

Abstract
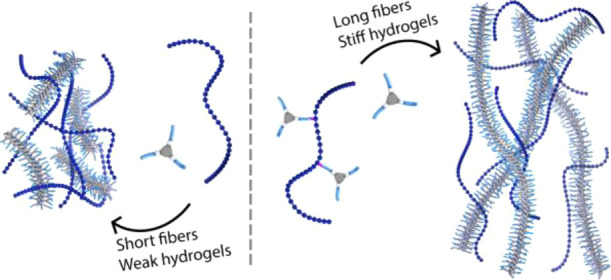
The use of supramolecular polymers to construct functional biomaterials is gaining more attention due to the tunable dynamic behavior and fibrous structures of supramolecular polymers, which resemble those found in natural systems, such as the extracellular matrix. Nevertheless, to obtain a biomaterial capable of mimicking native systems, complex biomolecules should be incorporated, as they allow one to achieve essential biological processes. In this study, supramolecular polymers based on water-soluble benzene-1,3,5-tricarboxamides (BTAs) were assembled in the presence of hyaluronic acid (HA) both in solution and hydrogel states. The coassembly of BTAs bearing tetra(ethylene glycol) at the periphery (BTA-OEG4) and HA at different ratios showed strong interactions between the two components that led to the formation of short fibers and heterogeneous hydrogels. BTAs were further covalently linked to HA (HA-BTA), resulting in a polymer that was unable to assemble into fibers or form hydrogels due to the high hydrophilicity of HA. However, coassembly of HA-BTA with BTA-OEG4 resulted in the formation of long fibers, similar to those formed by BTA-OEG4 alone, and hydrogels were produced with tunable stiffness ranging from 250 to 700 Pa, which is 10-fold higher than that of hydrogels assembled with only BTA-OEG4. Further coassembly of BTA-OEG4 fibers with other polysaccharides showed that except for dextran, all polysaccharides studied interacted with BTA-OEG4 fibers. The possibility of incorporating polysaccharides into BTA-based materials paves the way for the creation of dynamic complex biomaterials.
Introduction
Nature uses supramolecular interactions to construct complex hierarchical systems from smaller building blocks such as proteins, polysaccharides, nucleic acids, and lipids.1,2 Synthetic supramolecular materials have emerged as modular biomimetic platforms owing to their tunable dynamic behavior that resembles that of native systems.3 A unique example is provided by one-dimensional (1D) supramolecular polymers, which additionally mimic the fibrous structure of natural organizations such as the extracellular matrix (ECM).4 A range of scaffolds were found to assemble into 1D supramolecular polymers in water through hydrophobic effects, hydrogen bonding, hydrophilic and Coulomb interactions, and π–π stacking.5,6 Nevertheless, the simplicity of their biological active groups compared to that of natural systems remains an important challenge to address. Increasing the complexity in supramolecular polymers requires extensive fundamental studies on the assembly of multicomponent systems.7
Biofunctionalization of supramolecular polymers is usually achieved by the incorporation of simple biomolecules. Peptide amphiphiles assemble into 1D supramolecular polymers,8−10 and their intrinsic biofunctionality has been exploited for angiogenesis promotion,11 bone regeneration,12 and atherosclerosis treatment,13 or they have been exploited as responsive materials that mimic living organisms.14,15 In addition, ureidopyrimidinone-based supramolecular polymers have been functionalized with peptides for growth factor stabilization or, together with bis-urea, to provide cellular adhesion to the biomaterials.16,17 DNA has also been incorporated into supramolecular polymers as short nucleotides to improve structural control and modularity.18 Other supramolecular scaffolds have been decorated with small carbohydrates.19 Discotic supramolecular polymers bearing mannose moieties have, for example, been used for bacterial sensing.20 Nevertheless, these small biomolecules are not always sufficient to fulfill essential biological duties. A smaller portion of biofunctional supramolecular polymers contain more complex biomolecules, such as proteins that allow for a better biomimicry.21,22
The assembly of benzene-1,3,5-tricarboxamides (BTAs) into highly dynamic 1D supramolecular fibers in water through threefold hydrogen bonding of amides when surrounded by hydrophobic pockets has been thoroughly investigated in the past years.23−27 At higher concentrations of BTAs, the entanglement of fibers allowed one to obtain hydrogels.28 Recently, BTA-based supramolecular polymers have successfully been functionalized with charges for siRNA delivery,29 peptides,30 and small carbohydrates.31−33 A higher complexity was achieved by the introduction of DNA that was used for protein recruitment showing the potential of using BTA supramolecular polymers as modular biomaterials.34
Among the relevant complex native biomolecules, polysaccharides are widely found at the cellular surface and within the ECM, where they have both structural and functional roles. An important subclass of linear polysaccharides are glycosaminoglycans (GAGs), as they are involved in key biological processes ranging from cell adhesion, growth, and proliferation to regulation of signaling pathways.35 In particular, hyaluronic acid (HA) is a nonsulfated GAG consisting of alternating β-1,4-d-glucuronic acid and β-1,3-N-acetyl-d-glucosamine monosaccharides. HA is found throughout the body, as it is present in the ECM of all tissues.36 HA is known to bind CD44, a glycoprotein expressed on most cells that takes part in inflammation processes and is also responsible for ECM integrity and the HA-mediated motility receptors (RHAMM) that regulate cellular migration and proliferation.37,38 Due to its abundance in natural environments and key roles in biological processes, HA has been extensively incorporated into artificial biomaterials.39−42 Nevertheless, most of these HA-containing biomaterials are based on covalent chemistries and fail in reproducing the dynamics of natural systems.40,43 Some examples of dynamic HA-based supramolecular materials have been reported,44−46 and most of them assembled through host–guest interactions that have been applied in vivo;47−50 however, these materials lack the fibrous structure found in the ECM.
This study focusses on the incorporation of HA into dynamic, fiber-forming BTA-based materials with special emphasis on the mechanical properties of the materials obtained. For this purpose, the coassembly of water-soluble BTAs containing tetra(ethylene glycol) at the periphery (BTA-OEG4) and HA (10 kDa) was studied to assess the interference in the formation of 1D supramolecular fibers and hydrogels by the addition of this polysaccharide. Next, HA was functionalized with pendant BTAs (HA-BTA), and the coassembly with BTA-OEG4 resulted in the formation of 1D fibers, similar to those formed by BTA-OEG4 alone. Finally, the physical incorporation of four other polysaccharides (acetylated HA (10 kDa), HA (700 kDa), alginic acid (low viscosity), and dextran (10 kDa)) into BTA-OEG4 stacks is presented.
Experimental Section
Materials
All chemicals and reagents were purchased from commercial sources and used without further purification unless stated otherwise. Sodium hyaluronate (10 and 700 kDa) was obtained from Lifecore Biomedical, and dextran (10 kDa) and alginic acid (low viscosity) were obtained from Sigma. Solvents were dried using a MBRAUN solvent purification system (MB-SPS). Water was purified using an EMD Millipore Milli-Q Integral water purification system. Automated column chromatography was conducted with a Grace Reveleris X2 flash chromatography system using Reveleris silica flash cartridges. 1-Azido-3,6,9,12-tetraoxatetracosan-24-amine and BTA-OEG4 were synthesized according to previously published literature procedures.23,51
Synthesis and Assembly
Synthesis of HA-BTA
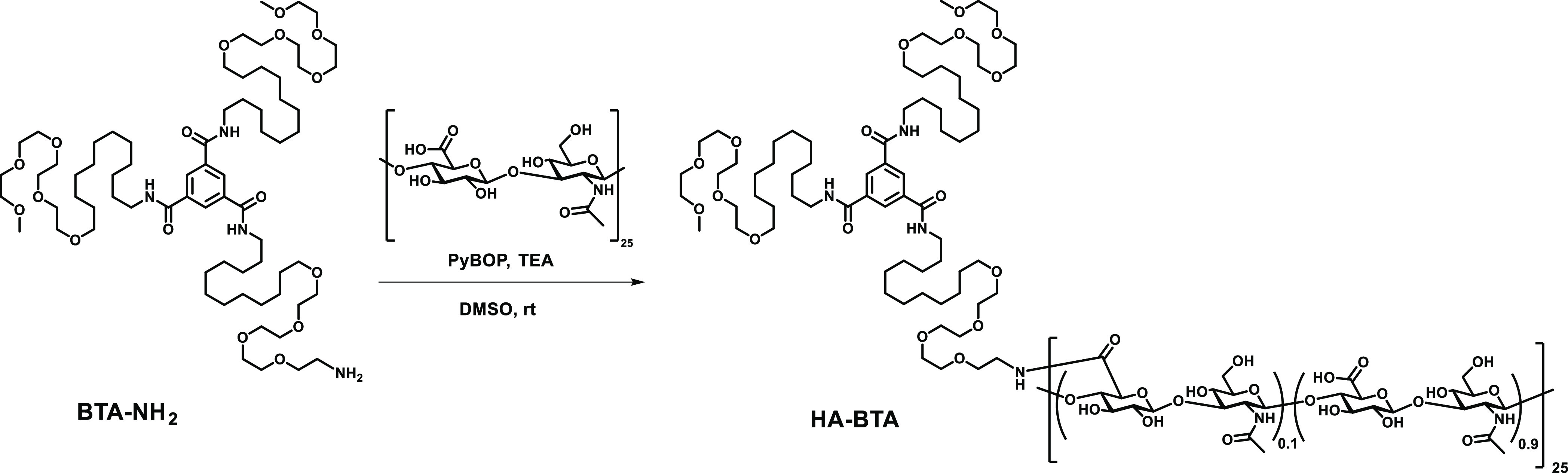
Sodium hyaluronate was protonated with amberlite-IR120 (H+) resin and lyophilized to obtain HA as a white powder. To a stirring solution of HA (50.0 mg, 6.63 μmol) in dimethyl sulfoxide (DMSO) (10 mL), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP, 65.6 mg, 125.9 μmol) and triethylamine (TEA, 17.6 μL, 125.9 μmol) were added followed by the addition of BTA-NH2 (24.9 mg, 18.9 μmol) (see the Supporting Information for synthesis details). The mixture was stirred overnight at room temperature (Scheme 1). The reaction mixture was further dialyzed (MWCO 6–8 kDa) overnight against DMSO and finally twice against water overnight. The purified product HA-BTA was lyophilized (45.5 mg), and the degree of substitution was 10% as determined by proton nuclear magnetic resonance (1H-NMR) (Figure S4).
Scheme 1. Synthesis of HA with Pendant BTAs (HA-BTA) with a Degree of Substitution of 10%.
Assembly of BTA Materials
Homoassembly. BTA-OEG4 and HA-BTA samples were prepared by weighing the solid material into a glass vial equipped with a magnetic stirring bar. MilliQ (MQ) water was added to obtain the desired concentration, and the sample was stirred at 90 °C for 15 min and vortexed for 15 s immediately afterward. All samples were left to equilibrate overnight at room temperature before being used for any measurements. Coassembly. BTA-OEG4 was coassembled with polysaccharides or HA-BTA by weighing both solid materials into a glass vial and adding MQ water to obtain the desired concentration. The sample was stirred at 90 °C for 15 min and vortexed for 15 s immediately afterward. All samples were left to equilibrate overnight at room temperature before being used for any measurements. Hydrogels. The same procedure as in the assembly of the BTA materials was performed to prepare the hydrogels, but an additional step of equilibration in an ice bath for 1 h was included after vortexing for 15 s. Hydrogels containing both HA-BTA and BTA-OEG4 did not require an equilibration step in ice, as the hydrogel was formed within a few seconds after the vortexing step.
Analytical Techniques.
Nuclear Magnetic Resonance (NMR)
NMR spectra were recorded on a Bruker 400 MHz Ultrashield spectrometer (400 MHz for 1H NMR). Deuterated solvents used are indicated in each case. Chemical shifts (δ) are expressed in ppm and are assigned to the residual peak of the solvent peak; multiplicity is abbreviated as follows: s, singlet; d, doublet; t, triplet; dt, doublet of triplets; ddt, doublet of doublets of triplets; td, triplet of doublets; tt, triplet of triplets; q, quartet; qd, quartet of doublets; and m, multiplet.
Matrix-Assisted Laser Absorption/Ionization-Time of Flight (MALDI-TOF)
Mass spectra were obtained with a Bruker Daltonic Autoflex spectrometer using α-cyano-4-hydroxycinnamic acid or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile as the matrix.
Liquid Chromatography-Mass Spectrometry (LCMS)
LCMS was performed using a system consisting of the following components: Shimadzu SCL-10A VP system controller with Shimadzu LC-10 AD VP liquid chromatography pumps with an Alltima C18 3 u (50 × 2.1 mm) reversed-phase column and gradients of water–acetonitrile supplemented with 0.1% formic acid, a Shimadzu DGU 20A3 prominence degasser, a Thermo Finnigan surveyor auto sampler, a Thermo Finnigan surveyor PDA detector, and a Thermo Scientific LCQ Fleet. Gradients were run from 5% MeCN to 100% MeCN over a 15 min period.
Ultraviolet–Visible Spectroscopy (UV–vis)
UV–vis absorbance spectra were recorded on a Jasco V-650 UV–vis spectrometer with a Jasco ETCT-762 temperature controller.
Cryogenic Transmission Electron Microscopy (CryoTEM)
Imaging was performed on samples with a 500 μM concentration of BTAs in water. Vitrified films were prepared using a “Vitrobot” instrument (FEI Vitrobot Mark IV, FEI Company) at 22 °C and a humidity of 100%. In the preparation chamber of the “Vitrobot,” a 3 μL sample was applied on a Quantifoil grid (R 2/2, Quantifoil Micro Tools GmbH), which was surface plasma treated just prior to use (Cressington 208 carbon coater operating at 5 mA for 40 s). The excess sample was removed by blotting using filter paper for 3 s with a blotting force of −1, and the thin film thus formed was shot (acceleration about 3 g) into liquid ethane just above its freezing point. Vitrified films were transferred into the vacuum of a CryoTITAN microscope equipped with a field emission gun that was operated at 300 kV, a postcolumn Gatan energy filter, and a 2048 × 2048 Gatan CCD camera. Vitrified films were observed using the CryoTITAN microscope at temperatures below −170 °C. Micrographs were taken under low-dose conditions, starting at a magnification of 6500 with a defocus setting of −40 μm and at a magnification of 24,000 with a defocus setting of −10 μm.
Total Internal Reflection Fluorescence Microscopy (TIRF)
TIRF images were acquired with a Nikon N-STORM system. Cy3 was excited using a 561 nm laser. Fluorescence was collected by means of a Nikon ×100, 1.4NA oil immersion objective and passed through a quad-band pass dichroic filter (97335 Nikon). Images were recorded with an EMCCD camera (ixon3, Andor, pixel size 0.17 μm). BTA fibers were prepared at a total concentration of 20 μM containing Cy3-labeled BTA-OEG4 (Cy3-BTA-OEG4) and further diluted to 2.5 μM and flown in a chamber between a glass microscope coverslip (Menzel-Gläser, no. 1.21 × 26 mm) and a glass slide, which were separated by double-sided tape.
Rheology
Rheological measurements were performed using a TA Instruments DHR-3 rheometer (TA Instruments). The hydrogels were deposited onto the rheometer stage and incubated for 10 s before all the measurements. A 20 mm aluminum cone with a 2.007° cone angle was used, and the gap height was set to a truncation height of 56 μm. The temperature was controlled strictly at 37 °C using a Peltier system, and a water trap prevented sample evaporation. Each measurement was repeated multiple times with different batches to ensure data reliability. The storage modulus G′ and loss modulus G″ were monitored under an applied strain of 0.1 to 1000% at a frequency of 1 rad s–1 for the strain sweep and a frequency of 600 to 0.1 rad s–1 at a strain of 1% for the frequency sweep. Alternating strains (1 or 1000%) with a fixed frequency of 1 rad s–1 were applied toward the hydrogels for six circles to investigate the self-healing behavior.
Results and Discussion
Coassembly of BTA-OEG4 Fibers with HA
In order to investigate whether HA can disrupt BTA-OEG4 stacks and destabilize the fiber formation, BTA fibers were assembled in MQ water in the presence of different concentrations of HA (Figure 1a). The concentration of BTA-OEG4 was fixed at 500 μM, and different amounts of HA were added prior to the assembly. Since the polysaccharide is a large molecule compared to the BTA-OEG4 monomer, the ratios between the two components were based on weight ranging from 0.5:2 to 3:2 (HA/BTA-OEG4). As previously reported, the signature UV profile of micrometer-long BTA-OEG4 fibers shows two maxima at 211 and 226 nm (Figure 1c, black line).23 Upon increasing the amount of HA, the intensity of the two maxima decreased and the signal at higher wavelengths, indicating scattering, increased (Figure 1c). This observation is explained by disruption of the BTA fibers and formation of precipitates due to destabilization and aggregation as a result of an interaction between the fibers and the HA polysaccharide that possibly acted as a chain stopper. CryoTEM images of samples containing HA/BTA-OEG4 (1:2) showed the formation of both short and long fibers (Figure 1b and Figure S5) with a diameter of around 5 nm. These coassemblies were significantly shorter than BTA-OEG4 analogues, as these fibers are several micrometers long and their ends are usually not visible by cryoTEM.23 The formation of shorter and polydisperse fibers was confirmed by TIRF microscopy when HA was coassembled with Cy3-labeled BTA-OEG4 (Cy3-BTA-OEG4) at a 2:2 ratio (Figure S6).
Figure 1.

Coassembly of BTA-OEG4 with HA in water. (a) Chemical structures of BTA-OEG4 and HA and schematic representation of the resulting assembly; (b) CryoTEM image of coassembled HA with BTA-OEG4 (500 μM) at a 1:2 weight ratio showed the formation of short polydisperse fibers (scale bar = 50 nm). The dark spherical objects are crystalline ice particles; (c) UV–vis spectra at 20 °C of the coassembly of different weight ratios of HA with BTA-OEG4 (500 μM) revealed that upon increasing the concentration of HA, the intensity of the typical bands corresponding to BTA-OEG4 (black) decreased; (d) rheology measurements of coassembled BTA-OEG4 (2 wt % in water) with different amounts of HA showed an increase in the storage modulus (G’) at 1 rad/s at an increasing concentration of HA and in the standard deviation between the measurements (n = 3).
As shown in previous studies,28 at increased concentrations of BTA-OEG4, the entanglement of fibers creates a network that results in a soft hydrogel by using the standard heating–cooling procedure following an equilibration step in ice for 1 h to form a more transparent hydrogel as compared to equilibration at room temperature.33 At 2 wt % in water, a BTA-OEG4 hydrogel has a storage modulus (G’) of 6 Pa (Figure 1d; Figure S7). At the same concentration of BTA-OEG4, addition of HA to the hydrogel resulted not only in an increase in G’ values but also in an increase in the standard deviation of the measurements. The latter was presumably due to high sample-to-sample variation caused by the solid content in the hydrogel state that led to heterogeneity in the rheology measurements. This phenomenon corroborated the UV–vis measurements and the microscopy imaging, as HA disrupted the fiber formation and led to heterogeneous samples.
Assembly of HA-BTA
Since the coassembly of HA with BTA-OEG4 fibers led to shorter and polydisperse fibers, we covalently functionalized HA with pendant BTAs. For this, asymmetric BTA-NH2 was synthesized, with an amine group at the periphery of one arm and methoxy end-capped tetra(ethylene glycol) units at the periphery of the other two arms to avoid the formation of esters during functionalization. BTA-NH2 was conjugated to the carboxylate groups of HA, yielding HA-BTA (Figure 2a) using PyBOP and triethylamine with a degree of substitution of 10% as confirmed by 1H-NMR (Figure S4), which corresponded to 2.5 BTAs per HA chain, considering that each HA polysaccharide consisted of 25 dimers. The assembly of HA-BTA in water was followed by UV–vis spectroscopy using a 500 μM concentration of BTA content and compared to that of BTA-OEG4 fibers. Since HA-BTA was soluble in water without following the heating–cooling procedure, the HA-BTA spectrum in water was recorded (Figure 2b, blue line), showing one maximum at 209 nm. This UV spectrum is usually observed in solvents such as methanol, typical for molecularly dissolved BTAs.23 After heating, cooling, and equilibrating the sample, HA-BTA showed a broader band at 210 nm and a second band with a maximum at 226 nm (Figure 2b, red line). The latter probably indicated the presence of some assembled BTAs. As a result, after the equilibration, a mixture of both assembled and molecularly dissolved states was obtained; apparently, the high hydrophilicity of HA-BTA hampers the pendant BTAs from being fully stacked. Moreover, the scattering signal at higher wavelengths indicated that the samples contained large aggregates. When compared to the assembly of BTA-OEG4 with noncovalently bound HA at the same ratio (Figure 2b, green line), the two maxima at 211 and 226 nm showed a much lower intensity, indicating reduced assembly for HA-BTA.
Figure 2.

Homoassembly of HA-BTA in water. (a) Chemical structure of HA-BTA and schematic representation of the resulting assembly; (b) UV–vis spectra at 20 °C of HA-BTA in water before (blue line) and after (red line) the heating–cooling procedure, compared to BTA-OEG4 fibers (black line) and BTA-OEG4 and HA at the same ratio as HA-BTA (green line) (BTA content was fixed to 500 μM in all cases), show no complete assembly for HA-BTA; (c) strain sweep oscillatory rheology (fixed angular frequency of 1 rad/s) of HA-BTA in water (5 wt %) reveals that no hydrogel was obtained (G’’ > G’), and the inverted vial test shows the formation of a viscous material; (d) schematic representation of the resulting mixture when coassembling HA and BTA-OEG4 at the same ratio as HA-BTA (5 wt %), and the inverted vial test shows the formation of a whitish liquid.
The assembly of HA-BTA at 5 wt % formed a transparent viscoelastic fluid as shown by both frequency sweep (Figure S8) and strain sweep oscillatory rheology (Figure 2c), as the loss modulus (G’’) was higher than the storage modulus (G’). In agreement with UV–vis measurements, HA-BTA did not completely assemble, and therefore, a hydrogel was not formed. However, when BTA-OEG4 and HA at the same ratio were coassembled as HA-BTA, a white-colored heterogeneous liquid was formed, as the BTA content was much lower than in the previous BTA-OEG4 and HA assembly (Figure 2d).
Coassembly of HA-BTA with BTA-OEG4
Since HA-BTA was not able to form a stiff network by itself, coassembly of BTA-OEG4 fibers and HA-BTA in water was explored, as the latter could act as the crosslinker between BTA fibers (Figure 3a). The two components were assembled following the previously described heating–cooling procedure at three different ratios based on weight. The total content of BTA in all the samples was fixed at 500 μM. When the ratio of HA-BTA/BTA-OEG4 is 0.5:2, this equals a weight ratio between the total amount of HA and BTA of 0.5 and 2, respectively. It is worth noting that lower concentrations of HA corresponded to lower concentrations of covalent HA-BTA in the samples. UV–vis spectroscopy revealed that the three samples assembled into 1D fibers as the two bands at 211 and 226 nm overlapped with the spectrum of BTA-OEG4 fibers alone (Figure 3b). This indicated that the three samples were able to assemble into elongated structures, similar to BTA-OEG4. In agreement with UV–vis, cryoTEM imaging of samples HA-BTA/BTA-OEG4 (0.5:2 = HA/BTA) and HA-BTA/BTA-OEG4 (2:2 = HA/BTA) confirmed the presence of long 1D fibers with a diameter of around 5 nm (Figure S9 and Figure 3c). However, these coassemblies displayed slightly shorter lengths compared to fibers formed with BTA-OEG4 alone, especially for sample (2:2) (Figure S9d–f). This is probably due to the fact that HA-BTA can act as a chain stopper, leading to shorter fibers.
Figure 3.
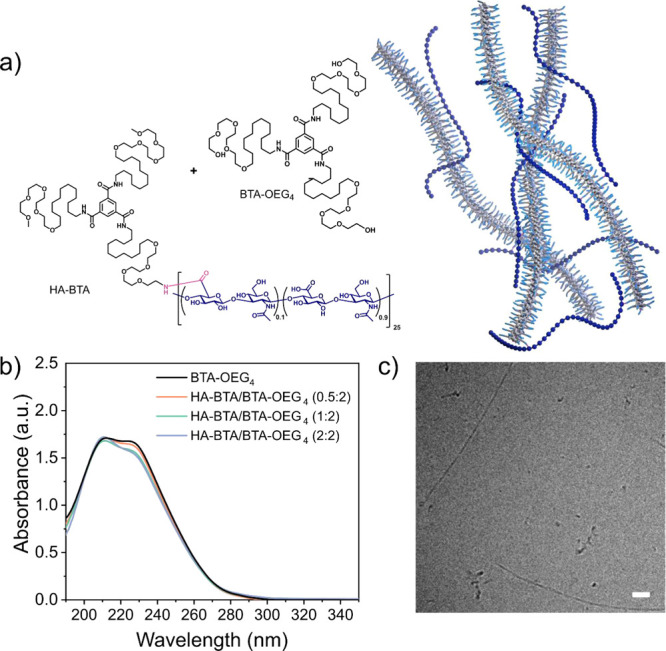
Coassembly of HA-BTA with BTA-OEG4 in water. (a) Chemical structure of BTA-OEG4 and HA-BTA for the assembly and scheme of the resulting network; (b) UV–vis spectra of coassembled HA-BTA and BTA-OEG4 at three different ratios (HA/BTA) based on weight (BTA content adjusted to 500 μM in all cases); (c) CryoTEM image of HA-BTA coassembled with BTA-OEG4 (HA-BTA/BTA-OEG4, 2:2 HA/BTA) shows fiber formation (scale bar = 50 nm).
The same three HA-BTA/BTA-OEG4 ratios used for spectroscopy measurements were selected at increased concentrations to form hydrogels. In this case, HA-BTA/BTA-OEG4 (0.5:2 = HA/BTA) contained 0.5 wt % of HA and 2 wt % of BTA, which corresponded to 2.5 wt % of total material, HA-BTA/BTA-OEG4 (1:2 = HA/BTA) contained 1 wt % of HA and 2 wt % of BTA, which corresponded to 3 wt % of total material, and HA-BTA/BTA-OEG4 (2:2 = HA/BTA) contained 2 wt % of HA and 2 wt % of BTA, which corresponded to 4 wt % of total material. This means that the three samples contained the same concentration of total BTA (2 wt %) that acts as the hydrogelator. The three samples were assembled following the heating–cooling procedure, but the equilibration step in ice for 1 h was not necessary as the hydrogels were formed within 1 min after removing the samples from the heating source. Compared to previous hydrogel mixtures of BTA-OEG4 and HA, these three samples showed a much faster crosslinking effect that did not require an equilibration step. Frequency sweep rheology measurements on the three samples showed an increase in stiffness in the coassembled samples of around 10-fold compared to that of BTA-OEG4 alone at 2 wt % (Figure 4a; Figure S12). Moreover, the stiffness could be tuned by changing the ratio between HA-BTA and BTA-OEG4. Within the investigated range of weight ratios, the lower the amount of HA-BTA, the higher the stiffness that ranged from 230 Pa for the sample containing a ratio 2:2 (HA/BTA, 4 wt % total material), 530 Pa for ratio 1:2 (HA/BTA, 3 wt % total material), and 700 Pa for ratio 0.5:2 (HA/BTA, 2.5 wt % total material). This drastic increase in storage moduli could be explained by the noncovalent crosslinking of BTA-OEG4 fibers by HA-BTA. Remarkably, the highest storage modulus was found for the hydrogel with the lowest wt % of HA-BTA, a phenomenon that has been seen before in binary hydrogels based on BTAs and ureidopyrimidinones.52,53 Strain sweep oscillatory rheology showed that the transition from gel-to-sol happened upon higher strain (γ) percentages compared to BTA-OEG4 alone, as the coassemblies displayed this transition at 500% strain (Figure S10) and BTA-OEG4 at 300% strain (Figure S7b). Thus, viscoelastic hydrogels formed by coassembly not only formed networks with a higher stiffness but also with an increased resilience compared to BTA-OEG4 hydrogels. The shear-thinning behavior of the three hydrogels was shown by a decrease in complex viscosity, as the strain rate increased (Figure S11). The self-healing behavior of the three samples was tested by alternating large amplitude and small amplitude oscillatory shear measurements revealing that the hydrogels were able to completely recover for at least 6 cycles (Figure 4b), similar to other BTA hydrogels.33
Figure 4.
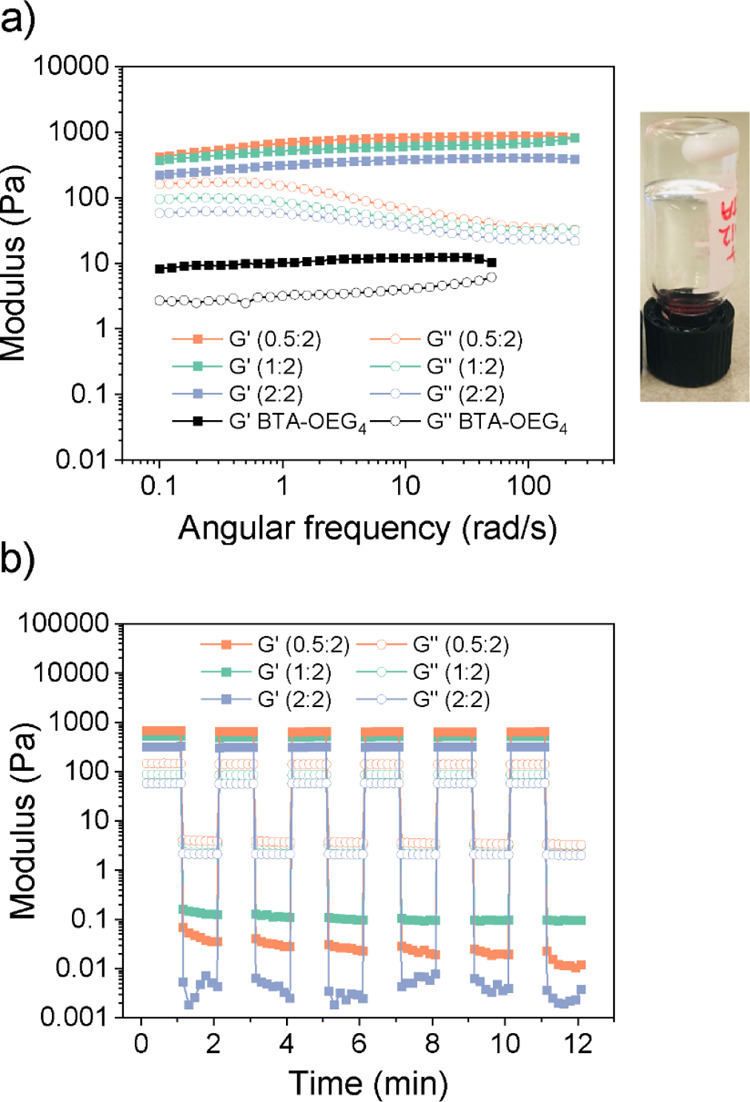
Rheological measurements of hydrogels from coassembled HA-BTA and BTA-OEG4 in water at three different weight HA/BTA ratios (0.5:2, 1:2, and 2:2). (a) Frequency sweep rheology measurements at 37 °C (1% fixed applied strain) reveal storage moduli between 250 and 700 Pa and the image of the inverted vial of the hydrogel (1:2). (b) Oscillatory rheology measurements alternating between 1 and 1000% strain for 30 s periods at 37 °C (1 rad/s fixed angular frequency) show self-healing properties for the three hydrogels during 6 cycles.
Incorporation of Other Polysaccharides into BTA Materials
Since HA shows a strong effect on the assembly of BTA-OEG4 fibers, other polysaccharides were selected to further study their influence on BTA fiber formation. Four polysaccharides were chosen that contained different functional groups and/or molecular weights (Scheme 2). The hydroxyl groups of HA (10 kDa) were acetylated (Acetyl-HA) in order to see whether these groups influence the assembly. HA with a molecular weight of 700 kDa (HA-700 k) rather than the originally used 10 kDa was in addition chosen to investigate the influence of the polysaccharide length during the assembly. Alginic acid (AA, low molecular weight) was selected as a linear polysaccharide composed of monomers of mannuronate and guluronate, both bearing carboxylate groups. Finally, in order to have a better understanding of the role of hydroxyl groups of polysaccharides in the BTA assembly, dextran was chosen as a polyglucose that does not contain carboxylates nor amide groups.
Scheme 2. Polysaccharides Used for the Coassembly with BTA-OEG4.
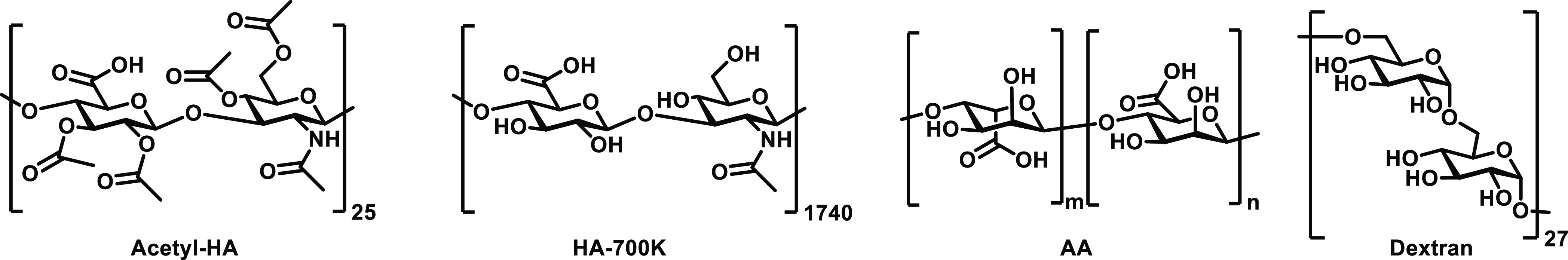
The BTA-OEG4 coassembly in water with the different polysaccharides was monitored by UV–vis spectroscopy, following the same procedure as that for HA (10 kDa). Two weight ratios of polysaccharide/BTA-OEG4 (500 μM) were selected (1:2 and 2:2) for the coassembly (Figure S13a and Figure 5a, respectively). Acetyl-HA showed a very strong effect on the BTA-OEG4 assembly, indicating that the acetylation of the hydroxyl groups did not reduce the interaction with BTA-OEG4 during the fiber formation. In contrast, coassembly of the fibers with a longer HA polymer (HA-700 k) seemed to have a lower effect compared to the low molecular weight analogue, as the absorbance of the bands for the assembled BTAs was higher. However, the scattering at higher wavelengths showed that the fibers were probably not well dispersed, and the samples were not homogeneous, as in HA 10 kDa. Interestingly, the UV–vis spectrum of AA coassembled with BTA-OEG4 showed a lower effect on the bands as compared to HA, which could also be observed by the lower scattering signal. Remarkably, dextran at these two ratios did not show any effect on the UV–vis spectra of BTA-OEG4, indicating that this glucose-based polysaccharide did not interfere in the BTA-OEG4 fiber formation at all. The lack of carboxylates and amides in its structure reduces the interaction of the polysaccharide with the fibers, suggesting that the hydroxyl groups did not affect fiber formation.
Figure 5.

Coassembly of BTA-OEG4 with different polysaccharides in water. (a) UV–vis spectra of BTA-OEG4 in water (500 μM) coassembled with Acetyl-HA, HA 700 kDa, AA, dextran, and HA 10 kDa at a 2:2 weight ratio. (b) Strain sweep oscillatory rheology at 37 °C (fixed angular frequency of 1 rad/s) of BTA-OEG4 in water (2 wt %) alone or coassembled with Acetyl-HA, HA-700 k, and dextran (1 wt %).
To further evaluate the possibility of incorporating polysaccharides into BTA materials, hydrogels in the presence of these polysaccharides were formed (Figure 5b). In order to maintain the same weight ratio as in UV–vis measurements, the BTA-OEG4 content in the hydrogels was 2 wt % and the polysaccharide concentration was 1 wt % in all cases. Strain sweep oscillatory rheology (Figure 5b) and frequency sweep rheology measurements (Figure S13b) showed that the hydrogel containing Acetyl-HA had similar mechanical properties to the hydrogel formed with HA (10 kDa), as it had a higher storage modulus (G’) compared to BTA-OEG4 alone (22 ± 4 Pa vs 6.0 ± 0.2 Pa, respectively). Hence, acetylation of the hydroxyl groups in the polysaccharide did not reduce the interaction with the fibers. In the presence of HA-700 k, the G’ value was higher compared to BTA-OEG4 (12 ± 1 Pa vs 6.0 ± 0.2 Pa, respectively), but also the loss modulus (G’’) was higher (9 ± 1 Pa vs 1.0 ± 0.1 Pa, respectively). Furthermore, the gel-to-sol transition occurred at 400% strain instead of 300%, as in the other polysaccharide coassembled hydrogels (Figure 5b). This indicates that having a longer HA polysaccharide during the assembly resulted in the formation of a different network compared to all other BTA-based hydrogels. Interestingly, BTA-OEG4 coassembled with AA did not lead to a hydrogel even if the typical BTA-OEG4 bands were observed by UV–vis spectroscopy. This result probably reflects an interaction of AA with BTA-OEG4 that was strong enough to affect the gelation probably because AA hampers fiber entanglement. Finally, BTA-OEG4 coassembled with dextran showed similar rheological properties to BTA-OEG4 alone (5.9 ± 0.5 Pa vs 6.0 ± 0.2 Pa, respectively), which agrees with the UV–vis experiments. This confirms that the interaction with polyglucose is lower than that with the other polysaccharides studied.
Conclusions
In summary, the incorporation of HA (10 kDa) into BTA-based supramolecular polymers and hydrogels was investigated by using three different approaches. First, BTAs bearing tetra(ethylene glycol) at the periphery (BTA-OEG4) were coassembled with HA in water. UV–vis spectroscopy and microscopy imaging revealed that the fibers assembled in the presence of this polysaccharide were shorter in length but formed insoluble aggregates. The mechanical properties of hydrogels formed in the presence of HA confirmed these results: these materials were very polydisperse with a high sample-to-sample variability. In the second approach, HA was covalently functionalized with BTAs (HA-BTA), and the homoassembly in water of this polymer was analyzed. UV–vis spectroscopy showed that HA-BTA was too hydrophilic to enable fiber formation. At 5 wt % of HA-BTA in water, a viscous material was obtained instead of a hydrogel. In order to improve the assembly of HA-containing supramolecular polymers and hydrogels, a third approach was performed that used the coassembly of HA-BTA and BTA-OEG4. UV–vis spectroscopy and cryoTEM imaging showed long 1D supramolecular fibers when these two components were coassembled in different ratios. The formation of hydrogels showed an overall increase in the storage modulus that could be tuned between 250 Pa (4 wt %) and 700 Pa (2.5 wt %), compared to the storage modulus of BTA-OEG4 that at the same concentration has a value of 6 Pa. Moreover, these HA-containing hydrogels were more resilient than BTA-OEG4 analogues, as the transition from gel-to-sol happened at 500% oscillatory strain instead of 300% (BTA-OEG4). Furthermore, the self-healing properties of these materials showed the potential of using them as biomaterials.
To understand the nature of the interactions of polysaccharides during the BTA-fiber formation, other polysaccharides containing different functional groups and having different lengths were selected for coassembly with BTA-OEG4. While acetylated HA (10 kDa), HA (700 kDa), and alginic acid (AA, low molecular weight) interfered with the fiber formation, dextran showed no interaction with the fibers, indicating that hydroxyl groups of polysaccharides do not likely affect the supramolecular polymerization. In correlation with the fiber assembly, hydrogels formed in the presence of these polysaccharides showed that only dextran produced materials with the exact same mechanical properties as BTA-OEG4 alone. It would therefore be interesting to analyze, in the future, the effect of having covalently attached BTAs on dextran for fiber and hydrogel formation. The possibility of introducing polysaccharides into BTA supramolecular polymers and hydrogels provides new insights into the design of dynamic and fibrous biomaterials bearing complex biofunctionalities that resemble native systems and can be used for a range of applications, such as regenerative medicine.
Acknowledgments
The authors acknowledge the ICMS Animation Studio for providing the artwork. S.V.-A. and G.M. acknowledge the funding received by Gravitation Program “Materials Driven Regeneration,” funded by the Netherlands Organization for Scientific Research (024.003.013). J.M. acknowledges a Marie Skłodowska-Curie postdoctoral fellowship (794016) for financial support. G.M. acknowledges the funding received by the Swiss National Science Foundation (SNSF “Early PostDoc Mobility” P2EZP2-178435). R.C. acknowledges TA Instruments for providing the DHR-3 rheometer under the Young Distinguished Rheologist Award instrument grant. S.S. and E.W.M acknowledge the European Research Council (H2020-EU.1.1., SYNMAT project, ID 788618).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.1c00927.
Synthetic details of BTA-NH2, NMR, TIRF imaging and additional cryoTEM imaging, rheology, and UV–vis spectra (PDF)
Author Present Address
# Centro de Investigacioìns Científicas Avanzadas (CICA), Universidade da Coruña, Rúa As Carballeiras, A Coruña 15071, Spain (J.M.)
Author Present Address
∥ R&D Department, Foresa Technologies, Av. Doña Urraca, 91, Caldas de Reis, Pontevedra 36650, Spain (S.V.-A.)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Gravitation Program “Materials Driven Regeneration,” funded by the Netherlands Organization for Scientific Research (024.003.013), Swiss National Science Foundation (SNSF “Early PostDoc Mobility” P2EZP2-178435), Marie Skłodowska-Curie postdoctoral fellowship (794016), and European Research Council Advanced Grant (788618 SYNMAT- ERC-2017-ADG 10025006).
The authors declare no competing financial interest.
Supplementary Material
References
- Liu Z.; Qiao J.; Niu Z.; Wang Q. Natural Supramolecular Building Blocks: From Virus Coat Proteins to Viral Nanoparticles. Chem. Soc. Rev. 2012, 41, 6178–6194. 10.1039/c2cs35108k. [DOI] [PubMed] [Google Scholar]
- Sanchez C.; Arribart H.; Giraud Guille M. M. Biomimetism and Bioinspiration as Tools for the Design of Innovative Materials and Systems. Nat. Mater. 2005, 4, 277–288. 10.1038/nmat1339. [DOI] [PubMed] [Google Scholar]
- Goor O. J. G. M.; Hendrikse S. I. S.; Dankers P. Y. W.; Meijer E. W. From Supramolecular Polymers to Multi-Component Biomaterials. Chem. Soc. Rev. 2017, 46, 6621. 10.1039/C7CS00564D. [DOI] [PubMed] [Google Scholar]
- Frantz C.; Stewart K. M.; Weaver V. M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200. 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular Polymerization. Chem. Rev. 2009, 109, 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Krieg E.; Bastings M. M. C.; Besenius P.; Rybtchinski B. Supramolecular Polymers in Aqueous Media. Chem. Rev. 2016, 116, 2414–2477. 10.1021/acs.chemrev.5b00369. [DOI] [PubMed] [Google Scholar]
- Vantomme G.; Meijer E. W. The Construction of Supramolecular Systems. Science 2019, 363, 1396–1397. 10.1126/science.aav4677. [DOI] [PubMed] [Google Scholar]
- Sato K.; Hendricks M. P.; Palmer L. C.; Stupp S. I. Peptide Supramolecular Materials for Therapeutics. Chem. Soc. Rev. 2018, 47, 7539. 10.1039/C7CS00735C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Webber M. J.; Stupp S. I. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers 2010, 94, 1–18. 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartgerink J. D.; Beniash E.; Stupp S. I. Peptide-Amphiphile Nanofibers: A Versatile Scaffold for the Preparation of Self-Assembling Materials. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5133–5138. 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammadov R.; Mammadov B.; Toksoz S.; Aydin B.; Yagci R.; Tekinay A. B.; Guler M. O. Heparin Mimetic Peptide Nanofibers Promote Angiogenesis. Biomacromolecules 2011, 12, 3508–3519. 10.1021/bm200957s. [DOI] [PubMed] [Google Scholar]
- Tansik G.; Kilic E.; Beter M.; Demiralp B.; Kiziltas Sendur G.; Can N.; Ozkan H.; Ergul E.; Guler M. O.; Tekinay A. B. A Glycosaminoglycan Mimetic Peptide Nanofiber Gel as an Osteoinductive Scaffold. Biomater. Sci. 2016, 4, 1328–1339. 10.1039/C6BM00179C. [DOI] [PubMed] [Google Scholar]
- Mansukhani N. A.; Peters E. B.; So M. M.; Albaghdadi M. S.; Wang Z.; Karver M. R.; Clemons T. D.; Laux J. P.; Tsihlis N. D.; Stupp S. I.; et al. Peptide Amphiphile Supramolecular Nanostructures as a Targeted Therapy for Atherosclerosis. Macromol. Biosci. 2019, 19, 1900066 10.1002/mabi.201900066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Iscen A.; Sai H.; Sato K.; Sather N. A.; Chin S. M.; Álvarez Z.; Palmer L. C.; Schatz G. C.; Stupp S. I. Supramolecular–Covalent Hybrid Polymers for Light-Activated Mechanical Actuation. Nat. Mater. 2020, 19, 900–909. 10.1038/s41563-020-0707-7. [DOI] [PubMed] [Google Scholar]
- Chin S. M.; Synatschke C. V.; Liu S.; Nap R. J.; Sather N. A.; Wang Q.; Álvarez Z.; Edelbrock A. N.; Fyrner T.; Palmer L. C.; Szleifer I.; Olvera de la Cruz M.; Stupp S. I. Covalent-Supramolecular Hybrid Polymers as Muscle-Inspired Anisotropic Actuators. Nat. Commun. 2018, 9, 2395. 10.1038/s41467-018-04800-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrikse S. I. S.; Spaans S.; Meijer E. W.; Dankers P. Y. W. Supramolecular Platform Stabilizing Growth Factors. Biomacromolecules 2018, 19, 2610–2617. 10.1021/acs.biomac.8b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaal R. C.; Buskermolen A. B. C.; Ippel B. D.; Fransen P.-P. K. H.; Zaccaria S.; Bouten C. V. C.; Dankers P. Y. W. Functional Peptide Presentation on Different Hydrogen Bonding Biomaterials Using Supramolecular Additives. Biomaterials 2019, 224, 119466 10.1016/j.biomaterials.2019.119466. [DOI] [PubMed] [Google Scholar]
- Wijnands S. P. W.; Meijer E. W.; Merkx M. DNA-Functionalized Supramolecular Polymers: Dynamic Multicomponent Assemblies with Emergent Properties. Bioconjugate Chem. 2019, 30, 1905–1914. 10.1021/acs.bioconjchem.9b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbianco M.; Bharate P.; Varela-Aramburu S.; Seeberger P. H. Carbohydrates in Supramolecular Chemistry. Chem. Rev. 2016, 116, 1693–1752. 10.1021/acs.chemrev.5b00516. [DOI] [PubMed] [Google Scholar]
- Müller M. K.; Brunsveld L. A Supramolecular Polymer as a Self-Assembling Polyvalent Scaffold. Angew. Chem., Int. Ed. 2009, 48, 2921–2924. 10.1002/anie.200900143. [DOI] [PubMed] [Google Scholar]
- Luo Q.; Dong Z.; Hou C.; Liu J. Protein-Based Supramolecular Polymers: Progress and Prospect. Chem. Commun. 2014, 50, 9997–10007. 10.1039/C4CC03143A. [DOI] [PubMed] [Google Scholar]
- Petkau-Milroy K.; Uhlenheuer D. A.; Spiering A. J. H.; Vekemans J. A. J. M.; Brunsveld L. Dynamic and Bio-Orthogonal Protein Assembly along a Supramolecular Polymer. Chem. Sci. 2013, 4, 2886–2891. 10.1039/c3sc50891a. [DOI] [Google Scholar]
- Leenders C. M. A.; Albertazzi L.; Mes T.; Koenigs M. M. E.; Palmans A. R. A.; Meijer E. W. Supramolecular Polymerization in Water Harnessing Both Hydrophobic Effects and Hydrogen Bond Formation. Chem. Commun. 2013, 49, 1963–1965. 10.1039/c3cc38949a. [DOI] [PubMed] [Google Scholar]
- Albertazzi L.; Van Der Zwaag D.; Leenders C. M. A.; Fitzner R.; Van Der Hofstad R. W.; Meijer E. W. Probing Exchange Pathways in One-Dimensional Aggregates with Super-Resolution Microscopy. Science 2014, 344, 491–495. 10.1126/science.1250945. [DOI] [PubMed] [Google Scholar]
- Leenders C. M. A.; Baker M. B.; Pijpers I. A. B.; Lafleur R. P. M.; Albertazzi L.; Palmans A. R. A.; Meijer E. W. Supramolecular Polymerisation in Water; Elucidating the Role of Hydrophobic and Hydrogen-Bond Interactions. Soft Matter 2016, 12, 2887–2893. 10.1039/C5SM02843D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafleur R. P. M.; Schoenmakers S. M. C.; Madhikar P.; Bochicchio D.; Baumeier B.; Palmans A. R. A.; Pavan G. M.; Meijer E. W. Insights into the Kinetics of Supramolecular Comonomer Incorporation in Water. Macromolecules 2019, 52, 3049–3055. 10.1021/acs.macromol.9b00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafleur R. P. M.; Herziger S.; Schoenmakers S. M. C.; Keizer A. D. A.; Jahzerah J.; Thota B. N. S.; Su L.; Bomans P. H. H.; Sommerdijk N. A. J. M.; Palmans A. R. A.; et al. Supramolecular Double Helices from Small C3-Symmetrical Molecules Aggregated in Water. J. Am. Chem. Soc. 2020, 142, 17644–17652. 10.1021/jacs.0c08179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders C. M. A.; Mes T.; Baker M. B.; Koenigs M. M. E.; Besenius P.; Palmans A. R. A.; Meijer E. W. From Supramolecular Polymers to Hydrogel Materials. Mater. Horiz. 2014, 1, 116–120. 10.1039/C3MH00103B. [DOI] [Google Scholar]
- Bakker M. H.; Lee C. C.; Meijer E. W.; Dankers P. Y. W.; Albertazzi L. Multicomponent Supramolecular Polymers as a Modular Platform for Intracellular Delivery. ACS Nano 2016, 10, 1845–1852. 10.1021/acsnano.5b05383. [DOI] [PubMed] [Google Scholar]
- Baker M. B.; Gosens R. P. J.; Albertazzi L.; Matsumoto N. M.; Palmans A. R. A.; Meijer E. W. Exposing Differences in Monomer Exchange Rates of Multicomponent Supramolecular Polymers in Water. ChemBioChem 2016, 17, 207–213. 10.1002/cbic.201500606. [DOI] [PubMed] [Google Scholar]
- Leenders C. M. A.; Jansen G.; Frissen M. M. M.; Lafleur R. P. M.; Voets I. K.; Palmans A. R. A.; Meijer E. W. Monosaccharides as Versatile Units for Water-Soluble Supramolecular Polymers. Chem. – Eur. J. 2016, 22, 4608–4615. 10.1002/chem.201504762. [DOI] [PubMed] [Google Scholar]
- Schoenmakers S. M. C.; Leenders C. M. A.; Lafleur R. P. M.; Lou X.; Meijer E. W.; Pavan G. M.; Palmans A. R. A. Impact of the Water-Compatible Periphery on the Dynamic and Structural Properties of Benzene-1,3,5-Tricarboxamide Based Amphiphiles. Chem. Commun. 2018, 54, 11128–11131. 10.1039/C8CC04818E. [DOI] [PubMed] [Google Scholar]
- Hendrikse S. I. S.; Su L.; Hogervorst T. P.; Lafleur R. P. M.; Lou X.; Van Der Marel G. A.; Codee J. D. C.; Meijer E. W. Elucidating the Ordering in Self-Assembled Glycocalyx Mimicking Supramolecular Copolymers in Water. J. Am. Chem. Soc. 2019, 141, 13877–13886. 10.1021/jacs.9b06607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnands S. P. W.; Engelen W.; Lafleur R. P. M.; Meijer E. W.; Merkx M. Controlling Protein Activity by Dynamic Recruitment on a Supramolecular Polymer Platform. Nat. Commun. 2018, 9, 65. 10.1038/s41467-017-02559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köwitsch A.; Zhou G.; Groth T. Medical Application of Glycosaminoglycans: A Review. J. Tissue Eng. Regen. Med. 2018, 12, e23–e41. 10.1002/term.2398. [DOI] [PubMed] [Google Scholar]
- Fraser J. R. E.; Laurent T. C.; Laurent U. B. G. Hyaluronan: Its Nature, Distribution, Functions and Turnover. J. Intern. Med. 1997, 242, 27–33. 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- de la Motte C. A. Hyaluronan in Intestinal Homeostasis and Inflammation: Implications for Fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G945–G949. 10.1152/ajpgi.00063.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman M. K.; Lee H.-G.; Schwertfeger K. L.; McCarthy J. B.; Turley E. A. The Content and Size of Hyaluronan in Biological Fluids and Tissues. Front. Immunol. 2015, 6, 261. 10.3389/fimmu.2015.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K. J.; Kumar S. Hyaluronic Acid: Incorporating the Bio into the Material. ACS Biomater. Sci. Eng. 2019, 5, 3753–3765. 10.1021/acsbiomaterials.8b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick J. A.; Prestwich G. D. Hyaluronic Acid Hydrogels for Biomedical Applications. Adv. Mater. 2011, 23, H41–H56. 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich G. D. Hyaluronic Acid-Based Clinical Biomaterials Derived for Cell and Molecule Delivery in Regenerative Medicine. J. Controlled Release 2011, 155, 193–199. 10.1016/j.jconrel.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Kirker K. R.; Prestwich G. D. Cross-Linked Hyaluronic Acid Hydrogel Films: New Biomaterials for Drug Delivery. J. Controlled Release 2000, 69, 169–184. 10.1016/S0168-3659(00)00300-X. [DOI] [PubMed] [Google Scholar]
- Highley C. B.; Prestwich G. D.; Burdick J. A. Recent Advances in Hyaluronic Acid Hydrogels for Biomedical Applications. Curr. Opin. Biotechnol. 2016, 40, 35–40. 10.1016/j.copbio.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Capito R. M.; Azevedo H. S.; Velichko Y. S.; Mata A.; Stupp S. I. Self-Assembly of Large and Small Molecules into Hierarchically Ordered Sacs and Membranes. Science 2008, 319, 1812–1816. 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- Helen Zha R.; Velichko Y. S.; Bitton R.; Stupp S. I. Molecular Design for Growth of Supramolecular Membranes with Hierarchical Structure. Soft Matter 2016, 12, 1401–1410. 10.1039/C5SM02381E. [DOI] [PubMed] [Google Scholar]
- Velichko Y. S.; Mantei J. R.; Bitton R.; Carvajal D.; Shull K. R.; Stupp S. I. Electric Field Controlled Self-Assembly of Hierarchically Ordered Membranes. Adv. Funct. Mater. 2012, 22, 369–377. 10.1002/adfm.201101538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. M.; Yang J. A.; Jung H.; Yeom J.; Park J. S.; Park K. H.; Hoffman A. S.; Hahn S. K.; Kim K. In Situ Supramolecular Assembly and Modular Modification of Hyaluronic Acid Hydrogels for 3D Cellular Engineering. ACS Nano 2012, 6, 2960–2968. 10.1021/nn204123p. [DOI] [PubMed] [Google Scholar]
- Rodell C. B.; MacArthur J. W. Jr.; Dorsey S. M.; Wade R. J.; Wang L. L.; Woo Y. J.; Burdick J. A. Shear-Thinning Supramolecular Hydrogels with Secondary Autonomous Covalent Crosslinking to Modulate Viscoelastic Properties In Vivo. Adv. Funct. Mater. 2015, 25, 636–644. 10.1002/adfm.201403550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodell C. B.; Dusaj N. N.; Highley C. B.; Burdick J. A. Injectable and Cytocompatible Tough Double-Network Hydrogels through Tandem Supramolecular and Covalent Crosslinking. Adv. Mater. 2016, 28, 8419–8424. 10.1002/adma.201602268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet A.; Forster R. A.; Parkins C. C.; Wu G.; Scherman O. A. Modulating Stiffness with Photo-Switchable Supramolecular Hydrogels. Polym. Chem. 2019, 10, 467–472. 10.1039/C8PY01554F. [DOI] [Google Scholar]
- Albertazzi L.; Martinez-Veracoechea F. J.; Leenders C. M. A.; Voets I. K.; Frenkel D.; Meijer E. W. Spatiotemporal Control and Superselectivity in Supramolecular Polymers Using Multivalency. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12203–12208. 10.1073/pnas.1303109110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereroudakis E.; Bantawa M.; Lafleur R. P. M.; Parisi D.; Matsumoto N. M.; Peeters J. W.; Del Gado E.; Meijer E. W.; Vlassopoulos D. Competitive Supramolecular Associations Mediate the Viscoelasticity of Binary Hydrogels. ACS Cent. Sci. 2020, 6, 1401–1411. 10.1021/acscentsci.0c00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieltyka R. E.; Pape A. C. H.; Albertazzi L.; Nakano Y.; Bastings M. M. C.; Voets I. K.; Dankers P. Y. W.; Meijer E. W. Mesoscale Modulation of Supramolecular Ureidopyrimidinone-Based Poly(Ethylene Glycol) Transient Networks in Water. J. Am. Chem. Soc. 2013, 135, 11159–11164. 10.1021/ja403745w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.