

Abstract

Water is one of the principal constituents by mass of living plant cell walls. However, its role and interactions with secondary cell wall polysaccharides and the impact of dehydration and subsequent rehydration on the molecular architecture are still to be elucidated. This work combines multidimensional solid-state 13C magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) with molecular dynamics modeling to decipher the role of water in the molecular architecture of softwood secondary cell walls. The proximities between all main polymers, their molecular conformations, and interaction energies are compared in never-dried, oven-dried, and rehydrated states. Water is shown to play a critical role at the hemicellulose–cellulose interface. After significant molecular shrinkage caused by dehydration, the original molecular conformation is not fully recovered after rehydration. The changes include xylan becoming more closely and irreversibly associated with cellulose and some mannan becoming more mobile and changing conformation. These irreversible nanostructural changes provide a basis for explaining and improving the properties of wood-based materials.
Introduction
For centuries, wood has been the ideal engineering material when low density coupled with high tensile strength was required. The use of softwood in construction has recently experienced a resurgence of popularity due to its high growth rate combined with renewability and carbon sequestration characteristics.1 Softwood dominates commercial forestry, e.g., Pinus radiata (D. Don) represents 90% of New Zealand planted production forest area and contributes 1.6% to its GDP.1 In a living tree, wood (never-dried and water-saturated) can be deformed beyond its yield point without breaking nor loss of stiffness.2 This unusual mechanical property can be explained by the stick-slip (Velcro) model, whereby cellulose can form transient bonds with the matrix (hemicelluloses plus lignin). While a living trunk can exceed its mechanical plastic point and fully recover, a dried trunk would suffer brittle failure.3 As the only compositional change is the reduction of water content, it implies that water plays a crucial mechano-structural role. However, water is absent from the stick-slip model. Therefore, an updated model that includes how water can influence cellulose–matrix interactions is required.
The amount of water in the woody xylem of a living tree can be up to twice its dried mass and generally consist of free water (found in the lumens of fluid conduction cells) and bound water (found mainly within the cell walls).4 The cell walls can contain water representing up to 30% of their dry mass.5 Bound water is confined within the secondary cell wall due to its strong interactions with polysaccharides.6 The equilibrium moisture content of wood after harvesting is mainly determined by its environment (relative humidity and temperature) and moisture history.7,8 Wood has a sigmoidal water sorption isotherm. The equilibrium moisture content of wood being higher during desorption than adsorption is an example of hysteresis. Wood exhibits a so-called “irreproducible hysteresis” between the never-dried and dry state.9 This hysteresis is due to irreversible structural alterations of the cell wall, reducing water binding sites through bond attrition.10 The hydrogen-bonding configuration changes could be due to the replacement of water-mediated hydrogen bonds by semipermanent hydrogen bonds between cellulose or hemicellulose polysaccharides.11
Cellulose, the major component of wood, is a β-(1,4)-linked polymer of glucose biosynthesized by the CesA protein complexes to form 18 cellulose chain microfibrils.12 Lignin, the other principal constituent, is a polyphenol that self-aggregates into hydrophobic nanodomains.13 In softwoods, the hemicellulose galactoglucomannan (GGM) makes up about 20% of the total mass and arabinoglucuronoxylan (AGX), the second most abundant hemicellulose, around 5–10%.14 Recent solid-state NMR studies focusing on secondary cell wall architecture revealed that a single softwood cellulose microfibril is associated with GGM and AGX.15 These hemicelluloses are themselves interacting with lignin via electrostatic contacts and α-ether or glucuronylester (of xylan) linkages.13,16−18 Moreover, the even patterning produced by the repeat of the XUXAXX pattern of AGX softwood allows it to bind cellulose in a two-fold screw configuration.19 A smaller proportion of three-fold screw AGX, possibly with two consecutive GlcA substitutions (XUUXX), may interact with the lignin nanodomain via electrostatic interactions.13,15,16,20−24 However, so far none of these studies considered the importance of the cell wall hydration status or its importance for the polymer conformations in their native state.
Due to their interactions, hemicelluloses have a regulator role of cell wall mechanics and are likely to determine the cellulose fibrils’ tertiary structure.25 Several models suggest that interactions between cellulose and hemicellulose are regulated by interstitial water present at their interface.26−28 The hydration level, pH of the aqueous environment, and the proximity between water and the various components influence the structure, conformation, dynamics, and intermolecular interactions of the cell wall polysaccharide network.29−31 The sum of those interactions directly impacts the cellulose microfibril crystal packing and the overall mechanical properties.32 Therefore, water should be considered as an integral part of the wood nanostructure. Understanding the effects of water absorption and desorption on cell wall nanostructure is of practical importance as the wood–water interactions regulate wood mechanical properties, durability, and dimensional stability during the drying process.33 Moreover, taking advantage of the hygroscopic properties of wood allows the creation of new functional materials.34,35
In this work, we have used solid-state NMR to characterize a fully 13C labeled P. radiata sapwood sample in never-dried, oven-dried, and rehydrated states. These observations were supported using a molecular dynamics simulation approach to present a coherent picture describing the presence of water at the cellulose–hemicellulose interface and both the reversible and the irreversible changes to the cell wall architecture contributing to the hysteresis of wood.
Experimental Section
Production and 13C-Labeling of P. radiata Cuttings
Small stem cuttings (∼5 cm) were produced by tree nursery Kools Sierteeltkwekerij (Deurne, The Netherlands) from 2-year-old P. radiata trees. The cuttings were placed on small pots (9 × 9 cm2) filled with a mixture of 1/3 vermiculite, 1/3 perlite, and 1/3 rockwool. After 7 months, callus tissue had developed, and the cuttings were then transferred to the labeling facility of IsoLife bv (Wageningen, The Netherlands) where they were labeled with 13C for 6 months in an atmosphere containing 13CO2 (99 atom % 13C).
Sample Preparation
After taking the P. radiata plantlets (10–15 cm) out of the labeling facility, the stems were cut underwater. Around 500 mg of cell walls containing sapwood was cut into small pieces and split into two samples. Half of the sample was stored in deionized water, and the other half was oven-dried at 105 °C to constant weight. Once the oven-dried sample was analyzed, the rotor was open, and deionized water was added to saturate the wood materials and left to equilibrate for 48 h. Once equilibrated, the excess surface water was carefully removed and the rotor was capped before analysis.
NMR
All pine samples were packed into a 3.2 mm Magic-Angle Spinning NMR rotor. The water-edited cross-polarization (CP) experiment, 30 ms CP-1H driven spin diffusion (PDSD), and 1000 ms CP-PDSDs were all performed on Bruker (Karlsruhe, Germany) Avance III solid-state NMR spectrometers operating at 1H and 13C Larmor frequencies of 500.1 MHz and 125.8 MHz, respectively. The direct polarization (DP) and CP-Incredible Natural Abundance DoublE QUAntum Transfer Experiments (INADEQUATE) for the hydrated pine samples were performed on Bruker (Karlsruhe, Germany) Avance III solid-state NMR spectrometers operating at 1H and 13C Larmor frequencies of 700.1 and 176.0 MHz, respectively. The remaining experiments were all performed on Bruker (Karlsruhe, Germany) Avance II+ solid-state NMR spectrometers operating at 1H and 13C Larmor frequencies of 600.1 and 150.9 MHz, respectively. Experiments were conducted at room temperature at MAS frequencies between 10 and 12.5 kHz unless otherwise stated. The 13C chemical shift was determined using the carbonyl peak at 177.8 ppm of l-alanine as an external reference with respect to tetramethylsilane (TMS); 90° pulse lengths were typically 3.0–3.5 μs (1H) and 3.0–4.0 μs (13C). Both 1H–13C CP with ramped (70–100%) 1H rf amplitude and a contact time of 1 ms and DP were used to obtain the initial transverse magnetization.36 The CP experiments emphasize the more rigid material, while a short, 2 s, recycle delay DP experiment was used to preferentially detect the mobile components and a 20 s delay was used for quantitative experiments. This delay was chosen after measuring the 13C T1 of the never-dried, oven-dried, and rehydrated pine samples (Table S1). SPINAL-64 decoupling was applied during acquisition at a 1H nutation frequency of 70–80 kHz.37 The proximity of water to different plant components was studied using a water-edited CP experiment.30,38 The CP parameters were as stated above, with the proton filter time being 3 ms and the recovery delay was varied from 1 to 100 ms. Intermolecular contacts were probed using 2D 13C–13C PDSD experiments with mixing times of 30 ms, 400 ms, and 1 s.39 The acquisition time in the indirect dimension (t1) of the CP-PDSD experiments was 6–8 ms. The sweep width in the indirect dimension was between 40 and 50 kHz with 48 acquisitions per t1 for CP-PDSD experiments, and the recycle delay was 2 s. Two-dimensional double quantum (DQ) correlation spectra were recorded using both a DP and a CP refocused INADEQUATE pulse sequence. This relies on scalar J coupling to correlate directly covalently bonded carbon nuclei.40−42 The acquisition time in t1 was between 5 and 7 ms and the carbon 90 and 180 pulse lengths were 4 and 8 μs, respectively. The 2τ spin-echo evolution times for a (π-τ-π)/2 spin echo of 4.66 ms and SPINAL-64 1H decoupling was applied during both the evolution and signal acquisition periods. The sweep width in the indirect dimension was 48–50 kHz with 160–192 acquisitions per t1 and recycle delay of 2 s. The 2D experiments were obtained by Fourier transformation into 4k (F2) × 2k (F1) points with an exponential line broadening of 50 Hz (CP) in F2 and squared sine bell processing in F1. All spectra obtained were processed and analyzed using Bruker Topspin version 3.6.
Modeling
Starting structures for mannan–cellulose and xylan–cellulose complexes were built using Cellulose-Builder.43 The 18-chain 2-3-4-4-3-2 fibril model of cellulose Iβ was considered with unit cell parameters of P. radiata (a = 0.795 nm, b = 0.847 nm, c = 1.034 nm, and γ = 99.2°) and a degree of polymerization of 24. A hemicellulose (mannan or xylan) chain with a degree of polymerization of 16 was placed on the (110) cellulose face. The xylan chain was parallel to the cellulose chains, whereas mannan and cellulose chains were antiparallel, as described elsewhere.44 The mannan chain was acetylated at oxygen O2 in residues 5, 8, 11, and 14 and at oxygen O3 in residues 3, 7, and 13. The xylan chain contained α-(1,2)-linked glucuronic acids at residues 3 and 9 and α-(1,3)-linked arabinoses at residues 1, 7, and 13. The axial hydroxyl groups and branches were adjusted using the CHARMM36 force field internal coordinates.45,46 Each system was simulated under fully hydrated conditions and in vacuum. A 16 Å thick water layer surrounded the carbohydrates in the fully hydrated system. To achieve charge neutrality in the xylan–cellulose complex, a calcium ion was added to the aqueous phase. For simulations in vacuum, two sodium ions were used instead.
Molecular dynamics (MD) simulations were performed using NAMD,47 with the CHARMM36 force field for carbohydrates (Guvench, 2009; Raman, 2010) and the TIP3P water model,48 under periodic boundary conditions. The mannose acetylation force field parameters were generated using CHARMM General Force Field (CGenFF) program.49,50 The electrostatic interactions were treated using the particle mesh Ewald method with a grid spacing of 1 Å and short-range interactions with a cutoff radius of 12 Å.51 The temperature was kept at 300 K with a Langevin thermostat, whereas the pressure was controlled at 1 bar with a Langevin piston for the hydrated systems.48 The vacuum simulations were performed at constant volume with a box length of 250 Å. The simulations were performed with a time step of 2 fs, and chemical bonds involving hydrogen atoms were constrained.
The systems were submitted to simulation stages: (1) 1000 steps of energy minimization and 100 ps of MD with all atoms from cellulose and C1, C2, C3, C4, C5, and O5 atoms from hemicellulose fixed and (2) 1000 steps of energy minimization and 1000 ps of MD with C1, C2, C3, C4, C5, and O5 atoms from cellulose and hemicellulose fixed. Finally, 50 and 90 ns for in vacuum and solvated systems, respectively, of MD were generated with only C1, C2, C3, C4, C5, and O5 cellulose atoms restrained with a force constant of 20 kcal mol–1 Å–2 to avoid distortions. In-house codes and VMD were used to perform the analyses.52
Results and Discussion
NMR 1D Spectra
Proximity of Cell Wall Components to Water in a Never-Dried Softwood
To determine which cell wall components are the closest to water, a water-edited cross-polarized (CP) NMR experiment was performed. In this, the signal coming from the magnetization of water protons diffuses to nearby carbons so that at short diffusion times the signal from carbons closest to water builds up first. Figure 1 shows a comparison of the spectra for a short (4 ms) and a long (100 ms) diffusion time together with the standard CP magic-angle spinning (MAS) spectrum of the cellulose region of the never-dried pine sample (141% moisture content (%MC)).
where %MC = moisture content, mwet = mass of wet wood, and mdry = mass of wood oven-dried to constant weight at 105 °C.
Figure 1.
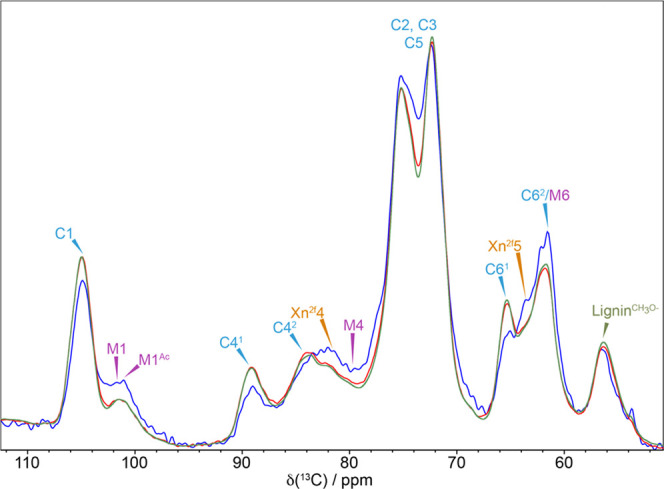
Proximity of secondary cell wall polysaccharides to water. A comparison of the water-edited 13C NMR spectrum of never-dried pine for two different diffusion times, 4 ms (blue) and 100 ms (red), together with a standard 13C CP-MAS NMR spectrum (green). The water-edited 4 ms spectrum have been multiplied by four to allow comparison. The polysaccharides that have a relatively enhanced signal at the short (4 ms) diffusion time are the components of the secondary cell wall that are closest to water. Spectra were recorded at a 13C Larmor frequency of 125.8 MHz and a MAS frequency of 10 kHz.
The major peaks are cellulose with the doubling of the C4 and C6 peaks reflecting two different glucose environments in the cellulose microfibril, which we refer to as domains 1 and 2.53 The standard CP-MAS and the water-edited 100 ms spectra are very similar. However, in the 4 ms spectrum, the xylan signals at ∼82 ppm (Xn2f4) and at ∼64 ppm (Xn2f5) and the mannan M4 peak at 79.9 ppm and the M1 peak at 101.0 ppm are very prominent compared to the CP-MAS spectrum indicating their proximity to water. There is also a more prominent signal at 61.5 ppm, which has contributions from M6 of mannan and a domain 2 C6 (C62) of cellulose.15 While much of this is likely to come from the mannan M6, the cellulose C62 is also likely to contribute as the C62 signal at ∼62.5 ppm is also enhanced. In contrast, the cellulose domain 1 is further from water than the hemicelluloses since the relative peak intensities of both the C41 at ∼89 ppm and C61 at ∼65 ppm are much reduced in the 4 ms spectrum, consistent with the view that cellulose within the fibril interior contributes to domain 1. The water-edited spectra highlight that hemicelluloses are closest to water and are therefore most likely to be significantly affected by the drying and rehydration processes.
Overview of Changes Induced by a Dehydration–Rehydration Cycle in a Never-Dried Softwood Cell Wall
To gain an overview of the major molecular changes that occur when a softwood secondary cell wall goes from a never-dried (141% moisture content) to an oven-dried state and is then rehydrated (157% moisture content), we compared quantitative 1D 13C DP-MAS NMR spectra of a never-dried pine sample, after complete drying and then after rehydration. The spectra, shown in Figure 2, were normalized to the same total integral area. The drying process significantly broadens the cellulose and hemicellulose region, while lignin (see Figures S1 and S2) appears to be less affected, with minimal broadening observed in the oven-dried spectrum. Broadening of the spectra is generally associated with less organized structures or decreased mobility. One of the key differences seen in the oven-dried pine sample spectrum is the mannan M1 peak at 101.0 ppm. This peak becomes significantly broader and shifts to ∼101.5 ppm such that it is no longer fully resolved becoming a shoulder on the ∼105 ppm cellulose C1 peak. The other major change visible in Figure 2 is that the composite C62/M6 peak shifts from 61.5 to 62.5 ppm for the oven-dried sample. While most of this change is likely to be due to the M6 of mannan, there could also be changes in the C6 cellulose domain 2 as the two peaks overlap.
Figure 2.
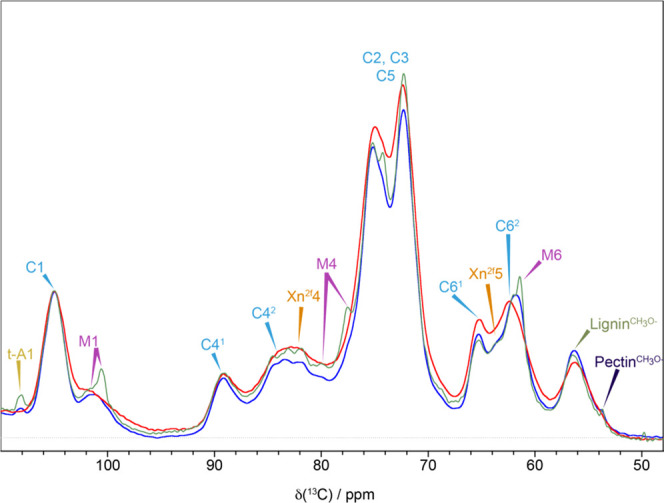
Differences in the quantitative 1D 13C NMR spectrum of pine in 3 different hydration states. A comparison of the polysaccharide region of the quantitative 1D 13C DP-MAS NMR spectra of never-dried pine (blue), oven-dried pine (red), and rehydrated pine (green). Spectra have been normalized to the same total integrated intensity and were recorded at a 13C Larmor frequency of 150.7 MHz and a MAS frequency of 12 kHz.
The main changes observed on drying appear to be reversed on rehydration of the pine sample. Indeed, after rehydration, most of the lines revert to their original never-dried width and position with the 1D CP-MAS spectra of the never-dried and rehydrated pine being almost identical (Figure S2). However, some significant differences can be seen in the 1D DP-MAS spectrum with, for example, the peaks from mannan becoming significantly narrower, indicating increased mobility of this component of the cell wall. The major difference appeared to be in the mannan and arabinose, with the most affected peaks being the mannan M1, M4, and M6 and the arabinose (Araf) A1 at ∼108 ppm. While the Araf peak only narrows on rehydration, the rehydrated mannan M1 shift changes from 101.0 ppm in the never-dried sample to 100.4 ppm. This clearly shows that some hemicellulose molecules are not fully reverting to their original conformation after the first dehydration and rehydration cycle.
NMR 2D Spectra
To obtain further insights on conformation and relative proximity between different components of the secondary cell walls, 2D NMR experiments were performed, both CP-PDSD with short (30 ms) and long (400 and 1000 ms) mixing times and CP and DP refocused INADEQUATE. The PDSD experiment reveals proximity between carbons since magnetization is transferred by the strongly distance-dependent dipole–dipole interaction. For short mixing times, cross-peaks are only between close carbons typically, in this case, within the same sugar residue, whereas for much longer mixing times, cross-peaks can be observed up to a maximum distance of ∼5–10 Å.
Molecular Changes in Cell Wall After Dehydration
Proximity Changes Between Molecules After Dehydration
Figure 3 shows an overlay of the 400 ms CP-PDSD spectrum of the never-dried and oven-dried pine together with slices extracted at the mannan M1, xylan Xn2f4, and lignin CH3O shifts. Slices of the mannan M1 at ∼101.0 ppm for the never-dried pine and oven-dried pine samples are compared in Figure 3a. All slices were normalized to the diagonal peak (i.e., the M1 peak for mannan in this case). The cross-peaks from M1 at 101.0 ppm to C1 at 105.0 ppm and to C42 at 83.0 ppm are larger in the oven-dried sample, showing that the oven-dried mannan is significantly closer to the cellulose than in the never-dried material. Similarly, there is a significant increase in the xylan cross-peaks from Xn2f4 (slice 82.2 ppm, Figure 3b) to M1 at 101 ppm and to the C41 and C42 peaks at 89.5 and 83.7 ppm, respectively. This indicates that xylan is closer to both mannan and cellulose in the oven-dried sample. Finally, a comparison of the slices taken at the lignin methoxy shift, 56.4 ppm, Figure 3c, shows a clear cross-peak at 105.0 ppm (the C1 position for cellulose) in the oven-dried lignin slice, whereas no peak is seen for the never-dried pine. In the never-dried pine sample, lignin–cellulose cross-peaks are only visible in a longer mixing time PDSD spectrum (e.g., 1000 ms Figure S3). This indicates that the lignin is significantly closer to the cellulose in the oven-dried sample. Overall, the decrease in distance between the matrix components surrounding the cellulose microfibril indicates a shrinkage of its structure at the molecular scale. It also suggests that interstitial water is present at the interface between cellulose and hemicelluloses. This shrinkage of the matrix-encapsulated microfibril is consistent with observations at nano and macroscale of drying wood using several different techniques (volumetric, atomic force microscopy, synchrotron, wide-angle X-ray scattering).32,54,55
Figure 3.

Relative proximities of the major components of the secondary cell wall. Left-hand side: A comparison of 400 ms 13C CP-PDSD NMR spectra of never-dried pine (blue) and oven-dried pine (red). Right-hand side: Slices are taken from the PDSD spectrum to highlight the key differences in cellulose, hemicellulose, and lignin proximities between never-dried and oven-dried softwood: (a) slice at the mannan M1 shift, 101 ppm; (b) slice at the xylan X4 shift, 82 ppm; and (c) slice at the lignin methoxy shift, 56 ppm. Spectra were recorded at a 13C Larmor frequency of 150.7 MHz and a MAS frequency of 12 kHz.
Conformational Changes After Dehydration
As all of the matrix elements surrounding the microfibrils are closer to the cellulose in the dehydrated sample, we investigated the impact on their conformation with a short mixing time experiment. Figure 4 shows the 30 ms CP-PDSD spectrum of the never-dried pine and oven-dried pine. There are small changes in the M1 and M6 shift of the mannan with the acetylated M1 and M6 mannan carbon shift increasing from 100.8 and 61.4 ppm to 101.1 and 62.0 ppm, respectively, on drying. The M4-acetylated mannan shift also changes slightly from 80.4 to 79.9 ppm, while the remaining mannan shifts appear unchanged by the drying process. We observed that the acetylated mannosyl residue conformation is more affected by the removal of water than the unacetylated residue conformation. Acetates are a bulky moiety, and they influence mannan backbone flexibility and can lead to aggregation upon drying.56,57 Small changes in the domain 2 cellulose carbon 6 region are also visible in the 30 ms CP-PDSD spectrum. However, a CP-refocused INADEQUATE spectrum (Figure 5a), which correlates carbons that are covalently bonded, provides a better resolution of the C6 cellulose environments and highlights the substantial changes between the many C6 environments of the never-dried and oven-dried pine. After drying, cellulose domain 2 (which is mainly surface cellulose) no longer has several resolved sites in the C6 region. Instead, they have merged into one large broad peak at ∼62.5 ppm. One of the main environments in the never-dried sample, labeled C62B at 61.7 ppm, almost wholly disappears, while the C62A environment at 62.0 ppm is significantly reduced. This is shown most clearly when comparing the sum of the 1D slices for the C5–C6 part of the spectrum shown in Figure 5b. The loss of the two distinct domain 2 cellulose environments coincides with an increase in the domain 1 cellulose signal. This implies that upon drying some of the domain 2 cellulose becomes more similar to domain 1.
Figure 4.

Conformational changes of the polysaccharides during drying. A comparison of 30 ms 13C CP-PDSD NMR spectra of never-dried pine (blue) and oven-dried pine (red). The 30 ms 13C CP-PDSD NMR spectra show the changes in the chemical shift of both mannan and the C6 of cellulose. The spectra were recorded at a 13C Larmor frequency of 125.8 MHz and a MAS frequency of 10 kHz.
Figure 5.

Key changes in the cellulose environments during the dehydration–rehydration cycle. (a) Comparison of the C5–C6 region of 13C CP-INADEQUATE NMR spectra of never-dried pine (blue) and oven-dried pine (red). The gray box indicates the region where the slices have been summed to give the line shapes shown on the right. (b) Comparison of the sum of slices taken in the C5–C6 of the 13C CP-refocused INADEQUATE NMR spectra for never-dried pine (blue), oven-dried pine (red), and rehydrated pine (green). The never-dried pine spectrum was recorded at a 13C Larmor frequency of 176.0 MHz and a MAS frequency of 12.5 kHz. The oven-dried and rehydrated pine spectra were recorded at a 13C Larmor frequency 150.7 MHz and a MAS frequency of 12 kHz. The spin-echo duration used was 2.2 ms.
Further evidence for some change in conformation can be seen in the INADEQUATE slices taken at double quantum shifts of 157.2 ppm (to emphasize C42) and 161.4 ppm (where both C41 and C42 signals can be seen) (Figure S4). On drying, the C42 peaks broaden significantly, and the ratio of C41 to C42 appears to change in favor of the C41 environment. Terminal Araf is also affected by drying as Figure 5a shows that the t-A4 signal at 85 ppm is absent and the t-A5 is scarcely visible. Thus, the arabinose conformation and mobility is also very sensitive to the absence of water. Recently, it was reported that xylan Araf signals in an INADEQUATE experiment were reduced by lyophilization and rehydration of sorghum.58
Molecular Changes in Cell Wall on Rehydration
Proximity Changes Between Molecules During Rehydration
The proximities between molecules in the rehydrated samples were compared to those obtained in the never-dried pine samples. The distance between cellulose and lignin increases compared to the dried state, as shown by comparing the 400 ms PDSD lignin slices (Figure S5a). For mannan (Figure S5c), the cross-peaks to cellulose are all weaker in the rehydrated sample compared to the never-dried sample. This suggests extra water being between the cellulose and some of the mannan. Therefore, at least a portion of the mannan moved further from the cellulose after rehydration than in the never-dried state.
In contrast, there were no significant changes in the proximity of xylan to cellulose between the oven-dried and the rehydrated sample. Therefore, the xylan remains closer to cellulose than in the never-dried pine sample since the cross-peaks to cellulose are all stronger (Figure S5b). These differences indicate that xylan is not as readily rehydrated as mannan. However, as the lignin regains its distance with the cellulose, it is expected that the overall matrix-encapsulated microfibril recovers its original dimension.
Conformational Changes During Rehydration
The changes in conformation seen in cellulose in the oven-dried sample appear to be mostly reversible upon rehydration. The rehydrated sample sees a return of the two domain 2 C6 cellulose environments (Figure 5b), although the change in the relative proportion of domain 1 and domain 2 cellulose indicates that cellulose does not fully return to its initial never-dried state (Figure 5b). Similarly, a comparison of the 30 ms CP-PDSD spectra of the never-dried and rehydrated pine (Figure S6) shows that most of the mannan molecules return to their original state upon rehydration. However, although the M1–M6 cross-peak at (∼101, ∼61.4 ppm) is again resolved, it is less intense. To further explore the changes happening upon rehydration, a 2D DP-INADEQUATE experiment with a short recycle time was performed. This DP spectrum, which allows the more mobile components to be investigated in more detail, reveals substantial changes. The spectrum (Figure S7) shows that a significant proportion of mannan became mobile. The shifts of this mannan fraction are similar to those of mannan in solution, with the largest shift change being for M4, which goes from 80.4 to 77.7 ppm (Table S2). This shift difference is similar, although smaller, to that observed for xylan in going from a two-fold screw conformation when bound to cellulose to a three-fold screw when unbound or in solution.20 It indicates that a fraction of the mannan becomes more mobile upon rehydration, allowing its backbone conformation to be altered. This would suggest that the rehydrated mannan has reduced interactions with the cellulose surface compared to its never-dried state. In contrast to mannan, no change is observed in any xylan carbon shift upon drying or rehydration of the pine samples, indicating that xylan conformation is independent of the surrounding water. In addition, as seen in the 1D DP spectrum of the rehydrated sample, the t-Araf signal narrows and it is again visible in both the DP-refocusedINADEQUATE (Figure S7) and the CP-refocused INADEQUATE (Figure S8) spectra.
When a fully dried softwood is rehydrated, most of the secondary cell wall changes are reversible, but a few are not. Mannan rehydrates, but a fraction of the mannan is no longer in its original position, close to the cellulose. Moreover, this fraction becomes more mobile with an altered backbone conformation. The hydration layer between cellulose and xylan is not able to reform. This indicates an irreversible aggregation of the xylan onto the cellulose surface.
Molecular Dynamics
Building the Cellulose–Hemicellulose Model
To support and help visualize the NMR results, we performed molecular dynamics (MD) simulations of softwood cellulose and hemicellulose environment in the presence and absence of water. To do so, the (110) surface of an 18-chain Iβ cellulose fibril with a 2-3-4-4-3-2 habit (Figure 6a) was complexed with a single hemicellulose (mannan or xylan) chain. Arabinoglucuronoxylans (AGX) consist of a β-(1,4)-linked-d-xylopyranosyl backbone with substitutions of α-(1,3)-l-arabinofuranose units and α-(1,2)-glucopyranosyl uronic acid (GlcA). Galactoglucomannans (GGM) have a mixed backbone of β-(1,4)-mannopyranosyl (Man) and β-(1,4)-glucopyranosyl residues (Glc). The mannose units can be substituted with α-(1,6)-linked galactopyranosyl (GalA) residues and O acetylated at the carbon 2 and 3 positions. Recent studies have provided a detailed picture of the structures, arrangements, and interactions of the major softwood hemicelluloses in the secondary cell wall.15 For AGX, at least two organizational patterns have been described. The main domain reveals an even spacing of GlcA and Araf side chains along the backbone. The GlcA are placed every 6th xylose residues and the Araf at -2 from the GlcA toward the nonreducing end. This substitution pattern allows xylan to strongly interact with the hydrophilic (and hydrophobic) cellulose surfaces in a two-fold screw conformation.19 The other less abundant AGX domain is decorated with GlcA on two consecutive xylose residues.59 This second domain might be preferentially interacting with lignin.59
Figure 6.

Cellulose–hemicellulose model used in the MD simulations. (a) 18-chain cellulose fibril with a 2-3-4-4-3-2 habit and a hemicellulose chain adsorbed at the (110) face. (b) Mannan chain model. (c) Xylan chain model.
So far, for GGM no molecular backbone nor acetylation patterns have been discerned. However, at least two populations of GGM have been described based on their recalcitrance and overall composition:59 a mannan population with low recalcitrance contains high mannose and a relatively low glucose content in the backbone, a high degree of acetylation, and a low degree of galactose substitutions (Man/Glc/Gal = 4:1:0.3 and DSAc = 0.4); while a GGM with low mannose acetylation, high glucose and galactose content (Man/Glc/Gal = 3:1:1.2 and DSAc = 0.1) is highly recalcitrant. Recently, the glucomannan backbone was described with a Man/Glc ratio of 2.16:1.60 The mannan backbone (Figure 6b) was therefore built with ∼30% of glucose (5 residues) and 70% of mannoses (11 residues) randomly distributed (Man/Glc = 2.2:1). Three mannosyl residues were acetylated at O3 atoms on the same side of the chain to avoid steric effects with the cellulose fibril, and four mannosyl residues were acetylated at O2 atoms, randomly distributed. No galactose side chain was added. This mannan configuration (Man/Glc/Gal = 2.2:1:0 and DSAc = 0.3) is close to the less recalcitrant fraction of mannan described by Martínez-Abad et al.59 To create a two-fold screw xylan compatible with cellulose, the chain model (Figure 6c) was decorated with two α-(1,2)-linked GlcA spaced by 6 xylosyl residues and three α-(1,3)-linked arabinoses placed at two xylosyl residues toward the nonreducing end from the GlcA.19
Hemicellulose Simulation in Solvated or Vacuum (Anhydrous) Systems
During the entire duration of the MD simulations, both mannan and xylan chains remain stably bound onto the cellulose surface. The root mean square displacements (RMSD) for the 10 center residues indicate that the hemicellulose chains maintain their conformation around the initial state (Figure S9a,b) and the sum of the dihedral torsions ϕ (C4-O4-C1-O5) and Ψ (C1-O4-C4-C5) for the 10 center residues confirmed that mannan and xylan adsorbed on the cellulose (110) surface are stretched and adopt a two-fold screw-like conformation in fully hydrated and anhydrous (vacuum) simulations (Figure S9c–f).19Figure 7a,b shows representative snapshots of the mannan–cellulose complex in water and in vacuum, respectively. The mannan chain remains in a similar conformation in both the hydrated and anhydrous systems, except for the O3 acetylations that increase their cellulose interactions in the absence of water. Figure 7c,d shows snapshots of the xylan–cellulose complex. In the anhydrous state, the entire xylan chain tilts ∼45° around its main axis and leans on the cellulose surface. In the anhydrous system relative to the hydrated system, while the normalized frequency of hydrogen bonds between xylan and cellulose increases, the hydrogen-bond frequency between mannan and cellulose remains similar (Figure S10a–d).
Figure 7.

Cross-sectional view of a representative simulation snapshot of the mannan–cellulose complex in (a) solvated and (b) anhydrous (vacuum) systems and the xylan–cellulose complex in (c) solvated and (d) anhydrous systems. (e) Interaction energy distribution between the hemicellulose chain and cellulose fibril for hydrated and anhydrous systems. (f) Interaction energy distribution between the O3 and O2 acetylations from the mannan chain and the cellulose fibril for hydrated and anhydrous systems. (g) Distribution of torsion angles ϕ + Ψ of the O3-acetylated Man residues in hydrated and anhydrous systems. Representative snapshots showing the interstitial water molecules in (h) mannan and (i) xylan for hydrated systems.
The mannan–cellulose interactions are little affected by the presence of water, except the interaction between the O3 acetylations and cellulose, which become stronger under anhydrous conditions (Figure 7b,f). The approximation of the O3 acetylation to the cellulose surface requires a local distortion of the main chain. This distortion led to a shift in the sum of torsion angles ϕ and Ψ of the O3-acetylated Man residues (Figure 7g), which peak around 145° and 156° in hydrated and anhydrous systems, respectively. No such shift is observed for the nonacetylated or O2-acetylated Man residues (Figure S9g). Xylan–cellulose interactions are stronger in the absence of water than in the hydrated system (Figure 7e). This reflects the leaning of the xylan chain against the cellulose surface in the absence of water (Figure 7d).
In the initial structures from which the simulations started, the backbone atoms of the hemicellulose chain occupied positions as if they were part of a larger cellulose Iβ crystal and the water molecules that comprised the hydration layer surrounded the hemicellulose–cellulose complex. As the simulations proceeded toward equilibrium, interstitial water molecules appeared between the hemicellulose and the neighboring cellulose chain and occupied a series of well-defined positions, bridging between hemicellulose and cellulose, as shown in Figure 7h. Interstitial water 1 is hydrogen bonding both hydroxymethyl groups from the mannan and cellulose chains, while interstitial water 2, 3, and 4 are hydrogen bonding the OH2 from cellulose and the axial O2, O2-acetyl, and equatorial OH2 from the mannan chain. Similarly, Figure 7i shows the interstitial water molecules between the xylan chain and cellulose. In this case, the interstitial water molecules play the role of the missing hydroxymethyl groups of the xylosyl residues. These water molecules are hydrogen bonding the O3 or both O3 and OH2 from cellulose and OH2 or O2 from the xylan chain. The interstitial waters are intermittent, so they exchange with the bulk continuously during simulations. The average water residence time at the interstitial positions is ∼0.5 ns for mannan–cellulose and 3 ns for xylan–cellulose complexes. These results indicate that the rate of interconversion between “bound” (interstitial) and “free” (bulk) states of water is roughly 1 order of magnitude slower for xylan–cellulose in comparison to mannan–cellulose. This difference can be explained by the fact that when a xylan chain is adsorbed onto a 110-cellulose face, the absence of hydroxymethyl groups on xylosyl residues creates spaces, which end up being occupied by a single water molecule migrating from the water bulk. This interstitial water is not hydrogen-bonded to the remaining water molecules. In contrast, the interstitial water molecules bridging the mannan–cellulose interfaces remain part of the hydrogen-bond network of the bulk water. However, both hydration layers are comparable to other biomolecular systems such as proteins and micelles and concentrated saccharide aqueous solutions.61,62
Discussion
The use of experimental solid-state NMR alongside MD simulations has provided a detailed and coherent picture of the effects of the first dehydration–rehydration cycle on the molecular architecture of green, never-dried wood. The 1D water-edited NMR experiments highlighted that the hemicelluloses were likely to be the focus of the changes due to their proximity to water, with 2D NMR demonstrating that water is an integral and key component of softwood molecular architecture. Based on these results, an updated model of the cellulose microfibril and hemicelluloses of softwood that includes water and the effect of the first drying and rehydration cycle on a never-dried cell wall is shown in Figure 8. The model based on that presented by Terrett et al.15 but with the inclusion of water displays a cellulose microfibril with a 2-3-4-4-3-2 habit surrounded by hemicelluloses in two-fold screw conformation and lignin nanodomains. The polysaccharide ratio is close to that reported in the literature.59,60 The never-dried model shows the hemicelluloses on the same microfibril face with interstitial water between them and the cellulose (Figure 8a). The MD simulations found almost an order of magnitude difference in the interstitial water molecules residence times between the hemicelluloses. This indicated that the water molecules are more tightly bound when at the xylan–cellulose interface and so are characterized as bound water in the model. The difference in the properties of the interstitial water might be reflected in the tighter adsorption of the two-fold screw xylan onto cellulose compared to the acetylated mannan observed during a mild acid hydrothermal subcritical water extraction.59
Figure 8.

Schematic model of a cellulose microfibril with a hydrated matrix and the changes occurring during the drying and rehydration processes as interpreted from the experimental solid-state NMR and MD simulations. (a) Transversal section of a cellulose microfibril with a 2-3-4-4-3-2 habit surrounded by the hemicellulose and lignin matrix. The cellulose is surrounded by a continuum of water molecules due to the presence of hydronium ions as the pH of the softwood cell wall is around 5.5. The water molecules present at the cellulose xylan interface (outlined in red) are tightly bound and exchange with the bulk of the water molecules at a slow rate. AGX and GGM molecules close to the cellulose microfibrils are in a two-fold screw configuration. GGM is divided into two categories (Man/Glc/Gal = 3:1:1.2 and DSAc = 0.1) and (Man/Glc/Gal = 4:1:0.3 and DSAc = 0.4).26 Glucuronic acid interacts with the lignin complex via electrostatic interactions. (b) Upon complete dehydration, a general collapse of the matrix surrounding the cellulose microfibril is observed. It leads to shrinkage in size with the lignin moving closer to the cellulose. The xylose now directly interacts with the cellulose surface and GGM moves closer to the cellulose surface. While the xylose residues of xylan are barely affected, the arabinose side chains start interacting with surrounding molecules in an aspecific manner. (c) Upon rehydration as the lignin–cellulose and hemicellulose–cellulose distances increase, the microfibril unit expands with its size becoming comparable to its original value. However, some changes are observed between the never-dried state and rehydrated state. The changes are highlighted with red dashed circles. ① The xylan–cellulose interface is not rehydrated. ② The rewetting of the cellulose is almost complete, but some of the cellulose does not revert to a domain 2 conformation. ③ The GGM regains its original position. However, a fraction of the GGM molecules becomes more mobile with a configuration similar to that in solution. ④ The arabinose side chains become more ordered and some arabinose becomes more mobile.
The PDSD NMR experiments showed clearly that upon dehydration both hemicelluloses became closer to each other and to cellulose and that lignin also became closer to both cellulose and xylan, indicating that the matrix encapsulating the microfibril has shrunk. Furthermore, the mannan conformation was affected more drastically than xylan upon removal of water since its chemical shift, especially that of the acetylated mannosyl M1 and M6 carbons, is significantly altered. Upon drying, the model shows the removal of all of the water and the collapse of the hemicelluloses and their side chains onto the cellulose microfibril. The removal of the interstitial water in the oven-dried sample also resulted in a significant change in the C62 cellulose environments and a change in the ratio of domain 1 and domain 2 of cellulose. The significant shift and broadening seen for the domain 2 cellulose combined with an increase in the relative amounts of domain 1 could result from the hemicelluloses and their side chains collapsing onto the cellulose microfibril surface. The increase in domain 1 cellulose could also be due to a change in the C6 conformation from gauche-trans (gt) to trans-gauche (tg) on drying since this conformation is known to affect both the C6 and C4 shifts significantly.63 This is illustrated in the model by changing the number of cellulose chains in the microfibril classed as domain 1 and domain 2 (Figure 8b).
Upon rehydration, the reversibility of the changes seen in mannan and xylan and the recovery of their original positions are very different. The MD shows that the interaction between cellulose and mannan does not change depending on the hydration status, while in an anhydrous environment, xylan interacts more strongly with the cellulose surface. This is consistent with the NMR results for the rehydrated pine, which shows that most of the mannan returns to its hydrated position. Thus, the interstitial water layer present at the mannan–cellulose interface can reform, whereas the xylan remains close to the cellulose as it is much harder for this water layer to reform, as illustrated in Figure 8c. A portion of the rehydrated mannan also became significantly more mobile than in the never-dried sample such that for this portion the mannan backbone adopts a conformation close to that of mannan in solution.64 This solution-like portion of the mannan has a significant change in mobility and conformation, indicating that it is no longer bound to the cellulose as it was in the never-dried sample. Thus, this mannan is shown surrounded by water and no longer as close to the cellulose microfibril face. The different behavior of xylan and mannan implies that the hemicelluloses have a different function in softwood. Considering water as an intrinsic structural element and not solely a regulator of the cell wall architecture is likely to impact various applications such as the pulping and timber industry. Indeed, it is traditionally known that the use of freshly cut (never-dried) wood is key to obtain an optimal pulp quality via a thermo-mechanical pulping process.65 The use of wood chip with a moisture content below fiber saturation point leads to an increase in specific energy consumption and negatively influences all important pulp strength and fiber properties. Moreover, the production of nanocellulose and regenerated cellulose aims to obtain a relatively pure cellulose (nano)fiber. If the wood chips have been dried enough to lead to the xylan molecules’ strong adsorption onto the cellulose microfibrils, harsher chemical treatments will be needed to dislodge the hemicellulose.
In addition, it is likely that the sigmoidal sorption and desorption observed in the water sorption kinetics finds part of its origin in the differential rate of the individual sorption/desorption kinetics of mannan and xylan. Moreover, the irreproducible hysteresis phase from never-dried to first dehydration is likely to be linked to the nondesorption of xylan upon rehydration. The main changes that occur during the first dehydration–rehydration cycle are highlighted in the model presented in Figure 8. The nonrecovery of the water layer at the xylan–cellulose interface may be the driver of the second main change, which is the conversion of cellulose domain 2 into domain 1. The final change incorporated in the model is related to the destabilization of a portion of the GGM becoming more labile.
Conclusions
Water is an integral component of wood ultrastructure. We have shown, using pine, that water is present at the cellulose–hemicellulose interface with xylan and mannan being bound differently to interstitial water and changes on dehydration being much more pronounced for mannan. Wood does not fully recover its original molecular architecture upon rehydration. Xylan becomes more closely associated with cellulose. Some of the mannan become more mobile and has a different conformation from the never-dried material. This irreversibility means that to obtain proper knowledge of cell wall structure future studies should be carried out on never-dried material. Fully understanding wood–water interactions is necessary to optimize the high-performance timber buildings currently being developed.66 We now have a molecular basis for explaining the influence of cell wall dehydration on timber mechanical properties.
Acknowledgments
The authors would like to acknowledge Elspeth McRae who helped them securing the MBIE proposal that funded this work. The authors are very thankful to Ton Gorissen from Isolife who made this project possible.
Glossary
Abbreviations
- AGX
arabinoglucuronoxylan
- Araf
arabinose
- CesA
cellulose synthase
- CP
cross-polarized
- DP
direct polarization
- GalA
galactopyranosyl
- GDP
gross domestic product
- GGM
galactoglucomannan
- Glc
glucopyranosyl
- INADEQUATE
Incredible Natural Abundance DoublE QUAntum Transfer Experiment
- Man
mannopyranosyl
- MAS
magic-angle spinning
- MD
molecular dynamics
- PDSD
proton driven spin diffusion
- ss-NMR
solid-state nuclear magnetic resonance
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.1c00937.
Solid-state NMR comparisons of never-dried, oven-dried, and rehydrated pine cell wall, model of the conformation of mannan and xylan chains bound to cellulose under hydrated and anhydrous conditions, table of 13C spin lattice relaxation times and table of mannan carbon shifts (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. The concept was developed by S.H. and M.S. The methodology was developed by S.H., R.C., R.D., and M.S.S. The ss-NMR was carried out by R.C. and R.D. The molecular dynamic modeling was performed by C.S.P., and M.S.S. P.D., S.P.B., M.S., R.D., and S.H. supervised the experimental part of the joint project. R.C., M.S., and R.D. wrote the first draft. R.D., M.S., P.D., S.H., and M.S.S. took part in the review of the manuscript.
The authors acknowledge the funding received from the New Zealand Ministry of Business, Innovation and Employment (MBIE) Endeavour Fund (Contract No. C04X1707, Fibre Grand Design) for supporting this work. M.S.S. and C.S.P. thank the Sao Paulo Research Foundation—FAPESP (Grants 2013/08293-7 and 2015/25031-1). The European Research Council (ERC Starting Grant 639907 awarded to Józef Lewandowski) and the University of Warwick funded the 700 MHz Bruker Avance III spectrometer used to obtain the data in Figures S7 and S8.
The authors declare no competing financial interest.
Notes
DATA AVAILABILITY Unprocessed NMR data files are available from http://wrap.warwick.ac.uk/159020/.
Supplementary Material
References
- New Zealand Plantation Forest Industry. Facts and Figures 2018/19; New Zealand Forest Owners Association Inc.: Wellington, New Zealand, 2019. [Google Scholar]
- Keckes J.; Burgert I.; Frühmann K.; Müller M.; Kölln K.; Hamilton M.; Burghammer M.; Roth S. V.; Stanzl-Tschegg S.; Fratzl P. Cell-wall recovery after irreversible deformation of wood. Nat. Mater. 2003, 2, 810–813. 10.1038/nmat1019. [DOI] [PubMed] [Google Scholar]
- Kifetew G.; Thuvander F.; Berglund L.; Lindberg H. The effect of drying on wood fracture surfaces from specimens loaded in wet condition. Wood Sci. Technol. 1998, 32, 83–94. 10.1007/BF00702589. [DOI] [Google Scholar]
- Ross R. J. E.Wood Handbook: Wood as an Engineering Material. General Technical Report FPL-GTR-190; U.S. Forest Products Laboratory, 2010; p 509.
- Hailwood A. J.; Horrobin S. Absorption of water by polymers: Analysis in terms of a simple model. Trans. Faraday Soc. 1946, 42, B084–B102. 10.1039/tf946420b084. [DOI] [Google Scholar]
- O’Neill H.; Pingali S. V.; Petridis L.; He J.; Mamontov E.; Hong L.; Urban V.; Evans B.; Langan P.; Smith J. C. Dynamics of water bound to crystalline cellulose. Sci. Rep. 2017, 7, 11840 10.1038/s41598-017-12035-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar C.Wood-Water Relations; Springer-Verlag: Berlin, 1988; p 283. [Google Scholar]
- Pearson H.; Gabbitas B.; Ormarsson S. Equilibrium moisture content of radiata pine at elevated temperature and pressure reveals measurement challenges. J. Mater. Sci. 2013, 48, 332–341. 10.1007/s10853-012-6750-2. [DOI] [Google Scholar]
- Shi J.; Avramidis S. Water sorption hysteresis in wood: I review and experimental patterns–geometric characteristics of scanning curves. Holzforschung 2017, 71, 307–316. 10.1515/hf-2016-0120. [DOI] [Google Scholar]
- Pearson H.; Ormarsson S.; Gabbitas B. Nonlinear tensile creep behavior of radiata pine at elevated temperatures and different moisture contents. Holzforschung 2015, 69, 915–923. 10.1515/hf-2014-0240. [DOI] [Google Scholar]
- Fratzl P.; Burgert I.; Gupta H. S. On the role of interface polymers for the mechanics of natural polymeric composites. Phys. Chem. Chem. Phys. 2004, 6, 5575–5579. 10.1039/b411986j. [DOI] [Google Scholar]
- Jarvis M. C. Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos. Trans. R. Soc. A 2018, 376, 20170045 10.1098/rsta.2017.0045. [DOI] [PubMed] [Google Scholar]
- Kang X.; Kirui A.; Dickwella Widanage M. C.; Mentink-Vigier F.; Cosgrove D. J.; Wang T. Lignin-polysaccharide interactions in plant secondary cell walls revealed by solid-state NMR. Nat. Commun. 2019, 10, 347 10.1038/s41467-018-08252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom E.Wood Chemistry: Fundamentals and Applications.; Gulf Professional Publishing, 1993. [Google Scholar]
- Terrett O. M.; Lyczakowski J. J.; Yu L.; Iuga D.; Franks W. T.; Brown S. P.; Dupree R.; Dupree P. Molecular architecture of softwood revealed by solid-state NMR. Nat. Commun. 2019, 10, 4978 10.1038/s41467-019-12979-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrett O. M.; Dupree P. Covalent interactions between lignin and hemicelluloses in plant secondary cell walls. Curr. Opin. Biotechnol. 2019, 56, 97–104. 10.1016/j.copbio.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Nishimura H.; Kamiya A.; Nagata T.; Katahira M.; Watanabe T. Direct evidence for α ether linkage between lignin and carbohydrates in wood cell walls. Sci. Rep. 2018, 8, 6538 10.1038/s41598-018-24328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Fernando L. D.; Kirui A.; Deligey F.; Wang T. Solid-state NMR of plant and fungal cell walls: A critical review. Solid State Nucl. Magn. Reson. 2020, 107, 101660 10.1016/j.ssnmr.2020.101660. [DOI] [PubMed] [Google Scholar]
- Busse-Wicher M.; Li A.; Silveira R. L.; Pereira C. S.; Tryfona T.; Gomes T. C.; Skaf M. S.; Dupree P. Evolution of xylan substitution patterns in gymnosperms and angiosperms: Implications for xylan interaction with cellulose. Plant Physiol. 2016, 171, 2418–2431. 10.1104/pp.16.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons T. J.; Mortimer J. C.; Bernardinelli O. D.; Pöppler A.-C.; Brown S. P.; deAzevedo E. R.; Dupree R.; Dupree P. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat. Commun. 2016, 7, 13902 10.1038/ncomms13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczakowski J. J.; Yu L.; Terrett O. M.; Fleischmann C.; Temple H.; Thorlby G.; Sorieul M.; Dupree P. Two conifer GUX clades are responsible for distinct glucuronic acid patterns on xylan. New Phytol. 2021, 231, 1720–1733. 10.1111/nph.17531. [DOI] [PubMed] [Google Scholar]
- Shimizu K.; Hashi M.; Sakurai K. Isolation from a softwood xylan of oligosaccharides containing two 4-O-methyl-d-glucuronic acid residues. Carbohydr. Res. 1978, 62, 117–126. 10.1016/S0008-6215(00)83383-2. [DOI] [Google Scholar]
- Yamasaki T.; Enomoto A.; Kato A.; Ishii T.; Shimizu K. Structural unit of xylans from sugi (Cryptomeria japonica) and hinoki (Chamaecyparis obtusa). J. Wood Sci. 2011, 57, 76–84. 10.1007/s10086-010-1139-9. [DOI] [Google Scholar]
- Martínez-Abad A.; Berglund J.; Toriz G.; Gatenholm P.; Henriksson G.; Lindström M.; Wohlert J.; Vilaplana F. Regular motifs in xylan modulate molecular flexibility and interactions with cellulose surfaces. Plant Physiol. 2017, 175, 1579–1592. 10.1104/pp.17.01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalla R.; Hackney J.; Uhlin I.; Thompson N. Hemicelluloses as structure regulators in the aggregation of native cellulose. Int. J. Biol. Macromol. 1993, 15, 109–112. 10.1016/0141-8130(93)90007-9. [DOI] [PubMed] [Google Scholar]
- Hill S. J.; Franich R. A.; Callaghan P. T.; Newman R. H. Nature’s nanocomposites: a new look at molecular architecture in wood cell walls. N. Z. J. For. Sci. 2009, 39, 251–257. [Google Scholar]
- Thomas L. H.; Martel A.; Grillo I.; Jarvis M. C. Hemicellulose binding and the spacing of cellulose microfibrils in spruce wood. Cellulose 2020, 27, 4249–4254. 10.1007/s10570-020-03091-z. [DOI] [Google Scholar]
- Khodayari A.; Thielemans W.; Hirn U.; Van Vuure A. W.; Seveno D. Cellulose-hemicellulose interactions-a nanoscale view. Carbohydr. Polym. 2021, 270, 118364 10.1016/j.carbpol.2021.118364. [DOI] [PubMed] [Google Scholar]
- Charlier L.; Mazeau K. Molecular modeling of the structural and dynamical properties of secondary plant cell walls: influence of lignin chemistry. J. Phys. Chem. B 2012, 116, 4163–4174. 10.1021/jp300395k. [DOI] [PubMed] [Google Scholar]
- White P. B.; Wang T.; Park Y. B.; Cosgrove D. J.; Hong M. Water–Polysaccharide Interactions in the Primary Cell Wall of Arabidopsis thaliana from Polarization Transfer Solid-State NMR. J. Am. Chem. Soc. 2014, 136, 10399–10409. 10.1021/ja504108h. [DOI] [PubMed] [Google Scholar]
- Phyo P.; Gu Y.; Hong M. Impact of acidic pH on plant cell wall polysaccharide structure and dynamics: insights into the mechanism of acid growth in plants from solid-state NMR. Cellulose 2019, 26, 291–304. 10.1007/s10570-018-2094-7. [DOI] [Google Scholar]
- Hill S. J.; Kirby N. M.; Mudie S. T.; Hawley A. M.; Ingham B.; Franich R. A.; Newman R. H. Effect of drying and rewetting of wood on cellulose molecular packing. Holzforschung 2010, 64, 421–427. 10.1515/hf.2010.065. [DOI] [Google Scholar]
- Thybring E. E.; Kymäläinen M.; Rautkari L. Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci.Technol. 2018, 52, 297–329. 10.1007/s00226-017-0977-7. [DOI] [Google Scholar]
- Keplinger T.; Cabane E.; Berg J. K.; Segmehl J. S.; Bock P.; Burgert I. Smart Hierarchical Bio-Based Materials by Formation of Stimuli-Responsive Hydrogels inside the Microporous Structure of Wood. Adv. Mater. Interfaces 2016, 3, 1600233 10.1002/admi.201600233. [DOI] [Google Scholar]
- Grönquist P.; Wood D.; Hassani M. M.; Wittel F. K.; Menges A.; Rüggeberg M. Analysis of hygroscopic self-shaping wood at large scale for curved mass timber structures. Sci. Adv. 2019, 5, eaax1311 10.1126/sciadv.aax1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz G.; Wu X.; Smith S. O. Ramped-amplitude cross polarization in magic-angle-spinning NMR. J. Magn. Reson., Ser. A 1994, 110, 219–227. 10.1006/jmra.1994.1208. [DOI] [Google Scholar]
- Fung B.; Khitrin A.; Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Luo W.; Hong M. Conformational Changes of an Ion Channel Detected Through Water– Protein Interactions Using Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 2378–2384. 10.1021/ja9096219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takegoshi K.; Nakamura S.; Terao T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001, 344, 631–637. 10.1016/S0009-2614(01)00791-6. [DOI] [Google Scholar]
- Lesage A.; Auger C.; Caldarelli S.; Emsley L. Determination of through-bond carbon– carbon connectivities in solid-state NMR using the INADEQUATE experiment. J. Am. Chem. Soc. 1997, 119, 7867–7868. 10.1021/ja971089k. [DOI] [Google Scholar]
- Fayon F.; Massiot D.; Levitt M. H.; Titman J. J.; Gregory D. H.; Duma L.; Emsley L.; Brown S. P. Through-space contributions to two-dimensional double-quantum J correlation NMR spectra of magic-angle-spinning solids. J. Chem. Phys. 2005, 122, 194313 10.1063/1.1898219. [DOI] [PubMed] [Google Scholar]
- Lesage A.; Bardet M.; Emsley L. Through-bond carbon– carbon connectivities in disordered solids by NMR. J. Am. Chem. Soc. 1999, 121, 10987–10993. 10.1021/ja992272b. [DOI] [Google Scholar]
- Gomes T. C.; Skaf M. S. Cellulose-Builder: A toolkit for building crystalline structures of cellulose. J. Comput. Chem. 2012, 33, 1338–1346. 10.1002/jcc.22959. [DOI] [PubMed] [Google Scholar]
- Yu L.; Lyczakowski J. J.; Pereira C. S.; Kotake T.; Yu X.; Li A.; Mogelsvang S.; Skaf M. S.; Dupree P. The patterned structure of galactoglucomannan suggests it may bind to cellulose in seed mucilage. Plant Physiol. 2018, 178, 1011–1026. 10.1104/pp.18.00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O.; Hatcher E.; Venable R. M.; Pastor R. W.; MacKerell A. D. Jr CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman E. P.; Guvench O.; MacKerell A. D. Jr. CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses. J. Phys. Chem. B 2010, 114, 12981–12994. 10.1021/jp105758h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Dupree R.; Simmons T. J.; Mortimer J. C.; Patel D.; Iuga D.; Brown S. P.; Dupree P. Probing the molecular architecture of Arabidopsis thaliana secondary cell walls using two-and three-dimensional 13C solid state nuclear magnetic resonance spectroscopy. Biochemistry 2015, 54, 2335–2345. 10.1021/bi501552k. [DOI] [PubMed] [Google Scholar]
- Adobes-Vidal M.; Frey M.; Keplinger T. Atomic force microscopy imaging of delignified secondary cell walls in liquid conditions facilitates interpretation of wood ultrastructure. J. Struct. Biol. 2020, 211, 107532 10.1016/j.jsb.2020.107532. [DOI] [PubMed] [Google Scholar]
- Schroeder H. A. Shrinking and swelling differences between hardwoods and softwoods. Wood Fiber Sci. 2007, 4, 20–25. [Google Scholar]
- Pauly M.; Ramírez V. New insights into wall polysaccharide O-acetylation. Front. Plant Sci. 2018, 9, 1210 10.3389/fpls.2018.01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J.; Kishani S.; Morais de Carvalho D.; Lawoko M.; Wohlert J.; Henriksson G.; Lindström M. E.; Wågberg L.; Vilaplana F. Acetylation and Sugar Composition Influence the (In) Solubility of Plant β-Mannans and Their Interaction with Cellulose Surfaces. ACS Sustainable Chem. Eng. 2020, 8, 10027–10040. 10.1021/acssuschemeng.0c01716. [DOI] [Google Scholar]
- Gao Y.; Lipton A. S.; Wittmer Y.; Murray D. T.; Mortimer J. C. A grass-specific cellulose–xylan interaction dominates in sorghum secondary cell walls. Nat. Commun. 2020, 11, 6081 10.1038/s41467-020-19837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abad A.; Jiménez-Quero A.; Wohlert J.; Vilaplana F. Influence of the molecular motifs of mannan and xylan populations on their recalcitrance and organization in spruce softwoods. Green Chem. 2020, 22, 3956–3970. 10.1039/D0GC01207F. [DOI] [Google Scholar]
- Berglund J.; Azhar S.; Lawoko M.; Lindström M.; Vilaplana F.; Wohlert J.; Henriksson G. The structure of galactoglucomannan impacts the degradation under alkaline conditions. Cellulose 2019, 26, 2155–2175. 10.1007/s10570-018-1737-z. [DOI] [Google Scholar]
- Bagchi B. Water dynamics in the hydration layer around proteins and micelles. Chem. Rev. 2005, 105, 3197–3219. 10.1021/cr020661+. [DOI] [PubMed] [Google Scholar]
- Sonoda M. T.; Skaf M. S. Carbohydrate clustering in aqueous solutions and the dynamics of confined water. J. Phys. Chem. B 2007, 111, 11948–11956. 10.1021/jp0749120. [DOI] [PubMed] [Google Scholar]
- Horii F.; Hirai A.; Kitamaru R. Solid-state 13C-NMR study of conformations of oligosaccharides and cellulose. Polym. Bull. 1983, 10, 357–361. 10.1007/BF00281948. [DOI] [Google Scholar]
- Whitney S. E.; Brigham J. E.; Darke A. H.; Reid J. G.; Gidley M. J. Structural aspects of the interaction of mannan-based polysaccharides with bacterial cellulose. Carbohydr. Res. 1998, 307, 299–309. 10.1016/S0008-6215(98)00004-4. [DOI] [Google Scholar]
- Tyrväinen J.Wood and Fiber Properties of Norway Spruce and Its Suitability for Thermomechanical Pulping; The Society of Forestry in Finland–The Finnish Forest Research Institute: Finland, 1995; Vol. 249, pp 1–155. [Google Scholar]
- Thybring E. E.; Glass S. V.; Zelinka S. L. Kinetics of water vapor sorption in wood cell walls: state of the art and research needs. Forests 2019, 10, 704 10.3390/f10080704. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.