

Abstract
Respiratory syncytial virus (RSV) infection in infancy is associated with increased risk of asthma, except in those with allergic disease at the time of infection. Using house dust mite allergen, we examined the effect of pre-existing atopy on post-viral airway disease using Sendai virus in mice, which models RSV infection in humans. Sendai virus drives post-viral airway disease in non-atopic mice, however, pre-existing atopy protected against the development of airway disease. This protection depended upon neutrophils, as depletion of neutrophils at the time of infection restored the susceptibility of atopic mice to post-viral airway disease.
Associated with development of atopy was an increase in PMN-DC hybrid cells that develop in Th2 conditions and demonstrated increased viral uptake. Systemic inhibition of IL-4 reversed atopic protection against post-viral airway disease, suggesting that increased virus uptake by neutrophils was IL-4 dependent. Finally, human neutrophils from atopic donors were able to reduce RSV infection of human airway epithelial cells in vitro, suggesting these findings could apply to the human. Collectively our data support the idea that pre-existing atopy derives a protective neutrophil response via potential interaction with IL-4, preventing development of post-viral airway disease.
Introduction:
The prevalence of asthma and other atopic diseases have increased in the U.S. and other modernized countries (1, 2). Further, allergic diseases represent a major health burden in the U.S, with over a third of children having at least one atopic disease (3). In fact, over 8% of the population has asthma with an estimated $81 billion per year of economic burden to U.S. economy (4, 5). While atopic disease has been increasing in the modernized world, the mechanisms driving this increased prevalence are unclear. One hypothesis is that respiratory viral infections may drive development of atopic disease (as well as exacerbating existing disease). Rhinovirus (RV) and respiratory syncytial virus (RSV) are the two main respiratory viruses associated with increased risk of developing asthma (6–8). RV is detected more commonly in older children that are wheezing, whereas RSV is a common cause of infant bronchiolitis in the first year of life, especially during the first 2-6 months of life (9). In fact, the risk of developing recurrent wheeze after RSV infection appears to be greatest in those infants and toddlers without pre-existing atopy; while with RV, the risk is greatest in those who have already developed atopy before the viral infection (9–12). Why pre-existing atopy would be a risk for one virus (RV) but potentially be protective for another (RSV) is not known.
Atopy involves the production of IgE. Others and we have demonstrated that respiratory viral infections (including RV and RSV) can drive IgE production against respiratory viruses (13–15). In fact, it is plausible to hypothesize that RV induced disease (requiring pre-existing atopy) may be a direct result of RV crosslinking anti-RV IgE on mast cells and subsequent degranulation and allergic cascade (13). Further, neutrophil extracellular traps (NETs) generated in atopic mice were found to exacerbate RV induced airway disease (16,17). While there are several potential mechanisms for pre-existing atopy driving RV induced disease, little is known about how pre-existing atopy affects post-RSV airway disease.
One study used ovalbumin (OVA) sensitized and challenged mice that were infected with RSV, and found increased severity of RSV induced airway disease with OVA sensitization and challenge (18). However, the relevance to humans is less clear due to the OVA model being dependent on intraperitoneal injections, and the fact that human clinical data suggest that a risk of post-RSV airway disease is greatest in those without pre-existing atopy. Further, RSV is not a natural rodent pathogen, and the resulting anti-viral immune response may be significantly different from that seen in mice infected by a rodent virus that naturally replicates in the rodent airway.
We have utilized a native rodent pathogen, Sendai virus (SeV), which has many similarities to RSV. Unlike RSV in the mouse, SeV replicates well in murine airways and induces long-standing airway hyper-reactivity (AHR) and mucous cell metaplasia (MCM). Using SeV we have characterized a mechanistic “pro-atopic” pathway that initiates with a respiratory viral infection and results in atopy, and post-viral airway hyper-reactivity with mucous cell metaplasia (together, hallmarks of “asthma”) (19–23). In this model, non-atopic mice infected with SeV results in accumulation of CD49d+ PMNs in the lung that drive FcεRI expression on conventional dendritic cells (cDC), and this FcεRI can then bind SeV specific IgE – all while SeV viral particles are still present. The SeV viral particles then crosslink the IgE leading to production of CCL28, a chemokine that drives development of post-viral airway disease. This model mimics RSV infection in humans, where a clinically severe infection early in life drives a markedly increased risk of asthma and allergic disease and is associated with a neutrophil dependent response (21).
To determine if pre-existing atopy has an effect on the development of post-viral airway disease, we have utilized the house dust mite (HDM) model of atopy and our SeV model. Similar to what has been suggested for humans with RSV, our data demonstrate that development of post-viral airway disease occurs only in those mice without pre-existing atopy. Surprisingly, atopy induced protection was dependent upon the innate immune response, with PMN from atopic mice being responsible for inducing protection against post-viral airway disease. In fact, these PMN appear to have taken up SeV particles through an IL-4 dependent process, leading to protection from post-viral airway disease in the mice with pre-existing atopy. This effect did not depend upon NETosis, as mice deficient in Mpo were protected from post-viral airway disease when made atopic before the viral infection. We also identified a unique PMN population in atopic mice that expressed some markers associated with antigen presenting cells (so-called PMN-DC hybrid cells), which was increased in the atopic mice. These trans-differentiated PMN-DCs have been observed in various inflammatory conditions and shown to provide increased protection against infections (24). Together, these data suggest that post-viral airway disease is prevented in mice with an atopic milieu via altered PMN function/phenotype with an increase in viral particle uptake, reduced peak SeV titers, and decreased CCL28 production.
Methods
Mice
All mice were obtained from Jackson Laboratory (Bar Harbor, ME). C57BL6 (WT) mice were purchased and bred in house, as were myeloperoxidase deficient mice (Mpo−/−; B6.129x1-Mpotm1Lus/J, Jackson Stock 004265).
Atopic model and viral inoculation
Mice were sensitized with 1 μg HDM extract (Cat#: XPB70 D3A Stallergenes Greer USA, Boston, MA) intranasally (i.n.) and challenged 7 days later with 10 μg HDM i.n. daily for five consecutive days (25). For controls, some mice were treated with PBS for the sensitization dose, challenge doses, or both sensitization and challenge doses. Three days after the last challenge dose, mice were inoculated with 2x105 pfu SeV or ultraviolet light inactivated SeV (UV-SeV), as we have published (21).
Measurement of airway hyper-reactivity
Airway hyper-responsiveness (AHR) was measured by invasive measurement of airway resistance to increasing methacholine (MCh) doses with doubling concentrations from 1.56 to 50 mg/ml using the FlexiVent system per manufacturer’s instructions (SCIREQ Inc) as we published (20). Respiratory system resistance (Rrs) was calculated using the FlexiVent system software.
Flow cytometry and Flow Activated Cell Sorting (FACS)
Cells were harvested from whole lungs and/or broncheoalveolar lavage and flow cytometry was performed as we have previously reported using standard techniques for labeling of cells (19, 22, 26). Stained cells were analyzed with a FACSCalibur flow cytometer (BD) and data analyzed with FlowJo software (Tree Star, Inc). For FACS, a FACSAria cell sorter (BD Biosciences, USA) was used to identify and collect neutrophils. The following antibodies were used for flow cytometry experiments: Ly6G (1A8); CD11c (N418); Gr-1 (RB6-8C5); all from BioLegend.
Neutrophil and PMN-DC hybrid were identified as follows by flow cytometry: PMN-DC hybrid (CD11c+ Ly6G+), PMN (CD11c− Ly6G+) and same gating also for sorting. Neutrophils for FACS were gated as (singlets, CD45.2+, Ly6G+).
Real-time PCR assay
Total RNA was isolated with either Trizol (Sigma-Aldrich) or the mirVana™ miRNA Isolation Kit (cat# AM1560, Thermo Fisher) that can isolate small RNA (10-mers) to several kilobases long. cDNA was generated with Maxima™ H Minus cDNA Synthesis Master Mix, with dsDNase, (cat# M1681, ThermoFisher) per manufacturer’s instructions, and qRT-PCR was performed using TaqMan fast master mix and a StepOne PlusPCR system (Applied Biosystems). The following primer and probe sets were utilized for qRT-PCR experiments:
SeV primer/probe set (SeV-1237F: 5’-GGC GGT GGT GCA ATT GAG-3’; SeV-1300R: 5’-CAT GAG CTT CTG TTT CTA GGT CGA T-3’; MGB TaqMan Probe SeV-1257T: 5’-AGC TCT AGA CAA TGC C-3’) was used for SeV qRT-PCR and copy number calculated based on a standard curve as we have published (27). TaqMan expression assay (ThermoFisher Scientific) was used to assess expression of Ccl28 (Mm00445039_m1), Muc5ac (Mm01276704_m1), Ifng (Mm99999071_m1), with data normalized to either Gapdh (4352339E) or Hprt1 (Mm03024075_m1). Sybr green assay was used to quantify green fluorescent protein (gfp) expression (eGFP-F: 5’-AAG GAC GAC GGC AAC TAC AA-3’; eGFP-R: 5’-CGA TGT TGT GGC GGA TCT TG-3’) and normalized to Hprt1 (ID# 7305155a1, Primer Bank Harvard). PMN-DC Hybrid and Neutrophil cells were sorted and RNA isolated by Arcturus Pico Pure RNA kit (cat#: KIT0204; Thermo Fisher Scientific) and SeV copy number/sorted cell determined by one step qRT-PCR performed using qScript 1-Step RT-qPCR kit (cat#: 95057-050; Quanta Bio).
Histochemistry
Mouse lung was fixed in 10% neutral buffer formalin at 25 cm H2O pressure, paraffin embedded, and then cut into 10 μm thick sections. Slides were then stained with Periodic acid-Schiff (PAS, Sigma Aldrich) stain. Quantification of mucous cells was performed by a blinded observer counting the number of PAS+ cells per mm of basement membrane using ImageJ software (NIH) as we have previously reported. (20)
PMN depletion
To deplete PMN, anti-Ly6G mAb (clone 1A8, BioXCell) 100 μg was given i.p. to C57BL6 mice on the indicated days. For controls, mice were treated with 100 μg of rat IgG2a mAb (clone 2A3, BioXCell) i.p. on the same days. Depletion of PMN in lungs was verified by flow cytometry staining with anti-Ly6G. anti-Gr1, or IgG isotype control (Supplemental Figure 3). Anti-Ly6G treatment resulted in a 76.67% reduction in lung PMN numbers 24h post injection relative to IgG treated controls (n≥2).
Phagocytosis
PMN phagocytosis was determined using PMN isolated from lungs and bone marrow of atopic or non-atopic mice, and purified by density gradient separation centrifugation (Histopaque-1.077; Sigma-Aldrich). RBC in the pellet were lysed using hypotonic solution as we have published (19). Phagocytosis was determined using the Abcam Phagocytosis assay kit (Cat # ab234053, Abcam). Briefly, ~2.5x 105 PMN were cultured with or without Green Zymosan slurry for 90 minutes. Cells were harvested, washed with the kit’s phagocytosis assay buffer, and mounted on glass slides (using a cytospin and DAPI with Vectashield anti-fade mounting media (Vector Laboratories)). Using a fluorescent microscope (Olympus Bx61), a blinded observer took five images of the cells using SlideBook 6 digital microscopy software with DAPI and GFP filter cubes (3i Intelligent Imaging Innovations, Inc). The zymosan positive cells were then counted using ImageJ and reported as percentage of total cell population (calculated from DAPI+ cells in each section). To assess the effect of IL-4 on phagocytosis, 2.5x 105 PMNs were isolated from naïve mouse bone marrow and treated with or without IL-4 (200 ng, cat# 214-14 PeproTech) for 90 mins with or without Zymosan and mounted on glass slides with DAPI for image analysis.
Air-liquid Interphase Culture
Development of a pseudostratified mouse airway epithelial cell (AEC) layer was performed as published (28). Briefly, murine tracheal epithelial cells were harvested and cultured in transwells at air-liquid interphase (ALI) for 4 to 6 weeks to obtain pseudostratified bronchoepithelial cell culture. These differentiated mouse ALI-AEC cultures (mTEC) appear similar to in vivo airway epithelium with goblet, ciliated, basal and other minor cell types.
Statistical analyses
Prism 7 (GraphPad Software) was used for statistical analyses; normally distributed data are presented as mean ± SEM and non-normally distributed data are presented as median ± interquartile range (IQR), unless stated otherwise. Airway hyperractivity was analyzed by two-way ANOVA with Tukey’s multiple comparisons test. Student’s t-test (for parametric data) or Mann Whitney U (for non-parametric data) were used to assess significant differences between two means (Student’s t-test) or medians (Mann Whitney U). For all tests, p < 0.05 was considered statistically significant.
Study approvals
All animal experimental protocols were approved by the institutional animal care and use committee (IACUC) of the Abigail Wexner Research Institute at Nationwide Children’s Hospital.
Human blood samples were obtained from de-identified volunteers so no IRB approval was required.
Results
Effect of atopy on post-viral airway disease
To examine the effect of pre-existing atopy in our well established SeV model we used HDM to make mice atopic. The HDM model was chosen because HDM is an allergen in human disease, and HDM is able to sensitize through the airways, making it more relevant than models that require i.p. injections for sensitization (e.g., the ovalbumin model). Sensitizing and challenging mice with HDM (but not sensitizing or challenging alone with HDM) led to significant airway disease, as evidenced by elevated AHR and MCM 3 days after the last challenge (Figure 1A–C).
Figure 1. Effect of house dust mite (HDM) model in C57BL6 mice.
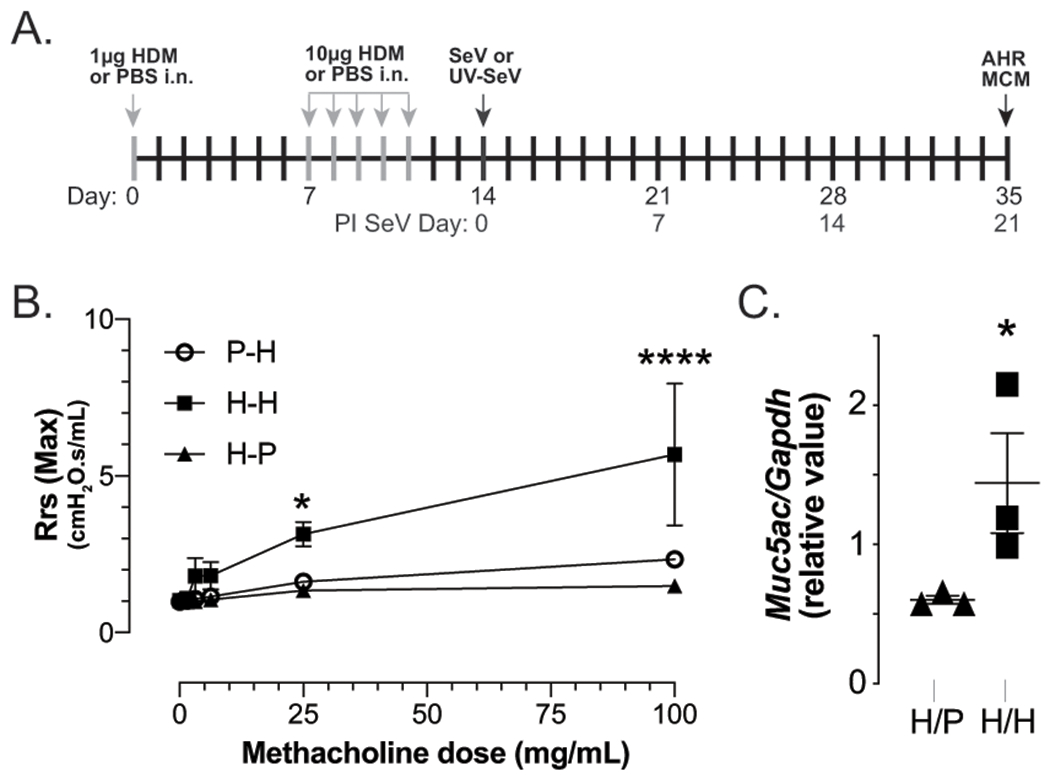
(A) Timeline for experiments. (B) Mice exposed to 1μg HDM extract i.n. followed 1wk later with 5 daily i.n. exposures to 10μg HDM without any viral infection developed increased airway hyper-responsiveness (AHR) on day 14 post HDM sensitization, as demonstrated by increased airways resistance with methacholine challenge, measured invasively with Flexivent (Scireq)). (C) Mucous cell metaplasia (MCM; measured by Muc5ac/Gapdh mRNA expression) is increased in atopic mice, as well. H/P, mice sensitized to HDM but challenged with PBS (H/P); P/H, mice “sensitized” to PBS and challenged with HDM; H/H, mice sensitized and challenged with HDM. n=3 mice per group.
Mice that had been sensitized or challenged with HDM still developed post-viral airway disease (i.e., AHR and MCM) 21 days after inoculation with 2x105 pfu SeV (35 days post HDM sensitization); however, mice that were both sensitized and challenged with HDM (i.e., made atopic) were protected from post-viral airway disease (Figure 2). Thus, in our model, pre-existing atopy prevents development of post-SeV airway disease.
Figure 2. Post-viral airway disease is prevented in atopic mice.
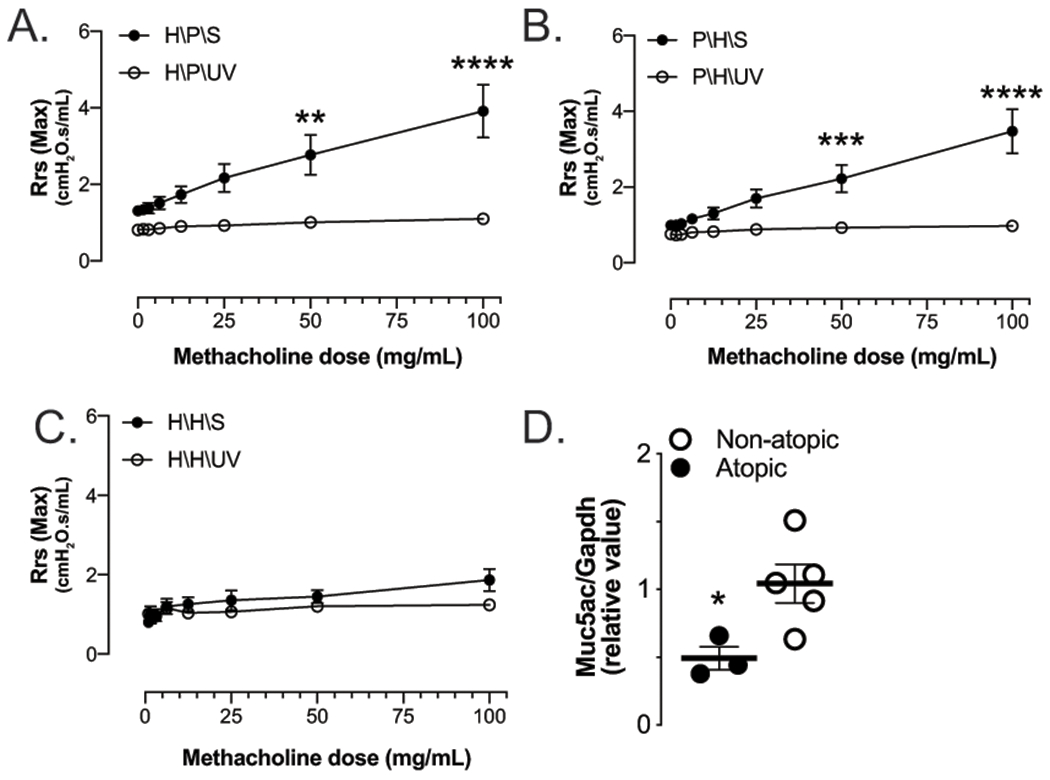
(A) Mice sensitized (H/P/S) or (B) challenged (P/H/S) with HDM develop significant AHR 21d PI SeV (closed circles) when compared to similar mice receiving UV-inactivated SeV (open circles). (C) Mice sensitized and challenged with HDM (H/H/S), however, fail to develop post-viral AHR or (D) mucous cell metaplasia with SeV infection (closed circles, SeV; open circles, UV-SeV). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 n≥3 mice per group.
Inflammatory response to SeV in atopic mice
Given that pre-existing atopy protected against development of post-viral airway disease, we examined whether pre-existing atopy reduced the overall burden of SeV induced illness. Normally infection with 2x105 pfu SeV leads to 20% weight loss (27); however, in mice with pre-existing atopy weight loss was significantly attenuated (Figure 3A). Since weight loss usually correlates with the viral inoculum, this suggested that atopy might limit viral replication leading to a less severe overall illness, and, in fact, we found that mice sensitized and challenged with HDM had SeV titers that were reduced by about a half-log compared to non-atopic mice (Figure 3B).
Figure 3. Pre-existing atopy reduces inflammatory response to SeV.
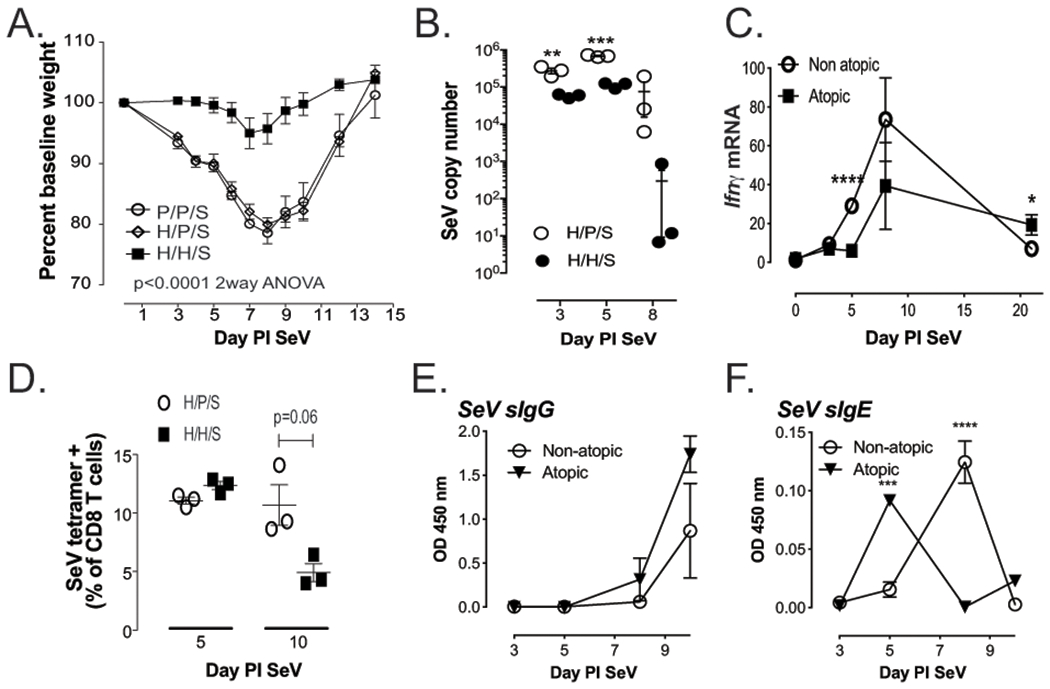
(A) Weight loss in atopic (H/H/S) was significantly attenuated (p<0.0001) when compared to non-atopic (H/P/S and P/P/S) mice after inoculation with regular dose SeV. (B) Peak SeV titers are reduced in atopic mice. (C) Ifnγ mRNA is reduced in atopic mice, while (D) peak SeV specific CD8 T cells are not affected. (E) SeV specific IgG in the serum is unaffected by atopic status, but (F) SeV specific IgE appears more rapidly in atopic mice. n≥3 mice per group/time point; **p<0.01, ***p<0.001, ****p<0.0001 atopic versus non-atopic
Given the reduction in SeV titers, we next wanted to examine the immune response to SeV to see if it was more robust in atopic mice. Interestingly, lung Ifnγ mRNA levels were actually decreased on day 5 PI SeV in atopic mice but rebounded by day 8 PI SeV (Figure 3C). This reduced Ifnγ mRNA may have been a result of the decreased viral titer, as we found no difference in SeV specific CD8 T cells (CD8+ cells that bound a tetramer specific for SeV NP324-332), in atopic and non-atopic mice (Figure 3D). Given the reduction in viral titer without an impairment in viral specific CD8 T cells, we next assessed whether the humoral immune response was altered in the atopic mice. As can be seen in figure 3E, levels of specific IgG against SeV were not different between atopic and non-atopic mice; however, anti-viral IgE (anti-SeV IgE) appeared more rapidly in the atopic mice (Figure 3F). Together, these data indicate that pre-existing atopy appears to lessen the viral titer and symptoms, while having minimal effects on the antiviral immune response.
We have previously demonstrated in our model a mechanistic pathway involving a PMN subset, the CD49d expressing PMN, which induces FcεRI expression on lung cDC, the FcεRI is then bound by anti-SeV IgE, and viral crosslinking of the cDC FcεRI leads to development of post-viral airway disease. Since atopic mice failed to develop post-viral airway disease, we next examined whether pre-existing atopy altered this well-characterized pathway. Pre-existing atopy did reduce the frequency of CD49d expressing PMN, but only at day 5 PI SeV, and this associated with a very modest reduction in the frequency of cDC expressing FcεRI (Supplemental Figure 1A,B). Further, SeV specific IgE actually appeared more rapidly in atopic mice compared to non-atopic mice but would still have been present to bind cDC FcεRI (Figure 3F). Individually, these minor variations between atopic and non-atopic mice do not seem as likely explanations for the loss of post-viral airway disease in atopic mice; however, combined they may have – in fact, message for the chemokine CCL28, which we have demonstrated is required for development of post-viral airway disease, was significantly reduced in atopic mouse lungs 21 d PI SeV (Supplemental Figure 1C). (22, 29).
Epithelial cell and viral infection
Since SeV replicates in the airway epithelium, and given the reduction in viral titer in mice with pre-existing atopy, we hypothesized that the airway epithelium of atopic mice might be resistant to SeV infection. To assess this, we took advantage of an ex vivo epithelial cell culture system with a SeV that included a cassette encoding green fluorescent protein (GFP) in the viral genome (gfp-SeV; (30). In a gfp-SeV infected cell gfp is transcribed, and subsequently translated and GFP expression can be quantified in infected cells by fluorescent microscopy (as well as gfp by qPCR). Importantly, the GFP is not packaged into the virus, but is simply free in the cytoplasm of infected cells. Murine tracheal epithelial cells (mTECs) were isolated from naive C57BL6 mice and differentiated at ALI. When these mTECs had developed into pseudostratified epithelial cells (after 4-6 weeks), 10 μg HDM was added daily to the apical surface of the cells for 3 days and inoculated with gfp-SeV (400 pfu) 24h after last HDM treatment. Using fluorescence microscopy we measured GFP+ cells daily over the following 2 days to determine the level of infection. As shown in Figure 4, HDM exposure had no effect on the frequency of mTECs that were GFP+. These data suggest that HDM exposure does not directly impair SeV replication in mTECs. Supporting this finding, is the fact that a recent publication showed that RSV viral load was not impacted by HDM treatment of human primary bronchial epithelial cells (31)
Figure 4. HDM treatment of mTECs does not impair SeV replication.
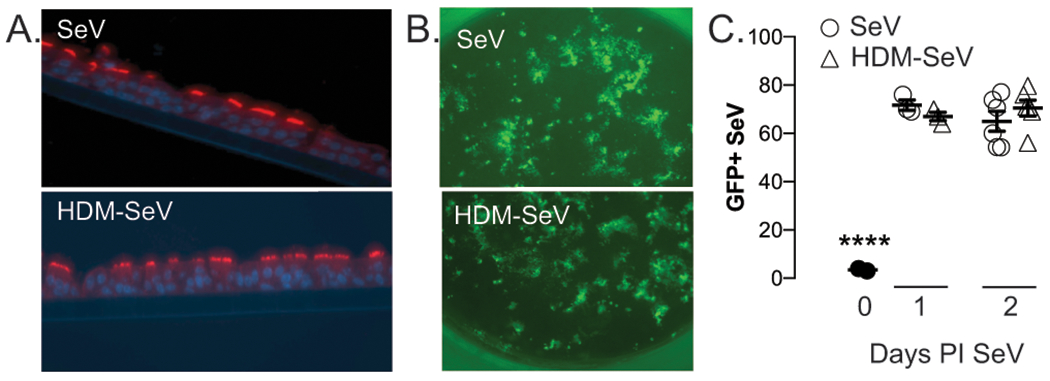
Murine tracheal epithelial cells (mTECs) were isolated and grown on transwell inserts for 4-6wks. (A) Cells differentiated into ciliated cells on the transwell at air-liquid interphase, as evidenced by Beta IV tubulin expression (red=anti-TUBB4 mAb; blue=dapi). Some cultures were exposed to 10μg HDM on the apical side for 4h daily for 3d, before all cultures were inoculated for 4h with 400pfu gfp-SeV. (B) Representative expression of GFP containing SeV in the cultures 2d PI (HDM-SeV, pretreated with HDM before SeV infection; SeV, no pretreatment). Images taken at 10x magnification using EVOS Cell imaging system. (C) Quantification of experiments shown in (B) by using ImageJ software. n=3 separate experiments, each from separate donors. ****p<0.0001 versus either treatment at day 1 or 2 PI.
Neutrophils are required to prevent post-viral airway disease in atopic mice
Given our previous work showing that PMN subsets are critical for development of post-viral airway disease, we asked whether PMN could be responsible for the protection against post-viral airway disease seen in atopic mice. Interestingly, depleting PMNs one day prior to viral inoculation slightly restored post-viral airway disease in atopic mice (compared to control IgG treated atopic mice, Figure 5A). However, depletion of PMNs one day post SeV inoculation (i.e., day 1 PI SeV), led to a much more robust restoration of post-viral disease in the atopic mice (Figure 5B & C). The development of post-viral airway disease in atopic mice depleted of PMNs one day prior to SeV was associated with evidence of increased SeV infection -- higher levels of SeV in whole lung at day 1 PI SeV (Figure 5D). These data suggest that PMNs are required for the protection against post-viral airway disease seen in atopic mice, and that the mechanism of this protection might involve impairment of viral particle production.
Figure 5. Neutrophils (PMNs) from atopic mice inhibit development of post-viral airway disease.
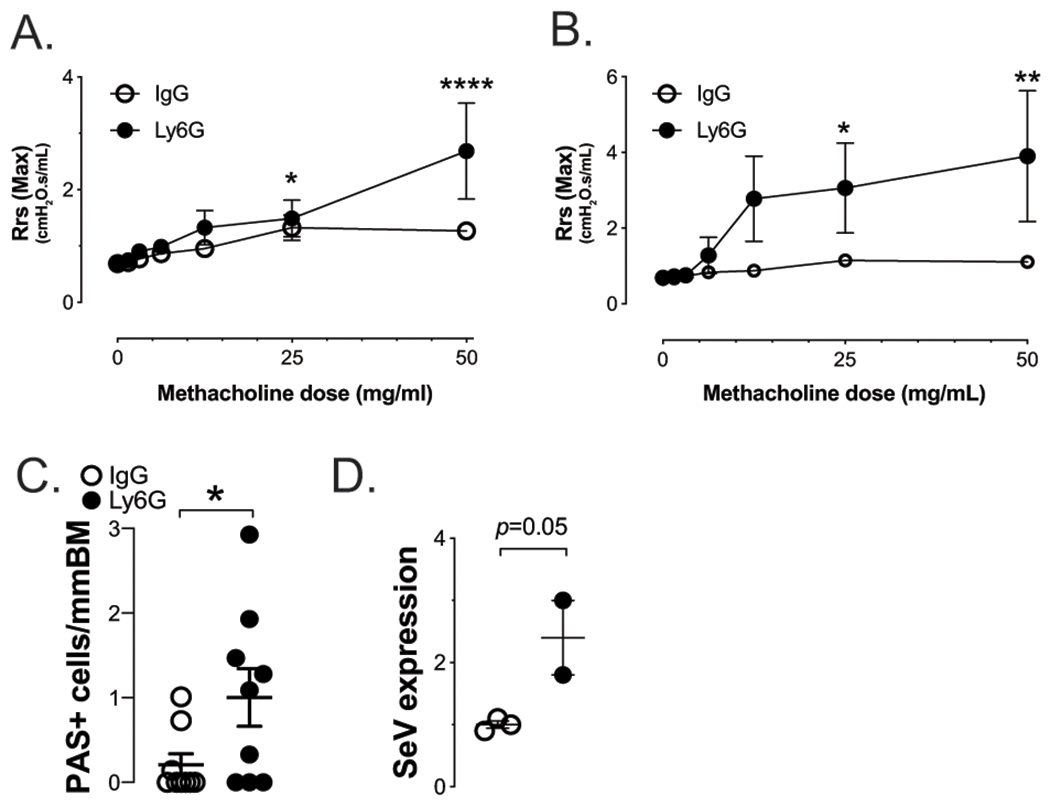
Depletion of PMNs with anti-Ly6G mAb 24h before (A) or 24h after (B&C) SeV infection restores post viral airway disease in atopic mice. In untreated mice, depletion of PMN with anti-Ly6G mAb 24h PI SeV had no effect on post viral airway disease (data not shown). (D) Gfp-SeV infection showed increased viral infection at d1 PI in mice depleted of Ly6G+ PMN 24 h before SeV inoculation (data are expressed as Gfp/Gapdh or SeV/Gapdh mRNA relative levels from whole lung). n≥3 mice per group.
PMNs are known to produce extracellular traps (NETs) that have been shown to be important for development of post-RV airway disease. We, therefore, wanted to assess whether PMN NETs might be what is driving the post-viral airway protection in atopic mice. To asses this, we took advantage of myeloperoxidase (MPO) deficient (Mpo−/−) mice. MPO is one of the major enzymes present in PMN, is protective against LPS-induce endotoxemia (32), and is a key component of neutrophil extracellular traps (NETs) (33). In fact, deficiency of Mpo is associated with an inability in PMNs to properly form NETs. Therefore, using Mpo−/− mice we assessed the effect of atopy on development of post-viral airway disease. As demonstrated in supplemental figure 2A&B, atopic Mpo−/− mice were still protected from post-viral airway disease, strongly indicating that the PMN dependent protection was not through NET production.
Neutrophils from atopic mice have increased viral particle uptake
Since PMNs were required for protection against post-viral airway disease, and pre-existing atopy was associated with decreased peak SeV titers without a significant role for NET production, we hypothesized that the PMN might be directly phagocytosing virus leading to reduced titer and subsequent prevention of post-viral airway disease. To test this hypothesis, we purified PMNs from the lungs of atopic and non-atopic mice and measured Zymosan uptake by phagocytosis. As shown in Figure 6A, PMN from atopic mice had significantly increased phagocytosis compared to PMN from non-atopic mice, suggesting increased viral uptake by PMN could explain the reduced viral titer in the atopic lung.
Figure 6. Atopic PMNs exhibit increased viral particle uptake.
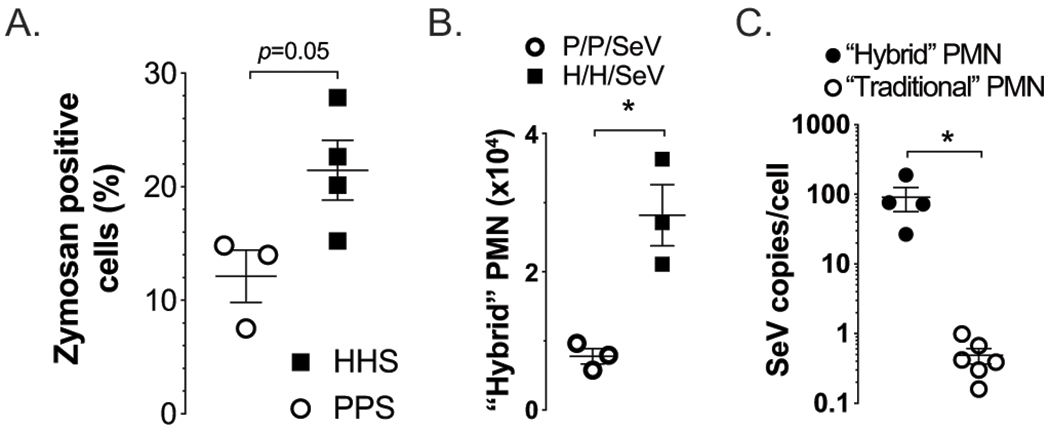
(A) PMN isolated three days PI SeV from HDM sensitized and challenged mice (HHS) demonstrated a greater level of phagocytosis, as indicated by Zymosan uptake. Flow cytometry at day 1 PI SeV demonstrates (B) an increase in the PMN-DC hybrid population in atopic mice (H/H/SeV) compared to non-atopic mice (P/P/SeV). (C) Hybrid cells had a proportionally higher SeV (gRNA) copy number per cell compared to traditional neutrophils. (A) and (B) n≥2 mice per group; (C) n≥4 mice per group; *p<0.05.
Recent studies have shown that PMNs can be differentiated into a relatively rare but unique neutrophil-DC hybrid population (PMN-DC) with surface markers seen in both PMNs (Ly6G) and DCs (CD11c); these PMN-DC hybrids potentially play a role in protection against infection in part through increased phagocytosis (24, 34). Interestingly, we found this PMN-DC hybrid increased by about 3-fold in the lungs of atopic mice 1 day PI SeV (Figure 6B), while traditional PMN numbers were not different between atopic and non-atopic mice (2.34±0.12x105 atopic and 2.62±0.86x105 non-atopic, n=3, mean±SEM) at this same timepoint. Further, when examined on a per-cell basis, the PMN-DC hybrid cells exhibited a nearly 100-fold higher uptake of SeV compared to traditional PMN (Figure 6C).
IL-4 is necessary for prevention of post-viral airway disease in atopic mice
Previously development of the hybrid PMN-DC population was reported to be dependent upon IL-4 (34). Therefore, we were interested in determining if the house dust mite model drove an IL-4 dependent process, as well. Initially, we assessed the cytokine response in the airways of atopic and non-atopic mice during the viral infection. Interestingly, by day 3 PI SeV most BAL cytokines were suppressed in atopic mice except for IL-4, which was significantly higher (Figure 7). These data supported the hypothesis that IL-4 drove production of the hybrid PMN-DC population, which then reduced viral titer and led to protection from post-viral airway disease.
Figure 7. Atopic mice have a skewed cytokine response to SeV infection.
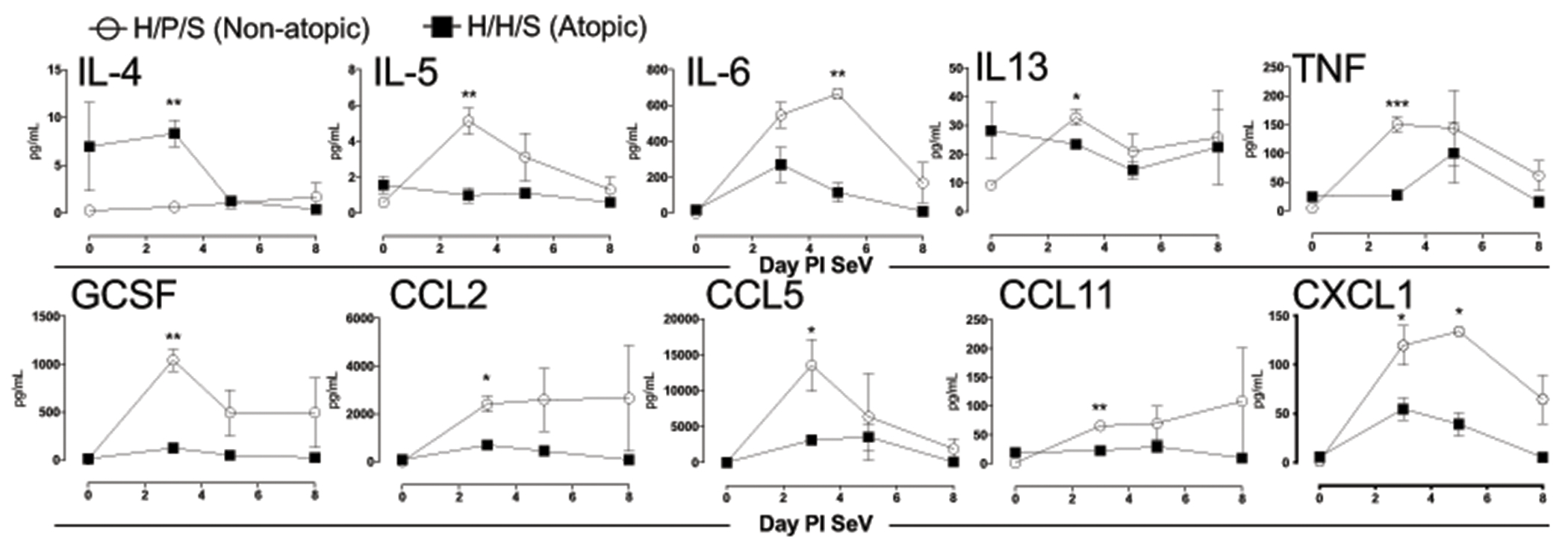
Atopic (filled squares) and non-atopic (open squares) mice were infected with SeV and BAL obtained at the indicated days PI. Cytokines and chemokines were detected using a multiplex array. No significant difference was seen between groups in terms of the concentrations of IL1α, IL1β, IL2, IL10, IL12p40, IL12p70, IL17A, GM-CSF, CCL3, and CCL4 (not shown). n≥3 mice per group/timepoint.
To specifically determine the role of IL-4 in the antiviral immune response, we used a blocking mAb to neutralize IL-4 in atopic mice 24h before SeV infection, and examined the effect on post-viral airway disease. Blockade of IL-4 early in the antiviral immune response trended to increase SeV titer 1 day PI SeV, but more importantly, inhibition of IL-4 led to a significant increase in AHR by day 21 PI SeV (Figure 8A & B). Although the PMN-DC hybrid cells are thought to require IL-4 for development, we did not see a reduction in the hybrid population in the anti-IL-4 treated mice (Figure 8C) (34). Why anti-IL-4 had no effect on these cells is not known, although the timing of inhibition, as well as the fact that multiple other cytokines including IL-13 and IL-3 have been reported to impact PMN-DC hybrid development, could explain the lack of an effect (34). Together, these data suggested to us that IL-4 might have been controlling PMN phagocytosis of viral particles. To test this, we purified PMNs from naïve mice and examined the effect of exogenous IL-4 administration on phagocytosis using a Zymosan assay. As shown in Figure 8D, PMNs cultured with IL-4 had significantly increased Zymosan uptake, suggesting that the effect of IL-4 was through the PMN, in support of our data with PMN depletion (Figure 5).
Figure 8: IL-4 is required for atopic protection from post-viral airway disease.
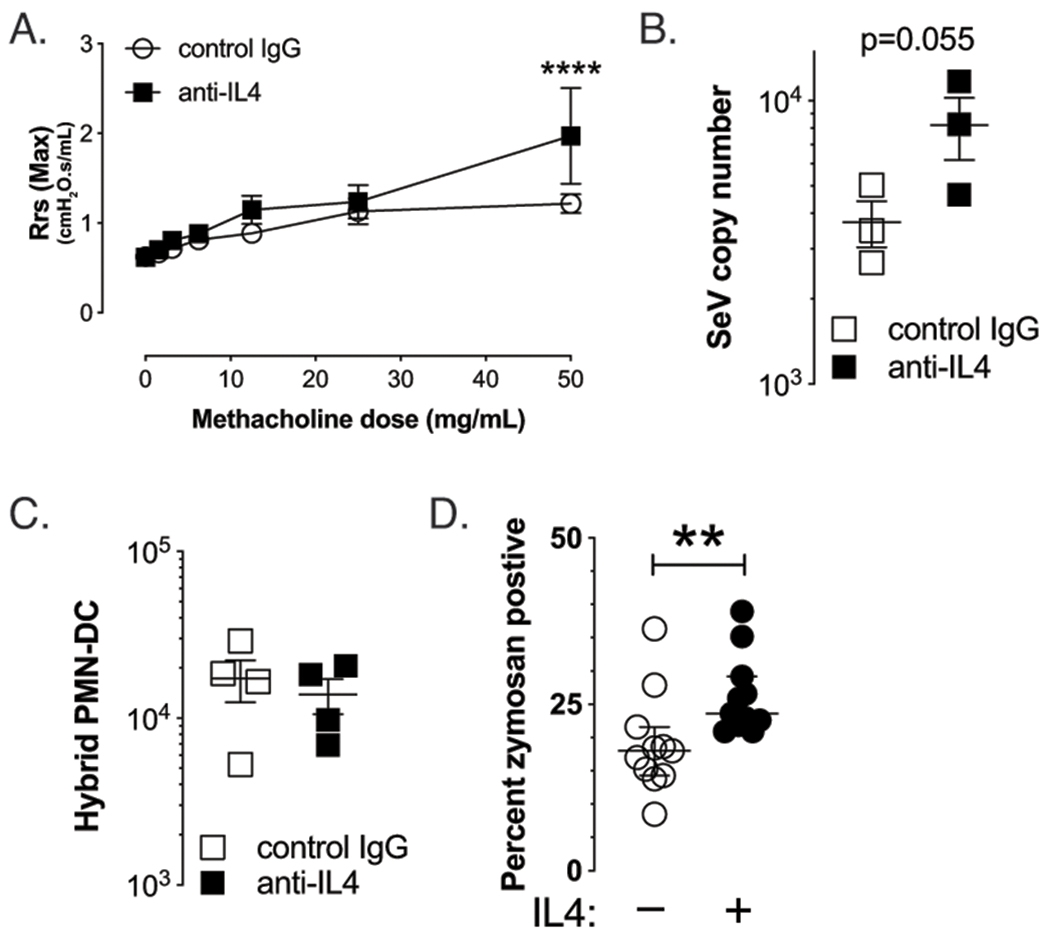
Atopic mice were treated with anti-IL-4 (clone 1B11) or control mAb (1.5 mg, i.p.) 24 hours before inoculation with SeV. (A) Blockade of IL-4 led to restoration of AHR by d21 PI SeV, n=3. (B) Restoration of post-viral airway disease was associated with a trend for increased SeV titer in whole lung of anti-IL-4 treated mice 1 day PI SeV. (C) However, no difference was seen in the quantity of hybrid PMN-DC cells with IL-4 blockade. (D) Culture of PMN with IL-4 for 90 min led to increased phagocytic activity in vitro, as evidenced by increased Zymosan uptake.
Human neutrophils demonstrate increased RSV phagocytosis
To determine if these findings had relevance to human disease, we isolated PMNs from de-identified peripheral blood of human subjects that self-reported to be atopic or non-atopic and added the PMNs to cultured A549 (human airway) cells. We then infected the A549 cells with gfp-RSV which, similar to gfp-SeV, leads to infected cells producing GFP that can be fluorescently quantified. As shown in Figure 9, addition of PMNs reduced GFP expression in A549 cells in a dose dependent fashion, with the greatest effect in those PMNs from the two subjects who self-identified as being atopic. This reduction in viral infection required live PMNs, as paraformaldehyde fixation of PMNs before addition to the A549 cells failed to reduce viral infection (i.e., GFP expression). These data suggest that our findings from the mouse model may be applicable to human disease, and may explain the protection against post-RSV asthma seen in allergic individuals.
Figure 9. Human PMNs prevent in vitro RSV viral infection of epithelial cells.
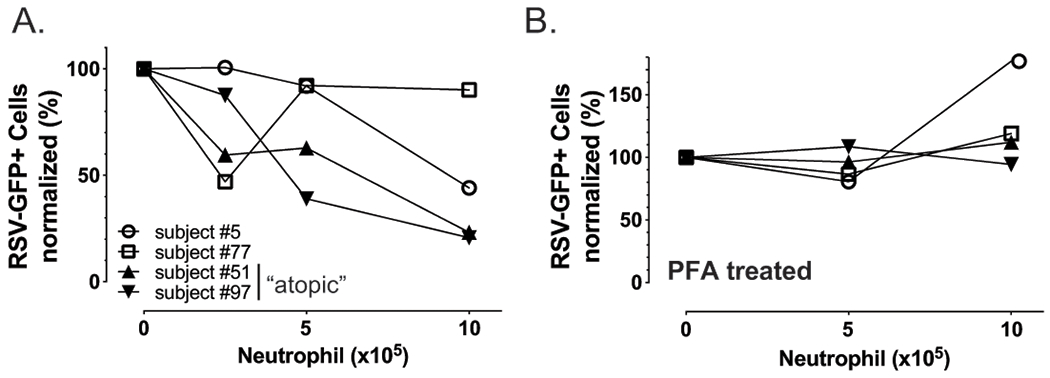
(A) Fresh human peripheral blood PMNs or (B) paraformaldehyde fixed PMNs from self-described atopic (triangles) or non-atopic donors were added at the indicated concentrations to A549 epithelial cells in culture. Two hours later 4000 pfu gfp-RSV was added to the cultures. After 48h cells were washed and GFP expressing A549 cells quantified by Tali Image-Based Cytometer (ThermoFisher).
Discussion
The role that respiratory viruses play in the development of wheezing and asthma continues to evolve with respiratory viral infections early in life being strongly correlated with increased risk of asthma and allergic disease (6, 35, 36). Utilizing a mouse model that has characteristics similar to human RSV infection, we and others have documented a mechanistic pathway that leads from viral infection of naïve mice to persistent post-viral airway disease (22, 37). Interestingly, unlike RV where pre-existing atopy increases the risk of post-viral wheezing and asthma, the risk for post-viral airway disease in RSV infected humans appears to be greatest in those without atopic disease before the viral infection (12). Using our model, we have found that the presence of pre-existing atopy (using HDM) prevented the development of post-viral airway disease. Thus, our model appears to replicate the epidemiologic data from humans.
Our studies suggest that PMN function acts as a double-edged sword in SeV infection. On one hand, CD49d expressing PMN are required for development of post-viral airway disease in non-atopic mice; however, as demonstrated in these studies, another subset of PMN appears to be required for protection against post-viral airway disease in atopic mice. From our data it appears that traditional PMN, which do not express CD49d and are present at the time of the viral inoculation, demonstrate increased phagocytic activity in an IL-4 dependent fashion Indeed, in human PMNs, IL-4 has been demonstrated to enhance phagocytosis and killing of opsonized bacteria (38). We also identified a PMN-DC hybrid, which did not appear to be IL-4 dependent, but likely also played a role in prevention of post-viral airway disease. It is worth noting that this PMN-DC hybrid population has recently been shown to provide protection against fungal infections both through direct killing via phagocytosis and induction of adaptive immunity and has been identified in other inflammatory conditions and bacterial infections; however, the role of this population in disease conditions remains poorly understood (24, 39). The fact that both anti-Ly6G and anti-IL4 led to increased post-viral airway disease suggests to us that the hybrid PMN-DC (and their viral uptake) remain quite relevant in the protection from post-viral airway disease in atopic mice. Indeed, it is likely a combination of both traditional and hybrid PMN populations that fully prevents development of post-viral airway disease in atopic mice.
Although we did not see a direct effect of HDM on viral replication in airway epithelial cells ex vivo, we cannot rule out the possibility that the HDM model drives resistance to viral infection in airway epithelial cells through an indirect pathway. However, this does not lessen the fact that the PMN-DC population was increased in atopic mice and was able to take up virus particles, and that traditional PMN had increased phagocytosis in an IL-4 dependent fashion. There could be both a reduction in total viral titer (as evidenced by whole lung SeV gRNA levels) due to effects on the airway epithelium, and a reduction in infectious SeV through phagocytosis of viral particles by the PMN subsets (demonstrated by the marked reduction in whole lung GFP expression in atopic mice). This mechanism would be consistent with our findings, that is, the requirement (but not sufficiency) for PMN in the protection from post-viral airway disease seen in atopic mice.
Our studies are predicated on the assumption that pre-existing atopy does not drastically alter the mechanistic pathway we have previously documented to drive post-viral airway disease in non-atopic mice. Pre-existing atopy appears to have a limited effect on antiviral IgE, CD49d expressing PMNs, and FcεRI expression by cDC; however, it is possible that when we treat atopic mice with anti-Ly6G, the subsequent development of post-viral airway disease is through different mechanisms from those seen in non-atopic mice. This is a possibility that will require further experiments beyond the scope of this report.
In summary, we have documented that development of atopy prior to SeV infection prevents development of post-SeV airway disease. This finding mimics human data suggesting that RSV is more likely to drive post-viral airway disease (asthma) in those without pre-existing atopy however, further studies are needed to establish a clear link between pre-existing atopy in humans and suppression of post-viral airway disease. Further, we have been able to demonstrate that this protection is associated with evidence of a reduction in productive viral infection, as well as increased viral particle uptake by PMN. In addition, the atopic milieu may ultimately prevent further tissue damage from the viral infection as PMNs with increased phagocytic activity have been shown to become unresponsive to further inflammatory stimuli (40). Depletion of PMN prior to or just after the viral infection restores development of post-viral airway disease in mice with pre-existing atopy, as does blockade of IL-4 early in the antiviral immune response. However, further research is needed to fully tie the IL-4 and PMN dependence of this process together. Importantly, we were also able to demonstrate that human PMN from atopic donors can reduce productive viral infection in human airway epithelial cells in vitro, suggesting our findings are relevant to human disease. This work demonstrates the importance of the PMN (and its subsets) in stopping the development of post-viral airway disease, and provides novel biomarkers and targets for potential targeted approaches in the future to prevent development of post-viral airway disease.
Supplementary Material
Key Points:
Pre-existing atopy protects against paramyxovirus induced airway disease.
Neutrophils and IL-4 are critical for protection against post-viral airway disease.
Acknowledgments
We thank Mr. Dave Dunaway of AWRI flowcytometry core for the technical assistance.
Financial information
This work was supported by R01 HL087778 and The Abigail Wexner Research Institute at Nationwide Children’s Hospital (to MHG).
References:
- 1.Centers for Disease, C., and Prevention. 2011. Vital signs: asthma prevalence, disease characteristics, and self-management education: United States, 2001--2009. MMWR Morb Mortal Wkly Rep 60: 547–552. [PubMed] [Google Scholar]
- 2.Asher MI, Montefort S, Bjorksten B, Lai CK, Strachan DP, Weiland SK, Williams H, and I. P. T. S. Group. 2006. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 368: 733–743. [DOI] [PubMed] [Google Scholar]
- 3.Silverberg JI, Simpson EL, Durkin HG, and Joks R. 2013. Prevalence of allergic disease in foreign-born American children. JAMA Pediatr 167: 554–560. [DOI] [PubMed] [Google Scholar]
- 4.Sly RM 1999. Changing prevalence of allergic rhinitis and asthma. Ann Allergy Asthma Immunol 82: 233–248; quiz 248–252. [DOI] [PubMed] [Google Scholar]
- 5.Nurmagambetov T, Kuwahara R, and Garbe P. 2018. The Economic Burden of Asthma in the United States, 2008-2013. Ann Am Thorac Soc 15: 348–356. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, and Grayson MH. 2009. The role of viruses in the development and exacerbation of atopic disease. Ann Allergy Asthma Immunol 103: 181–186; quiz 186,–187, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grayson MH, Feldman S, Prince BT, Patel PJ, Matsui EC, and Apter AJ. 2018. Advances in asthma in 2017: Mechanisms, biologics, and genetics. J Allergy Clin Immunol 142: 1423–1436. [DOI] [PubMed] [Google Scholar]
- 8.Hussain SA, Mejias A, Ramilo O, Peeples ME, and Grayson MH. 2019. Post-viral atopic airway disease: pathogenesis and potential avenues for intervention. Expert Rev Clin Immunol 15: 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikhail I, and Grayson MH. 2019. Asthma and viral infections: An intricate relationship. Ann Allergy Asthma Immunol 123: 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jartti T, Kuusipalo H, Vuorinen T, Soderlund-Venermo M, Allander T, Waris M, Hartiala J, and Ruuskanen O. 2010. Allergic sensitization is associated with rhinovirus-, but not other virus-, induced wheezing in children. Pediatr Allergy Immunol 21: 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jartti T, and Gern JE. 2017. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol 140: 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukkarinen M, Koistinen A, Turunen R, Lehtinen P, Vuorinen T, and Jartti T. 2017. Rhinovirus-induced first wheezing episode predicts atopic but not nonatopic asthma at school age. J Allergy Clin Immunol 140: 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tam JS, Jackson WT, Hunter D, Proud D, and Grayson MH. 2013. Rhinovirus specific IgE can be detected in human sera. J Allergy Clin Immunol 132: 1241–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welliver RC, Kaul TN, and Ogra PL. 1980. The appearance of cell-bound IgE in respiratory-tract epithelium after respiratory-syncytial-virus infection. N Engl J Med 303: 1198–1202. [DOI] [PubMed] [Google Scholar]
- 15.Welliver RC, Wong DT, Sun M, Middleton E Jr., Vaughan RS, and Ogra PL. 1981. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med 305: 841–846. [DOI] [PubMed] [Google Scholar]
- 16.Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, Glanville N, Choy KJ, Jourdan P, Burnet J, Tuthill TJ, Pedrick MS, Hurle MJ, Plumpton C, Sharp NA, Bussell JN, Swallow DM, Schwarze J, Guy B, Almond JW, Jeffery PK, Lloyd CM, Papi A, Killington RA, Rowlands DJ, Blair ED, Clarke NJ, and Johnston SL. 2008. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med 14: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, Radermecker C, Makrinioti H, Aniscenko J, Bartlett NW, Edwards MR, Solari R, Farnir F, Papayannopoulos V, Bureau F, Marichal T, and Johnston SL. 2017. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med 23: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto K, Graham BS, Ho SB, Adler KB, Collins RD, Olson SJ, Zhou W, Suzutani T, Jones PW, Goleniewska K, O’Neal JF, and Peebles RS Jr. 2004. Respiratory syncytial virus in allergic lung inflammation increases Muc5ac and gob-5. Am J Respir Crit Care Med 170: 306–312. [DOI] [PubMed] [Google Scholar]
- 19.Cheung DS, Ehlenbach SJ, Kitchens RT, Riley DA, Thomas LL, Holtzman MJ, and Grayson MH. 2010. Cutting edge: CD49d+ neutrophils induce FcepsilonRI expression on lung dendritic cells in a mouse model of postviral asthma. J Immunol 185: 4983–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung DS, Ehlenbach SJ, Kitchens T, Riley DA, and Grayson MH. 2010. Development of atopy by severe paramyxoviral infection in a mouse model. Ann Allergy Asthma Immunol 105: 437–443 e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung DS, Sigua JA, Simpson PM, Yan K, Hussain SA, Santoro JL, Buell EJ, Hunter DA, Rohlfing M, Patadia D, and Grayson MH. 2018. Cysteinyl leukotriene receptor 1 expression identifies a subset of neutrophils during the antiviral response that contributes to postviral atopic airway disease. J Allergy Clin Immunol 142: 1206–1217 e1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, Battaile JT, Alevy Y, Yan L, Agapov E, Kim EY, and Holtzman MJ. 2007. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med 204: 2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, Agapov E, and Holtzman MJ. 2007. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol 179: 1438–1448. [DOI] [PubMed] [Google Scholar]
- 24.Fites JS, Gui M, Kernien JF, Negoro P, Dagher Z, Sykes DB, Nett JE, Mansour MK, and Klein BS. 2018. An unappreciated role for neutrophil-DC hybrids in immunity to invasive fungal infections. PLoS Pathog 14: e1007073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, and Hammad H. 2012. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med 209: 1505–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grayson MH, Camarda LE, Hussain SA, Zemple SJ, Hayward M, Lam V, Hunter DA, Santoro JL, Rohlfing M, Cheung DS, and Salzman NH. 2018. Intestinal Microbiota Disruption Reduces Regulatory T Cells and Increases Respiratory Viral Infection Mortality Through Increased IFNgamma Production. Front Immunol 9: 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akk AM, Simmons PM, Chan HW, Agapov E, Holtzman MJ, Grayson MH, and Pham CT. 2008. Dipeptidyl peptidase I-dependent neutrophil recruitment modulates the inflammatory response to Sendai virus infection. J Immunol 180: 3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eenjes E, Mertens TCJ, Buscop-van Kempen MJ, van Wijck Y, Taube C, Rottier RJ, and Hiemstra PS. 2018. A novel method for expansion and differentiation of mouse tracheal epithelial cells in culture. Sci Rep 8: 7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas MA, Buelow BJ, Nevins AM, Jones SE, Peterson FC, Gundry RL, Grayson MH, and Volkman BF. 2015. Structure-function analysis of CCL28 in the development of post-viral asthma. J Biol Chem 290: 4528–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, and Garcin D. 2007. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J Virol 81: 12227–12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersson CK, Iwasaki J, Cook J, Robinson P, Nagakumar P, Mogren S, Fleming L, Bush A, Saglani S, and Lloyd CM. 2020. Impaired airway epithelial cell wound-healing capacity is associated with airway remodelling following RSV infection in severe preschool wheeze. Allergy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reber LL, Gillis CM, Starkl P, Jonsson F, Sibilano R, Marichal T, Gaudenzio N, Berard M, Rogalla S, Contag CH, Bruhns P, and Galli SJ. 2017. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med 214: 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, and Chong BH. 2019. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun 10: 1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geng S, Matsushima H, Okamoto T, Yao Y, Lu R, and Takashima A. 2013. Reciprocal regulation of development of neutrophil-dendritic cell hybrids in mice by IL-4 and interferon-gamma. PLoS One 8: e82929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sigurs N 2002. A cohort of children hospitalised with acute RSV bronchiolitis: impact on later respiratory disease. Paediatr Respir Rev 3: 177–183. [DOI] [PubMed] [Google Scholar]
- 36.Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, and Gustafsson PM. 2010. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65: 1045–1052. [DOI] [PubMed] [Google Scholar]
- 37.Sigua JA, Buelow B, Cheung DS, Buell E, Hunter D, Klancnik M, and Grayson MH. 2014. CD49d-expressing neutrophils differentiate atopic from nonatopic individuals. J Allergy Clin Immunol 133: 901–904 e905. [DOI] [PubMed] [Google Scholar]
- 38.Boey H, Rosenbaum R, Castracane J, and Borish L. 1989. Interleukin-4 is a neutrophil activator. J Allergy Clin Immunol 83: 978–984. [DOI] [PubMed] [Google Scholar]
- 39.Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, Kotol PF, Chojnacki BJ, Miyazaki T, Gallo RL, and Takashima A. 2013. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood 121: 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manfredi AA, Ramirez GA, Rovere-Querini P, and Maugeri N. 2018. The Neutrophil’s Choice: Phagocytose vs Make Neutrophil Extracellular Traps. Front Immunol 9: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.