

Abstract
MicroRNA (miRNA) is not a single sequence, but a series of multiple variants (also termed isomiRs) with sequence and expression heterogeneity. Whether and how these isoforms contribute to functional variation and complexity at the systems and network levels remain largely unknown. To explore this question systematically, we comprehensively analyzed the expression of small RNAs and their target sites to interrogate functional variations between novel isomiRs and their canonical miRNA sequences. Our analyses of the pan-cancer landscape of miRNA expression indicate that multiple isomiRs generated from the same miRNA locus often exhibit remarkable variation in their sequence, expression and function. We interrogated abundant and differentially expressed 5′ isomiRs with novel seed sequences via seed shifting and identified many potential novel targets of these 5′ isomiRs that would expand interaction capabilities between small RNAs and mRNAs, rewiring regulatory networks and increasing signaling circuit complexity. Further analyses revealed that some miRNA loci might generate diverse dominant isomiRs that often involved isomiRs with varied seeds and arm-switching, suggesting a selective advantage of multiple isomiRs in regulating gene expression. Finally, experimental validation indicated that isomiRs with shifted seed sequences could regulate novel target mRNAs and therefore contribute to regulatory network rewiring. Our analysis uncovers a widespread expansion of isomiR and mRNA interaction networks compared with those seen in canonical small RNA analysis; this expansion suggests global gene regulation network perturbations by alternative small RNA variants or isoforms. Taken together, the variations in isomiRs that occur during miRNA processing and maturation are likely to play a far more complex and plastic role in gene regulation than previously anticipated.
Keywords: microRNA (miRNA), isoforms, gene regulatory networks, gain of interactions, functional diversity, signaling specificity
INTRODUCTION
MicroRNAs (miRNAs) are a class of small endogenous non-coding RNAs, about 22 nt long, which are generated from single-stranded miRNA precursors (pre-miRNAs) that can fold into hairpin structures [1]. These non-coding RNAs contribute to multiple biological processes primarily by negatively regulating gene expression at the transcriptional or post-transcriptional level by cleaving transcripts or inhibiting their translation [2, 3]. Because of their flexible regulatory roles, miRNAs have been widely studied, particularly in the development of various diseases. In particular, miRNAs play an important role in the pathological processes of diverse cancers [4, 5], and several abnormally expressed miRNAs may be served as biomarkers for disease diagnosis or prognosis [6–8].
High-throughput sequencing techniques contribute significantly to miRNA studies, and an interesting phenomenon of multiple miRNA isoforms (also termed miRNA variants, or isomiRs) arising from a single genetic locus has been widely observed using deep sequencing data. These findings suggest miRNA sequence variability and the complexity of miRNA biogenesis and the dynamics of the miRNA-ome [9–12] (Figure 1A). These series of isomiRs have high sequence similarity but vary in length and expression levels, and some may be involved in carrying out functions different from those carried out by the canonical miRNA, by altering 5′ ends and miRNA ‘seed sequences’ (nucleotides 2–8) [13–15]. ‘Binarized’ isomiR profiles can be used to discriminate between different cancer types [16], and the most discriminatory isomiRs are also differentially expressed. On the basis of the contributions of alternative cleavage, RNA editing and modification events in the miRNA maturation process, isomiRs are prone to length and sequence variations, and some may contain altered 5′ ends and seed sequences and even target novel mRNAs. For example, miR-411 and its 5′-isomiR have distinct targets and functions in the vasculature under ischemia [17]; 5′-isomiR of miR-140-3p may be a potential target for prevention of triple negative breast cancer [18]; and 5′ isomiRs in rice also exhibit their probable biological activity [19]. Growing studies have surveyed the expression levels and possible functions of select isomiRs [20–23] and strongly support that the isomiRs could play roles in RNA interactome networks and that abnormally expressed isomiRs with varied seeds could serve as regulators in cancer cell signaling. However, the phenomenon of miRNA heterogeneity has not been systematically addressed, although the repertoire of isomiRs offers potential cancer biomarkers, particularly for isomiRs with novel seed sequences. Few studies have evaluated whether and how these isoforms contribute to functional variation and complexity at the systems level.
Figure 1 .
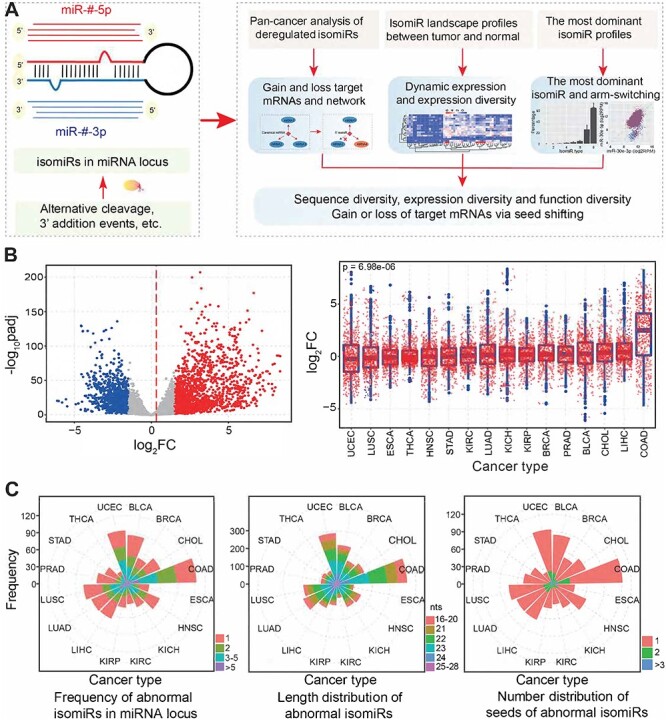
Systematic characterization of deregulated isomiRs across cancer types. (A) The phenomenon of multiple isomiRs in a miRNA locus. This study mainly includes three sections to explore isomiRs: pan-cancer analysis of deregulated isomiRs, isomiR landscape profiles compared between tumor and normal samples and the most dominant isomiR expression profiles. (B) The scatter plot shows the distributions of log2FC and –log10padj values based on differential expression analysis at the isomiR level (the baseMean value in DESeq analysis of each isomiR is more than 100 in one or more cancer types). The mean value of log2FC is shown using a red dashed line. Blue dots indicate significantly downregulated isomiRs, grey dots indicate normally expressed isomiRs and red dots indicate significantly up-regulated isomiRs. The box plots (right) indicate the detailed distribution of log2FC across cancer types. The P-value is estimated using a trend test based on the median of log2FC. FC, fold change in expression; padj, adjusted P-values. (C) Distributions of the frequency of differentially expressed isomiRs in miRNA loci, length of abnormal isomiRs and number of involved seeds in miRNA loci across cancers.
In this study, we investigated isomiR-mediated gene regulatory networks via a comprehensive analysis of a pan-cancer landscape of small RNA expression profiles and target sites based on data from thousands of cancer patients (Figure 1A). First, differentially expressed isomiR types were screened across cancer lineages at the isomiR level. IsomiRs with novel 5′ ends (5′ isomiRs) and seed sequences were analyzed to predict their target mRNAs. Second, the isomiR functional landscape was comprehensively charted across different cancer types, and isomiRs were further classified on the basis of canonical miRNA sequences. Specifically, compared with canonical miRNAs, 3′ and 5′ isomiR atlases were generated. Third, on the basis of each miRNA gene, the most dominant isomiR types were identified to explore potential variations between tumor and normal samples, especially the potential ‘arm-switching’ phenomenon between miR-#-5p and miR-#-3p. Finally, to validate the potential regulatory function of isomiRs with novel seeds, experimental validation was further performed in cancer cell lines. Taken together, our findings provide systematic evidence for widespread RNA regulatory network rewiring by 5′ isomiRs with novel seeds, which could elicit novel biological functions associated with perturbed targets at the global scale. These results suggest that isomiRs could serve as alternative biomarkers to explore regulatory roles of small RNAs compared to traditional canonical miRNAs.
MATERIALS AND METHODS
Deregulated isomiRs and mRNA expression profiles across cancer types
To perform a global analysis of isomiRs, we downloaded isomiR and mRNA expression datasets in 33 cancer types from The Cancer Genome Atlas (TCGA) website using the R package ‘TCGAbiolinks’ [24]. Deregulated isomiR landscape profiles were analyzed using DESeq2 [25] at the isomiR level: expression patterns were estimated for each specific isomiR sequence, although some of them were generated from the same miRNA locus. If an isomiR was not detected in more than 50% of total samples, it was removed from the DESeq2 analysis. As major targets of isomiRs, differential mRNA expression profiles were simultaneously estimated using DESeq2 package (if a gene was not detected in more than 50% of total samples, it was removed from the DESeq2 analysis). Abnormally expressed species were screened if they had higher enrichment levels (baseMean value more than 100) and simultaneously had significant expression divergence between tumor and normal samples. To understand sequence features among multiple isomiRs, a sequence logo was constructed using WebLogo [26].
Target mRNA prediction and validation
First, seed sequences were obtained from deregulated isomiRs according to their detailed location distribution on human chromosomes. 5′ isomiRs might involve novel seeds, and these abnormally expressed seeds were used to predict candidate target mRNAs as well as their canonical seeds using TargetScan 7.0 [27]. According to predicted target mRNAs of canonical seeds, the threshold value of context++_score was set by an integrative analysis of predicted targets, annotated targets of canonical miRNAs in TargetScan and the miRTarBase database [28]. Second, predicted targets of novel seeds and canonical seeds were further validated as to whether they were also differentially expressed in tumor samples. If candidate targets were deregulated and showed opposite differential expression patterns as compared to isomiRs, the targets would be further validated. Finally, co-expression analysis was performed in specific cancer types to identify potential isomiR-mRNA interactions. If the Spearman correlation coefficient of isomiR–mRNA was negative and the false discovery rate was <0.05 (each cancer type must contain at least 50 shared individual samples between tumor and normal samples), then the isomiR:mRNA pair was considered as verified and selected for further analysis.
For the obtained isomiR:mRNA pairs, we screened miRNA loci to compare targets between novel seeds and their canonical seeds if both novel seeds and canonical seeds were differentially and abundantly expressed. A hypergeometric test was used to estimate the significance of differences between target mRNAs of novel seeds and canonical seeds. The P-value was estimated using the following formula:
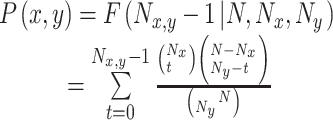 |
Where N was the largest total target mRNAs of canonical seeds and novel seeds in selected miRNA loci, and Nx and Ny indicated the number of target mRNAs that were regulated by novel seeds and canonical seeds, respectively. Nx,y represented the number of shared mRNAs regulated by canonical seeds and novel seeds. P < 0.05 indicated a significant difference between targets of canonical seeds and novel seeds.
Function analysis of novel seeds and canonical seeds
Based on the screened target mRNAs, functional enrichment analysis of deregulated isomiRs was performed using The Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.8 [29]. We also investigated the distributions of cancer hallmark genes [30] (http://software.broadinstitute.org/gsea/msigdb/), Cancer Gene Census (CGC) genes [31] (http://cancer.sanger.ac.uk/census), and core essential genes, or common essential genes from Hart et al. [32], Blomen et al. [33] and Wang et al. [34], between candidate targets of novel seeds and canonical seeds using the above-mentioned hypergeometric test (1−P(x,y)). In this specific hypergeometric test, N was the number of total human genes, and Nx and Ny indicated the number of target mRNAs of specific genes and total genes of cancer hallmark genes, CGC genes or essential genes, respectively. Nx,y represented the number of predicted cancer hallmark genes, CGC genes or essential genes. An adjusted P < 0.05 was considered significant.
IsomiR landscape profiles compared between tumor and normal samples
isomiR expression profiles were analyzed and compared between tumor and normal samples. First, isomiR profiles were obtained according to detailed locations on human chromosomes [for each isomiR, reads per million (RPM) was more than 50, and simultaneous analysis was also performed using a cutoff value of RPM of more than 10]. Second, isomiRs were classified according to miRNA loci and their seeds. Finally, length distributions, seed alterations, sequence variations and expression patterns were used to estimate potential divergence of small RNAs between tumor and normal samples at the isomiR level.
Moreover, the most dominant isomiR species were screened on the basis of every miRNA gene, and the median RPM value was used to estimate relative expression levels of the isomiRs. Only isomiRs with abundant enrichment levels (median RPM > 100) were included in this analysis. On the basis of miRNA loci, the most dominant isomiR species were compared to investigate potential divergence between tumor and normal samples in each cancer type, including sequence variation, seed shifting and arm-switching. We also identified the most dominant isomiR from each miRNA locus based on every individual sample to assess variation between the most dominant isomiR species. Finally, expression distributions of multiple isomiRs were estimated using relative expression percentages based on the total expression in a given miRNA locus in every individual sample to assess expression patterns among isomiRs.
Survival analysis
The clinical data in TCGA were obtained using the R package ‘TCGAbiolinks’ [24], including survival status, stage, grade, survival time and molecular subtype. The involved patients were divided into two groups based on the median expression of a specific isomiR type, and a log-rank test was used to compare the two groups. If the P-value was <0.05, it was considered statistically significant.
Cell culture and treatment
In order to further validate the potential regulatory function of isomiRs with novel seeds, we used SW480 (human colorectal carcinoma cell line). SW480 cells were cultured in Dulbecco’s Modified Eagle’s Medium (Wisent, St. Bruno, Canada) with 10% fetal bovine serum (Wisent, St. Bruno, Canada) and 1% penicillin–streptomycin in an incubator containing 5% CO2 at 37°C. Cell transfection experiments were performed using Lipofectamine 3000 (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Canonical miR-101-3p (mimic/nc, normal control) and its isomiR (novel-mimic/nc) were designed and synthesized. The sequence of each mimic (double-stranded) is as follows:
miR-101-3p-novel mimic sequence:
5′ GUACAGUACUGUGAUAACUGAA 3′
3′ CAUGUCAUGACACUAUUGACUU 5′
miR-101-3p-cano mimic sequence:
5′ UACAGUACUGUGAUAACUGAA 3′
3′ AUGUCAUGACACUAUUGACUU 5′
mimic NC sequence:
5′ UUUGUACUACACAAAAGUACUG 3′
3′ AAACAUGAUGUGUUUUCAUGAC 5′
Quantitative real-time RT-PCR
Total RNA was extracted from SW480 cells using TRIzol reagent (Life Technologies, Carlsbad, CA, USA). cDNA was generated from total RNA using a reverse transcription kit (Life Technologies, Carlsbad, CA, USA). RAB11FIP1 gene expression was measured by qPCR (Lightcycler96, Roche, Basel, Switzerland) using a SYBR green kit (Life Technologies, Carlsbad, CA, USA). RAB11FIP1 primers were: forward: 5′-AGCAAGAAGCCAGAGAGCAG-3′, reverse: 5′-CAAAGAGGTTCAGGGAGCTG-3′. GAPDH primers were forward: 5′-GTCAGTGGTGGACCTGACCT-3′, reverse: 5′-TGCTGTAGCCAAATTCGTTG-3′.
Western blotting
Harvested cells were suspended in RIPA buffer containing a protease inhibitor cocktail. Equal amounts of protein were loaded and separated by 10% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes (Millipore Sigma, USA). The membranes were then incubated overnight using appropriate antibodies against RAB11FIP1 and β-actin. The following antibodies and doses were used: Antibody RAB11FIP1 (Proteintech, Cat No. 16778–1-AP, Lot: 00025721, Dilution ratio:1:2000); Antibody β-actin (ORIGENE, Cat No. TA811000, Lot: V0104, Dilution ratio:1:10,000); Goat anti-mouse IgG-HRP (Santa Cruz, sc-2005, Lot: H2311, Dilution ratio:1:10,000) and Goat anti rabbit IgG-HRP (Santa Cruz, sc-2004, Lot: 12314, Dilution ratio: 1:10,000). These experiments were performed in triplicate. Quantitation of proteins was performed using the AlphaEasc FC system (Alpha InnotechFluorChem 8800, USA).
Statistical analysis
Wilcoxon rank-sum test, hypergeometric test, Kolmogorov–Smirnov test, paired t-test and trend test were used to perform hypothesis testing in relevant analyses, and all statistical analyses were performed using R programming language (version 3.4.3). Schematics of the coding–non-coding regulatory network were presented using Cytoscape 3.6.0 [35]. Venn distributions were estimated using a public tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Results
Deregulated isomiR profiles indicate sequence and expression diversity
Our previous studies have shown that isomiRs from a given miRNA locus may have varied expression patterns [9, 36], but the detailed differentially expressed isomiR profiles across cancer types are still unclear. To systematically investigate isomiR functional landscapes in tumor samples, we first performed a global analysis of differential expression from the isomiR level across cancer types in TCGA. Given that some cancer types had limited sample size in normal or tumor groups (<8), deregulated small RNAs were finally screened in 16 cancer types (Supplementary Table S1 available online at https://academic.oup.com/bib).
Differential expression profiles indicated that a series of isomiRs were deregulated in cancer (Figure 1B) and the distribution of fold-change values (log2FC) of these isomiRs significantly differed across cancer types (P = 6.98e−06 based on trend test, Figure 1B). Abnormal isomiR expression profiles suggested that multiple isomiRs could be produced from a single miRNA locus, and the isomiRs showed diverse length distributions, although most were 21–23 nt (Figure 1C). As indicated by abundantly expressed isomiRs, most miRNA loci had a single seed, but some loci appeared to generate two or more seeds that were also differentially expressed (Figure 1C). Specifically, these isomiRs had different length distributions compared with their canonical miRNAs. The most dominant length of deregulated isomiRs was 23 nt (25.78%), followed by 22 nt (21.56%), while the most dominant length of canonical miRNAs was 22 nt (47.60%), followed by 21 nt (17.87%) (Supplementary Figure S1A available online at https://academic.oup.com/bib). These findings support greater sequence diversity in isomiRs than in canonical miRNAs.
Clustering analysis revealed that total isomiR expression distribution varied across cancer types, and expression levels in related tissues, such as KIRP (Kidney Renal Papillary Cell Carcinoma), KIRC (Kidney Renal Clear Cell Carcinoma) and KICH (Kidney Chromophobe), in the kidney, were prone to cluster together (Figure 2A and Supplementary Figure S1B available online at https://academic.oup.com/bib). Deregulated isomiRs showed diverse expression patterns across cancer types, although isomiRs from a given miRNA locus had high sequence similarity and were more closely related at transcriptional levels (Figure 2B). For example, isomiRs in miR-10a-5p had diverse expression across different cancer types, and some were upregulated while others were downregulated within specific cancers (Figure 2B). This varied expression indicates that small RNAs may have tissue specificity, and indeed, some isomiRs are detected only in specific tissues. The expression diversity also indicates that isomiR generation is more complex than previously anticipated, and some mechanisms in maturation, such as processing, modification and regulation, may be critical factors that control isomiR repertoires and biological functions.
Figure 2 .
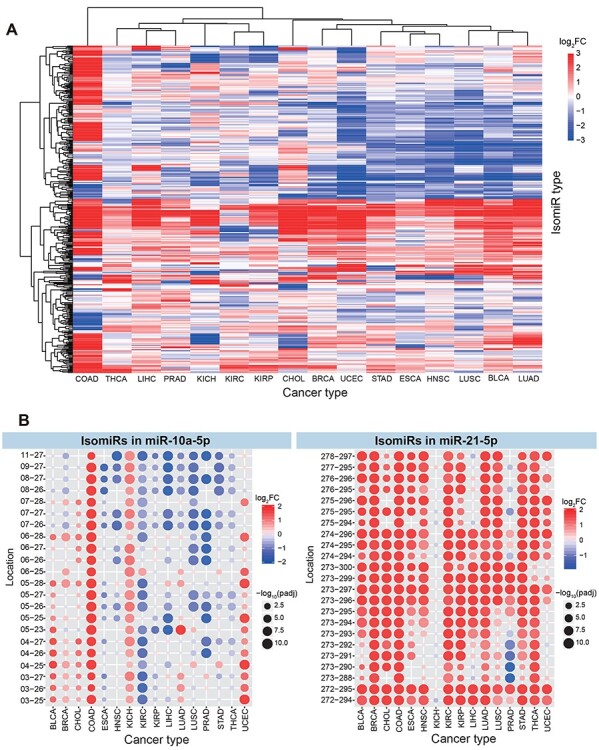
IsomiR expression patterns across different cancer types. (A) Clustering analysis of differentially expressed isomiRs indicates dynamic expression (these isomiRs are abundantly expressed species, and the baseMean value of each isomiR is more than 1000 in one or more cancer types). (B) Specific examples of differentially expressed isomiR profiles in miR-10a-5p and miR-21-5p loci. For example, miR-10a-5p, 03-25 indicates the locus hg38:chr17:48579903-48579925:− and miR-21-5p, 272-294 indicates the locus hg38:chr17:59841272-59841294:+.
Novel seeds via seed shifting events are prone to targeting novel mRNAs
Although 5′ isomiRs have been reported in multiple miRNA variants, knowledge of the 5′ isomiR landscape and function on a large scale is still lacking. Using differential isomiR expression profiles, we collected many types of deregulated isomiRs (Figure 2 and Supplementary Figure S1 available online at https://academic.oup.com/bib). IsomiRs exhibited diverse sequence and expression patterns, and some were involved in the alteration of 5′ ends that led to further variation in seed sequences (Supplementary Figure S1C available online at https://academic.oup.com/bib). These shifted seeds might involve targets different from those of their canonical seeds, including losing some target mRNAs or generating novel targets. These variants might have multiple isomiR types with distinct sequences, but they only contained limited seeds because some of them were 3′ isomiRs.
To further characterize potential variations in isomiR functions, candidate targets for novel seeds and their corresponding canonical seeds were predicted using TargetScan [27]. Multiple isomiR sequences with similar sequence relationships could be represented as larger miRNA families [9]. As expected, some common targets were shared between novel seeds and their canonical seeds (Supplementary Figure S1D available online at https://academic.oup.com/bib), and diverged targets derived mainly from sliding of the binding region of isomiR:mRNA or novel binding sites in the same targets (Supplementary Figure S1C available online at https://academic.oup.com/bib). Except for shared targets, specific targets for both canonical seeds and novel seeds were quite common (Supplementary Figures S1D–E andS2A–D available online at https://academic.oup.com/bib). Unique targets for novel seeds suggest novel biological functions of the shifted seeds. Although most isomiRs were shifted by only 1 nt, altered targets might further rewire circuits in coding–non-coding RNA regulatory networks and fine-tune signaling specificity (Supplementary Figures S1 and S2 available online at https://academic.oup.com/bib).
We collected candidate targets of 5′ isomiRs with novel seeds via three steps: (i) predicted and filtered targets of deregulated isomiRs via DESeq analysis (|log2FC| > 2 and adjusted P-value (Padj.) < 0.05); (ii) both isomiRs and candidate targets were differentially expressed, and they had opposite deregulated expression patterns and (iii) co-expression analysis was used to validate the expression correlation of the identified isomiRs and targets (correlation coefficient <0 and false discovery rate <0.05). On the basis of the collected targets, total isomiR–mRNA interactions were derived for function analysis (Supplementary Figure S2E–G available online at https://academic.oup.com/bib). Targets from three subsets (specific targets of canonical seeds, common targets of canonical and novel seeds and specific targets of novel seeds) showed consistent distribution of associated cancer hallmarks (Supplementary Figure S2E available online at https://academic.oup.com/bib). For example, miR-21-5p isomiRs appeared to markedly rewire networks, suggesting that the effects of a miRNA locus on network structure extend beyond those of the canonical miRNA (Supplementary Figure S2F available online at https://academic.oup.com/bib).
Interestingly, we found that seed sequences overlapped between independent miRNA loci (Supplementary Figure S3A and B available online at https://academic.oup.com/bib) and that these overlapping seeds could regulate the same target mRNAs. For instance, some isomiR types generated from miR-21-5p had the same seeds as isomiRs from the canonical miR-374a-3p, and isomiRs from miR-101-3p had the same seeds as isomiRs from miR-199a/b-3p (Supplementary Figure S3A available online at https://academic.oup.com/bib), although the involved miRNAs belonged to different miRNA gene families and had no sequence similarity. These seed overlaps could extend to more independent miRNA loci, which contribute to cross-talk at the isomiR level. This cross-talk would likely be more widespread among isomiR profiles (herein, we analyzed only deregulated isomiR profiles), although it is unclear whether this cross-talk would be cooperative or competitive in binding target mRNAs.
Rewired regulatory networks and novel pathways mediated by 5′ isomiRs
To validate the potential functional divergence of novel and canonical seeds from a specific miRNA locus, we analyzed 38 miRNA loci in which both canonical and isomiR sequences were differentially expressed at least in one cancer type (Supplementary Figure S3C and D available online at https://academic.oup.com/bib). These 38 loci generated 121 deregulated 3′ isomiRs that had seed sequences consistent with canonical seeds (containing 38 seeds) and 133 deregulated 5′ isomiRs that were involved in novel seeds via seed shifting (containing 59 novel seeds). A hypergeometric test was used to compare filtered targets of novel seeds and those of their canonical seeds, and we finally obtained 34 5′ isomiRs with novel seeds that were derived from 23 miRNA loci (hypergeometric test P < 0.05; Figure 3A). Among these, novel seeds tended to have distinct targets (Figure 3), although these seeds and their isomiRs had high sequence similarities. For example, in miR-21-5p locus, few common targets were shared by canonical seeds and novel seeds (about 7.60% of total targets were shared), and similar distributions were also observed among isomiRs from the miR-143-3p locus (Figure 3C). Both of these miRNA loci have been identified as important regulatory molecules that contribute to pathophysiological process of human diseases [37–40], but conventional analyses have considered only the canonical sequences. Our results indicate that novel seeds may have novel, distinct targets and gain new functions compared with their canonical seeds.
Figure 3 .
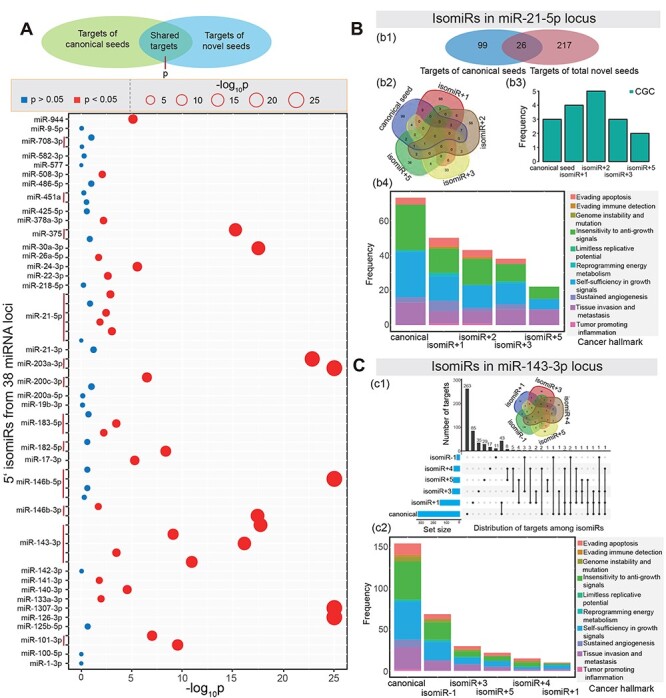
Functional analysis of novel seeds and their canonical seeds. (A) Some miRNA loci show significant differences between target mRNAs of novel seeds and their canonical seeds based on hypergeometric tests. (B) An example of functional analysis of isomiRs in miR-21-5p. b1: Distribution of candidate targets between total novel seeds and canonical seeds; b2: a Venn diagram of targets among detailed seeds; b3: number of CGC genes among different seeds; b4: distribution of cancer hallmarks among different seeds. (C) An example of functional analysis of isomiRs in miR-143-3p locus. c1: Distribution of targets among novel seeds and canonical seeds; c2: distribution of cancer hallmarks among different seeds.
Moreover, because of the critical roles of miRNAs in cancers [41–43], we explored whether targets of canonical and novel seeds were associated with cancer hallmarks, CGC genes, or core essential genes. Our analysis showed that targets of novel seeds were more likely to involve specific hallmarks of cancer than were targets of their canonical seeds (P < 0.05, hypergeometric test; Figure 4A and Supplementary Figure S4A available online at https://academic.oup.com/bib), although novel seeds might lose some functions of their canonical seeds. The cancer hallmarks of sustained angiogenesis and tissue invasion and metastasis were the most enriched, while a distinct difference could be detected between novel and canonical seeds. Gain or loss of targets for novel seeds was common, suggesting that novel functions were induced by these deregulated 5′ isomiRs, especially for multiple novel seeds from a given miRNA locus (such as the novel seeds in miR-183-5p; Figure 4A). On the other hand, except for miR-101-3p, miR-126-3p and miR-26a-5p loci, target mRNAs showed significant differences between novel seeds and canonical seeds in CGC gene enrichment analysis (Figure 4B), suggesting significant functional divergence between novel and canonical seeds despite their similar sequences. In contrast to the enrichment patterns for cancer hallmarks and CGC genes, only three novel seeds from the miR-143 locus were significantly enriched as essential genes (P < 0.05; Figure 4B), indicating that these isomiRs contribute to cell survival by regulating essential genes. These analyses collectively indicate functional divergence between novel seeds and their canonical seeds, while typical analyses based on canonical seeds often ignore these functions induced by novel seeds.
Figure 4 .
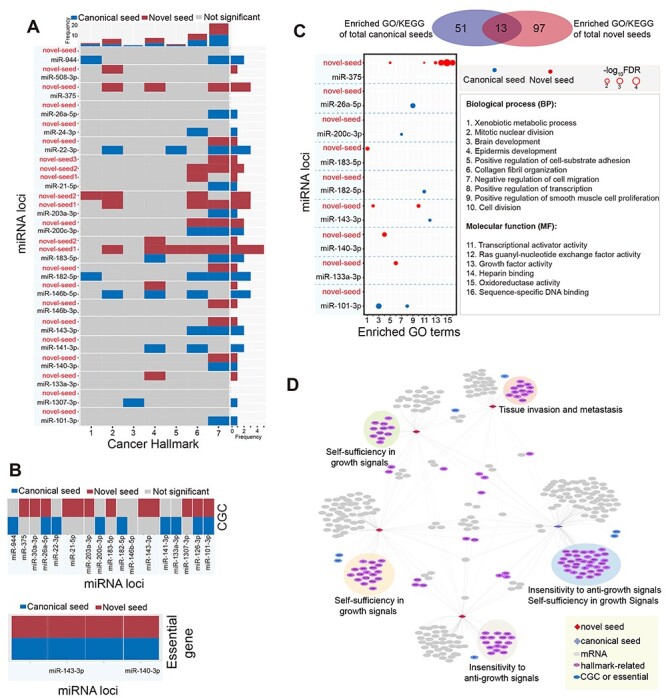
Rewired regulatory networks for novel seeds via seed shifting. (A) The heat map shows enrichment analysis of cancer hallmarks compared between novel seeds and their canonical seeds (if both canonical and novel seeds are not significant, the miRNA locus is removed). The x-axis indicates the seven cancer hallmarks: 1, evading apoptosis; 2, insensitivity to anti-growth signals; 3, limitless replicative potential; 4, reprogramming energy metabolism; 5, self-sufficiency in growth signals; 6, sustained angiogenesis; 7, tissue invasion and metastasis. (B) Enrichment analysis of CGC and essential genes between novel seeds and their canonical seeds (if both canonical and novel seeds are not significant, the miRNA locus is removed). (C) Distribution of enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and GO terms based on total novel seeds and canonical seeds as well as distribution of enriched GO terms compared between canonical seeds and novel seeds based on each miRNA locus (screened miRNA loci are derived from Figure 3A; if both canonical and novel seeds are not significant, the miRNA locus is removed). (D) An example of a rewired regulatory network and enriched cancer hallmarks in miR-21-5p. Five seeds are contained in this network, including one canonical seed (blue diamond) and four novel seeds (red diamond; isomiR+N, indicating that the canonical seed is shifted to 3′ ends by N nt). This network is constructed on the basis of seeds; indeed, every seed may involve multiple 3′ isomiR sequences. The most dominant enriched cancer hallmark is presented for specific targets.
In addition, functional enrichment analysis revealed that novel seeds were more likely to be involved in new functions than their canonical seeds (Figure 4C and Supplementary Figure S4B available online at https://academic.oup.com/bib). Enriched Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes pathways of total canonical and novel seeds showed a larger proportion of specific functions (only 13 were shared terms), indicating functional change between these deregulated isomiRs. For example, in miR-101-3p and miR-26a-5p loci, novel seeds lost functions compared with their canonical seeds, while in miR-133a-3p and miR-140-3p loci, novel seeds gained new functions compared with their canonical seeds (Figure 4C and Supplementary Figure S4B available online at https://academic.oup.com/bib). Of 14 analyzed miRNA loci, none showed the same enriched GO terms, and a significant difference was found between enriched function by targets of novel and canonical seeds. These functional changes strongly support that 5′ isomiRs are involved in new functions, although these isomiRs have been usually ignored in small RNA studies.
Finally, we constructed an isomiR–mRNA interaction network based on deregulated novel seeds and their canonical seeds (Figure 4D and Supplementary Figure S4C available online at https://academic.oup.com/bib), which can be used to simplify coding–non-coding regulatory networks, because many isomiRs are 3′ isomiRs that have the same seeds (Supplementary Figure S1C available online at https://academic.oup.com/bib). IsomiRs in the miR-21-5p locus were used as an example of a regulatory network (Figure 4D). Except for one annotated canonical seed, six novel seeds from 12 isomiR types were collected, and the isomiR–mRNA interaction network showed that each seed had specific targets that were clustered. Although various isomiR types increase the quantity of small RNAs, sequence diversity and function complexity, the critical issue is the potential functional relationships between isomiRs, particularly since they often share some targets. As discussed above, these isomiRs have multiple cross-talks, but whether their relationships are cooperative or competitive during binding of targets remains unknown (Supplementary Figure S4D available online at https://academic.oup.com/bib). Among the diverse and dynamic isomiR-ome, multiple isomiRs with sequence and expression diversity may have more correlations in targeting mRNAs, especially since some isomiRs may have the same seeds. Such relationships may substantially contribute to the complexity of interaction networks.
IsomiR landscape exhibits greater variation in tumor samples than in normal samples
To systematically survey the isomiR landscape in various cancers, we performed a global analysis to compare isomiR expression profiles between tumor and normal samples. We found that the phenomenon of multiple isomiRs was widespread in small RNAs, and more isomiR types were identified in tumor samples than in normal samples (Supplementary Figure S5A available online at https://academic.oup.com/bib). Nearly 30% (28.48%) of miRNA loci had two or more seeds based on RPM values of >50, and seed numbers significantly differed between tumor and normal samples (Kolmogorov–Smirnov test P = 2.50e−08). In normal samples, 24.00% of miRNA loci had two or more seeds, while in tumor samples, 31.00% of miRNA loci had varied seeds. These results reveal that larger variations in isomiRs are more likely to occur in tumor samples. These varied seeds may further rewire coding–non-coding RNA regulatory networks.
Indeed, except for isomiRs with varied seeds, more isomiR types were detected, especially for 3′ isomiRs (Supplementary Figure S5A available online at https://academic.oup.com/bib). Most miRNA loci had multiple isomiRs with sequence and expression diversity (the total mean number of isomiR types was 3.76, and the median number was 3.00), and the phenomenon was more pronounced in tumor samples (the mean number of isomiR types was 3.92 in tumor samples and 3.48 in normal samples). Numbers of isomiR types were associated with enrichment levels, and larger sample sizes of tumor types might also be a potential factor in more isomiR types. Furthermore, some cancer-specific isomiRs might also influence number of isomiR types. Different cancer types showed varied distributions in the number of isomiR types (Supplementary Figure S5A available online at https://academic.oup.com/bib), indicating variations in the isomiR-ome across various tissues.
Analysis of shifted seeds indicated that most were shifted to 3′ ends by 1 nt (Supplementary Figure S5B available online at https://academic.oup.com/bib), followed by shifting to 3′ ends by 2 nt and shifting to 5′ ends by 1 nt (P < 2.2e−16 for distribution of changed 5′ ends in tumor versus normal samples based on trend test). In general, both length distribution and seed shifting analyses showed similar distributions between tumor and normal samples (Supplementary Figure S5C available online at https://academic.oup.com/bib), indicating that generation of isomiRs is relatively stable, which further supports that isomiRs are not random events and are not simple by-products of miRNA maturation. These results provide further evidence of the roles of 5′ isomiRs in tuning the coding–non-coding RNA regulatory networks.
Dominant isomiR repertoire suggests a selective advantage of 5′ isomiRs
Although multiple isomiRs could be detected in each miRNA locus, our previous studies have shown that only several isomiRs were dominantly expressed, and others consistently had lower expression levels, although some also had unexpectedly high enrichment levels [9, 36]. In light of these findings, we further investigated the most dominant isomiR species based on each pre-miRNA and miRNA locus to understand the divergence of isomiRs between tumor and normal samples. The expression levels of most miRNA loci were different in tumor samples from those in normal samples, and different cancer types also showed diverse distributions of isomiR ratios (Figure 5A). The length distribution of dominant isomiRs in tumor samples showed greater variation than that in normal samples (Supplementary Figure S5D available online at https://academic.oup.com/bib), suggesting the greater variation in tumor samples via miRNA maturation process. Although most miRNA loci showed consistent dominant isomiR profiles, varied dominant sequences were detected for specific miRNA loci, and some were 5′ isomiRs with novel seeds (Figure 5A and B and Supplementary Figure S6A available online at https://academic.oup.com/bib). Among these, varied sequences in tumor samples comprised mainly three scenarios: 3′ isomiRs with the same seeds as the canonical miRNAs (only variation in 3′ ends); 5′ isomiRs with shifted seeds compared with the canonical miRNAs (variation in 5′ ends and potentially in 3′ ends) and a most dominant isomiR generated from another arm in pre-miRNA, owing to the arm-switching phenomenon. Therefore, some 5′ isomiRs were unexpectedly dominantly expressed, in some cases as the most dominant isomiR species (Figure 5A and B). For example, in the miR-30e locus, the 5p and 3p arms showed dominant enrichment levels, but different isomiRs were the most dominant species across different individual samples (Figure 5B and Supplementary Figure S6A available online at https://academic.oup.com/bib). Some of the most dominant isomiR species had novel seeds, indicating that isomiRs with novel seeds maybe dominantly expressed and contribute to further biological function. Thus, analyses based on the most dominant isomiRs strongly support a selective advantage of novel seeds that lead to novel biological functions.
Figure 5 .
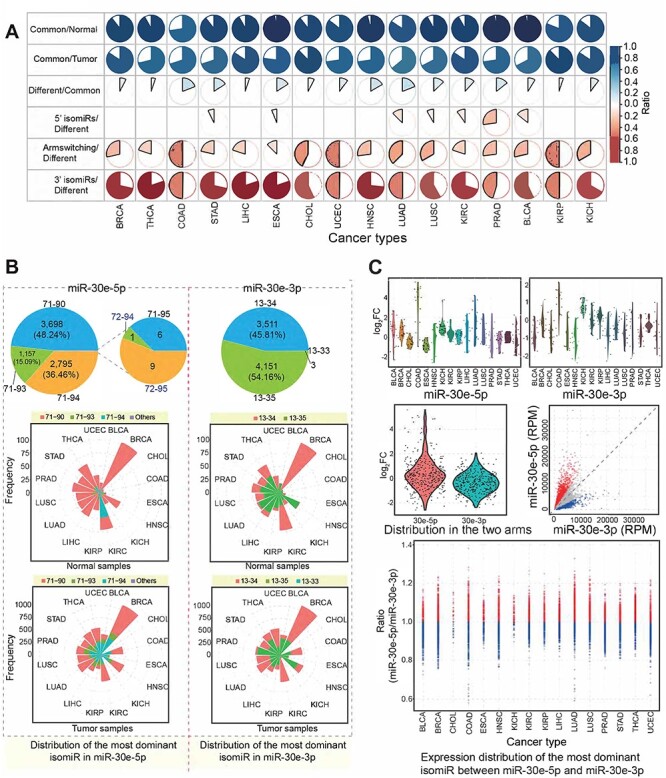
Dominant isomiR sequence analysis and arm-switching phenomenon based on pre-miRNA. (A) Pie charts indicate change in number of isomiRs across different cancer types. ‘Common’ indicates number of dominant isomiRs shared between tumor and normal samples, ‘normal’ indicates number of dominant isomiRs in normal samples, ‘tumor’ indicates number of dominant isomiRs in tumor samples and ‘different’ indicates number of dominant isomiRs differing between tumor and normal samples. (B) Frequency distributions of the most dominant isomiRs from miR-30e-5p and miR-30e-3p (based on frequencies of samples). These involved isomiRs were found to be the most dominant isomiRs in specific samples, and the detailed distribution across cancer types is also presented. 71-90 indicates a location of hg38:chr1:40754371-40754390:+, and 13-33 indicates hg38:chr1:40754413-40754433:+. (C) Distribution of abnormally expressed isomiRs from two arms of mir-30e. Scatter plot indicates expression variation between miR-30e-5p and miR-30e-3p across cancer types based on expression levels of the most dominant isomiRs generated from miR-30e-5p and miR-30e-3p.
Except for variation in 5′ ends and/or 3′ ends, the most dominant isomiR profiles also indicated arm-switching in two products of pre-miRNA, an important phenomenon in the small RNA world [44, 45]. The most dominant isomiR landscapes could give more direct evidence for arm-switching in miRNAs, and our analysis showed that this switching was not rare in some cancer samples compared with the corresponding normal samples (Figure 5C and Supplementary Figure S6B and C available online at https://academic.oup.com/bib). Although multiple isomiR types were detected, only a specific group of them were abundantly expressed and showed high expression percentages. In cholangiocarcinoma (CHOL) normal samples, the most dominant isomiR based on median in miR-30e-3p had a higher expression level compared with miR-30e-5p (ratio of expression of the most dominant isomiRs from 5p to those from 3p, 0.5744; P = 0.050), while in CHOL tumor samples, the divergence between miR-30e-5p and miR-30e-3p was more moderate (ratio of expression of the most dominant isomiRs from 5p to those from 3p, 1.2643; P = 0.002). The expression difference between 5p and 3p based on individual samples showed large variations (Figure 5C), suggesting that expression levels of miRNA gene are dynamic across different individual samples. Compared with expression distribution in normal samples, expression patterns in tumor samples showed greater expression variations (Supplementary Figure S6B available online at https://academic.oup.com/bib).
Varied clinical survival based on diverse isomiRs with length and sequence heterogeneity
To compare prognosis between diverse isomiRs, we performed survival analysis based on dominantly expressed isomiRs. We found divergences in survival in analyses of various isomiRs, although these isomiRs were generated from the same miRNA locus (Figure 6). IsomiRs with novel seeds predicted differences in clinical survival that were not associated with their canonical miRNA sequences or other 3′ isomiRs, although isomiRs with novel seeds are usually ignored in small RNA studies. In addition, dominantly expressed isomiRs (such as in miR-21-5p and miR-30e-5p) were also associated with differences in clinical survival (Figure 6A and B). Although these isomiRs showed high variations in expression, they may have the same 5′ ends and seed sequences (Figure 6). Indeed, many of these isomiRs were differentially expressed across cancer types, and they showed dynamic expression patterns (Figure 6).
Figure 6 .
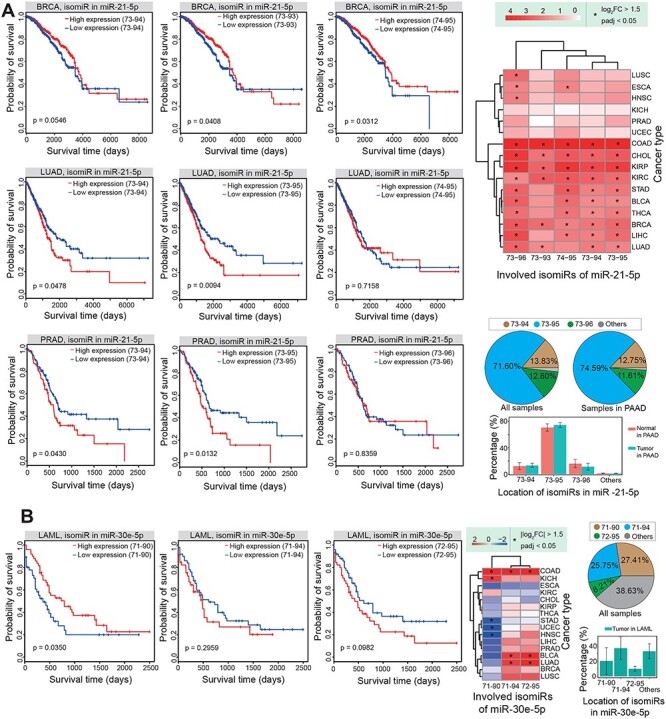
Survival analysis shows divergence among different isomiRs. (A) An example of survival analysis using different isomiRs in miR-21-5p and their expression patterns. Survival results vary between different isomiRs. 73-94 indicates a location of hg38:chr17:59841273-59841294:+. (B) Another example of survival analysis using different isomiRs in miR-30e-5p and their expression patterns. Survival results again vary between different isomiRs. In LAML, no normal samples are involved; ‘All samples’ represent samples involved from 33 cancer types. 71-90 indicates a location of hg38:chr1:40754371-40754390:+.
Experimental validation of biological function of isomiRs with novel seeds
To understand the potential biological function of isomiRs, especially 5′ isomiRs with novel seeds, we further performed relevant experiments in SW480 cell lines. As one of the top hits from our predictive model, miR-101-3p locus was selected to perform further analysis and experimental validation. This miRNA has been widely studied and plays important roles in diverse cancers, for example miR-101-3p can promote cell apoptosis in oral cancer by targeting BICC1 [46]; miR-101-3p can suppress proliferation and migration in hepatocellular carcinoma [47] and it also can lead to reduced cytokine production [48].
Our previous results have revealed dominantly expressed canonical miR-101-3p and its isomiR with novel seed. Paired analysis in diverse cancers showed that they were deregulated in some cancer types, especially in CHOL and COAD (colon adenocarcinoma), and consistent expression patterns were found between canonical miR-101-3p and its isomiR (Figure 7A). In some cancer types, canonical miRNAs were significantly downregulated, while its isomiR did not show abnormal expression, indicating that inconsistent expression patterns might exist among diverse isomiRs. We further performed experimental validation and showed that both mimic sequences of the canonical miRNA and its isomiR could significantly inhibit the expression of the target gene RAB11FIP1 at mRNA expression level (Figure 7B and Supplementary Figure S7 available online at https://academic.oup.com/bib). Moreover, our western blot results showed that overexpression of isomiR with novel seed could significantly inhibit the protein expression of target RAB11FIP1, while the canonical miR-101-3p did not show significant inhibitory effect (Figure 7C and Supplementary Figure S8 available online at https://academic.oup.com/bib), implying the regulatory role of isomiR. These results indicated that isomiR with the shifted seed can regulate the novel potential target mRNA that may not have significant interaction with the canonical miRNA. The potential varied target mRNAs would further rewire and fine-tune coding–non-coding RNA regulatory network.
Figure 7 .
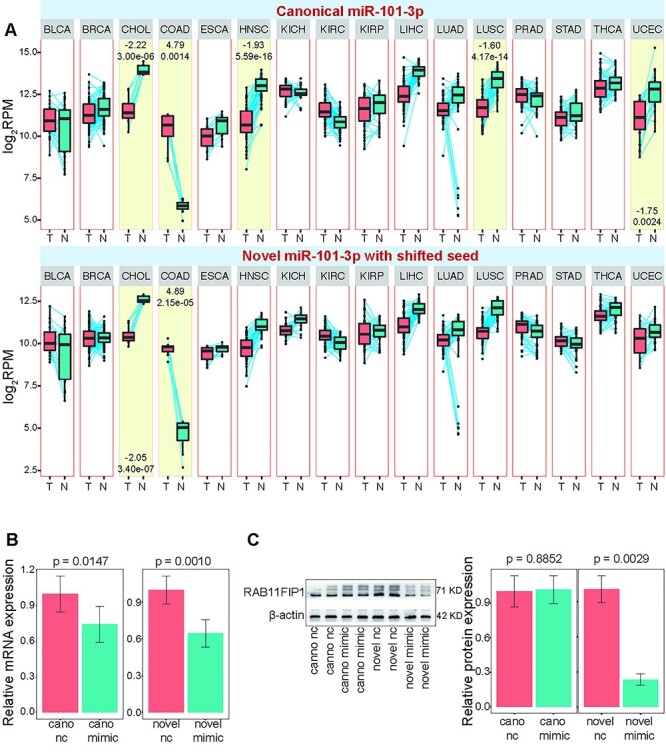
Experimental validation of biological function of isomiRs with novel seeds. (A) miR-101-3p locus is selected to perform further experimental validation. Paired analysis is performed on the canonical miRNA and its isomiR with a novel seed to show their specific expression patterns across diverse cancer types. (B) RAB11FIP1 mRNA levels are assessed after treatment with canonical miRNA or its isomiR mimics with RT-qPCR. nc, negative control. (C) Protein levels of RAB11FIP1 are evaluated after treatment with canonical miRNA or its isomiR mimics with western blotting.
DISCUSSION AND CONCLUSIONS
Here, our pan-cancer analysis provides diverse deregulated isomiR expression profiles across different tissues. Among the abundant isomiR types, 5′ isomiRs with novel seeds are common. Changing 5′ ends could lead to the generation of novel seeds through seed shifting of canonical seeds, while shifted seeds may bind the same target mRNAs, lose a target mRNA or gain a novel target, although they seem to obtain only one additional nucleotide via shifting to 5′ or 3′ ends and simultaneously lose a nucleotide in the opposite direction. Further experimental validation suggests that 5′ isomiRs could gain novel target mRNAs and then contribute to regulatory network rewiring. These novel seeds involve divergent functions through changing of targets and rewiring of the coding–non-coding RNA regulatory networks. Collectively, these findings strongly demonstrate that isomiRs are far more than auxiliary products and that deregulated species, particularly the 5′ isomiRs with novel seeds, are functional variants or isoforms in the small RNA world.
The phenomenon of multiple isomiRs generated from miRNA loci is not a new topic, but most traditional analyses still focus only on annotated and canonical miRNA sequences. However, a canonical miRNA sequence is only one sequence among multiple variants, and some isomiR types may involve varied 5′ ends and novel seeds through some molecular mechanisms, including alternative and imprecise cleavage by Drosha and Dicer, 3′ addition events and RNA editing [10, 11]. As a class of flexible, small regulatory molecules, variation of sequence, particularly variation in 5′ ends and seeds, may lead to a chain reaction affecting targets and biological functions, and a large amount of these variants could substantially increase the complexity of the miRNA-ome [11] and may even further rewire gene regulatory networks. Sequence, expression and function variations due to isomiRs may be pivotal in cancer studies. At the isomiR level, expression patterns show typical tissue specificity, as do canonical miRNAs, and some isomiR types are detected only in specific tissues [16, 23]. We found that divergence of isomiRs from a specific miRNA locus increases their flexible regulatory role in the coding–non-coding RNA regulatory networks, although the relationship (competition or cooperation) between these isomiRs remains unclear. Indeed, the phenomenon of multiple isomiRs is similar to that of homologous miRNAs, which are common in the miRNA-ome of humans [49] and other species such as plants [50, 51]. Many miRNA gene families, such as the let-7 gene family [52, 53], miR-30 gene family [54, 55] and miR-200 gene family [56, 57], have important roles in biological processes. A cooperative regulatory pattern exists among homologous miRNAs, and a similar cooperative regulatory pattern may exist among multiple isomiRs. However, it is unclear whether a competitive relationship also exists among isomiRs, especially 5′ isomiRs with shifted seeds that involve novel-specific targets compared with those of their canonical seeds. Further studies are needed to verify the potential regulatory relationships of isomiRs in the coding–non-coding RNA interaction networks.
Furthermore, on the basis of abnormal isomiR expression profiles, we explored the landscape of the most dominant isomiRs. Interestingly, although many miRNA loci generate consistent sequences, some show sequence divergence between tumor and normal samples, including changed 3′ ends, changed 5′ ends or generation of the most dominant isomiRs from different arms. If the most dominant isomiRs diverge only in 3′ ends, functions of 3′ isomiRs remain consistent, although we did not determine whether length divergence influences their biological activities. However, if the most dominant isomiRs are 5′ isomiRs with novel seeds, the dominant expression indicates their biological function and expression status. These findings strongly suggest that isomiRs are not simply random events in small RNA maturation and that regulatory networks rewired by these 5′ isomiRs produce novel contributions in biological pathways. Furthermore, another important phenomenon we observed, arm-switching at the isomiR level, further supports the critical roles of isomiRs in the coding–non-coding RNA regulatory networks. The expression distribution between these two arms may vary [44], and arm-switching may be detected in specific tissues. More importantly, our survival analysis shows variations in survival based on different isomiRs, even among isomiRs with the same seeds. However, conventional analyses only focus on annotated miRNA sequences, and isomiRs with sequence, length and expression diversities are often ignored, although they may represent potential biomarkers in diagnosis of diseases.
Although we performed a pan-cancer analysis to understand isomiRs, only isomiRs without nucleotide variations are involved in our study. Indeed, some isomiRs may be detected with varied nucleotides and more isomiR types would be found, which will largely contribute to various miRNA-ome and isomiR–mRNA interactions. Further studies should focus on biological function of isomiRs through experimental validation, especially those isomiRs with novel seeds and isomiRs with varied nucleotides to explore the potential roles in the pathogenesis of human diseases.
Taken together, our findings show that multiple isomiRs in a specific locus, ignored in conventional analyses, have biological functions that may contribute to specific biological pathways, especially isomiRs with shifted novel seeds. These variants have close relationships in sequence, expression and function. Their sequence and expression diversity can substantially enrich miRNA-ome and isomiR–mRNA interactome network. The potential of these variants to rewire networks may have a pivotal role in the development of cancers, and isomiRs may be a more suitable biomarker than canonical single miRNAs in cancer studies. As flexible, these length- and sequence-varied isoforms may be a new challenge for studying biological functions of small regulatory molecules, especially for isoform-mediated rewired gene regulatory networks.
Key Points
Many miRNA isoforms (isomiRs) exhibit divergence in their sequence, expression and function.
Novel targets of 5′ isomiRs alter interactions between small RNAs and mRNAs and rewire gene regulatory networks in complex regulatory patterns.
Regulatory networks rewired by 5′ isomiRs can result in novel biological functions.
Landscape of dominant isomiRs suggests varied repertoire of isomiR expression with selective advantage.
Systems-scale analysis at the miRNA isoform level provides insights into the complexity and plasticity of gene regulation in the small RNA world.
Supplementary Material
Conflicts of Interest
G.B.M. is on the SAB/consultant for Amphista, AstraZeneca, Chrysalis Biotechnology, GSK, Ellipses Pharma, ImmunoMet, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, Turbine and Zentalis Pharmaceuticals; G.B.M. has stock/options/financial at Catena Pharmaceuticals, ImmunoMet, Signalchem, and Tarveda; G.B.M. has licensed technology HRD assay to Myriad Genetics, DSP patents with Nanostring; G.B.M. has sponsored research from Nanostring Center of Excellence, and Ionis (provision of tool compounds). No conflicts of interest are disclosed by the other authors.
Acknowledgements
We are grateful to contributions from TCGA Research Network Analysis Working Group. We thank the MD Anderson high-performance computing core facility, Biomedical Research Computing Facility at UT Austin and Texas Advanced Computing Center (TACC) for computing assistance. We also acknowledge the Department of Scientific Publications at MD Anderson for editorial assistance.
Li Guo is a visiting faculty member at the Department of Systems Biology, MD Anderson Cancer Center.
Yongsheng Li is a senior research scientist at the Dell Medical School, University of Texas at Austin.
Kara M. Cirillo is a research assistant at the Department of Epigenetics and Molecular Carcinogenesis at MD Anderson Cancer Center.
Robert A. Marick is a graduate student at the Institute for Cellular & Molecular Biology, University of Texas at Austin.
Zhe Su is a postdoctoral research fellow at the Dell Medical School, University of Texas at Austin.
Xing Yin is a research engineering scientist associate at the Dell Medical School, University of Texas at Austin.
Xu Hua is a postdoctoral research fellow at the Department of Systems Biology, MD Anderson Cancer Center.
Gordon B. Mills is an endowed chair and a director of Precision Oncology and a professor in Cell, Development and Cancer Biology in the Knight Cancer Institute at Oregon Health Sciences University.
Nidhi Sahni is an assistant professor in the Department of Epigenetics and Molecular Carcinogenesis and Department of Bioinformatics and Computational Biology at MD Anderson Cancer Center and a regular faculty member of the QCB Graduate Program at Baylor College of Medicine.
S. Stephen Yi is a faculty member and director of Bioinformatics, Developmental Therapeutics Lab at Dell Medical School, Oden Institute for Computational Engineering & Sciences, Department of Biomedical Engineering and the Institute for Cellular & Molecular Biology, University of Texas at Austin.
Contributor Information
Li Guo, Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Yongsheng Li, Department of Oncology, Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX 78712, USA.
Kara M Cirillo, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA.
Robert A Marick, Department of Oncology, Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX 78712, USA.
Zhe Su, Department of Oncology, Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX 78712, USA.
Xing Yin, Department of Oncology, Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX 78712, USA.
Xu Hua, Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Gordon B Mills, Department of Cell, Developmental and Cancer Biology, School of Medicine, Oregon Health & Science University, Portland, OR 97201, USA; Precision Oncology, Knight Cancer Institute, Portland, OR 97201, USA.
Nidhi Sahni, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA; Program in Quantitative and Computational Biosciences (QCB), Baylor College of Medicine, Houston, TX 77030, USA; Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
S Stephen Yi, Department of Oncology, Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX 78712, USA; Oden Institute for Computational Engineering and Sciences (ICES), The University of Texas at Austin, Austin, TX 78712, USA; Interdisciplinary Life Sciences Graduate Programs (ILSGP), College of Natural Sciences, The University of Texas at Austin, Austin, TX 78712, USA; Department of Biomedical Engineering, Cockrell School of Engineering, The University of Texas at Austin, Austin, TX 78712, USA.
Funding
Susan G. Komen Foundation Grant (CCR19609287 to S.S.Y.); Pinnacle Research Award from American Association for the Study of Liver Diseases (to N.S.); Young Investigator Grant by Breast Cancer Alliance (to N.S.), Liz Tilberis Early Career Award by Ovarian Cancer Research Alliance (Grant# 649968 to N.S.); NCI grants P50CA217685, UO1CA217842, P50CA09851 (to G.B.M.). G.B.M. also received a kind gift from the Miriam and Sheldon Adelson Medical Research Foundation, and Breast Cancer Research Foundation.
References
- 1. Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 2013;14:475–88. [DOI] [PubMed] [Google Scholar]
- 2. Guo H, Ingolia NT, Weissman JS, et al. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010;466:835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010;11:597–610. [DOI] [PubMed] [Google Scholar]
- 4. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6:857–66. [DOI] [PubMed] [Google Scholar]
- 5. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 2006;6:259–69. [DOI] [PubMed] [Google Scholar]
- 6. Heverhagen AE, Legrand N, Wagner V, et al. Overexpression of microRNA miR-7-5p is a potential biomarker in neuroendocrine neoplasms of the small intestine. Neuroendocrinology 2018;106:312–7. [DOI] [PubMed] [Google Scholar]
- 7. Milger K, Gotschke J, Krause L, et al. Identification of a plasma miRNA biomarker signature for allergic asthma: a translational approach. Allergy 2017;72:1962–71. [DOI] [PubMed] [Google Scholar]
- 8. Stoicea N, Du A, Lakis DC, et al. The miRNA journey from theory to practice as a CNS biomarker. Front Genet 2016;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo L, Liang T. MicroRNAs and their variants in an RNA world: implications for complex interactions and diverse roles in an RNA regulatory network. Brief Bioinform 2018;19:245–53. [DOI] [PubMed] [Google Scholar]
- 10. Morin RD, O'Connor MD, Griffith M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 2008;18:610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neilsen CT, Goodall GJ, Bracken CP. IsomiRs--the overlooked repertoire in the dynamic microRNAome. Trends Genet 2012;28:544–9. [DOI] [PubMed] [Google Scholar]
- 12. Tan GC, Chan E, Molnar A, et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res 2014;42:9424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Babapoor S, Fleming E, Wu R, et al. A novel miR-451a isomiR, associated with amelanotypic phenotype, acts as a tumor suppressor in melanoma by retarding cell migration and invasion. PLoS One 2014;9:e107502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Llorens F, Banez-Coronel M, Pantano L, et al. A highly expressed miR-101 isomiR is a functional silencing small RNA. BMC Genomics 2013;14:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mercey O, Popa A, Cavard A, et al. Characterizing isomiR variants within the microRNA-34/449 family. FEBS Lett 2017;591:693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Telonis AG, Magee R, Loher P, et al. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res 2017;45:2973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Kwast R, Woudenberg T, Quax PHA, et al. MicroRNA-411 and its 5'-IsomiR have distinct targets and functions and are differentially regulated in the vasculature under ischemia. Mol Ther 2020;28:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhardwaj A, Singh H, Trinidad CM, et al. The isomiR-140-3p-regulated mevalonic acid pathway as a potential target for prevention of triple negative breast cancer. Breast Cancer Res 2018;20:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balyan S, Joseph SV, Jain R, et al. Investigation into the miRNA/5' isomiRNAs function and drought-mediated miRNA processing in rice. Funct Integr Genomics 2020;20:509–22. [DOI] [PubMed] [Google Scholar]
- 20. Mohammed J, Flynt AS, Panzarino AM, et al. Deep experimental profiling of microRNA diversity, deployment, and evolution across the Drosophila genus. Genome Res 2018;28:52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nejad C, Pepin G, Behlke MA, et al. Modified polyadenylation-based RT-qPCR increases selectivity of amplification of 3'-microRNA isoforms. Front Genet 2018;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nejad C, Pillman KA, Siddle KJ, et al. miR-222 isoforms are differentially regulated by type-I interferon. RNA 2018;24:332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trontti K, Vaananen J, Sipila T, et al. Strong conservation of inbred mouse strain microRNA loci but broad variation in brain microRNAs due to RNA editing and isomiR expression. RNA 2018;24:643–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crooks GE, Hon G, Chandonia JM, et al. WebLogo: a sequence logo generator. Genome Res 2004;14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chou CH, Chang NW, Shrestha S, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 2016;44:D239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 30. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hart T, Chandrashekhar M, Aregger M, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015;163:1515–26. [DOI] [PubMed] [Google Scholar]
- 33. Blomen VA, Majek P, Jae LT, et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015;350:1092–6. [DOI] [PubMed] [Google Scholar]
- 34. Wang T, Birsoy K, Hughes NW, et al. Identification and characterization of essential genes in the human genome. Science 2015;350:1096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo L, Yang Q, Lu J, et al. A comprehensive survey of miRNA repertoire and 3′ addition events in the placentas of patients with pre-eclampsia from high-throughput sequencing. PLoS One 2011;6:e21072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cui B, Liu W, Wang X, et al. Brucella Omp25 upregulates miR-155, miR-21-5p, and miR-23b to inhibit interleukin-12 production via modulation of programmed death-1 signaling in human monocyte/macrophages. Front Immunol 2017;8:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He Z, Yi J, Liu X, et al. MiR-143-3p functions as a tumor suppressor by regulating cell proliferation, invasion and epithelial-mesenchymal transition by targeting QKI-5 in esophageal squamous cell carcinoma. Mol Cancer 2016;15:51. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Jin YP, Hu YP, Wu XS, et al. miR-143-3p targeting of ITGA6 suppresses tumour growth and angiogenesis by downregulating PLGF expression via the PI3K/AKT pathway in gallbladder carcinoma. Cell Death Dis 2018;9:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lakhter AJ, Pratt RE, Moore RE, et al. Beta cell extracellular vesicle miR-21-5p cargo is increased in response to inflammatory cytokines and serves as a biomarker of type 1 diabetes. Diabetologia 2018;61:1124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Magri F, Vanoli F, Corti S. miRNA in spinal muscular atrophy pathogenesis and therapy. J Cell Mol Med 2018;22:755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mori M, Triboulet R, Mohseni M, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014;156:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009;138:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Griffiths-Jones S, Hui JH, Marco A, et al. MicroRNA evolution by arm switching. EMBO Rep 2011;12:172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo L, Zhang H, Zhao Y, et al. Selected isomiR expression profiles via arm switching? Gene 2014;533:149–55. [DOI] [PubMed] [Google Scholar]
- 46. Wang H, Guo Y, Mi N, et al. miR-101-3p and miR-199b-5p promote cell apoptosis in oral cancer by targeting BICC1. Mol Cell Probes 2020;52:101567. [DOI] [PubMed] [Google Scholar]
- 47. Liu Y, Tan J, Ou S, et al. MicroRNA-101-3p suppresses proliferation and migration in hepatocellular carcinoma by targeting the HGF/c-Met pathway. Investig New Drugs 2020;38:60–9. [DOI] [PubMed] [Google Scholar]
- 48. Huang T, Yang J, Zhang J, et al. MicroRNA-101-3p downregulates TLR2 expression, leading to reduction in cytokine production by Treponema pallidum-stimulated macrophages. J Invest Dermatol 2020;140:1566–1575 e1561. [DOI] [PubMed] [Google Scholar]
- 49. Guo L, Zhao Y, Zhang H, et al. Integrated evolutionary analysis of human miRNA gene clusters and families implicates evolutionary relationships. Gene 2014;534:24–32. [DOI] [PubMed] [Google Scholar]
- 50. Devi K, Dey KK, Singh S, et al. Identification and validation of plant miRNA from NGS data-an experimental approach. Brief Funct Genomics 2019;18:13–22. [DOI] [PubMed] [Google Scholar]
- 51. Fard EM, Moradi S, Salekdeh NN, et al. Plant isomiRs: origins, biogenesis, and biological functions. Genomics 2020;112:3382–95. [DOI] [PubMed] [Google Scholar]
- 52. Jiang X, Hawkins JS, Lee J, et al. Let-7 microRNA-dependent control of leukotriene signaling regulates the transition of hematopoietic niche in mice. Nat Commun 2017;8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kuppusamy KT, Jones DC, Sperber H, et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc Natl Acad Sci U S A 2015;112:E2785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan LW, Wang F, Meng F, et al. MiR-30 family potentially targeting PI3K-SIAH2 predicted interaction network represents a novel putative theranostic panel in non-small cell lung cancer. Front Genet 2017;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Heras V, Sangiao-Alvarellos S, Manfredi-Lozano M, et al. Hypothalamic miR-30 regulates puberty onset via repression of the puberty-suppressing factor, Mkrn3. PLoS Biol 2019;17:e3000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen X, Ling Y, Wei Y, et al. Dual regulation of HMGB1 by combined JNK1/2-ATF2 axis with miR-200 family in nonalcoholic steatohepatitis in mice. FASEB J 2018;32:2722–34. [DOI] [PubMed] [Google Scholar]
- 57. O'Brien SJ, Carter JV, Burton JF, et al. The role of the miR-200 family in epithelial-mesenchymal transition in colorectal cancer: a systematic review. Int J Cancer 2018;142:2501–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.