

Abstract
Cerebrosides (n-HexCer) and glycosphingosines (n-HexSph) constitute two sphingolipid subclasses. Both are comprised of a monosaccharide head group (glucose or galactose in mammalian cells) linked via either an alpha- or beta- glycosidic linkage to the sphingoid backbone (n = α or β, depending upon the nature of the linkage to the anomeric carbon of the sugar). Cerebrosides have an additional amide-bonded fatty acyl chain linked to the sphingoid backbone. While differentiating the multiple isomers (i.e., glucose vs. galactose, α- vs. β-linkage) is difficult, it is crucial for understanding their specific biological roles in health and disease states. Shotgun tandem mass spectrometry has been a powerful tool in both lipidomics and glycomics analysis but is often limited in its ability to distinguish isomeric species. This work describes a new strategy combining shotgun tandem mass spectrometry with gas-phase ion chemistry to achieve both differentiation and quantification of isomeric cerebrosides and glycosphingosines. Briefly, deprotonated cerebrosides, [n-HexCer−H]−, or glycosphingosines, [n-HexSph−H]−, are reacted with terpyridine(Terpy)-magnesium complex dications, [Mg(Terpy)2]2+ in the gas phase to produce charge-inverted complex cations, [n-HexCer−H+MgTerpy]+, or [n-HexSph−H+MgTerpy]+. The collision-induced dissociation (CID) of the charge-inverted complex cations leads to significant spectral differences between the two groups of isomers, α-GalCer, β-GlcCer, and β-GalCer for cerebrosides, and α-GlcSph, α-GalSph, β-GlcSph, and β-GalSph for glycosphingosines, which allows for isomer distinction. Moreover, we describe a quantification strategy with the normalized %Area extracted from selected diagnostic ions that quantify either three isomeric cerebroside or four isomeric glycosphingosine mixtures. Analytical performance was also evaluated in terms of accuracy, repeatability, and inter-day precision. Furthermore, CID of the product ions resulting from 443 Da loss from the charge-inverted complex cations ([n-HexCer−H+MgTerpy]+) is performed and demonstrated for localizing the double bond position on the amide-bonded monounsaturated fatty acyl chain in the cerebroside structure. The proposed strategy was successfully applied to the analysis of total cerebroside extracts from the porcine brain providing in-depth structural information of cerebrosides from a biological mixture.
Keywords: cerebrosides, glycosphingosines, tandem mass spectrometry, charge inversion, ion/ion reactions
Graphical Abstract
Introduction
Cerebrosides (HexCer) and glycosphingosines (HexSph) constitute two subclasses of glycosphingolipids. Both are comprised of a monosaccharide head group and a sphingoid base backbone, while HexCers also have a fatty acyl chain that links to the base with an amide bond.1 In mammalian systems, the de novo synthesis pathway generates most of the glycosphingolipids (GSLs) with either glucose or galactose as the head group. The sugars are linked to the sphingoid backbone via a beta-glycosidic linkage (β-linkage) or an alpha-glycosidic linkage (α-linkage), with the former dominating in mammalian cells.2–3 Due to the difficulty of differentiating isomers, they are usually reported as a single cerebroside, thereby preventing the recognition of possible differences in function of the isomers in a biological system.4–6
Advances in analytical techniques have led to some strategies for the differentiation of the diastereomerism between glucose and galactose head group on lipids, including liquid chromatography coupled with mass spectrometry (LC-MS)7–11 and shotgun mass spectrometry.12–13 The former methods usually require long separation times while the latter requires offline sample pretreatment steps. Recently, our group reported a shotgun mass spectrometry approach employing gas-phase ion/ion chemistry to differentiate and quantify diastereomeric pairs of glycosphingosines and cerebrosides in binary mixtures without recourse to condensed-phase derivatization. The reaction of deprotonated glycosphingolipids, [GSL−H]−, with magnesium-terpyridine complex dications, [Mg(Terpy)2]2+, leads to complex cations, [GSL−H + MgTerpy]+, that generate different fragmentation patterns upon ion-trap collisional-induced dissociation (CID), providing the ability to unambiguously identify diastereomeric pairs and their relative compositions in the mixtures.14
In recent decades, attention has been drawn to α-linked cerebrosides for their roles in mediating the immune system in various models.15 Alpha-galactosylceramides (α-GalCer), for example, have been demonstrated to be a substrate for type I natural killer T cells that could be used for regulating innate immunity by activating the costimulatory signals with both NKT cells and dendritic cells.16–17 This makes α-GalCer a potential drug motif for the immunotherapy of different autoimmune diseases and cancers.18 Therefore, while the natural de novo synthetic pathway of GSLs in mammals strongly disfavors α-glycosidic linked GSLs,19 differentiation of the anomeric α- and β- glycosidic linkages may be needed for various biomedical studies. We show below how an α-glycosidic linked GSL can be used as an internal standard for the absolute quantitation of isomeric β-glycosidic linked GSLs. Most of the studies that have involved α-GalCer identification relate to synthesis, wherein nuclear magnetic resonance (NMR) was used for the characterization of the products.20–22 However, it is often a challenge for NMR to analyze complex matrices often associated with biological samples (e.g., cell extracts). Recently, cryogenic gas-phase infrared (IR) action spectroscopy has been demonstrated to be able to distinguish anomeric glycosidic linkages in glycolipids. However, this requires both incorporation of specialized IR techniques with mass spectrometry and post-acquisition spectra fitting.23 Therefore, a simple mass spectrometry based strategy that can directly probe the anomeric glycosidic bond is needed.
Several strategies to differentiate the anomericity of the glycosidic bonds via mass spectrometry have been described. Most of the methods include cationization by different metal ions, such as alkali metal ions,24–25 alkaline earth metal ions,26 and transition metal ions,27–28 in which the fragmentation patterns yield different results after the activation of the metal ion-saccharide complexes. Salpin et al., for example, reported a lead ion adduction method in which the CID spectrum of [Pb(disaccharide)−H]+ gave different base peaks and fragmentation fingerprints from different glycosidic linkages.29 The specific ion type, [Pb(disaccharide)−H]+, is analogous to the charge-inverted complex cations from the gas-phase ion/ion reaction described herein. However, addition of metal salts to the sample solution often leads to variations in yields and increased complexity of the mass spectrum, which may cause additional analytical problems. Moreover, none of the above methods demonstrated the ability to identify different glycosidic linked isomers in a mixture.
Quantifying isomeric mixture components presents a further challenge. To our knowledge, there are no reported methods for differentiating and quantifying both α- and β-glycosidic linkages and their different head groups simultaneously for cerebrosides and glycosphingosines with shotgun mass spectrometry. Therefore, in this work we describe a shotgun mass spectrometry strategy using ion chemistry to chemically modify both cerebroside and glycosphingosine ions in the gas phase to differentiate stereoisomers and achieve both relative and absolute quantification with a single spiking test that requires less use of analytical standards.
In addition to the structural complexities associated with the saccharide head groups in glycosphingolipids, the amide-bonded fatty acyl chain can add to the overall challenge of fully characterizing the lipid species. For example, reports have suggested that signaling functions of sphingolipids are related to fatty acyl chain length.30–31 In the case of unsaturated fatty acyl chains, locating the double bond can be particularly challenging. The different fatty acyl side chains (i.e., chain length and degree and location of unsaturation) have also been shown to influence the effect of α-GalCer on immune response.32 Previous reports have demonstrated the use of reversed-phase LC-MS to differentiate the fatty acyl chain, but the poor ability for differentiating hydrophilic groups hinders the distinction of the monosaccharide head group.33–35 Besides orthogonal separation techniques, various dissociation methods including ultraviolet photodissociation (UVPD)36 and ozone induced dissociation (OzID)37–38 have been demonstrated to identify the double bond position of the unsaturated fatty acyl chain in glycosphingolipids. Using gas-phase ion chemistry coupled with ion-trap CID, our group has also presented the ability to identify the double positions in various lipid classes, including fatty acids,39–40 glycerolphospholipids,41–42 and fatty acid esters of hydroxy fatty acids.43 Therefore, in this work we also demonstrate that gas-phase ion chemistry coupled with tandem mass spectrometry (MS3) can differentiate the double bond position on the amide-bonded monounsaturated fatty acyl chain in cerebrosides. Overall, we present a shotgun strategy that couples gas-phase ion/ion chemistry with ion-trap CID to provide in-depth structural information (Figure 1) and the relative composition of cerebrosides and glycosphingosines in mixtures.
Figure 1.
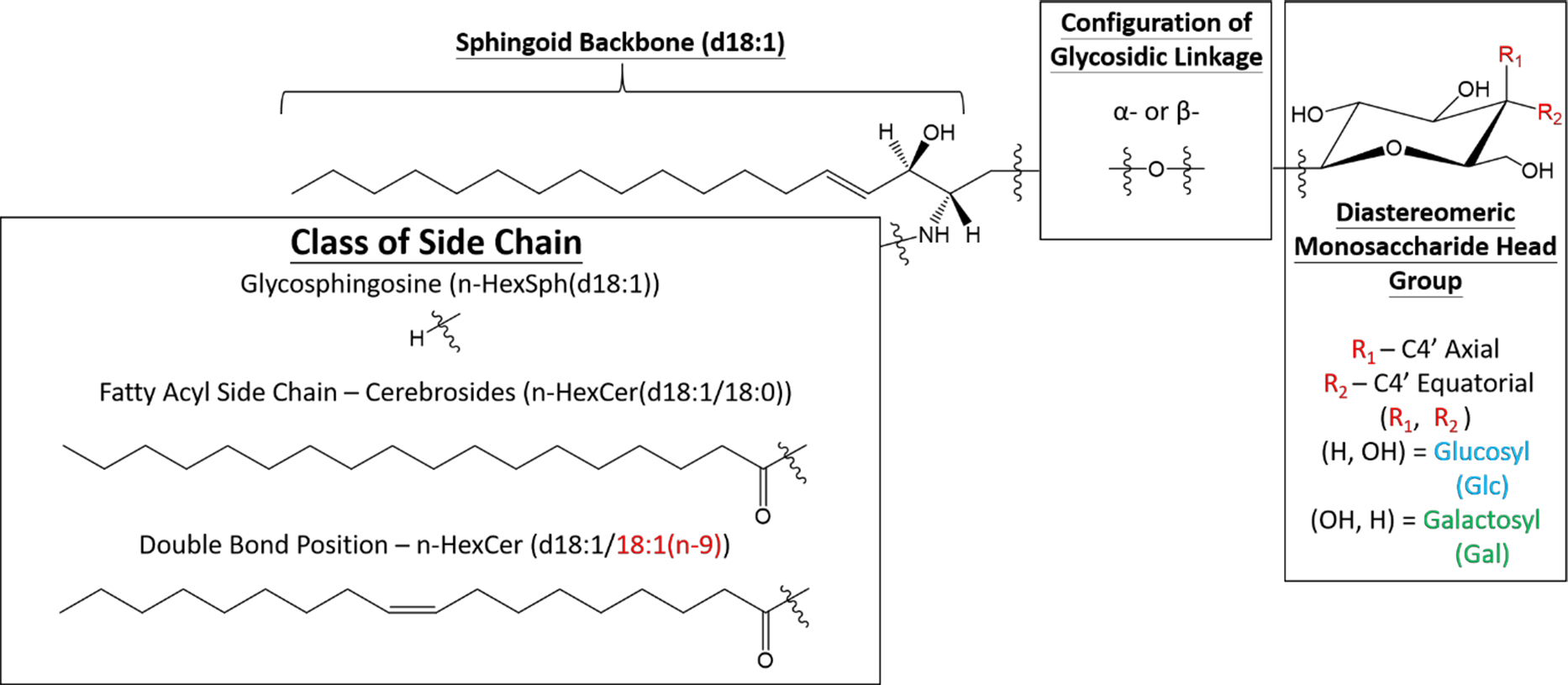
The general structures of glycosphingosines and cerebrosides with the possible isomeric positions within the structure.
Experimental Section
Materials.
All lipid standards and total cerebrosides extract (porcine brain) were purchased from Avanti Polar Lipids, Inc (Alabaster, AL). Magnesium chloride (MgCl2) and 2,2’;6’,2”-terpyridine (Terpy) were purchased from Sigma-Aldrich (St. Louis, MO). MS-grade water and methanol (MeOH) were purchased from Fisher Scientific (Pittsburgh, PA).
Sample Preparation.
Solutions of cerebrosides and glycosphingosine standards were prepared in MeOH to a final concentration of 0.01 mg/mL. MgCl2 and Terpy were mixed in methanolic solution with 1:1 (molar ratio) to a final concentration of ~50 μM for the metal-ligand complex.20 For relative quantification, different ratios of isomeric cerebrosides or glycosphingosine solutions were prepared, holding the final lipid concentration at 0.01 mg/mL. For total cerebrosides extract analysis, 1 mg of purified extract powder was dissolved in 1 mL of methanol as the stock solution and stored at −20°C before use. Prior to analysis, the lipid extract was diluted with MS grade MeOH to a final concentration of 0.01 mg/mL.
Mass Spectrometry.
All experiments were performed on a TripleTOF 5600 quadrupole time-of-flight mass spectrometer (SCIEX, Concord, ON, Canada) that has been modified for ion/ion reactions.44 Alternately pulsed dual nano-electrospray ionization (nESI) allows for the sequential injection of anions and cations.45 The experimental procedures were similar to reported previously.14 In short, lipid anions ([GSL−H]−) and metal-ligand reagent dications ([Mg(Terpy)2]2+) were alternately generated via nESI, mass-selected in Q1, and transferred to q2 for mutual storage (10–30 ms). Sequential resonance ejection ramps in q2 were used to mass-select targeted ion/ion reaction product ions for MSn experiments.46 Ion-trap CID was performed under the following conditions: q=0.2, AC amplitudes = 0.115V (n-HexCer complex); 0.078V (n-HexSph complex), activation time = 150 ms, and the CAD gas pressure set at 8 (estimated to be 8 mtorr). Mass analysis was performed via orthogonal acceleration time-of-flight (TOF).
Absolute Quantification.
To achieve absolute quantification, a single amount of α-GalCer(d18:1/16:0) was spiked into an aliquot of the total cerebrosides solution. In brief, a total of 1μL of 0.001 mg mL−1 of α-GalCer(d18:1/16:0) was added to a 99 μL aliquot of the cerebrosides solution. Three replicates were used to calculate the percentage of the all cerebroside isomers in the samples, and further back-calculated the absolute concentration in the total cerebrosides extracts solution using the known spiked amount of α-GalCer(d18:1/16:0) via equations 1 and 2:
| (eq. 1) |
| (eq. 2) |
where all the concentrations were expressed in μg mL−1, and can further back-calculate the amount of the cerebrosides in the total extract in the unit of ng mg−1. %α-GalCer, %β-GlcCer, and %β-GlcCer were the calculated percentages from the relative quantification.
Results and Discussion
Differentiation of glycosidic linkages and monosaccharide head group of cerebrosides via gas-phase ion chemistry.
We previously reported an ion/ion reaction shotgun tandem mass spectrometry strategy to identify the diastereomeric pairs of cerebrosides in binary mixtures.14 In summary, after producing the cerebroside complex cation, [HexCer−H + MgTerpy]+, via ion/ion reaction and subjecting it to ion-trap CID, specific diagnostic product ion spectra from both [GlcCer−H + MgTerpy]+ and [GalCer−H + MgTerpy]+ were generated allowing us to differentiate and quantify the diastereomeric pair in the sample. In this work, we further extend this strategy to investigate the ability of gas-phase chemistry (i.e., ion/ion reaction followed by CID) to differentiate the anomericity of the glycosidic linkages in cerebrosides. Figure 2 shows the gas-phase ion/ion reaction results and the mass spectra after ion-trap CID of the charge-inverted cerebroside complex cations, [α-GalCer(d18:1/16:0)–H + MgTerpy]+ (m/z 955.6), [β-GlcCer(d18:1/16:0)−H + MgTerpy]+ (m/z 955.6), and [β-GalCer(d18:1/16:0)–H + MgTerpy]+ (m/z 955.6). We note that an α-glucosylceramide analytical standard was not commercially available at the time of this report, and therefore, our data only examines α-galactosylceramide. Paralleling previous observations, we observed that product ions with neutral loss (NL) of a single Terpy ligand (m/z 740.6, NL = 233 Da) and its subsequent water-adducted product (m/z 722.6, NL = 215 Da) show the highest abundances in the CID spectrum of [β-GalCer(d18:1/16:0)–H + MgTerpy]+ (Figure 2(d)), and neutral loss of water (m/z 937.6) and neutral loss of 443 Da (Figure S1, m/z 512.3) are more prominent upon CID of [β-GlcCer(d18:1/16:0)–H + MgTerpy]+ (Figure 2(c)). It was also noticeable that neutral losses associated with the sugar (NL = 162 Da and NL = 180 Da) are more significant in the CID spectrum of [β-GlcCer(d18:1/16:0)–H + MgTerpy]+ than that of [β-GalCer(d18:1/16:0)–H + MgTerpy]+. Figure 2(b) shows an even more significant loss of sugar in the CID spectrum of [α-GalCer(d18:1/16:0)–H + MgTerpy]+, and there is almost no neutral loss of Terpy observed in the α-linked cerebroside spectrum.
Figure 2.

The comparison of the CID spectra among cerebrosides after gas-phase ion/ion reaction. (a) The post-ion/ion reaction spectrum of cerebroside anion with [Mg(Terpy)2]2+ cation. (b) The CID spectrum of the [α-GalCer–H+MgTerpy]+ (m/z 955.6). (c) The CID spectrum of the [β-GlcCer–H+MgTerpy]+ (m/z 955.6). (d) The CID spectrum of the [β-GalCer–H+MgTerpy]+ (m/z 955.6). The values inside the parenthesis indicate the neutral loss. The lightning bolt (🗲) signifies the collisionally activated precursor ion. The solid circle (●) indicates the mass selection in the negative ion mode analysis and the black and white squares (■/□) indicate the positive ion mode analysis with and without mass selection, respectively.
The ease with which the Terpy ligand is lost from a given complex ion is related to how well the cerebroside can stabilize the Mg2+ ion and therefore reflects the interactions of the cerebroside with the MgTerpy2+ adduct. We previously noted that the different orientations of the hydroxyl groups on the C3’ and C4’ positions on the sugar head group leads to differences in the stabilization of Mg2+, and therefore propensities for the loss of the Terpy ligand for the two β-isomers (compare Figures 2(c) and 2(d)).14 The α-galactosylceramide has a similar C3’-C4’ orientation to that of the β-galactosylceramide but no Terpy loss was observed from the α-galactosylceramide complex (compare Figure 2(b) to Figure 2(d)). Therefore, it is apparent that the two anomeric anions (viz., [α-GalCer−H]− and [β-GalCer−H]−) interact with the MgTerpy2+ adduct in distinct ways.
We measured the dissociation kinetics of the two charge-inverted galactosylceramide complex cations under a common set of activation conditions to determine their relative kinetic stabilities (see SI for a description of the dissociation rate measurement). Figure 3 shows that the [β-GalCer(d18:1/16:0)–H + MgTerpy]+ complex is significantly less stable (i.e. it fragments at a 4–5x greater rate) than the [α-GalCer(d18:1/16:0)–H + MgTerpy]+ complex. Furthermore, the comparison of Figures 2(b) and 2(d) shows that the two complexes differ dramatically in the Terpy loss fragmentation pathway (i.e., Terpy loss dominates for CID of [β-GalCer(d18:1/16:0)–H + MgTerpy]+ whereas it is absent in the case of [α-GalCer(d18:1/16:0)–H + MgTerpy]+). The β-GalCer(d18:1/16:0) anion clearly stabilizes the Mg2+ ion more than does the α-GalCer(d18:1/16:0) anion thereby facilitating Terpy loss. This observation reflects the fact that cation interaction effects can play a major role in the dissociation of carbohydrate ions.47 The distinct product ion spectra from the ion-trap CID of the charge-inverted cerebroside complex cations allow us to empirically differentiate the anomeric configuration of the glycosidic linkages between α- and β-, as well as the diastereomerism from the monosaccharide head group, glucose and galactose.
Figure 3.

The dissociation kinetic plot of isomeric charge-inverted galactosylceramide complex cations. Error bars are express with standard deviation (n=3). The p-value between the two slope is < 0.01 indicating the significantly different rate constant between the two complex cations. (The procedure for the dissociation rate measurement is provided in Supporting Information.)
Differentiation of glycosidic linkages and monosaccharide head group of glycosphingosines via gas-phase ion chemistry.
All four analytical standards, α-glucosylsphingosine (d18:1) (α-GlcSph), α-galactosylsphingosine (d18:1) (α-GalSph), β-glucosylsphingosine (d18:1) (β-GlcSph), and β-galactosylsphingosine (d18:1) (β-GalSph), were commercially available. Therefore, we performed the gas-phase ion/ion reaction with the deprotonated standards ([n-HexSph−H]−) and [Mg(Terpy)2]2+, followed by ion-trap CID (Figure 4). Analogous to the cerebrosides, the charge-inverted α-linked glycosphingosine complex cations show almost no Terpy loss upon ion-trap CID (Figures 4(a) and 4(b)). For [α-GalSph−H + MgTerpy]+ (Figure 4(b)), the prominent product ion at m/z 418 is consistent with sphingoid backbone loss (NL = 299 Da). In the case of [α-GlcSph−H + MgTerpy]+ (Figure 4(a)), the m/z 418 product ion is less abundant and an ion at m/z 400, likely a water loss following loss of the sphingoid backbone, is observed to be slightly more abundant than the m/z 418 product. Overall, using the sugar loss, Terpy loss, sphingoid base loss, and water loss following sphingoid base loss, it is possible to differentiate the four isomers experimentally via the gas-phase ion/ion reaction coupled with ion-trap CID.
Figure 4.
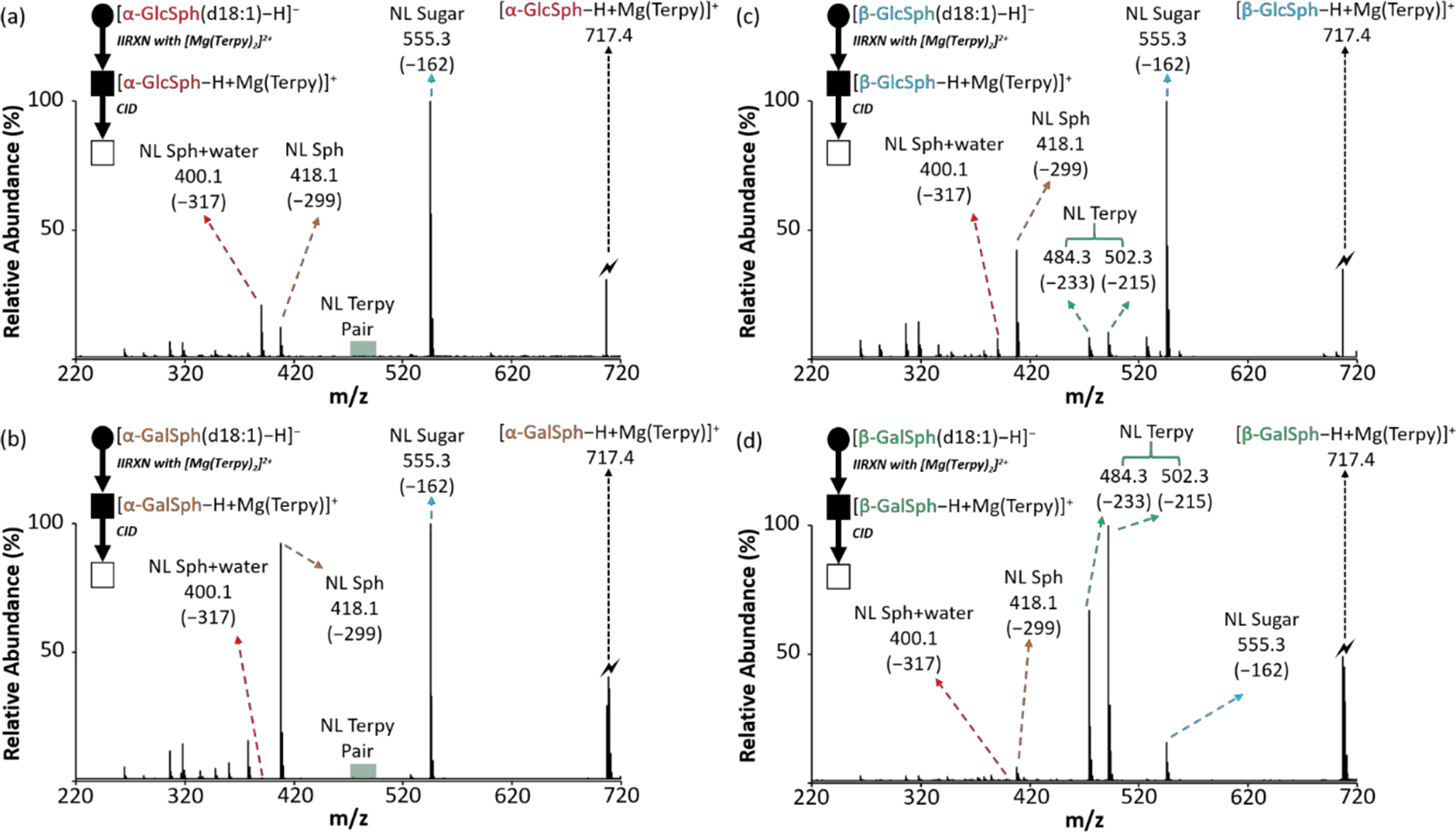
The comparison of the CID spectra among glycosphingosines after gas-phase ion/ion reaction. (a) The CID spectrum of the [α-GlcSph–H + MgTerpy]+ (m/z 717.4). (b) The CID spectrum of the [α-GalSph–H + MgTerpy]+ (m/z 717.4). (c) The CID spectrum of the [β-GlcSph–H + MgTerpy]+ (m/z 717.4). (d) The CID spectrum of the [β-GaSph–H + MgTerpy]+ (m/z 717.4). The values inside the parenthesis indicate the neutral loss. The symbols represent as same as those in Figure 2.
Relative quantification of the cerebroside and glycosphingosine isomers in a mixture.
It is typically challenging to do quantitative analysis with shotgun lipidomics, and it becomes more complicated with multiple isomers (e.g., more than two isomers). In our previous work, we demonstrated the use of a gas-phase ion/ion reaction combined with tandem mass spectrometry to quantify the relative composition of the diastereomeric pairs of both cerebrosides and glycosphingosines in binary mixtures.14 Here, we extend the strategy to quantify three and four commercially available isomers from cerebrosides and glycosphingosines, respectively, in a mixture.
First, the charge-inverted cerebroside complex cations are produced as described above, followed by ion-trap CID (Figure 2). In order to differentiate three isomers in the mixture, diagnostic product ions mentioned from the previous section were pooled into three classes; Terpy loss (NL = 233 Da and NL = 215 Da, NL 233 + NL 215), the ions associated with water and 443 Da loss (NL = 18 and NL = 443 Da, NL 18+NL 443), and the ions related to glycosidic bond cleavage (neutral loss of sugar, NL = 162 Da and NL = 180 Da, NL 162+NL 180). Commercially available isomers, α-GalCer(d18:1/16:0), β-GlcCer(d18:1/16:0), and β-GalCer(d18:1/16:0), were mixed to prepare various molar ratios among the isomers in the mixtures (Experimental section). The CID spectra of the fully dissociated precursor cation mixtures are provided in Figure S2.
To quantify the relative compositions of the isomers in the mixtures, the areas of the monoisotopic peaks from the diagnostic product ions were extracted and normalized to the total extracted peaks from the pure-component CID results. Table 1 shows the normalized %area (%A) of the three groups of product ions from fifteen replicates (five replicates per day for three days). The %A are placed in the following equations to calculate the percentage of three isomers in unknown samples:
| (eq. 3) |
| (eq. 4) |
| eq.5) |
Table 1.
The normalized %area from the pure components of cerebroside isomers.
| 100% CB | NL of Terpy (%, NL 215 + NL 233) | NL of water+443 (%, NL 18 + NL 443) | NL of sugar (%, NL 162 + NL 180) | SD* |
|---|---|---|---|---|
| α-GalCer(d18:1/16:0) | 0.2 | 56.3 | 43.5 | 0.03 |
| β-GlcCer(d18:1/16:0) | 41.6 | 49.1 | 9.3 | 0.2 |
| β-GalCer(d18:1/16:0) | 90.7 | 7.3 | 2.0 | 0.1 |
Standard deviation (SD) is obtained from the %A group with the lowest percentage.
The analytical performance in terms of accuracy, repeatability, and inter-day precision were also evaluated at various molar ratio of the isomeric mixtures (procedures can be found in the supporting information). Table S1 summarizes the relative quantification results with the analytical performance. The accuracies for relative quantification of the cerebrosides ranged from 94.9 to 105.2% from different molar ratios, with the highest SD around 3.2%. The results suggest the relative quantitation is achieved with the applied strategy. In addition, the inter-day precisions of the platform from various molar ratios are all below 5.4% RSD, indicating the relative quantification results obtained from different days were comparable.
As mentioned at the beginning of this section, increasing the number of isomers in a mixture complicates quantification with shotgun mass spectrometry. To our knowledge, no examples of quantifying more than three isomers simultaneously with shotgun lipidomics have been reported.39, 48–50 Therefore, we explored the ability of this strategy to quantify four glycosphingosine isomers, α-GlcSph(d18:1), α-GalSph(d18:1), β-GlcSph(d18:1), and β-GalSph(d18:1), in the mixture. A similar strategy from the cerebrosides section was applied and both the details and the equation set are shown in the supporting information. Table S2 shows the normalized %area and Table S3 summarizes the relative quantification results along with the analytical performances from the analysis of various mixtures. The accuracies for relatively quantifying the four isomeric glycosphingosines ranged from 84.2 to 114.0%, with the highest SD around 8.0%, and the inter-day precisions are all below 13.5% RSD. We note that it is crucial to have an accurate measurement of the relative abundances of the diagnostic ions to obtain accurate quantitative results. There is a higher variation for the quantification of the four isomers, which is most likely due to the low percentage from several ion groups in the pure component table (e.g., NL = 317 Da for both α-GalSph and β-GalSph), leading to an approximately 5% error when there is no other isomer in the sample. Overall, the above results show the applicability of the strategy to achieve relative quantification of three cerebroside isomers and four glycosphingosine isomers.
Identification of the double bond position on the fatty acyl side chain of cerebrosides via gas-phase ion chemistry.
In addition to the isomerism arising from the monosaccharide head group, the amide-bonded fatty acyl chain on the cerebrosides (Figure 1) adds to the structural diversity of this lipid class. A single platform that could provide structural information from both the monosaccharide head group and the fatty acyl chain would comprehensively cover the isomerism in cerebrosides. We previously reported a charge switching gas-phase ion/ion reaction strategy to identify the double bond position(s) on unsaturated fatty acids by reacting a deprotonated fatty acid with tris-phenanthroline magnesium dications followed by ion-trap CID.40 In brief, the charge-inverted fatty acid complex cations generate a spectral gap with 12 Da spacing at the corresponding double bond position upon ion-trap CID.40 This approach, in some cases with some variation in the overall workflow, has been extended to various classes of lipids containing fatty acyl chains including glycerolphospholipids,41 ether glycerolphospholipids,42 and fatty acid esters of hydroxy fatty acids.43
Instead of the tris-phenanthroline magnesium dication, we used the terpyridine magnesium dication as the charge switching reagent in the current work because, in the former case, ligand loss (180 Da) could be confused with one of the sugar neutral loss channels. After CID of the charge-inverted cerebroside complex cations (Figure 2), no abundant product ions directly reveal fatty acyl chain structural information. We previously proposed the structure of the ion generated by 443 Da loss (Figure S1)14 to be similar in structure to a charge-inverted fatty acid complex cation.40 Therefore, another round of ion isolation and CID was performed on the first generation product ions formed from 443 Da loss. Figure 5 shows the MS3 result derived from both β-GlcCer(d18:1/18:0) and β-GlcCer(d18:1/18:1). The product ion spectra of 443 Da loss ions reveal the informative spectral pattern previously noted for monounsaturated fatty acyl chains. An informative spectral gap and a 12 Da spacing at the double bond position can be observed in Figure 5(b) whereas saturated a fatty acyl chain only shows 14 Da spacings among the fragmented ions (Figure 5(a)). The standard obtained from the vendor suggests a double bond at the n-9 position on the fatty acyl chain, which agrees with our result. As expected, there is no significant difference between the MS3 spectra from analytical standards of β-GlcCer(d18:1/18:1(n-9)) and β-GalCer(d18:1/18:1(n-9)) (data not shown). The above results demonstrate the ability to identify double bond position from the amide-bonded monounsaturated fatty acyl side chain on cerebrosides using the gas-phase charge inversion ion/ion reaction with deprotonated cerebroside anions and terpyridine-magnesium dication.
Figure 5.
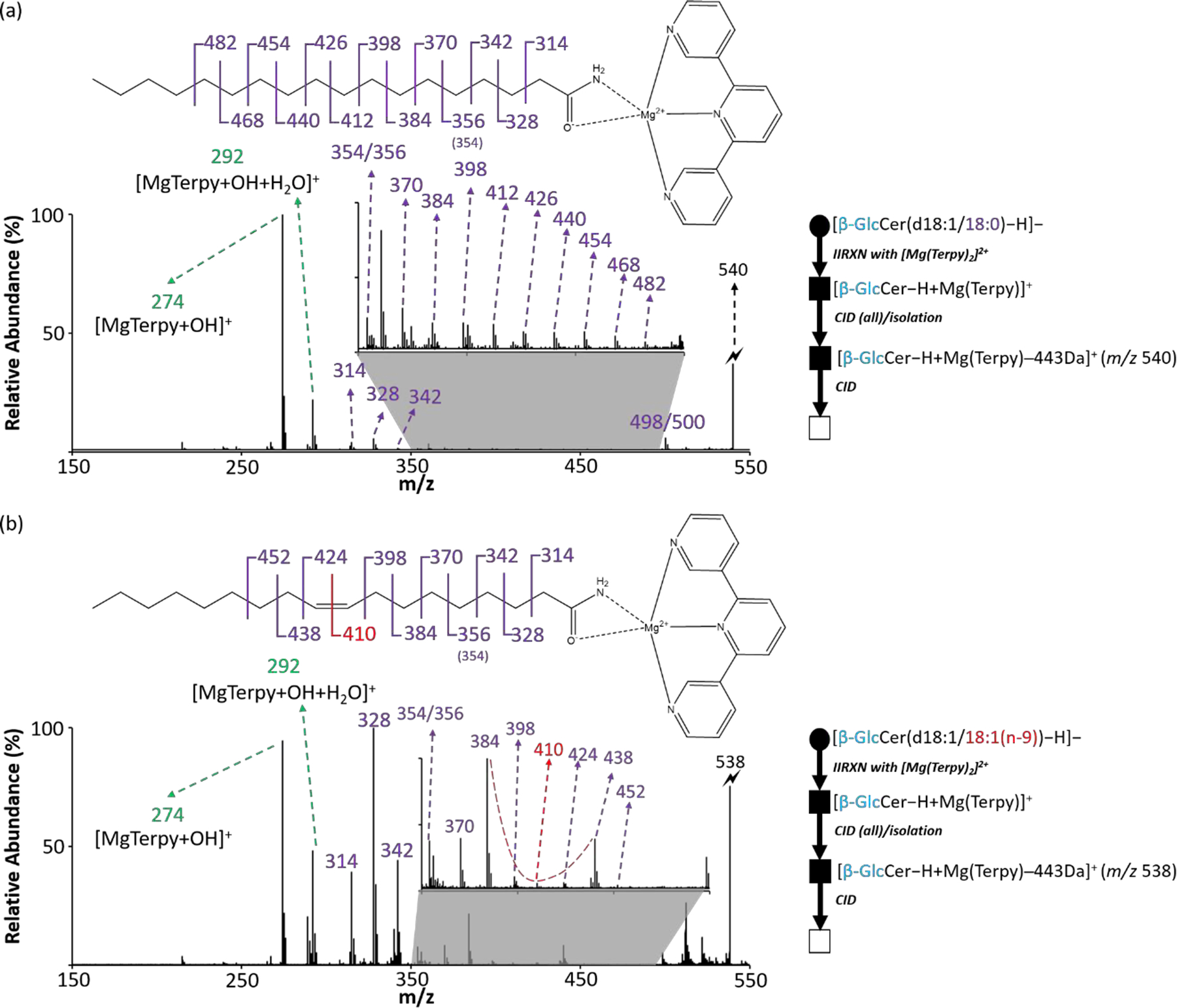
The identification of double bond position from the monounsaturated fatty acyl side chain on cerebrosides. (a) The CID spectrum of 443 Da loss ion from [β-GlcCer(d18:1/18:0)−H + MgTerpy]+. (b) The CID spectrum of 443 Da loss ion from [β-GlcCer(d18:1/18:1 (n-9))−H + MgTerpy]+. The inserts are the zoom-in spectra of m/z region ranged from 350 to 500. The red dashed line signifies the special spectral gap pointing the double bond position. The symbols represent as same as those in Figure 2.
Analysis of total cerebroside extract from porcine brain.
The strategy described here was applied to total cerebroside extracts from porcine brain. We previously profiled 14 different cerebrosides in the extracts.14 As indicated above, α-linked cerebrosides are present in bacteria or can be synthetic products but they are not prominent in mammalian systems.19 We therefore focused on the only available commercial α-linkage cerebroside standard, α-GalCer(d18:1/16:0), and its corresponding isomers in the brain extracts. Figure S4 shows the CID spectra of the m/z 955.6 precursor ion before and after spiking α-GalCer(d18:1/16:0) into the sample, and the top panel from Table 2 summarizes the relative quantities of the isomers in the sample. The pre-spiked results of the relative compositions of β-GlcCer(d18:1/16:0) and β-GalCer(d18:1/16:0), which were 11.9% and 89.3%, respectively, agreed with our previous report (10.9% and 89.1%). After spiking α-GalCer(d18:1/16:0) into the sample, the percentages among the three isomers change but the molar ratio for the two non-spiked isomers, β-GlcCer(d18:1/16:0) and β-GalCer(d18:1/16:0), remained the same, which is 0.133 (β-GlcCer(d18:1/16:0) to β-GalCer(d18:1/16:0)), suggesting that the spiking test would still be able to reflect their original relative composition, and to accurately quantify as low as 2% of the isomer. The result demonstrates the applicability of using the gas-phase ion chemistry to differentiate and quantify the three cerebroside isomers in the biological extract.
Table 2.
The analytical results of the cerebroside isomers in total brain extract.
| Quantification of Cerebroside Isomers | ||||
| n-HexCer (d18:1/16:0) | Relative Quantification | |||
| Test (N=5) | Calculated α-GalCer (%) * | Calculated β-GlcCer (%) | Calculated β-GalCer (%) | |
| Non-spiked test | ND** | 11.9 ± 2.8 | 89.3 ± 2.1 | |
| Spiked test | 80.1 ± 1.6 | 2.2 ± 0.6 | 16.60 ± 1.6 | |
| Absolute Quantification (N=5) | ||||
| Calculated β-GlcCer ng mg −1 *** | Calculated β-GalCer ng mg −1 | |||
| 27.9 ± 6.6 | 209.8 ± 24.2 | |||
| Profiled Cerebrosides with Monounsaturated Fatty Acyl Chain | ||||
| Profiled β-GalCer (total carbon number) | Fatty acyl chains (sphingoid backbone/side chain) | Double bond position (amide bonded side chain) | ||
| 36:2 | d18:1/18:1 | n−9 | ||
| 42:2 | d18:1/24:1 | n−8 and n−9 | ||
| 44:2 | d18:1/26:1 | n−6 | ||
Mean ± SD.
Not-detectable.
The concentrations of cerebrosides are expressed as per mg brain extract.
In addition to relative quantification, we also attempted to perform absolute quantification with this isomer species. By applying equations 3 and 4, we can back-calculate the absolute quantity of β-GlcCer(d18:1/16:0) and β-GalCer(d18:1/16:0) after spiking the known amount of α-GalCer(d18:1/16:0). Table 2 also shows the results of absolute quantification. This approach uses α-GalCer(d18:1/16:0) as an internal standard that is absent in the non-spiked sample. If all three isomers were to be present in the original sample, modification of the strategy would be needed (e.g., two-point spiking test with a revised equation set.) The proposed strategy has the advantage that it requires only a single analytical standard (i.e., α-GalCer(d18:1/16:0)) for the isomeric cerebroside group to achieve absolute quantification, thereby avoiding the need for a calibration curve for each isomer.
We also probed double bond position of selected cerebrosides from the porcine brain extract. Three cerebrosides that we profiled with a monounsaturated fatty acyl chain, including β-GalCer(d18:1/18:1), β-GalCer(d18:1/24:1), and β-GalCer(d18:1/26:1), were further subjected to MS3 experiments for the identification of the double bond position. Figures S5 and S6 show the CID spectra, and the bottom panel from Table 2 summarizes the results. Two of the cerebrosides, β-GalCer(d18:1/18:1) and β-GalCer(d18:1/26:1), proved to be dominated by a single component with unsaturation at the n-9 and n-6 positions, respectively. In the case of β-GalCer(d18:1/24:1), de novo spectral interpretation suggests the presence of two major components with unsaturation at either the n-8 or n-9 positions. The difference between the CID spectra from the standard of β-GalCer(d18:1/24:1(n-9)) (Figure S6(a)) and the corresponding isomer(s) from the brain extract sample (Figure S6(b)) suggest that, while the the n-9 isomer is present, another isomer with unsaturation at n-8 is also there. However, there is no analytical standard for β-GalCer(d18:1/24:1(n-8)) to allow for a clear validation of this result. We also did not profile any cerebrosides from the brain extracts or find standards with more than one double bond on the amide-bonded fatty acyl chain.
Conclusions
In this work, we demonstrated a shotgun tandem mass spectrometry approach involving gas-phase ion/ion chemistry and ion trap CID to provide in-depth structural information from both cerebrosides and glycosphingosines. The gas-phase ion/ion reaction between deprotonated cerebrosides ([n-HexCer−H]−) and [Mg(Terpy)2]2+ leads to charge-inverted complex cations, [n-HexCer−H + Mg(Terpy)]+. Ion trap CID of these ions yields distinctive product ion spectra for the three isomers, α-GalCer, β-GlcCer, and β-GalCer. The same [Mg(Terpy)2]2+ reaction with deprotonated glycosphingosines derived from four isomers, α-GlcSph, α-GalSph, β-GlcSph, and β-GalSph, also forms charge-inverted complex cations. Subsequent CID of these cations allows for each isomer to be identified and profiled. This strategy enables the distinction of monosaccharide head group diastereomers and anomeric glycosidic linkage to the sphingoid backbone. Moreover, relative quantification of three isomeric cerebrosides and four isomeric glycosphingosines mixtures is achieved by analyzing the normalized %area from the diagnostic product ions. The analytical performance for the quantification in terms of accuracy, repeatability, and inter-day precision is also reported.
The ion/ion reaction followed byion trap CID strategy has been extended to locate the double bond position on the cerebroside amide-bonded fatty acyl chain. The site of unsaturation in a cerebroside’s fatty acyl chain can be identified via an informative spectral gap and a characteristic 12 Da spacing at the double bond in an MS3 experiment on the 443 Da loss ion generated from the MS2 experiment of the cerebroside. A total cerebroside extract from porcine brain was subjected to these approaches. By spiking a known amount of α-GalCer(d18:1/16:0) into the extract as an internal standard, it is possible to generate both relative and absolute quantities of the β-GlcCer(d18:1/16:0) and β-GalCer(d18:1/16:0) isomers in the extract. Finally, we identified the sites of double bond location in four cerebrosides in the porcine brain extract with monounsaturated fatty acyl chains, including β-GalCer(d18:1/18:1(n-9)), β-GalCer(d18:1/24:1(n-8)), β-GalCer(d18:1/24:1(n-9)), and β-GalCer(d18:1/26:1(n-6)). However, a lack of calibration standards is a complication for quantifying cerebrosides with isomeric monounsaturated amide-bonded fatty acyl chains, and for identifying sites of unsaturation when multiple double bonds are present.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) under Grants GM R37-45372 and GM R01-118484.
Footnotes
Supporting Information
The Supporting Information is available free of charge at the ACS website.
The proposed structures of the fragment ions; CID spectra of the charge inversion complexes; MS3 spectra for double bond location identification (Figure S1 to S6); Relative quantification and analytical performance (Table S1 to S3) and supporting experimental procedures and equations.
References
- 1.Merrill AH, Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem Rev 2011, 111 (10), 6387–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breton C; Šnajdrová L; Jeanneau C; Koča J; Imberty A, Structures and mechanisms of glycosyltransferases. Glycobiology 2005, 16 (2), 29R–37R. [DOI] [PubMed] [Google Scholar]
- 3.Schnaar RL; Kinoshita T, Glycosphingolipids. In Essentials of Glycobiology [Internet], 3rd edition ed.; Varkim A; Cummings RD; Esko JD; Stanley P; Hart GW; Aebi M; Darvill AG; Kinoshita T; Packer NH; Prestegard JH; Schnaar RL; Seeberger PH, Ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2017. [PubMed] [Google Scholar]
- 4.Han X; Gross RW, Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev 2005, 24 (3), 367–412. [DOI] [PubMed] [Google Scholar]
- 5.Hakomori S. i., Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochim Biophys Acta Gen Subj 2008, 1780 (3), 325–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufresne M; Patterson NH; Norris JL; Caprioli RM, Combining Salt Doping and Matrix Sublimation for High Spatial Resolution MALDI Imaging Mass Spectrometry of Neutral Lipids. Anal Chem 2019, 91 (20), 12928–12934. [DOI] [PubMed] [Google Scholar]
- 7.Shaner RL; Allegood JC; Park H; Wang E; Kelly S; Haynes CA; Sullards MC; Merrill AH, Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res 2009, 50 (8), 1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scherer M; Leuthäuser-Jaschinski K; Ecker J; Schmitz G; Liebisch G, A rapid and quantitative LC-MS/MS method to profile sphingolipids. J Lipid Res 2010, 51 (7), 2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M; Tong X; Lv P; Feng B; Yang L; Wu Z; Cui X; Bai Y; Huang Y; Liu H, A not-stop-flow online normal-/reversed-phase two-dimensional liquid chromatography–quadrupole time-of-flight mass spectrometry method for comprehensive lipid profiling of human plasma from atherosclerosis patients. J Chromatogr A 2014, 1372, 110–119. [DOI] [PubMed] [Google Scholar]
- 10.Boutin M; Sun Y; Shacka JJ; Auray-Blais C, Tandem Mass Spectrometry Multiplex Analysis of Glucosylceramide and Galactosylceramide Isoforms in Brain Tissues at Different Stages of Parkinson Disease. Anal Chem 2016, 88 (3), 1856–1863. [DOI] [PubMed] [Google Scholar]
- 11.von Gerichten J; Schlosser K; Lamprecht D; Morace I; Eckhardt M; Wachten D; Jennemann R; Gröne H-J; Mack M; Sandhoff R, Diastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals. J Lipid Res 2017, 58 (6), 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han X; Cheng H, Characterization and direct quantitation of cerebroside molecular species from lipid extracts by shotgun lipidomics. J Lipid Res 2005, 46 (1), 163–175. [DOI] [PubMed] [Google Scholar]
- 13.Pham HT; Julian RR, Characterization of glycosphingolipid epimers by radical-directed dissociation mass spectrometry. Analyst 2016, 141 (4), 1273–1278. [DOI] [PubMed] [Google Scholar]
- 14.Chao H-C; McLuckey SA, Differentiation and Quantification of Diastereomeric Pairs of Glycosphingolipids Using Gas-Phase Ion Chemistry. Anal Chem 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama H; Nagafuku M; Suzuki A; Iwabuchi K; Inokuchi J-I, The regulatory roles of glycosphingolipid-enriched lipid rafts in immune systems. FEBS Lett 2018, 592 (23), 3921–3942. [DOI] [PubMed] [Google Scholar]
- 16.Kawano T; Cui J; Koezuka Y; Toura I; Kaneko Y; Sato H; Kondo E; Harada M; Koseki H; Nakayama T; Tanaka Y; Taniguchi M, Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Vα14 NKT cells. Proc Natl Acad Sci U.S.A. 1998, 95 (10), 5690–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossjohn J; Pellicci DG; Patel O; Gapin L; Godfrey DI, Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol 2012, 12 (12), 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar A; Suryadevara N; Hill TM; Bezbradica JS; Van Kaer L; Joyce S, Natural Killer T Cells: An Ecological Evolutionary Developmental Biology Perspective. Front Immunol 2017, 8, 1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W; Guillaume J; Baqi Y; Wachsmann I; Gieselmann V; Van Calenbergh S; Müller CE, Synthesis and structure-activity relationships of cerebroside analogues as substrates of cerebroside sulphotransferase and discovery of a competitive inhibitor. J Enzyme Inhib Med Chem 2020, 35 (1), 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plettenburg O; Bodmer-Narkevitch V; Wong C-H, Synthesis of α-Galactosyl Ceramide, a Potent Immunostimulatory Agent. J Org Chem 2002, 67 (13), 4559–4564. [DOI] [PubMed] [Google Scholar]
- 21.Du W; Gervay-Hague J, Efficient Synthesis of α-Galactosyl Ceramide Analogues Using Glycosyl Iodide Donors. Org Lett 2005, 7 (10), 2063–2065. [DOI] [PubMed] [Google Scholar]
- 22.Fan G-T; Pan Y.-s.; Lu K-C; Cheng Y-P; Lin W-C; Lin S; Lin C-H; Wong C-H; Fang J-M; Lin C-C, Synthesis of α-galactosyl ceramide and the related glycolipids for evaluation of their activities on mouse splenocytes. Tetrahedron 2005, 61 (7), 1855–1862. [Google Scholar]
- 23.Kirschbaum C; Greis K; Mucha E; Kain L; Deng S; Zappe A; Gewinner S; Schöllkopf W; von Helden G; Meijer G; Savage PB; Marianski M; Teyton L; Pagel K, Unravelling the structural complexity of glycolipids with cryogenic infrared spectroscopy. Nat Comm 2021, 12 (1), 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z; Ogden S; Leary JA, Linkage position determination in oligosaccharides: mass spectrometry (MS/MS) study of lithium-cationized carbohydrates. J Org Chem 1990, 55 (20), 5444–5446. [Google Scholar]
- 25.Cerda BA; Wesdemiotis C, Thermochemistry and structures of Na+ coordinated mono- and disaccharide stereoisomers. Int J Mass spectrom 1999, 189 (2), 189–204. [Google Scholar]
- 26.Fura A; Leary JA, Differentiation of calcium(2+)- and magnesium(2+)-coordinated branched trisaccharide isomers: An electrospray ionization and tandem mass spectrometry study. Anal Chem 1993, 65 (20), 2805–2811. [DOI] [PubMed] [Google Scholar]
- 27.Smith G; Leary JA, Differentiation of stereochemistry of glycosidic bond configuration: Tandem mass spectrometry of diastereomeric cobalt-glucosyl-glucose disaccharide complexes. J Am Soc Mass Spectrom 1996, 7 (9), 953–957. [DOI] [PubMed] [Google Scholar]
- 28.Smith G; Leary JA, Mechanistic Studies of Diastereomeric Nickel(II) N-Glycoside Complexes Using Tandem Mass Spectrometry. J Am Chem Soc 1998, 120 (50), 13046–13056. [Google Scholar]
- 29.Firdoussi AE; Lafitte M; Tortajada J; Kone O; Salpin JY, Characterization of the glycosidic linkage of underivatized disaccharides by interaction with Pb(2+) ions. J Mass Spectrom 2007, 42 (8), 999–1011. [DOI] [PubMed] [Google Scholar]
- 30.Grösch S; Schiffmann S; Geisslinger G, Chain length-specific properties of ceramides. Prog Lipid Res 2012, 51 (1), 50–62. [DOI] [PubMed] [Google Scholar]
- 31.Chao H-C; Lee T-H; Chiang C-S; Yang S-Y; Kuo C-H; Tang S-C, Sphingolipidomics Investigation of the Temporal Dynamics after Ischemic Brain Injury. J Proteome Res 2019, 18 (9), 3470–3478. [DOI] [PubMed] [Google Scholar]
- 32.Iwabuchi K; Nakayama H; Oizumi A; Suga Y; Ogawa H; Takamori K, Role of Ceramide from Glycosphingolipids and Its Metabolites in Immunological and Inflammatory Responses in Humans. Mediators Inflamm 2015, 2015, 120748–120748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitaker BD, Analysis of Plant Cerebrosides by C18 and C6 HPLC. In Physiology, Biochemistry and Molecular Biology of Plant Lipids, Williams JP; Khan MU; Lem NW, Eds. Springer Netherlands: Dordrecht, 1997; pp 143–145. [Google Scholar]
- 34.Merrill AH; Sullards MC; Allegood JC; Kelly S; Wang E, Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 2005, 36 (2), 207–224. [DOI] [PubMed] [Google Scholar]
- 35.Sullards MC; Liu Y; Chen Y; Merrill AH Jr., Analysis of mammalian sphingolipids by liquid chromatography tandem mass spectrometry (LC-MS/MS) and tissue imaging mass spectrometry (TIMS). Biochim Biophys Acta 2011, 1811 (11), 838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan E; Nguyen CQN; Shiea C; Reid GE, Detailed Structural Characterization of Sphingolipids via 193 nm Ultraviolet Photodissociation and Ultra High Resolution Tandem Mass Spectrometry. J Am Soc Mass Spectrom 2017, 28 (7), 1406–1419. [DOI] [PubMed] [Google Scholar]
- 37.Thomas MC; Mitchell TW; Harman DG; Deeley JM; Nealon JR; Blanksby SJ, Ozone-Induced Dissociation: Elucidation of Double Bond Position within Mass-Selected Lipid Ions. Anal Chem 2008, 80 (1), 303–311. [DOI] [PubMed] [Google Scholar]
- 38.Barrientos RC; Vu N; Zhang Q, Structural Analysis of Unsaturated Glycosphingolipids Using Shotgun Ozone-Induced Dissociation Mass Spectrometry. J Am Soc Mass Spectrom 2017, 28 (11), 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Randolph CE; Foreman DJ; Blanksby SJ; McLuckey SA, Generating Fatty Acid Profiles in the Gas Phase: Fatty Acid Identification and Relative Quantitation Using Ion/Ion Charge Inversion Chemistry. Anal Chem 2019, 91 (14), 9032–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Randolph CE; Foreman DJ; Betancourt SK; Blanksby SJ; McLuckey SA, Gas-Phase Ion/Ion Reactions Involving Tris-Phenanthroline Alkaline Earth Metal Complexes as Charge Inversion Reagents for the Identification of Fatty Acids. Anal Chem 2018, 90 (21), 12861–12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Randolph CE; Blanksby SJ; McLuckey SA, Toward Complete Structure Elucidation of Glycerophospholipids in the Gas Phase through Charge Inversion Ion/Ion Chemistry. Anal Chem 2020, 92 (1), 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Randolph CE; Shenault DSM; Blanksby SJ; McLuckey SA, Structural Elucidation of Ether Glycerophospholipids Using Gas-Phase Ion/Ion Charge Inversion Chemistry. J Am Soc Mass Spectrom 2020, 31 (5), 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Randolph CE; Marshall DL; Blanksby SJ; McLuckey SA, Charge-switch derivatization of fatty acid esters of hydroxy fatty acids via gas-phase ion/ion reactions. Anal Chim Acta 2020, 1129, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Y; Chrisman PA; Erickson DE; Liu J; Liang X; Londry FA; Yang MJ; McLuckey SA, Implementation of Ion/Ion Reactions in a Quadrupole/Time-of-Flight Tandem Mass Spectrometer. Anal Chem 2006, 78 (12), 4146–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia Y; Liang X; McLuckey SA, Pulsed Dual Electrospray Ionization for Ion/Ion Reactions. J Am Soc Mass Spectrom 2005, 16 (11), 1750–1756. [DOI] [PubMed] [Google Scholar]
- 46.McLuckey SA; Goeringer DE; Glish GL, Selective ion isolation/rejection over a broad mass range in the quadrupole ion trap. J Am Soc Mass Spectrom 1991, 2 (1), 11–21. [DOI] [PubMed] [Google Scholar]
- 47.Bythell BJ; Abutokaikah MT; Wagoner AR; Guan S; Rabus JM, Cationized Carbohydrate Gas-Phase Fragmentation Chemistry. J Am Soc Mass Spectrom 2017, 28 (4), 688–703. [DOI] [PubMed] [Google Scholar]
- 48.Hsu F-F, Mass spectrometry-based shotgun lipidomics - a critical review from the technical point of view. Anal Bioanal Chem 2018, 410 (25), 6387–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han X; Yang K; Gross RW, Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev 2012, 31 (1), 134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen IØ; Vidas Olsen A; Dicroce-Giacobini J; Papaleo E; Andersen KK; Jäättelä M; Maeda K; Bilgin M, Comprehensive Evaluation of a Quantitative Shotgun Lipidomics Platform for Mammalian Sample Analysis on a High-Resolution Mass Spectrometer. J Am Soc Mass Spectrom 2020, 31 (4), 894–907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.