

ABSTRACT
Cultured Myxococcota are predominantly aerobic soil inhabitants, characterized by their highly coordinated predation and cellular differentiation capacities. Little is currently known regarding yet-uncultured Myxococcota from anaerobic, nonsoil habitats. We analyzed genomes representing one novel order (o__JAFGXQ01) and one novel family (f__JAFGIB01) in the Myxococcota from an anoxic freshwater spring (Zodletone Spring) in Oklahoma, USA. Compared to their soil counterparts, anaerobic Myxococcota possess smaller genomes and a smaller number of genes encoding biosynthetic gene clusters (BGCs), peptidases, one- and two-component signal transduction systems, and transcriptional regulators. Detailed analysis of 13 distinct pathways/processes crucial to predation and cellular differentiation revealed severely curtailed machineries, with the notable absence of homologs for key transcription factors (e.g., FruA and MrpC), outer membrane exchange receptor (TraA), and the majority of sporulation-specific and A-motility-specific genes. Further, machine learning approaches based on a set of 634 genes informative of social lifestyle predicted a nonsocial behavior for Zodletone Myxococcota. Metabolically, Zodletone Myxococcota genomes lacked aerobic respiratory capacities but carried genes suggestive of fermentation, dissimilatory nitrite reduction, and dissimilatory sulfate-reduction (in f_JAFGIB01) for energy acquisition. We propose that predation and cellular differentiation represent a niche adaptation strategy that evolved circa 500 million years ago (Mya) in response to the rise of soil as a distinct habitat on Earth.
IMPORTANCE The phylum Myxococcota is a phylogenetically coherent bacterial lineage that exhibits unique social traits. Cultured Myxococcota are predominantly aerobic soil-dwelling microorganisms that are capable of predation and fruiting body formation. However, multiple yet-uncultured lineages within the Myxococcota have been encountered in a wide range of nonsoil, predominantly anaerobic habitats, and the metabolic capabilities, physiological preferences, and capacity of social behavior of such lineages remain unclear. Here, we analyzed genomes recovered from a metagenomic analysis of an anoxic freshwater spring in Oklahoma, USA, that represent novel, yet-uncultured, orders and families in the Myxococcota. The genomes appear to lack the characteristic hallmarks for social behavior encountered in Myxococcota genomes and displayed a significantly smaller genome size and a smaller number of genes encoding biosynthetic gene clusters, peptidases, signal transduction systems, and transcriptional regulators. Such perceived lack of social capacity was confirmed through detailed comparative genomic analysis of 13 pathways associated with Myxococcota social behavior, as well as the implementation of machine learning approaches to predict social behavior based on genome composition. Metabolically, these novel Myxococcota are predicted to be strict anaerobes, utilizing fermentation, nitrate reduction, and dissimilarity sulfate reduction for energy acquisition. Our results highlight the broad patterns of metabolic diversity within the yet-uncultured Myxococcota and suggest that the evolution of predation and fruiting body formation in the Myxococcota has occurred in response to soil formation as a distinct habitat on Earth.
KEYWORDS: fruiting body formation, genome resolved metagenomics, Myxobacteria, predation
INTRODUCTION
The “Myxobacteria” represent a phylogenetically coherent lineage within the domain Bacteria (1). Originally assigned to the class Deltaproteobacteria (2, 3), they have recently been recognized as a separate phylum (Myxococcota) based on phylogenomic assessment, a proposal empirically supported by their distinct metabolic and structural characteristics (4). The Myxococcota are highly social organisms, displaying specific behaviors (predation and fruiting body formation) that require a high level of kin recognition, cell-to-cell communication, and intercellular coordination (5). Indeed, cellular differentiation in the Myxococcota has aptly been described as the most successful foray for a prokaryotic organism into multicellularity (5).
Both predation and fruiting body formation processes involve differential gene activation and expression in seemingly equivalent cells, leading to distinct cellular differentiation and disparate fates in response to external environmental stimuli. As predators, model Myxococcota utilize an epibiotic strategy, where swarms of motile cells surround and lyse prey cells via the production of secondary metabolites and extracellular enzymes (6). Significant coordination of motility and lytic agent production between individual cells has been proposed as a means to enhance the efficiency of Myxococcota predator swarms (see reference 7 but also see reference 8). Cellular differentiation in the Myxococcota entails the formation of elaborate multicellular structures (fruiting bodies) in response to nutrient depletion (9). The process involves cell aggregation and subsequent differentiation of a subset of cells into resistant myxospores, another subset into peripheral rods on the outside the fruiting body adapted to rapidly respond to reappearing nutrients, and a third subset undergoing programmed cell death (9, 10).
An extensive body of literature, spanning decades, on the mechanistic basis of social behavior in model Myxococcota is available (5, 6, 9, 11–13). Predation is enabled by two types of gliding motility (individual adventurous [A] motility and cooperative social [S] motility), exopolysaccharide production, secretion of secondary metabolites and extracellular enzymes (proteases and carbohydrate active enzymes [CAZymes]), and mechanisms for kin/self-recognition and cheater elimination (5) (the latter is also important during aggregation and fruiting body formation). Starvation-induced cellular differentiation is mediated by four modules of highly interconnected and signal-responsive cascades of signal transduction networks that act sequentially and cooperatively to coordinate and time aggregation, fruiting body formation, and sporulation (11). Simultaneously, a starvation-induced stringent response prompts the production of extracellular signals (most importantly A-signal, and C-signal) that activate a wide range of transcription factors to facilitate aggregation, regulate the onset of sporulation, and complete the development process (11).
Most cultured Myxococcota, henceforth referred to as “model Myxococcota,” are aerobic soil dwellers, known to inhabit the top layers of agricultural, forest, and even desert soils (14). This strong niche preference pattern attests to the contribution of their unique capacities to their success in soil ecosystems. The dual saprophytic/predatory capacities allow Myxococcota to utilize live microbial cells as well as microbial, floral, and faunal detritus as food sources (14). Their gliding motility allows them not only to get in close proximity with their prey, but also to access their insoluble substrates in soils (14). Their social behavior allows the sharing of resources (especially exoenzymes), enabling a more efficient process in which higher enzymatic activity is achieved compared to individual cells (14). Fruiting body formation guarantees long-term survival under adverse and highly fluctuating conditions in soil, as well as faster recovery and propagation under more favorable circumstances.
However, while Myxococcota appear to be most successful and prevalent in soils, members of this phylum have also been isolated from nonsoil habitats (e.g., Nannocystaceae and Haliangiaceae from aerobic marine sediments [15, 16], and the facultative anaerobic Anaeromyxobacter from contaminated soils and sediments [17]). Further, multiple amplicon-based diversity surveys have identified Myxococcota-affiliated sequences in nonsoil habitats, many of which represent anoxic/hypoxic settings (18–24). Recently, the implementation of genome-resolved metagenomic approaches has resulted in the recovery of Myxococcota genomes from a wide range of nonsoil habitats, almost invariably constituting a minor fraction of the population (25–30). Interestingly, many of these yet-uncultured lineages identified using 16S rRNA gene amplicon or metagenomic surveys appear to represent distinct novel, yet-uncultured lineages within the Myxococcota.
Given these observed strong niche adaptation patterns, as well as the predicted correlation between Myxococcota predation and cellular differentiation capacities, and successful propagation in soil, we hypothesized that novel, yet-uncultured Myxococcota recovered from anaerobic nonsoil habitats will display distinct metabolic, physiological, and lifestyle capacities compared to their soil counterparts. Here, we analyzed multiple genomes representing two novel Myxococcota lineages recovered from the completely anoxic, sulfide-laden (8 to 10 mM) source sediment in Zodletone Spring, an anaerobic sulfide-and sulfur-rich spring in Oklahoma. We investigated the metabolic capacities, physiological preferences, structural features, and potential social behavior of these lineages and compared their predicted capacities to model social aerobic Myxococcota. Our results suggest that nonsoil Myxococcota possess severely curtailed pathways for the typical social behavior of soil Myxococcota, potentially utilize fermentation and/or sulfate reduction for energy generation as opposed to aerobic respiration, and show preferences to polysaccharide and sugars, rather than proteins and amino acids, as carbon and energy sources. We argue that such differences provide important clues to the evolution of social behavior in the Myxococcota in light of our understanding of the history of soil formation and oxygen accumulation in the atmosphere.
RESULTS
Novel Myxococcota in Zodletone Spring sediments.
Six genomes were recovered from the anoxic black sediment sources (3 metagenome-assembled genomes [MAGs]) and the water column (3 MAGs) of Zodletone Spring, with estimated completion and contamination percentages ranging from 89.08 to 96.01% and from 1.94 to 3.87%, respectively. Phylogenomic analysis (Fig. 1), as well as amino acid identity (AAI) and relative evolutionary divergence (RED) values (Table 1), placed these genomes into one novel order (order JAFGXQ01; n = 5) and one novel family (family JAFGIB01, order Polyangiales; n = 1) within the class Polyangia. Intraorder AAI values assigned the five genomes in novel order JAFGXQ01 to two families (novel families JAFGVO01 and JAFGXQ01) and three genera (novel genera JAFGVO01, JAFGQN01, and JAFGXQ01). Names were assigned based on the assembly accession number of the most complete genome within each lineage (Table 1; see Table S1 in the supplemental material).
FIG 1.
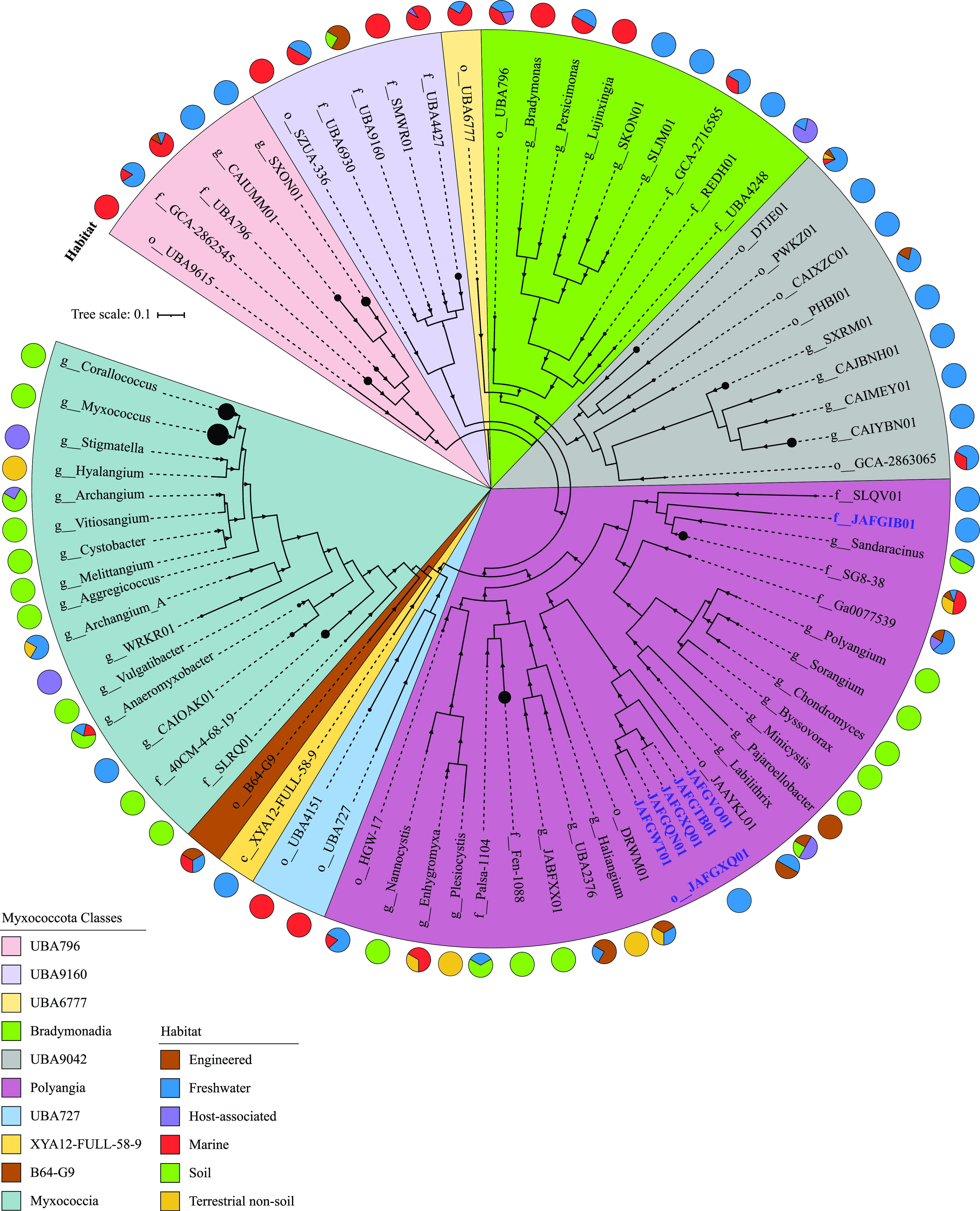
Phylogenomics of the Myxococcota, including novel lineages from Zodletone Spring. The maximum likelihood trees were constructed in RAxML (79) using all Myxococcota genomes available from the GTDB r95 database based on the concatenated alignments of 120 housekeeping genes obtained from GTDB-Tk (78). The tree was rooted (root not shown) with the two Bdellovibrionota genomes Halobacteriovorax marinus (GenBank assembly accession number GCF_000210915.2) and Bdellovibrio bacteriovorus (GenBank assembly accession number GCF_000196175.1). The tree is wedged (shown as black circles at the end of branches) to represent genus level taxonomy (g__), unless the number of available genomes per genus is less than 5, in which case the family level (f__), or order level (o__) taxonomy is shown instead. The size of the wedge is proportional to the number of genomes. Bootstrap support values based on 100 replicates are shown as triangles for nodes with >70% support. Class-level taxonomy is color-coded as shown in the legend. The track around the tree represents the ecosystem classification of the habitat from which the genomes originated. Zodletone genomes are labeled in blue bold text with their GenBank assembly accession number.
TABLE 1.
Similarity statistics and GTDB classification of the MAGs analyzed in this study
| Bin name | WGS Project (Assembly) accession no. | Completeness (%) | Contamination (%) | GTDB classification |
Similarity statistics |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Class | Order | Family | Genus | REDa value | AAIb | SGCc | ||||
| ZodW_Metabat.174 | JAFGVO01 (GCA_016937935.1) | 91.24 | 3.26 | Polyangia | JAFGXQ01 | JAFGVO01 | JAFGVO01 | 0.490 | 38.1 ± 0.9 | 40.1 ± 5.62 |
| Zod_Metabat.76 | JAFGQN01 (GCA_016935665.1) | 94.84 | 3.87 | JAFGXQ01 | JAFGQN01 | 0.493 | ||||
| ZodW_Metabat.284 | JAFGWT01 (GCA_016937015.1) | 89.08 | 3.87 | 0.489 | ||||||
| Zod_Metabat.947 | JAFGTB01 (GCA_016934495.1) | 94.19 | 3.23 | JAFGXQ01 | 0.491 | |||||
| ZodW_Metabat.4 | JAFGXQ01 (GCA_016936575.1) | 96.01 | 3.23 | 0.491 | ||||||
| Zod_Metabat.169 | JAFGIB01 (GCA_016929805.1) | 90.7 | 1.94 | Polyangiales | JAFGIB01 | JAFGIB01 | 0.660 | 40.1 ± 1.7 | 40.7 ± 5.07 | |
RED (relative evolutionary divergence) values are based on placing the genomes in the GTDB backbone bacterial tree (available at https://data.gtdb.ecogenomic.org/releases/release95/95.0/). RED values were used to confirm the novelty of the taxonomic lineage to which the genomes are assigned. Genomes are assigned to novel orders if their RED values fall between 0.62 and 0.46, and novel families if their RED values fall between 0.62 and 0.77.
AAI (average amino acid identity) is calculated using the AAI calculator (http://enve-omics.ce.gatech.edu/). The arbitrary AAI cutoffs used were 49%, 52%, 56%, and 68% for class, order, family, and genus, respectively.
SGC, shared gene content.
Comparative genomic analysis between Zodletone Myxococcota and model Myxococcota.
Comparative genomic analysis between Zodletone Myxococcota MAGs and genomes of all described type species in the phylum Myxococcota (n = 27) was conducted. These genomes belong to classes Myxococcia (n = 12), Polyangia (n = 12), and Bradymonadia (n = 3), 20 of which exhibit the distinct social behavior of the Myxococcota (5–9, 11–13, 15–17, 31). We utilized only genomes from type species to ensure the availability of experimental data regarding various aspects of their lifestyle. Compared to model Myxococcota genomes (i.e., those shown to exhibit predation behavior and to form fruiting bodies, n = 20); Zodletone genomes were significantly smaller (6.15 ± 1.28 Mb versus 11.44 ± 2.52 Mb), with a lower gene count (5,129 ± 1,005 versus 9.461 ± 1,611) and GC content (49.53 ± 5.67 versus 69.68 ± 1.74) (Student’s t test P < 0.00001) (Fig. 2). Further, multiple additional differences were observed in gene families previously implicated in mediating Myxococcota social lifestyle between Zodletone MAGs and genomes of social Myxococcota. Extracellular proteolytic enzymes are crucial components of the predatory machinery in Myxococcota, aiding in degrading prey-released proteins and/or inducing prey lysis (6). Zodletone genomes carried a significantly lower number of proteases/peptidases compared to model Myxococcota (58 ± 3.4 versus 130 ±17) (Fig. 2; Table S2). Of note is the absence of representatives of MEROPS family M15 (peptidoglycan endopeptidases) specifically implicated in prey cell lysis (Table S2). Further, model Myxococcota also secrete a plethora of secondary metabolites, such as pigments, siderophores, bacteriocins, and antibiotics, that attack and lyse their prey (6). Zodletone Myxococcota genomes encoded a significantly lower number of biosynthetic gene clusters (BGCs, 8 ± 3), mostly belonging to the nonribosomal peptide synthetase (NRPS)-polyketide synthase (PKS) type. By comparison, model Myxococcota encoded a larger number of BGCs (38 ± 16), belonging to a wider range (NRPS, PKS, terpenes, siderophores, and phenazines) of BGC classes (Fig. 2; Table S3).
FIG 2.

Comparative genomics of Zodletone novel Myxococcota genomes to the genomes of 27 type species belonging to the classes Myxococcia, Polyangia, and Bradymonadia. The species and their GenBank assembly accession numbers are Anaeromyxobacter dehalogenans 2CP-1, GCF_000022145.1; Haliangium ochraceum DSM 14365, GCF_000024805.1; Plesiocystis pacifica SIR-1, GCF_000170895.1; Corallococcus coralloides DSM 2259, GCF_000255295.1; Cystobacter fuscus DSM 2262, GCF_000335475.2; Hyalangium minutum, GCF_000737315.1; Sandaracinus amylolyticus, GCF_000737325.1; Archangium gephyra, GCF_001027285.1; Chondromyces crocatus, GCF_001189295.1; Vulgatibacter incomptus, GCF_001263175.1; Labilithrix luteola, GCF_001263205.1; Minicystis rosea, GCF_001931535.1; Melittangium boletus DSM 14713, GCF_002305855.1; Nannocystis exedens, GCF_002343915.1; Bradymonas sediminis, GCF_003258315.1; Lujinxingia litoralis, GCF_003260125.1; Polyangium fumosum, GCF_005144585.1; Persicimonas caeni, GCF_006517175.1; Myxococcus fulvus, GCF_007991095.1; Pyxidicoccus fallax, GCF_012933655.1; Stigmatella aurantiaca, GCF_900109545.1; Vitiosangium, GCF_003044305.1; Aggregicoccus, GCA_009659535.1; Pajaroellobacter abortibovis, GCF_001931505.1; Byssovorax cruenta, GCA_001312805.1; Enhygromyxa salina, GCF_002994615.1; and Sorangium cellulosum B, GCF_000067165.1. Zodletone genomes are labeled in blue bold text with their GenBank assembly accession number. Class-level taxonomy is color-coded as shown in the legend. The tracks underneath the tree show the ecosystem classification of the habitat from which the genomes originated, the assembly genome size (gray bars), GC content (yellow bars), total number of genes in the genome (cyan bars), and coding density (pink bars). The number of biosynthetic gene clusters (BGCs, purple), proteases (yellow), CAZymes (green), and transcription factors (blue) encoded in each genome are shown as a heatmap with the color tones explained in the legend. Under the heatmaps, the two outermost tracks denote the presence (filled squares)/absence (empty squares) of the Myxococcota typical social lifestyle as evidenced by experimental pure culture work (blue), and/or the machine learning approach (green) we used for lifestyle prediction based on the informative set of KO numbers provided in Data Set S1. BGCs, biosynthetic gene clusters; GH, glycosyl hydrolases; PL, polysaccharide lyases; CE, carbohydrate esterase; OCSs, one-component systems; TFs, transcription factors; RR, response regulator; SF, sigma factor; TCSs, two-component systems; HK, histidine kinases; PP, phospho-relay proteins.
Predation in model soil Myxococcota is also associated with secretion of extracellular or outer membrane CAZymes for targeting prey cell walls. While the overall numbers of CAZymes encoded in Zodletone genomes were not significantly different from those encountered in model soil Myxococcota genomes (Fig. 2), the CAZyme families were significantly different between the two groups (Table S4). Specifically, model Myxococcota genomes were enriched in two glycosyl hydrolase (GH) families, GH23 (peptidoglycan lyases, consistent with their ability to target prey cell walls) and GH13 amylases (Student’s t test P < 0.02). Instead, Zodletone genomes were significantly (Student’s t test P < 0.02) enriched in GH and polysaccharide lyase (PL) families targeting polysaccharide degradation, e.g., GH12, GH5, GH45, GH8, GH9 endoglucanases and cellobiohydrolases for cellulose degradation; GH10, GH11 xylanases for hemicellulose backbone degradation, and GH43 and GH54 xylosidases for hemicellulose side chain sugar removal; and PL1 and PL11 pectin/pectate/rhamnogalacturonan lyases for pectin degradation.
Finally, the collective social behavior in model soil Myxococcota is underpinned by an expanded arsenal of transcriptional factors. These include signal transduction one-component systems (OCS) (with a sensory domain and a response effector domain present in the same gene) and two-component systems (TCS) (with a sensor histidine kinase [HK], a partner response regulator [RR], and occasionally a phosphotransfer protein [PP]), as well as other transcriptional factors (TFs), including transcriptional regulators (TRs), and alternative sigma factors (SFs) (32). Model soil Myxococcota genomes encode 241 ± 87 OCS genes, 329 ± 95 TCS genes, and 127 ± 56 TF genes. In contrast, Zodletone Myxococcota genomes encoded significantly lower numbers of OCSs, TCSs, and TFs (65 ± 14, 198 ± 58, and 69 ± 18, respectively) (Student’s t test P < 0.05) (Fig. 2; Table S5). This pattern of curtailed transcription factor repertoire in Zodletone genomes was pronounced in OCS and TCS systems (Fig. 2), specifically OCS families AraC, ArsR, GntR, LysR, MarR, TetR, and Xre and TCS-RR belonging to the families CheY, NarL, OmpR, and FrzZ (Table S5) (Student’s t test P < 0.05).
Comparative genomics analysis of predation and cellular differentiation genes/pathways in the Myxococcota.
We assessed the distribution patterns of pathways implicated in Myxococcota social behavior in Zodletone Myxococcota and compared them to Myxococcus xanthus, the model myxobacterium extensively studied for its social behavior, as well as to Anaeromyxobacter dehalogenes, Vulgatibacter incomptus, and Labilithrix luteola. The latter three isolates share many genomic features with social Myxococcota (large genome size, high GC content, large number of genes), but lack predation and fruiting body formation capacities (33, 34). The following 13 pathways were examined: 4 gene regulatory networks governing sporulation, aggregation, and fruiting body formation; exopolysaccharide production genes; 2 extracellular signals production gene clusters (A-signal and C-signal); aggregation, sporulation, and fruiting body formation genes; chemosensory pathways; developmental timers; two motility gene clusters (A-motility and S-motility); and outer membrane exchange genes (Fig. 3 and Table 2; Table S6, supplemental text). Detailed analysis of the distribution patterns of genes in these pathways, as well as a background on their known functions is presented in the text of the supplemental material. Collectively, the analysis clearly demonstrates that social behavior pathways were severely curtailed in Zodletone Myxococcota genomes (Table 2; Table S6, supplemental text), where homologues of genes specific for the model Myxococcota social lifestyle (e.g., sporulation, extracellular signal production, motility, and outer membrane exchange) were missing from Zodletone genomes. Specifically, the most notable deficiencies were (i) absence of homologues for extracellular signal production that control early events in aggregation (Table 2; Table S6, supplemental text), (ii) absence of homologues for the two transcription factors FruA and MrpC, which work cooperatively to control the start of sporulation (35–37), although we acknowledge that this absence has also been noted outside the suborder Cystobacterineae (38), (iii) absence of homologues for sporulation-specific genes (11, 35, 37, 39, 40), motility-specific genes, and the outer membrane exchange receptor TraA, which recognizes kin and allows membrane fusion (12). Such a pattern was also observed in the genomes of Anaeromyxobacter dehalogenes, Labilithrix luteola, and Vulgatibacter incomptus, all of which have been experimentally shown to lack the capacity to aggregate into mounds, form fruiting bodies, or sporulate in pure culture. Further, for pathways with homologues identified in Zodletone genomes, the majority of such homologues carried genes that are widely distributed in bacterial genomes and not specific to the Myxococcota, e.g., transcriptional response regulators, serine/threonine kinases, peptidase domains, guanylate cyclase domains, chemotaxis-associated domains, or type IV pili.
FIG 3.

A cartoon depicting the 13 pathways associated with Myxococcota social lifestyle examined in detail in this study. Myxococcus cells are shown as blue rods, while prey cells are depicted as gray cocci and rods. Pathways active during nutrient availability are shown above the dotted line, while those induced by starvation are shown below the dotted line. Each of the pathways is shown in bold text within a color-coded outline. The same color code is used for the group of genes in each pathway and in Table S6. For each pathway, a pie chart for the number of gene homologues identified in Zodletone genomes as a percentage of the total number of genes in the pathway is shown in blue, while the percentage of gene homologues absent is shown in orange. The size of the pie chart is proportional to the number of genes in each pathway and ranges from 1 (FruA module) to 35 (the aggregation/sporulation/fruiting body formation module). Arrowheads depict the effect where activation is shown as triangular arrowheads, and inhibition is shown as horizontal line arrowheads. FruA* denotes the active form of FruA. EPS, exopolysaccharide; OME, outer membrane exchange.
TABLE 2.
The 13 different pathways/processes examined in this study as determinants of the social behavior in model Myxococcota genomes, along with the number of genes in each module, and the percentage of genes identified in Zodletone genomes as well as the genomes of three Myxococcota type species known to exhibit nonsocial behaviora,b
| Major function in the social lifestyle | Process/pathway | No. of genes in module | Zodletone lineages (%) |
Type species (nonsocial) (%) |
Notes on homologues identified/missing in Zodletone genomes |
||||
|---|---|---|---|---|---|---|---|---|---|
| O__JAFGXQ01 | F__JAFGIB01 | Anaeromyxobacter dehalogenes | Labilithrix luteola | Vulgatibacter incomptus | Homologues identified | Homologues missing | |||
| Gene regulatory networks governing early events of aggregation and mound formation prior to sporulation | Enhancer-binding protein (EBP) module | 9 | 88.9 | 88.9 | 88.9 | 100 | 88.9 | 8 total: 6 DNA-binding transcriptional response regulator domains of the NtrC family, 1 serine/threonine kinases, and 1 peptidase | ActC (one of the Act group of proteins) |
| MrpC module | 8 | 75 | 62.5 | 75 | 75 | 87.5 | 2 catalytic domains of serine/threonine kinases, 2 component system sensor histidine kinases, an oligopeptide transporter, and a DNA-binding transcriptional response regulator of the NtrC family | MrpC, a transcription factor that works cooperatively to control the start of sporulation | |
| Nla24 module | 2 | 100 | 100 | 100 | 100 | 100 | A DNA-binding transcriptional response regulator of the NtrC family and a diguanylate-cyclase (DGC) or GGDEF domain | ||
| FruA module | 1 | 0 | 0 | 0 | 0 | 0 | FruA, a transcription factor that works cooperatively to control the start of sporulation | ||
| Exopolysaccharide production necessary for the formation of fruiting bodies | EPS production | 8 | 12.5 | 12.5 | 50 | 100 | 62.5 | 1 homologue identified with a sugar transporter domain | 7 out of 8 homologues missing |
| Extracellular signal production | C-signal | 3 | 0 | 0 | 0 | 0 | 0 | All proteins in this module: CsgA encoding the actual C-signal protein, the activating protease PopC, and its inhibitor PopD | |
| A-signal | 8 | 25 | 25 | 75 | 75 | 75 | 2 DNA-binding transcriptional response regulators of the NtrC family | A-signal production-specific homologues | |
| Gene regulatory networks governing sporulation | Aggregation, sporulation, fruiting body formation | 35 | 11.4 | 11.4 | 40 | 22.9 | 42.7 | 1 sugar transporter and 3 transcriptional regulators | The majority of sporulation-specific genes The dev operon CRISPR-Cas genes The Exo and Nfs genes (both involved in forming the spore polysaccharide coat) The spore coat protein Tps The putative FAD-binding monooxygenase MXA2872 FruA-MrpC-regulated genes (Fmg) mcu operon encoding a chaperone/usher secretion system important for spore-coat assembly |
| Coordination of the two motility systems (Frz) and induction of Nla24 module (Dif) | Chemosensory pathways | 17 | 35.3 | 35.3 | 40 | 25.7 | 31.4 | 1 Dif gene and 3 Frz genes identified, mainly due to their similarity to chemotaxis protein histidine kinases, chemotaxis methyltransferase, and methylesterase Two of the three cell-polarity regulatory proteins were identified, a small GTPase, and its cognate GTPase activator |
5 of the 6 Dif chemosensory network homologues The cytoplasmic receptor for the Frz chemosensory network FrzCD, the 2 coupling proteins FrzA and FrzB, the dual response regulator protein FrzZ that localizes to the cell poles and interact with the 3 cell-polarity regulatory proteins 1 of the 3 cell-polarity regulatory proteins (RomR) |
| Fine tuning and optimizing the onset of aggregation and sporulation | Development timers | 6 | 50 | 50 | 83.3 | 83.3 | 83.3 | RodK and 2 of the RedCDEF, a 4-component system necessary for controlling aggregation timing Homologues are similar to sensor histidine kinase (n = 2), and a helix-turn-helix-containing response regulator |
2 of the RedCDEF, a 4-component system necessary for controlling aggregation timing. |
| Motility | Adventurous (A) motility | 18 | 33.3 | 27.8 | 77.8 | 38.9 | 88.9 | 3 Glt complex genes and 2 Agl complex genes (similarity to TolQR/ExbBD/MotAB-like channel) | AglZ that contributes to the assembly of the inner membrane complex CglB, the lipoprotein interacting with the substratum at focal adhesion sites GltA, B, H, K (outer membrane beta barrel structures that transport the lipoprotein CglB to the external surface) AglQS (inner membrane complex) |
| Social (S) motility | 14 | 57.1 | 57.1 | 71.4 | 64.3 | 64.3 | S motility is mediated by type IV pili (T4P) The majority of T4P genes were identified |
PilA (the major pilin), PilI, and PilO | |
| Sharing of resources, kin-recognition for exclusion of cheaters | Outer membrane exchange (OME) | 2 | 50 | 50 | 50 | 100 | 50 | TraA partner, TraB, was identified mainly because it contains an outer membrane OmpA domain | TraA, the cell surface receptor, was not identified in Zodletone genomes. |
More details are in the supplementary text and Table S6.
The cells with percentage of genes identified are color coded as follows: dark gray with white font, >50% of the pathway was identified in the genome; light gray, 25 to 50% of the pathway was identified in the genome; white, <25% of the pathway was identified in the genome.
Machine learning approaches suggest the absence of social behavior in Zodletone Myxococcota.
It is important to note that most of the physiological, mutational, and transcriptomic studies of soil Myxococcota were conducted on the model organism M. xanthus, (class Myxococcia), a relatively distant relative of Zodletone Myxococcota (class Polyangia). Further, while genes for exopolysaccharide production, adventurous motility, extracellular signal production, and sporulation are highly specific, a large proportion of the gene regulatory network and developmental timing proteins governing aggregation, sporulation, and fruiting body formation are homologues to signal transduction proteins involved in various cellular processes in a wide swath of lineages and are hence universally distributed within the bacterial world. Similarly, chemosensory network proteins in Myxococcota are homologues to a wide range of chemotaxis proteins. Indeed, in the genomes of the nonsocial Anaeromyxobacter dehalogenes, Labilithrix luteola, and Vulgatibacter incomptus, homologues for the highly specific extracellular signal production and sporulation genes were not identified, but homologues for chemosensory networks and gene regulatory networks were found, attesting to the above-mentioned caveats with the approach.
Therefore, as a complementary approach, we employed a machine learning technique to predict social behavior potential in Zodletone Myxococcota. The approach depends on first identifying, from the initial set of all genes in the genomes, a group of genes with assigned KEGG orthology (KO) numbers in the genomes of known social Myxococcota that are absent from the genomes of known nonsocial Myxococcota. These candidate genes are then used for model training using the random forest algorithm, and the constructed model is then employed to predict the social behavior based on the genomic content of Zodletone Myxococcota genomes. The occurrence pattern of the list of 634 KOs (see Data Set S1 in the supplemental material) selected for model training predicted nonsocial behavior for Zodletone Myxococcota lineages with a Matthew’s correlation coefficient of +1, confirming the patterns observed with the comparative genomics approach detailed above.
Structural features and metabolic capacities.
Structurally, Zodletone Myxococcota MAGs encoded determinants of Gram-negative cell walls (lipopolysaccharide [LPS] biosynthesis and G-peptidoglycan structure), motility (flagellar assembly and type IV pili), pigmentation (carotenoid biosynthesis), chemotaxis, and rod shape (MreBCD and RodA). The genomes also encoded type III (partial) and type VI secretion systems. Such characteristics are similar to those displayed by vegetative cells of cultured Myxococcota (Figure S1a, Table S7).
Genomic analysis predicted key differences in anabolic capacities between Zodletone Myxococcota and cultured Myxococcota. Zodletone MAGs did not encode the capacity for glycogen or trehalose biosynthesis, both of which are biosynthesized and used as storage molecules by cultured Myxococcota and have been shown to be essential for sporulation (41). Additionally, evidence for a glyoxylate shunt was missing from Zodletone MAGs. The glyoxylate shunt is employed by cultured Myxococcota to bypass CO2 loss and NADH production during the tricarboxylic acid (TCA) cycle and drive the metabolism toward oxaloacetate in preparation for gluconeogenesis (40). Further, key differences in levels of amino acid auxotrophy were predicted, where Zodletone Myxococcota MAGs encoded capacities for biosynthesis of almost all amino acids, compared to the observed auxotrophy for branched-chain amino acids (in 9 type species) and aromatic amino acids (in 5 type species) in cultured Myxococcota (42, 43). Such a pattern reflects the dependence of cultured Myxococcota on proteins and amino acids as substrates (and hence their ready availability for biosynthetic purposes) as opposed to the lack of such capacity in Zodletone genomes, necessitating amino acid biosynthesis from metabolic precursors. Finally, while cultured Myxococcota incorporate sulfur from sulfate and organic sources (e.g., taurine, alkane sulfonate, and dimethyl sulfone in 6 type species) and incorporate N from ammonia and organic sources (e.g., urea in 11 type species), such capacities for S or N incorporation from organic sources were not encoded in Zodletone Myxococcota MAGs (Table S7). Additionally, order JAFGXQ01 genomes encoded the capability to fix atmospheric nitrogen (Figure S1a, Table S7).
Genomic analysis also demonstrated multiple key differences in catabolic processes (substrate utilization patterns, respiratory capacities, electron recycling pathways, and ATP generation mechanisms) between Zodletone and model Myxococcota genomes. While most model Myxococcota (with the exception of Sorangium cellulosum) rely on amino acids and lipids as substrates and are poor carbohydrate consumers (41), Zodletone genomes encode a much lower number of amino acid degradation pathways (only 9, compared to 15 in type species), consistent with their observed limited proteolytic capabilities (Table S2). Instead, Zodletone Myxococcota appear to possess a more extensive carbohydrate degradation capacity (Table 3; Table S7, Fig. S1a), with pathways enabling the degradation of nine different sugars, sugar alcohols, sugar amines, and uronic acids encoded in their genomes. This is consistent with the possession of a wide range of polysaccharide-degrading CAZymes, as described above (Table S4). Finally, Zodletone order JAFGXQ01 genomes encoded an incomplete beta-oxidation pathway for long-chain fatty acid degradation (Table 3; Fig. S1a), a pathway commonly occurring in cultured Myxococcota to enable fatty acid consumption as the main carbon and energy source.
TABLE 3.
Catabolic capabilities deduced from genomic analysis of Zodletone Myxococcota in comparison to type species genomes available from the KEGG organism databasea
| Carbon sources | JAFGXQ01 | JAFGIB01 | Haliangium ochraceum | Anaeromyxobacter dehalogenans 2CP-C | Melittangium boletus | Archangium gephyra | Labilithrix luteola | Cystobacter fuscus | Chondromyces crocatus | Persicimonas caeni | Sorangium cellulosum so ce56 | Bradymonas sediminis | Corallococcus coralloides | Sandaracinus amylolyticus | Stigmatella aurantiaca | Minicystis rosea | Myxococcus xanthus | Vulgatibacter incomptus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sugar catabolism to central metabolites | |||||||||||||||||||
| Glucose | Y | P | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Mannose | Y | N | N | Y | Y | Y | N | Y | N | N | Y | N | Y | N | Y | N | Y | Y | |
| Galactose | Y | N | N | N | N | N | N | N | N | N | N | N | N | Y | N | N | N | N | |
| Fructose | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | N | |
| Hexosamines | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Uronic acids | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Mannitol | Y | N | N | N | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | |
| Arabinose | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Xylose | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Amino acid catabolism | |||||||||||||||||||
| Alanine | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Aspartate | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Asparagine | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Glutamate | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Glutamine | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | |
| Glycine | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Cysteine | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | |
| Methionine | N | N | Y | N | N | Y | Y | N | N | Y | N | N | N | N | Y | N | N | Y | |
| Serine | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Threonine | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Valine | N | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | |
| Leucine | N | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | |
| Isoleucine | N | Y | P | P | P | P | Y | P | P | Y | P | Y | P | P | P | P | P | P | |
| Lysine | N | N | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | |
| Proline | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Arginine | N | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Histidine | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Tyrosine | N | N | P | P | P | P | P | P | P | P | P | P | P | P | P | Y | P | P | |
| Tryptophan | N | N | P | P | N | P | N | N | N | P | N | N | P | N | P | N | N | N | |
| Fatty acid degradation | |||||||||||||||||||
| Long chain | P | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Lactate | N | N | Y | Y | N | Y | Y | N | N | Y | N | N | N | N | N | N | N | N | |
| Propionate | Y | Y | N | Y | N | N | Y | N | Y | Y | N | Y | N | N | N | Y | N | N | |
| Butanoate | N | N | N | Y | N | N | N | N | N | Y | N | N | N | N | N | N | N | N | |
| Fermentation and respiration | |||||||||||||||||||
| Fate of pyruvate | |||||||||||||||||||
| Pyruvate to acetyl-CoA | |||||||||||||||||||
| Pyruvate dehydrogenase (EC 1.2.4.1) | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Pyruvate:ferrodoxin oxidoreductase (EC 1.2.7.1) | Y | Y | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | Y | |
| Acetyl-CoA to acetate | |||||||||||||||||||
| Acetyl-CoA synthetase (EC 6.2.1.1) | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Acetate---CoA ligase (ADP-forming) subunit alpha (EC 6.2.1.13) | Y | Y | Y | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | N | |
| Succinyl-CoA:acetate CoA-transferase (EC 2.8.3.18) | N | N | N | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Phosphate acetyltransferase and acetate kinase (EC 2.3.1.8 and EC 2.7.2.1) | N | N | N | Y | N | N | N | N | N | Y | N | N | N | N | N | N | N | N | |
| Ethanol production from acetate | Y | Y | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Ethanol production from acetyl-CoA | N | N | Y | N | N | N | N | N | N | Y | N | N | N | N | N | Y | N | N | |
| Formate production from pyruvate | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| d-lactate production from pyruvate | Y | Y | Y | N | N | N | N | N | Y | Y | N | N | N | N | N | N | Y | Y | |
| l-lactate production from pyruvate | N | N | N | N | Y | Y | N | Y | N | N | Y | N | N | Y | Y | Y | N | N | |
| Acetoin production from pyruvate (via acetolactate) | N | Y | N | Y | N | N | N | N | N | N | Y | N | N | N | N | N | N | N | |
| Butanediol production from acetoin | N | Y | N | Y | N | N | N | N | N | N | Y | N | N | N | N | N | N | N | |
| TCA cycle | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Respiration (aerobic) | |||||||||||||||||||
| NADH dehydrogenase (complex I) | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Succinate dehydrogenase (complex II) | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Cytochrome c reductase (complex III) | N | N | Y | N | N | N | Y | N | N | N | Y | N | N | N | N | N | N | N | |
| Alternate complex III | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Complex IV | |||||||||||||||||||
| Cytochrome bd respiratory O2 reductase (high O2 affinity, complex IV) | Y | Y | N | Y | N | Y | Y | N | Y | Y | Y | N | Y | N | N | Y | Y | Y | |
| Cytochrome c oxidase aa3 type (low O2 affinity) | N | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Cytochrome c oxidase cbb3 type (high O2 affinity) | N | N | N | Y | N | Y | Y | N | N | Y | N | Y | N | Y | N | Y | Y | Y | |
| ATP synthase (complex V) | |||||||||||||||||||
| F-type | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Dissimilatory nitrite reduction to ammonia (DNRA) | |||||||||||||||||||
| Nitrate reductase | |||||||||||||||||||
| Periplasmic NapAB (EC 1.9.6.1) | N | N | N | N | N | Y | N | N | N | Y | Y | Y | N | N | N | N | N | N | |
| Membrane-bound NarGHI (EC 1.7.5.1) | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Nitrite reductase | |||||||||||||||||||
| NirBD; NADH (EC 1.7.1.15) | N | N | N | N | N | N | N | Y | N | N | Y | N | N | Y | N | N | N | N | |
| NrfAH; cytochrome; ammonia-forming (EC 1.7.2.2) | Y | Y | N | Y | N | Y | N | N | N | N | Y | N | Y | N | N | N | Y | Y | |
| Denitrification | N | N | N | P | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Dissimilatory sulfate reduction | |||||||||||||||||||
| Sulfate adenylyltransferase | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Adenylylsulfate reductase | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Quionone interacting membrane-bound oxidoreductase complex (QmoABC) | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Dissimilatory sulfite reductase | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Tetrathionate respiration | |||||||||||||||||||
| Octaheme tetrathionate reductase (Otr) | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Thiosulfate disproportionation | |||||||||||||||||||
| Thiosulfate reductase (PhsABC) | N | Y | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Thiosulfate oxidation | |||||||||||||||||||
| SOX complex | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Sulfite oxidation | |||||||||||||||||||
| SoeABC | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | N | N | N | N | |
| Sulfide oxidation | |||||||||||||||||||
| Sulfide:quinone oxidoreductase (EC 1.8.5.4) | N | N | Y | Y | Y | Y | N | Y | Y | Y | N | N | Y | N | Y | N | Y | N | |
Y, full pathway identified; N, full pathway missing; P, partial pathway identified.
All cultured Myxococcota (with the exception of the genus Anaeromyxobacter) are aerobic microorganisms. In contrast, Zodletone Myxococcota genomes lack key genes for aerobic respiration, specifically, homologues for either complex III or alternative complex III, and lack homologues for the low-affinity cytochrome oxidase aa3. High-affinity cytochrome bd ubiquinol oxidase is encoded in Zodletone genomes but could possibly be employed in detoxification of trace amounts of O2 that are present. Instead, the genomes encode genes enabling the utilization of nitrite as a terminal electron acceptor via the cytochrome-linked nitrite reductase NrfAH (Table 3; Fig. S1a). Further, the single genome representative of novel family JAFGIB01 encodes a full dissimilatory sulfate reduction machinery, including sulfate adenylyltransferase (Sat; EC 2.7.7.4) for sulfate activation to APS, adenylylsulfate reductase (AprAB; EC:1.8.99.2) for adenylyl sulfate reduction to sulfite, QmoABC for electron transfer, dissimilatory sulfite reductase (DsrAB; EC:1.8.99.5) and its cosubstrate DsrC for dissimilatory sulfite reduction to sulfide, and the sulfite reduction-associated membrane complex DsrMKJOP for linking cytoplasmic sulfite reduction to energy conservation (Table 3, Fig. S1a). The JAFGIB01 genome also encoded octaheme tetrathionate reductase (otr) and thiosulfate reductase phsABC, suggesting the capability to utilize tetrathionate and thiosulfate as terminal electron acceptors in addition to sulfate (Fig. S1a). To our knowledge, sulfur species respiration capability in the Myxococcota has not been reported and could possibly be a reflection of the sulfur and sulfide-rich Zodletone Spring environment from which the MAG was binned. Phylogenetically, JAFGIB01 DsrAB were most closely affiliated with DsrAB sequences encountered in Acidobacteria genomes (Fig. S1b) (44).
Besides respiration, additional pathways for electron disposal were identified in Zodletone Myxococcota genomes. These include fermentative processes for acetate, ethanol, and lactate production from pyruvate (Table 3; Fig. S1a). In addition, the genomes encoded a full Wood Ljungdahl pathway (WLP), probably acting as an electron sink mechanism for reoxidizing reduced ferredoxin, as previously noted in “Candidatus Bipolaricaulota” and Desulfobacterota genomes (45, 46). Finally, a possible additional mechanism for ATP production in Zodletone Myxococcota is the utilization of the Rhodobacter nitrogen fixation (RNF) complex for reoxidizing reduced ferredoxin at the expense of NAD, with the concomitant export of protons to the periplasm, generating a proton motive force that can drive ATP production via oxidative phosphorylation via the encoded F-type ATP synthase. Consistent with encoding RNF complex components, the genomes also encoded elements for electron carrier recycling, including the cytoplasmic electron bifurcating mechanism, HydABC. Analysis of Myxococcota type species genomes revealed the absence of RNF complex components, HydABC electron bifurcation system, and the WLP, consistent with a strictly aerobic mode of metabolism.
DISCUSSION
Multiple notable differences were observed between Zodletone Myxococcota and soil Myxococcota. Of these, the observed variation in GC content was most notable. Within the Bacteria, GC content has been observed to be positively correlated with genome size (47), temperature (48, 49), and salinity preferences (50). Further, higher GC content is associated with aerobic life style (51). As such, we speculate that the notable difference in GC content between Zodletone Myxococcota and soil Myxococcota could be a reflection of their smaller genome size and aerobic lifestyle. In addition, it is possible that a lower rate of intragenic recombination could be occurring in Zodletone Myxococcota, leading to a less pronounced effect of GC-biased gene conversion on genome GC content (52) and a more pronounced effect of spontaneous cytidine deamination on GC to AT mutations (53, 54), collectively leading to a reduction of genomic GC content over evolutionary times from a possible common ancestor with higher GC content, as previously speculated (52).
Further, our comparative genomics and machine learning analyses revealed severely curtailed machineries for predation and cellular differentiation in Zodletone Myxococcota (Fig. 2 and 3; Table 2; Table S6). Such a conclusion is in agreement with a recent study that predicted the absence of predation potential in MAGs/single amplified genomes (SAGs) encompassing most of the publicly available, yet-uncultured Myxococcota (4). As such, a clear delineation exists between two phylogenetically and behaviorally distinct groups within the Myxococcota. The first encompasses aerobic top soil dwellers in classes Myxococcia and Polyangia that are characterized by possessing a highly sophisticated machinery enabling predation and cellular differentiation behaviors. Few freshwater and marine strains possessing such capacities have been reported, but their presence has been attributed to air and dust transport from neighboring soils (15, 16). Members of this group were readily obtained in pure cultures. The second group encompasses phylogenetically distinct families and orders within the classes Myxococcia and Polyangia (including Zodletone MAGs), as well a few yet-uncultured Myxococcota classes. These lineages are almost invariably encountered within nonsoil habitats (e.g., freshwater, marine, host-associated, and engineered ecosystems) and appear to lack the capacity for predation and social differentiation. Most of these lineages are currently uncultured, with the exception of members of the genus Anaeromyxobacter (55).
We argue that such patterns could provide important clues about the evolution of social behavior in the Myxococcota when considered in light of our understanding of the history of soil formation and the rise of atmospheric oxygen in the atmosphere. Soil formation and transition from barren crusts to current soil orders through organic matter deposition and transformation has been enabled by the evolution of lichen associations, plant terrestrialization, formation of mycorrhizal association, and subsequent colonization by soil microfauna. All such processes are mediated by aerobic organisms (algae, fungi, plants, and fauna) and hence were possible only after the accumulation of oxygen to levels comparable to current values in the atmosphere (approximately 500 to 600 million years ago [Mya] [56]). The formation of soil structures as a new and organic-rich habitat has certainly spurred multiple evolutionary processes for enabling terrestrial adaptation within the microbial world. Various processes have been reported in multiple soil-prevalent lineages, from CAZyme and BGC acquisition in the Acidobacteria (44, 57) to acquisition of stress tolerance, adherence, and regulatory genes in the ammonia-oxidizing archaea (58). Here, it appears that the development of predation and cellular differentiation machineries has enabled the Myxococcota to assume an apex predator niche and imparted them with strong survival capacities in soil, respectively. Indeed, as previously noted, the ecological success of social Myxococcota in soils appears to be in stark contrast to the rarity and low relative abundance of nonsocial Myxococcota in other habitats (59).
The evolution of beneficial trait(s) in a single lineage of Myxococcota in soil could be propagated to the broader soil Myxococcota community through intraclade, habitat-specific horizontal gene transfer (HGT), resulting in the observed checkered distribution pattern, where social behavior is observed only in specific families within the classes Myxococcia and Polyangia. HGT between closely related taxa is a well-established phenomenon (60) that has been widely documented, e.g., in mediating the spread of antibiotic resistance in related clinical strains (61, 62). The barrier for HGT within closely related taxa is predictably lower, given the expected similarity in codon usage pattern, GC content, restriction enzyme machinery, and overall genome architecture between donor and recipient strains. Similarly, physical proximity in the same habitat is seen as a facilitator of genetic exchange through HGT (63–65).
In conclusion, our results strongly indicate that anaerobic Myxococcota do not possess the capacity for typical social behavior and display distinct structural, anabolic, and catabolic differences compared to model aerobic Myxococcota. We document their dependence on fermentation and/or nitrite or sulfate reduction for energy generation, as well as their preference for polysaccharide metabolism over protein, amino acids, and lipid metabolism. We further propose that such differences strongly underscore the importance of niche differentiation in shaping the evolutionary trajectory of the Myxococcota and suggest soil formation as a strong driver for developing social behavior in this lineage.
MATERIALS AND METHODS
Site description and geochemistry.
Zodletone Spring is located in the Anadarko Basin of western Oklahoma (34.99562°N, 98.68895°W). The site geochemistry has been previously described in detail (66–68). Briefly, at the spring source, sulfide and gaseous hydrocarbon-saturated waters are slowly (8 liters/min) ejected, along with sediments that deposit at the source of the spring. High (8 to 10 mM) sulfide concentrations maintain complete anoxic conditions (oxygen levels, <0.1 μM) in the spring sediments. Oxygen concentrations in the 50-cm water column overlying the sediments vary from 2 to 4 μM at 2 mm above the source to complete oxygen exposure on the top of the water column (66).
Sampling and nucleic acid extraction.
The sampling and DNA extraction processes have been previously described in detail (45, 69). Briefly, 10 different sediment samples (≈50 g each) were collected at a 5-cm depth, as well as from the standing overlaid water in sterile containers. DNA was extracted from 0.5 g of source sediments from each replicate sample. For water samples, 10 liters of water was filtered in 0.2-μm sterile filters, and DNA was directly extracted from the filters. Extraction was conducted using the DNeasy PowerSoil kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocols.
Metagenome sequencing, assembly, and binning.
All extractions from sediment or water samples were pooled, and the pooled DNA was used for the preparation of sequencing libraries using the Nextera XT DNA library prep kit (Illumina, San Diego, CA, USA) as per the manufacturer’s instructions. DNA sequencing was conducted using two lanes on the Illumina HiSeq 2500 platform and 150-bp paired-end technology for each of the water and sediment samples using the services of a commercial provider (Novogene, Beijing, China). Metagenomic sequencing of the sediments and water samples yielded 281 Gbp and 323 Gbp of raw data, respectively. Reads were assessed for quality using FastQC, followed by quality filtering and trimming using Trimmomatic v0.38 (70) using a sliding window size of 4 bases and a sliding window average minimum quality of 15, a leading and trailing minimum quality of 3, and a minimum read length of 36. High-quality reads were assembled into contigs using MEGAHIT v1.1.3 (71) with a minimum Kmer of 27, maximum kmer of 127, Kmer step of 10, and minimum contig length of 1,000 bp. Bowtie2 was used to calculate the percentage of reads that assembled into contigs and the sequencing coverage for each contig. Contigs of >1 Kbp were binned into draft metagenome-assembled genomes (MAGs) using MetaBAT (72) and MaxBin v2 (73), followed by selection of the highest-quality bins using DasTool (74). CheckM (75) was used for the estimation of genome completeness, strain heterogeneity, and contamination by employing the lineage-specific workflow (lineage_wf flag). Quality designation of draft genomes was based on the criteria set forth using MIMAGs (Minimum Information about a Metagenome-Assembled Genome) (76).
Genome classification.
Taxonomic classifications followed the Genome Taxonomy Database (GTDB) release r95 (77) and were carried out using the classify_workflow in GTDB-Tk v1.3.0 (78). Phylogenomic analysis utilized the concatenated alignment of a set of 120 single-copy bacterial genes (77) generated using GTDB-Tk. A maximum-likelihood phylogenomic tree was constructed in RAxML using the PROTGAMMABLOSUM62 model and default parameters (79) and members of the Bdellovibrionota as an outgroup. To further assign genomes to putative families and genera, average amino acid identity (AAI) and shared gene content (SGC) were calculated using the AAI calculator (http://enve-omics.ce.gatech.edu/). The arbitrary AAI cutoffs used were 49%, 52%, 56%, and 68% for class, order, family, and genus, respectively (80, 81). Further, relative evolutionary divergence (RED) values, based on placement in the GTDB backbone tree (available at https://data.gtdb.ecogenomic.org/releases/release95/95.0/), were used to confirm the novelty of lineages to which the genomes are assigned. Values between 0.62 and 0.46 are indicative of a novel order, and values between 0.62 and 0.77 are indicative of a novel family.
Annotation and genomic analysis.
Protein-coding genes were predicted using Prodigal (82). GhostKOALA (83) was used for the annotation of every predicted open reading frame in bins and to assign protein-coding genes to KEGG orthologies (KOs), followed by metabolic pathway visualization in KEGG Mapper (84). In addition, all genomes were queried with custom-built HMM profiles for alternate complex III components and hydrogenases. To construct HMM profiles, a representative protein was queried against the KEGG gene database using BLASTP, and hits with e Values of <1e−80 were downloaded, aligned, and used to construct an HMM profile using the hmmbuild function of HMMER v3.1b2 (85). Hydrogenase HMM profiles were built using alignments downloaded from the Hydrogenase Database (HydDB) (86). The hmmscan function of HMMER (85) was used with the constructed profiles and a thresholding option of -T 100 to scan the protein-coding genes for possible hits. Further confirmation was achieved through phylogenetic assessment and tree-building procedures. The 5S, 16S, and 23S rRNA sequences were identified using Barrnap 0.9 (https://github.com/tseemann/barrnap). tRNA sequences were identified using tRNAscan-SE v2.0.6 (May 2020) (87). Genomes were mined for CRISPR and Cas proteins using the CRISPRCasFinder (88). Proteases, peptidases, and protease inhibitors were identified using BLASTP against the MEROPS database (89), while carbohydrate active enzymes (CAZymes) were identified by searching all open reading frames (ORFs) from all genomes against the dbCAN hidden Markov models v9 (90) (downloaded from the dbCAN Web server in September 2020). AntiSMASH 3.0 (91) was used with default parameters to predict biosynthetic gene clusters in the genomes. Metabolic reconstruction of reference Myxococcota type species genomes was obtained from the KEGG genomes database (https://www.genome.jp/kegg/genome/) and used for comparative genomics to Zodletone Myxococcota genomes.
Phylogenetic analysis of dissimilatory sulfite reductase DsrAB.
Predicted dissimilatory sulfite reductase subunits A and B were compared to reference sequences for phylogenetic placement by first aligning them to corresponding subunits from sulfate-reducing taxa using MAFFT (92). DsrA and DsrB alignments were concatenated in Mega X (93) and used to construct maximum-likelihood phylogenetic trees using FastTree v 2.1.10 (94).
Machine learning approaches.
The genomes of type species of cultured social (i.e., experimentally verified to be involved in predation and observed to undergo cellular differentiation and fruiting body formation) Myxococcota lineages (n = 24) and all cultured nonsocial Myxococcota lineages (n = 13) were downloaded from GenBank (June 2021). Lineages were assigned their “social” status using prior culture-based observations (15–17, 31). Genomes were annotated with KO numbers using GhostKOALA (83) using default parameters, and gene counts were assembled into a matrix. Informative KOs (n = 634) were selected using indicator analysis with the R package indicspecies (95) using the multipattern function and were used to build a predictive model. Data were then centered, scaled, and Box-Cox transformed using the R package caret (https://topepo.github.io/caret/). Random forest classification training was performed in Python 3 with the ensemble method of Scikit-learn v 0.24.1 (96). The data were randomly divided, with 75% of the data selected to serve as the training set and the remaining 25% reserved for model verification. Model training was performed with default parameters and 1,000 estimators. The model successfully predicted the social behavior (with 100% accuracy) in the 25% data subset reserved for model verification. Social abilities of novel Myxococcota lineages were then predicted using the constructed model. Matthew’s correlation coefficient (97) was used to quantify classification accuracy. Including only pure cultures genomes in our model with experimentally verified social behavior ensured that the model was accurately trained on possession of social behavior rather than arbitrary genomic artifacts due to phylogenetic relatedness.
Data availability.
The individual assembled Myxococcota MAGs analyzed in this study have been deposited at DDBJ/ENA/GenBank under the accession numbers JAFGVO000000000, JAFGQN000000000, JAFGWT000000000, JAFGTB000000000, JAFGXQ000000000, and JAFGIB000000000.
ACKNOWLEDGMENTS
We thank David W. Waite at the Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, University of Queensland, for helpful discussions regarding utilization of machine learning approaches for trait prediction.
This work was supported by NSF grant 2016423 to N.H.Y. and M.S.E.
Footnotes
Supplemental material is available online only.
Contributor Information
Noha H. Youssef, Email: noha@okstate.edu.
Pablo Ivan Nikel, Novo Nordisk Foundation Center for Biosustainability.
REFERENCES
- 1.Shimkets LJ, Dworkin M, Reichenbach H. 2006. The Myxobacteria, p 31–115. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The Prokaryotes, vol 7. Proteobacteria: delta, epsilon subclass. 10.1007/0-387-30747-8_3. Springer, New York, NY. [DOI] [Google Scholar]
- 2.Shimkets L, Woese CR. 1992. A phylogenetic analysis of the myxobacteria: basis for their classification. Proc Natl Acad Sci USA 89:9459–9463. 10.1073/pnas.89.20.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spröer C, Reichenbach H, Stackebrandt E. 1999. The correlation between morphological and phylogenetic classification of myxobacteria. Int J Syst Bacteriol 49:1255–1262. 10.1099/00207713-49-3-1255. [DOI] [PubMed] [Google Scholar]
- 4.Waite DW, Chuvochina M, Pelikan C, Parks DH, Yilmaz P, Wagner M, Loy A, Naganuma T, Nakai R, Whitman WB, Hahn MW, Kuever J, Hugenholtz P. 2020. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int J Syst Evol Microbiol 70:5972–6016. 10.1099/ijsem.0.004213. [DOI] [PubMed] [Google Scholar]
- 5.Cao P, Dey A, Vassallo CN, Wall D. 2015. How myxobacteria cooperate. J Mol Biol 427:3709–3721. 10.1016/j.jmb.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiery S, Kaimer C. 2020. The predation strategy of Myxococcus xanthus. Front Microbiol 11:2. 10.3389/fmicb.2020.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berleman JE, Kirby JR. 2009. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev 33:942–957. 10.1111/j.1574-6976.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall RC, Whitworth DE. 2019. Is “wolf-pack” predation by antimicrobial bacteria cooperative? Cell behaviour and predatory mechanisms indicate profound selfishness, even when working alongside kin. Bioessays 41:e1800247. 10.1002/bies.201800247. [DOI] [PubMed] [Google Scholar]
- 9.Muñoz-Dorado J, Marcos-Torres FJ, García-Bravo E, Moraleda-Muñoz A, Pérez J. 2016. Myxobacteria: moving, killing, feeding, and surviving together. Front Microbiol 7:781. 10.3389/fmicb.2016.00781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muñoz-Dorado J, Moraleda-Muñoz A, Marcos-Torres FJ, Contreras-Moreno FJ, Martin-Cuadrado AB, Schrader JM, Higgs PI, Pérez J. 2019. Transcriptome dynamics of the Myxococcus xanthus multicellular developmental program. Elife 8:e50374. 10.7554/eLife.50374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroos L. 2017. Highly signal-responsive gene regulatory network governing Myxococcus development. Trends Genet 33:3–15. 10.1016/j.tig.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sah GP, Wall D. 2020. Kin recognition and outer membrane exchange (OME) in myxobacteria. Curr Opin Microbiol 56:81–88. 10.1016/j.mib.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zusman DR, Scott AE, Yang Z, Kirby JR. 2007. Chemosensory pathways, motility and development in Myxococcus xanthus. Nat Rev Microbiol 5:862–872. 10.1038/nrmicro1770. [DOI] [PubMed] [Google Scholar]
- 14.Reichenbach H. 1999. The ecology of the myxobacteria. Environ Microbiol 1:15–21. 10.1046/j.1462-2920.1999.00016.x. [DOI] [PubMed] [Google Scholar]
- 15.Garcia R, Müller R. 2014. The family Nannocystaceae, p 213–229. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. 10.1007/978-3-642-39044-9_305. Springer, Berlin, Germany. [DOI] [Google Scholar]
- 16.Garcia R, Müller R. 2014. The family Haliangiaceae, p 173–181. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. 10.1007/978-3-642-39044-9_271. Springer, Berlin, Germany. [DOI] [Google Scholar]
- 17.dos Santos DFK, Kyaw CM, De Campos TA, Miller RNG, Noronha EF, Bustamante MMDC, Kruger R. 2014. The family Cystobacteraceae, p 19–40. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. 10.1007/978-3-642-39044-9_304. Heidelberg, Berlin, Germany. [DOI] [Google Scholar]
- 18.Ji Y, Angel R, Klose M, Claus P, Marotta H, Pinho L, Enrich-Prast A, Conrad R. 2016. Structure and function of methanogenic microbial communities in sediments of Amazonian lakes with different water types. Environ Microbiol 18:5082–5100. 10.1111/1462-2920.13491. [DOI] [PubMed] [Google Scholar]
- 19.Iizuka T, Tokura M, Jojima Y, Hiraishi A, Yamanaka S, Fudou R. 2006. Enrichment and phylogenetic analysis of moderately thermophilic myxobacteria from hot springs in Japan. Microb Environ 21:189–199. 10.1264/jsme2.21.189. [DOI] [Google Scholar]
- 20.Tian F, Yu Y, Chen B, Li H, Yao Y-F, Guo X-K. 2009. Bacterial, archaeal and eukaryotic diversity in Arctic sediment as revealed by 16S rRNA and 18S rRNA gene clone libraries analysis. Polar Biol 32:93–103. 10.1007/s00300-008-0509-x. [DOI] [Google Scholar]
- 21.Kandel PP, Pasternak Z, van Rijn J, Nahum O, Jurkevitch E. 2014. Abundance, diversity and seasonal dynamics of predatory bacteria in aquaculture zero discharge systems. FEMS Microbiol Ecol 89:149–161. 10.1111/1574-6941.12342. [DOI] [PubMed] [Google Scholar]
- 22.Li SG, Zhou XW, Li PF, Han K, Li W, Li ZF, Wu ZH, Li YZ. 2012. The existence and diversity of myxobacteria in lake mud - a previously unexplored myxobacteria habitat. Environ Microbiol Rep 4:587–595. 10.1111/j.1758-2229.2012.00373.x. [DOI] [PubMed] [Google Scholar]
- 23.Kou W, Zhang J, Lu X, Ma Y, Mou X, Wu L. 2016. Identification of bacterial communities in sediments of Poyang Lake, the largest freshwater lake in China. Springerplus 5:401. 10.1186/s40064-016-2026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohr KI. 2018. Diversity of myxobacteria: -we only see the tip of the iceberg. Microorganisms 6:84. 10.3390/microorganisms6030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC, Finn RD. 2021. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol 39:105–114. 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Royo-Llonch M, Sánchez P, Ruiz-González C, Salazar G, Pedrós-Alió C, Labadie K, Paoli L, Chaffron S, Eveillard D, Karsenti E, Sunagawa S, Wincker P, Karp-Boss L, Bowler C, Acinas SG. 2020. Ecogenomics of key prokaryotes in the arctic ocean. bioRxiv 10.1101/2020.06.19.156794:2020.06.19.156794. [DOI]
- 27.Nobu MK, Narihiro T, Mei R, Kamagata Y, Lee PKH, Lee P-H, McInerney MJ, Liu W-T. 2020. Catabolism and interactions of uncultured organisms shaped by eco-thermodynamics in methanogenic bioprocesses. Microbiome 8:111–111. 10.1186/s40168-020-00885-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye L, Mei R, Liu W-T, Ren H, Zhang X-X. 2020. Machine learning-aided analyses of thousands of draft genomes reveal specific features of activated sludge processes. Microbiome 8:16. 10.1186/s40168-020-0794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robbins SJ, Song W, Engelberts JP, Glasl B, Slaby BM, Boyd J, Marangon E, Botté ES, Laffy P, Thomas T, Webster NS. 2021. A genomic view of the microbiome of coral reef demosponges. ISME J 15:1641–1654. 10.1038/s41396-020-00876-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jégousse C, Vannier P, Groben R, Glöckner FO, Marteinsson V. 2021. A total of 219 metagenome-assembled genomes of microorganisms from Icelandic marine waters. PeerJ 9:e11112. 10.7717/peerj.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia R, Müller R. 2014. The family Myxococcaceae, p 191–212. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. 10.1007/978-3-642-39044-9_303. Springer, Berlin, Germany. [DOI] [Google Scholar]
- 32.Whitworth DE, Zwarycz A. 2020. A genomic survey of signalling in the Myxococcaceae. Microorganisms 8:1739. 10.3390/microorganisms8111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanford RA, Cole JR, Tiedje JM. 2002. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl Environ Microbiol 68:893–900. 10.1128/AEM.68.2.893-900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto E, Muramatsu H, Nagai K. 2014. Vulgatibacter incomptus gen. nov., sp. nov. and Labilithrix luteola gen. nov., sp. nov., two myxobacteria isolated from soil in Yakushima Island, and the description of Vulgatibacteraceae fam. nov., Labilitrichaceae fam. nov. and Anaeromyxobacteraceae fam. nov. Int J Syst Evol Microbiol 64:3360–3368. 10.1099/ijs.0.063198-0. [DOI] [PubMed] [Google Scholar]
- 35.Mittal S, Kroos L. 2009. Combinatorial regulation by a novel arrangement of FruA and MrpC2 transcription factors during Myxococcus xanthus development. J Bacteriol 191:2753–2763. 10.1128/JB.01818-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogawa M, Fujitani S, Mao X, Inouye S, Komano T. 1996. FruA, a putative transcription factor essential for the development of Myxococcus xanthus. Mol Microbiol 22:757–767. 10.1046/j.1365-2958.1996.d01-1725.x. [DOI] [PubMed] [Google Scholar]
- 37.Robinson M, Son B, Kroos D, Kroos L. 2014. Transcription factor MrpC binds to promoter regions of hundreds of developmentally-regulated genes in Myxococcus xanthus. BMC Genomics 15:1123. 10.1186/1471-2164-15-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huntley S, Wuichet K, Søgaard-Andersen L. 2014. Genome evolution and content in the myxobacteria, p 31–50. In Yang Z, Higgs PI (ed), Myxobacteria: genomics, cellular and molecular biology, vol 1. Caister Academic Press, Poole, UK. [Google Scholar]
- 39.Boysen A, Ellehauge E, Julien B, Søgaard-Andersen L. 2002. The DevT protein stimulates synthesis of FruA, a signal transduction protein required for fruiting body morphogenesis in Myxococcus xanthus. J Bacteriol 184:1540–1546. 10.1128/JB.184.6.1540-1546.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller FD, Treuner-Lange A, Heider J, Huntley SM, Higgs PI. 2010. Global transcriptome analysis of spore formation in Myxococcus xanthus reveals a locus necessary for cell differentiation. BMC Genomics 11:264. 10.1186/1471-2164-11-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtis PD, Shimkets LJ. 2007. Metabolic pathways relevant to predation, signaling, and development, p 241–258. In Whitworth DE (ed), Myxobacteria: multicellularity and differentiation. ASM Press, Washington, DC. 10.1128/9781555815677.ch14. [DOI] [Google Scholar]
- 42.Bretscher AP, Kaiser D. 1978. Nutrition of Myxococcus xanthus, a fruiting myxobacterium. J Bacteriol 133:763–768. 10.1128/jb.133.2.763-768.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dworkin M. 1962. Nutritional requirements for vegetative growth of Myxococcus xanthus. J Bacteriol 84:250–257. 10.1128/jb.84.2.250-257.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yadav A, Borrelli JC, Elshahed MS, Youssef NH. 2021. Genomic analysis of family UBA6911 (group 18 Acidobacteria) expands the metabolic capacities of the phylum and highlights adaptations to terrestrial habitats. bioRxiv 10.1101/2021.04.09.439258:2021.04.09.439258. [DOI] [PMC free article] [PubMed]
- 45.Murphy CL, Biggerstaff J, Eichhorn A, Ewing E, Shahan R, Soriano D, Stewart S, VanMol K, Walker R, Walters P, Elshahed MS, Youssef NH. 2021. Genomic characterization of three novel Desulfobacterota classes expand the metabolic and phylogenetic diversity of the phylum. Environ Microbiol 23:4326–4343. 10.1111/1462-2920.15614. [DOI] [PubMed] [Google Scholar]
- 46.Youssef NH, Farag IF, Rudy S, Mulliner A, Walker K, Caldwell F, Miller M, Hoff W, Elshahed M. 2019. The Wood-Ljungdahl pathway as a key component of metabolic versatility in candidate phylum Bipolaricaulota (Acetothermia, OP1). Environ Microbiol Rep 11:538–547. 10.1111/1758-2229.12753. [DOI] [PubMed] [Google Scholar]
- 47.Almpanis A, Swain M, Gatherer D, McEwan N. 2018. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb Genom 4:e000168. 10.1099/mgen.0.000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musto H, Naya H, Zavala A, Romero H, Alvarez-Valín F, Bernardi G. 2006. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem Biophys Res Commun 347:1–3. 10.1016/j.bbrc.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 49.Zheng H, Wu H. 2010. Gene-centric association analysis for the correlation between the guanine-cytosine content levels and temperature range conditions of prokaryotic species. BMC Bioinformatics 11(Suppl 11):S7. 10.1186/1471-2105-11-S11-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul S, Bag SK, Das S, Harvill ET, Dutta C. 2008. Molecular signature of hypersaline adaptation: insights from genome and proteome composition of halophilic prokaryotes. Genome Biol 9:R70. 10.1186/gb-2008-9-4-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naya H, Romero H, Zavala A, Alvarez B, Musto H. 2002. Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J Mol Evol 55:260–264. 10.1007/s00239-002-2323-3. [DOI] [PubMed] [Google Scholar]
- 52.Lassalle F, Périan S, Bataillon T, Nesme X, Duret L, Daubin V. 2015. GC-content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet 11:e1004941. 10.1371/journal.pgen.1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ely B. 2021. Genomic GC content drifts downward in most bacterial genomes. PLoS One 16:e0244163. 10.1371/journal.pone.0244163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildebrand F, Meyer A, Eyre-Walker A. 2010. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet 6:e1001107. 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas SH, Wagner RD, Arakaki AK, Skolnick J, Kirby JR, Shimkets LJ, Sanford RA, Löffler FE. 2008. The mosaic genome of Anaeromyxobacter dehalogenans strain 2CP-C suggests an aerobic common ancestor to the delta-proteobacteria. PLoS One 3:e2103. 10.1371/journal.pone.0002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Canfield DE. 1998. A new model for Proterozoic ocean chemistry. Nature 396:450–453. 10.1038/24839. [DOI] [Google Scholar]
- 57.Eichorst SA, Trojan D, Roux S, Herbold C, Rattei T, Woebken D. 2018. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ Microbiol 20:1041–1063. 10.1111/1462-2920.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abby SS, Kerou M, Schleper C. 2020. Ancestral reconstructions decipher major adaptations of ammonia-oxidizing archaea upon radiation into moderate terrestrial and marine environments. mBio 11:e02371-20. 10.1128/mBio.02371-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou X-w, Li S-g, Li W, Jiang D-m, Han K, Wu Z-h, Li Y-z. 2014. Myxobacterial community is a predominant and highly diverse bacterial group in soil niches. Environ Microbiol Rep 6:45–56. 10.1111/1758-2229.12107. [DOI] [PubMed] [Google Scholar]
- 60.Adato O, Ninyo N, Gophna U, Snir S. 2015. Detecting horizontal gene transfer between closely related taxa. PLoS Comput Biol 11:e1004408. 10.1371/journal.pcbi.1004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Donnenberg MS. 2000. Pathogenic strategies of enteric bacteria. Nature 406:768–774. 10.1038/35021212. [DOI] [PubMed] [Google Scholar]
- 62.Pallen MJ, Wren BW. 2007. Bacterial pathogenomics. Nature 449:835–842. 10.1038/nature06248. [DOI] [PubMed] [Google Scholar]
- 63.Beiko RG, Harlow TJ, Ragan MA. 2005. Highways of gene sharing in prokaryotes. Proc Natl Acad Sci USA 102:14332–14337. 10.1073/pnas.0504068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moliner C, Fournier PE, Raoult D. 2010. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol Rev 34:281–294. 10.1111/j.1574-6976.2010.00209.x. [DOI] [PubMed] [Google Scholar]
- 65.Shterzer N, Mizrahi I. 2015. The animal gut as a melting pot for horizontal gene transfer. Can J Microbiol 61:603–605. 10.1139/cjm-2015-0049. [DOI] [PubMed] [Google Scholar]
- 66.Buhring SI, Sievert SM, Jonkers HM, Ertefai T, Elshahed MS, Krumholz LR, Hinrichs K-U. 2011. Insights into chemotaxonomic composition and carbon cycling of phototrophic communities in an artesian sulfur‐rich spring (Zodletone, Oklahoma, USA), a possible analog for ancient microbial mat systems. Geobiology 9:166–179. 10.1111/j.1472-4669.2010.00268.x. [DOI] [PubMed] [Google Scholar]
- 67.Senko JM, Campbell BS, Henriksen JR, Elshahed MS, Dewers TA, Krumholz LR. 2004. Barite deposition resulting from phototrophic sulfide-oxidizing bacterial activity. Geochim Cosmochim Acta 68:773–780. 10.1016/j.gca.2003.07.008. [DOI] [Google Scholar]
- 68.Spain AM, Elshahed MS, Najar FZ, Krumholz LR. 2015. Metatranscriptomic analysis of a high-sulfide aquatic spring reveals insights into sulfur cycling and unexpected aerobic metabolism. Peer J 3:e1259. 10.7717/peerj.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yadav A, Borrelli JC, Elshahed MS, Youssef NH. 2021. Genomic analysis of family UBA6911 (group 18 Acidobacteria) expands the metabolic capacities of the phylum and highlights adaptations to terrestrial habitats. Appl Environ Microbiol 87:e0094721. 10.1128/AEM.00947-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 72.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu YW, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 74.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. 2018. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 3:836–843. 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, Genome Standards Consortium , et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. 2020. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 38:1079–1086. 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 78.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36:1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konstantinidis KT, Rosselló-Móra R, Amann R. 2017. Uncultivated microbes in need of their own taxonomy. ISME J 11:2399–2406. 10.1038/ismej.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. 2017. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol 2:1533–1542. 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 82.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428:726–731. 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 84.Kanehisa M, Sato Y. 2020. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci 29:28–35. 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41:e121. 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Søndergaard D, Pedersen CN, Greening C. 2016. HydDB: a Web tool for hydrogenase classification and analysis. Sci Rep 6:34212. 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol 1962:1–14. 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C. 2018. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 46:W246–W251. 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. 2018. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46:D624–D632. 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang L, Zhang H, Wu P, Entwistle S, Li X, Yohe T, Yi H, Yang Z, Yin Y. 2018. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res 46:D516–D521. 10.1093/nar/gkx894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346. 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nakamura T, Yamada KD, Tomii K, Katoh K. 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34:2490–2492. 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35:1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2: approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Cáceres M, Legendre P. 2009. Associations between species and groups of sites: indices and statistical inference. Ecology 90:3566–3574. 10.1890/08-1823.1. [DOI] [PubMed] [Google Scholar]
- 96.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. 2011. Scikit-learn: machine learning in Python. J Machine Learning Res 12:2825–2830. [Google Scholar]
- 97.Chicco D, Jurman G. 2020. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics 21:6. 10.1186/s12864-019-6413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text, Fig. S1, Tables S1 to S7. Download aem.01706-21-s0001.pdf, PDF file, 1.1 MB (1.1MB, pdf)
Data Set S1. Download aem.01706-21-s0002.xlsx, XLSX file, 0.04 MB (38.2KB, xlsx)
Data Availability Statement
The individual assembled Myxococcota MAGs analyzed in this study have been deposited at DDBJ/ENA/GenBank under the accession numbers JAFGVO000000000, JAFGQN000000000, JAFGWT000000000, JAFGTB000000000, JAFGXQ000000000, and JAFGIB000000000.