

Abstract
The early promise of gene-based therapies is currently being realized at an accelerated pace with over 155 active clinical trials and multiple FDA-approvals for therapeutic oligonucleotides, the vast majority of which contain modified phosphate linkages. These unnatural linkages have desirable biological and physical properties but are often accessed with difficulty using phosphoramidite chemistry. Here we report a flexible and efficient [P(V)]-based platform that can install a wide variety of phosphate linkages at will into oligonucleotides. This approach uses readily accessible reagents and can efficiently install not only stereodefined or racemic thiophosphates, but can install any combination of (S, R or rac)-PS with native phosphodiester (PO2) and phosphorodithioate (PS2) linkages into DNA and other modified nucleotides. Importantly this platform easily accesses this diversity under a standardized coupling protocol with sustainably prepared, stable, P(V) reagents.
One Sentence Summary:
In Nature, oligonucleotides are synthesized using a P(V)-based, enzymatically assisted process whereas P(III)-based reagents have dominated the mainstream laboratory production protocol for the past 50 years. Harnessing the innate reactivity of P(V) in the laboratory in this context has the potential to simplify and democratize this process especially with regards to non-canonical P-linkages. This work presents a unified, P(V)-based reagent platform amenable to automated oligonucleotide synthesis that enables streamlined access to previously challenging chimeric sequences bearing phosphodiester (PO2), either diastereomer of phosphorothioate (PS), and phosphorodithioate (PS2) linkages.
In traditional small molecule drug discovery, the features that determine target specificity and pharmacokinetics [DMPK (distribution, metabolism, pharmacokinetics)] are usually inextricably linked (1). Oligonucleotide-based therapeutics, on the other hand, have been referred to as “informational drugs” wherein the pharmacophore and PK properties can, in theory, be separately optimized since the nucleoside sequence directly determines the former while the unifying chemistry (the phosphate linkages used to couple the nucleosides) largely affects the latter (2). Advances in organic synthesis have had a profound impact on the ability of modern medicinal chemistry to target and rapidly access increasingly complex small molecule leads. In contrast, as the range of oligonucleotide sequences and conceivable phosphate linkages has expanded, the fundamental chemistry used to enable their synthesis has largely remained unchanged despite numerous refinements and improvements. The incorporation of varied phosphate linkages has been documented to have a profound impact on both the properties and efficacy of the resulting structures (3). The hypothetical chimeric sequence (1) illustrated in Figure 1, adorned with four different phosphorus-based linkages and multiple sugar backbones, tests the limits of the scope of existing methods. The impact of judiciously designing backbone chemical modifications in therapeutic oligonucleotides is only beginning to be realized (4–7). The opportunity to install broader combinations and variations, in at will, in any order, thus presents a new step in the evolution of oligonucleotide therapeutics and requires the invention of enabling science; one such step is described herein. The commercialization of oligonucleotides and, more specifically, phosphorothioate antisense oligonucleotides (PS-ASOs) faces significant challenges (8–14). The ability to easily make single-isomer species from stable species/fragments will aid control over product quality and has the potential to enable commercialization of these novel therapeutics sustainably (i.e., through enhanced regulatory control of these complex molecules). Thus, improved methods of oligonucleotide synthesis, amenable to standard methods of automation, would have a near-immediate translational impact, enabling both the interrogation of greater linker permutations in drug discovery and better, more sustainable commercialization [see SI for process mass intensity (PMI) analysis] (3).
Fig. 1.

Introduction and background (A) Inspiration, (B) Challenges facing P(V) based synthesis (C) Recent developments and this work. i-Pr, iso-propyl; Me, methyl; Bz, benzoyl; [O], oxidation; [S], sulfurization
We recently demonstrated a practical approach to address the synthesis of stereopure phosphorothioates (R-PS and S-PS, mainly in the context of DNA), with the disclosure of the phosphorus sulfur incorporation (dubbed PSI or ψ) reagents. However, this initial study did not address the installation of other linkages such as phosphorodithioates and native phosphate diesters (15), other sugar chemistries (such as LNA), or applicability to a modern automated synthesizer protocol.
With these goals in mind, several nontrivial challenges were apparent—with significant lore and reactivity concerns to address when using a P(V)-based approach (16–20). For example, the rates associated with P(V)- reagents have historically been viewed as too sluggish to ever compete with the P(III)-manifold (21–23). Such approaches also had chemoselectivity challenges in that the guanine and thymidine bases would interfere in sequential couplings, reacting with the coupling reagents (24). Employing P(III)-based reagents to install phosphorodithioate (PS2) linkages is less than ideal. For instance, Caruthers reported a protocol based on protected thiophosphoramidites that requires discreet oxidation and deprotection steps (25) and suffers from the formation of a nearly inseparable phosphorothioate byproduct in ca. 5–10% yield. Despite this serious issue, this method is still commonly used as there are simply no viable alternatives (26). Finally, chimeric sequences with multiple modifications could be desired by medicinal chemists, but only certain combinations (PS/PO, PS/PS2, PO/PS2) are present in the literature. Even a hybrid synthetic approach that merges P(III)-based phosphoramidite chemistry with P(V) reagents is unworkable as it suffers from a lack of chemoselectivity (vide infra) as oxidation of P(III) to P(V) requires a protecting group on exposed PS/PS2 linkages to avoid desulfurization. The use of organic oxidants was ruled out due to the extreme number of equivalents required and their extended reaction times (27).
In this disclosure, three new reagent systems are described [ψ2 (3), rac-ψ (4), and ψO (5)] that, when combined with the previously reported [(+)-ψ, (+)-2] and [(−)-ψ, (−)-2], provide a unified P(V) approach that entirely departs from the rubric of P(III)-based oligonucleotide synthesis and enables the at will and controlled synthesis of specific chimeric oligonucleotides (Figure 1C). Along with these new reagents are protocols for their unified application in commercial synthesizers using a single coupling protocol which spans all coupling types. This redox-neutral platform, based on the native P(V) oxidation state, challenges past assumptions of sluggish reactivity and enables access to five relevant P-linkages across a range of sugar backbones (DNA and LNA) and bases (A, T, G, mC) in one oligonucleotide construct. Aside from enabling straightforward access to a wide range of chimeric oligonucleotides, the implementation of this new protocol benefits from a reduced reliance on protecting group chemistry (which eliminates the labile cyanoethyl group and therefore acrylonitrile production upon deprotection) (28, 29), bespoke additives (30), and redox fluctuations. It is also of note that this new P(V)-platform eliminates one full step in the standard SPOS protocol (namely the phosphorous oxidation).
Figure 2 outlines the synthesis of ψ2 and ψO, reagents for the incorporation of phosphorodithioate and native phosphodiester linkages, respectively. The development of these reagents required extensive experimentation, whereas the synthesis of rac-ψ was relatively trivial and proceeded by analogy to ψ using cyclohexene oxide (see SI). Fully sulfurized versions of diphosphate esters, known as phosphorodithioate linkages, are isopolar and isostructural analogs of phosphates that are completely stable to nucleases (31) while maintaining the ability to form stable duplexes and elicit desirable mRNA cleavage by RNase H without the complexity of chirality at phosphorus (32, 33). The pioneering work of Stec on phospholane heterocycles, which inspired the development of (+)-ψ and (−)-ψ, was an essential precedent for the present work (19). In 1995 it was disclosed that dithiaphospholanes could be installed onto nucleosides and coupled to afford dinucleotides incorporating a phosphorodithioate linkage, albeit requiring a separate oxidation step to install sulfur, and a toxic and unstable (explosive) reagent (Figure 2A) (18). Our P(V)-centric study thus built on the lessons of these two precedents with the goals of eliminating the extraneous oxidation step and dangerous reagents. Upon identification of the optimal leaving group (21 evaluated, see SI for Hammett correlations of leaving groups) and ring size (two evaluated), inexpensive P2S5 could be combined with pentafluorophenol followed by capping of P(V) intermediate (6) with thiirane to generate ψ2 on large scale (>100 g). In this way, unsafe P(III) chemistry was avoided, and a stable and viable P(V) reagent was developed.
Fig. 2.

Development of a fully P(V) platform for oligonucleotide synthesis (A) Reagent development, (B) Overcoming chemoselectivity challenges (C) Overcoming historical P(V) rate challenges. PFP, pentafluorophenol; TFA, trifluoroacetic acid; DCA, dichloroacetic acid; DBP, dibutylphosphate; [S], sulfurization; DDTT, 3-((Dimethylamino-methylidene)amino)-3H-1,2,4-dithiazole-3-thione; DCI, 4,5-dicyanoimidazole; DBU, 1,8-Diazabicyclo[5.4.0]undec-7-ene.
Oligonucleotides with native phosphodiester linkages have poor pharmacokinetics and are rapidly degraded by nucleases, but their use is invaluable in routine molecular biology and diagnostic settings (34). In addition, limited PO linkages are incorporated into current antisense oligonucleotides. To the best of our knowledge, there are no known P(V)-based reagents for achieving P(III)-competitive reactivity and chemoselectivity that are stable and amenable to automated synthesis (34). Although the design of such a reagent benefited from the lessons of our previous studies, it was repeatedly thwarted by the challenge of identifying both a highly reactive and stable entity upon loading to a monomer. We evaluated nearly 30 different backbones along with three different leaving groups before arriving at ψO (see SI for comprehensive summary). The backbone optimization systematically evaluated ring size, substituents, electronic effects, and stereochemistry to probe effects on loading, coupling, and overall stability with ψO emerging as the only viable candidate. The synthesis outlined in Figure 2A requires three simple, scalable steps (>50 g). In a similar fashion to ψ2, inexpensive P2S5 was reacted with 4-bromothiophenol yielding P(V) intermediate (7) which, when combined with hydrogenated cis-limonene oxide (8), yields the PS reagent (9); desulfurization with SeO2 yields ψO.
With these new reagents in hand, a side-by-side comparison with state-of-the-art P(III) chemistry was conducted (Figure 2B). For the installation of the PS2 linkage, the three-step P(III) approach to access simple dimer (10) via P(III) adduct (12) resulted in ca. 7% of the PS impurity resulting from desulfurization during the deprotection event. In contrast, utilizing ψ2, dimer (10) was cleanly accessed in two steps via P(V) adduct (11) in >99% purity. In order to field-test and contrast the synthesis of mixed PO/PS backbones by the two platforms, PS dimer (13) was subjected to a standard phosphoramidite coupling with (18) to yield protected trimer (14) which, upon oxidation of P(III) to the requisite P(V), resulted in rapid desulfurization [see ratio of (15)/(16) over time]. On the other hand, the redox-neutral P(V) approach employing P(V) adduct (17) cleanly delivered the unprotected mixed PS/PO trimer (19) without any loss of sulfur.
The final challenge that P(V)-based reagents face is the longstanding perception that their diminished coupling rates preclude them from being employed in traditional automated oligonucleotide synthesis regimes. With a full suite of reagents in hand based on P(V), their coupling performance was evaluated side-by-side with canonical P(III) chemistry through kinetics studies (Figure 2C). In addition, the original P(V)-based coupling using phosphotriester chemistry was included as this was the initial benchmark. Temporal reaction progress monitoring was performed by taking aliquots of the mixture coupling P-loaded-T with AZT and monitoring product formation by HPLC/MS using an internal standard. Full kinetic profiles are included in the Supporting Information, and here we present the main trends by considering relative rates calculated from these profiles. Consistent with the literature, the classic P(V)-based phosphotriester approach was extremely sluggish, as revealed in the orange bar of Figure 2C. However, as also shown in Figure 2C, the P(V) reagent suite detailed herein performed equally well to the industry standard P(III) protocol with all reactions reaching full conversion in under two minutes.
Extensive optimization of the SPOS methodology for phosphoramidite-based synthesis has occurred over the course of the last 30+ years. While some of these methods could be leveraged in this new context, there were areas where existing solutions were not compatible with the P(V) synthesis protocol (Figure 3A). Notably, existing universal supports afforded insufficient stability toward DBU, prompting the development of a universal support (20) with significantly improved base stability. Guided by Stec’s earlier work, Pya protecting groups were employed instead of the standard amide protecting groups (Figure 3B) (18). Improved results were also obtained when a Pom protection was employed for thymidine (35). With all P(V) reagents in hand and the chemoselectivity and relative coupling rates established, a systematic interrogation was undertaken to test the efficiency of this redox-neutral P(V)-platform on automated solid-phase oligonucleotide synthesis (SPOS, Figure 3A). The cycle commences with the deblocking of the DMT protecting group of a resin-bound nucleoside to afford a free 5′-alcohol that is primed to react with any P(V)-loaded nucleotide in the subsequent coupling step. This key step was carefully optimized for all reagents through systematic reactivity and hydrolysis studies using UV and 31P NMR analyses to enable a robust double coupling protocol for each P(V) reagent. The subsequent capping and deblocking steps complete the solid-phase cycle and set the stage for the next coupling (Figure 3A). The utility of any new reagent system for an oligonucleotide synthesis platform is wedded to its fidelity and robustness in the context of preparing diverse sequences with a single protocol. Thus, a matrix was designed to incorporate all possible nucleobase (A, C, G, T) and sugar (DNA, LNA) combinations templated onto a 3-10-3 DNA/LNA gapmer scaffold, the current state of the art in RNase H activating ASOs (Figure 3C) (3). LNA modifications that have a dramatic effect on binding affinity were coupled in near quantitative yields (36). A single protocol was employed regardless of the P(V) monomers used to assess the generality of the method versus sequence-specific optimization. First, the general method was field-tested to produce homogeneous, chiral PS-ASOs with both alternating (21, 22) and continuous stereochemical patterns (23–26). This represents the second industrially viable platform to produce stereopure PS-ASOs, and the first to employ redox-neutral, sustainably derived P(V)-based reagents (37). With this milestone achieved, the incorporation of PO2 linkages into these constructs was pursued. These chimeric sequences (27–30) could be accessed in high purity with no significant loss of sulfur during synthesis. Next, sequences bearing both PS and PS2 (31–34) linkages were prepared. Finally, constructs containing all four possible linkages were cleanly produced (35, 36). Thus, these protocols describe a convenient single platform for probing linkage SAR that could enable systematic tuning of physical and biological properties (38, 39). Given the differences in scale, chemical sequence, method of purification and quantification, a direct comparison between yields of this P(V)-platform and those of other stereopure methods is outside of the scope of this communication. In its present manifestation, these novel, homogeneous, chimeric oligonucleotides (21–38) were produced in 12-27% isolated yield using a sequence- and linkage- agnostic protocol. When compared directly to the stereorandom constructs (produced using state-of-the-art chemistry) that were produced in 30-60% yield, and the ability to prepare unique chimeric systems, this represents a relatively small gap to fill to bring homogeneous, stereopure oligonucleotides on par with their stereorandom counterparts. From a pragmatic perspective the observed yields even in this first disclosure are more than enough to progress a medicinal chemistry program.
Fig. 3.
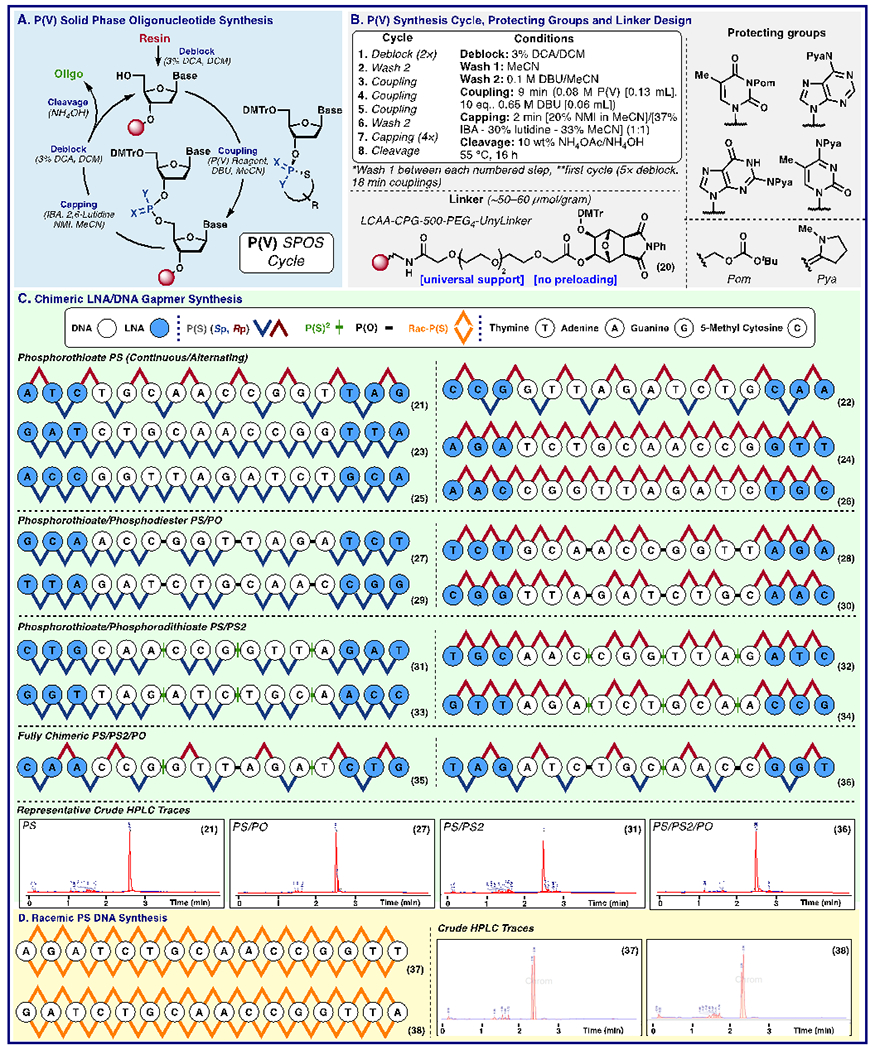
Automated P(V) oligonucleotide synthesis (A) P(V) Solid-Phase oligonucleotide synthesis cycle (B) Synthetic cycle conditions, protecting groups and linker chemistry (C) 3-10-3 LNA/DNA gapmer synthesis (D) racemic phosphorothioate oligonucleotide synthesis. DCA, dichloroacetic acid; IBA, isobutryic anyhydride, NMI, N-methyl imidazole.
Current methods to produce PS oligonucleotides generate a statistical mixture of isomers depending on the specific conditions used (40). While homogeneous, well-defined ASOs are of value, no true P(V) oligonucleotide synthesis platform would be complete without rapid access to these stereorandom variants (41). Thus, a final need was to produce rac-ψ (Figure 1), a derivative of ψ that retains high reactivity and provides mixtures of diastereomers comparable to those obtained with P(III) methodology. rac-ψ was loaded onto DNA cores, and the corresponding monomers were cleanly implemented (Figure 3D) into the aforementioned workflow, thus enabling racemic PS oligos to be produced on this platform (37, 38).
Oligonucleotide therapeutics, so-called informational drugs, target essentially every level of the central dogma. Given the immense chemical space conceivable, the number of nucleic acid modifications that have been investigated to date is quite narrow, with even fewer represented in the clinic. Although P(III)-based phosphoramidites have revolutionized access to vast swaths of this space, new linkages and chemical modifications have pushed the limits of what is currently accessible. Oligonucleotide synthesis originated with P(V)-based reagents as Nature only uses this oxidation state to craft its building blocks of life, but it was cast away shortly thereafter due to perceptions of their reduced reactivity and selectivity. This work provides a compelling justification for renewed research in this field as it can enable democratized access to a wide range of medicinally promising chimeric sequences. Finally, this new approach offers a realistic opportunity to connect the exploration of new chemical space directly to patients through a platform containing improved environmental sustainability — enhancing our ability to discover, develop and commercialize this promising class of therapeutics.
Supplementary Material
Acknowledgments.
Financial support for this work was provided by Bristol Myers Squibb, NIGMS (GM106210), NIH (grant number GM-118176), Marie Skłodowska-Curie Global Fellowships (749359-EnanSET, N.M.P) within the European Union research and innovation framework programme (2014-2020) and NSF SURF Program (TSRI, R.N). We thank D.-H. Huang and L. Pasternack (Scripps Research) for assistance with NMR spectroscopy; J. Chen (Automated Synthesis Facility, Scripps Research) for purification of compounds and acquisition of HRMS data; A. L. Rheingold, C. E. Moore, and M. Gembicky (UCSD) for X-ray analysis and Subha Mukherjee (BMS) for calculating the PMI. Current affiliations: N.M.P, Institute of Molecular Science (ICMol), Universitat de València, Paterna 46980, Spain; J.L.O, Clarochem Ireland Ltd. Damastown, Mulhuddart, Dublin 15, D15 DP73, Ireland.
Footnotes
Competing Interests. M.A.S., B.Z., K.W.K., M.D.E., P.S.B., R.E.O and I.M.M. are listed as inventors on US Patent Application US 2019/0322694, “Novel phosphorous (V)-based reagents, processes for the preparation thereof, and their use in making stereo-defined organophosphorous (V) compounds.” K.W.K and P.S.B are co-founders and shareholders of Elsie Biotechnologies.
Data Availability.
The data that support the findings of this study are available within the paper and its Supplementary Materials. Crystallographic data is available free of charge from the Cambridge Crystallographic Data Centre under reference CCDC 2093530.
References.
- 1.Hughes JP, Rees S, Kalindjian SB, Philpott KL, Principles of early drug discovery. Br. J. Pharmacol 162, 1239–1249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khvorova A, Watts JK, The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol 35, 238–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wan WB, Seth PP, The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem 59, 9645–9667 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Byrne M et al. , Stereochemistry enhances potency, efficacy, and durability of Malat1 antisense oligonucleotides in vitro and in vivo in multiple species. Transl. Vis. Sci. Technol 10, 23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y et al. , Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun 12, 847 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bleicher K et al. , Gapmer oligonucleotides comprising a phosphorodithioate internucleoside linkage and therapeutic use. WO/2019/122282 (2019).
- 7.Bethge L, Hauptmann J, F rauendorf C, Weingärtner A. Nucleic acids for inhibiting expression of a target gene comprising phosphorodithioate linkages. WO/2019/092280 (2019).
- 8.Tredenick T, “Oligonucleotides: Opportunities, pipeline and challenges” (2016); https://www.pharmamanufacturing.com/articles/2016/oligonucleotides-opportunities-pipeline-and-challenges/.
- 9.Eckstein F, Armstrong VW, Sternbach H, Stereochemistry of polymerization by DNA-dependent RNA-polymerase from Escherichia coli: An investigation with a diastereomeric ATP-analogue. Proc. Natl. Acad. Sci 73, 2987–2990 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilk A, Stec WJ, Analysis of oligo(deoxynucleoside phosphorothioate)s and their diastereomeric composition. Nucleic Acids Res. 23, 530–534 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews BI et al. , Sustainability challenges and opportunities in oligonucleotide manufacturing. J. Org. Chem 86, 49–61 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimizu M, Wada T, Asymmetric auxiliary group. US/10696711 (2020).
- 13.Oka N, Yamamoto M, Sato T, Wada T, Solid-phase synthesis of stereoregular oligodeoxyribonucleoside phosphorothioates using bicyclic oxazaphospholidine derivatives as monomer units. J. Am. Chem. Soc 130, 16031–16037 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Suchsland R, Appel B, Virta P, Müller S, Synthesis of fully protected trinucleotide building blocks on a disulphide-linked soluble support. RSC Adv. 11, 3892–3896 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knouse KW et al. , Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 361, 1234–1238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stec WJ, Grajkowski A, Koziolkiewicz M, Uznanski B, Novel route to oligo(deoxyribonucleoside phosphorothioates). Stereocontrolled synthesis of P-chiral oligo(deoxyribonucleoside phosphorothioates). Nucleic Acids Res. 19, 5883–5888 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stec WJ et al. , Diastereomers of nucleoside 3′-O-(2-thio-1,3,2-oxathia(selena)phospholanes): Building blocks for stereocontrolled synthesis of oligo(nucleoside phosphorothioate)s. J Am. Chem. Soc 117, 12019–12029 (1995). [Google Scholar]
- 18.Okruszek A, Sierzcha-la A, Fearon KL, Stec WJ, Synthesis of oligo(deoxyribonucleoside phosphorodithioate)s by the dithiaphospholane approach. J. Org. Chem 60, 6998–7005 (1995). [Google Scholar]
- 19.Stec WJ et al. , Deoxyribonucleoside 3’-O-(2-thio- and 2-oxo-“spiro”-4,4-pentamethylene-1,3,2-oxathiaphospholane)s: Monomers for stereocontrolled synthesis of oligo(deoxyribonucleoside phosphorothioate)s and chimeric PS/PO oligonucleotides. J. Am. Chem. Soc 120, 7156–7167 (1998). [Google Scholar]
- 20.Guga P, Stec WJ, Synthesis of phosphorothioate oligonucleotides with stereodefined phosphorothioate linkages. Curr. Protoc. Nucleic Acid Chem 14, 4.17.11–14.17.28 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Eckstein F, Rizk I, Synthesis of oligonucleotides by use of phosphoric triesters. Angew. Chem., Int. Ed. Engl 6, 695–696 (1967). [DOI] [PubMed] [Google Scholar]
- 22.Reese CB, Saffhill R, Oligonucleotide synthesis via phosphotriester intermediates: The phenyl-protecting group. Chem. Commun. (London), 767–768 (1968). [Google Scholar]
- 23.Letsinger RL, Lunsford WB, Synthesis of thymidine oligonucleotides by phosphite triester intermediates. J. Am. Chem. Soc 98, 3655–3661 (1976). [DOI] [PubMed] [Google Scholar]
- 24.Reese CB, Skone PA, The protection of thymine and guanine residues in oligodeoxyribonucleotide synthesis. J. Chem. Soc., Perkin Trans 1, 1263–1271 (1984). [Google Scholar]
- 25.Wiesler WT, Caruthers MH, Synthesis of phosphorodithioate DNA via sulfur-linked, base-labile protecting groups. J. Org. Chem 61, 4272–4281 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Solid-phase synthesis of RNA analogs containing phosphorodithioate linkages. Curr. Protoc. Nucleic Acid Chem 70, 4.77.71–74.77.13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radzikowska E, Baraniak J, Synthesis of PS/PO-chimeric oligonucleotides using mixed oxathiaphospholane and phosphoramidite chemistry. Organic & Biomolecular Chemistry 13, 269–276 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Guo X, Stolee JA, Fillon YA, Zou L, Trace-level determination of acrylonitrile generated in the manufacturing process of oligonucleotides by static headspace gas chromatography with an electron impact(+) mass detector. Org. Process Res. Dev 25, 318–326 (2021). [Google Scholar]
- 29.Capaldi DC et al. , Synthesis of high-quality antisense drugs. Addition of acrylonitrile to phosphorothioate oligonucleotides: Adduct characterization and avoidance. Org. Process Res. Dev 7, 832–838 (2003). [Google Scholar]
- 30.Oka N, Wada T, Saigo K, An oxazaphospholidine approach for the stereocontrolled synthesis of oligonucleoside phosphorothioates. J. Am. Chem. Soc 125, 8307–8317 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Porritt GM, Reese CB, Use of the 2,4-dinitrobenzyl protecting group in the synthesis of phosphorodithioate analogues of oligodeoxyribonucleotides. Tetrahedron Lett. 31, 1319–1322 (1990). [Google Scholar]
- 32.Caruthers MH et al. , Chemical and biochemical studies with dithioate DNA. Nucleosides Nucleotides 10, 47–59 (1991). [Google Scholar]
- 33.Marshall WS, Caruthers MH, Phosphorodithioate DNA as a potential therapeutic drug. Science 259, 1564–1570 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Caruthers MH, The chemical synthesis of DNA/RNA: Our gift to science. J. Biol. Chem 288, 1420–1427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Koning MC et al. , Simple and efficient solution-phase synthesis of oligonucleotides using extractive work-up. Org. Process Res. Dev 10, 1238–1245 (2006). [Google Scholar]
- 36.Singh SK, Koshkin AA, Wengel J, Nielsen P, LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun 455–456 (1998). [Google Scholar]
- 37.Iwamoto N et al. , Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechno l 35, 845–851 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Crooke ST, Liang X.-h., Baker BF, Crooke RM, Antisense technology: A review. J. Biol. Chem 296, 100416 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crooke ST, Baker BF, Crooke RM, Liang XH, Antisense technology: An overview and prospectus. Nat. Rev. Drug Discovery, 10.1038/s41573-021-00162-z (2021). [DOI] [PubMed] [Google Scholar]
- 40.Jahns H et al. , Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat. Commun 6, 6317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostergaard ME et al. , Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 48, 1691–1700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sekine M, Ohkubo A, Seio K, Proton-block strategy for the synthesis of oligodeoxynucleotides without base protection, capping reaction, and P-N bond cleavage reaction. J Org Chem 68, 5478–5492 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Ravikumar VT et al. , UnyLinker: An efficient and scaleable synthesis of oligonucleotides utilizing a universal linker molecule: A novel approach to enhance the purity of drugs. Organic Process Research & Development 12, 399–410 (2008). [Google Scholar]
- 44.Brooks JL, Olsen P, Chen L, Rodriguez AA, UnyLinker dimer impurity characterization and process improvement. Tetrahedron Letters 58, 1050–1052 (2017). [Google Scholar]
- 45.McBride LJ, Kierzek R, Beaucage SL, Caruthers MH, Nucleotide chemistry. 16. Amidine protecting groups for oligonucleotide synthesis. Journal of the American Chemical Society 108, 2040–2048 (2002). [Google Scholar]
- 46.Shimizu MW, Takeshi;. (Chiralgen, LTD., 2014), chap WO 2014/010250 A1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the paper and its Supplementary Materials. Crystallographic data is available free of charge from the Cambridge Crystallographic Data Centre under reference CCDC 2093530.