldrich) for 4 h at 37°C. The cells were fed every 3-4 days by first removing 400 μl of the media and adding 500 μl of freshly made growth factor media. The cells were fixed on day 28 of differentiation with 4% paraformaldehyde, blocked in 10% Normal Goat Serum, 2% BSA, permeabilized with 0.3% Triton X-100 and then immunostained with primary antibodies overnight at 4°C. An immunofluorescence approach was used to identify different retinal cell types in which the differentiated spheres were incubated with antibodies specific to rhodopsin (MAB5316, RRID:AB_2156055), cone arrestin (AB15282, RRID:AB_301818), RPE65 (MAB5428, RRID:AB_571111), PAX6 (AB2237, RRID:AB_2270373), and GFAP (G3893, RRID:AB_477010) from MilliporeSigma; PKCα (MA1-157, RRID:AB_2536865) from Thermo Fisher Scientific; MiTF (ab12039, RRID:AB_298801) and CRX (ab193448) from Abcam; Chx10 (sc-365519, RRID:AB_10842442) from Santa Cruz Biotechnology.
Immunostaining was also performed on 10-14 μm eye or 10 μm RSC sphere sections on Superfrost Plus glass slides with the sections outlined to form a well using a hydrophobic barrier PAP pen (H-4000, Vector Labs). Antibodies specific to Cnr1 (PA1-743, RRID:AB_2736149), Il15ra (PA5-79467, RRID:AB_2746583), Nptxr (PA5-25954, RRID:AB_2543454), and Grm7 (PA5-77422, RRID:AB_2736149) were obtained from Thermo Fisher Scientific; ABCG2 (orb155559) from Biorbyt Ltd; Notch1 (bs-1335R, RRID:AB_10854452) from Bioss and Frizzled-1 (MAB11201, RRID:AB_357466) from R&D Systems.
Secondary antibody staining was carried out with AF488-labeled goat anti-rabbit [A-11034, RRID:AB_2576217], AF488-labeled goat anti-mouse [A-11001, RRID:AB_2534069], AF568-labeled goat anti-rabbit [A-11011, RRID:AB_143157], and AF568 goat anti-mouse antibodies [A-11004] (RRID:AB_141371) (1:400, Thermo Fisher Scientific). Secondary antibodies were incubated directly with differentiated cells without primary antibodies as negative controls. Nuclei were stained using 10 ng/ml of 4’,6-diamidino-2-phenylindole (DAPI, P36931, Prolong Gold, thermo Fisher Scientific) nuclear stain or bisbenzimide/Hoechst (B1155, Sigma-Aldrich).
Cell imaging
Immunostained RSCs within the microfluidic devices or differentiated retinal cells within the tissue culture plates were imaged with a Nikon Ti-E Eclipse microscope with an automated stage controller and a CMOS camera (Andor Neo). The blue channel was used for DAPI staining, with a typical exposure time of 10–20 ms. The green channel was used for the AF488- and FITC-conjugated antibody staining, with a typical exposure time of 40–60 ms. The orange channel was used for the AF568-conjugated antibody staining, with a typical exposure time of 100–120 ms. The red channel was used for the AF647- and APC-conjugated antibody staining, with a typical exposure time of 200–300 ms. The exposure time was set individually for each sample and kept constant while scanning. The imaging was qualitative in nature and hence the variation of exposure time did not affect the results. Cells were imaged by overlaying the fluorescent images. In addition, immunofluorescence of indicated proteins (ABCG2, Notch1, Frizzled-1, Cnr1, Il15ra, Nptxr, and Grm7) was measured by Zeiss AxioObserver D1 and their localizations were confirmed with confocal image analysis using Olympus Fluoview (FV1000).
Single-cell RNA sequencing
Primary ciliary epithelial cells were incubated with a mixture of 1:1:1 Fz1, ABCG2, and Notch1 antibodies modified with magnetic nanoparticles and then sorted using the microfluidic device. The single cells were picked from high-enrichment FAN+ fraction (combined high and medium zones) under a microscope at 4°C and placed individually in two 96-well PCR plates on a −20°C block. The plates were sealed and kept at −80°C. The rest of the cells were counted using a hemocytometer and then plated in 24-well plates under sphere-forming conditions and then re-counted visually within 16 hours. The number of spheres was determined after 7 days in vitro. Once the spheres were counted and 1:2 frequency of sphere formation was achieved, the frozen cells were delivered to the Princess Margaret Genomics facility for sequencing. Single cell cDNA was isolated using the SMART-Seq v4 Ultra Low Input RNA Kit for sequencing. Direct-sort single-cell RNA sequencing was carried out using Illumina NextSeq500 (Illumina Inc.). The data set were then aligned to GRCm38 using HiSAT2, transcripts assembly with Stringtie (known genes only) and raw read count with HTSeq file format. The cluster assessment and visualization were performed using scClustVis software: (https://f1000research.com/articles/7-1522/v2). Further evaluation of the dataset for pathway analysis and differentially expressed transcriptional factors were performed using Enrichr software.
Knockdown of transcription factors
Using Accell siRNAs at 1 μM concentrations (Dharmacon), we assessed whether the candidate differentially expressed transcription factor genes identified using the Enrichr software were functionally relevant in retinal stem cells or their progeny using the sphere-forming assay. The negative controls were untransfected cells and cells treated with pooled non-targeting (NT) siRNA in Accell delivery media containing FGF2 and heparin. Primary CE cells from CD1 mice were first sorted using the microfluidic device into either Cnr1+ or Grm7+ enriched populations or unsorted controls. The cells were then mixed with siRNAs in the media to a final concentration of 1 μM siRNA in 100 μl media with 1000 cells/well in a 96-well Nunc plate. Sphere number was compared between our serum-free media and the Accell delivery media and the number of spheres was unaffected by the different media conditions, so we only used the Accell delivery media for these experiments. The positive controls were cells treated with Gapdh siRNA and Ppib siRNA. Half of the samples were then assessed for gene knockdown using qPCR to measure the RNA levels of the knocked down genes on day 3, and the transfection efficiency was measured using green, fluorescent NT siRNA, which was also measured on day 3. For the sphere assays, the cells were only treated with the Accell siRNAs at the time of plating which lasts for 3 days, and the number of spheres were assessed on day 7 after plating.
Monolayer siRNA Experiment
Primary CE cells were plated for 5 days in 6-well Nunc plates at 100,000 cells/well on laminin in the presence of FGF2 and heparin. 1 nM Hdac10 siRNAs or NT siRNAs were added at time of plating. On day 5, the cells were incubated in TrypLE enzymes for 5 min at 37°C, diluted with SFM, spun down at 1500 RPM, resuspended in 400μl of SFM and counted. The cells were then divided and sorted into Cnr1+ or Grm7+ populations. The cells collected from the high capture zone, or the effluent were counted and replated for retinal sphere assays at clonal density. The spheres were counted 7 days after plating in the clonal assay.
Statistical Analyses
All statistical analyses were performed using the GraphPad Prism software. The specifics of the statistical tests and number of replicates are stated in the figure legends. Statistical significance was defined as follows: ns (p > 0.05), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001).
Supplementary Material
ACKNOWLEDGMENTS
We thank the families of the eye donors for their generosity, and the medical fellows of the Eye Bank of Canada for the management of the human eye gifts. Research reported in this publication was supported by the Canadian Institutes of Health Research (Grant #s FDN-148415 and FDN-501558), the Krembil Foundation, the Medicine by Design CFREF program, the Natural Sciences and Engineering Research Council of Canada (Grant #2016-06090), the Province of Ontario though the Ministry of Research, Innovation and Science (Grant #RE05-009), and the National Cancer Institute of the National Institutes of Health (Grant # 1R33CA204574). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the other funding agencies.
Footnotes
The authors declare no competing financial interests. All data and materials are within the paper, supplementary figures and the scRNA sequencing data will be made available online (shiny.baderlab.org).
Safety Statement. No unexpected or unusually high safety hazards were encountered with the reported work.
Additional data and figures, including characterization of ANF+ CE populations, validation of identified cell-surface markers, validation of identified transcription factors.
REFERENCES
- 1.da Cruz L, Fynes K, Georgiadis O, Kerby J, Luo YH, Ahmado A, Vernon A, Daniels JT, Nommiste B, Hasan SM, Gooljar SB, Carr AF, Vugler A, Ramsden CM, Bictash M, Fenster M, Steer J, Harbinson T, Wilbrey A, Tufail A, Feng G, Whitlock M, Robson AG, Holder GE, Sagoo MS, Loudon PT, Whiting P and Coffey PJ, Nat. Biotechnol, 2018, 36, 328–337. [DOI] [PubMed] [Google Scholar]
- 2.Tropepe V, Coles BL, Chiasson BJ, Horsford DJ, Elia AJ, McInnes RR and van der Kooy D, Science, 2000, 287, 2032–2036. [DOI] [PubMed] [Google Scholar]
- 3.Coles BL, Angenieux B, Inoue T, Del Rio-Tsonis K, Spence JR, McInnes RR, Arsenijevic Y and van der Kooy D, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 15772–15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballios BG, Clarke L, Coles BL, Shoichet MS and Van Der Kooy D, Biol. Open, 2012, 1, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cicero SA, Johnson D, Reyntjens S, Frase S, Connell S, Chow LM, Baker SJ, Sorrentino BP and Dyer MA, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 6685–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belanger MC, Robert B and Cayouette M, Dev Cell, 2017, 40, 137–150. [DOI] [PubMed] [Google Scholar]
- 7.Ramsden CM, Powner MB, Carr AJ, Smart MJ, da Cruz L and Coffey PJ, Development, 2013, 140, 2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomis S, Labib M, Coles BLK, van der Kooy D, Sargent EH and Kelley SO, ACS Appl. Mater. Interfaces, 2018, 10, 34811–34816. [DOI] [PubMed] [Google Scholar]
- 9.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A and Sanes JR, Cell, 2016, 166, 1308–1323 e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark BS, Stein-O’Brien GL, Shiau F, Cannon GH, Davis-Marcisak E, Sherman T, Santiago CP, Hoang TV, Rajaii F, James-Esposito RE, Gronostajski RM, Fertig EJ, Goff LA and Blackshaw S, Neuron, 2019, 102, 1111–1126 e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Wu M, Han X, Wang P and Qin L, Angew. Chem. Int. Ed. Engl, 2015, 54, 10838–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiu FWY, Bagci H, Fisher AG, deMello AJ and Elvira KS, J. Chem. Technol. Biotechnol, 2016, 91, 16–24. [Google Scholar]
- 13.Zhang J, Wei X, Zeng R, Xu F and Li X, Future Sci OA, 2017, 3, FSO187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavakoli H, Zhou W, Ma L, Perez S, Ibarra A, Xu F, Zhan S and Li X, TrAC Trends Anal. Chem, 2019, 117, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segaliny AI, Li G, Kong L, Ren C, Chen X, Wang JK, Baltimore D, Wu G and Zhao W, Lab Chip, 2018, 18, 3733–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhang W and Qin L, Angew. Chem. Int. Ed. Engl, 2014, 53, 2344–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han X, Ma Y, Zhang K, Zhang P, Shao N and Qin L, Proteomics, 2020, 20, 1900223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, deMello AJ and Elvira KS, Lab Chip, 2016, 16, 1197–1205. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Ling V and Li PC, Anal. Chem, 2008, 80, 4095–4102. [DOI] [PubMed] [Google Scholar]
- 20.Zhao W, Cui CH, Bose S, Guo D, Shen C, Wong WP, Halvorsen K, Farokhzad OC, Teo GS, Phillips JA, Dorfman DM, Karnik R and Karp JM, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 19626–19631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA and Toner M, Nature, 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C, Lin J, Ramnath N, Wicha MS, Hayes DF, Simeone DM and Nagrath S, Nat. Nanotechnol, 2013, 8, 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Santana SM, Liu H, Bander NH, Hawkins BG and Kirby BJ, Electrophoresis, 2013, 34, 2970–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamza B, Ng SR, Prakadan SM, Delgado FF, Chin CR, King EM, Yang LF, Davidson SM, DeGouveia KL, Cermak N, Navia AW, Winter PS, Drake RS, Tammela T, Li CM, Papagiannakopoulos T, Gupta AJ, Shaw Bagnall J, Knudsen SM, Vander Heiden MG, Wasserman SC, Jacks T, Shalek AK and Manalis SR, Proc. Natl. Acad. Sci. U. S. A, 2019, 116, 2232–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimmerling RJ, Lee Szeto G, Li JW, Genshaft AS, Kazer SW, Payer KR, de Riba Borrajo J, Blainey PC, Irvine DJ, Shalek AK and Manalis SR, Nat. Commun, 2016, 7, 10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang JH, Miettinen TP, Chen L, Olcum S, Katsikis G, Doyle PS and Manalis SR, Nat. Methods, 2019, 16, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poudineh M, Aldridge PM, Ahmed S, Green BJ, Kermanshah L, Nguyen V, Tu C, Mohamadi RM, Nam RK, Hansen A, Sridhar SS, Finelli A, Fleshner NE, Joshua AM, Sargent EH and Kelley SO, Nat. Nanotechnol, 2017, 12, 274–281. [DOI] [PubMed] [Google Scholar]
- 28.Durmus NG, Tekin HC, Guven S, Sridhar K, Arslan Yildiz A, Calibasi G, Ghiran I, Davis RW, Steinmetz LM and Demirci U, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, E3661–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stroock AD, Dertinger SK, Ajdari A, Mezic I, Stone HA and Whitesides GM, Science, 2002, 295, 647–651. [DOI] [PubMed] [Google Scholar]
- 30.Mair B, Aldridge PM, Atwal RS, Philpott D, Zhang M, Masud SN, Labib M, Tong AHY, Sargent EH, Angers S, Moffat J and Kelley SO, Nat. Biomed. Eng, 2019, 3, 796–805. [DOI] [PubMed] [Google Scholar]
- 31.Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP Jr., Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA and Toner M, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balenci L, Wonders C, Coles BL, Clarke L and van der Kooy D, Stem Cells, 2013, 31, 2218–2230. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya S, Das A, Mallya K and Ahmad I, J. Cell Sci, 2007, 120, 2652–2662. [DOI] [PubMed] [Google Scholar]
- 34.Ding XW, Wu JH and Jiang CP, Life Sci., 2010, 86, 631–637. [DOI] [PubMed] [Google Scholar]
- 35.Balenci L and van der Kooy D, Stem Cells Dev., 2014, 23, 230–244. [DOI] [PubMed] [Google Scholar]
- 36.Dvoriantchikova G, Perea-Martinez I, Pappas S, Barry AF, Danek D, Dvoriantchikova X, Pelaez D and Ivanov D, PLoS One, 2015, 10, e0131054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jadhav AP, Mason HA and Cepko CL, Development, 2006, 133, 913–923. [DOI] [PubMed] [Google Scholar]
- 38.Giordano F, De Marzo A, Vetrini F and Marigo V, Mol. Vis, 2007, 13, 1842–1850. [PubMed] [Google Scholar]
- 39.Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G and Sandberg R, Nat. Methods, 2013, 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- 40.Innes BT and Bader GD, F1000Research, 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW and Ma'ayan A, Nucleic Acids Res., 2016, 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walters CL and Blendy JA, J. Neurosci, 2001, 21, 9438–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda M, Tanaka Y, Omodaka K, Nishiguchi KM, Nakamura O, Tsuda S and Nakazawa T, Sci. Rep, 2016, 6, 28736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dworkin S, Malaterre J, Hollande F, Darcy PK, Ramsay RG and Mantamadiotis T, Stem Cells, 2009, 27, 1347–1357. [DOI] [PubMed] [Google Scholar]
- 45.Tong JJ, Liu J, Bertos NR and Yang XJ, Nucleic Acids Res., 2002, 30, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Velthoven CTJ, de Morree A, Egner IM, Brett JO and Rando TA, Cell Rep., 2017, 21, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E and Doetsch F, Neuron, 2014, 82, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coles BL and van der Kooy D, J. Vis. Exp, 2010, 43, 2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.