

Abstract
Chloroplast genomes in land plants include approximately 20 intron-containing genes. Most of the introns are similar to the group II introns found in fungi, algae and some bacteria, but no self-splicing has been reported. To analyze splicing reactions in chloroplasts, we developed a tobacco (Nicotiana tabacum) chloroplast-based in vitro system. We optimized the splicing reaction using atpF precursor messenger RNA (pre-mRNA). Our system requires a high ATP concentration, whereas ATP is not necessary for self-splicing group II introns. Self-splicing group II introns possess two exon-binding sites (EBS1 and 2) complementary to two intron-binding sites (IBS1 and 2) in the 3′ end of 5′ exons, which are involved in 5′ splice-site selection. Using our in vitro system and atpF pre-mRNA, we analyzed short sequences corresponding to the above EBSs and IBSs. Mutation analyses revealed that EBS1–IBS1 pairing is essential, while EBS2–IBS2 pairing is important but not crucial for splicing. The first 3′ exon nucleotide determines the 3′ splice sites of self-splicing introns. However, mutations to this nucleotide in atpF pre-mRNA did not affect splicing. This result suggests that the mechanism underlying chloroplast pre-mRNA splicing differs partly from that mediating the self-splicing of group II introns.
Keywords: Chloroplast •, Gene •, Intron •, In vitro •, Pre-mRNA •, Splicing
Introduction
Chloroplast genomes in land plants contain approximately 120 genes, of which about 20 have introns (Daniell et al. 2016, Nguyen and Kang 2017, Dobrogojski et al. 2020). For example, the tobacco (Nicotiana tabacum) chloroplast genome includes 12 protein-coding genes and six transfer RNA genes with introns (Wakasugi et al. 2001). Genetic approaches used to investigate higher plants have revealed that dozens of nucleus-encoded proteins are involved in splicing (e.g. Stern et al. 2010, de Longevialle et al. 2010, Barkan and Small 2014). This is also the case for the green alga Chlamydomonas reinhardtii (Perron et al. 2004). The chloroplast-encoded maturase also contributes to splicing (Zoschke et al. 2010, Barthet et al. 2020). However, the splicing process in chloroplasts remains relatively uncharacterized.
Eukaryotic nuclear genes contain many introns, and large ribonucleoprotein complexes (i.e. spliceosomes) mediate the splicing of precursor messenger RNAs (pre-mRNAs). The development of in vitro splicing systems from mammalian and yeast cells has enabled researchers to precisely analyze the splicing process (Krainer et al. 1984, Lin et al. 1985). Analyses of nuclear splicing have identified a two-step trans-esterification for removing introns (Fabrizio and Lührmann 2012). First, the 2ʹ OH of a branch site A carries out a nucleophilic attack on the 5ʹ splice site, breaking the phosphodiester bond and simultaneously forming a 2ʹ-5ʹ phosphodiester linkage between the branch site and the 5ʹ end of the intron, forming the free 5ʹ exon and the intron-3ʹ exon intermediates. In the second step, the 3ʹ OH of the 5ʹ exon attacks the 3ʹ splice site, breaking the phosphodiester bond there and forming a new phosphodiester bond between the 5ʹ exon and the 3ʹ exon. The 5′ and 3′ splice sites are defined by short consensus sequences. The RNA and protein components of small nuclear ribonucleoproteins are crucial for splice-site selections (Fabrizio and Lührmann 2012).
The group II–type introns are present in many fungal mitochondria, algae, plant organelles and some bacteria (Michel et al. 1989, Bonen 2008). Group II introns consist of highly structured sequences organized into six distinct domains (DI to DVI, see Fig. 1) (Pyle 2016, Smathers and Robart 2019). Some of the group II introns can self-splice in vitro under non-physiological conditions (unusually high Mg2+ and salt concentrations and high temperatures) (Pyle 2016). Specifically, trans-acting proteins and RNAs are not involved in this self-splicing. A previous analysis of self-splicing proved that these introns are excised in a mechanism similar to that used for spliceosomal introns (Pyle 2016). In the first step, the 5′ splice site is recognized via the base pairings of the two exon-binding sites (EBS1 and EBS2) located in DI and the two intron-binding sites (IBS1 and IBS2) in the 5′ exon. In the second step, the recognition of the 3′ splice site involves the base pairing between a specific intron nucleotide (EBS3 or δ) in DI and the first 3′ exon nucleotide (IBS3 or δ′). Other splicing pathways are similar to those in the spliceosome and produce ligated exons and intron lariats.
Fig. 1.
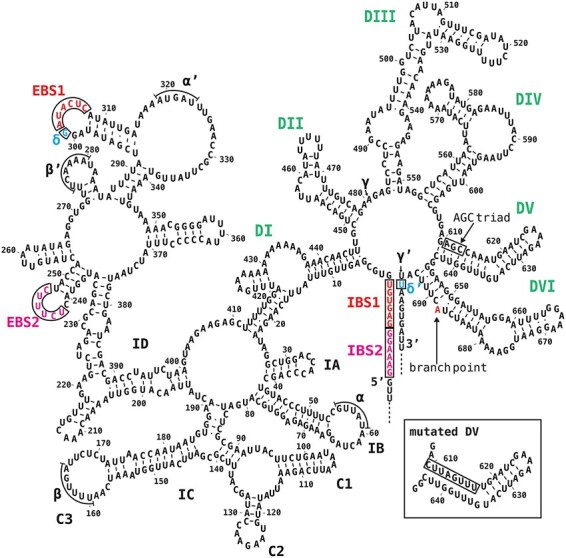
Model of the tobacco atpF intron secondary structure. Domains I–VI are labeled in bold uppercase letters (green). EBSs and IBSs (red, boxed) and δ and δ′ (blue, boxed) are presented. Additional possible interactions (α–α′, β–β′ and γ–γ′) are related to intron compaction. The conserved AGC triad in DV is boxed. The branch point A (red) in DVI is indicated. The inset presents the mutated DV (mDV). The intron and exon sequences are from accession Z00044.2.
There are no reports of self-splicing in higher plant chloroplasts. Therefore, the development of in vitro systems is crucial for investigating specific details of the splicing process. We herein describe the establishment of a chloroplast extract-based system that accurately performs splicing reactions in vitro. Using this system, we analyzed the splicing of the tobacco atpF pre-mRNA. We first demonstrated that the EBS1–IBS1 pairing is essential for splicing, whereas the EBS2–IBS2 pairing is necessary for efficient splicing but is not required. We then proved that the first 3′ exon nucleotide (δ′) is not involved in efficient splicing and likely does not contribute to the 3′splice-site selection. Our results suggest that the mechanism underlying splicing in higher plant chloroplasts differs partly from that mediating the self-splicing of group II introns.
Results
Models of atpF intron secondary structures
The atpF gene encodes the ATP synthase CF0 subunit I and has a 695-bp intron (Bird et al. 1985). The group II introns can be divided into two main subgroups, group IIA and IIB, based on specific structural differences (Michel et al. 1989, Pyle 2016). The atpF intron belongs to group IIA. A model of its secondary structure based on intron alignments (Michel et al. 1989), models of the maize atpF intron (Ostersetzer et al. 2005) and the Lactococcus lactis Ll. LtrB group IIA intron (Dai et al. 2008) was constructed with Mfold (Zuker 2003).
The model consists of six domains (DI to DVI) that radiate from a central wheel (Fig. 1). Moreover, DI has been further divided into subdomains IA to ID. The EBS1 and EBS2 sequences (red) in DID are complementary to IBS1 and IBS2 sequences (red) at the 3′ terminus of the 5′ exon. DIV is too short (49 nt) to encode a protein such as a reverse transcriptase and hence the pre-mRNA lost intron mobility. DV is the most highly conserved region of the entire intron and is absolutely required for splicing. One of the most conserved sequences in DV is the AGC triad (boxed), which is critical for splicing. DVI contains a bulged adenosine (red) that serves as a branch point. The first G of the excised 5′ exon forms a 2′-5′ bond with this A, resulting in a lariat intron. Lariat introns were detected in vivo in atpF pre-mRNAs in higher plant chloroplasts (Kim and Hollingsworth 1993, Vogel and Börner 2002). The interaction between the G (blue, δ, boxed) preceding EBS1 and the first U (blue, δ′, boxed) of the 3′ exon is important for efficient splicing and the recognition of the 3′ splice-site of self-splicing group IIA introns (Dai et al. 2008). The overall secondary structure model is similar to that of the well-characterized L. lactis Ll. LtrB group IIA intron. We then constructed models of atpF introns from spinach (Spinacia oleracea), pea (Pisum sativum), Arabidopsisthaliana and rice (Oryza sativa) because spinach and pea have long been used for analyses of chloroplast proteins, Arabidopsis has been used for the analysis of nuclear genes encoding chloroplast proteins and rice has a long history for breeding and genetic analyses (Supplementary Fig. S1). The eudicot models are similar to each other, whereas the monocot rice and maize (Zea mays) models are approximately 100 bp longer than the eudicot models because of extra sequences in the DI stems. Unlike the tobacco model, the other four models have A in the δ position, resulting in an A–U pair in the δ–δ′ interaction. The A–U pair has been detected in many of the other examined species, from flowering plants to mosses. However, the G–U pair is also present in at least 19 Solanaceae species (e.g. tomato and potato) that have been investigated to date.
In vitro splicing system
Sensitive assays are a prerequisite for developing in vitro RNA splicing systems. Although 32P-labeled pre-RNA substrates have been widely used, we failed to detect splicing in vitro when using these substrates because of the substantial background. Consequently, we designed several different assays, among which the following reverse transcription polymerase chain reaction (RT-PCR)-based method is appropriate for our chloroplast system.
Vector for preparing pre-mRNA substrates.
We modified the pBluescript II SK+ vector to clone chloroplast genes containing introns (Fig. 2). We first surveyed several restriction sites in all 12 tobacco protein-coding genes containing introns. For the short coding regions (petB, petD and rpl16), 200- to 400-bp 5′ untranslated regions (UTRs) were included. For the trans-splicing of 5′ rps12 and 3′ rps12 genes, 800 bp of the flanking intron sequences were added. All of these sequences lack BssHII, ApaI, SacII and NotI sites. Therefore, we designed a cassette containing these restriction enzyme sites as well as SacI and EcoRI sites (Fig. 2A). Because chloroplast extracts contain diverse RNA species, including spliced mRNAs (Supplementary Fig. S3A), we added an artificial sequence (KS/T3, see Fig. 2C) to the gene fragments to distinguish in vitro spliced mRNAs from endogenous mRNAs (Supplementary Fig. S3B). A 173-bp fragment (box, see Fig. 2 legend) in pBluescript II SK+ was removed via a digestion with BssHII (Fig. 2B), after which the above cassette was ligated to produce a modified pBluescript II SK+, which we designated pIVS (Fig. 2C). A variety of chloroplast gene fragments can be inserted between the ApaI and SacII sites of this vector (red triangles).
Fig. 2.
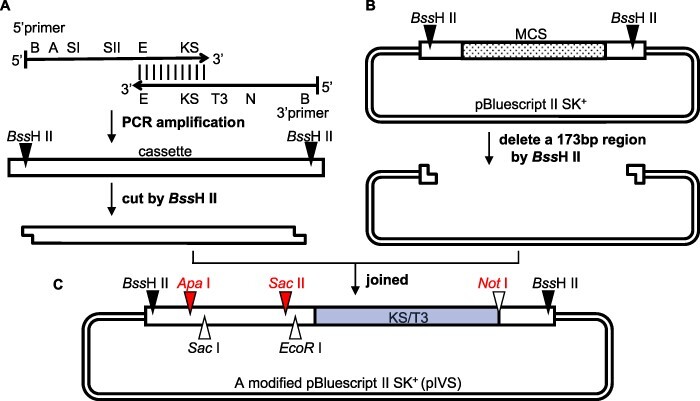
Construction of the pIVS vector for preparing pre-mRNAs. (A) A 95-bp cassette was prepared by a PCR amplification involving the 5′ primer containing the BssHII (B) ApaI (A), SacI (SI), SacII (SII) and EcoRI (E) sites and a KS sequence (KS) as well as the 3′ primer with the (E) KS/T3 sequence (KS/T3), NotI (N) and BssHII (B) sites. The KS/T3 sequence consists of a 17-bp KS primer-binding site and a 20-bp T3 promoter from pBluescript II SK + . The primers are complementary between E and KS, and their full sequences are provided in Supplementary Fig. S2. (B) A 173-bp region [box, including the T7 promoter, the multiple cloning sites (MCS, dots), KS/T3] in pBluescript II SK+ was deleted following digestion with BssHII. (C) Schematic of the pIVS vector. The SacI site (triangle) is an alternative site for inserting specific gene fragments (rpl16, rps16, rpoC1, atpF and petB, which have a 5′ UTR with a single SacI site). The NotI (red) is used to linearize the plasmid for transcription, whereas EcoRI is used to digest the templates.
Pre-mRNA substrates.
Fig. 3 presents the steps involved in preparing substrates, with the tobacco atpF gene used as an example. The gene fragment was prepared via two successive PCR amplifications of the tobacco chloroplast DNA (Fig. 3A). The first PCR involved the atpF 5′ primer (containing the T7 promoter and part of the atpF 5′ exon) and the atpF 3′ primer (consisting of part of the atpF 3′ exon and a SacII site). The second PCR was completed with the ApaI/T7 primer (containing an ApaI site and the T7 promoter) and the pre-existing 3′ primer. The atpF gene fragment was digested with ApaI and SacII and then inserted into the pIVS vector digested with the same enzymes. The circular plasmid DNA (atpF.pIVS) was linearized by NotI and transcribed by T7 RNA polymerase (Fig. 3C). The resulting pre-mRNA was purified after digesting the template DNA with EcoRI and DNase I. We prepared 16 pre-mRNA substrates from 10 genes (two pre-mRNAs each from five genes, one pre-mRNA each from four genes and two pre-mRNAs from clpP-1 as it has two introns, see Supplementary Table S1).
Fig. 3.
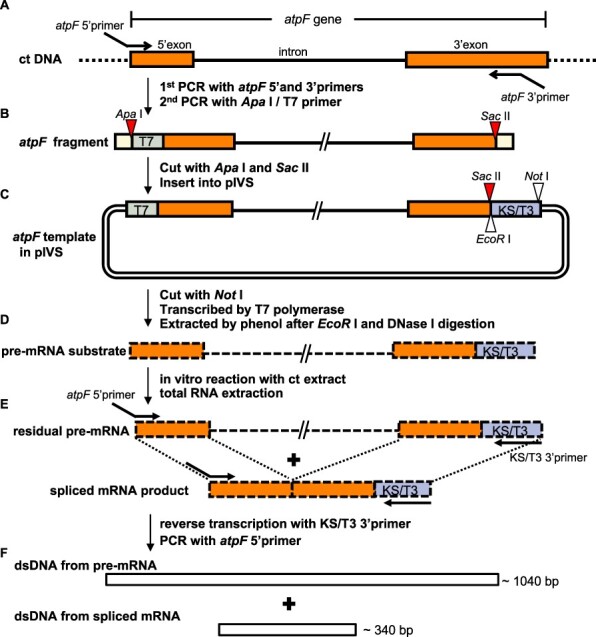
Schematic of the in vitro splicing procedures. (A) The atpF fragment was prepared from chloroplast (ct) DNA by a PCR amplification with the atpF 5′ primer (containing the 21-bp T7 promoter and the first 23 bp of the 5′ exon) and the atpF 3′ primer [containing a mid-3′ exon sequence (positions 100–137) and a SacII site]. The ApaI/T7 5′ primer was added and the PCR was continued to attach an ApaI site to the 5′ end. (B) The atpF fragment contains an ApaI site (red triangle), T7 (green), 145-bp full 5′ exon (orange), 695-bp intron (bold line), the first 137 bp of the 3′ exon (orange) and a SacII site (red triangle). Both ends (yellow) include either the full ApaI or SacII recognition sequence flanked by 4 nt (from the primers). The atpF fragment was digested with ApaI and SacII and then inserted into pIVS digested with the same enzymes. (C) The circular DNA (atpF.pIVS) was linearized by NotI and transcribed by T7 RNA polymerase. The resulting pre-mRNA was purified after digesting the template DNA with EcoRI and DNase I. (D) The pre-mRNA with a 37-bp KS/T3 sequence (blue) was incubated with a ct extract, after which the total RNA was extracted. (E) A RT was completed by adding the KS/T3 3′ primer (30 nt, complementary to KS/T3). A PCR amplification was then completed with the atpF 5′ primer. (F) The PCR products were separated by 2% agarose gel electrophoresis and stained with ethidium bromide. The expected sizes of the pre-mRNA (approximately 1,040 bp) and the spliced mRNA (approximately 340 bp) corresponded to the sequence from the end of the atpF 5′ primer to the end of the atpF 3′ primer. Primers are in Supplementary Fig. S8.
Chloroplast extracts.
Intact chloroplasts were prepared from green tobacco leaves (see the ‘Materials and Methods’ section). On the basis of published procedures for preparing active chloroplast extracts for in vitro RNA editing (Hirose and Sugiura 2001), translation (Hirose and Sugiura 1996), transcription (Orozco et al. 1986) and coupled transcription–translation systems (Bard et al. 1986) (Supplementary Table S2A,B), we evaluated various combinations of chloroplast lysis methods, extraction buffers and dialysis buffers. The isolated chloroplasts were lysed with Triton X-100 and a high KCl concentration (2M), after which the lysate was ultra-centrifuged and dialyzed. We prepared more than 50 chloroplast extracts.
Reaction conditions.
We simultaneously prepared various reaction mixtures based on published in vitro reaction conditions (Supplementary Table S2C) as well as HeLa cell and yeast systems (Lin et al. 1985, Mayeda and Krainer 2012). The reaction mixture components and their concentrations were systematically varied. For example, we tested reaction mixtures containing 1–50 fmol pre-mRNA, with reaction temperatures and times of 28–37°C and 30–120 min, respectively. We then tried to detect spliced mRNAs using different combinations of pre-mRNA substrates, chloroplast extracts and reaction conditions. Finally, of the 16 pre-mRNAs, the spliced products of the expected sizes were clearly detected for the atpF and petD pre-mRNAs and weakly detected for the clpP-1 and ndhA pre-mRNAs (Supplementary Fig. S5). Notably, we used short and long atpF pre-mRNAs (5′ exon only and 5′ exon + UTR, see Supplementary Table S1), but spliced mRNA was produced from only the short atpF pre-mRNA. The reason for this is unclear. We used the short atpF pre-mRNA (hereafter referred to as atpF pre-mRNA) for further analyses. Its full sequence is provided in Supplementary Fig. S4.
Detection of spliced mRNAs.
A schematic of our assay is provided in Fig. 3D–F. After the reactions, the total RNA was extracted and reverse-transcribed with the KS/T3 3′ primer corresponding to KS/T3 (blue) for a PCR amplification involving the atpF 5′ primer. The resulting PCR products (Fig. 3F) were separated by 2% agarose gel electrophoresis (see the gel pattern in Fig. 5A).
Fig. 5.
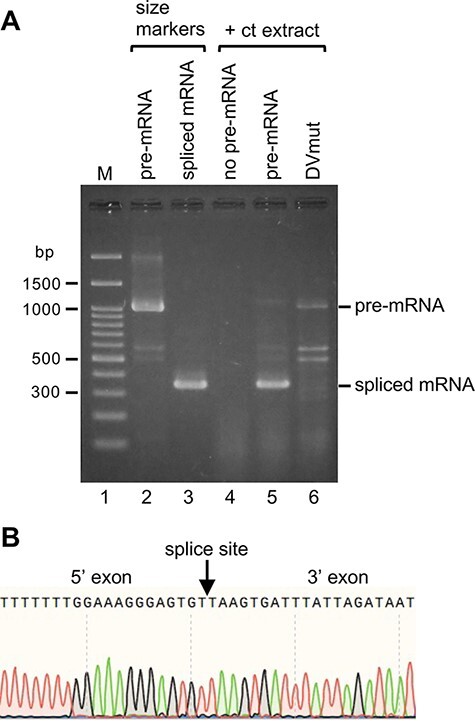
Detection of spliced atpF mRNA products after the in vitro splicing reactions. (A) Gel patterns of spliced mRNA products after the 2-h incubation at 28°C. The bands represent the double-stranded DNA produced from the respective mRNAs by RT-PCR amplification (see Fig. 3F). The pre-mRNA (approximately 1,040 nt) and the synthesized spliced mRNA (approximately 340 nt) were used as size markers (lanes 2 and 3). The reaction without the pre-mRNA (lane 4). The spliced mRNA after the reaction (lane 5). No splicing for the pre-mRNA with a mutated Domain V (DVmut, lane 6). (B) Sequencing of the DNA reverse-transcribed from the spliced mRNA (lane 5). A portion of the sequence is presented. The splice site is indicated with an arrow.
Optimization of splicing reactions.
After detecting splicing activity, we optimized the reaction parameters using the atpF pre-mRNA. The effects of increasing the Mg2+, NH4+ and K+ concentrations are presented in Fig. 4A–C. The optimal conditions were provided by 10 mM Mg acetate, 100 mM NH4 acetate and 70 mM K acetate. Additionally, the optimal ATP concentration was 6 mM, with higher ATP concentrations substantially inhibiting splicing reactions (Fig. 4D, see the ‘Discussion’ section). Splicing was observed over a broad range of pre-mRNA concentrations (Fig. 4E). The reaction proceeded linearly for up to 2 h after a 30 min time lag and then plateaued after 4 h (Fig. 4F, see the ‘Discussion’ section). A typical reaction mixture included the four abovementioned components as well as dithiothreitol, polyethylene glycol, glycerol and an RNase inhibitor in 30 mM HEPES-KOH (pH 7.7). The reaction mixture (10 μl) contained 10 fmol atpF pre-mRNA and 4 μl chloroplast extract (5–15 μg protein). The reaction was completed during a 2-h incubation at 28°C. The optimal reaction conditions varied slightly between chloroplast extracts.
Fig. 4.
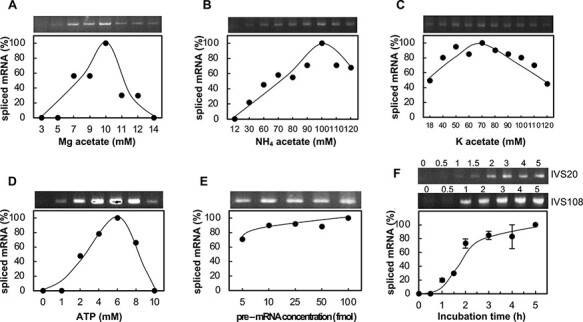
Optimization of reaction conditions. (A) Mg acetate concentration. (B) NH4 acetate concentration. (C) K acetate concentration. (D) ATP concentration. (E) Pre-mRNA concentration. (F) Time courses of two independent splicing reactions. Mean values were calculated based on two experiments.
Analysis of in vitro spliced products
The products of the atpF pre-mRNA splicing reactions were analyzed by gel electrophoresis (Fig. 5). A spliced mRNA of the expected size (approximately 340 nt) was detected (lane 5 of Fig. 5A). The pre-mRNA bands (approximately 1,040 nt) were faint or almost undetectable, likely because they were rapidly degraded during the 2-h incubation. Spliced mRNA bands were not detected for the reaction lacking the chloroplast extract (lane 2). Similarly, a band corresponding to spliced mRNA was also missing for the reaction mixture lacking pre-mRNA (lane 4). Because DV comprises the main active site for splicing, we mutated DV in the atpF pre-mRNA (see Fig. 1 inset) and used the resulting sequence as a control (i.e. non-splicing pre-mRNA). As expected, no spliced product was detected when this mutated pre-mRNA was included in the reaction mixture (lane 6, DVmut). These results clearly indicate that the approximately 340-nt band (lane 5) was not from an endogenous spliced mRNA (see Supplementary Fig. S3A) but was generated from the exogenous pre-mRNA substrate. The band (corresponding to the spliced mRNA) was gel-purified (lane 5) and the DNA was then directly sequenced, which confirmed the accuracy of the splicing (Fig. 5B).
Splice-site selections
As in the secondary structure of the atpF intron (see Fig. 1), the predicted EBS1 and EBS2 in the DI are complementary to IBS1 and IBS2 in the 5′ exon, respectively (Fig. 6A). We first examined whether these sequences are required for the atpF pre-mRNA splicing. We mutated these elements to disrupt possible EBS–IBS interactions (Fig. 6B, red and lowercase letters indicate mutated nucleotides), after which the mutated pre-mRNA substrates were included in the splicing reactions. We first mutated EBS1 to mEBS1 and then IBS1 to mIBS1. As indicated in Fig. 6D, no obvious bands corresponding to the spliced mRNA product (approximately 340 nt) were detected for mEBS1 (lane 4) or mIBS1 (lane 5). Next, we mutated EBS2 to mEBS2 (Fig. 6C). A spliced product was weakly detected (Fig. 6D, lane 6). We then deleted the first 5 nt from EBS2 (i.e. the loop of the EBS2 hairpin) to produce dEBS2, which also resulted in a faint band for the spliced product (lane 7). An analysis with Mfold revealed that dEBS2 did not induce any significant changes to the intron secondary structure. We also mutated IBS2 to mIBS2. A very faint band was again observed (Fig. 6D, lane 8). The two spliced mRNA bands in lanes 6 and 7 were gel-purified, and the corresponding reverse-transcribed DNA fragments were directly sequenced to confirm that both products were correctly spliced mRNAs (Supplementary Fig. S6A). We repeated the splicing reactions with different chloroplast extracts and obtained similar results (Supplementary Fig. S7). Accordingly, we experimentally confirmed that the EBS1–IBS1 pairing is essential for splicing and is likely crucial for selecting the appropriate 5′ splice site. Additionally, the EBS2–IBS2 pairing is important for efficient splicing but may not always be required for splicing.
Fig. 6.
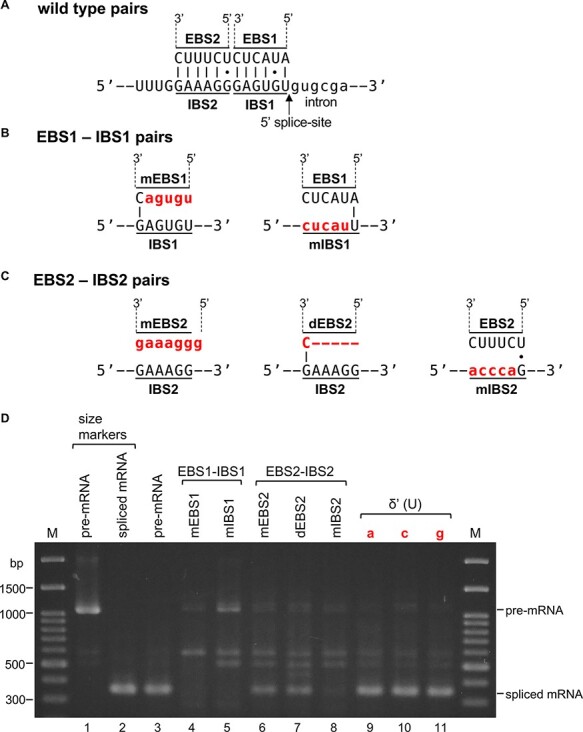
Effects of mutations to the EBS–IBS pairings and to δ′ (U) on the splicing of the pre-mRNA substrate. (A) Pairings between wild-type EBS1 and IBS1 (top) and wild-type EBS2 and IBS2 (bottom). (B) EBS1 mutated to mEBS1 and IBS1 mutated to mIBS1 (red letters indicate mutated nucleotides). (C) EBS2 mutated to mEBS2 (left), deletion of EBS2 to dEBS2 (middle, red bars indicate deleted nucleotides) and IBS2 mutated to mIBS2 (right). (D) Gel patterns of spliced products from atpF pre-mRNA. Lane 1: pre-mRNA, lane 2: synthetic spliced mRNA, lane 3: wild-type pairing, lane 4: mEBS1–IBS2 pairing, lane 5: EBS1–mIBS1 pairing, lane 6: mEBS2–IBS2 pairing, lane 7: dEBS2–IBS2 pairing, lane 8: EBS2–mIBS2 pairing, lane 9: U to a (red), lane 10: U to c (red), and lane 11: U to g (red) at the δ′ nucleotide (U). M: size markers.
Next, we replaced the terminal U (δ′, blue) of the 3′ exon (see Fig. 1) with A, C or G. All three mutations had no apparent effects on splicing reactions (Fig. 6D, lanes 9–11). The sequencing of these spliced products confirmed that all were accurately spliced (Supplementary Fig. S6B). These results imply that the U nucleotide (δ′) minimally influences the splicing efficiency and the selection of the 3′ splice site. In self-splicing group IIA introns, δ′ interacts with the δ (G) preceding EBS1, and the δ–δ′ interaction is critical for the 3′ splice-site selection. Hence, our finding may reflect a novel process in the splicing of chloroplast group II–type introns.
Discussion
In this study, we developed a method for the accurate splicing of atpF pre-mRNA with a chloroplast extract. A long lag period (approximately 45 min) was observed in an earlier study regarding the application of a HeLa cell nuclear extract for in vitro splicing (Krainer et al. 1984). In the current study, our chloroplast extract system also included a long lag period (>30 min) (see Fig. 4F). However, our previously developed in vitro translation system (Hirose and Sugiura 1996) and in vitro RNA editing system (Hirose and Sugiura 2001) did not have a similar time lag. Hence, this lag seems to be unique to splicing and may reflect the time required for the splicing complexes to assemble. The in vitro self-splicing of group II introns does not require ATP (Pyle 2016). In contrast, ATP is essential for our in vitro system (see Fig. 4D). The optimal ATP concentrations in our in vitro translation and RNA editing systems are reportedly 1–2 mM (Hirose and Sugiura 1996, 2001). Mammalian in vitro splicing systems require 0.04–0.5 mM ATP (Krainer et al. 1984, Mayeda and Krainer 2012). Similarly, the yeast system needs at least 0.5 mM ATP (Lin et al. 1985). Comparatively, our in vitro chloroplast system requires an unusually high ATP concentration. However, in our system, splicing was essentially undetectable in the presence of 10 mM ATP. Such a sharp inhibition was not observed in HeLa cell and yeast systems, but the reason for this is unknown.
In group IIA self-splicing introns, EBS1–IBS1 and EBS2–IBS2 interactions are necessary for defining the 5′ splice site, and δ–δ′ interactions are important for selecting the 3′ splice site (Pyle 2016). Our mutation analyses indicate that the EBS1–IBS1 interaction is essential for splicing and selecting the 5′ splice site, whereas EBS2–IBS2 ensures efficient splicing but is not always crucial for splicing (see Fig. 6). We then observed that mutations to the δ′ nucleotide, which disrupt the expected δ–δ′ interaction, do not influence the splicing efficiency or the selection of the 3′ splice site. These results imply that the mechanism underlying chloroplast splicing varies from those mediating the self-splicing of specific group II introns. We speculate that trans-acting factors, such as proteins, RNAs and/or RNA–protein complexes, are involved in the 3′ splice-site selection in chloroplasts.
A major advantage of the in vitro splicing system is that it enables a systematic analysis of the predicted cis-elements. Generally, mutations to exons (i.e. protein-coding regions) lead to amino acid changes in the encoded proteins and often produce premature stop codons in spliced mRNAs. For example, replacing the atpF δ′ (U) with one of three other nucleotides changes the original UUA (leucine) codon to UAA (stop), UCA (serine) and UGA (stop). Thus, mutations to IBSs and δ′ need to be carefully considered. Therefore, our in vitro system may be a viable option for such assays. The results of this study may form the basis of future research aimed at elucidating the biochemical mechanism underlying chloroplast pre-mRNA splicing. Because relatively large amounts of chloroplasts can easily be extracted from tobacco leaves, further elaborations of our system may help researchers search for protein and RNA factors required for the various steps of the splicing process.
Materials and Methods
Isolation of chloroplasts and preparation of extracts
Tobacco (Nicotiana tabacum var. Bright Yellow 4) plants in soil were grown for 6 weeks in a growth room at approximately 26°C with a 16-h white light/8 h dark cycle. Intact chloroplasts were isolated as previously described (Sugiura et al. 1986), with minor modifications. Leaves were collected during the light period. About 40 g expanded leaves (10–13 cm long) were added to 100 ml MCB1 [0.3 M mannitol, 50 mM HEPES-NaOH, pH 8.0, 2 mM ethylenediaminetetraacetic acid (EDTA), 10 mM β-mercaptoethanol, 0.1% (w/v) bovine serum albumin and 0.6% polyvinylpyrrolidone (MW approximately 40,000)] and then disrupted with the TML14 household mixer (Tescom, Tokyo, Japan) or the Polytron PT10-35 homogenizer (Kinematica, Luzern, Switzerland). The homogenate was filtered through four layers of cotton gauze, after which the filtrate was centrifuged at 900 × g for 3 min at 4°C. The resulting green pellet was suspended in 6 ml MCB1 and then added to two discontinuous Percoll gradients (20% 6 ml, 50% 12 ml and 80% 12–18 ml Percoll in MCB1). After a centrifugation at 8,000 × g for 9 min at 4°C, the lower green band was collected and mixed with 3 volumes of MCB2 (0.32 M mannitol, 50 mM HEPES-NaOH, pH 8.0, 2 mM EDTA and 7–10 mM β-mercaptoethanol). Intact chloroplasts were precipitated by centrifugation at 1,000 × g for 2–3 min at 4°C and then washed once with 35 ml MCB2. The pellet was suspended in 2.5 volumes of chilled extraction buffer (30 mM HEPES-KOH, pH 7.7, 10 mM Mg acetate, 2 M KCl, 0.2% Triton X-100 and 10 mM dithiothreitol). After a 15-min incubation on ice with gentle agitation, the viscous lysate was transferred to a 2-ml microtube and centrifuged at 16,000 × g for 30 min at 4°C. The supernatant was dialyzed twice (1.5 h each) against 500 ml dialysis buffer [30 mM HEPES-KOH, pH 7.7, 3 mM Mg acetate, 45 mM K acetate, 30 mM ammonium acetate, 10% (v/v) glycerol and 8 mM dithiothreitol] at 4°C. The dialysis was completed with the Slide-A-Lyzer Dialysis cassette (10 K MWCO) (Pierce Biotechnology, Rockford, IL, USA). The resulting chloroplast extracts were rapidly frozen in liquid nitrogen and stored at −80°C. The chloroplast extracts remained active for at least for 6 months.
Construction of a modified vector
The pBluescript II SK+ vector (Stratagene, La Jolla, CA, USA) was modified for cloning chloroplast genes (see Fig. 2). The cassette was first prepared by PCR with the 5′ primer and its partially complementary 3′ primer (sequences in Supplementary Fig. S2) and the Pyrobest DNA polymerase kit (including the buffer and a deoxynucleotide triphosphates (dNTP) mixture, TaKaRa Bio, Kusatsu, Shiga, Japan). The amplified sequence was digested with BssHII (Fig. 2A). The pBluescript II SK+ vector was also digested with BssHII to replace the 173-bp fragment from positions 620 to 792 (see Fig. 2B), with the abovementioned cassette to produce the pIVS vector (Fig. 2C). The resulting construct was amplified in Escherichia coli XL-1 Blue and isolated by Wizard Plus SV Minipreps DNA Purification System (Promega, Madison, WI, USA). The clone was sequenced to confirm it was correctly generated. (Similarly, atpF.pIVS and its mutant constructs in the next paragraphs were also amplified.)
Preparation of atpF pre-mRNAs
The atpF gene fragment was amplified from tobacco chloroplast DNA (Sugiura et al. 1986) by PCR as described in the ‘Results’ section using the above DNA polymerase kit (Fig. 3A, B). The PCR fragment was ligated to the pIVS vector as mentioned in the ‘Results’ section (Fig. 3C). The resulting clone (atpF.pIVS) was sequenced to confirm it was correctly generated. The plasmid was linearized with NotI. The pre-mRNA was synthesized from the linearized DNA with the AmpliScribe T7-Flash Transcription Kit (Epicentre Biotechnologies, Madison, WI, USA) (Fig. 3C). The resulting reaction mixture was treated with EcoRI and RNase-free DNase I (TURBO DNA-free™, Ambion, Austin, TX, USA) at 37°C for 30 min. The total RNA was extracted with a phenol/chloroform solution and then precipitated with sodium acetate/ethanol (Fig. 3D). The RNA pellets were dissolved in RNase-free water and stored at −80°C.
Mutagenesis of atpF.pIVS
The plasmid DNA was denatured and annealed with a pair of primers (i.e. a primer with the desired mutation and the complementary primer) (see Supplementary Fig. S8). After completing a PCR, the wild-type plasmid was digested with DpnI, whereas the mutated plasmid was inserted into competent cells. The atpF.pIVSs with mutagenized EBS1, EBS2 and DV were prepared with the Pyrobest DNA Polymerase kit. The atpF.pIVSs with mutagenized IBS1, IBS2 and U at the δ′ nucleotide were prepared with Phusion High-Fidelity DNA polymerase (Thermo Scientific, Waltham, MA, USA). The buffer and dNTP mixture were prepared according to the manufacturer’s protocol. Part of EBS2 was deleted with a forward primer (atpF.EBS2 del.F) and a reverse primer (atpF.EBS2 del.R) flanking the EBS2 sequence and the Pyrobest DNA Polymerase kit. The mutant constructs were checked by DNA sequencing.
In vitro RNA splicing reaction
The 10-µl reaction mixture contained 30 mM HEPES-KOH, pH 7.7, 10 mM Mg acetate, 70 mM K acetate, 100 mM NH4 acetate, 6 mM ATP, 3.2 mM dithiothreitol, 3.25% polyethylene glycol 8000, 4% glycerol, 12 U RNase inhibitor HPRI (TaKaRa Bio), 10 fmol atpF pre-mRNA and 4 µl chloroplast extract (5–15 µg protein). After a 2-h incubation at 28°C, 90 µl water was added to the mixture to stop the reaction.
Detection of pre-mRNAs and spliced mRNAs
To extract the total RNA, 100 µl phenol/chloroform was added to the reaction mixtures, which were then vigorously vortexed (Fig. 3D). The suspension was centrifuged at 16,000 × g for 5 min at room temperature. The aqueous phase was transferred to a new tube, to which 10 µl 3 M Na acetate and 250 µl 99.5% ethanol were added. The mixture was incubated at −20°C for 5 min. The RNA was pelleted by centrifugation at 16,000 × g for 15 min at 4°C and then washed once with 70% ethanol. The RNA pellet was dried briefly and dissolved in 1.25 µl water and 5 µl 2× Bca 1st Buffer from the BcaBEST™ RNA PCR Kit (version 1.1) (TaKaRa Bio). The resulting RNA solution was used as the substrate for amplifying pre-mRNAs and spliced mRNAs with the BcaBEST™ RNA PCR kit (version 1.1) (Fig. 3E). The KS/T3 3′ primer was used for RT. The atpF 5′ and KS/T3 3′ primers were used for the PCR amplification. The 10 µl RT reaction mixture comprised 1–10 fmol RNA, 25 pmol KS/T3 3′ primer and 8.25 µl kit RT components. The RT conditions were as follows: 65°C for 1 min, 30°C for 5 min, gradual increase to 65°C for 15 min, 65°C for 15 min, 98°C for 5 min and 5°C for 5 min. After the RT reaction, 0.2 µM atpF 5′ primer and 40 µl PCR solution (1× Bca 2nd Buffer, 2.5 mM MgSO4, 1.25 U/50 µl Bca-Optimized Taq) were added to the RT mixture. The PCR conditions were as follows: 30 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 50 s. ReverTra Ace (TOYOBO) and Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific) were also used for RT-PCR. Twenty-four cycles yielded sufficient products (Figs. 5, 6, Supplementary Figs. S6, S7). After the PCR amplification, 10 µl PCR products were separated on a 2% agarose gel and stained with ethidium bromide (Fig. 3F). To exclude potential artifacts, we followed the kit-attached instructions.
Details regarding the primers are provided in Supplementary Fig. S8.
Supplementary Material
Acknowledgements
This work was performed in part in Sugiyama Jogakuen University, Nagoya. We thank Edanz Group for editing a draft of this manuscript.
Contributor Information
Keiko Inaba-Hasegawa, Center for Gene Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan; Laboratory of Phage Biologics, Graduate School of Medicine, Gifu University, Yanagido 1-1, Gifu 501-1194, Japan.
Ayumi Ohmura, Center for Gene Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan.
Masayo Nomura, Center for Gene Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan.
Masahiro Sugiura, Center for Gene Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan.
Supplementary Data
Supplementary data are available at PCP online.
Data Availability
No new datasets were generated in this study.
Funding
Japan Society for the Promotion of Science (JSPS) KAKENHI [17K0441 to M.S., 04J06115 to K.I-H.].
Disclosures
The authors have no conflicts of interest to declare.
References
- Bard J.D.J., Bourque D.P. and Zaitlin D. (1986) Coupled transcription-translation in chloroplast lysates. Meth. Enzymol. 118: 270–282. [DOI] [PubMed] [Google Scholar]
- Barkan A. and Small I. (2014) Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 65: 415–442.doi: 10.1146/annurev-arplant-050213-040159. [DOI] [PubMed] [Google Scholar]
- Barthet M.M., Pierpont C.L. and Tavernier E.-K. (2020) Unraveling the role of the enigmatic MatK maturase in chloroplast group IIA intron excision. Plant Direct. 4: 1–17.doi: 10.1002/pld3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird C.R., Koller B., Auffret A.D., Huttly A.K., Howe C.J., Dyer T.A., et al. (1985) The wheat chloroplast gene for CF0 subunit I of ATP synthase contains a large intron. EMBO J. 4: 1381–1388.doi: 10.1002/j.1460-2075.1985.tb03790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen L. (2008) Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion 8: 26–34.doi: 10.1016/j.mito.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Dai L., Chai D., Gu S.-Q., Gabel J., Noskov S.Y., Blocker F.J.H., et al. (2008) A three-dimensional model of a group II intron RNA and its interaction with the intron-encoded reverse transcriptase. Mol. Cell 30: 472–485.doi: 10.1016/j.molcel.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell H., Lin C.-S., Yu M. and Chang W.-J. (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17: 134–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Longevialle A.F., Small I.D. and Lurin C. (2010) Nuclearly encoded splicing factors implicated in RNA splicing in higher plant organelles. Mol. Plant 3: 691–705.doi: 10.1093/mp/ssaq025. [DOI] [PubMed] [Google Scholar]
- Dobrogojski J., Adamiec M. and Lucinski R. (2020) The chloroplast genome: a review. Acta Physiol. Plant 42: 98.doi: 10.1007/s11738-020-03089-x. [DOI] [Google Scholar]
- Fabrizio P., Lührmann R. (2012) The spliceosome in constitutive splicing. InAlternative pre-mRNA Splicing: Theory and Protocols. Edited by Stamm, S., Smith, C. and Lührmann, R. pp. 49–64. Wiley-VCH, Weinheim. [Google Scholar]
- Hirose T. and Sugiura M. (1996) Cis-acting elements and trans-acting factors for accurate translation of chloroplast psbA mRNAs: development of an in vitro translation system from tobacco chloroplasts. EMBO J. 15: 1687–1695.doi: 10.1002/j.1460-2075.1996.tb00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T. and Sugiura M. (2001) Involvements of a site-specific trans-acting factor and a common RNA-binding protein in the editing of chloroplast mRNAs: development of a chloroplast in vitro RNA editing system. EMBO J. 20: 1144–1152.doi: 10.1093/emboj/20.5.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-K. and Hollingsworth M.J. (1993) Splicing of group II introns in spinach chloroplasts (in vivo): analysis of lariat formation. Curr. Gent. 23: 175–180.doi: 10.1007/BF00352018. [DOI] [PubMed] [Google Scholar]
- Krainer A.R., Maniastis T., Ruskin B. and Green M.R. (1984) Normal and mutant human β-Globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell 36: 993–1005.doi: 10.1016/0092-8674(84)90049-7. [DOI] [PubMed] [Google Scholar]
- Lin R.-J., Newman A.J., Cheng S.-C. and Abelson J. (1985) Yeast mRNA splicing in vitro. J. Biol. Chem. 260: 14780–14792.doi: 10.1016/S0021-9258(17)38641-6. [DOI] [PubMed] [Google Scholar]
- Mayeda A., Krainer A.R. (2012) In vitro splicing assays. InAlternative pre-mRNA Splicing: Theory and Protocols. Edited by Stamm, S., Smith, C. and Lührmann, R. pp. 321–329. Wiley-VCH, Weinheim. [Google Scholar]
- Michel F., Umesono K. and Ozeki H. (1989) Comparative and functional anatomy of group II catalytic introns – a review. Gene 82: 5–30.doi: 10.1016/0378-1119(89)90026-7. [DOI] [PubMed] [Google Scholar]
- Nguyen D.S. and Kang H. (2017) Comprehensive analysis of chloroplast intron-containing genes and conserved splice sites in dicot and monocot plants. Sci. Technol. Dev. J. Nat. Sci. 1: 60–68.doi: 10.32508/stdjns.v1iT1.435. [DOI] [Google Scholar]
- Orozco E.M. Jr., Mullet J.E., Hanley-Bowdoin L. and Chua N.-H. (1986) In vitro transcription of chloroplast protein genes. Meth. Enzymol. 118: 232–253. [DOI] [PubMed] [Google Scholar]
- Ostersetzer O., Cooke A.M., Watkins K.P. and Barkan A. (2005) CRS1, a chloroplast group II intron splicing factor, promotes intron folding through specific interactions with two intron domains. Plant Cell 17: 241–255.doi: 10.1105/tpc.104.027516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron K., Goldschmidt-Clermont M. and Rochix J.-D. (2004) A multiprotein complex involved in chloroplast group II intron splicing. RNA 10: 704–711.doi: 10.1261/rna.5237804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A.M. (2016) Group II intron self-splicing. Annu. Rev. Biophys. 45: 183–205.doi: 10.1146/annurev-biophys-062215-011149. [DOI] [PubMed] [Google Scholar]
- Smathers C.M. and Robart A.R. (2019) The mechanism of splicing as told by group II introns: ancestors of the spliceosome. BBA-Gene Regul. Mech. 1862: 19439. [DOI] [PubMed] [Google Scholar]
- Stern D.B., Goldschmidt-Clermont M. and Hanson M.R. (2010) Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 61: 125–155.doi: 10.1146/annurev-arplant-042809-112242. [DOI] [PubMed] [Google Scholar]
- Sugiura M., Shinozaki K., Zaita N., Kusuda M. and Kumano M. (1986) Clone bank of the tobacco (Nicotiana tabacum) chloroplast genome as a set of overlapping restriction endonuclease fragments: mapping of eleven ribosomal protein genes. Plant Sci. 44: 211–216.doi: 10.1016/0168-9452(86)90093-2. [DOI] [Google Scholar]
- Vogel J. and Börner T. (2002) Lariat formation and a hydrolytic pathway in plant chloroplast group II intron splicing. EMBO J. 21: 3794–3803.doi: 10.1093/emboj/cdf359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakasugi T., Tsudzuki T. and Sugiura M. (2001) The genomics of land plant chloroplasts: gene content and alteration of genomic information by RNA editing. Photosynth. Res. 70: 107–118.doi: 10.1023/A:1013892009589. [DOI] [PubMed] [Google Scholar]
- Zoschke R., Nakamura M., Liere K., Sugiura M., Börner T. and Schmitz-Linneweber C. (2010) An organellar maturase associates with multiple group II introns. Proc. Natl. Acad. Sci. USA 107: 3245–3250.doi: 10.1073/pnas.0909400107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31: 3406–3415.doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new datasets were generated in this study.