

Abstract
Our ability to quickly detect and respond to harmful environmental stimuli is vital for our safety and survival. This inherent acute pain detection is a “gift” because it both protects our body from harm and allows healing of damaged tissues1. Damage to tissues from trauma or disease can result in distorted or amplified nociceptor signaling and sensitization of the spinal cord and brain (Central Nervous System; CNS) pathways to normal input from light touch mechanoreceptors. Together, these processes can result in nagging to unbearable chronic pain and extreme sensitivity to light skin touch (allodynia). Unlike acute protective pain, chronic pain and allodynia serve no useful purpose and can severely reduce the quality of life of an affected person. Chronic pain can arise from impairment to peripheral neurons, a phenomenon called “peripheral neuropathic pain.” Peripheral neuropathic pain can be caused by many insults that directly affect peripheral sensory neurons, including mechanical trauma, metabolic imbalance (e.g., diabetes), autoimmune diseases, chemotherapeutic agents, viral infections (e.g., shingles). These insults cause “acquired” neuropathies such as small-fiber neuropathies, diabetic neuropathy, chemotherapy-induced peripheral neuropathy, and post herpetic neuralgia. Peripheral neuropathic pain can also be caused by genetic factors and result in hereditary neuropathies that include Charcot-Marie-Tooth disease, rare channelopathies and Fabry disease. Many acquired and hereditary neuropathies affect the skin, our largest organ and protector of nearly our entire body. Here we review how cutaneous nociception (pain perceived from the skin) is altered following diseases that affect peripheral nerves that innervate the skin. We provide an overview of how noxious stimuli are detected and encoded by molecular transducers on subtypes of cutaneous afferent endings and conveyed to the CNS. Next, we discuss several acquired and hereditary diseases and disorders that cause painful or insensate (lack of sensation) cutaneous peripheral neuropathies, the symptoms and percepts patients experience, and how cutaneous afferents and other peripheral cell types are altered in function in these disorders. We highlight exciting new research areas that implicate non-neuronal skin cells, particularly keratinocytes, in cutaneous nociception and peripheral neuropathies. Finally, we conclude with ideas for innovative new directions, areas of unmet need, and potential opportunities for novel cutaneous therapeutics that may avoid CNS side effects, as well as ideas for improved translation of mechanisms identified in preclinical models to patients.
Keywords: cutaneous, neuropathic pain, keratinocytes, skin, nociceptor, neuropathy
I. Introduction: Landscape of our review
A living organism’s ability to quickly detect and respond to harmful environmental stimuli is vital for their safety and survival. This reflexive process functions to protect our body from harm and allow healing of damaged tissues. The first step in the perception of pain is called nociception, the process of detecting and encoding noxious stimuli. Nociception is mediated by a network of peripheral sensory nerve endings (nociceptors) which innervate the skin, muscle, tendons, fascia and visceral organs2,3 to enable the detection of potentially damaging environmental and endogenous stimuli. These nociceptors synapse onto central neurons (located in the dorsal spinal cord for the body or the brainstem for the head) which further process the nocifensive information and relay the signal to the brain. Here we will focus on cutaneous nociceptors and peripheral neuropathies that affect the skin.
Damage to tissues from trauma or disease can result in distorted or amplified nociceptor signaling. This amplified signaling is typically protective, preventing further damage and resolving once the underlying condition has healed. Sometimes however, pain can persist long after the initial tissue insult, serving no obvious beneficial purpose and severely reducing the quality of life of those affected. This is chronic pain, or pain that typically lasts longer than 6 months, sometimes for years. Chronic pain can be driven by the amplified nociceptor signaling itself, or by the secondary sensitization of CNS circuits that now in an amplified state, respond to light touch-sensitive afferents to signal pain (tactile allodynia). Chronic pain is especially common in states of injury and disease that directly affect the somatosensory nervous system, such as painful peripheral neuropathies. Neuropathic pain can result from a variety of common injury and disease states, including mechanical trauma, metabolic imbalance (e.g., diabetes), viral infections (e.g., shingles, AIDS), bacterial infections (e.g., leprosy which is caused by Mycobacterium leprae), alcoholism, and chemotherapy treatment (e.g., Taxol). Peripheral neuropathic pain involves multiple distressing sensory dysfunctions, including spontaneous pain (shooting, burning or stabbing pain; “pins and needles” feeling) and enhanced evoked pain responses to touch and cold stimuli. Despite the high prevalence of neuropathic pain (7–8% of the general population), treatment options are limited owing the diversity and complexity of the underlying biological mechanisms4.
The focus of this review is on how cutaneous nociception (pain perceived from the skin) is altered following diseases that affect peripheral nerves. Whereas many types of common acute and chronic pain are associated with deeper target tissues such as muscle, bone, fascia and visceral organs, the mechanisms and processes underlying deep tissue pain are reviewed elsewhere3,5–10. Importantly, the spinal cord, brainstem, cortex and descending pain faciliatory and inhibitory pathways are extensively involved in central sensitization11 and altered circuit signaling in painful peripheral neuropathies. For example, ectopic activity in sensitized sensory neurons drives enhanced input to the spinal cord; this ectopic activity occurs not only in nociceptive Aδ and C fibers, but can also occur in Aβ fibers12–19. The enhanced input to the spinal cord can then amplify spinal cord circuits which subsequently respond to normal inputs from low threshold, light touch myelinated (Aβ) afferents that gain access to pain pathways and convey signals to the brain that are interpreted as tactile allodynia (extreme touch sensitivity)20,21. Further details of the complex CNS sensitization circuits and their sensitization mechanisms are beyond the scope of this review and are reviewed elsewhere by CNS experts22–26. Here, we will focus on peripheral mechanisms and review cutaneous nociception in peripheral nerve diseases. We will highlight common and rare diseases that cause painful peripheral neuropathies and exciting new research that implicate non-neuronal skin cells in cutaneous nociception and neuropathic pain.
II. Cutaneous nociception primer:
Anatomy, detection, encoding, conveying stimuli to CNS.
The process of cutaneous nociception is mediated by peripheral nociceptor endings in the skin. Both hairy skin (covers most of the body) and glabrous skin (covers the palms of the hands, feet and lips) are densely innervated by nociceptors as well as other somatosensory afferent endings27–30. Nociception begins when these terminals detect a noxious physical or chemical stimulus of sufficient intensity to induce action potential firing. These action potentials are propagated toward central terminals in the spinal cord or brainstem where this sensory information is encoded into CNS processing centers, ultimately mediating the perception of pain.
Anatomically, nociceptors that innervate the skin have their cell bodies contained within the dorsal root ganglia (DRG) which are located outside the spinal cord, within the bony vertebral foramen. One DRG contains the somata of nociceptors as well as other sensory afferent fibers (e.g., low threshold, light touch mechanoreceptors). Each nociceptor soma is pseudounipolar in that a single process projects and bifurcates, making a “T-junction” such that the peripheral axon innervates the periphery, whereas the central axon projects to and synapses in the spinal cord. In general, one entire DRG innervates one cutaneous dermatome on one side of the body and the central axons of the DRG nociceptors project to a given spinal cord level. Amazingly, a single DRG nociceptor peripheral axon that innervates the skin of a human foot can be 1.2 meters in length, while the central axon that innervates the spinal cord can measure 30 cm31. Furthermore, most of the nociceptor’s protein synthesis and energy metabolism occur in the soma, which comprises only ~0.2 % of the cell’s total cytoplasm, making the soma a tiny factory that must support the enormous peripheral and central processes31. The face and head’s counterpart to the DRG is the trigeminal ganglia, where one large trigeminal ganglia is located on either side of the head under the brain in Meckel’s cave of the dura matter; the trigeminal ganglia contain the somata of nociceptors (and other sensory afferents) that give rise to the trigeminal nerve which branches peripherally to innervate the skin of the face and head. The central process of trigeminal neurons project to the brainstem and enter the CNS at the level of the pons.
Many functional subtypes
Many cutaneous nociceptors that innervate the body and the head have unmyelinated axons (C fibers) or thinly myelinated axons (Aδ fibers); however, some thick myelinated (Aβ) cutaneous nociceptors, normally associated with non-painful touch sensation, also have been identified in species from rodents to primates32,33 suggesting that the contributions of Aβ nociceptors to cutaneous pain should not be overlooked. The speed of action potential transmission is directly correlated with the amount of myelin and the diameter of the axon; C fibers conduct action potentials slowly and mediate slow-onset, persistent pain, whereas Aδ fibers conduct more rapidly and mediate fast-onset, protective, reflexive pain32.
Myelinated and unmyelinated cutaneous nociceptors are extremely heterogeneous. In addition to being classified by their conduction velocities, they are categorized by their responsiveness to noxious thermal and mechanical stimuli. The most common type of C fibers are polymodal, meaning that they respond to many modes including heat, cold, mechanical and chemical stimuli9. Other C fibers respond to a smaller repertoire of stimuli such as C-mechano heat, C-mechano cold, C-mechano heat cold. Some C fibers sense warming stimuli34, whereas other low threshold C fibers mediate gentle, pleasant skin touch35. A fiber nociceptors are not usually chemically sensitive but can be categorized into subtypes including A-mechano, A-mechano heat and A-heat; some of these A fibers are also cold sensitive9. Furthermore, a class of C fiber nociceptors that are normally insensitive to mechanical stimuli (“silent nociceptors”) can become responsive to mechanical stimuli following peripheral skin tissue injury. Whereas silent nociceptors are relatively rare in rodent skin36, they are common in human and pig skin37–39.
Transducers that detect and encode
Thermal heat:
A variety of molecular transducers have been identified that convert thermal, chemical and mechanical stimuli into action potentials in the nerve terminal. The most is known about cutaneous heat transducers, in part because the pioneer molecular heat transducer was identified a quarter of a century ago in 1997. The discovery of the capsaicin (the spicy chemical in hot chili peppers) and heat-sensitive channel Transient Receptor Potential Vanilloid 1 (TRPV140) launched the pain and somatosensory fields into the molecular transduction sphere. TRPV1 is a member of the Transient Receptor Potential (TRP) family, which in mammals encompasses around 30 members. All TRP channels are made of four subunits, where each subunit has six transmembrane domains and intracellular N- and C-termini. TRPV1 is expressed in C fiber and some Aδ fiber nociceptors and mediates all responsiveness to capsaicin41,42. When expressed in heterologous cells, TRPV1 confers responsiveness to noxious heat (42°C) to the cell40,41. However, in vivo, genetic deletion of TRPV1 in mice only modestly reduces the noxious behavioral and sensory neuron responses to heat42, indicating that other noxious heat transducers exist. In line with this finding, the Transient Receptor Melastatin 3 (TRPM3) channel has recently been identified to be a noxious heat sensor in mammalian nociceptors and to function independent of TRPV143. Subsequent studies have reported that noxious heat sensation in rodents is mediated by three channels (TRPV1, TRPM3 and TRPA1 (Transient Receptor Potential Ankyrin 1) 44. Such functional redundancy in channels suggests that there may be a fail-safe mechanism to avoid acute burn injury. Mammals are also sensitive to non-noxious warm temperatures (30–42°C), and as such three TRP channels, TRPV3, TRPV4 and TRPM2 are activated by warm temperatures in this range (25–42°C) 45–51 and likely encode our perception of warmth to the skin. Whereas TRPV3 and TRPV4 have been found in sensory neurons, they are far more highly expressed in skin keratinocytes45,47,48,52–54. Indeed, heat activates TRPV3 in keratinocytes which signal to sensory neurons via ATP55. Growing evidence (see section below on keratinocytes) indicates that keratinocytes are intimately involved in how we detect cutaneous temperature and touch, including in the noxious range.
Thermal cold:
On the opposite side of physiological zero (temperature at which our skin feels neither warm nor cold) are transducers for cold temperature. Two TRP channels mediate cold transduction in mammals. First, Transient Receptor Potential Melastatin 8 (TRPM8) was identified by its responsiveness to the cold-mimetic chemical menthol47,56. TRPM8 can be activated by modest decreases in ambient temperature (~26°C) and mice that lack TRPM8 have severe deficits in behavioral detection of both cooling and noxious cold (10°C) temperatures57–60. That some responsiveness to noxious cold remains in these mice spurred the search for additional noxious cold sensors. Indeed, another TRP channel, Transient Receptor Potential Ankyrin 1 (TRPA1) channel (aka “the wasabi receptor), has been shown to respond to intensely cold temperatures (<18°C) when expressed in heterologous cells61. Some in vivo studies support this finding as mice lacking TRPA1 were reported to have decreased withdrawal responses to cold surfaces or acetone-mediated cooling62 and decreased cold-induced nociceptive behaviors and trigeminal neuron responses63. However, other studies have reported normal cold-evoked behavioral and afferent responses in TRPA1 null mice64,65. It is possible that the disparate findings may be due to TRPA1 acting as a cellular sensor following cold-induced tissue damage66,67. Recently, the metabotropic G protein-coupled glutamate receptor GluK2 has been identified in mammalian sensory neurons and shown to be sensitive to noxious cold (~18 C68); although behavioral studies in rodents have not yet determined its contribution to cold detection in vivo. Alternatively, some two-pore potassium channel family members (KCNK4 or TRAAK) together with KCNK2 (TREK-1) have been shown to contribute to noxious cold-induced behaviors and neuronal activity69.
Mechanical:
Most cutaneous sensory neurons of all conduction velocities (Aβ, Aδ and C fibers) respond to mechanical stimuli. For cutaneous sensory neurons to respond to mechanical stimuli and initiate action potentials, they must express force-sensitive molecules that somehow convert physical energy into receptor potentials. Sophisticated studies in model organisms including bacteria, fruit flies (Drosophila melanogaster) and nematodes (Caenorhabditis elegans) have discovered a number of intrinsically mechanically-gated ion channels70–72. However, in mammals, the identification of bona fide mechanotransduction channels that transduce skin touch into action potentials remained elusive until 10 years ago. A major breakthrough in somatosensory mechanosensation occurred with the discovery of the novel, mechanically-gated ion channels Piezo1 and Piezo273. Piezo channels are the largest identified ion channels and have the greatest number of transmembrane domains (24–3674). Piezo channels exist as a three-blade-propeller-like homotrimer in naïve cells75–78. Both Piezo1 and Piezo2 channels form an ion pore that is directly gated by membrane stretch79–81. Piezo channels are found in nearly every organ of the body including bladder, colon, lung, bone, vasculature, skin and even red blood cells, reflecting on a molecular level the physiological fact that most organs rely on mechanical signaling73,82; as a result, Piezo channels are essential for survival83,84. Patients that have mutations in Piezo2 suffer from profound mechanosensory and proprioceptive deficits85–87. Interestingly, patients with Piezo2 mutations also fail to develop sensitization and painful reactions to touch after skin inflammation88. Complete deletion of Piezo2 from sensory neurons in mice reduces nociceptor responses to force and eliminates mechanical allodynia following skin inflammation and peripheral nerve injury89. Together these data suggest that Piezo2 may serve as a drug target for the cutaneous mechanical allodynia associated with skin inflammation and perhaps peripheral neuropathies in patients. The finding that cutaneous nociceptors retain some responsiveness to acute noxious mechanical stimuli suggests that there are other noxious mechanotransduction channels to discover. Other channels that have been validated to be inherently mechanically-activated include OSCA/TMEM63 and members of the K2P family including TREK-1, TREK-2 and TRAAK90–92 and further studies will need to determine their relevance to noxious mechanosensation in skin. Another protein TACAN (Tmem120A) was recently identified and found expressed extensively in nociceptive sensory neurons93. Although recent studies have not supported the premise that TACAN is a bona fide mechanotransduction ion channel94,95, it may serve a modulatory role in nociceptive mechanosensation after tissue injury96. Key questions are whether cutaneous nociceptors use a repertoire of mechanically-activated channels to transduce mechanical pain97 and whether different subtypes of nociceptors utilize different combinations of these and yet undiscovered noxious mechanotransducers97.
Transmission of nociceptor signals to CNS:
Once transduction of a noxious stimulus occurs, the membrane of the sensory neuron depolarizes (known as a generator potential), and, if the membrane potential rises above threshold, an action potential is generated that transmits the signal to the spinal cord or brainstem. Voltage-gated sodium (Na+; NaV) channels are critical for the upstroke or rapid depolarization phase of the action potential. Voltage-gated Na+ channels are comprised of one α-subunit and two β-subunits98. The α-subunit contains all components necessary for a functional voltage-gated channel, including the ion pore, voltage sensor and inactivation gate, whereas the β-subunits influence channel gating, inactivation and trafficking99,100. Voltage-gated Na+ channels are key regulators of the excitability of sensory neurons in that they are critical for amplification of the generator potential following stimulus transduction, electrogenesis of the action potential and release of neurotransmitters from sensory neuron terminals. The subtypes Nav1.7, Nav1.8 and Nav1.9 are preferentially expressed in nociceptive afferent neurons and have been linked to pain disorders in humans, including small-fiber neuropathies101–103, whereas nociceptors and non-nociceptive afferents both express Nav1.1 and Nav1.6104. On a functional level, the subunits Nav1.9 and Nav1.7 set the threshold and excitability of the nociceptor and amplify subthreshold stimuli, Nav1.7 and 1.6 contribute to the rising phase of the action potential, and Nav1.8 is the main contributor to the rising phase and mediates the shape and duration of the action potential in the nociceptor soma104–110.
Other families of voltage-gated ion channels mediate the action potential downstroke in nociceptors. Once voltage-gated Na+ channels inactivate at peak depolarization, delayed rectifier voltage-gated K+ channels and Ca2+ modulated K+ channels activate and mediate membrane repolarization111,112. Action potentials in the soma of nociceptors are followed by a long-duration after hyperpolarization where the membrane potential falls below the resting potential for a period of time113–115. The after hyperpolarization can be mediated by multiple channel types including voltage-gated K+ channels, Ca2+-activated K+ channels, and HCN (Hyperpolarization-activated cyclic nucleotide–gated) channels; the physiological role of the afterhyperpolarization phase is thought to be in regulating inter-spike intervals and limiting input from fast-firing nociceptors116. The central terminals of C fiber and Aδ fiber nociceptors innervate the superficial layers of the dorsal spinal cord or brainstem. For example, C fibers innervate the most superficial lamina I and II, whereas Aδ nociceptors innervate lamina I and V117. Once the action potential reaches the central terminal of the nociceptor in the spinal cord or brainstem, voltage-gated Ca2+ channels are activated, allowing Ca2+ influx that elicits release of classic neurotransmitters including glutamate that activates AMPA and NMDA receptors on spinal cord or brainstem neurons, and peptide neurotransmitters such as Substance P and CGRP that activate and modulate CNS neurons to relay the signal to toward higher brain centers117.
III. Cutaneous peripheral neuropathies from common to rare
Peripheral neuropathy:
Peripheral neuropathies encompass a broad class of heterogeneous disorders that stem from direct damage, disease or impairment of peripheral sensory neuron terminals, axons or even pathology that originates within the DRG, such as altered NaV channel function, that drives the cutaneous pain21,118,119. The perceptions associated can range from painful to insensate where skin is numb. Their underlying causes are multifarious, ranging from metabolic, traumatic, toxic, infectious and hereditary. Over 100 types of peripheral neuropathies exist, with each type having its own symptoms and prognosis. As a group, peripheral neuropathies are common, affecting 1–12% of individuals of all age and up to 30% of those 65 and older120. Peripheral neuropathies have variable etiologies and are classified according to the deficits they cause and the mechanisms that cause the neuropathy. Peripheral neuropathies can be generally classified into either mononeuropathy if a single peripheral nerve is affected, or polyneuropathy if multiple peripheral nerves are affected. Mononeuropathies and polyneuropathies can affect cutaneous sensory, motor and autonomic nerve fibers or a combination of these nerve types, and thereby elicit symptoms in skin, motor control or autonomic nervous system function. For the purposes of this review, we will focus on peripheral neuropathies that elicit cutaneous symptoms and deficits. Cutaneous symptoms can include sensory loss, hypersensation, pain or a mixture of all of these.
Mononeuropathies:
Physical injury or trauma from an accident is the most common cause of mononeuropathies. Additionally, prolonged pressure on a nerve, caused by extended periods of being sedentary such as lying in bed or sitting in a wheelchair or continuous, repetitive motions, can trigger a mononeuropathy. The most common mononeuropathy is carpal tunnel syndrome, which results from overuse and strain injury of the wrist. Carpel tunnel affects 4–5% of the general population ages 40–60 years and is more common in women121–123. It frequently occurs in assembly-line workers who perform repetitive wrist motions (fish and meat processing factories report incidences up to 73%124) and those who work with computer keyboards (repetitive data entry) for prolonged periods of time. The median nerve courses through the carpel tunnel passageway in the wrist to innervate the thumb and first three fingers of the hand. Pinching or compressing the median nerve often results in numbness or unpleasant tingling percepts (“paresthesias”), burning and pain in the hand, and the pain can radiate proximally to the arm and shoulder. The pathophysiological mechanisms that cause the pain in carpel tunnel syndrome are complex and multifactorial. Entrapment of the nerve causes compression and traction which results in malfunction in the intraneural microcirculation, ischemic vascular injury and breakdown of the blood nerve barrier. This results in edema, hypoxia, lesions in the myelin and damage to the nerve axons. In addition to spontaneous pain, some mononeuropathies can manifest as allodynia (pain due to a stimulus that does not normally provoke pain) and hyperalgesia (enhanced sensitivity to a normally painful stimulus) to cutaneous applied stimuli; mechanical and thermal allodynia have been reported in animal models of traumatic neuropathy, as well as in patients suffering traumatic nerve injuries125–127.
Small-fiber neuropathies:
Small-fiber neuropathy (SFN) is a disorder that selectively affects thinly myelinated Aδ and unmyelinated C fibers where the endings of fibers are damaged and die back (Wallerian degeneration) into the peripheral nerve trunk, where they continue to fire aberrantly128. Clinically SFN is characterized by neuropathic pain, most frequently described as burning, stabbing, shooting or prickling, but can also include numbness or tingling. It typically involves length-dependent or stocking-glove decrease in sensation to light-touch or pinprick129,130. SFN is typically associated with autoimmune diseases, diabetes and vitamin B deficiencies and toxicity (e.g. alcohol), although in over 50% of patients, no underlying etiology can be identified (idiopathic)131. As autonomic nerves also contain small fibers, some patients also experience autonomic dysfunction including altered sweating or thermoregulation, bowel dysfunction, dizziness or dry eyes or mouth. SFN severely affects patients quality of life. It can be diagnosed by skin punch biopsy and is evident by reduced or disrupted intraepidermal nerve fiber density. Recently, gain-of-function variants in the genes that encode for the NaV1.7, NaV1.8 and NaV1.9 sodium channel subunits have been identified in patients with SFN131.
Diabetic Neuropathies:
The most common cause of peripheral polyneuropathy is diabetic neuropathy. A staggering 463 million individuals world-wide have diabetes132, making diabetes the largest global epidemic of the 21st century105,133. Diabetic neuropathy affects nearly 30% of individuals with diabetes over their lifespan134,135. If the rate of diabetes continues unabated, estimates indicate that among the 9.5 billion people living across the world in 2045, 10.9% of the population (>700 million individuals) will have diabetes and approximately half of these individuals will experience diabetic neuropathy136. Diabetic neuropathy occurs in both patients with type 1 diabetes (when the body’s immune system attacks pancreatic beta cells which stop producing insulin) and type 2 diabetes (insufficiency in response to insulin and decreased insulin production). Diabetic neuropathies are heterogeneous in etiology where the most common type is distal sensory neuropathy which affects the distal ends of large myelinated Aβ fibers, more often sensory than motor axons, and the next most common form is distal small fiber neuropathy which largely affects the unmyelinated C fibers and is associated with burning feet syndrome137. However, recent evidence suggests that the selective involvement of large or small fibers is uncommon and most patients with pain have a mixed-fiber polyneuropathy that affects Aβ, Aδ and C fibers similarly138. Distal sensory neuropathy is a symmetric, primarily sensory neuropathy that affects the distal regions of the lower limbs and can spread proximally. It is caused by high circulating blood glucose and triglyceride levels that, if not well controlled, over time result in damage to the long-projecting peripheral nerves in the feet and hands.
Several systemic metabolic disturbances cause diabetic neuropathy. The prolonged hyperglycemia results in excess glucose moving into the polyol pathway where it is converted to sorbitol and fructose, the accumulation of which leads to structural breakdown of nerve axons139. Furthermore, the excess glucose reacts with proteins, nucleotides and lipids resulting in production of advanced glycation end products which disrupt peripheral nerve metabolism and result in peripheral neuropathy140. Additionally, oxidative stress and the production of free radicals damage blood vessels, leading to ischemia in tissue around peripheral nerves, produce advanced glycation end products and collectively, damage the peripheral nerve fibers141. Diabetic neuropathy targets peripheral sensory axons, autonomic axons and over time, motor axons. As it progresses, the sensory nerve terminals degenerate or die back into the nerve trunk but the somata remain relatively unaffected anatomically. It is a length-dependent neuropathy whereby the longest sensory axons that innervate the skin of the distal limbs (such as skin of the toes) die back first, followed by loss of cutaneous innervation of more proximal limbs. This process results in the glove and stocking pattern of sensory deficits. The increased serum glucose also causes damage to a range of cells beyond neurons, including Schwann cells, endothelial cells, fibroblasts and keratinocytes (see below for more on keratinocytes and cutaneous nociception). For example, the supportive Schwann cells, which are intimately involved in peripheral nerve function and myelination, can be damaged by severe chronic hyperglycemia, leading to demyelination of peripheral nerves and sensory deficits142.
Approximately 30–50% of patients with diabetic neuropathy develop neuropathic pain or parasthesias105,143–145; the remainder have insensate diabetic neuropathy. The most common symptoms of painful diabetic neuropathy include paresthesias (unpleasant, abnormal sensations), tingling, numbness, burning and lancinating pain in the feet and hands (distal “stocking-and-glove” distribution of deficits). Cutaneous hypersensitivity or hyperesthesia that results from contact with clothing or sheets at night is particularly bothersome to these patients and can interrupt their sleep quality, which can further amplify the pain sequela. Patients often have a mixture of cutaneous pain but also regions of sensory loss. Paradoxically, spontaneous pain can often even be perceived in numb, denervated skin (anesthetic dolorosa). A likely mechanistic explanation for this paradox is that hyperexcitable cutaneous afferent endings die back into the nerve trunk (but not all the way to the DRG somata) where they continue to fire spontaneously, perhaps via activation of aberrantly-expressed ion channels in the nerve ending as an “ectopic pacemaker”146. This mechanisms also accounts for Tinel’s sign when a nerve trunk is tapped and elicits a tingling, “pins and needles” sensation. Mechanistically, the spontaneous burning pain is correlated with spontaneous activity in sensory neurons. The spontaneous activity may result from increased expression of Nav1.8 channels in sensory neurons which will result in increased action potential transmission to the CNS and neuropathic pain147. Furthermore, the chronic hyperglycemia in diabetes induces changes in cellular oxidative status, increase in free radicals and oxidative damage to tissues. The changes in cellular oxidative status has been shown to modulate the activities of a variety of redox-sensitive TRP channels including TRPA1, TRPC5, TRPM2, TRPM7 and TRPV1148. Therefore, targeting redox TRPs has potential in treating painful diabetic neuropathy.
Conversely, insensate diabetic neuropathy is even more common than painful diabetic neuropathy149. While not painful, it can have equally or worse negative effects on quality of life because the numbness or loss of mechanical sensation in the feet can lead to dangerous, even life-threatening falls. Patients also often lose their sensation of heat and cold in the feet. Since patients can’t feel physical stimuli, blisters and calluses in the skin of the toes, feet and legs develop. A common and serious complication of diabetic neuropathy is cutaneous ulcers of the foot resulting in major decreases in quality of life due to the prolonged immobilization required to heal the ulcers. If inadequately treated, the ulcers can become infected, worsen to gangrene, result in foot deformation and ultimately require amputation. As the distal axons are affected first and most severely, insensate diabetic neuropathy is more common in individuals who are taller than average150. The causes underlying insensate neuropathy include demyelination and slowed conduction velocities of large sensory and motor conduction velocities, demyelination and depletion of small sensory nerves in the skin (and cornea). Although there can be regeneration of fibers in clusters151, this is not sufficient to overcome the distal degeneration of fibers149.
Chemotherapy-induced peripheral neuropathy (CIPN):
Many modern first-line chemotherapy drugs used to treat cancer cause both acute and chronic peripheral polyneuropathy (incidence ranges from 19–85%; 152,153). Multiple classes of chemotherapeutics used include long established compounds platinum agents (e.g., oxaliplatin), taxanes (e.g., paclitaxel) and vinca alkaloids (e.g., vincristine). Newer compounds include bortezomid, eribulin and ixabepilone154,155. While each chemotherapeutic combats cancer cell proliferation through different anti-mitotic mechanisms, they all can induce peripheral neuropathies by inadvertently damaging sensory neurons156. While CIPN often resolves after cessation of treatment, approximately 30% of patients suffer from persistent neuropathic pain that affects their function and quality of life157. Further, the severity of acute neuropathy during treatment may limit the dose of chemotherapeutic that can be administered, or even require termination of treatment. As many common cancers such as breast, ovarian and colorectal are prevalent and are treated with chemotherapeutics, CIPN affects several million patients globally each year. Chronic CIPN can persist for months or years after cessation of treatment. Treatment options are extremely limited and often ineffective; there are currently no FDA approved therapeutics for prevention or treatment of CIPN157.
CIPN presents as a glove and stocking peripheral neuropathy where patients describe symptoms including numbness, paresthesia, spontaneous pain, and cutaneous hypersensitivity to mechanical and cold stimuli. Severe cases can also involve loss of vibration sensation. The long axons of toes and feet are typically affected first, followed by impairment in the fingers and hands. Quantitative sensory testing in patients with CIPN has demonstrate sensory deficits in thermal and touch sensation, suggesting that CIPN is associated with deficits in multiple sensory afferent subtypes158–160. Changes in epidermal nerve fiber density in the skin inversely correlate with pain severity; the lowest nerve counts are found in the most painful, distal areas, while higher densities are in more proximal regions where there is numbness or no sensory deficit161. Recently, it was demonstrated using ex vivo skin nerve recording that chemotherapeutic treatment can directly sensitize Aδ and C fiber afferents to mechanical stimulation in mice162. Thus, direct sensitization of primary afferents may contribute to the development of CIPN mechanical allodynia and hyperalgesia.
Chemotherapeutic treatment also appears to affect sensory neuron mitochondrial function; both myelinated A fiber and C fiber peripheral axons and the dorsal root ganglia somata exhibit swollen, vacuolated mitochondria in rodent models of CIPN163–167. Mitochondrial dysfunction can increase the production of reactive oxygen species (ROS), which can damage sensory neurons and lead to the development of CIPN pain162,168,169. Additionally, a variety of alterations to neuronally expressed ion channels have been associated with CIPN pain, including several members of the TRP family of ion channels (see170 for an in-depth review). For example, both antagonism of TRPV4 and TRPA1 inhibited paclitaxel-induced mechanical allodynia, while TRPA1 antagonism attenuate paclitaxel-induced cold allodynia171. Thus, treatments utilizing drugs that can reduce oxidative stress, as well as drugs that can modulate CIPN associated ion channels, may be useful for alleviating CIPN pain.
Post herpetic neuralgia (PHN): Infection: Example:
The varicella-zoster virus (VZV) is a highly virulent neurotropic virus that causes varicella (chickenpox) upon initial infection and, upon later reactivation, herpes zoster (shingles). Following the initial infection, the virus is transported retrogradely along the axons of sensory neurons in the skin to establish a latent infection within the dorsal root or trigeminal ganglia172–174. Because senescence of cellular immunity occurs as individuals advance in age, the virus can be reactivated, particularly after a stressful life event, transported anterogradely down axons to the skin and present as the painful blistering rash characteristic of herpes zoster172,175. While the pain associated with herpes zoster typically resolves within 2–4 weeks, a common complication (10% of patients) is the development of post herpetic neuralgia (PHN), a condition characterized by excruciating chronic neuropathic pain that can last at least 3 months after healing of the acute skin lesions176. This neuropathic pain is characterized as ongoing spontaneous burning or stabbing pain, as well as touch and thermal allodynia, which persist in the same location of the herpetic rash. The mechanisms underlying the development PHN pain are not fully understood, although one likely locus for the ongoing (ectopic) activity is the dorsa root ganglion146. A second likely locus is the endings of peripheral sensory neurons that die back from the epidermis146. Additionally, peripheral sensitization of nociceptors by inflammation and direct viral damage, as well as the central reorganization of normally innocuous touch Aβ afferent input to dorsal horn pain pathways, have been proposed177,178. Rat models of post herpetic neuralgia have been developed and have sensory deficits, including long-lasting cutaneous mechanical allodynia and hyperalgesia, that is similar to those observed in patients with post herpetic neuralgia. However, rodents are not fully permissive hosts to VZV infection, which complicates small animal model studies of post herpetic neuralgia179. Skin nerve recordings in rodents or microneurography recordings in humans could reveal important information about how sensory afferent subtypes are sensitized in postherpetic neuralgia.
Hereditary neuropathy:
Hereditary neuropathies are a group of inherited disorders that affect the peripheral nervous system. They can affect any combination of motor, sensory and autonomic nerves. The most common type is Charcot-Marie-Tooth disease, a motor and sensory neuropathy that includes weakness or paralysis of the foot and lower leg muscles, inability to sense heat, cold and touch, shortening of muscles or tendons that causes cramping and pain. Another category of hereditary sensory neuropathies include channelopathies, a heterogeneous group of disorders resulting from the dysfunction of ion channels, most often involving mutations in transient receptor potential family members involved in stimulus transduction, or in voltage-gated channels involved in action potential transmission180,181. For example, NaV 1.7 channels set the excitability of nociceptors, and either gain- or loss of function mutations profoundly affect pain sensation. Gain of function mutations underlie inherited erythromelalgia or paroxysmal extreme pain disorder whereas in contrast, loss of function mutations result in congenital insensitivity to pain182.
Specific example of hereditary neuropathy: Fabry disease:
Fabry disease is an X-linked recessive lysosomal storage disorder caused by mutations the gene encoding for α-Galactosidase A (α-Gal A), a lysosomal hydrolase that catalyzes the removal of the terminal α-galactose residue from glycosylated molecules in the lysosome. Dysfunction or absence of α-Gal A causes accumulation of glycosylated products such as globotriaosylceramide (Gb3), lyso-globotriaosylceramide (lyso-Gb3). The glycosphingolipid products build up and form aggregates in lysosomes of many cell types183,184. Whereas many tissue types and organs are affected including kidney, heart, eye and gut, peripheral neurons are particularly susceptible to the substrate buildup since neurons do not replicate. The dorsal root ganglia neurons and their peripheral axons are a particular site of substrate buildup as they do not turnover and cannot reduce the substrate on their own. This results in small fiber neuropathy where nerve fibers are lost or damaged, resulting in sensory abnormalities and severe pain185. Pain begins in childhood and persists throughout life, disrupting work, daily activities and social interactions. Patients have high rates of anxiety and depression, which also decrease their quality of life. Treatment of the underlying disease with enzyme replacement therapy is costly, complicated to receive, and does not adequately alleviate patient’s pain186,187. While Fabry Disease is categorized as a rare hereditary neuropathy, with a historic estimated prevalence of 1:40,000–117,000188,189, recent newborn genetic screening suggests that it may be far more prevalent and as frequent as 1:1,400190. Fabry patients experience a plethora of pain symptoms that affect the skin and that severely impact daily function. These include hypersensitivity to mechanical stimuli, heat and cold intolerance, and intense episodic pain episodes that can be triggered by environmental heat, fever or stress185. Dorsal root ganglia and axons of peripheral nerves are an epicenter of Fabry disease and may be an example of the DRG being the key driver of the pain pathology since DRGs accumulate Gb3 and lyso-Gb3 and exhibit profound sensitization185146,191–193. Peripheral neuropathy affects over 25% of patients with Fabry disease and is characterized by loss of small myelinated and unmyelinated fibers particularly in the lower extremities and skin of the feet194–196. The fiber loss is significantly greater in the skin than in the peripheral nerve trunk195 and although this has not been directly documented for Fabry disease, this may be due to length-dependent dying back of the peripheral nerves into the nerve trunk, as occurs with diabetic neuropathy. Mechanistically, it is not yet known whether the accumulation of Gb3 leads to metabolic changes through mitochondrial dysfunction, whether long-term high concentrations of Gb3 have neurotoxic effects, or whether Gb3 accumulation can lead to activation of inflammatory states within the neuron that cause distal axon degeneration at the molecular level. Several sensory neuron expressed ion channels have been implicated in the pathology of Fabry peripheral neuropathy, including NaV1.7, TRPV1, and inward-rectifying potassium channels197,198. In particular, it was recently demonstrated that TRPA1 is sensitized in Fabry rat sensory neurons and that antagonism of TRPA1 reduces in vivo mechanical hypersensitivity in Fabry rats193. Future studies into how sensory neuron ion channel function is altered in Fabry disease could lead to the discovery of novel therapeutic targets for treating Fabry pain.
IV. Beyond sensory neurons: are non-neuronal cells involved in cutaneous peripheral neuropathic pain?
Where are the peripheral sites of the dysfunction in cutaneous neuropathic pain? First, nociceptive sensory neurons are critical for the transduction of noxious stimuli under normal conditions, and the cutaneous terminals and axons of nociceptors become sensitized after tissue or nerve injury through a plethora of molecular and cellular mechanisms of nociceptor sensitization (reviewed in 117,199–207). Second, large myelinated Aβ fibers that normally transduce non-painful tactile stimuli can become sensitized17,19. Third, the DRG itself is a key site for ectopic activity and sensitization146,185,193. Exciting translational studies of DRGs excised from patients with either chemotherapy-induced neuropathy pain or radicular/neuropathic pain demonstrate that ectopic (spontaneous) activity and hyperexcitability occurs in DRGs that innervate dermatomes associated with pain208,209. Future studies on human DRGs and other human tissues in the pain pathway will be invaluable for translating preclinically-identified pain targets to the clinic.
Growing evidence indicates that a number of non-neuronal cell types that are beneficial and protective under normal, healthy conditions can be dysregulated in pathological conditions and contribute to persistent cutaneous neuropathic pain by releasing cytokines, chemokines, growth factors and other biologically active small molecules. These non-neuronal cells include monocytes, macrophages, Schwann cells, satellite glial cells, astrocytes, mast cells and T cell lymphocytes, microglia and their intricate roles in sensory neuron sensitization are reviewed elsewhere210–212. Moreover, what is often underappreciated is that substances circulating in blood can access and thereby sensitize sensory neurons and glial cells at the level of the DRG because the DRGs are densely vascularized with capillaries protected by a permeable blood-nerve barrier. In some pathological conditions associated with neuropathic pain, this blood-nerve barrier can be further disrupted through impaired peripheral pericyte function31,213–215.
Here we will focus attention on keratinocytes. Keratinocytes are the predominant epidermal cell type, comprising 95% of the epidermis and forming the key barrier to external pathogens; vice versa, they are the key barrier to loss of water and solutes from within the body216. All subclasses of cutaneous primary afferent neurons, including nociceptors, terminate within or in close proximity to keratinocytes and keratinocytes are often the first point of contact between the body and external stimuli33,217–220. Thus, keratinocytes are in a prime position to modify the firing of cutaneous afferents. However, this intimate association between keratinocytes and sensory neurons, as well as the fact that keratinocytes and sensory neurons express many of the same sensory relevant receptors, complicates research efforts to dissect the effects of individually stimulating keratinocytes or sensory neurons in awake behaving animals. This problem was solved in a landmark study by the Caterina group, who re-expressed the TRPV1 capsaicin receptor specifically in the keratinocytes of TRPV1 global knockout mice, allowing for the selective stimulation of keratinocytes with capsaicin. Remarkably, application of capsaicin to the skin of these animals resulted nocifensive paw-attending responses221. The sole way this behavioral response is elicited is via capsaicin activation of keratinocytes, which subsequently transmits the signal to nociceptive afferent endings to convey the signal to the spinal cord. Successive studies utilizing optogenetics to selectively activate keratinocytes demonstrated that keratinocytes can shape the responses of primary afferents to cutaneously-applied stimuli222. Moreover, optogenetic inhibition of keratinocytes in vivo dampens responses to noxious mechanical and thermal stimuli223,224.
Mechanistically, keratinocytes depolarize in response to mechanical stimuli and signal to sensory nerve endings by releasing ATP, which activates sensory neuron P2X4 channels225. This release of ATP from keratinocytes appears to be voltage-dependent and may occur through vesicular mechanisms and/or connexin hemichannels223,226,227. Recent evidence shows that keratinocyte and sensory neurons are more intimately associated than previously thought. Structurally, sensory neurons have been shown to tunnel directly through the keratinocyte cytoplasm and be entirely encapsulated by keratinocytes220. Exciting new data from Talagas and colleagues shows that human keratinocytes make en-passant synapse-like contacts with fine sensory neuron fibers (C and Aδ type) as the fibers course through the epidermis. These keratinocytes contain synaptic vesicles that express synaptophysin and synaptotagmin 1 and communicate with sensory neurons through SNARE-mediated vesicle release228. These findings are intriguing since keratinocytes turn over from basal stem cells to superficial stratum corneum every 40–56 days in humans229 (8–10 days in mice230). This suggests that keratinocyte to sensory neuron associations are transient, perhaps with the keratinocyte sliding up along or around the sensory fiber as they migrate from the basal epithelial layer superficially228. The en passant synapse like contacts may allow keratinocytes to concentrate signaling molecules and modulators at the nerve membrane and tune sensory detection228. Therefore, the long-used textbook terminology of “free nerve endings” is not likely correct, as many cutaneous afferent fibers are connected to keratinocytes228 or Schwann cells231. All together, these exciting data indicate that keratinocytes are essential peripheral participants in the transmission of acute noxious cutaneous mechanical, thermal and chemical stimuli to the CNS.
Keratinocytes are the first site of cutaneous injury and are anatomically well positioned at the cellular and tissue levels to modulate signaling in sensory neuron fibers. A key question is whether keratinocytes contribute to the cutaneous hypersensitivity, allodynia and pain following skin injuries. This exciting hypothesis remains to be tested but the potential for keratinocytes involvement in painful skin is high. Indeed, alterations in keratinocyte function and signaling are known to contribute to inflammatory skin disorders such as dermatitis and psoriasis232,233. Keratinocytes can function as primary transducers of itch through the ion channel TRPV4; TRPV4 expression in skin keratinocytes is required for histaminergic itch and cholestatic itch induced by the lipid lysophosphatidylcholine234,235Keratinocyte-expressed TRPV4 has also been implicated in inflammatory sunburn pain, as selective knockout of TRPV4 from keratinocytes protects against the development of UVB induced mechanical and thermal hyperalgesia236. Furthermore, keratinocytes from patients with fibromyalgia display increased expression of the pain mediators interleukin-10 (IL-10) and ephrin receptor-A4 (EPHA4)237. Keratinocytes may also contribute to persistent neuropathic pain, as human keratinocytes transplanted into a ligated and transected peripheral nerve in rodents lead to hyperexcitability of sensory neurons and chronic pain behavior in vivo238. Additionally, “sensitive skin syndrome” is a clinical condition that often occurs with small fiber neuropathies and is characterized by the occurrence of unpleasant sensations such as burning, stinging, tingling, pricking or itching in response to innocuous thermal, physical or chemical stimuli239. Paradoxically, in small fiber neuropathies, the density of fine nerve endings is decreased indicating that factors other than increased nerve ending density mediate the cutaneous pain. Intriguingly, in skin from patients with small fiber neuropathy, keratinocytes associated with fine nerve endings express a higher density of synaptic vesicles, suggesting that these en passant synapses between keratinocyte and nerve membranes might possibly contribute to hypersensitivity in cutaneous painful conditions by tuning or amplifying nerve ending responsiveness228. Additionally, Keratinocytes from patients with neuropathic pain have changes in gene expression profiles of signaling molecules and ion channels known to be involved in pain conditions. For example, keratinocytes from patients with complex regional pain syndrome and post-herpetic neuralgia exhibit increased expression of the pro-algesic factor CGRP240 and a number of voltage-gated sodium channels including NaV1.1, NaV1.2, NaV1.6, NaV1.7, and NaV1.8241. In patients with SFN, the density of synaptic vesicles in keratinocytes is increased228, suggesting that despite the decreased density of nerve endings in SFN, the enhanced synaptic-like contacts may contribute to the cutaneous pain in this neuropathy. Furthermore, keratinocytes from patients with SFN display increased expression of interleukin-8, C-X-C motif chemokine 3, endothelin receptor type A, NTN1 and transforming growth factor-β1237,242. These data suggest that keratinocytes might be peripheral drivers of nociceptor signaling in injury states, potentially through increased keratinocyte transduction of cutaneous stimuli and through the release of neuroactive factors that modulate nociceptor action potential firing (Figure 1).
Figure 1.
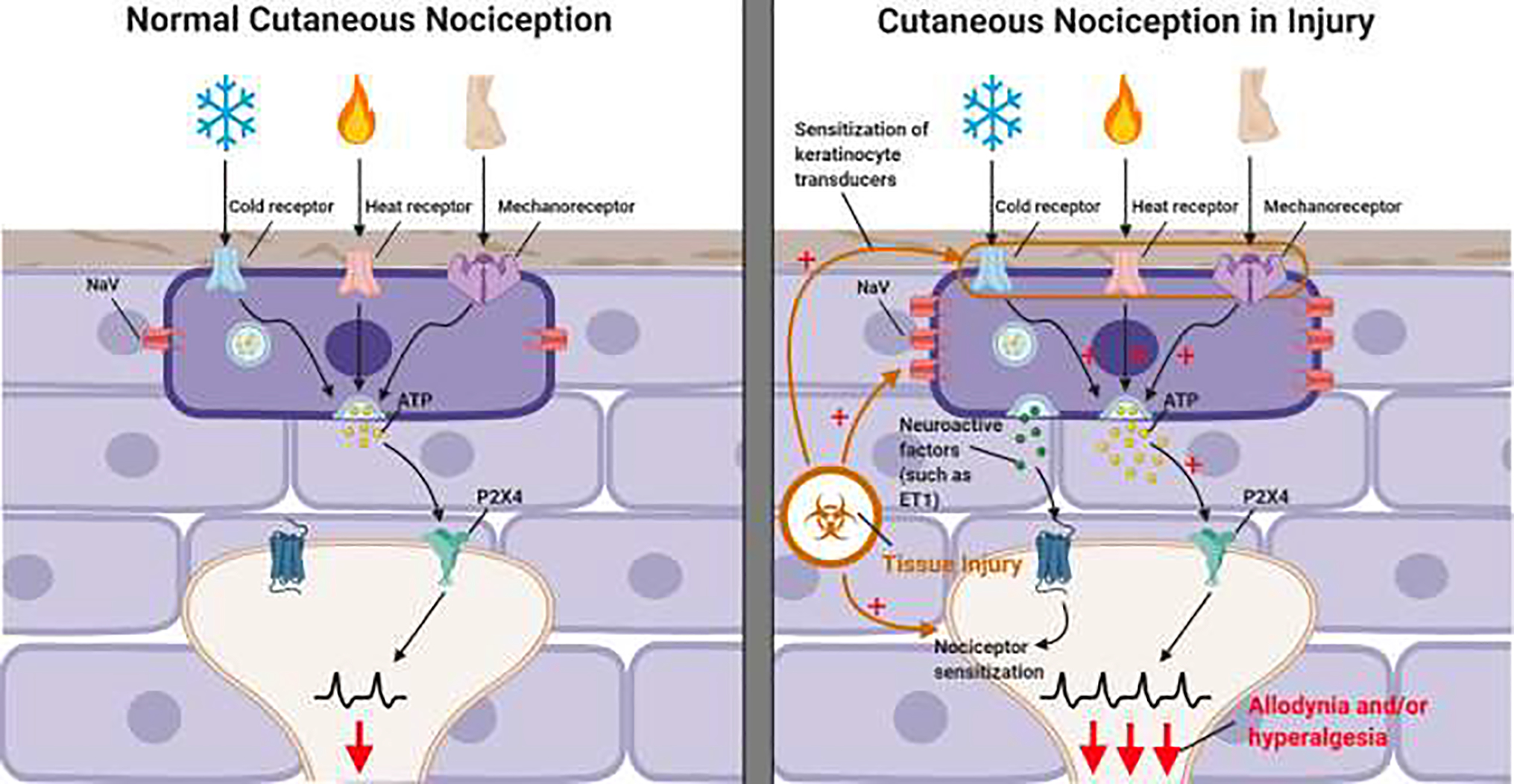
Under normal healthy conditions, keratinocytes contribute to the detection of innocuous and noxious cutaneously applied cold, heat, and mechanical stimuli. This is mediated by keratinocyte to sensory neuron ATP-P2X4 signaling but also likely involves other unidentified signaling pathways224,256. The molecular machinery that enables keratinocytes to detect heat, cold and mechanical stimuli have not been definitively identified, though keratinocytes do express the cold receptor TRPM8, the warm and heat activated receptors TRPV1, TRPV3 and TRPV4, as well as the mechanoreceptor Piezo145,47,257–259.
Keratinocyte to nociceptor signaling may be enhanced following injury, contributing to the sensitization of nociceptors and the development of allodynia and hyperalgesia following skin injury. A potential mechanism through which keratinocytes could enhance nociception is through the injury induced sensitization of keratinocyte expressed receptors. Keratinocyte also express a variety of voltage gated sodium channels (NaV) and the expression of many of these are upregulated in states of neuropathic injury241. Sensitization of keratinocyte expressed ion channels could lead to increased activation of keratinocytes in response to cutaneous applied stimuli and a subsequent increase in the release of ATP and other keratinocyte-expressed neuroactive factors onto nociceptor terminals. Figure 1 was Created with BioRender.com
Keratinocytes may not be the only cutaneous cell capable of modulating nociceptor firing. A recent study has shown that cutaneous nociceptive C fiber endings, long thought to be “free” unencapsulated nerve endings, are actually ensheathed by a novel type of glial cell called “nociceptive Schwann cells,” which form a mesh-like sensory organ that extends from the dermis into the epidermis. These Schwann cells appear to participate in detecting noxious stimuli applied to the skin and conveying nociceptive signals to the sensory terminals of nociceptors231. Like keratinocytes, nociceptive Schwann cells may be sensitized following tissue injury and release factors that modulate nociceptor firing, enhancing the perception of allodynia and hyperalgesia in cutaneous pain conditions.
V. The Horizon: What can we see and seek in the future?
For many neuropathic pain states, there are few effective treatments, leading to suffering and low quality of life in patients. Developing better therapeutics will require increased understanding of how peripheral and central pain circuits are altered in neuropathic injury. While current animal models of neuropathic pain are useful for studying evoked pain (allodynia and hyperalgesia), many patients primarily complain of spontaneous shooting or burning pains, as well as paresthesia like “pins and needles” sensations. The development of better “face-valid” and quantitative assays for measuring spontaneous pain in peripheral neuropathies would aid in investigating the mechanisms behind these devastating symptoms. Some assays that may be useful for studying spontaneous neuropathic pain include conditioned place preference, measurements of facial grimace, and automated behavior recognition software, such as HomeCageScan, that can monitor animals for ongoing pain behaviors243–245. The development of more clever behavioral assays that evaluate spontaneous pain in rodents would boost the field. Additionally, there are still relatively few studies that employ single nerve fiber recordings of identified afferent subtypes, either through ex or in vivo skin-nerve fiber recordings in animal models of cutaneous neuropathic pain, or microneurography studies in human patients suffering from peripheral neuropathies17,110,246–251. Future research utilizing these techniques could provide critical information regarding how specific fiber types are modulated in neuropathic injury in an in vivo or ex vivo setting.
Future directions must investigate the roles of keratinocytes, as well as nociceptive Schwann cells, in cutaneous pain and neuropathic disease states. This investigation could be done in a variety of animal models of inflammatory and neuropathic pain. Pairing these animal models with optogenetic, chemogenetic, and gene editing techniques that enable the selective activation and modulation of keratinocytes, nociceptive Schwann cells, or sensory neurons, could potentially reveal how these cutaneous cell types function together to encode noxious stimuli to the CNS in painful disease states. Research into bidirectional, or even tridirectional, signaling between keratinocytes, nocifensive Schwann cells, and sensory neurons, could reveal novel signaling pathways that drive cutaneous pain. The ability to garner human skin tissue samples provides an easy translational advantage to the field since human keratinocytes, Schwann cells, and other non-neuronal cell types can be directly investigated220,224. Transcriptomics analyses of keratinocytes, Schwann cells and other skin cell types can be performed on skin samples from human patients, potentially patients with a variety of peripheral neuropathies. The development of iPSC nociceptors, keratinocytes and nociceptive Schwann cells from stem cells derived from patients with cutaneous neuropathies is an exciting reality252–255. In vitro co-cultures of animal and/or human skin cells and sensory neurons could reveal important signaling interactions between these cell types, perhaps even how these interactions are altered upon in vitro exposure to neuropathy-inducing agents like paclitaxel. An exciting therapeutic opportunity is the possibility to develop novel, topically-administered cutaneous therapeutics that affect skin cells and sensory endings only while avoiding CNS side effects. For example, lidocaine gels and patches target voltage-gated NaV channels and block cutaneous nerves from firing action potentials; when applied to painful skin, these are routinely used to treat postherpetic cutaneous neuropathy. Additional topical patches, gels that rapidly penetrate skin and even injectable or micro dermal abrasion applied analgesics for painful skin could be developed that target novel keratinocyte pain targets. For example, Piezo1 and TRPV4 are extensively expressed in keratinocytes and involved in cutaneous sensitization. Perhaps these channels could be topically targeted to alleviate mechanical pain without anesthetizing the affected skin from all sensation.
Highlights:
Cutaneous nociceptors exist in a plethora of diverse subtypes based on their anatomy, sensitivity to external and internal stimuli, molecular transducers and receptors, and signaling molecules within.
Many peripheral neuropathies have prominent cutaneous deficits including pain, sensory loss, hypersensation, or a mixture of deficits.
The focus of this review is on how cutaneous nociception (pain perceived from the skin) is altered following diseases that affect peripheral nerves.
We discuss mechanisms underlying acquired neuropathies (small-fiber neuropathies, diabetic neuropathy, chemotherapy-induced peripheral neuropathy, post herpetic neuralgia) and hereditary neuropathies (channelopathies, Fabry disease).
Sensory neurons are not the sole transduction cell type in skin. Keratinocytes and nociceptive Schwann cells are critical players in somatosensation and pain transduction. Much remains to be discovered about the roles of these non-neuronal skin cells in painful and insensate peripheral neuropathies.
Acknowledgements:
The authors thank the National Institutes of Health, National Institute of Neurological Disorders and Stroke for the following grants that supported this review: R37NS108278, R01NS070711 and R01NS040538 to C.L.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited:
- 1.Yancey P & Brand PW The gift of pain: why we hurt and what we can do about it. (Zondervan, 1997). [Google Scholar]
- 2.Dubner R Basic mechanisms of pain associated with deep tissues. Canadian journal of physiology and pharmacology 69, 607–609 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Graven-Nielsen T Fundamentals of muscle pain, referred pain, and deep tissue hyperalgesia. Scandinavian journal of rheumatology. Supplement 122, 1–43 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Torrance N, Smith BH, Bennett MI & Lee AJ The Epidemiology of Chronic Pain of Predominantly Neuropathic Origin. Results From a General Population Survey. Jounal of Pain 7, 281–289 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Graven-Nielsen T & Arendt-Nielsen L Assessment of mechanisms in localized and widespread musculoskeletal pain. Nature reviews. Rheumatology 6, 599–606 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Staud R Peripheral Pain Mechanisms in Chronic Widespread Pain. Best Pract Res Clin Rheumatol 25, 155–164 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaible HG & Grubb BD Afferent and spinal mechanisms of joint pain. Pain 55, 5–54 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Cervero F & Laird JMA Visceral pain. Lancet 353, 2145–2148 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Raja SN, Meyer RA & Campbell JN Peripheral Mechanisms of Somatic Pain. Anesthesiology 68, 571–590 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Mayer EA & Gebhart GF Basic and clinical aspects of visceral hyperalgesia. Gastroenterology 107, 271–293 (1994). [DOI] [PubMed] [Google Scholar]
- 11.Woolf CJ Evidence for a central component of post-injury pain hypersensitivity. Nature 306, 686–688 (1983). [DOI] [PubMed] [Google Scholar]
- 12.Raja SN, Ringkamp M, Guan Y & Campbell JN John J. Bonica Award Lecture : Peripheral neuronal hyperexcitability: the “ low-hanging” target for safe therapeutic strategies in neuropathic pain. Pain 161, S14–S26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh M, Donaldson LF & Lumb BM Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. Pain 156, 1074–1083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hulse RP Identification of mechano-sensitive C fibre sensitization and contribution to nerve injury-induced mechanical hyperalgesia. European Journal of Pain 20, 615–625 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Forstenpointer J et al. Sensitized vasoactive C-nociceptors : key fibers in peripheral neuropathic pain. Pain 4, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devor M Ectopic discharge in Aβ afferents as a source of neuropathic pain. Experimental Brain Research 196, 115–128 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Djouhri L et al. Cutaneous Aβ-Non-nociceptive, but Not C-Nociceptive, Dorsal Root Ganglion Neurons Exhibit Spontaneous Activity in the Streptozotocin Rat Model of Painful Diabetic Neuropathy in vivo. Frontiers in Neuroscience 14, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu YF & Henry JL Excitability of A-beta sensory neurons is altered in an animal model of peripheral neuropathy. BMC Neuroscience 13, 1–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Djouhri L L5 spinal nerve axotomy induces sensitization of cutaneous L4 Abeta-nociceptive dorsal root ganglion neurons in the rat in vivo. Neuroscience Letters 624, 72–77 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Cervero F & Laird JMA Mechanisms of touch-evoked pain (allodynia): a new model. Pain 68, 13–23 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Liu CN et al. Tactile allodynia in the absence of C-fiber activation: Altered firing properties of DRG neurons following spinal nerve injury. Pain 85, 503–521 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Baron R, Hans G & Dickenson AH Peripheral input and its importance for central sensitization. Annals of Neurology 74, 630–636 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Kuner R Central mechanisms of pathological pain. Nature Medicine 16, 1258–1266 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Latremoliere A & Woolf CJ Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. Journal of Pain 10, 895–926 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woolf CJ Central sensitization: Implications for the diagnosis and treatment of pain. Pain 152, S2–S15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woolf CJ & Salter MW Neuronal Plasticity: Increasing the Gain in Pain. Science 288, 1765–1768 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Weddell G The pattern of cutaneous innervation in relation to cutaneous sensibility. Journal of anatomy 75, 346–34667 (1941). [PMC free article] [PubMed] [Google Scholar]
- 28.Weddell G The anatomy of cutaneous sensibility. British medical bulletin 3, 167–172 (1945). [DOI] [PubMed] [Google Scholar]
- 29.Arthur R & Shelley W The innervation of human epidermis. The Journal of investigative dermatology 32, 397–411 (1959). [DOI] [PubMed] [Google Scholar]
- 30.Joong Woo Leem, Willis WD & Jin Mo Chung. Cutaneous sensory receptors in the rat foot. Journal of Neurophysiology 69, 1684–1699 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Devor M Unexplained peculiarities of the dorsal root ganglion. Pain 82, 27–35 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Djouhri L & Lawson SN Aβ-fiber nociceptive primary afferent neurons: A review of incidence and properties in relation to other afferent A-fiber neurons in mammals. Brain Research Reviews 46, 131–145 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Paré M et al. The Meissner corpuscle revised: A multiafferented mechanoreceptor with nociceptor immunochemical properties. Journal of Neuroscience 21, 7236–7246 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hallin RG, Torebjork HE & Wiesenfeld Z Nociceptors and warm receptors innervated by C fibres in human skin. J Neurophysiology 44, 313–319 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olausson H, Wessberg J, Morrison I, Mcglone F & Vallbo A The neurophysiology of unmyelinated tactile afferents. Neuroscience and Biobehavioral Reviews 34, 185–191 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Wetzel C et al. A stomatin-domain protein essential for touch sensation in the mouse. Nature 445, 206–209 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Schmidt R et al. Novel classes of responsive and unresponsive C nociceptors in human skin. Journal of Neuroscience 15, 333–341 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prato V et al. Functional and Molecular Characterization of Mechanoinsensitive “Silent” Nociceptors. Cell Reports 21, 3102–3115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rukwied R et al. Slow depolarizing stimuli differentially activate mechanosensitive and silent C-nociceptors in human and pig skin. Pain 161, (2020). [DOI] [PubMed] [Google Scholar]
- 40.Caterina MJ et al. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Tominaga M et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Caterina MJ et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Vriens J & Voets T Sensing the heat with TRPM3. Pflugers Archiv European Journal of Physiology 470, 799–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vandewauw I et al. A TRP channel trio mediates acute noxious heat sensing. Nature 555, 662–666 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Guler A et al. Heat-Evoked Activation of the Ion Channel, TRPV4. Journal of Neuroscience 22, 6408–6414 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe H et al. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. Journal of Biological Chemistry 277, 47044–47051 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Peier AM et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 296, 2046–2049 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Smith GD et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418, 186–190 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Xu H et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418, 181–186 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Togashi K et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO Journal 25, 1804–1815 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan CH & McNaughton PA The TRPM2 ion channel is required for sensitivity to warmth. Nature 536, 460–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung M-KK, Lee H, Mizuno A, Suzuki M & Caterina MJ TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. Journal of Biological Chemistry 279, 21569–21575 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Moqrich A et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307, 1468–1472 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Liedtke W & Friedman JM Abnormal osmotic regulation in trpv4−/− mice. Proceedings of the National Academy of Sciences of the United States of America 100, 13698–13703 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mandadi S et al. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflügers Archiv : European journal of physiology 458, 1093–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKemy DD, Neuhausser WM & Julius D Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Bautista DM et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204–208 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Dhaka A et al. TRPM8 Is Required for Cold Sensation in Mice. Neuron 54, 371–378 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Knowlton WM et al. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. Journal of Neuroscience 33, 2837–2848 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Milenkovic N et al. A somatosensory circuit for cooling perception in mice. Nature Neuroscience 17, 1560–1566 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Story GM et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Kwan KY et al. TRPA1 Contributes to Cold, Mechanical, and Chemical Nociception but Is Not Essential for Hair-Cell Transduction. Neuron 50, 277–289 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Karashima Y et al. TRPA1 acts as a cold sensor in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America 106, 1273–1278 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jordt SE et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Kwan KY, Glazer JM, Corey DP, Rice FL & Stucky CL TRPA1 modulates mechanotransduction in cutaneous sensory neurons. Journal of Neuroscience 29, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aubdool AA et al. TRPA1 is essential for the vascular response to environmental cold exposure. Nature Communications 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viana F TRPA1 channels: molecular sentinels of cellular stress and tissue damage. The Journal of Physiology 594, 4151–4169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gong J et al. A Cold-Sensing Receptor Encoded by a Glutamate Receptor Gene. Cell 178, 1375–1386.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noël J et al. The mechano-activated K± channels TRAAK and TREK-1 control both warm and cold perception. EMBO Journal 28, 1308–1318 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong K & Driscoll M A transmembrane domain of the putative channel subunit MEC-4 influences mechanotransduction and neurodegeneration in C. elegans. Nature 367, 470–473 (1994). [DOI] [PubMed] [Google Scholar]
- 71.Kernan M, Cowan D & Zuker C Genetic dissection of mechanosensory transduction: Mechanoreception-defective mutations of drosophila. Neuron 12, 1195–1206 (1994). [DOI] [PubMed] [Google Scholar]
- 72.Sukharev SI, Blount P, Martinac B, Blattnert FR & Kung C A large-conductance mechanosensitive channel in E. coli endoced by mscL alone. Nature 368, 265–268 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Coste B et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Volkers L, Mechioukhi Y & Coste B Piezo channels: from structure to function. Pflugers Archiv European Journal of Physiology 467, 95–99 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Coste B et al. Piezo1 ion channel pore properties are dictated by C-terminal region. Nature Communications 6, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Q, Zhou H, Li X & Xiao B The mechanosensitive Piezo1 channel: a three-bladed propeller-like structure and a lever-like mechanogating mechanism. FEBS Journal 286, 2461–2470 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Wang L et al. Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 573, 225–229 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Zhao Q et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature 554, 487–492 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Cox CD, Bavi N & Martinac B Biophysical Principles of Ion-Channel-Mediated Mechanosensory Transduction. Cell Reports 29, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Syeda R et al. Piezo1 Channels Are Inherently Mechanosensitive. Cell Reports 17, 1739–1746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taberner FJ et al. Structure-guided examination of the mechanogating mechanism of PIEZO2. Proceedings of the National Academy of Sciences of the United States of America 116, 14260–14269 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu J, Lewis AH & Grandl J Touch, Tension, and Transduction – The Function and Regulation of Piezo Ion Channels. Trends in Biochemical Sciences 42, 57–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ranade SS et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proceedings of the National Academy of Sciences of the United States of America 111, 10347–10352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Woo S-H et al. Piezo2 is the principal mechanotransduction channel for proprioception. Nature Neuroscience 18, 1756–1762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chesler AT et al. The Role of PIEZO2 in Human Mechanosensation. New England Journal of Medicine 375, 1355–1364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delle Vedove A et al. Biallelic Loss of Proprioception-Related PIEZO2 Causes Muscular Atrophy with Perinatal Respiratory Distress, Arthrogryposis, and Scoliosis. American Journal of Human Genetics 99, 1206–1216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mahmud AA et al. Loss of the proprioception and touch sensation channel PIEZO2 in siblings with a progressive form of contractures. Clinical Genetics 91, 470–475 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Szczot M et al. PIEZO2 mediates injury-induced tactile pain in mice and humans. Science Translational Medicine 10, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murthy SE et al. The mechanosensitive ion channel Piezo2 mediates sensitivity to mechanical pain in mice. Science Translational Medicine 10, eaat9897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Murthy SE et al. OSCA/TMEM63 are an evolutionarily conserved family of mechanically activated ion channels. eLife 7, 1–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brohawn SG, Su Z & MacKinnon R Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K + channels. Proceedings of the National Academy of Sciences 111, 3614–3619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kefauver JM, Ward AB & Patapoutian A Discoveries in structure and physiology of mechanically activated ion channels. Nature 587, 567–576 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beaulieu-Laroche L et al. TACAN Is an Ion Channel Involved in Sensing Mechanical Pain. Cell 180, 956–967.e17 (2020). [DOI] [PubMed] [Google Scholar]
- 94.Niu Y et al. Analysis of the Mechanosensor Channel Functionality of TACAN Laboratory of Molecular Neurobiology and Biophysics, Laboratory of Mass Spectrometry and Gaseous Ion Chemistry, 3 Proteomics Resource Center, Rockefeller University, 4 Howard Hughes Medical. bioRxiv (2021). [Google Scholar]
- 95.Xue, Zeng W et al. TMEM12A is a coenzyme A-binding membrane protein with structural similarities to ELOVL fatty acid elongase. eLife doi: 10.75, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bonet IJM, Araldi D, Bogen O & Levine JD Involvement of TACAN, a Mechanotransducing Ion Channel, in Inflammatory But Not Neuropathic Hyperalgesia in the Rat. The journal of pain 22, 498–508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arenas OM & Lumpkin EA Touching Base with Mechanical Pain. Cell 180, 824–826 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Catterall WA Cellular and molecular biology of voltage-gated sodium channels. Physiological Reviews 72, (1992). [DOI] [PubMed] [Google Scholar]
- 99.Isom LL Sodium channel β subunits: Anything but auxiliary. Neuroscientist 7, 42–54 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Grieco TM, Malhotra JD, Chen C, Isom LL & Raman IM Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45, 233–244 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Djouhri L et al. The TTX-resistant sodium channel Nav 1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. Journal of Physiology 550, 739–752 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Djouhri L et al. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel α subunit protein. Journal of Physiology 546, 565–576 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fang X et al. The presence and role of the tetrodotoxin-resistant sodium channel Nav1.9 (NaN) in nociceptive primary afferent neurons. Journal of Neuroscience 22, 7425–7433 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bennett DL, Clark XAJ, Huang J, Waxman SG & Dib-Hajj SD The role of voltage-gated sodium channels in pain signaling. Physiological Reviews 99, 1079–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 105.Feldman EL et al. Diabetic neuropathy. Nature Reviews Disease Primers 5, (2019). [DOI] [PubMed] [Google Scholar]
- 106.Blair NT & Bean BP Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. Journal of Neuroscience 22, 10277–10290 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nassar MA et al. Nociceptor-specific gene deletion reveals a major role for Nav 1.7 (PN1) in acute and inflammatory pain. Proceedings of the National Academy of Sciences of the United States of America 101, 12706–12711 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.King GF & Vetter I No Gain, No Pain: Na1.7 as an Analgesic Target. ACS Chemical Neuroscience 5, 749–751 (2014). [DOI] [PubMed] [Google Scholar]
- 109.Gold MS Sodium channels and pain therapy. Current Opinion in Anaesthesiology 13, 565–572 (2000). [DOI] [PubMed] [Google Scholar]
- 110.McDermott LA et al. Defining the Functional Role of Na V 1.7 in Human Nociception. Neuron 101, 905–919.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fontaine B, Plassart-Schiess E & Nicole S Diseases caused by voltage-gated ion channels. Molecular Aspects of Medicine 18, 415–463 (1997). [DOI] [PubMed] [Google Scholar]
- 112.Wallner M, Meera P & Toro L Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: A transmembrane β-subunit homolog. Proceedings of the National Academy of Sciences of the United States of America 96, 4137–4142 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Djouhri L, Bleazard L & Lawson SN Association of somatic action potential shape with sensory receptive properties in guinea-pig dorsal root ganglion neurones. Journal of Physiology 513, 857–872 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fang X, McMullan S, Lawson SN & Djouhri L Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. Journal of Physiology 565, 927–943 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Villière V & McLachlan EM Electrophysiological properties of neurons in intact rat dorsal root ganglia classified by conduction velocity and action potential duration. Journal of Neurophysiology 76, 1924–1941 (1996). [DOI] [PubMed] [Google Scholar]
- 116.Boada MD, Ririe DG & Eisenach JC Post-discharge hyperpolarization is an endogenous modulatory factor limiting input from fast-conducting nociceptors (AHTMRs). Molecular Pain 13, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Basbaum AI, Bautista DM, Scherrer G & Julius D Cellular and molecular mechanisms of pain. Cell 139, 267–84 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zimmermann M Pathobiology of neuropathic pain. European Journal of Pharmacology 429, 23–37 (2001). [DOI] [PubMed] [Google Scholar]
- 119.Rush AM et al. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579.1, 1–14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lehmann HC, Wunderlich G, Fink GR & Sommer C Diagnosis of peripheral neuropathy. Neurological Research and Practice 2, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.S AM Pathophysiology of carpal tunnel syndrome. Neurosciences 20, 4–9 (2015). [PMC free article] [PubMed] [Google Scholar]
- 122.Treaster DE & Burr D Gender differences in prevalence of upper extremity musculoskeletal disorders. Ergonomics 47, 495–526 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Gasparotti R, Padua L, Briani C & Lauria G New technologies for the assessment of neuropathies. Nature Reviews Neurology 13, 203–216 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Kim JY, Kim J Il, Son JE & Yun SK Prevalence of carpal tunnel syndrome in meat and fish processing plants. Journal of Occupational Health 46, 230–234 (2004). [DOI] [PubMed] [Google Scholar]
- 125.Bennett GJ & Xie YK A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107 (1988). [DOI] [PubMed] [Google Scholar]
- 126.Decosterd I & Woolf CJ Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Campbell JN & Meyer RA Mechanisms of Neuropathic Pain. Neuron 52, 77–92 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gaudet AD, Popovich PG & Ramer MS Wallerian degeneration : Gaining perspective on inflammatory events after peripheral nerve injury. Jounal of Neuroinflammation 8, 1–13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Saperstein DS Small Fiber Neuropathy. Neurologic Clinics 38, 607–618 (2020). [DOI] [PubMed] [Google Scholar]
- 130.Tesfaye S et al. Diabetic Neuropathies: Update on Definitions, Diagnostic Criteria, Estimation of Severity, and Treatments. Diabetes Care 33, 2285–2293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sopacua M et al. Small-fiber neuropathy: Expanding the clinical pain universe. Journal of the Peripheral Nervous System 24, 19–33 (2019). [DOI] [PubMed] [Google Scholar]
- 132.IDF Atlas-9th edition. International Diabetes Federation. (2019). [PubMed] [Google Scholar]
- 133.Tabish SA Is Diabetes Becoming the Biggest Epidemic of the Twenty-first Century? International journal of health sciences 1, V–VIII (2007). [PMC free article] [PubMed] [Google Scholar]
- 134.Acker K. Van, Bouhassira D, Bacquer D. De, Weiss S & Matthys K Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes and Metabolism 35, 206–213 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Truini A et al. A cross-sectional study investigating frequency and features of definitely diagnosed diabetic painful polyneuropathy. Pain 159, 2658–2666 (2018). [DOI] [PubMed] [Google Scholar]
- 136.Boyle JP, Thompson TJ, Gregg EW, Barker LE & Williamson DF Projection of the year 2050 burden of diabetes in the US adult population: Dynamic modeling of incidence, mortality, and prediabetes prevalence. Population Health Metrics 8, 1–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sasaki H et al. Spectrum of diabetic neuropathies. Diabetology International 11, 87–96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Galosi E et al. Differential involvement of myelinated and unmyelinated nerve fibers in painful diabetic polyneuropathy. Muscle and Nerve 63, 68–74 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Carrington A & Litchrifeld J The Aldose Reductase Pathway and Non-enzymatic Glycation in the Pathogenesis of Diabetic Neuropathy: A Critical Review for the End of the 20th Century. Diabetes Review 7, 275–299 (1999). [Google Scholar]
- 140.Ryle C & Donaghy M Non-enzymatic glycation of peripheral nerve proteins in human diabetics. Journal of the Neurological Sciences 129, 62–68 (1995). [DOI] [PubMed] [Google Scholar]
- 141.Figueroa-Romero C, Sadidi M & Feldman EL Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Reviews in Endocrine and Metabolic Disorders 9, 301–314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gumy LF, Bampton ETW & Tolkovsky AM Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Molecular and Cellular Neuroscience 37, 298–311 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Abbott CA, Malik RA, Van Ross ERE, Kulkarni J & Boulton AJM Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 34, 2220–2224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Frank T, Nawroth P & Kuner R Structure-function relationships in peripheral nerve contributions to diabetic peripheral neuropathy. Pain 160, S29–S36 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Harding JL, Pavkov ME, Magliano DJ, Shaw JE & Gregg EW Global trends in diabetes complications: a review of current evidence. Diabetologia 62, 3–16 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Devor M Rethinking the causes of pain in herpes zoster and postherpetic neuralgia: The ectopic pacemaker hypothesis. Pain Reports 3, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun W et al. Reduced conduction failure of the main axon of polymodal nociceptive C-fibres contributes to painful diabetic neuropathy in rats. Brain 135, 359–375 (2012). [DOI] [PubMed] [Google Scholar]
- 148.Adhya P & Sharma SS Redox TRPs in diabetes and diabetic complications: Mechanisms and pharmacological modulation. Pharmacological Research 146, 104271 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Calcutt NA Diabetic neuropathy and neuropathic pain: a (con)fusion of Pathogenic Mechanisms? Pain 161, S65–S86 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cheng YJ et al. Peripheral insensate neuropathy - A tall problem for US adults? American Journal of Epidemiology 164, 873–880 (2006). [DOI] [PubMed] [Google Scholar]
- 151.Malik RA et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 48, 578–585 (2005). [DOI] [PubMed] [Google Scholar]
- 152.Fallon MT & Colvin L Neuropathic pain in cancer. British Journal of Anaesthesia 111, 105–111 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Cavaletti G et al. Chemotherapy-induced peripheral neurotoxicity: A multifaceted, still unsolved issue. Journal of the Peripheral Nervous System 24, S6–S12 (2019). [DOI] [PubMed] [Google Scholar]
- 154.Cavaletti G & Marmiroli P Chemotherapy-induced peripheral neurotoxicity. Current Opinion in Neurology 28, 500–507 (2015). [DOI] [PubMed] [Google Scholar]
- 155.Grisold W, Cavaletti G & Windebank AJ Peripheral neuropathies from chemotherapeutics and targeted agents: Diagnosis, treatment, and prevention. Neuro-Oncology 14, 45–54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Starobova H & Vetter I Pathophysiology of chemotherapy-induced peripheral neuropathy. Frontiers in Molecular Neuroscience 10, 1–21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Flatters SJL, Dougherty PM & Colvin LA Clinical and preclinical perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): A narrative review. British Journal of Anaesthesia 119, 737–749 (2017). [DOI] [PubMed] [Google Scholar]
- 158.Cata JP et al. Quantitative Sensory Findings in Patients With Bortezomib-Induced Pain. Journal of Pain 8, 296–306 (2007). [DOI] [PubMed] [Google Scholar]
- 159.Boyette-Davis JA et al. Follow-up psychophysical studies in bortezomib-related chemoneuropathy patients. Journal of Pain 12, 1017–1024 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Argyriou AA et al. Neurophysiological, nerve imaging and other techniques to assess chemotherapy-induced peripheral neurotoxicity in the clinical and research settings. Journal of Neurology, Neurosurgery and Psychiatry 90, 1361–1369 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Boyette-Davis JA et al. Persistant chemotherapy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer Chemother Pharmacol 71, 619–626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shim HS et al. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Molecular Pain 15, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Flatters SJL & Bennett GJ Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: Evidence for mitochondrial dysfunction. Pain 122, 245–257 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Krukowski K, Nijboer CH, Huo X, Kavelaars A & Heijnen CJ Prevention of chemotherapy-induced peripheral neuropathy by the small-molecule inhibitor pifithrin-μ. Pain 156, 2184–2192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Barrire DA et al. Paclitaxel therapy potentiates cold hyperalgesia in streptozotocin-induced diabetic rats through enhanced mitochondrial reactive oxygen species production and TRPA1 sensitization. Pain 153, 553–561 (2012). [DOI] [PubMed] [Google Scholar]
- 166.Xiao WH, Zheng H & Bennett GJ Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 203, 194–206 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zheng H, Xiao WH & Bennett GJ Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Experimental Neurology 238, 225–234 (2012). [DOI] [PubMed] [Google Scholar]
- 168.Kim HK, Zhang YP, Gwak YS & Abdi S Phenyl N-tert-butylnitrone, a free radical scavenger, reduces mechanical allodynia in chemotherapy-induced neuropathic pain in rats. Anesthesiology 112, 432–439 (2010). [DOI] [PubMed] [Google Scholar]
- 169.Fidanboylu M, Griffiths LA & Flatters SJL Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PLoS ONE 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aromolaran KA & Goldstein PA Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy: Cause and effect? Molecular Pain 13, 1–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Materazzi S et al. TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflugers Archiv European Journal of Physiology 463, 561–569 (2012). [DOI] [PubMed] [Google Scholar]
- 172.Kennedy PGE, Grinfeld E & Gow JW Latent varicella-zoster virus in human dorsal root ganglia. Virology 258, 451–454 (1999). [DOI] [PubMed] [Google Scholar]
- 173.Gershon AA et al. Varicella zoster virus infection. Nature Reviews Disease Primers 1, 1–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gershon AA et al. Latency of varicella zoster virus in dorsal root, cranial, and enteric ganglia in vaccinated children. Transactions of the American Clinical and Climatological Association 123, 17–35 (2012). [PMC free article] [PubMed] [Google Scholar]
- 175.Kennedy PGE Key issues in varicella-zoster virus latency. Journal of neurovirology 8 Suppl 2, 80–84 (2002). [DOI] [PubMed] [Google Scholar]
- 176.Dworkin RH & Portenoy RK Proposed classification of herpes zoster pain. Lancet (London, England) 343, 1648 (1994). [DOI] [PubMed] [Google Scholar]
- 177.Fields HL, Rowbotham M & Baron R Postherpetic neuralgia: Irritable nociceptors and deafferentation. Neurobiology of Disease 5, 209–227 (1998). [DOI] [PubMed] [Google Scholar]
- 178.Hadley GR et al. Post-herpetic Neuralgia: a Review. Current Pain and Headache Reports 20, 1–5 (2016). [DOI] [PubMed] [Google Scholar]
- 179.Guedon JMG et al. Neuronal changes induced by Varicella Zoster Virus in a rat model of postherpetic neuralgia. Virology 482, 167–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gross F & Üçeyler N Mechanisms of small nerve fiber pathology. Neurosicence Letters 737, 1–6 (2020). [DOI] [PubMed] [Google Scholar]
- 181.Cregg R, Momin A, Rugiero F, Wood JN & Zhao J Pain channelopathies. J Physiology 588, 1897–1904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dib-hajj SD, Geha P & Waxman SG Sodium channels in pain disorders: pathophysiology and prospects for treatment. Pain 158, S97–S107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bernardes TP, Foresto RD & Kirsztajn GM Fabry disease: genetics, pathology, and treatment. Revista da Associacao Medica Brasileira (1992) 66Suppl 1, s10–s16 (2020). [DOI] [PubMed] [Google Scholar]
- 184.Miller JJ, Kanack AJ & Dahms NM Progress in the understanding and treatment of Fabry disease. Biochimica et Biophysica Acta - General Subjects 1864, 129437 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Üçeyler N, Ganendiran S, Kramer D & Sommer C Characterization of Pain in Fabry Disease. The Clinical Journal of Pain 30, 915–920 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Arends M et al. Phenotype, disease severity and pain are major determinants of quality of life in Fabry disease: results from a large multicenter cohort study. Journal of Inherited Metabolic Disease 41, 141–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rombach SM, Hollak CE, Linthorst GE & Dijkgraaf MG Cost-effectiveness of enzyme replacement therapy for Fabry disease. Orphanet Journal of Rare Diseases 8, 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Meikle PJ Prevalence of Lysosomal Storage Disorders. Jama 281, 249 (1999). [DOI] [PubMed] [Google Scholar]
- 189.Zarate YA & Hopkin RJ Fabry’s disease. The Lancet 372, 1427–1435 (2008). [DOI] [PubMed] [Google Scholar]
- 190.Elliott S et al. Dataset and standard operating procedure for newborn screening of six lysosomal storage diseases: By tandem mass spectrometry. Data in Brief 8, 915–924 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Burand A & Stucky C Fabry disease pain: patient and preclinical parallels. Pain doi: 10.10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yatziv S & Devor M Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain 160, 2105–2114 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Miller JJ et al. Neuropathic pain in a Fabry disease rat model. 3, 1–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ohnishi A & Dyck PJ Loss of small peripherial sensory neurons in fabry disease. Archives of Neurology 31, 120–127 (1974). [DOI] [PubMed] [Google Scholar]
- 195.Scott LJ et al. Quantitative analysis of epidermal innervation in Fabry disease. Neurology 52, 1249–1254 (1999). [DOI] [PubMed] [Google Scholar]
- 196.Torvin Møller A et al. Functional and structural nerve fiber findings in heterozygote patients with Fabry disease. Pain 145, 237–245 (2009). [DOI] [PubMed] [Google Scholar]
- 197.Hofmann L et al. Characterization of small fiber pathology in a mouse model of fabry disease. eLife 7, 1–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Geevasinga N, Tchan M, Sillence D & Vucic S Upregulation of inward rectifying currents and Fabry disease neuropathy. Journal of the peripheral nervous system : JPNS 17, 399–406 (2012). [DOI] [PubMed] [Google Scholar]
- 199.Basso L & Altier C Transient Receptor Potential Channels in neuropathic pain. Current Opinion in Pharmacology 32, 9–15 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Ellis A & Bennett DLH Neuroinflammation and the generation of neuropathic pain. British Journal of Anaesthesia 111, 26–37 (2013). [DOI] [PubMed] [Google Scholar]
- 201.Gold MS & Gebhart GF Nociceptor sensitization in pain pathogenesis. Nature Medicine 16, 1248–1257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Julius D TRP channels and pain. Annual review of cell and developmental biology 29, (2013). [DOI] [PubMed] [Google Scholar]
- 203.Lolignier S, Eijkelkamp N & Wood JN Mechanical allodynia. Pflugers Archiv : European journal of physiology 467, 133–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Luo J, Feng J, Liu S, Walters ET & Hu H Molecular and Cellular Mechanisms that Initiate Pain and Itch. Cellular Molecular Life Sciences 72, 3201–3223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Stein C et al. Peripheral mechanisms of pain and analgesia. Brain Research Reviews 60, 90–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stucky CL et al. Roles of transient receptor potential channels in pain. Brain Research Reviews 60, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Woolf CJ & Ma Q Nociceptors-Noxious Stimulus Detectors. Neuron 55, 353–364 (2007). [DOI] [PubMed] [Google Scholar]
- 208.North RY et al. Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142, 1215–1226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li Y et al. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. The Journal of Neuroscience 38, 1124–1136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ji R, Chamessian A & Zhang Y Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wei Z, Fei Y, Su W & Chen G Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Frontiers in Cellular Neuroscience 13, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kocot-Kepska M et al. Peripheral Mechanisms of Neuropathic Pain — The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain. Pharmaceuticals 14, 1–22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Abram SE, Yi J, Fuchs A & Hogan QH Permeability of injured and intact peripheral nerves and dorsal root ganglia. Anesthesiology 105, 146–153 (2006). [DOI] [PubMed] [Google Scholar]
- 214.Jimenez-Andrade JM et al. Vascularization of the Dorsal Root Ganglia and Peripheral Nerve of the Mouse: Implications for Chemical-Induced Peripheral Sensory Neuropathies. Molecular Pain 4, 1744–8069-4–10 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jacobs JM, Macfarlane RM & Cavanagh JB Vascular leakage in the dorsal root ganglia of the rat, studied with horseradish peroxidase. Journal of the Neurological Sciences 29, 95–107 (1976). [DOI] [PubMed] [Google Scholar]
- 216.Fuchs E Keratins and the skin. Annual Review of Cell and Developmental Biology 11, 123–153 (1995). [DOI] [PubMed] [Google Scholar]
- 217.Abraira VE & Ginty DD The sensory neurons of touch. Neuron 79, 618–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kruger L, Perl ER & Sedivec MJ Fine Structure of Myelinated Mechanical Nociceptor Endings in Cat Hairy Skin. J Comp Neurol 198, 137–154 (1981). [DOI] [PubMed] [Google Scholar]
- 219.Owens DM & Lumpkin EA Diversification and specialization of touch receptors in skin. Cold Spring Harbor perspectives in medicine 4, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Talagas M et al. Intra-epidermal nerve endings progress within keratinocyte cytoplasmic tunnels in normal human skin. Experimental Dermatology 29, 387–392 (2020). [DOI] [PubMed] [Google Scholar]
- 221.Pang Z et al. Selective keratinocytes stimulation is sufficient to evoke nociception in mice. Pain (2015). [DOI] [PubMed] [Google Scholar]
- 222.Baumbauer KM et al. Keratinocytes can modulate and directly initiate nociceptive responses. eLife 4, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Moehring F et al. Keratinocytes mediate innocuous and noxious touch via ATP-P2X4 signaling. eLife 7, 1–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sadler KE, Moehring F & Stucky CL Keratinocytes are required for normal cold and heat sensation. 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Moehring F, Halder P, Seal RP & Stucky CL Uncovering the Cells and Circuits of Touch in Normal and Pathological Settings. Neuron 100, 349–360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Inoue K et al. Mechanism underlying ATP release in human epidermal keratinocytes. Journal of Investigative Dermatology 134, 1465–1468 (2014). [DOI] [PubMed] [Google Scholar]
- 227.Barr TP et al. Air-Stimulated ATP Release from Keratinocytes Occurs through Connexin Hemichannels. PLoS ONE 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Talagas M et al. Keratinocytes Communicate with Sensory Neurons via Synaptic-like Contacts. Annals of Neurology 88, 1205–1219 (2020). [DOI] [PubMed] [Google Scholar]
- 229.Halprin KM Epidermal ‘turnover time’--a re-examination. The British journal of dermatology 86, 14–19 (1972). [DOI] [PubMed] [Google Scholar]
- 230.Potten CS, Saffhill R & Maibach HI Measurement of the transit time for cells through the epidermis and stratum corneum of the mouse and guinea-pig. Cell and tissue kinetics 20, 461–472 (1987). [DOI] [PubMed] [Google Scholar]
- 231.Abdo H et al. Specialized cutaneous Schwann cells initiate pain sensation. Science (New York, N.Y.) 365, 695–699 (2019). [DOI] [PubMed] [Google Scholar]
- 232.Martin SF Immunological mechanisms in allergic contact dermatitis. Current Opinion in Allergy and Clinical Immunology 15, 124–130 (2015). [DOI] [PubMed] [Google Scholar]
- 233.McCormick T, Ayala-Fontanez N & Soler D Current knowledge on psoriasis and autoimmune diseases. Psoriasis: Targets and Therapy 7 (2016). doi: 10.2147/ptt.s64950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chen Y et al. Transient Receptor Potential Vanilloid 4 Ion Channel Functions as a Pruriceptor in Epidermal Keratinocytes to Evoke Histaminergic Itch *. Journal Biological Chemistry 291, 10252–10262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Chen Y et al. Epithelia-sensory neuron crosstalk underlies cholestatic itch induced by lysophosphatidylcholine. Gastroenterology in press, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Moore C et al. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proceedings of the National Academy of Sciences 110, 15502–15502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kreß L et al. Differential impact of keratinocytes and fibroblasts on nociceptor degeneration and sensitization in small fiber neuropathy. Pain 162, 1262–1272 (2020). [DOI] [PubMed] [Google Scholar]
- 238.Radtke C, Vogt PM, Devor M & Kocsis JD Keratinocytes acting on injured afferents induce extreme neuronal hyperexcitability and chronic pain. Pain 148, 94–102 (2010). [DOI] [PubMed] [Google Scholar]
- 239.Talagas M & Misery L Role of Keratinocytes in Sensitive Skin. Frontiers in Medicine 6, 1–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hou Q et al. Keratinocyte expression of calcitonin gene-related peptide β: Implications for neuropathic and inflammatory pain mechanisms. Pain 152, 2036–2051 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhao P et al. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: Evidence for a role in pain. Pain 139, 90–105 (2008). [DOI] [PubMed] [Google Scholar]
- 242.Karl F et al. Fibromyalgia versus small fiber neuropathy: diverse keratinocyte transcriptome signature. Pain (2021). doi: 10.1097/j.pain.0000000000002249 [DOI] [PubMed] [Google Scholar]
- 243.Gregory NS et al. An overview of animal models of pain: Disease models and outcome measures. Journal of Pain 14, 1255–1269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sperry MM, Yu YH, Welch RL, Granquist EJ & Winkelstein BA Grading facial expression is a sensitive means to detect grimace differences in orofacial pain in a rat model. Scientific Reports 8, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Roughan JV, Wright-Williams SL & Flecknell PA Automated analysis of postoperative behaviour: Assessment of homecagescan as a novel method to rapidly identify pain and analgesic effects in mice. Laboratory Animals 43, 17–26 (2009). [DOI] [PubMed] [Google Scholar]
- 246.Ochoa JL, Campero M, Serra J & Bostock H Hyperexcitable polymodal and insensitive nociceptors in painful human neuropathy. Muscle & nerve 32, 459–472 (2005). [DOI] [PubMed] [Google Scholar]
- 247.Serra J et al. Hyperexcitable C nociceptors in fibromyalgia. Annals of Neurology 75, 196–208 (2014). [DOI] [PubMed] [Google Scholar]
- 248.Serra J et al. Effects of a T-type calcium channel blocker, ABT-639, on spontaneous activity in C-nociceptors in patients with painful diabetic neuropathy: A randomized controlled trial. Pain 156, 2175–2183 (2015). [DOI] [PubMed] [Google Scholar]
- 249.Kleggetveit Petter I et al. High spontaneous activity of C-nociceptors in painful polyneuropathy. 153, 2040–2047 (2012). [DOI] [PubMed] [Google Scholar]
- 250.Jankowski MP et al. Sensitization of cutaneous nociceptors after nerve transection and regeneration: Possible role of target-derived neurotrophic factor signaling. Journal of Neuroscience 29, 1636–1647 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Boada MD et al. Nerve injury induces a new profile of tactile and mechanical nociceptor input from undamaged peripheral afferents. Journal of Neurophysiology 113, 100–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Nickolls AR et al. Transcriptional Programming of Human Mechanosensory Neuron Subtypes from Pluripotent Stem Cells Resource Transcriptional Programming of Human Mechanosensory Neuron Subtypes from Pluripotent Stem Cells. Cell reports 30, 932–946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Rostock C, Schrenk-Siemens K, Pohle J & Siemens J Human vs. Mouse Nociceptors – Similarities and Differences. Neuroscience 387, 13–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Schrenk-Siemens K et al. HESC-derived sensory neurons reveal an unexpected role for PIEZO2 in nociceptor mechanotransduction. bioRxiv (2020). [Google Scholar]
- 255.Mittal K & Schrenk-Siemens K Lessons from iPSC research: Insights on peripheral nerve disease. Neuroscience Letters 738, 135358 (2020). [DOI] [PubMed] [Google Scholar]
- 256.Moehring F et al. Keratinocytes mediate innocuous and noxious touch via ATP-P2X4 signaling. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Denda M et al. Immunoreactivity of VR1 on epidermal keratinocyte of human skin. Biochemical and biophysical research communications 285, 1250–2 (2001). [DOI] [PubMed] [Google Scholar]
- 258.Denda M, Tsutsumi M & Denda S Topical application of TRPM8 agonists accelerates skin permeability barrier recovery and reduces epidermal proliferation induced by barrier insult: role of cold-sensitive TRP receptors in epidermal permeability barrier homoeostasis. Experimental dermatology 19, 791–5 (2010). [DOI] [PubMed] [Google Scholar]
- 259.Inoue K et al. Functional vanilloid receptors in cultured normal human epidermal keratinocytes. Biochemical and biophysical research communications 291, 124–9 (2002). [DOI] [PubMed] [Google Scholar]