

SUMMARY
The haploinsufficiency of C9orf72 is implicated in the most common forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), but the full spectrum of C9orf72 functions remains to be established. Here we report that C9orf72 is a mitochondrial inner membrane-associated protein regulating cellular energy homeostasis via its critical role in the control of oxidative phosphorylation (OXPHOS). The translocation of C9orf72 from the cytosol to the inter-membrane space is mediated by the redox-sensitive AIFM1/CHCHD4 pathway. In mitochondria, C9orf72 specifically stabilizes translocase of inner mitochondrial membrane domain containing 1 (TIMMDC1), a crucial factor for the assembly of OXPHOS Complex I. C9orf72 directly recruits the prohibitin complex to inhibit the m-AAA protease-dependent degradation of TIMMDC1. The mitochondrial Complex I function is impaired in C9orf72-linked ALS/FTD patient-derived neurons. These results reveal a previously unknown function of C9orf72 in mitochondria and suggests that defective energy metabolism may underlie the pathogenesis of relevant diseases.
Keywords: Mitochondrion, oxidative phosphorylation, protein transport, ALS, FTD
eTOC blurb
Wang et al. show that C9orf72, whose haploinsufficiency contributes to the pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia, is imported into mitochondria, where it regulates energy metabolism by maintaining the assembly and stability of specific protein complexes. The function of C9orf72 in the mitochondria highlights the importance of energy homeostasis in the pathogenesis of ALS and related neurodegenerative diseases.
Graphical Abstract
INTRODUCTION
The hexanucleotide repeat expansion in a noncoding region of the C9orf72 gene is the most common genetic cause of the neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (DeJesus-Hernandez et al., 2011; Renton et al., 2011), which primarily affect motor neurons and the frontotemporal lobes of the brain, respectively, supporting the notion that ALS and FTD are two conditions within a continuous clinical spectrum. The repeat expansion in C9orf72 has also appeared in rare cases of other neurological conditions such as Alzheimer’s disease (Harms et al., 2013; Majounie et al., 2012). The proposed mechanisms of pathogenesis involving the genetic anomaly in the C9orf72 gene include RNA toxicity (Donnelly et al., 2013), repeat-associated non-ATG-dependent translation (Ash et al., 2013; Mori et al., 2013), and loss of C9orf72 functions. These mechanisms have not been fully understood, although they may potentially act together to contribute to the neurodegenerative diseases (Taylor et al., 2016).
Multiple lines of evidence suggest that loss of C9orf72 function plays a role in ALS/FTD pathogenesis. A reduction in C9orf72 transcript and protein levels has consistently been found in the cells and tissues of ALS/FTD patients (DeJesus-Hernandez et al., 2011; Donnelly et al., 2013; Haeusler et al., 2014; Mori et al., 2013; Waite et al., 2014). Loss of C9orf72 hypersensitizes cells to stress (Maharjan et al., 2017), and C9orf72 deficiency exacerbates motor deficits in two mouse models expressing the repeat expansion (Shao et al., 2019; Zhu et al., 2020). Furthermore, haploinsufficiency of C9orf72 drives neurodegeneration in human motor neuronal cultures from patients (Shi et al., 2018). However, studies of contributions of loss of C9orf72 to the disease have been hampered by a limited understanding of its physiological functions. C9orf72 is a DENN-like protein that has been implicated in the regulation of autophagy and immunity (McCauley et al., 2020; O’Rourke et al., 2016; Sellier et al., 2016; Sivadasan et al., 2016; Su et al., 2020; Sullivan et al., 2016; Ugolino et al., 2016; Yang et al., 2016); however, the full spectrum of C9orf72 functions remains to be established.
Clinical and epidemiological studies indicate that dysregulated energy metabolism is closely related to ALS/FTD (Dupuis et al., 2011). The central organelle in the regulation of cellular energy metabolism is the mitochondrion, a double-membrane organelle where ATPs are generated through a metabolic process known as oxidative phosphorylation (OXPHOS), which requires five complexes (I to V) located on the mitochondrial inner membrane. Here, we describe findings that C9orf72 is a mitochondrial protein and that it plays a key role in maintaining respiratory chain complex integrity and cellular energy homeostasis.
RESULTS
C9orf72 is Imported to the Mitochondrial Intermembrane Space by AIFM1/CHCHD4
In a previous quantitative proteomic analysis for C9orf72 interactors co-immunoprecipitated from whole cell lysates (Liu et al., 2018), we found that 13 out of the 39 top binding partners of C9orf72 were mitochondrial proteins (Figure S1A; Table S1), raising the possibility that C9rof72 is located to the mitochondrion. To examine the mitochondrial association of C9orf72, we performed subcellular fractionation of mouse embryonic fibroblasts (MEFs) isolated from C9orf72-knockout (C9KO) and wild-type (WT) littermate mice. Endogenous C9orf72 was detected by immunoblotting in the mitochondrial fraction from only the WT MEFs (Figure S1B and S1C). To determine where C9orf72 resides within mitochondria, we subjected isolated mitochondria to osmotic stress together with proteinase K digestion (Figure S1B). When the mitochondria were intact, C9orf72 was protected from proteinase K digestion (Figure 1A, left panels); however, when the outer mitochondrial membrane (OMM) was broken by the osmotic stress treatment, C9orf72 was sensitive to the proteinase K digestion, a pattern identical to that for the inner mitochondrial membrane (IMM) protein marker TIM23 (Figure 1A, right panels), indicating that C9orf72 is exposed to the inter-membrane space (IMS). Furthermore, when the mitochondria were sonicated or treated with sodium carbonate (pH 11.5) to extract peripherally membrane-associated proteins from the membrane-containing pellet (Figure S1B) (Vogtle et al., 2017), a portion of C9orf72 was still present in the pellet fraction, in a pattern similar to that of a tight IMM-binding protein (MB) TIM44, suggesting that C9orf72 is an IMM-binding protein (Figure 1B). In comparison, the transmembrane proteins (M) TOM70 and TIM23, located on OMM and IMM, respectively, and the soluble protein (S) SOD1 showed different fractionation patterns than that of C9orf72 (Figure 1B). Furthermore, endogenous C9orf72 was shown to colocalized with the IMM marker TIM23 by immunostaining in WT MEFs, as compared to the negative control C9KO MEFs (Figure 1C). To verify the sub-mitochondrial location of C9orf72, we performed immuno-electron microscopy (immune-EM) analysis using isolated mitochondria (Figure S1B) and demonstrated that endogenous C9orf72 is localized to the IMM (Figure 1D).
Figure 1. C9orf72 is an Inner Mitochondrial Membrane Binding Protein.

(A) Isolated mitochondria (Mito) from WT and C9KO MEFs were subjected to digestion with proteinase K (PK). C9orf72, an IMM protein TIM23, a matrix protein NDUFB9, and OMM proteins TOM70 and TOM20 were used as controls.
(B) Mitochondria isolated from WT and C9KO MEFs were either sonicated or treated with sodium carbonate to extract peripheral membrane-binding proteins from the pellet fraction (P) to the supernatant fraction (S).
(C) Representative confocal images and quantification of colocalization of endogenous C9orf72 and TIM23 in WT and C9KO MEFs (n = 40 cells). Scale bars, 10 μm.
(D) C9orf72 immuno-electron microscopy (EM) analysis of WT and C9KO mitochondria isolated from mouse brain cortex tissues (n = 14 images). Red arrows point to immunogold-labeled C9orf72. Scale bars, 100nm.
(E and F) The gel-shift AMS-modification assay was used to measure the oxidation states of C9orf72 in cytosolic and mitochondrial fractions of HEK293 cells. R: Reduced. O(p): Partially oxidized. O: Oxidized. The oxidation states of native C9orf72 are quantified (n = 4) (F).
(G) HEK293 cells with knockdown of CHCHD4 or AIFM1 were subjected to subcellular fractionation and immunoblotting of indicated proteins (n = 4).
(H) Representative autoradiographs and quantitative analysis of [35S]C9orf72 imported into mitochondria isolated from HEK293 cells treated with the indicated siRNAs and proteinase K (n = 3).
(I) Kinetic analysis of the C9orf72 import reaction as shown in (H). A representative gel image and quantification are shown (n = 3).
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S1 and S2, and Table S1.
Our proteomic analysis and subsequent co-immunoprecipitation experiments indicate that one of the binding partners of C9orf72 is AIFM1 (Figure S1D; Table S1). The AIFM1/CHCHD4 oxidoreductase is responsible for importing a number of proteins containing a cysteine-rich region into mitochondrial IMS through a disulfide relay system (Hangen et al., 2015; Modjtahedi and Kroemer, 2016). We noted that six of the nine cysteines in C9orf72 are clustered in a small region and conserved across species (Figure S1E and S1F), suggesting that the mitochondrial localization of C9orf72 may be mediated by AIFM1/CHCHD4. Typically, substrates that utilize this mechanism are oxidized by the AIFM1/CHCHD4 complex in the IMS to form intramolecular disulfide bonds, which lock the substrate proteins into a folded conformation and trap them in the IMS (Modjtahedi et al., 2016). To test this model, we examined the formation of disulfide bonds by using the 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) shift assay (Kloppel et al., 2011), in which AMS modification of reduced cysteine residues results in a change in protein migration on the gel. We found that C9orf72 from the mitochondrial fraction was partially oxidized, as compared to the more reduced form of the protein from the cytosolic fraction isolated from MEFs (Figure 1E and 1F), indicating that endogenous C9orf72 localized to the mitochondria has more disulfide bonds than does C9orf72 localized to the cytosol. Similarly, exogenously introduced C9orf72 showed a higher level of oxidization in the mitochondria than in the cytosol of HEK293 cells (Figure S1G).
Next, we assessed whether the mitochondrial import of C9orf72 depends on the AIFM1/CHCHD4 complex. When AIFM1 or CHCHD4 was knocked down by siRNAs, the relative level of C9orf72 in the mitochondrial fraction was significantly decreased (Figure 1G). This change was similar to the decrease observed in a previously established substrate of AIFM1/CHCHD4, NDUFA8 (Hangen et al., 2015) (Figure 1G). We also performed in vitro mitochondrial import analysis of cell-free metabolically labeled C9orf72 protein. After incubation with radiolabeled C9orf72 protein, the mitochondria were treated with proteinase K to digest the proteins outside the organelle, and then the mitochondrial C9orf72 level was quantified by radiography. When AIFM1 or CHCHD4 was knocked down by siRNAs, the level of mitochondrial C9orf72 and the rate of its mitochondrial import were significantly decreased (Figure 1H and 1I). Conversely, loss of C9orf72 did not affect the mitochondrial import of the AIFM1/CHCHD4 substrate NDUFA8 (Figure S1H). Together, our data suggested that C9orf72 is a substrate of the AIFM1/CHCHD4 complex.
Since AIFM1/CHCHD4-dependent mitochondrial import is mediated by the disulfide relay system, we examined the cysteine residues involved in the IMS-targeting of C9orf72. We generated a series of cell lines stably expressing C9orf72 with one to four mutations over the six cysteines of the cysteine-rich (C-rich) region. None of the single or double cysteine mutants affected the IMS localization of C9orf72; however, either of the triple mutations, C3S-1 (C215/231/233S) or C3S-2 (C252/262/266S), covering the left or right half of the six cysteines, abolished the mitochondrial localization of the protein (Figure S2A and S2B). This result suggests that the C-rich region is critical for the import of C9orf72 into IMS and that, within the left or right half segment of the C-rich region, the proximate cysteines can compensate each other in the formation of the disulfide bonds required for mitochondrial retention (Figure S2C and S2D). The quadruple mutant C4S (C231/233/262/266S) also showed defective mitochondrial localization, suggesting that the four cystines are necessary for the disulfide bonds formation (Figure S2B–S2D). Collectively, these data demonstrate that C9orf72 is imported into IMS through the AIFM1/CHCHD4 pathway and that the cysteines in the C-rich region are critical for this process.
C9orf72 is Crucial for Complex I-mediated OXPHOS Activity under Energetic Stress
Since the mitochondrion is the major metabolic hub of the cell, we hypothesized that mitochondrial C9orf72 may play a role in controlling cellular metabolism. To understand the metabolic processes controlled by the C9orf72 protein, we performed a new whole-proteome tandem mass tag (TMT)-labeling quantitative mass spectrometry analysis and, in parallel, a global metabolite profiling of C9KO MEFs using the WT MEFs as controls. The proteomic analysis detected a total of 5576 proteins, among which 252 proteins were upregulated and 548 proteins downregulated by more than 1.5-fold in the C9KO MEFs (Figure S3A; Table S2). The metabolomic analysis revealed that 54 metabolites were significantly altered in C9KO MEFs (Figure S3B; Table S3). When the proteomic and metabolomic datasets were combined using a multi-Omics platform, we found through pathway enrichment analysis that the most significantly down-regulated pathway in the C9KO cells was OXPHOS (Figure S3C). Consistently, multiple proteins of OXPHOS Complex I (CI, NADH-ubiquinone reductase) were down-regulated and the substrates of CI, such as NADH, were accumulated (Figure S3B and S3E; Table S2 and S3). Accordingly, glycolysis, the other major cellular ATP-generating pathway, was enhanced in the C9KO MEFs (Figure S3D and S3F; Table S2 and S3). These data suggested that cells lacking C9orf72 exhibit dysregulated OXPHOS and stressed energy homeostasis.
We then examined how the loss of C9orf72 affected the activities of the mitochondrial OXPHOS pathway. First, we subjected C9KO and WT MEFs to a galactose sensitivity assay. Cells are forced to produce ATP through OXPHOS when glucose in the medium is replaced with galactose (Robinson et al., 1992; Whitfield et al., 1981). Hence, cells deficient in OXPHOS are less viable in the galactose medium. The survival of C9KO and WT MEFs grown in either glucose or galactose medium was measured by staining for annexin V, a marker for cell death, followed by microscopic examination (Figure 2A) or flow cytometry analysis (Figure S4A and S4B). Only minimal cell death occurred in either the WT or C9KO MEFs grown under normal glucose conditions. While the galactose treatment was toxic to all cells, the C9KO MEFs underwent a significantly higher rate of cell death than did the WT MEFs (Figure 2A and Figure S4A and S4B). Similar results were obtained in C9KO human HAP1 cells or HEK293 cells with C9orf72 knockdown (Figure S4C and S4D). Moreover, when the growth rates were normalized to those under the glucose condition, C9KO MEFs exhibited decreased cell viability compared to WT MEFs under the galactose condition (Figure 2B). To confirm the specificity of this finding, we generated two independent C9KO MEF lines that stably expressed human C9orf72 protein, which shares 98% identity with its mouse ortholog, to near physiological levels (Figure S4E). The restoration of C9orf72 significantly rescued the galactose-induced cell death in C9KO MEFs (Figure S4F), confirming that the loss of C9orf72 was responsible for the galactose sensitivity.
Figure 2. Loss of C9orf72 Decreases Complex I-associated Mitochondrial Function.

(A) Cell death of WT and C9KO MEFs, with or without galactose treatment for the indicated time periods, was detected by annexin V staining and microscopy (n = 4).
(B) Cell viability of WT and C9KO MEFs in galactose medium was examined and normalized to that under normal glucose condition (n = 3).
(C) MAPR of mitochondria isolated from WT and C9KO MEFs, with or without galactose treatment, by using different substrates that enter the respiratory chain at either CI or CII (n = 3). GM: glutamate plus malate. SR: succinate plus rotenone.
(D) Analysis of mitochondrial OCR in WT and C9KO MEFs. Cells were maintained in glucose or galactose medium for 16 h before OCR detection (n = 8). The dotted lines indicate the time of adding oligomycin (O), FCCP (F), and rotenone and antimycin A (R&A).
(E) Intracellular ATP concentrations in MEFs, with or without galactose treatment, were determined (n = 3).
(F) Representative pseudocolor ratiometric images and quantification of the ATP/ADP ratio in WT and C9KO MEFs expressing PercevalHR (n = 30 cells). Scale bars, 5 μm.
(G and H) Cell death (n = 3–5) and intracellular ATP concentrations (n = 3) in WT and C9KO MEFs, after being cultured under normal or low-glucose conditions for 72 h, was analyzed.
(I) Glycolysis in WT and C9KO MEFs was analyzed by measuring the concentration of its end product, L-lactate, secreted outside the cell, after culturing for 24 or 48 h (n = 3).
(J) Representative images and quantification of mitochondrial membrane potential (ΔΨm) in WT and C9KO MEFs by TMRM staining (n > 60 cells). Scale bars, 5 μm.
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S3 and S4, and Table S2 and S3.
Next, to further assess the C9orf72-associated OXPHOS activities, we measured the mitochondrial ATP production rate (MAPR) and oxygen consumption rates (OCR) in vitro using mitochondrial isolated from WT and C9KO MEFs. By using glutamate and malate (GM) as substrates, which are converted into NADH and thereby activate OXPHOS through CI, we found that MAPR was consistently decreased in the C9KO cells (Figure 2C). Furthermore, the CI-associated OXPHOS activity in the C9KO cells was decreased substantially when cells were preconditioned with the galactose medium. In contrast, the Complex II (CII, succinate dehydrogenase)-mediated OXPHOS activity was not altered in the C9KO cells, when succinate and rotenone (SR) were present to completely block CI activity and stimulate electron flow through CII (Figure 2C). Consistently, a significant reduction in OCR was observed in the galactose-stressed C9KO mitochondria when CI-associated OXPHOS was activated (Figure S4G). Lastly, by using inhibitors against each of the five OXPHOS complexes, we found that loss of C9orf72 specifically sensitized the cells to the toxicity induced by inhibition of CI but not other complexes under the galactose condition (Figure S4H). Together, our data suggest that C9orf72 is specifically required for CI-mediated OXPHOS functions.
To further understand the functional role of C9orf72 in CI-related mitochondrial respiration in cellulo, we used Seahorse analysis to monitor the OCRs in intact WT and C9KO MEFs. When grown in glucose medium, the WT and mutant cells did not show a significant difference in OCR (Figure 2D), suggesting that the perturbation by loss of C9orf72 can be compensated when the energetic demand is low under the basal condition. However, once cells were stressed by the galactose treatment, there was a significant decrease in both basal and maximal OCRs in the C9KO MEFs, as compared to WT MEFs (Figure 2D), in consistence with the decreased cell survival in the C9KO MEFs (Figure 2A). This result indicates that C9orf72 plays a critical role in the regulation of mitochondrial respiration under the stress conditions. Moreover, restoration of C9orf72 in the C9KO MEFs led to a significant increase in the basal and maximal OCR levels under galactose conditions (Figure S4I). Similar reductions in basal and maximal OCRs were observed in C9KO human HAP1 cells or HEK293 cells with C9orf72 knockdown after the galactose treatment (Figure S4J and S4K). Also, similar to the observation that loss of C9orf72 led to a mitochondrial defect that only manifested under the galactose stress condition (Figure 2D), we found that a moderate reduction of CI, achieved by selective knockdown of one of its subunits, showed defects in the mitochondrial OCR only under the galactose stress condition but not under the normal glucose condition, whereas a severe reduction of CI caused significant defects in OCR under both normal and stress conditions (Figure S4L and S4M), consistent with notion that C9orf72-related CI functions are crucial for maintaining sufficient mitochondrial respiration when cells undergo energetic stress.
In further support of the role of C9orf72 in OXPHOS under stress, the C9KO MEFs exhibited significant deficits in intracellular ATP levels as compared to WT MEFs in the galactose medium (Figure 2E). Moreover, restoration of C9orf72 expression in C9KO MEFs significantly increased the ATP levels (Figure S4N). In addition, we measured the cellular ATP/ADP ratio using a genetically encoded fluorescent ratiometric probe, PercevalHR (Tantama et al., 2013), and found a significant decrease in C9KO MEFs when compared to WT MEFs under the galactose-treatment conditions (Figure 2F). Moreover, in addition to the galactose treatment, we also observed that the C9KO MEFs exhibited reduced ATP levels and cell survival compared to WT MEFs when cells were stressed by glucose starvation (Figure 2G and 2H). The survival defect in the C9KO MEFs could be rescued by restoration of C9orf72 expression (Figure S4O). Notably, C9orf72 mutants with defective mitochondrial localization (C3S-1/C3S-2) failed to rescue the ATP production and cell survival phenotypes in the C9KO MEFs (Figure S4P and S4Q). Together, these data indicate that mitochondrial C9orf72 plays a critical role in the maintenance of mitochondrial OXPHOS activity under stress conditions.
Since impaired OXPHOS could lead to compensatory activation of glycolysis (Everts et al., 2012), we analyzed the glycolytic activity by measuring production of extracellular lactate in the cells lacking C9orf72. Consistent with the proteomic and metabolomic analyses (Figure S3D and S3F), the glycolytic activity was significantly enhanced in the C9KO MEFs when compared to WT MEFs (Figure 2I). This phenotype in C9KO MEFs could be rescued by C9orf72 restoration, confirming its dependence on C9orf72 (Figure S4R). This data suggests that glycolysis may serve as a compensatory energy source for cells with reduced C9orf72 functions under stress conditions.
We analyzed additional indicators of mitochondrial integrity and morphology in order to confirm the role of C9orf72 in regulating mitochondrial function. Mitochondrial membrane potential (Δψm) generated by OXPHOS complexes (I, III and IV) is an essential component for mitochondrial ATP synthesis. Using tetramethylrhodamine methyl ester (TMRM), a cell-permeant dye that accumulates in polarized mitochondria and thus measures Δψm, we found that the mitochondria in C9KO MEFs were significantly depolarized when compared to WT MEFs under both glucose and galactose-treatment conditions (Figure 2J), consistent with the compromised OXPHOS function in the absence of C9orf72. Treatment with an uncoupler, carbonyl cyanide m-chlorophenyl hydrazone (CCCP), significantly diminish the fluorescence signals. The general mitochondrial morphology, including total mitochondrial area and branch lengths, remained largely unaltered in the absence of C9orf72 (Figure S4S). Together, these data indicate that C9orf72 plays a critical role in the maintenance of mitochondrial CI-associated OXPHOS activity, energy homeostasis, and cellular health.
C9orf72 Regulates OXPHOS Complex I Assembly by Stabilizing Assembly Factor TIMMDC1
Next we investigated the molecular mechanism through which loss of C9orf72 alters CI-mediated OXPHOS functions. By immunoblotting for molecular markers of the mitochondrial respiratory chain complexes, we confirmed the specific downregulation of the CI in C9KO MEFs, whereas the markers of the other complexes remained unchanged (Figure 3A). Similar results were obtained using purified mitochondria from the brains and spinal cords of C9KO mice (Figure 3B).
Figure 3. C9orf72 is Required for the Effective Assembly of Mitochondrial Complex I.

(A) Relative abundance of OXPHOS complexes in WT and C9KO MEFs was determined by immunoblotting against their respective makers (n = 3).
(B) Western blotting analysis of the protein markers of OXPHOS complexes in isolated mitochondria of brain cortices and spinal cords from C9KO mice and WT littermates (n = 3).
(C) BN-PAGE of the OXPHOS complexes in the isolated mitochondria from WT and C9KO MEFs (n = 4). Mature supercomplex forms of CI (SCs) were quantified by measuring the levels of NDUFB9 and NDUFB11. The CI-bound supercomplex of CIII [CIII (SCs)] or that of CIV [CIV (SCs)] as well as the free forms of CIII (CIII2) or CIV (CIVn) were examined and quantified using the marker for CIII or CIV, UQCRC1 or MTCO1, respectively. TIM23 was used as a loading control.
(D) The rates for the assembly of the CI and CIII complexes in MEFs were analyzed by BN-PAGE after the cells were treated with chloramphenicol (CAM) to deplete CI and CIII, which were then allowed to recover (n = 4).
(E) The in vitro CI assembly assay was performed by incubating the isolated mitochondria and 35S-labeled CI subunit NDUFS8 for the indicated times in the presence or absence of CCCP (n = 4). The mature CI level was measured by BN-PAGE. [35S]NDUFS8 import into the mitochondria, measured by SDS-PAGE, served as an internal control.
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001. See also Figures S5 and S6, and Table S4.
CI, the first enzymatic complex of the respiratory chain, is composed of 44 subunits in humans (Guerrero-Castillo et al., 2017). It can be functionally divided into three distinct modules: NADH dehydrogenase (N module), ubiquinone reduction (Q module) and proton translocation (P module). Mature CI mainly exists in supercomplex forms (SCs) together with OXPHOS Complexes III and IV (I1+III2+IVn) (Acin-Perez et al., 2008). To ascertain whether C9orf72 plays a role in maintaining the higher-order integrity of the CI complexes, we utilized Blue-native (BN)-PAGE to visualize the intact complexes by immunoblotting to detect its subunits NDUFB9 and NDUFB11. CI SCs were significantly decreased in C9KO MEFs (Figure 3C). Consistently, immunoblots for the Complex III (CIII) subunit UQCRC1 revealed that the CI-bound form of CIII supercomplexes [CIII (SCs)] were significantly decreased in C9KO MEFs (Figure 3C). In contrast, the complex composed of CIII itself (CIII2) was increased in C9KO MEFs, when compared to WT MEFs. Likewise, loss of C9orf72 led to reduced Complex IV supercomplexes [CIV (SCs)], while the free forms of Complex IV (CIVn) were not changed in the C9KO mitochondria (Figure 3C). These data indicate that C9orf72 is required for the maintenance of the mature CI and its SCs. The decrease in CI components upon reduction of C9orf72 was confirmed in other cell types, including HEK293 and HAP1 cells (Figure S5A–S5C). The low levels of CI components in C9KO MEFs could be increased by restoration of the expression of C9orf72 but not its mutants with defective mitochondrial localization (Figure S5D–S5F).
Next, we determined whether the decrease in the levels of CI components in C9KO MEFs leads to reduced CI enzymatic activity. By determining the NADH oxidation rate or the NADH-ubiquinone reduction rate, we found that the CI NADH-dehydrogenase and NADH-ubiquinone reductase activities were decreased by approximately 25% in C9KO MEFs when compared to WT MEFs (Figure S5G and S5H). Furthermore, the impairment of CI activities in C9KO MEFs was alleviated by restoration of C9orf72 expression (Figure S5I and S5J), confirming the specific role of C9orf72 in maintaining CI activity.
Next, we investigated the mechanism through which C9orf72 regulates CI levels. While a previous transcriptome study suggested that loss of C9orf72 does not affect the transcription of genes encoding CI subunits (O’Rourke et al., 2016), it has been reported that CI assembly blockage is sufficient to induce a fast turnover of multiple CI subunits, leading to significantly reduction in the protein levels of almost all CI subunits (Guerrero-Castillo et al., 2017; Stroud et al., 2016; Zurita Rendon and Shoubridge, 2012). Therefore, we hypothesized that mitochondrial C9orf72 is necessary for normal assembly of CI, and loss of C9orf72 causes impaired CI assembly, leading to reduced CI subunits levels. CI assembly is a stepwise process consisted of a set of intermediates. When the mature and intermediate CI complexes were examined by two-dimensional (2D) gel electrophoresis, using NDUFB9 as the CI marker, we noted that, while the level of mature CI was decreased in the mitochondria from C9KO MEFs as compared to WT MEFs, CI intermediates with lower molecular weights accumulated in the absence of C9orf72 (Figure S6A). Such decrease of mature CI and the concurrent accumulation of its intermediates indicate an impaired CI assembly, which was also observed in cells treated with chloramphenicol (CAM), a chemical known to inhibit mitochondrial translation and completely block CI assembly (Figure S6B). To gain an overview of CI assembly as regulated by C9orf72, we conducted an SILAC-based complexome profiling of mitochondria isolated from WT and C9KO MEFs (Figure S6C) (Guarani et al., 2014). Consistent with the immunoblotting data, the quantitative mass spectrometry analysis indicated that most CI/CIII/CIV subunits exhibited decreased levels in mature SCs, accompanied by increased CIII2, in C9KO cells (Figure S6D–S6F; Table S4), while as controls the CII and CIV levels remained unchanged (Figure S6F; Table S4). Notably, in C9KO cells, CI subunits belonging to N module and distal P (PD) modules were accumulated in the gel slices corresponding to the lower-molecular-weight intermediates (Figure S6G and S6H; Table S4), suggesting that the assemblies of N and PD modules were impaired upon loss of C9orf72. Together, these data indicated that C9orf72 is required for the assembly of mature CI from its intermediates.
To determine the efficiency of CI assembly, we first treated the C9KO and WT MEFs with CAM to deplete mature OXPHOS complexes including CI and CIII (Ugalde et al., 2004). After removal of CAM, both CI and CIII assembly restarts and reaches its normal level in 24 h (Figure S6I). We found that the newly formed CI became significantly less in C9KO MEFs than in WT MEFs after 9 h of assembly recovery, suggesting a decrease in CI assembly in C9KO MEFs (Figure 3D). In contrast, the CIII assembly rate was not affected in C9KO MEFs. Next, taking advantage of the fact that CI subunits can be imported into mitochondria and assembled into mature CI in vitro (Stroud et al., 2013), we performed cell-free metabolic labeling to monitor the fate of a CI subunit, NDUFS8, during its importation into mitochondria and assembly into CI. The assembly rate of radiolabeled NDUFS8 into CI was decreased in the absence of C9orf72, as evidenced by significantly less accumulation of radiolabeled NDUFS8 in CI without C9orf72 in BN-PAGE analysis (Figure 3E, BN-PAGE panel). Meanwhile, the process of importing radiolabeled NDUFS8 into the mitochondria was unaffected, as shown by the SDS-PAGE analysis (Figure 3E, SDS-PAGE panel), indicating that the reduced incorporation of NDUFS8 into CI was a result of impaired assembly. Furthermore, treatment with CCCP eliminated the radiolabeled signals in the mature CI complex, confirming the specificity of the CI assembly (Figure 3E, BN-PAGE panel). Therefore, our data indicate that loss of C9orf72 reduces CI assembly efficiency and leads to the reduction in the level and function of mature CI complexes.
CI assembly requires multiple assembly factors (Guerrero-Castillo et al., 2017; Stroud et al., 2016). Since mitochondrial C9orf72 is found to be localized to the IMM, we asked whether C9orf72 interacts with any CI assembly factors and regulates the assembly process at this compartment. We found that TIMMDC1, one of the two known IMM-localized CI assembly factors (Guarani et al., 2014; Stroud et al., 2016), interacts with C9orf72, as shown by co-immunoprecipitation (co-IP) assays in both HEK293 cells and WT MEFs (Figure 4A and 4B). By using 2D gel electrophoresis, we observed that TIMMDC1 was associated with the mature CI and three intermediate complexes (subcomplex 1–3, Sub-C1–3) in MEFs (Figure 4C and 4D), consistent with a previous report of the association of TIMMDC1 with these subcomplexes (Guerrero-Castillo et al., 2017). Importantly, mitochondrial C9orf72 specifically co-migrated with the second TIMMDC1-containing subcomplex (Sub-C2) (Figure 4C and 4D). Together, these data indicate that C9orf72 interacts with TIMMDC1 in the CI-assembly intermediate Sub-C2.
Figure 4. C9orf72 Stabilizes CI Assembly Factor TIMMDC1 from Degradation.

(A and B) co-IP of TIMMDC1 with anti-C9orf72 antibody in isolated mitochondria from HEK293 cells (A), or from WT or C9KO MEFs (B).
(C and D) Two-dimensional BN/SDS–PAGE analysis of isolated mitochondria from WT and C9KO MEFs, followed by immunoblotting against TIMMDC1 and C9orf72 (C). The histograms of TIMMDC1- or C9orf72-containing complexes are shown in (D), indicating the migration distance and relative abundance of the complexes. Sub-C1–3: TIMMDC1-containing subcomplex 1–3. The gray dashed line marks the comigration of C9orf72 and TIMMDC1 in the position of subcomplex 2 (Sub-C2).
(E) Quantification of the relative abundance of TIMMDC1 in Sub-C1–3 and mature CI as shown in (C) (n = 5).
(F) TIMMDC1 levels in isolated mitochondria from WT and C9KO MEFs were analyzed by immunoblotting (n = 3).
(G) TIMMDC1 levels in isolated mitochondria from C9orf72-restored C9KO MEFs and controls were analyzed by immunoblotting (n = 3).
(H) CHX chase analysis of TIMMDC1 stability in WT and C9KO MEFs (n = 3). Mitochondrial lysates were collected and TIMMDC1 levels were determined.
(I and J) Two-dimensional BN/SDS–PAGE analysis of OXPHOS complexes from the mitochondria isolated from C9KO MEFs with or without restored C9orf72 protein expression (I). The relative abundance of TIMMDC1 in Sub-C1–3 and mature CI was quantified (J) (n = 3).
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S6.
We found that the TIMMDC1 level was significantly decreased in the absence of C9orf72 when isolated mitochondria were analyzed by immunoblotting (Figure 4F), and that the low level of TIMMDC1 could be rescued by restoration of the expression of C9orf72 but not its mutants with defective mitochondrial localization (Figure 4G and S6J). Furthermore, the half-life of the TIMMDC1 protein was significantly decreased in C9KO MEFs when compared to WT MEFs, as shown by cycloheximide (CHX) chase analysis (Figure 4H), indicating that C9orf72 is crucial for maintaining TIMMDC1 protein stability. Moreover, when protein complexes were examined by 2D-PAGE, the reduction in TIMMDC1 was found to be specifically restricted to the mature CI and Sub-C1/2, but not in the lower-molecular-weight upstream intermediate Sub-C3 (Figure 4C–4E). The restoration of C9orf72 expression in C9KO MEFs increased the level of TIMMDC1 in the mature CI and Sub-C1/2 complexes but not in the more upstream Sub-C3 complex (Figure 4I and 4J). These data support the finding that C9orf72 interacts with TIMMDC1 and acts as a cofactor to maintain its stability specifically at Sub-C2.
TIMMDC1 Sub-C2, containing CI subunits from the Q and proximal P (PP) modules, is crucial for the addition of PD and N module at the subsequent stages towards the final assembly of mature CI (Guerrero-Castillo et al., 2017). Thus, as a consequence of loss of C9orf72, the reduction in TIMMDC1 at Sub-C2 leads to decreased assembly of this and subsequent complexes including the Sub-C1 and mature CI, accompanied by the accumulation of N and PD modules intermediates, as observed in our analysis (Figure S6G and S6H). Furthermore, the CI assembly defects observed in C9KO cells are similar to those observed in cells lacking TIMMDC1. It was reported that loss of TIMMDC1 leads to accumulation of the components of the N and PD modules in their assembly intermediates of low molecular weights (Guarani et al., 2014). Notably, most of the CI subunits that we found accumulated in the assembly intermediates in C9KO cells were also reportedly increased in the assembly intermediates in the TIMMDC-deficient cells (Figure S6G and S6H, marked in red). In addition, when CI assembly in WT MEFs cells lacking TIMMDC1 was examined by 2D-PAGE, similar accumulations of NDUFB9 intermediates were observed when compared with C9KO MEFs (Figure S6A, S6K and S6L). Taken together, our data demonstrate that C9orf72 interacts with TIMMDC1 and stabilizes it specifically at the Sub-C2 to facilitate the CI assembly (Figure S6M).
C9orf72 Recruits PHBs to Inhibit TIMMDC1 Degradation by Mitochondrial mAAA Protease AFG3L2
To further understand the mechanism underlying the regulatory role C9orf72 on TIMMDC1 turnover, we performed an additional TMT labeled quantitative proteomic screen for C9orf72 binding partners specifically in mitochondria from both human (HAP1) and mouse (MEF) cells (Figure S7A and S7B; Table S5). Among the potential mitochondrial partners of C9orf72 identified in both human and mouse cells, PHB1 and PHB2, the two subunits of the prohibitin (PHB) complex, were the strongest interactors and readily visible on silver-stained electrophoresed gels (Figure S7A and S7B). Immunoblot analysis was used to confirm the pull-down of both PHB2 and PHB1 in the C9orf72 immunoprecipitates from WT but not C9KO MEFs (Figure 5A). Reciprocal co-immunoprecipitation experiments in HEK293 cells confirmed the interaction between C9orf72 and PHB2 (Figure 5B and 5C). C9orf72 specifically interacted with PHB2 but not with PHB1 in vitro (Figure 5D and S7C), indicating that C9orf72 interacts with the PHB complex through its direct binding to PHB2.
Figure 5. C9orf72 Recruits the PHB Complex to Stabilize TIMMDC1.

(A) Co-IP of PHB1 and PHB2 with anti-C9orf72 antibody was analyzed with isolated mitochondria from WT and C9KO MEFs
(B) Mitochondria isolated from HEK293 cells were solubilized and subjected to immunoprecipitation with the anti-C9orf72 antibody, followed by immunodetection of PHB2.
(C) Mitochondria lysates from HEK293 cells were subjected to immunoprecipitation with the anti-PHB2 antibody, followed by immunodetection of C9orf72.
(D) The direct interaction of purified His-SUMO-C9orf72 and GST-PHB2 was determined by the Ni-NTA pull-down of C9orf72, followed by immunoblot analysis of the proteins.
(E) WT and C9KO MEFs were transduced with lentiviral PHB2 shRNAs and the knockdown efficiency of PHBs was determined by immunoblotting. CTRL: non-targeting control shRNA.
(F) CHX chase analysis of the turnover of TIMMDC1 in WT and C9KO MEFs, with or without the knockdown of PHB2. Quantifications of the turnover rates of TIMMDC1 are shown (n = 4).
Data are means ± s.d., analyzed by one-way analysis of variance (ANOVA). ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S7 and Table S5.
To study the role of PHB complex in the regulation of TIMMDC1 stability, we generated MEF lines with stable knockdown of PHB2 or PHB1, either of which is required for the stability of the other subunit and therefore the function of the PHB complex (Kasashima et al., 2006; Merkwirth et al., 2008), with or without the deletion of C9orf72 (Figure 5E and S7D). The reduction of either PHB1 or PHB2 significantly increased the turnover rate of TIMMDC1 in WT MEFs (Figure 5F and S7E). The steady-state level of TIMMDC1 was unchanged, probably due to an increase in the synthesis of protein as evidenced by the upregulation of its mRNA level upon knockdown of PHBs (Figure S7F and S7G). Thus, our data indicate that the PHB complex suppresses the degradation of TIMMDC1. Furthermore, the turnover rate of TIMMDC1 in WT MEFs with the PHB knockdown was comparable to that observed in C9KO MEFs with the normal level of PHB, and the PHB knockdown had no further effect on the stability of TIMMDC1 in the C9KO MEFs, indicating that the regulation of TIMMDC1 stabilization by C9orf72 is dependent on the PHB complex (Figure 5F and S7E).
PHB forms a multimeric ring structure in the IMM, where it partners with the IMM-embedded m-AAA protease AFG3L2 in a larger complex that inhibits the proteolysis mediated by AFG3L2 (Merkwirth and Langer, 2009; Osman et al., 2009; Steglich et al., 1999). AFG3L2 is the catalytic subunit of one of the two mitochondrial IMM-embedded AAA+ metalloproteases, which are the major proteolytic machines responsible for maintaining protein homeostasis in the IMM (Patron et al., 2018). Given that C9orf72 regulates the turnover of IMM-located TIMMDC1 by recruiting PHB complex, we asked whether AFG3L2 is the proteolytic machine of this process. To test this hypothesis, we knocked down AFG3L2 and observed the accumulation of TIMMDC1 (Figure 6A). We found that the CAM treatment reduced the level of TIMMDC1; however, the knockdown of AFG3L2 blocked the CAM-induced reduction in TIMMDC1, indicating that AFG3L2 is required for the degradation of TIMMDC1 (Figure 6B).
Figure 6. Mitochondrial AAA Protease AFG3L2 Mediates the C9orf72-related degradation of TIMMDC1.

(A) AFG3L2 and TIMMDC1 were analyzed by immunoblotting using isolated mitochondria from HEK293 cells transfected with AFG3L2 shRNAs or non-targeting control shRNAs (n = 3).
(B) HEK293 cells were transfected with AFG3L2 shRNAs or non-targeting controls, followed by treatment with increasing concentrations of chloramphenicol (CAM), and TIMMDC1 levels were analyzed in whole cell lysates (n = 3).
(C) Co-IP of AFG3L2 with anti-TIMMDC1 antibody in isolated mitochondria from WT and C9KO HAP1 cells (n = 3).
(D) Co-precipitation of endogenous AFG3L2 by the pull-down of TIMMDC1-His with Ni-NTA agarose beads in the presence or absence of C9orf72-Flag expressed in HEK293 cells. The expression of C9orf72-Flag decreased the interaction of AFG3L2 with TIMMDC1-His (solid arrowhead) while increasing the level of TIMMDC1-His in the input (open arrowhead), with the quantifications shown alongside (n = 3).
(E) Co-IP of AFG3L2 with anti-TIMMDC1 antibody was analyzed in WT and C9KO HAP1 cells with or without PHB2 depletion (n = 3).
(F) Immunoblot analysis of TIMMDC1 in mitochondria isolated from WT and C9KO MEFs expressing shRNAs against AFG3L2 or non-targeting controls (n = 3).
(G and H) Two-dimensional BN/SDS–PAGE analysis of the TIMMDC1-containing subcomplexes and mature CI in isolated mitochondria from WT and C9KO MEFs expressing shRNAs against AFG3L2 (G). The relative abundance of TIMMDC1 in Sub-C1–3 and mature CI was quantified (n = 3) (H).
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S7.
Then we established that AFG3L2 interacts with TIMMDC1 in co-immunoprecipitation analysis (Figure 6C and 6D). Notably, the interaction between AFG3L2 and TIMMDC1 was increased by loss of C9orf72 (Figure 6C), whereas overexpression of C9orf72 led to a reduced interaction between AFG3L2 and TIMMDC1 accompanied by an increase in TIMMDC1 level (Figure 6D), indicating that C9orf72 controls the access to TIMMDC1 by its protease AFG3L2. Moreover, we found that the interaction between AFG3L2 and TIMMDC1 was significantly enhanced when the PHB complex was depleted in the WT cells (Figure 6E), indicating that, like C9orf72, PHB also controls the access to TIMMDC1 by AFG3L2. Furthermore, the effects on the interaction between AFG3L2 and TIMMDC1 were comparable as a result of the depletion of PHB versus the loss of C9orf72, and the depletion of PHB had no further effect on enhancing the interaction between AFG3L2 and TIMMDC1 in the absence of C9orf72 (Figure 6E). Together, these results suggest that C9orf72 partners with PHB to inhibit the proteolytic activity of AFG3L2 by limiting its access to the substrate TIMMDC1.
Furthermore, AFG3L2 depletion resulted in equally enhanced levels of TIMMDC1 in WT and C9KO MEFs, indicating that the fraction of TIMMDC1 that was destabilized as a result of the loss of C9orf72 is mainly degraded through AFG3L2 (Figure 6F). In contrast, depletion of the other IMM AAA protease YME1L failed to rescue TIMMDC1 protein level in C9KO cells to the same degree as in WT cells, suggesting a specific role of AFG3L2 in C9orf72-mediated regulation of TIMMDC1 stability (Figure S7H). When the CI complexes were analyzed by 2D-PAGE, the accumulation of TIMMDC1 as a result of AFG3L2 depletion was found only in the mature CI and Sub-C1/2, but not in the upstream Sub-C3 (Figure 6G and 6H). Moreover, the differences in the levels of these complexes between WT and C9KO MEFs were abolished when AFG3L2 was stably knocked down, indicating that the AFG3L2 depletion fully rescued the defects in the TIMMDC1-dependent CI assembly caused by loss of C9orf72 (Figure 6G and 6H). Thus, these results suggest that C9orf72 stabilizes TIMMDC1 specifically at the second TIMMDC1-containing subcomplex (Sub-C2) by repressing the targeting of TIMMDC1 to AFG3L2 for degradation.
Mitochondrial C9orf72 Function in C9-ALS Patient Cells
Since the level of C9orf72 was found to be reduced in C9-ALS patients, we examined whether C9orf72-dependent mitochondrial functions were dysregulated in the relevant patients’ cells. We first confirmed that C9orf72 was decreased in the induced motor neurons (MNs) generated from C9-ALS patients’ induced pluripotent stem cells (iPSCs), when compared to non-ALS controls (Figures 7A; Table S6). We also found that the mitochondrial CI levels were reduced in patients’ iPSC-MNs (Figures 7A). Similar results were obtained in the human postmortem spinal cord tissue samples from ALS/FTD patients (Figure 7B; Table S6). To analyze the intact CI levels in C9-ALS patients, we performed the BN-PAGE using four independent lines of B lymphocytes from ALS patients carrying the C9orf72 mutants. The mature CI levels in the isolated mitochondria were significantly lower in the patients’ cells, in accordance with the reduced C9orf72 protein levels (Figure 7C; Table S6). In addition, we observed a significant reduction in the CI activity and the TIMMDC1 level in the mitochondria from the patients’ cells (Figure 7C and 7D). Furthermore, the interactions between TIMMDC1 and AFG3L2 in the patients’ B lymphocytes were significantly enhanced (Figure 7E).
Figure 7. Dysregulated OXPHOS Activity and C9orf72 Functions in C9-ALS Patients.

(A) Immunoblot analysis and quantification of OXPHOS proteins and C9orf72 in iPSC-MNs from C9orf72-linked ALS patients (C9-ALS) and healthy controls (controls, n = 3; patients, n = 3).
(B) Immunoblot analysis and quantification of OXPHOS proteins and C9orf72 in spinal cord tissues from C9-ALS patients and controls (controls, n = 5; patients, n = 3).
(C) BN-PAGE analysis of intact CI levels in B lymphocytes derived from C9-ALS patients and controls via NDUFB9 immunoblotting. C9orf72 and TIMMDC1 levels were analyzed by SDS-PAGE (controls, n = 4; patients, n = 4).
(D) CI NADH-ubiquinone reductase activities in the isolated mitochondria from B lymphocytes derived from healthy controls and C9-ALS patients were determined (n = 3).
(E) The interaction between TIMMDC1 and AFG3L2 in mitochondria isolated from B lymphocytes of C9-ALS patients and controls was analyzed by co-immunoprecipitation with the anti-TIMMDC1 antibody (n = 3).
(F and G) Analysis of cell death (F) and intracellular ATP concentrations (G) of B lymphocytes from C9-ALS patients and healthy controls, cultured in either glucose or galactose medium for 24 h (n = 3).
(H) Intracellular ATP content of four iPSC-MN lines (two control lines and two C9-ALS lines) maintained under glucose conditions or treated with galactose for 24 h (n = 4).
(I) Control and C9-ALS iPSC-MN lines were maintained in glucose or galactose medium for 5 days before analysis of cell viability by calcein-AM staining (n = 3–4). Representative images of calcein-AM-stained MNs (CTRL 1 and C9-ALS 1) and the quantification of the cell viability are shown.
(J) Intracellular ATP concentrations of four iPSC-MN lines (two from C9-ALS patients and two from healthy controls) treated with excess glutamate for 12 h (n = 4).
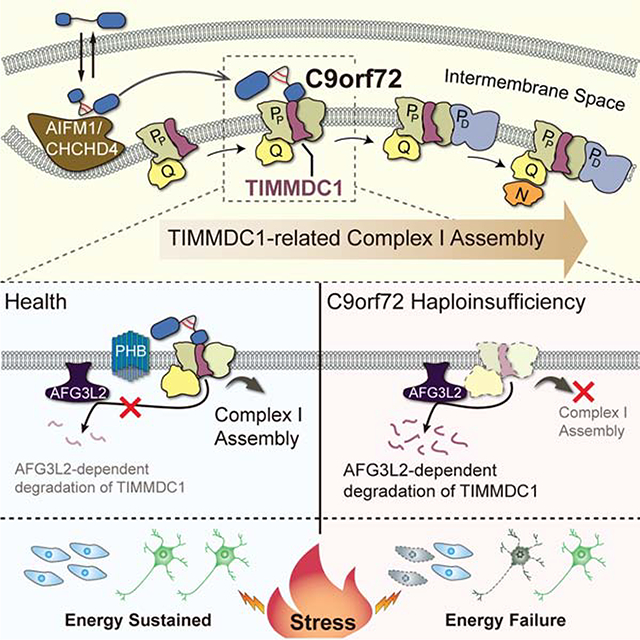
(K) Graphical summary of C9orf72-regulated CI assembly in mitochondria. C9orf72 is imported into the mitochondrial IMS, via an AIFM1/CHCHD4 oxidase-dependent process, during which C9orf72 forms intramolecular disulfide bonds in the cysteine-rich region. The IMS-located C9orf72 binds to TIMMDC1, an essential assembly factor of CI, specifically at the second assembly subcomplex (Sub-C2). C9orf72 promotes the stability of TIMMDC1 by recruiting PHB, which blocks the access of the AFG3L2 (m-AAA) protease to TIMMDC1. In C9-ALS patients, the haploinsufficiency of C9orf72 led to reduced CI assembly and function.
Data are means ± s.d., analyzed by unpaired two-sided Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant. See also Figures S7 and Table S6.
Next, we examined the mitochondrial OXPHOS activities in C9orf72 patients’ cells. We found that cell death induced by galactose treatment was significantly increased in patients’ B lymphocytes (Figure 7F). Consistently, the cellular ATP levels in the patients’ B lymphocytes were significantly reduced after galactose treatment (Figure 7G). We further validated the decreased ATP content and cell viability in the C9-ALS MNs after treatment with galactose (Figure 7H and 7I). To confirm whether loss of C9orf72 protein directly contributes to the CI/OXPHOS dysregulation in C9-ALS patients, we expressed C9orf72 in C9-ALS MNs and observed that the elevated levels of C9orf72 significantly rescued the phenotypes of reduced CI and intracellular ATP levels (Figure S7I and S7J). In neurons, OXPHOS is crucial for meeting the augmented energy demand posed by neuronal stimulation, whereas glycolysis is necessary for maintaining the basal energy supply (Hall et al., 2012; Rangaraju et al., 2014; Rangaraju et al., 2019). Thus, we tested whether increased neuronal activity could expose the potential OXPHOS defect in C9-ALS MNs. Indeed, a short-term glutamate exposure significantly reduced the ATP content in the C9-ALS MNs when compared to controls (Figure 7J). Since such a glutamate pulse leads to later C9-ALS MNs death (Shi et al., 2018), our data suggest that the ATP depletion resulting from C9orf72-related OXPHOS deficiency precedes and may contribute to the later degenerative response of C9-ALS MNs.
DISCUSSION
The present study has established C9orf72 as a critical mitochondrial regulator of cellular energy homeostasis that acts by promoting the assembly of CI, whose integrity is key to energy homeostasis (Figure 7K). We found that C9orf72 is imported into the mitochondrial IMS through the AIFM1/CHCHD4 pathway, which was previously shown to be crucial for the assembly and function of OXPHOS complexes (Modjtahedi and Kroemer, 2016; Modjtahedi et al., 2016). In line with the notion that AIFM1/CHCHD4 oxidoreductase-dependent mitochondrial import is mediated by a disulfide relay system (Hangen et al., 2015; Modjtahedi and Kroemer, 2016), C9orf72 localized to the mitochondria has more disulfide bonds than does C9orf72 in the cytosol, and the cysteine residues of C9orf72 are critical for its import into mitochondria. The fact that these cysteines are evolutionally conserved among vertebrates suggests that the mitochondrial import of C9orf72 are fundamentally important for cellular fitness.
The assembly of CI is an intricate process involving the formation of distinct subcomplexes that are sequential intermediates containing multiple subunits and assembly factors, prior to the formation of the mature CI complex. TIMMDC1 is a key assembly factor required for the formation of the intermediate and mature CI complexes. There are three TIMMDC1-containing subcomplexes formed during CI assembly: Sub-C3 (Q/PP-a modules), Sub-C2 (Q/PP modules), and Sub-C1 (Q/P modules) (Guerrero-Castillo et al., 2017). We found that C9orf72 is specifically located in the second subcomplex and promotes the assembly of the mature CI. Loss of C9orf72 protein impairs the subsequent assembly of N and PD modules into mature CI, leading to the accumulation of N and PD intermediates (Figure S6G and S6H). The influence of C9orf72 on these complexes is mediated through the regulation of TIMMDC1 stability. The degradation of TIMMDC1 is mediated by the m-AAA protease, whose catalytic subunit AFG3L2 interacts with TIMMDC1 and controls its turnover. We propose that C9orf72 acts by directly recruiting the PHB complex to inhibit the proteolytic activity of AFG3L2 locally in the complex containing C9orf72 and TIMMDC1. The mitochondrial PHB complex was previously shown to form a supercomplex with AFG3L2 through interaction with its IMS domain and negatively regulate its protease activity (Steglich et al., 1999), likely owing to the large ring structure of the PHB complex that may limit the accessibility of AFG3L2 to its substrates (Merkwirth and Langer, 2009; Osman et al., 2009). Consistently, we found that the PHB complex suppresses the interaction between AFG3L2 and its substrate TIMMDC1 and inhibits the degradation of TIMMDC1. Loss of C9orf72 relieves the suppression of AFG3L2 by PHB and thereby activates the degradation of TIMMDC1. Our results suggest that the interplay between PHB and AFG3L2 is actively regulated, rather than kept in a quiescent state, potentially allowing for regulated control of the substrate selectivity of the AFG3L2 protease, as exemplified by the inhibitory effect of C9orf72 on the AFG3L2-mediated turnover of TIMMDC1.
The newly identified mitochondrial function of C9orf72 suggests that it plays a role in energetic homeostasis especially under stress conditions. Loss of C9orf72 resulted in approximately 30–50% reduction in the CI levels and 25% reduction in the CI activities. Under basal conditions, such medium reduction in CI level does not have a major impact on the OXPHOS output or cell viability, likely due to the spared capacity of CI and increased CI substrates that resulted from concomitantly upregulated glycolysis (Yadava and Nicholls, 2007). However, under the conditions of energetic stresses, such as low glucose supply or excitatory stimulation, loss of C9orf72 is sufficient to induce cell death, indicating that the energy buffering capacity of the cell is significantly damaged in the absence of C9orf72. Thus, the present study demonstrates that mitochondrial C9rof72 acts as a safeguarding factor to ensure the full function of CI, which is especially crucial for energy homeostasis and cellular health under stress conditions.
Our findings further implicate mitochondrial defects in the pathogenesis of ALS and related neurodegenerative diseases. For example, SOD1 could form intermolecular disulfide bonds in mitochondria and damage electron transport chain complex functions (Deng et al., 2006; Jung et al., 2002). TDP-43 could enter mitochondria and cause CI disassembly or mitochondrial DNA into the cytoplasm in pathological conditions (Wang et al., 2016; Yu et al., 2020). FUS has also been implicated in energy metabolism regulation and been observed to inhibit the ATP synthase function (Deng et al., 2018; Wang et al., 2015). In the present study, we identified a distinct function of C9orf72 protein in the IMM that regulates the quality control and assembly of the CI complex. An estimated 6% of total C9orf72 is imported into the mitochondrial IMS. The fact that IMS has a small volume relative to the cytosol implies that the local concentration of C9orf72 in mitochondria could be relatively high and C9orf72 may play an important role in mitochondrial biology. Indeed, our study demonstrates that the majority of mitochondrial C9orf72 is in complex with TIMMDC1 (Sub-C2), and this interaction is crucial for cellular energy homeostasis under stress conditions. Although DPR products from the repeat expansion of the C9orf72 gene have been reported to damage mitochondria and increase ROS levels (Dafinca et al., 2016; Lopez-Gonzalez et al., 2016), our study has revealed a distinct mechanism through which C9orf72 haploinsufficiency impairs mitochondrial functions. Notably, elevated C9orf72 expression was able to rescue the CI dysregulation in C9-ALS motor neurons. Mitochondrial dysfunctions have been implicated in other major neurodegenerative conditions including Parkinson’s and Huntington’s diseases (Costa and Scorrano, 2012; Lin and Beal, 2006). Given that mitochondrial damages are associated with aging, a major risk factor for neurodegenerative diseases, our study suggests that the C9orf72 haploinsufficiency may exacerbate mitochondrial dysfunctions during the aging process and thereby contribute to the pathogenesis of its relevant diseases.
Limitations of Study
Although we have demonstrated an important role of mitochondria-located C9orf72 protein in regulating mitochondria Complex I function, these studies are not designed to exclude other mitochondria-regulatory mechanisms that controlled by this protein. For example, given our observation of the interaction between C9orf72 and AIFM1, a regulatory effect of C9orf72 on the AIFM1/CHCHD4 pathway remains a possibility. Additionally, while the cultured cells, including human or mouse C9KO cells and the patient-derived B lymphocytes or iPSC-motor neurons, provide powerful model systems to study the molecular mechanisms underlying energy dysregulation and mitochondrial C9orf72 protein function, the relevance of this mechanism to ALS/FTD remains to be further characterized in animal models.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jiou Wang (jiouw@jhmi.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The published article includes all datasets generated during this study and are available in the associated Supplemental Tables. This study did not generate new code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
The generation of female murine embryonic fibroblasts (MEFs) from WT and C9orf72 knockout littermates was described previously (Ugolino et al., 2016). Briefly, ES cells lacking C9orf72 with a strain background of C57BL/6N-Atm1Brd were microinjected into blastocysts, and the derived mice were maintained on the C57BL/6 background. Male mice bearing the targeted allele were crossed with the Sox2-Cre recombinase transgenic female mice (Jackson Laboratory) maintained on the C57BL/6 background, to remove the LoxP-flanked neomycin selection cassette. The offspring heterozygous for the C9KO allele were also maintained on the C57BL/6 background by backcrossing to C57BL/6 wild-type animals for more than ten generations. Immortalized MEF lines were isolated from Day 13 embryos by trypsin digestion, followed by transfecting cells with the SV40-T antigen-expressing plasmid pSG5 Large T using Lipofectamine 2000. Both the WT and C9KO MEFs were verified to carry the short allele of the COX7A2L gene, excluding its potential influence on mitochondrion-related phenotypes. MEFs and HEK293 cells were cultured in DMEM (Thermo Fisher) supplemented with 10% FBS and 1% penicillin (10,000 units/ml)/streptomycin (10 mg/ml). Human B lymphocytes were obtained from Coriell Cell Repositories (Table S6) and maintained on RPMI 1640 medium containing 15% FBS. The male C9orf72 knockout HAP1 cells were created at Horizon Genomics (Vienna) by using CRISPR/Cas9 and maintained in IMDM (Thermo Fisher) supplemented with 10% FBS. All cell lines were cultured in a humidified cell incubator at 37°C in a 5% CO2 atmosphere.
The iPSCs were obtained from the National Institutes of Health Cell Repositories (Table S6), with informed consent obtained in accordance with the guidance of source institutions. The iPSCs were maintained on Matrigel (BD)-coated plates in StemFlex medium (Thermo Fisher). Cells were cultured at 37°C in a hypoxic (5% O2) incubator.
Tissue Samples
Brian and spinal cord tissue used for subcellular fractionation assay were harvest from 12-week-old male wild-type and C9KO mice. Mice were bred and maintained under conventional conditions in a temperature- and humidity-controlled mouse facility, according to the protocol approved by the Johns Hopkins Animal Care and Use Committee following the National Research Council’s guidance of the Care and Use of Laboratory Animals.
Human postmortem spinal cord tissue samples from ALS/FTD patients and healthy controls were obtained from the Johns Hopkins Medical Institute Brain Bank and Department of Veterans Affairs Biorepository Brain Bank (Table S6), with informed consent obtained in accordance with the guidance of source institutions. Tissues were homogenized in pre-chilled Dounce glass homogenizers in modified RIPA buffer (50 mM Tris at pH 7.5, 150 mM NaCl, 1% NP40, 0.1% SDS, 100 mM NaF, 17.5 mM β-glycerophosphate, 2.5% sodium deoxycholate, and 10% glycerol) supplemented with 1X phosphatase inhibitors (Sigma), 1 mM PMSF, 2 mM NaVO4, and 1X protease inhibitor cocktail. Samples were further sonicated for 15 min on ice, followed by centrifugation at 12,000 g at 4°C for 10 min, and supernatants were collected for western blot analysis.
METHOD DETAILS
Plasmids
The Flag-tagged human C9orf72 construct was generated using the Gateway cloning system (Thermo Fisher) with a C-terminal 3xFlag tag. Complementary DNAs (cDNAs) of wild-type human C9orf72 and TIMMDC1 were amplified by PCR and subcloned into the pLenti-CMV-Puro-DEST (w118–1) vector (Campeau et al., 2009) using the Gateway cloning system (Thermo Fisher). A panel of Cys->Ser point mutations of C9orf72 were generated by PCR-based site-directed mutagenesis and cloned into the pLenti-CMV-Puro-DEST (w118–1) vector. For in vitro translation used in the mitochondrial import assay, human C9orf72, NDUFS8, or NDUFA8 cDNA was cloned into pGEM-7Zf(+) (Promega) using Gibson assembly (New England Biolabs). Recombinant plasmid for the expression of human C9orf72 in bacteria was generated by cloning C9orf72-coding sequence fused to C-terminal of 6xHis-SUMO tag into the pSATl vector. GW1-PercevalHR was a gift from Gary Yellen (Addgene) (Tantama et al., 2013). The C9orf72 shRNAs were constructed as described previously (Ugolino et al., 2016). Briefly, the shRNA sequence 5′cttccacagacagaacttagtttctacct 3′ was cloned into the pRFP-C-RS vector (Origene).
Cell Transfection, and Treatment
To knock down C9orf72 in HEK293 cells, the plasmid encoding an shRNA targeting C9orf72 or a scrambled control was transfected into HEK293 cells using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instructions. To knock down AIFM1 or CHCHD4 in HEK293 cells, siRNAs were prepared at a final concentration of 20 nM and transfected using Lipofectamine RNAiMax (Thermo Fisher) according to the manufacturer’s instructions. The transfected cells were analyzed 72–96 h after transfection.
For the preparation of lentivirus, HEK293 cells were co-transfected with a lentiviral vector plus two packaging plasmids, pSPAX2 and pMD2.G, using Lipofectamine 3000. Forty-eight hours after transfection, lentiviral supernatant was centrifuged at 1,000 g for 5 min and subsequently filtered through a 0.45-μm cellulose acetate filter. For lentivirus infection, cells were plated at 5 X 104 cells per well in 6-well plates and then supplemented with viral medium for 72 h. Subsequently, cells were selected with puromycin for 72 h at a final concentration of 1.5 μg/ml. After multiple rounds of puromycin selection, cells were maintained with 0.5μg/ml puromycin and subjected to further analysis.
To replace the glucose in the medium with galactose, MEF, HEK293 or HAP1 cells were washed with PBS and cultured in glucose-free DMEM/IMDM (Thermo Fisher) containing 10% dialyzed FBS (Thermo Fisher) and 4.5 g/L galactose (Sigma). To reduce the glucose concentration in the medium, MEF cells were washed with PBS and cultured in glucose-free DMEM (Thermo Fisher) containing 10% dialyzed FBS (Thermo Fisher) and 1.5 g/L glucose (Sigma).
To completely delete the fully assembled CI, mitochondrial translation in MEFs was inhibited by treatment with 50 μg/ml chloramphenicol for 5 days. After the treatment, cells were either collected or washed twice with PBS and maintained in normal medium to recover for up to 24 h. To stimulate the degradation of endogenous TIMMDC1, HEK293 cells were treated with increasing concentrations of chloramphenicol for 24 h. To measure the degradation rate of endogenous TIMMDC1, MEFs were treated with 20 μg/ml cycloheximide for up to 4 h before harvesting.
Motor Neuron Generation
The method for converting human iPSCs into motor neurons is based on a previous report with minor modification (Du et al., 2015). In brief, on day 0, iPSC clones were suspended and transferred into one Matrigel-coated well of a 6-well plate with 10 μM Rock Inhibitor (Stemgent). On day 1, the medium was changed to neuronal medium [a mixture of 50% Neurobasal medium (Thermo Fisher) and 50% DMEM/F12 medium (Thermo Fisher) with 1X Glutamax (Thermo Fisher), 0.5X N2 and B27 supplement (Thermo Fisher) and 0.1mM ascorbic acid (Sigma)], supplemented with 3 μM CHIR99021 (Stemgent), 2 μM SB431542 (Stemgent), and 2 μM DMH-1 (Torcris) for 6 d. Cells were then dissociated and seeded into Matrigel-coated 10-cm dishes in neuronal medium plus 1 μM CHIR99021, 2 μM DMH-1, 2 μM SB431542, 0.1 μM RA (Sigma) and 0.5 μM purmorphamine (Stemgent). On day 13, cells were dissociated with 1 U/mL dispase (Stemcell) and placed in 10-cm ultra-low adhesion plates (Corning) in neural medium plus 0.5 μM RA and 0.2 μM purmorphamine. On day 21, cells were dissociated with 1X Accutase (Thermo Fisher) and seeded at a density of 2 X 106 cells per well into PDL/Laminin (Sigma)-coated 6-well plates with neuronal medium plus 0.5 μM RA, 0.2 μM purmorphamine, and 0.1 μM Compound E (Sigma). Cells were differentiated for 36–40 days before experimental analysis. To express C9orf72 protein in the C9-ALS iPSCs-derived motor neurons, on day 28, cells were transduced with C9orf72-expressing lentivirus for 12 h. Cells were then cultured for 12 days before experimental analysis.
Cell Survival Assay
Cell death in MEF, HAP1, HEK293, or human B lymphocytes treated with galactose or low-glucose medium was measured by staining cells with annexin V AlexaFluor-488 conjugate (Thermo Fisher). In brief, 2 X 105 cells from each group were incubated with 200 μl of stain for 30 min at room temperature in the dark, then examined with flow cytometer (BD LSR II) or a fluorescence microscope (Nikon Eclipse TS100). For microscopy, annexin V-positive cells were counted in three randomly selected fields of view using a 20X objective. For each group, at least 500 cells from three or four independent experiments were analyzed.
To determine the cell viability in MEFs, cells were grown in a 96-well plate with either glucose or galactose medium for 4 days. 10 μl of the cell counting kit-8 (CCK-8) (Abcam) solution were added to each well of the plate. The plate was incubated for 1 hour and the absorbance of each well was measured at 460 nm using a microplate reader (BioTek, Synergy H1). To determine the sensitivity of MEFs to OXPHOS inhibitors, cells in a 96-well plate were cultured in the galactose medium with the indicated inhibitors for 12 h, and the cell survival was quantified by crystal violet staining followed by the measurement of absorbance at 590 nm using the plate reader.
To analyze the viability of iPSC-derived motor neurons after galactose treatment, on day 21 of culturing, cells were seeded at a density of 0.5 X 106 cells per well into PDL/laminin-coated 24-well plates with complete medium. After 14 days, the cells were washed with PBS and maintained in galactose-containing neuronal medium [glucose-free neurobasal medium (Thermo Fisher) supplemented with 4.5 g/L galactose, 1X Glutamax, 0.5X N2 and B27 supplement, 0.1 mM ascorbic acid, 0.5 μM RA, 0.2 μM purmorphamine, and 0.1 μM Compound E] for indicated times. Cells then were gently washed with 1X HBSS with calcium and magnesium (Thermo Fisher) and stained with 2 μM calcein AM (Thermo Fisher), followed by measurement of fluorescence with a plate reader according to the manufacturer’s instructions. Images of randomly selected fields of view were captured by a fluorescence microscope (Nikon Eclipse TS100).
Immunofluorescent and Other Cell Staining
For immunofluorescence analysis of cultured cells, cells were washed three times with PBS and then fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Fixed cells were then permeabilized with 0.25% Triton X-100 in PBS and blocked by blocking buffer (1X PBS, 3% BSA, and 0.1% Tween-20) for 1 h at room temperature. Cells were incubated with primary antibody overnight at 4°C and fluorescently conjugated secondary antibody for 1 h at room temperature. Coverslips were mounted with ProLong Diamond with DAPI (Thermo Fisher). For mitochondrial morphology measurements, immunofluorescence images of TOM20 were captured randomly from each group using a TSC SP8 confocal microscope (Leica) and quantified via the Mitochondrial Network Analysis (MiNA) toolset for Fiji software (Valente et al., 2017). In brief, images were pre-processed for binarizing and skeletonizing, followed by measurements of mitochondrial coverage area (mitochondrial footprint) and branch lengths. The colocalization of C9orf72 and TIM23 was analyzed using the colocalization tool in Image-Pro Plus software. Mitochondrial membrane potential (Δψm) was measured with tetramethylrhodamine (TMRM). In brief, MEFs in glass bottom dishes were stained with TMRM (100 nM) in phenol-free culture medium, with or without 20 μM CCCP, at 37°C in the dark for 30 min. Live images of the cells were captured with a SP8 Confocal Microscope (Leica) and analyzed using Fiji software. The mean intensity of the TMRM signal for each cell was measured and then normalized to the cell size.
To visualize the ATP levels inside individual living cells, MEF cells were plated on glass-bottom dishes and transfected with the plasmid expressing the fluorescent sensor PercevalHR using Lipofectamine2000 according to the manufacturer’s instructions. After 24 h, cells were maintained in phenol-free culture medium, and live cell images were acquired using a SP8 Confocal Microscope (Leica). PervecalHR was imaged using the excitation wavelength of 405 nm or 488 nm and a window of emission wavelength of 494–553 nm. The mean pixel intensity of the whole cell was acquired, and the PercevalHR ratio was calculated (ATP:ADP=F488 nm / F405 nm) and displayed in a pseudo-colored image using the Fiji software.
Proteomic Analysis
To compare the whole proteomes of WT and C9KO MEF cells, as illustrated in Figure S3A, cells were washed with ice-cold PBS and lysed in lysis buffer (8 M urea / 50 mM tetraethylammonium bicarbonate). The lysates were diluted to a lower concentration of 1 M urea, and then digested with trypsin/Lys-C. Proteins were incubated overnight and then acidified, desalted, and lyophilized sequentially. After being resuspended in 0.2% formic acid and subsequent quantification, peptides were then lyophilized and labeled with 6-plex tandem mass tagging (TMT) reagents (Thermo Scientific). Labeled peptides were then mixed in 1:1 ratio and desalted consequently. The LC-MS/MS was analyzed with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher). The resulting spectra were analyzed by Proteome Discoverer 2.1 (database used: mouse RefSeq version 78) following standard procedures. For all searches, carbamidomethylation on cysteine residues and TMT on Lys and peptide N termini were set as fixed modifications, and oxidation on methionine was set as a dynamic modification. Up to a 1% false discovery rate was allowed for both peptide and protein levels.
To analyze the interactome of the mitochondrial C9orf72 protein, as shown in Figure S7A and S7B, mitochondria were isolated from MEFs or HAP1 cells, with or without C9orf72, and subjected to immunoprecipitation using an anti-C9orf72 antibody. The immunoprecipitated samples in elution buffer (0.1 M glycine, 0.25 M Tris-HCl) were digested on a 30-kDa molecular weight cut-off membrane in 80 mM triethylammonium bicarbonate buffer, using the standard filter-aided sample preparation (FASP) protocol (Wisniewski et al., 2009). After digestion, the peptides were labeled by 6-plex tandem mass tagging (TMT) reagents (Thermo Scientific). Labelled mouse or human samples were then combined, respectively, and cleaned up on HLB Oasis plates. Combined samples were analyzed by LC-MS on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher). The resulting data were searched against the SwissProt mouse or human database using MASCOT software.
For the SILAC-based proteomic analysis of mitochondrial complexome profiling, WT and C9KO MEFs were grown in light medium (12C6,14N4 L-Arginine, 12C6,14N2 L-Lysine) and heavy medium (13C6,15N4 L-Arginine, 13C6,15N2 L-Lysine), respectively, and verified for near-completion of labeling by mass spectrometry. Cells were collected and subjected for mitochondrial isolation, followed by solubilization with 5 g digitonin (Sigma-Aldrich) per gram of protein. Equal amounts of lysates were mixed prior to BN-PAGE analysis as described below. Each gel lane was cut into 45 slices with a width of 0.75 mm, which were then subjected to reduction, alkylation, and trypsin in-gel digestion. The peptides were analyzed on an Orbitrap Fusion Lumos Tribrid Mass Spectrometer coupled with the Ultimate3000 RSLCnanl nano-flow liquid chromatography system (Thermo Fisher). Peptides were quantified using MaxQuant software (v1.6.10.43).
Metabolomic Analysis
After being washed with ice-cold PBS buffer, cultured WT or C9KO MEFs were immediately incubated with dry ice-cold 80:20 methanol/water solution. Cells were scraped and incubated at −80°C for at least 2 h to precipitate the proteins. After centrifugation at 20,000 g for 5 min at 4°C, supernatants were dried and reconstituted with 60% methanol with 0.1% formic acid, and then subjected to LC/MS analysis as described previously (Krug et al., 2014). In brief, for chromatographic separation, 5 μl samples were loaded onto an Agilent 1260 high performance liquid chromatography system (Agilent). A Cogent Diamond Hydride (MicroSol) column (150 X 2.1mm i.d., 4 μm particle size, 100 Å pore size) was used. The mass spectrometry was conducted on a 6520 accurate-mass Q-TOF LC-MS system (Agilent) equipped with a dual electrospray (ESI) ion source. The mass spectrometer was operated in the negative mode, and the gas temperature was held at 325°C. The drying gas flow was set to 10 l/min. The nebulizer pressure was set to 45 psig. The capillary voltage was 4.0 kV. The MS data acquisition was performed in the range of 70–1100 m/z, with acquisition rate at 1.5 spectra/s. Data were acquired with MassHunter Acquisition software (Agilent), and then processed with MassHunter Qualitative Analysis software (Agilent, version 5.0). Putative metabolites were identified by matching the accurate m/z values of the peaks with an in-house-built database derived from HMDB, KEGG, METLIN, and other public databases. Data files were then imported into Mass Profiler Professional software (Agilent, version 12.1) for further processing. The parameters were set as follows: minimum absolute abundance, 5000 counts; retention time range, 0–25 min; mass range, 70–1100 m/z; minimum of ions, 2; multiple charge state forbidden; retention time window, 1 min; mass window, 10 p.p.m. + 2.0 mDa. A significant metabolite list was generated after Student’s t-test, and plotted using the MetaboAnalyst platform (https://www.metaboanalyst.ca/).
Protein Purification and Binding Assays
Recombinant C9orf72 was prepared as described previously (Liu et al., 2018). Briefly, human C9orf72 fused with a 6xHis-SUMO tag was cloned into the pSATL vector and transformed into E. coli BL21 (DE3) cells (Agilent Technologies). Cells were lysed in lysis buffer (20 mM Tris-HCl at pH 8.0, 250 mM NaCl, 100 mM glycine, and 1 mM TCEP) supplemented with 10 mM imidazole, 1 M urea, 1 mM ATP, 1 mM MgCl2, 1X bestatin, pepstatin A, E-64, and phenylmethylsulfonyl fluoride, followed by disruption with a French press. The expressed C9orf72-His-SUMO fusion protein was purified by immobilized metal affinity chromatography (IMAC, Bio-Rad) charged with nickel sulfate, and eluted with 250 mM imidazole-containing lysis buffer. The C9orf72 protein was then desalted in low-salt buffer (20 mM Tris-HCl at pH 8.0, 75 mM NaCl, 100 mM glycine, and 1 mM TCEP) using an Amicon centrifugal filter unit (molecular weight cutoff 30 kDa).
For the in vitro protein interaction assay, 2 μg of purified C9orf72 proteins was mixed with 1 μg of GST-tagged PHB1/2 proteins or GST control protein in 300 μl binding buffer (20 mM Tris-HCl at pH 8.0, 250 mM NaCl, and 20 mM imidazole) and incubated overnight at 4°C with gentle agitation. His-tagged C9orf72 was then precipitated with Ni-NTA beads (Qiagen). Beads were washed five times with wash buffer (20 mM Tris-HCl at pH 8.0, 250 mM NaCl, 35 mM imidazole, and 1% Tween-20) and then boiled in 1X SDS sample loading buffer for 5 min. The precipitated proteins were analyzed and quantified via SDS-PAGE and immunoblotting.
Immunoprecipitation
For the co-IP assay of C9orf72 and TIMMDC1 in MEF or HEK293 cells, or C9orf72 and PHB2 in MEF cells, isolated mitochondrial from MEFs and HEK293 cells were lysed in IP buffer (25 mM Tris at pH 8.0, 150 mM NaCl, 1mM EDTA, and 1X protease inhibitor cocktail) supplemented with 1% digitonin. For detection of PHB2 and C9orf72 interaction in HEK293 cells, isolated mitochondria were lysed in IP buffer in the presence of 0.3% sodium deoxycholate, as described previously (Wei et al., 2017). For the co-IP assay of TIMMDC1 and AFG3L2, cells were collected and lysed in NP40 buffer (50 mM Tris at pH 8.0, 150 mM NaCl, 1% NP40, and 1X protease inhibitor cocktail). For the co-IP assay of C9orf72 and AIFM1, isolated mitochondria from HEK293 cells were collected and lysed in NP40 buffer. After centrifugation at 20,000 g for 20 min, the supernatants were collected for protein concentration assay and immunoprecipitation. The supernatants were pre-cleared with protein A/G beads (Thermo Fisher) plus rabbit IgG. After the centrifugation, supernatants were incubated with either the indicated polyclonal primary antibody or a rabbit IgG control plus protein A/G beads overnight at 4°C with gentle inversion. After incubation, the beads were washed three times with wash buffer (25 mM Tris at pH 8.0, 150 mM NaCl, 1mM EDTA, and 1% Tween-20), and then eluted with IgG elution buffer (pH 2.0; Thermo Fisher) at room temperature for 15 min. The resulting eluates were boiled with 1X SDS sample loading buffer before being analyzed by SDS-PAGE and immunoblotting.
Immunoblotting
For SDS-PAGE, both whole-cell and mitochondrial fractions were collected and lysed in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP40, and 1% sodium deoxycholate) plus protease inhibitor cocktail and 1mM PMSF on ice. After centrifugation (22,000 g, 20 min at 4°C), supernatants were saved, and protein concentrations were determined by the BCA analysis (Thermo Fisher) before SDS-PAGE analysis. Actin, GAPDH, or Tubulin was used as a loading control for the whole-cell samples; TOM20 was used as a loading control for the purified mitochondrial samples.
Blue-native gel electrophoresis was performed with the NativePAGE Bis-Tris Gel system (Thermo Fisher) according to the manufacturer’s instructions. Isolated mitochondria were first quantified by measuring the protein concentration using the BCA analysis. Equal amounts of mitochondria from each group were then resuspended in mitochondrial buffer and solubilized by 5 g digitonin per gram of protein in the presence of 1 mM PMSF and protease inhibitor cocktail for 30 min on ice. After centrifugation (22,000 g, 20 min at 4°C), the supernatants were collected, and 5% G-250 sample additive was added, before the samples were loaded on 4–16% gradient NativePAGE Bis-Tris gels (Thermo Fisher). Electrophoresis was started at 150 V for 30 min using dark running buffer (1X NativePAGE running buffer and 1X NativePAGE cathode buffer additive, Thermo Fisher) at 4°C, and continued at 300 V in light running buffer (1X NativePAGE running buffer, 0.1X NativePAGE cathode buffer additive) for 90 min. After electrophoresis, proteins were transferred to polyvinylidene difluoride (PVDF) membranes and then probed with the indicated antibodies. For second-dimension gel electrophoresis, lanes excised from the first BN-PAGE gel were boiled for 15 min in 1X SDS sample loading buffer (50 mM Tris, pH8.0, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 2% SDS, 1% β-mercaptoethanol, and 12.5 mM EDTA). The gel strip was loaded onto 4–20% SDS-PAGE gels for second-dimension electrophoresis followed by immunoblot analysis.
Cytosolic and Mitochondrial Fractionation
Cells were washed and collected in ice-cold PBS. After three cycles of freeze-thaw to lyse the cells, cell pellets were resuspended in mitochondrial buffer [200 mM mannitol, 68 mM sucrose, 10 mM HEPES-KOH at pH 7.4, 10 mM KCl, 1mM EDTA, 1 mM EGTA, and 0.1% bovine serum albumin (BSA)] plus protease inhibitor cocktail (Sigma) and subjected to homogenization (20 strokes) in pre-chilled Dounce glass homogenizers (Sigma). The homogenates were centrifuged at 700 g at 4°C for 10 min to remove unbroken cells and nuclei. Subsequently, the mitochondrial fractions were pelleted by centrifugation at 3,000 g at 4°C for 10 min. The supernatants were saved as cytosolic fractions, whereas the mitochondrial pellets were washed twice and stored in mitochondrial buffer. To further purify the mitochondria for the immune-EM assay, the mitochondrial fraction was overlaid on top of 8 ml Percoll medium (225 mM mannitol, 25 mM HEPES at pH 7.4, 1 mM EGTA, and 30% Percoll) and subjected to ultra-centrifugation at 100,000 g for 30 min at 4°C in an SW41Ti rotor. The pellet was suspended in mitochondrial buffer again, followed by centrifugation at 10,000 g for 5 min at 4°C to obtain purified mitochondria.
To analyze the sub-mitochondrial localization of C9orf72, the isolated mitochondria were subjected to alkaline extraction in freshly prepared 0.1 M Na2CO3 (pH 11.0). Membranes were pelleted at 100,000 g for 1 h. For sonication, isolated mitochondria were sonicated at low power for 3 min on ice and subsequently centrifuged at 4°C for 1 h at 100,000 g. Pellets from both procedures were saved, and supernatants were precipitated with 10% trichloroacetic acid (TCA) and washed four times with ice-cold acetone. Both soluble (S, supernatant) and insoluble (P, pellet) fractions were subjected to immunoblot analysis.
For the proteinase K protection assay, freshly isolated mitochondria were split into two equal aliquots: one resuspended in osmotic buffer (25 mM sucrose, 10 mM MOPS-KOH, pH 7.2) to disrupt the outer mitochondrial membrane, and the other in the regular mitochondrial buffer. Samples were treated with proteinase K (0.1 – 10 μg/ml) for 30 min on ice to digest surface-exposed proteins. The reaction was stopped by adding PMSF (2 mM), and samples were analyzed by immunoblotting.
Complex I Activity Assay
Mitochondrial CI NADH-dehydrogenase activities in WT and C9KO MEFs were assessed by using the CI activity microplate assay (Abcam). In brief, MEF cells were lysed, and protein concentrations were determined by BCA assay. After immunocapturing the CI from 200 μg of total protein lysate, the rate of oxidation of NADH to NAD+ was calculated on the basis of the change in absorbance (450 nm) per minute (mOD/min). To analyze CI NADH-ubiquinone reductase activities in WT and C9KO MEFs, isolated mitochondria were analyzed using the Mitochondrial CI colorimetric assay (Biovision).
qPCR
RNA extraction, cDNA generation, and qPCR were performed as described previously (Liu et al., 2018). Briefly, total RNAs were extracted by using TRIzol reagent (Thermo Fisher). For quantitative PCR, 1 μg RNA was reverse transcribed (Qiagen), followed by PCR amplification with iQ SYBR Green PCR mix (Bio-Rad). Gene expression data were calculated by the ΔΔCt method. Actin was used as the reference control.
ATP Measurements
ATP was measured using a luciferase-based luminescence assay (Thermo Fisher). MEFs or iPSC-derived motor neurons were washed with PBS twice and subjected to ATP extraction by 0.5% TCA. After incubation for 15 min at room temperature, the ATP-containing supernatants were transferred to a fresh tube and neutralized by 0.1 M Tris-HCl buffer (pH 7.75), and the remaining proteins were dissolved in RIPA buffer and the protein concentration measured by BCA assay. The ATP concentration was determined using a standard curve for the luciferase assay and then normalized to the protein concentration.
MAPR was determined based on the luminometric kit described above (Wibom and Hultman, 1990). Briefly, MEFs were grown under normal glucose or galactose medium for the indicated times. Mitochondria were prepared freshly as described above. Equal aliquots of the mitochondrial suspensions were incubated with the luminometric reaction mix, as well as substrates for oxidation and 35μM ADP. Substrates added (final concentration) were as follows: CI, 10 mM glutamate plus 1 mM malate; CII, 20 mM succinate plus 0.1 mM rotenone. The MAPR were determined using an ATP standard curve and normalized to the protein concentration.
Mitochondrial Respiration Assay
To monitor the mitochondrial OXPHOS activity in cellulo, MEF, HAP1, or HEK293 cells were seeded onto a Seahorse XF96 Cell Culture Microplate (Agilent) at a density of 1.0 (MEF or HAP1) or 2.0 (HEK293) X 104 cells per well and allowed to adhere for 6 h in complete medium at 37°C and 5% CO2. Subsequently, the cells were washed with PBS twice and maintained in either complete medium or galactose medium for 16 h. The cells were then washed and incubated in the XF base medium (Agilent) with 4 mM L-glutamine in the absence (galactose group) or presence (control group) of 1 mM sodium pyruvate and 4.5 g/L glucose. Mitochondrial OCRs were determined using the Seahorse XF96 Analyzer (Agilent). The following drugs were injected at the final concentrations given: for MEFs, oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM), and rotenone (0.5 μM) and antimycin A (0.5 μM); for HAP1 cells, oligomycin (2 μM), FCCP, (2 μM), and rotenone (0.9 μM) and antimycin A (0.9 μM); for Hela or HEK293 cells, oligomycin (2 μM), FCCP, (1 μM), and rotenone (0.75 μM) and antimycin A (0.75 μM). Basal OCR was determined by subtracting the OCR measured after the addition of rotenone/antimycin A from the value prior to the addition of oligomycin. Maximal OCR was calculated by subtracting the OCR measured after the addition of rotenone/antimycin A from the value in the presence of FCCP. Total protein was isolated from each well and quantified by BCA assay for normalization.
To measure the OCR in isolated mitochondria in vitro, MEFs were grown under the glucose or galactose medium for the indicated times. Mitochondria were freshly isolated and supplemented with substrates of CI or CII, as described in the method of MAPR measurement. The OCRs were analyzed by Extracellular Oxygen Consumption Assay (Abcam) following the manufacturer’s protocol.
In vitro Import and Assembly Assay
Human C9orf72, NDUFA8, or NDUFS8 cDNA in the pGEM-7Zf(+) vector (Promega) was transcribed and translated in vitro using the TNT-coupled reticulocyte lysate system (Promega) in the presence of L-[35S]methionine (PerkinElmer). Following translation, the mixture containing [35S]methionine-labeled C9orf72, NDUFA8, or NDUFS8 protein was incubated with equal amounts of mitochondria that were freshly isolated from indicated cells at 25°C for 30 min. Unimported proteins were removed by adding proteinase K (10 μg/ml) at 4°C for 10 min. After adding PMSF (2 mM) to stop the digestion, mitochondria were collected by centrifugation at 10,000 g at 4°C for 10 min and solubilized in 1X SDS sample loading buffer. The imported C9orf72, NDUFA8, or NDUFS8 protein was analyzed by SDS-PAGE. To measure the CI assembly in vitro, following the mitochondrial importation of [35S] NDUFS8, with or without 50 μM CCCP, and proteinase K treatment as mentioned above, the isolated mitochondria were solubilized by adding digitonin, and the intact CI was analyzed by BN-PAGE. The radioactive signals on the transferred membrane were visualized by exposure to X-ray film and quantified by Quantity One software.
AMS Modification Assay
Cytosolic or mitochondrial fractions prepared from 1.0 X 107 MEF cells were lysed in modified RIPA buffer with 1% SDS plus protease inhibitor cocktail and subsequently sonicated for 5 min. For the DTT-treated controls, samples were supplemented with 100 mM DTT and boiled at 95°C for 5 min. Samples then were precipitated with 10% TCA on ice for 10 min and centrifuged at 20,000 g at 4°C for 15 min. The TCA pellets were washed with ice-cold acetone, dried, and resuspended in 6 M urea buffer (50 mM Tris-HCl at pH 7.5, 6 M urea, and 1% SDS) containing 10 mM AMS. Samples were incubated at 30°C in the dark for 30 min, and proteins were precipitated by 10% TCA. Protein pellets were washed by with ice-cold acetone, dried, and dissolved in 1X SDS sample loading buffer and denatured for 15 min at 70°C. Proteins were separated by SDS-PAGE, followed by immunoblot analysis.
Immune Electron Microscopy
For immune-EM, pure mitochondrial fractions from WT and C9KO mouse brain cortices were fixed in 0.1 M HEPES containing 4% formaldehyde and 0.1% glutaraldehyde at room temperature for 45 min. Samples were dehydrated in ethanol and embedded in LR White resin. Thin sections were blocked for 15 min with blocking buffer (PBS containing 1% BSA, 1% normal goat serum, and 0.01% Tween-20). Grids were incubated with a polyclonal anti-C9orf72 antibody (1:20) in blocking buffer at 4°C for 12 h. After five washes with PBS containing 5 % normal goat serum, grids were incubated for 2 h in 10-nm gold-conjugated goat anti-rabbit IgG (British BioCell International) diluted 1:20 in blocking buffer. Grids were then rinsed with PBS and fixed with glutaraldehyde to stabilize the gold particles. Sections were then stained with 2% uranyl acetate solution and the Sato lead solution. Images were captured using an AMT XR80 8-megapixel CCD camera and a Philips/FEI BioTwin CM120 transmission electron microscope (operated at 80 kV). The specificity of the gold signals in the mitochondria from WT MEFs was further confirmed by the controls without the primary antibody.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the data are presented as means ± s.d. The band intensity in immunoblots was determined using Bio-Rad Quantity One software. For single comparisons, the statistical significance was analyzed using Student’s t-test. For multiple means of comparison, one-way analysis of variance (ANOVA) was performed, and the statistical significance was analyzed by the Bonferroni post-hoc test. For enrichment analysis, Fisher’s test was performed. The sample size (n) represents biological replicates per experiment. * P < 0.05; ** P < 0.01; *** P < 0.001 and statistical parameters can be found in the figure legends. No method was used to test for normal distribution of data.
Supplementary Material
Table S2. Differentially Regulated Proteins Identified by TMT Labeling Quantitative Mass Spectrometry in WT and C9KO MEFs, Related to Figure 2.
Table S3. Metabolomic Data of WT and C9KO MEFs, Related to Figure 2.
Table S4. SILAC-based Complexome Analysis of Mitochondrial Complexes Separated on BN-PAGE, Related to Figure 3.
Table S5. The Potential Interactors of Mitochondrial C9orf72 Determined by TMT-based Quantitative Proteomic Analysis, Related to Figure 5.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Total OXPHOS antibody cocktail | Abcam | ab110413 |
| Mouse monoclonal anti-C9orf72 | Bio-Rad | VMA00065 |
| Rabbit polyclonal anti-C9orf72 | Proteintech Group | 22637-1-AP |
| Mouse monoclonal anti-TIMMDC1 | Santa Cruz | sc-514927 |
| Rabbit polyclonal anti-TIMMDC1 | Sigma Aldrich | HPA053214 |
| Rabbit monoclonal anti-NDUFB9 | Abcam | ab200198 |
| Rabbit monoclonal anti-NDUFB11 | Abcam | Ab183716 |
| Mouse monoclonal anti-UQCRC1 | Abcam | ab110252 |
| Mouse monoclonal anti-MTCO1 | Abcam | ab14705 |
| Mouse monoclonal anti-NDUFA8 | Santa Cruz | sc-398097 |
| Mouse monoclonal anti-AIFM1 | Santa Cruz | sc-13116 |
| Mouse monoclonal anti-CHCHD4 | Santa Cruz | sc-365137 |
| Rabbit polyclonal anti-AFG3L2 | Abcam | ab154990 |
| Mouse monoclonal anti-PHB1 | Santa Cruz | sc-377037 |
| Mouse monoclonal anti-PHB2 | Santa Cruz | sc-133094 |
| Rabbit monoclonal anti-PHB2 | Cell Signaling | 14085 |
| Rabbit polyclonal anti-TOM70 | Sigma Aldrich | HPA014589 |
| Mouse monoclonal anti-TIM44 | Santa Cruz | sc-390755 |
| Rabbit polyclonal anti-SOD1 | Enzo Life Sciences | ADI-SOD-100 |
| Rabbit polyclonal anti-YME1L | Proteintech Group | 11510-1-AP |
| Rabbit polyclonal anti-TIM23 | Proteintech Group | 11123-1-AP |
| Mouse monoclonal anti-TIM23 | Santa Cruz | sc-514463 |
| Rabbit monoclonal anti-TOM20 | Cell Signaling | 42406 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling | 2118 |
| Mouse monoclonal anti-His tag | Santa Cruz | sc-8036 |
| Mouse monoclonal anti-Flag tag | Sigma Aldrich | F1804 |
| Mouse monoclonal anti-GST tag | Santa Cruz | sc-138 |
| Mouse monoclonal anti-β-actin | Santa Cruz | sc-47778 |
| Rabbit monoclonal anti-β-tubulin | Cell Signaling | 2128 |
| Bacterial and Viral Strains | ||
| Stbl3 competent E. coli | Thermo Fisher Scientific | C737303 |
| BL21(DE3) competent E. coli | Agilent | 200131 |
| Biological Samples | ||
| Human spinal cord tissue | See Table S6 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rock inhibitor Y27632 | Stemgent | 04-0012 |
| N2 supplement | Thermo Fisher Scientific | 17502048 |
| B27 supplement | Thermo Fisher Scientific | 17504044 |
| Neurobasal medium | Thermo Fisher Scientific | 21103049 |
| Glucose-free neurobasal medium | Thermo Fisher Scientific | A2477501 |
| Fetal bovine serum | Thermo Fisher Scientific | 10438026 |
| Dialyzed fetal bovine serum | Thermo Fisher Scientific | 26400044 |
| CHIR99021 | Stemgent | 04-0004 |
| SB431542 | Stemgent | 04-0010 |
| DMH-1 | Tocris | 4126 |
| Retinoic acid | Sigma Aldrich | R2625 |
| Purmorphamine | Stemgent | 04-0009 |
| Dispase | Stemcell | 07923 |
| Compound E | Sigma Aldrich | 530509 |
| Galactose | Sigma Aldrich | G5388 |
| Ascorbic acid | Sigma Aldrich | A4544 |
| Calcein AM | Thermo Fisher Scientific | C3099 |
| Image-iT TMRM | Thermo Fisher Scientific | I34361 |
| CCCP | Sigma Aldrich | C2759 |
| 13C6,15N4 L-Arginine | Cambridge Isotope Laboratories | CNLM-539-H |
| 13C6,15N2 L-Lysine | Cambridge Isotope Laboratories | CNLM-291-H |
| Tandem mass tag (TMT) reagents | Thermo Fisher Scientific | 90061 |
| Digitonin | Sigma Aldrich | D141 |
| cOmplete protease inhibitor cocktail | Roche | 11873580001 |
| Protein A/G beads | Thermo Fisher Scientific | 88803 |
| IgG elution buffer | Thermo Fisher Scientific | 21028 |
| TRIzol reagent | Thermo Fisher Scientific | 15596026 |
| EasyTag L-[35S]-Methionine | PerkinElmer | NEG709A500UC |
| AMS | Thermo Fisher Scientific | A485 |
| Cycloheximide | Sigma Aldrich | C1988 |
| Recombinant human C9orf72 protein | (Liu et al., 2018) | N/A |
| Recombinant human PHB1 protein | Abnova | H00005245-P01 |
| Recombinant human PHB2 protein | Abnova | H00011331-P02 |
| Recombinant SUMO protein | MyBioSource | MBS2553243 |
| Critical Commercial Assays | ||
| Seahorse XF cell mito stress test kit | Agilent | 103708-100 |
| NativePAGE Novex Bis-Tris gel system | Thermo Fisher Scientific | BN2001; BN2002; BN1002BOX; BN2008; LC0725 |
| Gibson assembly cloning kit | New England Biolabs | E5510S |
| Cell counting kit-8 (CCK-8) | Abcam | Ab228554 |
| TNT coupled reticulocyte lysate systems | Promega | L4611 |
| Complex I enzyme activity microplate assay kit | Abcam | ab109721 |
| Mitochondrial Complex I activity colorimetric assay kit | Biovision | K968 |
| QuantiTect reverse transcription kit | Qiagen | 205313 |
| iQ SYBR green PCR mix | Bio-Rad | 1708880 |
| ATP determination kit | Thermo Fisher Scientific | A22066 |
| Extracellular oxygen consumption assay | Abcam | ab197243 |
| Experimental Models: Cell Lines | ||
| Human HEK293 | ATCC | CRL-1573 |
| Mouse embryonic fibroblasts control | (Ugolino et al., 2016) | N/A |
| Mouse embryonic fibroblasts C9orf72 KO | (Ugolino et al., 2016) | N/A |
| Human HAP1 control | Horizon Discovery | C631 |
| Human HAP1 C9orf72 KO | Horizon Discovery | HZGHC003729c002 |
| Human B lymphocytes | See Table S6 | N/A |
| Human iPSCs | See Table S6 | N/A |
| Experimental Models: Organisms/Strains | ||
| B6.Tg(Sox2-cre)1Amc/J mice | The Jackson Laboratory | 008454 |
| C57BL/6J mice | (Ugolino et al., 2016) | N/A |
| C57BL/6J C9orf72 KO mice | (Ugolino et al., 2016) | N/A |
| Oligonucleotides | ||
| Human AIFM1 siRNA GGGCAAAAUCGAUAAUUCU | (Hangen et al., 2015) | N/A |
| Human CHCHD4 siRNA GCAUGGAUUGAUACUGCCATT | (Hangen et al., 2015) | N/A |
| Mouse TIMMDC1 qPCR FWD AGTACTCCGGTGAGACGGTT | This study | N/A |
| Mouse TIMMDC1 qPCR REV TGCAGACTGCTTCTCCACTC | This study | N/A |
| Mouse Actin qPCR FWD GGCTGTATTCCCCTCCATCG | This study | N/A |
| Mouse Actin qPCR REV CCAGTTGGTAACAATGCCATGT | This study | N/A |
| Recombinant DNA | ||
| Plasmid: mouse TIMMDC1 shRNA | Sigma Aldrich | TRCN0000202057 |
| Plasmid: human NDUFS5 shRNA 1# | Sigma Aldrich | TRCN0000036639 |
| Plasmid: human NDUFS5 shRNA 2# | Sigma Aldrich | TRCN0000036643 |
| Plasmid: mouse PHB1 shRNA 1# | Sigma Aldrich | TRCN0000088457 |
| Plasmid: mouse PHB1 shRNA 2# | Sigma Aldrich | TRCN0000088455 |
| Plasmid: mouse PHB2 shRNA 1# | Sigma Aldrich | TRCN0000054428 |
| Plasmid: mouse PHB2 shRNA 2# | Sigma Aldrich | TRCN0000054429 |
| Plasmid: human AFG3L2 shRNA | Sigma Aldrich | TRCN0000051489 |
| Plasmid: mouse AFG3L2 shRNA | Sigma Aldrich | TRCN0000241172 |
| Plasmid: mouse YME1L shRNA | Sigma Aldrich | TRCN0000031200 |
| Plasmid: human C9orf72 shRNA | (Ugolino et al., 2016) | N/A |
| Plasmid: pLenti-CMV-Puro-DEST | (Campeau et al., 2009) | Addgene # 17452 |
| Plasmid: pLenti-CMV-Puro-C9orf72 | This Study | N/A |
| Plasmid: GW1-PercevalHR | (Tantama et al., 2013) | Addgene #49082 |
| Plasmid: pGEM-7Zf(+)-C9orf72 | This Study | N/A |
| Plasmid: pGEM-7Zf(+)-NDUFS8 | This Study | N/A |
| Plasmid: pGEM-7Zf(+)-NDUFA8 | This Study | N/A |
| Plasmid: pSATL-C9orf72-His-SUMO | (Liu et al., 2018) | N/A |
| Software and Algorithms | ||
| Quantity One | Bio-Rad | https://www.bio-rad.com/en-us/product/quantity-one-1-d-analysis-software |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| Fiji | N/A | https://fiji.sc/ |
| Image-Pro Plus | Media Cybernetics | https://www.mediacy.com/imageproplus |
Highlights.
C9orf72 is imported into mitochondrial IMS through the AIFM1/CHCHD4 pathway
Mitochondrial C9orf72 is crucial for OXPHOS functions and energy metabolism
C9orf72 enables effective Complex I (CI) assembly by stabilizing TIMMDC1
C9orf72 haploinsufficiency impairs CI and OXPHOS functions in ALS/FTD patient cells
ACKNOWLEDGEMENTS
This work was supported by grants from NIH (NS074324, NS089616, NS110098, GM123266, S10OD021844), Packard Center for ALS Research at Johns Hopkins, Muscular Dystrophy Association, and the U.S. Department of Defense. We thank NINDS and NIGMS Cell Repository, Johns Hopkins Brain Bank, and VA Biorepository Brain Bank for providing patient cells and tissues, Johns Hopkins Mass Spectrometry Core for proteomic analysis, Tricia Nilles for flow cytometry analysis, and members of Wang lab for discussion.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, and Enriquez JA (2008). Respiratory active mitochondrial supercomplexes. Molecular cell 32, 529–539. [DOI] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, and Scorrano L (2012). Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. The EMBO journal 31, 1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, Vowles J, Douglas AG, Fletcher-Jones A, Browne C, et al. (2016). C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 34, 2063–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, et al. (2006). Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proceedings of the National Academy of Sciences of the United States of America 103, 7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Wang P, Chen X, Cheng H, Liu J, Fushimi K, Zhu L, and Wu JY (2018). FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proceedings of the National Academy of Sciences of the United States of America 115, E9678–E9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du ZW, Chen H, Liu H, Lu J, Qian K, Huang CL, Zhong X, Fan F, and Zhang SC (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nature communications 6, 6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Pradat P-F, Ludolph AC, and Loeffler J-P (2011). Energy metabolism in amyotrophic lateral sclerosis. The Lancet Neurology 10, 75–82. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, Pearce EL, and Pearce EJ (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarani V, Paulo J, Zhai B, Huttlin EL, Gygi SP, and Harper JW (2014). TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Molecular and cellular biology 34, 847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, and Nijtmans L (2017). The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell metabolism 25, 128–139. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Klein-Flugge MC, Howarth C, and Attwell D (2012). Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 8940–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, Lam NV, Zhu C, Godin I, Muller K, et al. (2015). Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Molecular cell 58, 1001–1014. [DOI] [PubMed] [Google Scholar]
- Harms M, Benitez BA, Cairns N, Cooper B, Cooper P, Mayo K, Carrell D, Faber K, Williamson J, Bird T, et al. (2013). C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA neurology 70, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Higgins CM, and Xu Z (2002). Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. Journal of neurochemistry 83, 535–545. [DOI] [PubMed] [Google Scholar]
- Kasashima K, Ohta E, Kagawa Y, and Endo H (2006). Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. The Journal of biological chemistry 281, 36401–36410. [DOI] [PubMed] [Google Scholar]
- Kloppel C, Suzuki Y, Kojer K, Petrungaro C, Longen S, Fiedler S, Keller S, and Riemer J (2011). Mia40-dependent oxidation of cysteines in domain I of Ccs1 controls its distribution between mitochondria and the cytosol. Molecular biology of the cell 22, 3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug AK, Gutbier S, Zhao L, Poltl D, Kullmann C, Ivanova V, Forster S, Jagtap S, Meiser J, Leparc G, et al. (2014). Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP(+). Cell death & disease 5, e1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, and Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang T, Ji YJ, Johnson K, Liu H, Bailey S, Suk Y, Lu YN, Liu M, and Wang J (2018). A C9orf72-CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes & development 32, 1380–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, and Gao FB (2016). Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 92, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan N, Kunzli C, Buthey K, and Saxena S (2017). C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Molecular neurobiology 54, 3062–3077. [DOI] [PubMed] [Google Scholar]
- Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, Troncoso JC, Hardy J, Singleton AB, and Traynor BJ (2012). Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med 366, 283–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley ME, O’Rourke JG, Yanez A, Markman JL, Ho R, Wang X, Chen S, Lall D, Jin M, Muhammad A, et al. (2020). C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 585, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, and Langer T (2008). Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes & development 22, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, and Langer T (2009). Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochimica et biophysica acta 1793, 27–32. [DOI] [PubMed] [Google Scholar]
- Modjtahedi N, and Kroemer G (2016). CHCHD4 links AIF to the biogenesis of respiratory chain complex I. Molecular & cellular oncology 3, e1074332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modjtahedi N, Tokatlidis K, Dessen P, and Kroemer G (2016). Mitochondrial Proteins Containing Coiled-Coil-Helix-Coiled-Coil-Helix (CHCH) Domains in Health and Disease. Trends in biochemical sciences 41, 245–260. [DOI] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. [DOI] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Merkwirth C, and Langer T (2009). Prohibitins and the functional compartmentalization of mitochondrial membranes. Journal of cell science 122, 3823–3830. [DOI] [PubMed] [Google Scholar]
- Patron M, Sprenger HG, and Langer T (2018). m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell research 28, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, and Ryan TA (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Lauterbach M, and Schuman EM (2019). Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 176, 73–84 e15. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, and Wallace DC (1992). Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochemical medicine and metabolic biology 48, 122–126. [DOI] [PubMed] [Google Scholar]
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, OuladAbdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. The EMBO journal 35, 1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, Liang C, Chang Q, Zhang W, Yang M, and Chen JF (2019). C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta neuropathologica communications 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nature medicine 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A, Lojewski X, Sterneckert J, Hermann A, Shaw PJ, et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nature neuroscience 19, 1610–1618. [DOI] [PubMed] [Google Scholar]
- Steglich G, Neupert W, and Langer T (1999). Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Molecular and cellular biology 19, 3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Formosa LE, Wijeyeratne XW, Nguyen TN, and Ryan MT (2013). Gene knockout using transcription activator-like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. The Journal of biological chemistry 288, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, et al. (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126. [DOI] [PubMed] [Google Scholar]
- Su MY, Fromm SA, Zoncu R, and Hurley JH (2020). Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 585, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, and Hu F (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta neuropathologica communications 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M, Martinez-Francois JR, Mongeon R, and Yellen G (2013). Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nature communications 4, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr., and Cleveland DW (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, and Nijtmans L (2004). Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Human molecular genetics 13, 2461–2472. [DOI] [PubMed] [Google Scholar]
- Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, Brady NR, Hamacher-Brady A, and Wang J (2016). Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS genetics 12, e1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente AJ, Maddalena LA, Robb EL, Moradi F, and Stuart JA (2017). A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta histochemica 119, 315–326. [DOI] [PubMed] [Google Scholar]
- Vogtle FN, Burkhart JM, Gonczarowska-Jorge H, Kucukkose C, Taskin AA, Kopczynski D, Ahrends R, Mossmann D, Sickmann A, Zahedi RP, et al. (2017). Landscape of submitochondrial protein distribution. Nature communications 8, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, and Blake DJ (2014). Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiology of aging 35, 1779 e1775–1779 e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jiang X, Chen G, and Xu J (2015). Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiology of aging 36, 527–535. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, Jiang S, Ma X, Jiang Z, da Rocha EL, et al. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nature medicine 22, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr., Mishra P, and Levine B (2017). Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168, 224–238 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield CD, Bostedor R, Goodrum D, Haak M, and Chu EH (1981). Hamster cell mutants unable to grow on galactose and exhibiting an overlapping complementation pattern are defective in the electron transport chain. The Journal of biological chemistry 256, 6651–6656. [PubMed] [Google Scholar]
- Wibom R, and Hultman E (1990). ATP production rate in mitochondria isolated from microsamples of human muscle. The American journal of physiology 259, E204–209. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nature methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Yadava N, and Nicholls DG (2007). Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 7310–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, and Chen JF (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Science advances 2, e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, De Nardo D, et al. (2020). TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, Diaz Garcia S, Ghosh Dastidar S, Rodriguez MJ, King P, et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nature neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita Rendon O, and Shoubridge EA (2012). Early complex I assembly defects result in rapid turnover of the ND1 subunit. Human molecular genetics 21, 3815–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Differentially Regulated Proteins Identified by TMT Labeling Quantitative Mass Spectrometry in WT and C9KO MEFs, Related to Figure 2.
Table S3. Metabolomic Data of WT and C9KO MEFs, Related to Figure 2.
Table S4. SILAC-based Complexome Analysis of Mitochondrial Complexes Separated on BN-PAGE, Related to Figure 3.
Table S5. The Potential Interactors of Mitochondrial C9orf72 Determined by TMT-based Quantitative Proteomic Analysis, Related to Figure 5.
Data Availability Statement
The published article includes all datasets generated during this study and are available in the associated Supplemental Tables. This study did not generate new code.