

Abstract
This cross-sectional study examines the feasibility of using real-world data, such as billing, claims, and electronic health records, to emulate US Food and Drug Administration–required confirmatory clinical trials for the 50 new therapeutic agents that received accelerated approval between 2009 and 2018.
Introduction
Under the accelerated approval pathway of the US Food and Drug Administration (FDA), therapeutic agents targeting serious or life-threatening diseases can receive approval on the basis of surrogate markers that are reasonably likely to predict clinical benefit conditional on the conduct of postapproval confirmatory trials.1 As required by the 21st Century Cures Act of 2016, the FDA has developed guidance on the use of observational methods and real-world data (RWD) (eg, billing, claims, and electronic health record [EHR] data) to generate clinical evidence to fulfill postapproval study requirements.2 Prior evaluations have suggested important limitations to feasibly emulating trials using RWD because of difficulties in reliably ascertaining interventions, indications, trial inclusion and exclusion criteria, and primary end points from claims and/or structured EHR data.3
A better understanding of the feasibility of emulating FDA-required postapproval trials conducted to verify clinical benefit is critical because these studies often face recruitment challenges, continue to focus on surrogate markers as end points,1 and are delayed for years after approval.1,4 Therefore, we conducted this cross-sectional study to examine the feasibility of using RWD to emulate FDA-required postapproval confirmatory trials for all new therapeutic agents that received accelerated approval between 2009 and 2018.
Methods
This study followed the Strengthening the Reporting of Observational studies in Epidemiology (STROBE) reporting guideline. This study did not require institutional review board approval because it was based on publicly available information, in accordance with 45 CFR §46. Informed consent was not needed because no patient data were used.
We used the Drugs@FDA database to identify all FDA-required postapproval confirmatory trials for new molecular entity drugs and biologics that were regulated by the FDA Center for Drug Evaluation and Research and granted accelerated approval between 2009 and 2018.5 For all postapproval confirmatory trials designed to verify efficacy, we reviewed study descriptions, ClinicalTrials.gov information, and abstracts of publications to determine the proportion of trials for which the (1) clinical indication, (2) at least 80% of the clinical inclusion and exclusion criteria, (3) the comparator, and (4) the primary end point(s) could be routinely ascertained from RWD using previously described methods (eTable in the Supplement).3 These characteristics were considered unlikely to be routinely ascertained from observational data if researchers would find it difficult to develop a computable phenotype using available RWD sources.3 The data were analyzed in Microsoft Excel.
Results
Between 2009 and 2018, the FDA approved 41 new therapeutic agents via the accelerated approval pathway, requiring a total of 50 postapproval confirmatory trials (Table). Twenty postapproval confirmatory trials (40%) were ongoing at the time of accelerated approval.
Table. Characteristics of New Therapeutic Agents Granted Accelerated Approval and FDA-Required Postapproval Confirmatory Trials, 2009-2018.
| Characteristic | No. (%) |
|---|---|
| Therapeutic agent characteristics | 41 (100) |
| Class | |
| Drug | 30 (73) |
| Biologic | 11 (27) |
| Therapeutic area | |
| Cancer and hematology | 33 (80) |
| Other | 8 (20) |
| Priority review | |
| Yes | 37 (90) |
| No | 4 (10) |
| Fast track | |
| Yes | 24 (59) |
| No | 17 (41) |
| Breakthrough therapy | |
| Yes | 21 (51) |
| No | 12 (29) |
| NAa | 8 (20) |
| Orphan drug designation | |
| Yes | 38 (93) |
| No | 3 (7) |
| FDA-required postapproval confirmatory trial characteristics | 50 (100) |
| Type of postapproval confirmatory trial | |
| New clinical trial | 27 (54) |
| Complete or submit results | 20 (40) |
| Otherb | 3 (6) |
| Trial focus | |
| Efficacy | 32 (64) |
| Efficacy and safety | 18 (36) |
| Randomization | |
| Yes | 43 (86) |
| No | 1 (2) |
| NA (single group assignment) | 6 (12) |
| Allocation concealment | |
| Double or higher | 18 (36) |
| Single | 0 |
| None | 29 (58) |
| Unclearc | 3 (6) |
| No. of study patients, median (IQR) | 424 (295-545) |
Abbreviations: FDA, US Food and Drug Administration; NA, not applicable.
Therapeutic agents approved before the origination of the breakthrough therapy designation on July 9, 2012.
Includes postmarketing requirements for longer-duration trial follow-up, revisions of ongoing trials, and submission of results from new and completed trials.
All 3 trials with unclear allocation concealment were randomized trials.
Among the 50 postapproval confirmatory trials, 12 (24%) had a clinical indication that could be routinely ascertained from claims and/or structured EHR data, whereas 38 (76%) required nonroutinely ascertainable disease severity or treatment-related qualifiers (Figure). Of the 46 trials for which clinical inclusion and exclusion criteria were available on ClinicalTrials.gov, 2 (4%) had at least 80% of their criteria that could be routinely ascertained. Of the 41 trials with a comparator arm, 22 (54%) used active comparators, all of which could be ascertained using RWD. Of the 49 trials for which primary end point information was available, 20 (40%) had at least 1 end point that could be routinely ascertained from RWD. Overall, none of the FDA-required postapproval confirmatory trials had all of the following: (1) a clinical indication, (2) at least 80% of the clinical inclusion and exclusion criteria, (3) a comparator, and (4) at least 1 primary end point that could be routinely ascertained from RWD.
Figure. Study Flowchart.
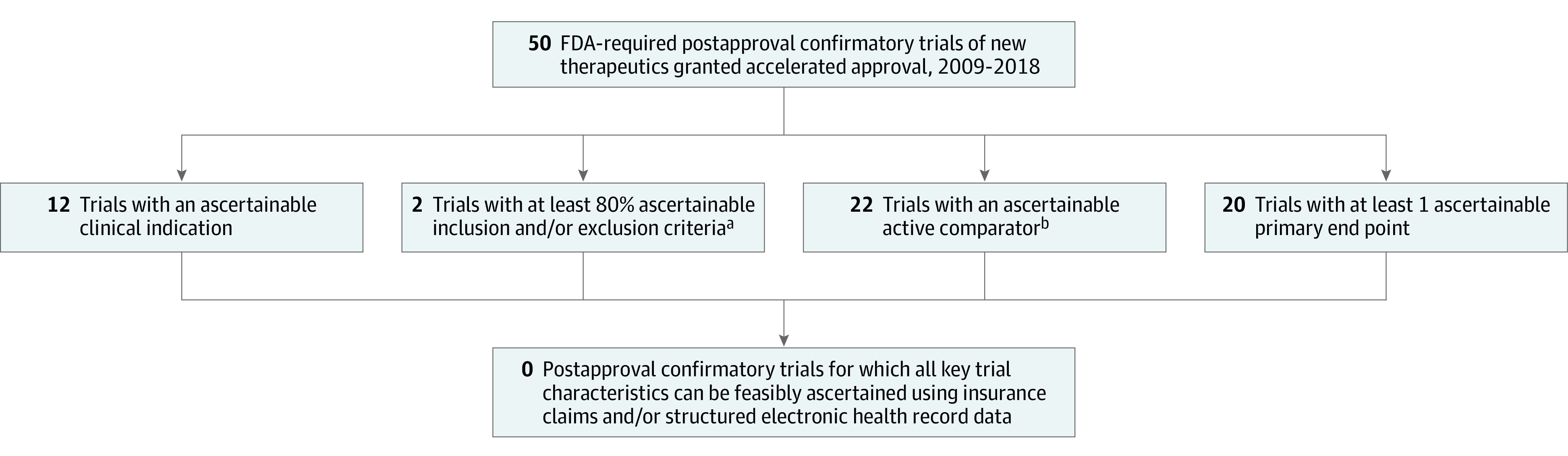
Flowchart demonstrates the feasibility of using real-world data to emulate 50 US Food and Drug Administration (FDA)–required postapproval confirmatory trials of new therapeutic agents granted accelerated approval between 2009 and 2018.
aThere were 4 trials without clearly defined inclusion and exclusion criteria (no ClinicalTrials.gov registration).
bThere were 41 trials with a comparator arm. The “active” designation includes a standard of care comparator, an active comparator, and a standard of care with an active comparator.
Discussion
The findings of this cross-sectional study suggest that none of the 50 FDA-required postapproval confirmatory trials for therapeutic agents granted accelerated approval between 2009 and 2018 could have been feasibly emulated using currently available claims and/or structured EHR data. In particular, the narrowly defined indications and strict inclusion and exclusion criteria of the FDA-required postapproval confirmatory trials precluded emulation using RWD. Although RWD can be used to ascertain clinical outcomes for real-world populations receiving therapeutic agents, our findings suggest that current observational methods and RWD can complement, but are unlikely to replace, postapproval confirmatory trial requirements.
This study had some limitations. Certain determinations were subjective, we did not consider the relative importance of an individual criterion, and we did not consider accelerated approvals for supplemental indications. Our findings suggest that to use RWD for regulatory evaluations, initiatives are necessary to standardize data elements across different facilities, clinicians, and EHRs.6
eTable. Postapproval Confirmatory Trial Emulation Methods
References
- 1.Gyawali B, Ross JS, Kesselheim AS. Fulfilling the mandate of the US Food and Drug Administration’s accelerated approval pathway: the need for reforms. JAMA Intern Med. 2021. doi: 10.1001/jamainternmed.2021.4604 [DOI] [PubMed] [Google Scholar]
- 2.US Food and Drug Administration . Use of real-world evidence to support regulatory decision-making for medical devices: guidance for industry and Food and Drug Administration staff. Published August 20, 2017. Accessed October 1, 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices
- 3.Bartlett VL, Dhruva SS, Shah ND, Ryan P, Ross JS. Feasibility of using real-world data to replicate clinical trial evidence. JAMA Netw Open. 2019;2(10):e1912869. doi: 10.1001/jamanetworkopen.2019.12869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallach JD, Egilman AC, Ross JS, Woloshin S, Schwartz LM. Timeliness of postmarket studies for new pharmaceuticals approved between 2009 and 2012: a cross-sectional analysis. J Gen Intern Med. 2019;34(4):492-495. doi: 10.1007/s11606-018-4779-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skydel JJ, Zhang AD, Dhruva SS, Ross JS, Wallach JD. US Food and Drug Administration utilization of postmarketing requirements and postmarketing commitments, 2009-2018. Clin Trials. 2021;18(4):488-499. doi: 10.1177/17407745211005044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertagnolli MM, Anderson B, Quina A, Piantadosi S. The electronic health record as a clinical trials tool: opportunities and challenges. Clin Trials. 2020;17(3):237-242. doi: 10.1177/1740774520913819 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable. Postapproval Confirmatory Trial Emulation Methods