

Abstract
Background
Neonatal arrhythmia is a common complication that might be life-threatening or serious, but the genetic causes are unclear in most cases. The aim of this study is to investigate the genetic causes of neonatal arrhythmia in a NICU in China.
Methods
Newborns who were diagnosed with arrhythmia during the neonatal period were enrolled from Children’s Hospital of Fudan University between January 1st 2016, and December 31st, 2019. A neonatal gene panel was performed for each infant.
Results
In total, 98 neonatal infants with arrhythmia were enrolled. Fourteen genes and a copy number change were identified and classified as pathogenic/likely pathogenic in 22 patients (22.4%), including 4 genes related to syndrome, 4 related to conduction, 2 related to metabolism, 2 related to structure, 2 related to respiration and immunity, respectively, and trisomy 21. Altogether, 6 genes (6/14, 42.9%) caused original heart structure or conduction abnormalities, leading to arrhythmia. Infants with ventricular tachycardia or fibrillation, atrioventricular block and long-QT syndrome all had positive gene results. The gene positive rate among arrhythmic infants with congenital heart disease or severe heart failure was higher than that of infants without congenital heart disease or severe heart failure.
Conclusions
The genetic disorders associated with neonatal arrhythmia could be syndrome-, conduction-, metabolism-, and structure-related. Infants with non-benign arrhythmia, especially ventricular tachycardia or fibrillation, long-QT syndrome, or high-grade atrioventricular block, have a higher rate of genetic abnormalities and should undergo genetic sequencing. Neonates with hereditary arrhythmias may have a higher risk of congenital heart disease or heart failure.
Keywords: Arrhythmia, neonate, genetic counseling, precise treatment
Introduction
Arrhythmia is one of the most common manifestations of cardiovascular disease other than structural heart disease in the neonatal period. The incidence of neonatal arrhythmia is reported to be 1–5% among live births (1-3). Some types of arrhythmia, such as ventricular flutter and fibrillation, grade III atrioventricular block and long-QT syndrome, are life-threatening. Over previous decades, molecular genetics research has established a link between a number of inherited, lethal cardiac arrhythmias and disease-causing variants in genes encoding ion channels or other membrane components in adults (4-7). Some studies have reported that neonatal arrhythmias, such as long-QT syndrome, are related to genetic abnormalities (3,6), but the incidence of genetic abnormalities has not been reported for newborns with arrhythmia. Arrhythmias in the neonatal period may occur in newborns with or without cardiac structural abnormalities. Some serious diseases in the neonatal period can also lead to cardiac failure or electrolyte disturbances, which can lead to arrhythmias. Therefore, it is important to explore the genetic abnormalities associated with arrhythmia in the neonatal period and identify life-threatening arrhythmia and complications.
In this study, we performed whole exome sequencing of samples from 98 newborns with arrhythmia from the NICU (neonatal intensive care unit) and level II neonatal ward of Children’s Hospital of Fudan University, and the study was conducted over four years. We aimed to explore the common types of arrhythmia in newborns and the relationships between different arrhythmia types and inherited diseases and then focused on the prognosis of arrhythmias caused by genetic defects in newborns. These findings may help neonatologists in the clinical management of neonatal arrhythmias carry out genetic testing as early as possible for highly genetically related arrhythmias and may improve prognoses. We present the following article in accordance with the STROBE reporting checklist (available at https://dx.doi.org/10.21037/tp-21-233).
Methods
Patient cohort
Patients were enrolled between January 1st, 2016, and December 31st, 2019, per the following inclusion criteria: (I) newborns were diagnosed with neonatal arrhythmia (ICD 10, P29.100) according to 12-lead electrocardiogram (ECG) or 24-hour dynamic ECG, and (II) parents agreed to inclusion and signed informed consent forms for whole-exome sequencing. Perinatal history, clinical manifestation, laboratory, electrocardiography, Holter monitoring, echocardiogram, and application of antiarrhythmic and cardiotonic drugs were reviewed. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by the Ethics Committee of Children’s Hospital, Fudan University (No. 2015[98]) and individual consent for this retrospective analysis was waived.
Next-generation sequencing
Genomic DNA fragments of patients were enriched for exome sequences using the Agilent (Santa Clara, CA, USA) SureSelectXT Human All Exon 50 Mb kit. After enrichment by PCR, the DNA libraries were sequenced on a HiSeq2000/2500 sequencer according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). The average on-target sequencing depth was 120X, and fraction of official target covered with at least 20X is higher than 96.5%.
Annotation and Validation of SNVs and CNVs
The annotation and filtrations of both SNVs and CNVs followed the pipeline established in our hospital (8). SNVs were annotated by ANNOVAR and VEP software and compared computationally with ESP, ExAC, gnomAD, HGMD and ClinVar. Pathogenicity of SNVs was defined based on American College of Medical Genetics criteria (9). Meanwhile, DGV, DECIPHER, ClinVar, and ClinGen genome dosage maps were utilized in the assessment of CNVs. The detected SNVs were confirmed using PCR, and PCR-amplified DNA products were subjected to Sanger sequencing (3500XL Genetic Analyzer, Applied Biosystems) according to the manufacturer’s specifications. The detected CNVs were confirmed by karyotype or chromosomal microarray.
Statistical analysis
The data were analyzed with SPSS version 18.0 software. Independent-sample t-tests were used to compare the continuous parametric variables between the two groups. A continuous-calibration chi-square test was used to compare the rate between the two groups. P values <0.05 were used to indicate statistical significance.
Results
Demographics and clinical characteristics
The total number of inpatients with an age at admission of <28 days after birth in the NICU and neonatal ward of Children’s Hospital of Fudan University was 12,697 between January 2016 and December 2019. During this period, the total number of patients with a discharge diagnosis of neonatal arrhythmia was 121 (1.0%). Eighteen infants were excluded because their parents declined exome sequencing. Five infants were excluded due to incomplete ECG data. Finally, 98 neonates were enrolled in this study.
There were 60 males (61.2%) and 38 females (38.8%), with a mean gestational age of 36.6±3.9 weeks and a mean birth weight of 2,739.7±867.1 g. There were 38 preterm infants, 7 small-for-gestational-age infants and 9 infants who were one of a set of twins. The detailed demographic and basic clinical characteristics are shown in Table 1. Eighteen infants (18/98, 18.4%) were delivered by emergent cesarean section because of suspected fetal distress based on an abnormal fetal heart rhythm, while none of them were diagnosed with neonatal asphyxia after birth. Fourteen of eighteen infants had fetal tachycardia arrhythmia [2 with an atrial, 3 with a junctional and 1 with a ventricular premature beat; 4 with atrial tachycardia; 2 with fetal supraventricular tachycardia (SVT); and 2 with atrial flutter]. Four of eighteen infants had fetal bradyarrhythmia (1 persistent sinus bradycardia, 3 atrioventricular block).
Table 1. Characteristics of neonates with arrhythmia.
| Characteristics | Cases (N=98) (%) |
|---|---|
| Sex (male) | 60 (61.2) |
| Preterm | 38 (38.8) |
| Small for gestational age | 7 (7.1) |
| Twin | 9 (9.2) |
| Mean gestational age (weeks) | 36.6±3.9 |
| Mean birth weight (g) | 2,739.7±867.1 |
| Abnormal fetal heart rhythm | 18 (18.4) |
Arrhythmias among these infants were divided into three subgroups, including premature contraction, tachyarrhythmia and bradyarrhythmia based on the most severe or frequent arrhythmia in the infant. Premature contraction, including atrial, junctional and ventricular premature beats, accounted for the highest proportion (44.9%) of arrhythmias. SVT was the most common type of tachyarrhythmia. Atrioventricular conduction block and bundle branch block made up the majority of bradyarrhythmias. Table 2 shows the proportions of the different types of arrhythmia in this study.
Table 2. Proportions of different type of arrhythmias.
| Arrhythmia types | Cases (N=98) | % |
|---|---|---|
| Premature contraction (n=44, 44.9%) | ||
| Atrial premature beat | 19 | 19.4 |
| Junctional premature beat | 14 | 14.3 |
| Ventricular premature beat | 11 | 11.2 |
| Tachyarrhythmia (n=36, 36.7%) | ||
| Atrial tachycardia | 9 | 9.2 |
| Supraventricular tachycardia | 15 | 15.3 |
| Atrial flutter | 5 | 5.1 |
| Atrial fibrillation | 2 | 2.0 |
| Ventricular tachycardia | 2 | 2.0 |
| Ventricular fibrillation | 3 | 3.1 |
| Bradyarrhythmia (n=18, 18.4%) | ||
| Sinus bradycardia | 4 | 4.1 |
| First-degree atrioventricular block | 4 | 4.1 |
| Second-degree atrioventricular block | 3 | 3.1 |
| Third-degree atrioventricular block | 2 | 2.0 |
| Left bundle branch block | 1 | 1.0 |
| Right bundle branch block | 3 | 3.1 |
| Long-QT syndrome | 1 | 1.0 |
Overview of the genetic spectrum
Overall, the genetic diagnostic yield was 22.4% (22/98) in this cohort, which included 20 probands with single gene defects and 2 patients with chromosome abnormality. Pathogenic/likely pathogenic variants in 14 genes and a copy number change were identified in 22 patients (22.4%) (Tables S1,S2). Among the 14 genes, 4 were syndrome-related genes (PTPN11, TSC2, TBX5 and EFTUD2), 4 were ion channel-related genes (SCN2A, SCN5A, KCNQ1 and KCNH2), 2 were metabolism-related genes (SLC25A20 and CPT2), 2 were cardiomyopathy-related genes (DSG2 and PKP2) and 2 were either respiratory- or immune-related genes (PHOX2B and IL10RA). A copy number change, trisomy 21, was identified in two patients. Altogether, 6 genes (6/14, 42.9%) caused original heart structure or conduction abnormalities and then led to arrhythmia. The other genes caused metabolic or other systemic diseases and secondary arrhythmia (Table S3).
Genetic findings in three subgroups of arrhythmia
In this cohort, premature contraction accounted for the highest proportion of cases (44.9%), with a molecular diagnostic yield of only 11.4% (5/44) (Figure 1). Infants with premature beats had disease-causing variants in syndromic, respiration-, and immune-related genes, though these genes had no direct relationship with their arrhythmias (Figure 2).
Figure 1.
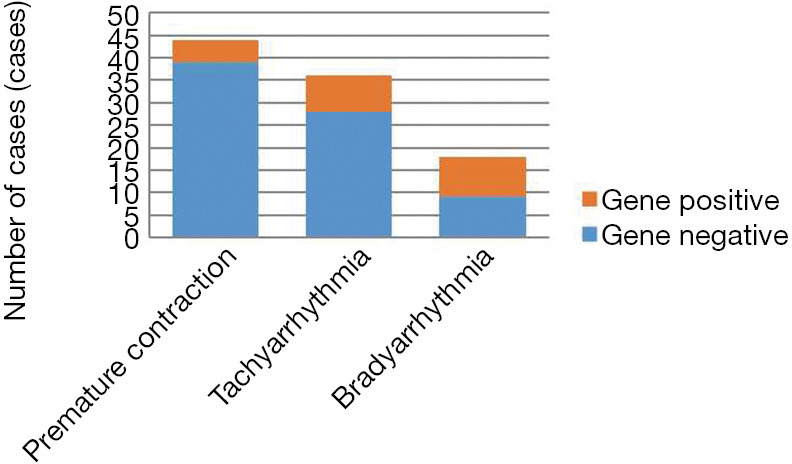
Account of gene positive in different type of arrhythmia.
Figure 2.
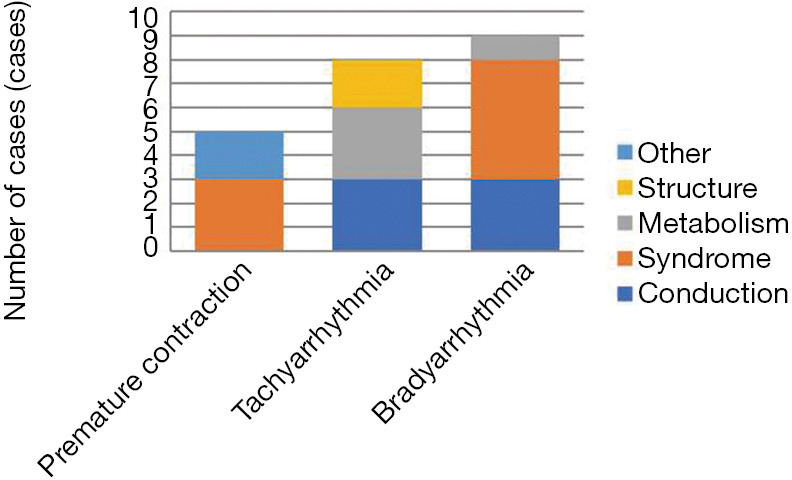
Account of different gene positive in different type of arrhythmia.
Infants with tachyarrhythmia accounted for 36.7% of cases, and their molecular diagnostic yield was 22.2% (8/36) (Figure 1). Among infants with tachyarrhythmia, 5 newborns had ventricular tachycardia and ventricular fibrillation, and all of them had genetic defects, including defects in 4 genes (KCNH2, DSG2, CPT2 and SLC25A20).
Bradyarrhythmia accounted for only 18.4% of all infants but had the highest molecular diagnostic yield (50%, 9/18) (Figure 1). For bradyarrhythmia, persistent sinus bradycardia, Grade I atrioventricular block and long-QT syndrome had higher genetic positive rates (50%, 100% and 100%), including mainly syndrome- and conduction-related genes (Figure 2).
Ion channel-related genes were found only in infants with either tachyarrhythmia or bradyarrhythmia and not in infants with premature contraction. Newborns with ventricular tachycardia and fibrillation, atrioventricular block and long-QT syndrome should be highly suspected of having molecular defects.
Relationship between clinical complications and genetic results in infants with neonatal arrhythmia
Table 3 showed the molecular diagnostic yields among neonates with arrhythmia and congenital heart disease. These yields were much higher than those among neonates without heart disease, and the difference was statistically significant (P<0.01). The most common congenital heart diseases were hypertrophic cardiomyopathy and ventricular septal defects. Four infants with hypertrophic cardiomyopathy had conduction abnormalities (3 grade I atrioventricular blocks and 1 right branch bundle block). Three of them had severe heart failure, and one infant died at 3 days after birth.
Table 3. Genetic results for neonatal arrhythmia with or without CHD and heart failure.
| Classification | Newborns with genetic findings (n=22) |
Newborns without genetic findings (n=76) | Pearson chi-square test | P |
|---|---|---|---|---|
| Arrhythmia with CHD | 7 | 5 | 7.902 | <0.01 |
| Arrhythmia without CHD | 15 | 71 | ||
| Arrhythmia with heart failure | 6 | 6 | 5.962 | <0.05 |
| Arrhythmia without heart failure | 16 | 70 |
CHD, congenital heart disease.
The molecular diagnostic yield for neonates with arrhythmia and heart failure was higher than that for infants without heart failure in this study, and the difference was statistically significant (P<0.05). All four infants diagnosed with carnitine palmityl transferase II deficiency showed acute and severe heart failure and died within 5 days after birth. Three of them had ventricular fibrillation, and one had atrioventricular block. In this study, infants with atrial flutter (8 infants) and ventricular fibrillation (3 infants) were treated with electric defibrillation. Infants with malignant arrhythmias were treated with anti-arrhythmic drug and/or heart failure drugs.
Discussion
The onset of neonatal arrhythmia could occur during fetal development or be secondary to organic disease during the neonatal period. As reported, fetal arrhythmia occurs in as many as 1–3% of all pregnancies and is benign in most cases (10,11). The most common fetal arrhythmia in our study was tachycardia, particularly atrial and junctional premature beats. Although 18 infants were delivered by emergent cesarean section for suspected fetal distress due to an abnormal fetal heart rhythm, only one of them with fetal SVT had true neonatal asphyxia. In addition to identification by fetal ECG, fetal arrhythmia can be found by fetal auscultation, electrocardiography and magnetocardiography (12,13). Although most fetal arrhythmias are benign, approximately 10% of them, such as SVT, atrial flutter, high-degree atrioventricular block and long-QT syndrome, may be life-threatening. Therefore, further evaluation of all fetuses with irregular rhythms or inappropriate heart rates is necessary, especially if there is a family history of premature sudden cardiac arrest or perinatal loss.
The incidence of neonatal arrhythmia is reported to be 1–5% among all neonates (1-3). The incidence of neonatal arrhythmia in this study was 1%, which might be lower than the actual incidence because not all inpatients accepted ECG screening. SVT was the second most common arrhythmia in this cohort, after premature beats. As reported, SVT is the most common arrhythmia in infants, and the incidence of SVT requiring inpatient care ranges from 1:4,500–1:5,000 in the first year of life (14-16). Persistent SVT can cause heart failure, so it should be identified and treated as soon as possible.
The genetic diagnostic rate in this cohort was 21.0%, and syndrome-related and ion channel-related genetic defects were the most common. Two of three infants with Noonan syndrome had first-degree atrioventricular block and one of them had hypertrophic cardiomyopathy during the neonatal period. The PTPN11 gene is the most common disorder in Noonan syndrome and covers nearly half (42.5%) of the reported disease-causing variants (17,18). Hypertrophic cardiomyopathy can be mild or severe, can present from the prenatal period to late childhood and is observed in 20% of infants with Noonan syndrome (18). In this study, ion channel-related genes included mainly sodium channels (SCN2A and SCN5A) and potassium channels (KCNQ1 and KCNH2). Among the 6 infants with ion channel-associated gene abnormalities, 4 had bradyarrhythmia, and 2 had tachyarrhythmia. According to the analysis report, positivity for any of the approximately 40 genes encoding cardiac sodium, potassium and calcium ion channels could lead to an arrhythmogenic substrate in a structurally normal heart (19). This could result in some life-threatening arrhythmias, such as long-QT syndrome, short-QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia and even sudden infant death syndrome (19-22). In our cohort, four infants with malignant arrhythmia died early after birth, ultimately they were diagnosed with carnitine palmityl transferase II deficiency based on genetic sequencing reports. This reminds us that in addition to primary ECG disorder and arrhythmia-causing cardiomyopathy, some severe diseases may also cause life-threatening arrhythmia, and on these occasions, gene detection is necessary.
Atrial, supraventricular, and ventricular premature contractions are thought to be benign (3). In healthy newborns, these occasional ectopic beats will not cause clinical symptoms and do not require therapy, and they are most often self-limiting and disappear when the infant grows older, often within weeks of their appearance (3,23). In this study, the genetic diagnostic rate among infants with only premature contraction was lower than that among infants with tachyarrhythmia or bradyarrhythmia. SVT, ventricular tachycardia, atrioventricular conduction abnormalities and long-QT syndrome are classified as non-benign arrhythmia (3). In this study, infants with ventricular tachycardia or fibrillation, atrioventricular block and long-QT syndrome all had positive gene results, which indicates that neonates with non-benign arrhythmia had a higher risk of gene abnormalities. Therefore, those arrhythmias should be treated as soon as possible, and genetic testing should be performed as soon as possible.
Analysis of the relationship between clinical complications and genetic findings showed that newborns with arrhythmia and congenital heart disease or severe heart failure had a higher risk of genetic disorder. Therefore, for those infants, genetic sequencing should be performed as early as possible so that the etiology behind the arrhythmia can be more precisely determined.
Conclusions
Premature beat and SVT are the most common arrhythmias in the neonatal period. SVT, ventricular tachycardia, atrioventricular block and long-QT syndrome are highly related to genetic abnormalities. Genetic testing is recommended for newborns with these kinds of arrhythmia and severe complications. Neonates with hereditary arrhythmias may have a higher risk of congenital heart disease or heart failure.
Supplementary
The article’s supplementary files as
Acknowledgments
We thank the patients and their families for their participation in this study. The authors would like to express their gratitude to the participating families.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by the Ethics Committee of Children’s Hospital, Fudan University (No. 2015[98]) and individual consent for this retrospective analysis was waived.
Footnotes
Reporting Checklist: The authors have completed the STROBE reporting checklist. Available at https://dx.doi.org/10.21037/tp-21-233
Data Sharing Statement: Available at https://dx.doi.org/10.21037/tp-21-233
Peer Review File: Available at https://dx.doi.org/10.21037/tp-21-233
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/tp-21-233). The authors have no conflicts of interest to declare.
References
- 1.Badrawi N, Hegazy RA, Tokovic E, et al. Arrhythmia in the neonatal intensive care unit. Pediatr Cardiol 2009;30:325-30. 10.1007/s00246-008-9355-4 [DOI] [PubMed] [Google Scholar]
- 2.Kundak AA, Dilli D, Karagöl B, et al. Non benign neonatal arrhythmias observed in a tertiary neonatal intensive care unit. Indian J Pediatr 2013;80:555-9. 10.1007/s12098-012-0852-3 [DOI] [PubMed] [Google Scholar]
- 3.Ban JE. Neonatal arrhythmias: diagnosis, treatment, and clinical outcome. Korean J Pediatr 2017;60:344-52. 10.3345/kjp.2017.60.11.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arndt AK, MacRae CA. Genetic testing in cardiovascular diseases. Curr Opin Cardiol 2014;29:235-40. 10.1097/HCO.0000000000000055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helm BM, Freeze SL, Spoonamore KG, et al. The Genetic Counselor in the Pediatric Arrhythmia Clinic: Review and Assessment of Services. J Genet Couns 2018;27:558-64. 10.1007/s10897-017-0169-5 [DOI] [PubMed] [Google Scholar]
- 6.Wilde AAM, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol 2018;4:569-79. 10.1016/j.jacep.2018.03.006 [DOI] [PubMed] [Google Scholar]
- 7.Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J 2014;10:29-33. 10.14797/mdcj-10-1-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong X, Liu B, Yang L, et al. Clinical exome sequencing as the first-tier test for diagnosing developmental disorders covering both CNV and SNV: a Chinese cohort. J Med Genet 2020;57:558-66. 10.1136/jmedgenet-2019-106377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan SM. Fetal arrhythmias: diagnosis and treatment. J Matern Fetal Neonatal Med 2020;33:2671-8. 10.1080/14767058.2018.1555804 [DOI] [PubMed] [Google Scholar]
- 11.Maeno Y, Hirose A, Kanbe T, et al. Fetal arrhythmia: prenatal diagnosis and perinatal management. J Obstet Gynaecol Res 2009;35:623-9. 10.1111/j.1447-0756.2009.01080.x [DOI] [PubMed] [Google Scholar]
- 12.Wacker-Gussmann A, Strasburger JF, Cuneo BF, et al. Diagnosis and treatment of fetal arrhythmia. Am J Perinatol 2014;31:617-28. 10.1055/s-0034-1372430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bravo-Valenzuela NJ, Rocha LA, Machado Nardozza LM, et al. Fetal cardiac arrhythmias: Current evidence. Ann Pediatr Cardiol 2018;11:148-63. 10.4103/apc.APC_134_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu MH, Chen HC, Kao FY, et al. Postnatal cumulative incidence of supraventricular tachycardia in a general pediatric population: A national birth cohort database study. Heart Rhythm 2016;13:2070-5. 10.1016/j.hrthm.2016.06.006 [DOI] [PubMed] [Google Scholar]
- 15.Turner CJ, Wren C. The epidemiology of arrhythmia in infants: a population-based study. J Paediatr Child Health 2013;49:278-81. 10.1111/jpc.12155 [DOI] [PubMed] [Google Scholar]
- 16.Bjeloševič M, Illíková V, Tomko J, et al. Supraventricular tachyarrhythmias during the intrauterine, neonatal, and infant period: A 10-year population-based study. Pacing Clin Electrophysiol 2020;43:680-6. 10.1111/pace.13964 [DOI] [PubMed] [Google Scholar]
- 17.El Bouchikhi I, Belhassan K, Moufid FZ, et al. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med 2016;3:133-42. 10.1016/j.ijpam.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome. Lancet 2013;381:333-42. 10.1016/S0140-6736(12)61023-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernández-Falgueras A, Sarquella-Brugada G, Brugada J, et al. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology (Basel) 2017;6:7. 10.3390/biology6010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Niekerk C, Van Deventer BS, du Toit-Prinsloo L. Long QT syndrome and sudden unexpected infant death. J Clin Pathol 2017;70:808-13. 10.1136/jclinpath-2016-204199 [DOI] [PubMed] [Google Scholar]
- 21.Tester DJ, Wong LCH, Chanana P, et al. Cardiac Genetic Predisposition in Sudden Infant Death Syndrome. J Am Coll Cardiol 2018;71:1217-27. 10.1016/j.jacc.2018.01.030 [DOI] [PubMed] [Google Scholar]
- 22.Sarquella-Brugada G, Cesar S, Zambrano MD, et al. Electrocardiographic Assessment and Genetic Analysis in Neonates: a Current Topic of Discussion. Curr Cardiol Rev 2019;15:30-7. 10.2174/1573403X14666180913114806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaeggi E, Öhman A. Fetal and Neonatal Arrhythmias. Clin Perinatol 2016;43:99-112. 10.1016/j.clp.2015.11.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as