

Abstract
Engineering flavin-free NAD(P)+-dependent dehydrogenases to reduce biomimetic nicotinamide analogues (mNAD+s) is of importance for eliminating the need for costly NAD(P)+ in coenzyme regeneration systems. Current redox dye-based screening methods for engineering the mNAD+ specificity of dehydrogenases are frequently encumbered by a background signal from endogenous NAD(P) and intracellular reducing compounds, making the detection of low mNAD+-based activities a limiting factor for directed evolution. Here, we develop a high-throughput screening method, NAD(P)-eliminated solid-phase assay (NESPA), which can reliably identify mNAD+-active mutants of dehydrogenases with a minimal background signal. This method involves (1) heat lysis of colonies to permeabilize the cell membrane, (2) colony transfer onto filter paper, (3) washing to remove endogenous NAD(P) and reducing compounds, (4) enzyme-coupled assay for mNADH-dependent color production, and (5) digital imaging of colonies to identify mNAD+-active mutants. This method was used to improve the activity of 6-phosphogluconate dehydrogenase on nicotinamide mononucleotide (NMN+). The best mutant obtained after six rounds of directed evolution exhibits a 50-fold enhancement in catalytic efficiency (kcat/KM) and a specific activity of 17.7 U/mg on NMN+, which is comparable to the wild-type enzyme on its natural coenzyme, NADP+. The engineered dehydrogenase was then used to construct an NMNH regeneration system to drive an ene-reductase catalysis. A comparable level of turnover frequency and product yield was observed using the engineered system relative to NADPH regeneration by using the wild-type dehydrogenase. NESPA provides a simple and accurate readout of mNAD+-based activities and the screening at high-throughput levels (approximately tens of thousands per round), thus opening up an avenue for the evolution of dehydrogenases with specific activities on mNAD+s similar to the levels of natural enzyme/coenzyme pairs.
Keywords: high-throughput screening, directed evolution, biomimetic nicotinamide analogues, NAD(P)+-dependent dehydrogenases, coenzyme regeneration
Graphical Abstract
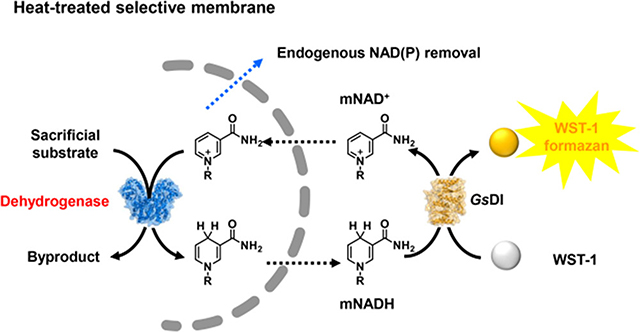
INTRODUCTION
All living organisms depend on redox chemistry catalyzed by thousands of oxidoreductases that utilize only a few coenzymes.1,2 In particular, the coenzymes nicotinamide adenine dinucleotide (NAD+/NADH) and its phosphorylated form, nicotinamide adenine dinucleotide phosphate (NADP+/NADPH), are used by two-thirds of oxidoreductases3 for the transport and storage of hydride, which can later be incorporated into a myriad of valuable biochemicals.3–8 The molecular structure of NAD(P) consists of two parts. While the nicotinamide portion provides the essential chemical function (i.e., accepting or donating hydride),9 the adenosine dinucleotide moiety confers the receptor-ligand recognition and can be replaced by other base dinucleotides10 or even simpler motifs,4 such as sugar, alkyl, or benzyl groups,5 to form biomimetic nicotinamide analogues (mNAD+/mNADH). The potential for lower cost and higher stability of the simpler mNAD+s relative to NAD(P)9,11,12 allows for use of high mNAD+ concentrations to overcome their often increased KM, and a number of mNADH-dependent production enzymes (e.g., ene-reductases,9 cytochrome P450 BM3 mutants,13 a DT-diaphorase,5,14 and an NADH-oxidase15) have been reported. Additionally, mNADs containing adenine-replaced dinucleotides have been used to create bio-orthogonal redox systems,2,16 by exploiting the mNAD-preferred engineered dehydrogenases to disconnect the reaction of interest from NAD(P)-dependent metabolism.
Significant improvements in the utility of mNADH-dependent enzymes for preparative applications could be achieved using a regeneration system comprising a regeneration enzyme that catalyzes mNAD+ reduction using a sacrificial reducing agent (Scheme 1) to eliminate the need (and cost) of (super)stoichiometric quantities of mNAD+.11,12 Importantly, the most efficient mNADH-dependent production enzymes developed to date contain a flavin prosthetic group5,17 that facilitates hydride transfers involving mNAD(H).11 Despite important advances,4,5,11,12 a majority of NAD(P)H-regeneration dehydrogenases (e.g., glucose dehydrogenase,11 phosphite dehydrogenase,18 and 6-phosphogluconate dehydrogenase19) are flavin-free enzymes and do not accept these small-size mNADs. For example, the specific activities of two previously characterized flavin-free dehydrogenases on a biomimetic coenzyme, nicotinamide mononucleotide (NMN+), were less than 0.05 U/mg, which are one-to-four orders of magnitude lower than on the natural coenzyme NAD(P)+ (Table S1). These results raise the question of whether the activity of flavin-free dehydrogenases on these mNAD+s can be enhanced to the levels of natural enzyme/coenzyme pairs.
Scheme 1. Enzyme-Coupled mNADH Regeneration Systema.
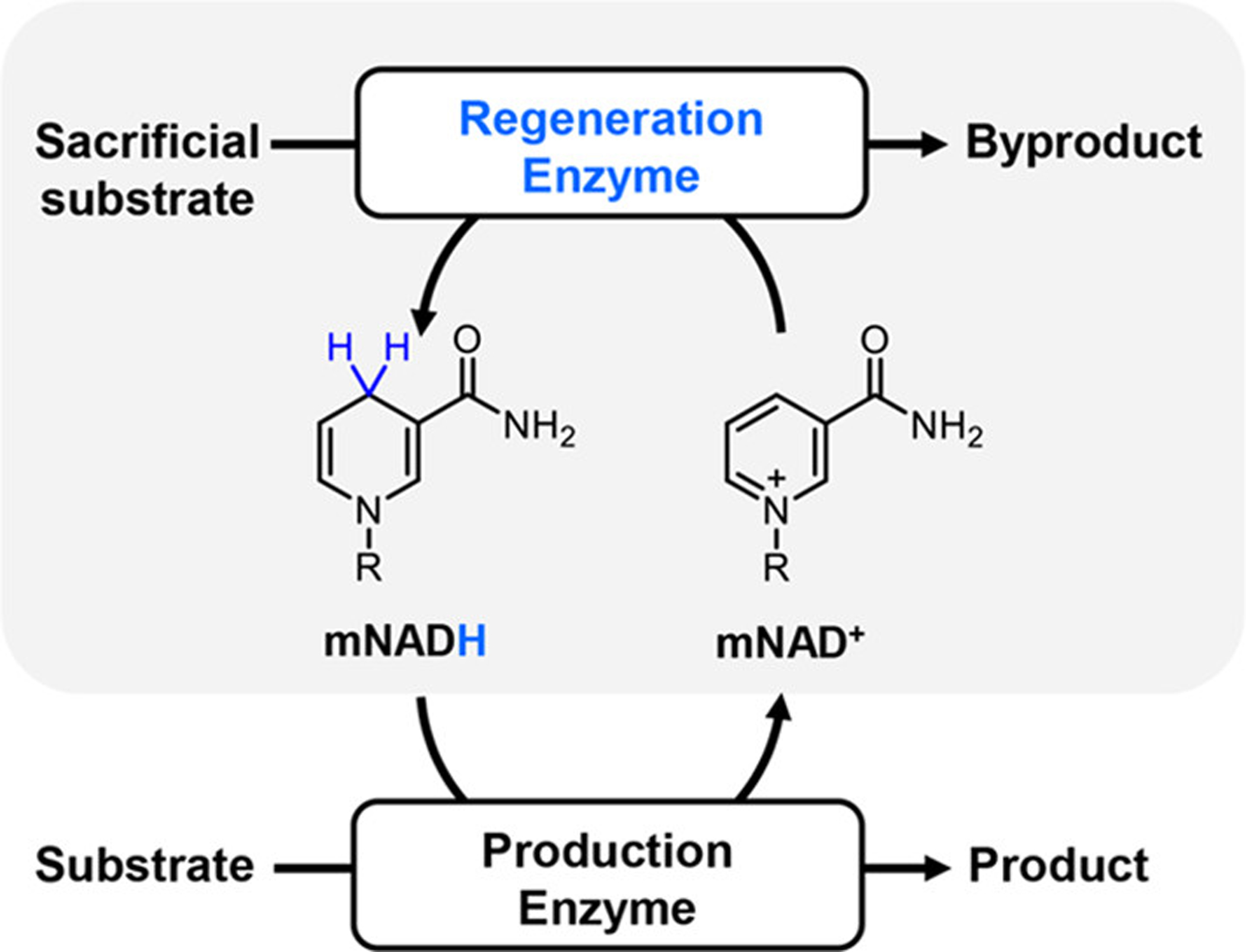
aReactions catalyzed by mNADH-dependent production enzymes require an efficient enzyme-coupled mNADH regeneration system (highlighted) comprising a regeneration enzyme for in situ mNADH regeneration and a sacrificial substrate.
Directed evolution can be used to rapidly create protein mutants with desired properties via iterative rounds of mutagenesis and functional screening/selection. We envisioned that this technique could be used to improve dehydrogenase activities on mNAD+s, which are poor coenzymes for native dehydrogenases. The success of directed evolution campaigns requires high-throughput screening (HTS) methods that can correctly identify rare positive hits from diverse mutant libraries.20 Previous attempts to improve dehydrogenase activity on mNAD+s have been carried out using the UV absorbance of mNADH in a microtiter plate format.11,21 These methods are encumbered by the low molar absorptivity of mNADHs and the high background of cell lysate,22,23 and they typically limit library sizes to thousands of mutants. Redox dye-based colorimetric assays2,23,24 resolve many of these limitations and are amenable to HTS. The throughput levels can be increased from 103 (microwell plate screening2,16,23,25) to 105 (filter assay26–28 or Petri dish-based double-layer screening29) to 106 mutants (absorbance-activated droplet sorting screening30) with smaller sample volumes and faster sorting efficiency. These methods cannot be applied to efficiently screen dehydrogenase mutants on nucleotide-free mNAD+s, however, due to the high background signal that arises from enzyme activity on endogenous NAD(P)27 or the cross-reactivity of redox dyes31,32 and the intermediate electron carrier (IEC, either enzymatic or chemical) with intracellular reducing compounds.
In this study, we describe the development of a new HTS method, NAD(P)-eliminated solid-phase assay (NESPA), which can identify mNAD+-active mutants of dehydrogenases. NESPA was designed to overcome limitations in our previous method,29 namely, the failure to detect low mNAD+-based activities due to the substantial background signal. Through a combination of colony heat treatment, buffer wash, a unique redox-dye-based assay, and digital imaging, NESPA enabled to detect dehydrogenase activities on diverse mNAD+s with a screening capacity of up to 50,000 mutants without sophisticated equipment or labor-intensive processes. To demonstrate the feasibility of this method, we used NESPA to improve the specific activity of NADP+-dependent 6-phosphogluconate dehydrogenase from Thermotoga maritima (T. maritima) (6PGDH, EC 1.1.1.44)33 on NMN+, resulting in a mutant with comparable specific activity and identical regeneration relative to wild-type 6PGDH acting on NADP+. This study highlights the utility of NESPA for improving the activity of NAD(P)+-dependent dehydrogenases for mNADH regeneration.
RESULTS
Overview of NESPA Screening.
NESPA was developed to identify active mutants of NAD(P)+-dependent dehydrogenases on mNAD+s (Figure 1). This approach involves first plating Escherichia coli cells harboring a dehydrogenase mutant library on LB agar medium. Colonies are heat-treated at 70 °C for 1 h to permeabilize the cell membrane toward small molecules (e.g., NADP and NAD), deactivate mesophilic redox enzymes of the host, and oxidize the reduced compounds (e.g., NAD(P)H). Colonies are subsequently transferred onto the surface of filter paper, which is placed in a Buchner funnel and washed to remove endogenous NAD(P) and other metabolites. To detect the mNAD+-based activity by the redox dye-based assay, the filter paper is then placed in a Petri dish and overlaid with agarose solution containing the substrate (e.g., 6-phosphogluconate, 6PG), mNAD+, a redox dye, and an IEC. Reduced mNAD (mNADH) reacts with the oxidized dye in the enzyme-coupled reaction to generate a colorimetric response proportional to the mutant activity. Finally, the colonies are photographed and the color intensities are evaluated by digital imaging24,34 to identify positive mutants. Individual plasmids containing the mutated gene are then extracted from the colonies for DNA recovery (see Figure S1 for detailed step-by-step operations).
Figure 1.
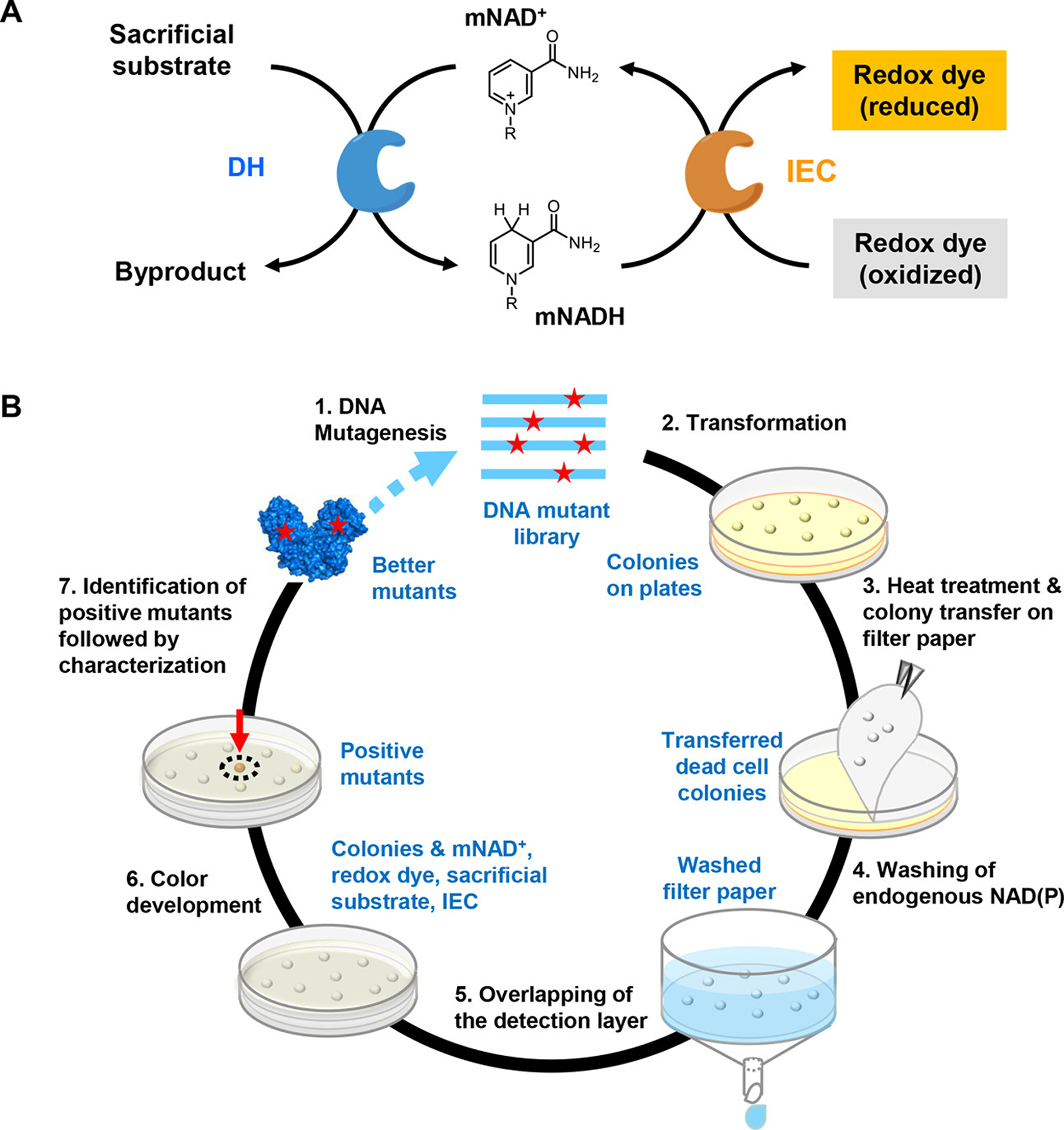
Schematic presentation of NESPA for NAD(P)+-dependent dehydrogenases evolution on mNAD+. (A) Colorimetric assay to detect the dehydrogenase activity on mNAD+. (B) Overview of NESPA for the evolution of mNAD+-active dehydrogenase.
NESPA Optimization.
Because the colorimetric assay for mNAD+-based activity tends to be masked by the background signal from endogenous NAD(P) and other reducing compounds, several variables in the general protocol outlined above were optimized to ensure specific detection of the mNADH signal. Most importantly, the sensitivity of NESPA depends directly on the molar absorptivity of the redox dye used. The correlation between colorimetric intensity and the mNADH concentration, however, can be seriously impeded by the cross-reactivity of the dye to oxidants or reductants. Therefore, it was necessary to select a redox dye that has a sensitive readout but minimal cross-reactivity. Among 24 candidates, three dyes were chosen for further evaluation based on their possessing (1) a reduction potential less than 0.5 V to minimize non-specific reduction, (2) high oxygen tolerance for irreversible color development, (3) high molar absorptivity for improved sensitivity, and (4) IEC-selective dye reduction (Table S2 and Figure 2A). These compounds, 3,3′-[3,3′-dimethoxy-(1,1′-biphenyl)-4,4′-diyl]-bis[2-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (NBT), 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT), and 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1) (Figure 2B), were then used to detect the activity of a dehydrogenase, 6PGDH, on NMN+. Figure 2C compares the color development of these tetrazolium dye-based assays. In these experiments, diaphorase (DI) from Geobacillus stearothermophilus (GsDI) was used as the IEC and no buffer wash was used. WST-1 was found to be the best dye because it had the highest ratio of the NMNH signal to the background signal from the dye cross-reactivity (~8.5) while NBT was the worst (~2.1) (Figure 2D and Figure S2A).
Figure 2.
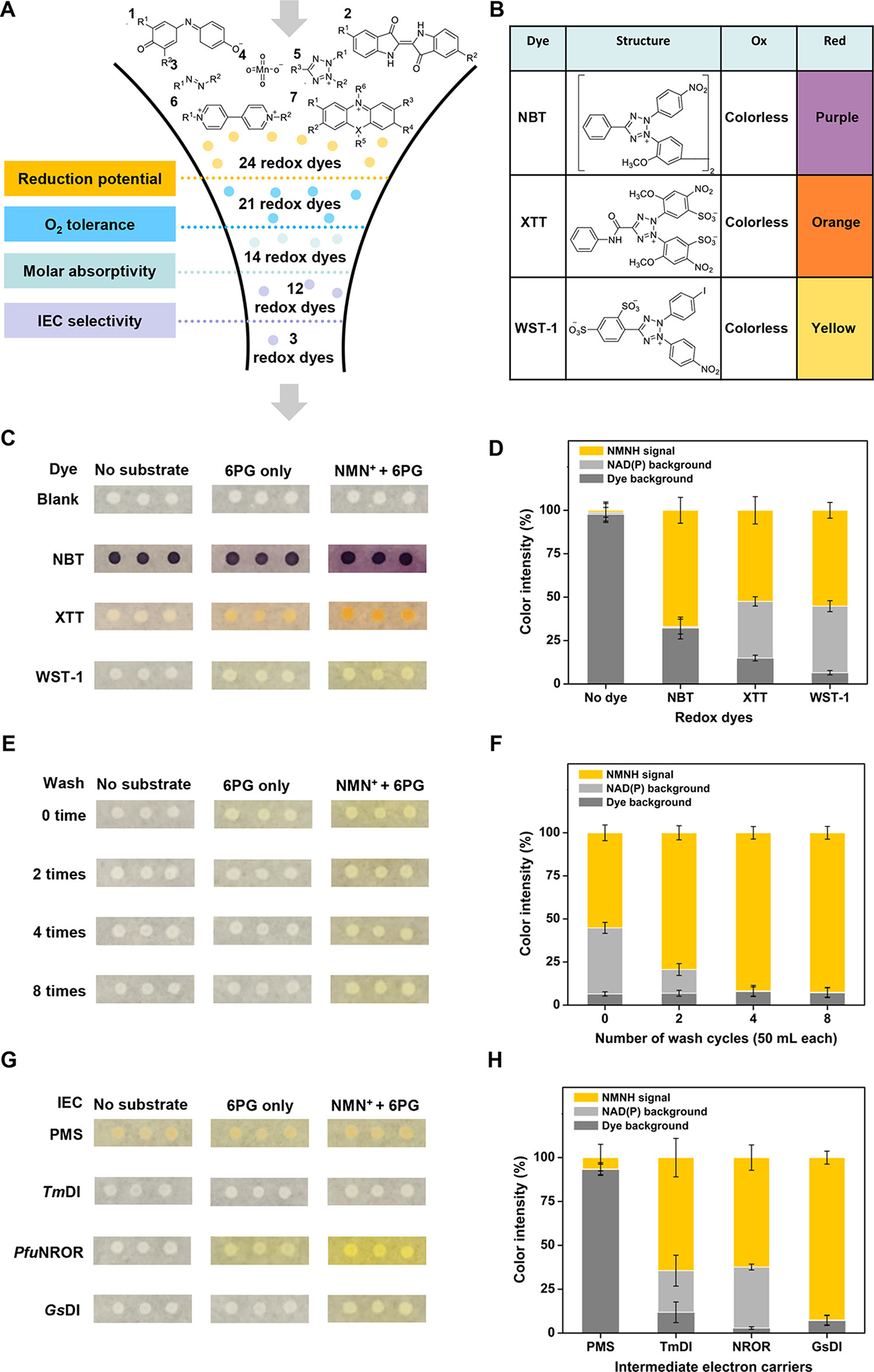
Optimization of redox dye, buffer washing volume, and IEC for NESPA screening. (A) Down-selection results for 24 redox dyes based on reduction potential, O2 tolerance, molar absorptivity, and IEC selectivity. (B) Chemical structures of selected tetrazolium dyes and their colorimetric changes for mNADH detection. (C, D) Optimization of the redox dye for NESPA. Heat-treated colonies were overlaid by melted agarose solution containing all reagents for color development (NMN+ + 6PG). The percentages of NMNH signal, NAD(P) background, and dye background of the total signal are presented in the stacked bar chart. (E, F) Optimization of the buffer washing volume for NESPA. (G, H) Optimization of the IEC for NESPA. Heat-treated colonies were washed and overlaid by melted agarose solution containing enzymatic IEC (i.e., GsDI, PfuNROR, or TmDI) or PMS to identify the most selective IEC.
Next, different methods for cell lysis and buffer wash were investigated. These two parameters are crucial for minimizing the background signal from endogenous NAD(P), leading to increased accuracy of mNAD+-based activity and decreased identification of false-positive mutants active exclusively on NAD(P)+. A partially-disrupted cell membrane that can release NAD(P) but retain cytoplasmic dehydrogenases is the prerequisite for NESPA. Cell lysis using lysozyme2,11 and various chemical agents26 (e.g., EDTA and Triton X-100) were examined, and both liberated a large amount of intracellular 6PGDH. This resulted in a significant loss of 6PGDH for color development in colonies (lanes 7 and 8, Figure S3A) and a high NAD(P) background in lysate (Figure S4). Heat treatment, on the other hand, was found to render colonies permeable to NAD(P) (MW, <1 kD), while retaining most dehydrogenase (MW, >55 kD) and plasmid DNA (MW, >5000 kD). Eight rounds of buffer wash removed nearly all background signal from endogenous NAD(P) (Figure 2E,F), while maintaining more than 73% of 6PGDH based on SDS-PAGE analysis and 55% of plasmid based on comparison of the number of plated transformants (Figure S3C). The ratio of the NMNH signal to the background signal from NAD(P) and dye cross-reactivity was enhanced 10-fold from 1.2 to 11.4 (Figure 2F and Figure S2B).
Finally, a range of different IECs were evaluated. To maximize the ratio of the NMNH signal to the background signal, a suitable electron carrier should efficiently transfer hydride from NMNH to the redox dye (i.e., WST-1) but be less active on other reducing compounds. Three thermophilic enzymatic IECs, including GsDI, rubredoxin oxidoreductase from Pyrococcus furiosus (PfuNROR),35 and DI from T. maritima (TmDI),36 and the abiotic IEC phenazine methosulfate (PMS)23 (Figure 2G) were compared. GsDI, which provided up to 177-fold higher signal-to-background (on NMNH) compared to other IECs, was selected (Figure S2C). It was noted that chloramphenicol and sodium azide are crucial to inhibit the cell regrowth of heat-treated colony (data not shown), thus minimizing the nonspecific reduction of WST-1 for the long-term color development (i.e., longer than 12 h). Together, the optimization efforts outlined above greatly decreased the background signal from intracellular reducing compounds and endogenous NAD(P). The screening reliability coefficient Z-factor37 of the final assay is 0.88, illustrating the assay efficacy for detecting NMN+-based activity (a Z-factor higher than 0.5 indicates a high-quality assay; 1.0 is the maximum value).
Establishment of NESPA Screening.
After reliable detection of dehydrogenase activity on NMN+ using NESPA was established, the scope of this method was explored by investigating its ability to detect the 6PGDH activity on three other mNAD+s: nicotinamide guanine dinucleotide (NGD+),10 nicotinamide hypoxanthine dinucleotide (NHD+),10 and 1-benzyl-3-carbamoylpyridinium chloride (BNA+).11 As shown in Figure S5A, the dehydrogenase activity on these mNAD+s (i.e., NHD+, NGD+, and BNA+) could be distinguished from the background signal by fine-tuning the coenzyme concentration and the reaction time. The Z-factors of assays on NGD+, NHD+, NMN+, and BNA+ were 0.83, 0.69, 0.88, and 0.55, respectively (Figure S5B). The sensitivity of the setup was tested by measuring the color intensities of colonies stained with 0 to 50 μM of reduced WST-1 formazan. The absorbance calibration plot exhibited good linearity over a wide range of concentrations (2.5 to 50 μM) (Figure S6). The detection limit of WST-1 formazan was thus determined to be 2.5 μM. It is noted that this value is 20-fold higher than those of UV absorbance based microtiter-plate screening22 and 3-fold higher than that of AADS.30 This high sensitivity allows NESPA to detect enzymes with low activity on mNAD+s, which is expected early in evolutionary campaigns.
To validate the general utility of NESPA for identifying mutations that improve dehydrogenase activity toward mNAD+, two NADP+-dependent dehydrogenases, 6PGDH and glucose-6-phosphate dehydrogenase from T. maritima (G6PDH, EC 1.1.1.49) were selected, and residues in the predicted NADP-binding pocket were identified based on homology modeling and substrate docking. NESPA was then used to screen for NMN+-active mutants from single iterative saturation mutagenesis (ISM)38,39 libraries targeting residues that bind the 2′-phosphate of NADP+ (N32/R33I/T34I for 6PGDH; A64/R65I/T66I for G6PDH). After color development and imaging analysis, the two positive mutants showing the greatest color intensity were isolated from each library. Because the color intensities of colonies under the screening condition were far below the maximum value (0.1–0.2 of positive mutants vs 0.62 of 50 μM WST-1 formazan), the conversion of WST-1 formazan was expected to correlate with mutant activity on NMN+. The color intensities of the positive mutants (A1 and A2 for the 6PGDH library and B1 and B2 for the G6PDH library) and another three mutants from each mutant library (A3–A5 for the 6PGDH library and B3–B5 for the G6PDH library) were then measured. Gratifyingly, the color intensities of colonies positively correlated with the specific activities of the purified dehydrogenases on NMN+ (Figure 3). The two identified 6PGDH mutants (A1:N33Y/R33I/T34I and A2:R33I/T34I) exhibited 1.1-fold higher specific activities on NMN+ and the two identified G6PDH mutants (B1: A64S/R65I/T66I and B2: A64D/R65I/T66I) exhibited 1.8 and 1.2-fold higher activities on NMN+ compared with the wild-type enzymes. These results validated the proof of concept of this approach for engineering of NAD(P)+-dependent dehydrogenases on mNAD+s.
Figure 3.
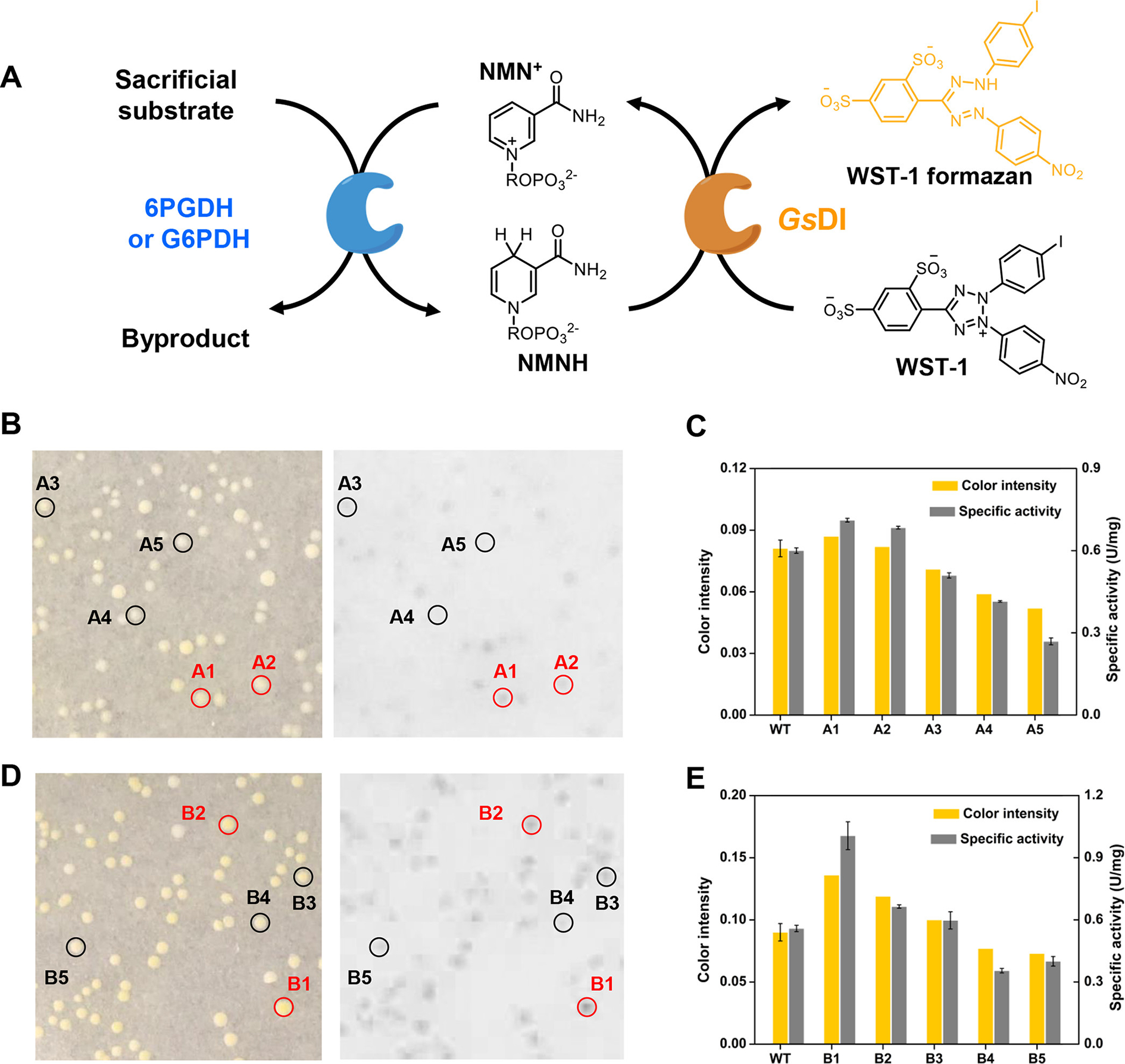
Validation of NESPA for accurate identification of NMN+-active mutants. (A) Assay scheme used to detect 6PGDH or G6PDH activity on NMN+. Petri dish results of (B) 6PGDH and (D) G6PDH libraries were photographed under white light, where the inverted saturation slices were used for intensity analysis. The two identified positive mutants and three randomly selected mutants under white light are indicated by the red and black circles, respectively. (C, E) Comparison of the color intensities of the selected mutants and their specific activities. The color intensity of wild type (WT) was estimated based on average of 10 colonies from the control petri dish.
Engineering of 6PGDH on NMN+.
Using NESPA, we evolved 6PGDH into a highly NMN+-active enzyme. NMN+ was selected because its cost is less than that of NAD(P)+, its half-life is ~2× that of NAD(P)+ at 60 °C (Figure S7), and it has high aqueous solubility in both its oxidized and reduced form. 6PGDH, which can reduce NAD(P)+ to NAD(P)H by concurrent oxidation of 6PG, has been used for coenzyme regeneration in different in vitro biosystems.19,33,36,40 The development of an NMN+-compatible 6PGDH could therefore be important for decreasing the use of costly NAD(P) in the in vitro biosystems with enhanced commercial feasibility.9,12,41,42
Six rounds of directed evolution were completed to obtain 6PGDH mutants with increased activity on NMN+. In most rounds, approximately 20,000 mutants were screened (vide infra) and up to 10 colonies showing highest color intensities were identified and characterized (see the analysis of a typical screening plate in Figure S8). The mutant showing the highest NMN+-based activity in each round was used as the template for the next round of evolution in which more stringent screening conditions (i.e., a shorter reaction time window and/or a lower concentration of NMN+ for color development) were applied. This cycle was repeated until further improvement was not obtained (~50,000 colonies were screened in the seventh round).
Two approaches were applied to create mutant libraries for the evolution of 6PGDH. In stage 1, SWISS-MODEL was used to generate a structural model of 6PGDH. Eight residues within 5.5 Å of the ADP motif of bound NADP+ (Figure 4A) were identified based on docking simulations. These residues were classified into three sites for constructing ISM libraries (library A: N32/R33I/T34I; library B: A11/V12; library C: D82/T83/Q86). Screening progressed from library A to C with the best mutant from the previous round serving as the parent for the next round. Following this approach, the specific activity of 6PGDH on NMN+ was increased 15-fold from 0.6 (WT) to 9.19 U/mg (Mut 3–1) (Figure 4B). In stage 2, random mutagenesis of the entire gene (i.e., error-prone PCR with an average of 4 mutations per gene) was used. Through three sequential rounds of evolution, the 6PGDH was further evolved to Mut 6–1. Its specific activity on NMN+ is 17.7 U/mg, which was comparable to that of the WT on NADP+ (18.0 U/mg) and 2-fold increase as compared to that of Mut 3–1. The color development of WT and the six mutants on NADP+ and NMN+ was also compared based on the WST-1/GsDI microplate assay (Figure 4C). The specific activities of the mutants on NMN+ showed an increasing trend from the WT to Mut 6–1, while all mutants except Mut 1–1 retained high activities on NADP+.
Figure 4.
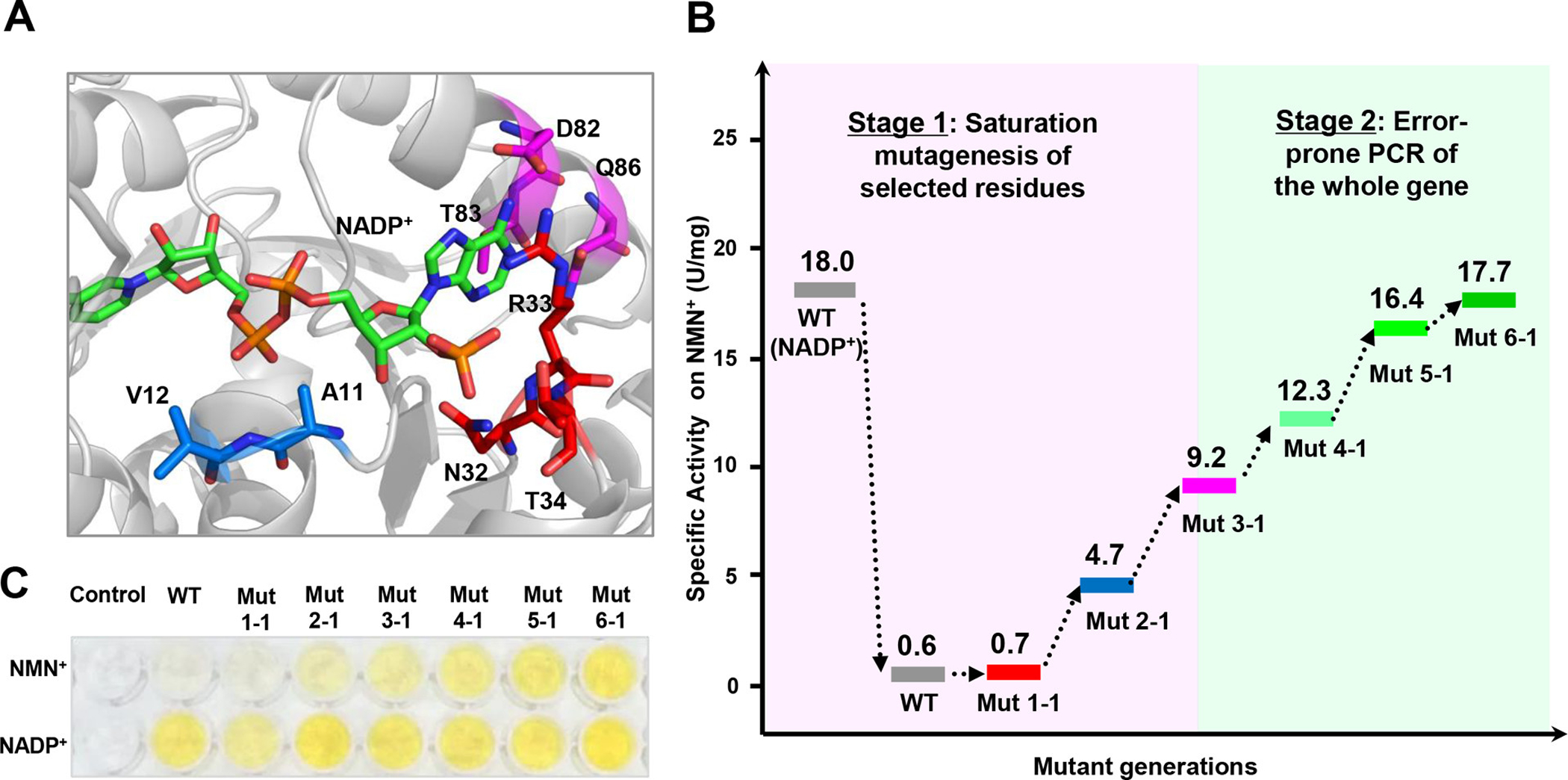
Directed evolution to improve 6PGDH activity on NMN+. (A) Structural model of wild-type 6PGDH (WT) in complex with NADP+. Residues within <5.5 Å of the 2′ phosphate, pyrophosphate, or adenine moieties of NADP+ are colored red, blue, and magenta, respectively. (B) Evolutionary progress of mutant activities on NMN+. (C) Images of activity measurement of the WT and mutants on NADP+ and NMN+ based on the WST-1/GsDI assay.
The specific activities and apparent kinetic constants of the WT and mutants on NMN+ and NADP+ are presented in Table 1. The WT is highly specific for NADP+, showing a kcat of 15.9 s−1, a KM of 0.0012 mM, and a catalytic efficiency of 13,395 mM−1 s−1 but having a catalytic efficiency of 0.043 mM−1 s−1 on NMN+. In contrast to previous reports,43 the addition of 5 mM adenosine or adenosine 5-phosphate to the 6PGDH reaction did not result in improved activity on NMN+. Each of the new mutants showed enhanced catalytic efficiency with NMN+ compared to their parental enzyme. The kcat/KM of the 6PGDH mutants on NADP+, however, first decreased by a factor of 10,000 to 1.3 mM−1 s−1 in Mut 1–1 and then recovered to 113.0 mM−1 s−1 from Mut 2–1 to Mut 5–1. The final mutant, Mut 6–1, is highly active on NMN+. This mutant had a kcat of 27.4 s−1 on NMN+, which is 20-fold higher than that of the WT. Remarkably, this value is also 1.7-fold higher than that of WT on NADP+ (15.9 s−1). Such an improvement on nucleotide-free mNAD+s has never been reported in flavin-free natural/engineered dehydrogenases; kcat values for all such dehydrogenases acting on mNAD+s were 100–10,000 times lower than those of the WT enzymes on NAD(P)+.9,11,44 With the decreased KM (13.5 mM) on NMN+, the catalytic efficiency of Mut 6–1 on NMN+ increased to 2.04 mM−1 s−1, a greater 50-fold improvement over the WT but still three orders of magnitude lower than that of WT on NADP+ (i.e., 13,394 mM−1 s−1). On NADP+, the Mut 6–1 exhibited a 90-fold lower catalytic efficiency (148 mM−1 s−1) than WT, which was caused by the 158-fold increased KM but minor improved kcat. Importantly, because high concentrations of NMN+ (20 mM) can be used to offset the high KM of Mut 6–1, specific activities comparable to that of WT can be observed.
Table 1.
Specific Activities and Apparent Kinetic Constants of 6PGDHs for NMN+ and NADP+a
| NADP+ | NMN+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| enzyme | mutations | sp. act. (U/mg) | kcat (S−1) | KM (mM) | kcat/KM (mM−1 s−1) | sp. act. (U/mg) | kcat (S−1) | KM (mM) | kcat/KM (mM−1 s−1) |
| wild-type | - | 18.0 ± 0.8 | 15.9 ± 0.2 | 0.0012 ± 0.0001 | 13394.5 | 0.60 ± 0.01 | 1.3 ± 0.1 | 30.6 ± 1.7 | 0.042 |
| Mut 1–1 | R33I/T34I | 1.2 ± 0.2 | 14.8 ± 0.7 | 11.5 ± 1.1 | 1.3 | 0.68 ± 0.01 | 1.7 ± 0.1 | 37.9 ± 3.6 | 0.046 |
| Mut 2–1 | A11G/R33I/T34I | 10.2 ± 0.7 | 28.3 ± 0.6 | 2.1 ± 0.1 | 13.2 | 4.66 ± 0.02 | 10.2 ± 0.2 | 20.7 ± 0.7 | 0.49 |
| Mut 3–1 | A11G/R33I/T34I/D82L/T83L/Q86L | 16.4 ± 0.1 | 21.1 ± 0.3 | 0.39 ± 0.02 | 53.7 | 9.19 ± 0.09 | 19.3 ± 0.4 | 27.5 ±1.2 | 0.70 |
| Mut 4–1 | A11G/K27R/R33I/T34I/D82L/T83L/Q86L/I120F/D294V/Y383C/N387S/A447V | 20.0 ± 0.3 | 21.9 ± 0.2 | 0.22 ± 0.01 | 101.0 | 12.31 ± 0.14 | 18.9 ± 0.4 | 15.1 ± 0.6 | 1.25 |
| Mut 5–1 | A11G/K27R/R33I/T34I/F60Y/D82L/T83L/Q86L/K118N/I120F/D294V/F326S/Y383C/N387S/A447V | 20.6 ± 0.4 | 23.6 ± 0.3 | 0.21 ± 0.01 | 113.0 | 16.40 ± 0.40 | 25.8 ± 0.8 | 16.0 ± 1.1 | 1.62 |
| Mut 6–1 | A11G/K27R/R33I/T34I/F60Y//D82L/T83L/Q86L/K118N/I120F/D2S1E/D294V/F326S/F329Y/Y383C/N387S/V390G/A447V | 27.1 ± 0.8 | 28.9 ± 0.3 | 0.19 ± 0.01 | 148.6 | 17.70 ± 0.35 | 27.4 ± 0.5 | 13.5 ± 0.5 | 2.04 |
New introduced mutations relative to their parent enzyme are highlighted in bold; each value represents the average ± standard deviation of triplicate independent measurements
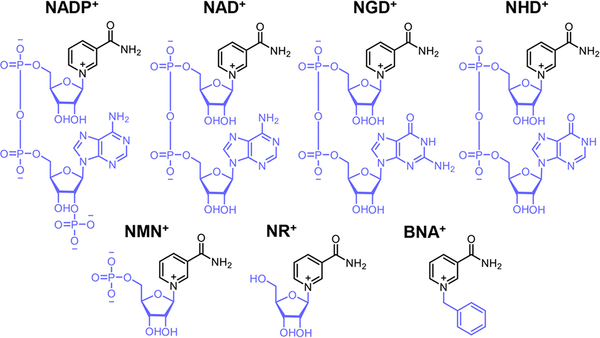
We also characterized the changes in the specific activities of the WT and Mut 6–1 on a series of coenzymes (Table 2). Intriguingly, Mut 6–1 showed improved activities toward all coenzymes examined. The specific activities of Mut 6–1 on NADP+, NAD+, NGD+, NHD+, NMN+, NR+, and BNA+ were 1.5, 5.8, 4.1, 6.3, 29.5, 7.0, and 8.9-fold higher than those of the WT, respectively. The increased promiscuous activity on these coenzymes highlights the capability of NESPA to generate more active mutants on other mNAD+s.
Table 2.
Specific Activities of the WT and Mut 6–1 on NAD(P)+ and mNAD+sa
| |||
|---|---|---|---|
| specific activity (U/mg) | |||
| coenzyme | WT | Mut 6–1 | fold change (Mut 6–1/wild-type) |
| NADP+ | 18.0 ± 0.8 | 27.1 ± 0.8 | 1.5 |
| NAD+ | 4.9 ± 0.2 | 28.5 ± 0.2 | 5.8 |
| NGD+ | 6.9 ± 0.5 | 28.7 ± 2.8 | 4.1 |
| NHD+ | 6.4 ± 0.1 | 40.4 ± 0.1 | 6.3 |
| NMN+ | 0.60 ± 0.01 | 17.7 ± 0.35 | 29.5 |
| NR+ | (1.8 ± 0.1) × 10−3 | (1.3 ± 0.1) × 10−2 | 7 |
| BNA+ | (3.5 ± 0.6) × 10−4 | (3.1 ± 0.2) × 10−3 | 8.9 |
Each value represents the average ± standard deviation of triplicate independent measurements.
Structural Basis for the Improved Activity of 6PGDH on NMN+.
To shed light on specific enzyme-NMN+ interactions that might have led to observed improvement, we built protein homology models of WT and Mut 6–1 using SWISS-MODEL docked with both substrates (i.e., NMN+ and 6PG). Modeling comparison suggested substantial changes in both the hydrophobicity and the volume of the mutated coenzyme binding pocket, which might be associated with the increase in NMN+-based activity. For example, the terminal phosphate of NMN+ was bound proximal to A11. The mutation A11G was found to improve catalysis with the NMN+, which could be due to a decreased steric clash from the phosphate group (Figure S9). Additionally, while the binding of NMN+ in the adenosine-binding pocket was not observed in the docking results, if such nonreactive binding did occur during catalysis, it could be hindered by mutations R33I/T34I/D82L/T83L/Q86L that create a smaller, more hydrophobic binding site to improve activity (Figure S10). Finally, because the full activity of 6PGDH requires a homodimer involving a coenzyme-binding subunit threading through the neighboring 6PG-binding subunit, the mutation A447V, which improves hydrophobic interactions at the dimer interface (Figure S11), could lead to the enhanced electron transfer from 6PG to NMN+. Together, these mutations might give rise to better accommodation of the NMN+ in the active site, enabling the small biomimetic coenzyme to adopt the reactive orientation required for efficient catalysis. To discover better mutants (e.g., higher kcat and lower KM) on NMN+, future efforts could focus on site saturation mutagenesis of residues identified in the current work,45 obtaining the crystal structure of Mut 6–1 with NMN+ to enable structure-guided mutagenesis and further engineering based on computational modeling.25
NMNH Regeneration for Enzyme-Coupled Conversion.
The applicability of the engineered 6PGDH for NMNH regeneration was tested using a flavin-containing ene-reductase from Thermus scotoductus (TsER, EC 1.3.1.31) that can react with numerous mNADHs.11,46 In these reactions, 6PGDH reduces NMN+ to NMNH, which is then used by TsER to convert the 2-methyl-2-cyclohexen-1-one (1a) to 2-methylcyclohexanone (1b) (Figure 5A). Mutant 6–1 was compared to the WT in terms of turnover frequency (TOF) and yield (determined by gas chromatography, GC) to demonstrate its improved effectiveness for NMN+ reduction.
Figure 5.
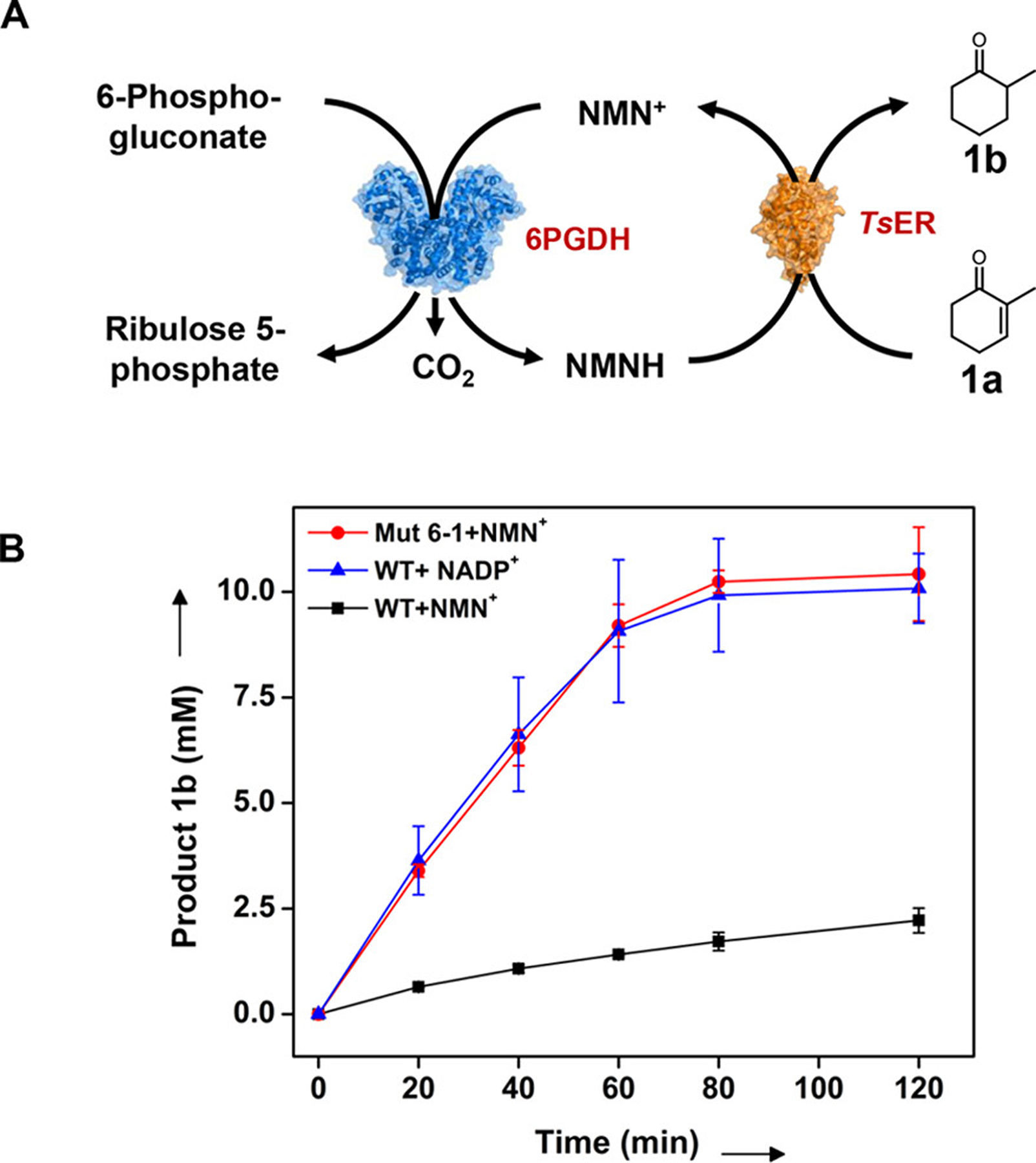
Conversion of 2-methyl-2-cyclohexen-1-one (1a) to 2-methylcyclohexanone (1b) with the NMN+ or NADP+-based regeneration system using wild-type or engineered 6PGDH. (A) Schematic of the synthetic enzymatic pathway. (B) Profiles of production of 2-methylcyclohexanone (1b) based on regeneration system of NMNH or NADPH. Average yields of three independent trials as determined by GC relative to the internal standard are shown.
Figure 5B shows the production of 2-methylcyclohexanone over 120 min reaction with three different coenzyme regeneration systems. The use of WT/NADP+ and Mut 6–1/NMN+ led to >98% yield of 2-methylcyclohexanone at 80 min, whereas the use of WT/NMN+ reached less than 24% conversion at 120 min. The average TOF of the Mut 6–1/NMN+ pair over the linear range (i.e., 20 min) was ~18.5 s−1, approximately 5.2-fold higher than that of the WT/NMN+ (TOF = 3.5 s−1). This TOF, however, was comparable to the value with WT/NADP+ (TOF = 19.8 s−1, less than 8% difference), suggesting that NMN+ can be regenerated as fast as the natural coenzyme using engineered 6PGDH. Notably, the blank control samples without the added coenzyme resulted in less than 4% conversion (data not shown), which indicated negligible activity from residual enzyme-bound NAD(P) in the in vitro bioconversion.11,47 Together, these results demonstrated that NESPA can generate readily active dehydrogenase mutants to reduce mNAD+ and thus drive the naturally NAD(P)+-dependent enzyme cascade reaction using biomimetic nicotinamide analogues.
DISCUSSION
In this study, a HTS method, NESPA, was developed and used to identify mNAD+-active mutants of NAD(P)+-dependent dehydrogenases for cofactor regeneration. The combination of a heat-induced membrane permeabilization with colony buffer washes and the careful design of the WST-1/GsDI assay addresses two intrinsic limitations in other state-of-the-art approaches: the background signal from the endogenous NAD(P) and from intracellular reducing compounds. These treatments also improve the individual parameters responsible for detection sensitivity: (i) the WST-1 formazan and digital imaging lower the detection limit of assay, (ii) the diverse coenzyme scope and thermostability48 of GsDI enable the measurement of slowly regenerated mNADHs, (iii) the whole-cell screening provided concentrated dehydrogenase for color development, and (iv) the heat treatment and the cell regrowth inhibitor minimize the nonspecific dye reduction, increasing the possible screening time window (see more comparisons in Table S3). As a consequence, NESPA enables the determination of dehydrogenase activities on diverse mNAD+s (e.g., NHD+, NGD+, NMN+, and BNA+), allowing various enzyme and mNAD+ panels accessible for directed evolution. Unlike previous methods that depend on sophisticated equipment and/or labor-intensive screening processes, NESPA requires only simple instruments and can identify active hits from 1,000 mutants in less than 1 min using digital imaging. In practice, NESPA can be used to screen up to 50,000 mutants per round of directed evolution and each round requires only 4 days, significantly faster than typical 96-well plate methods.
NESPA is a general screening platform that allows the identification of mNAD+-active mutants from other NAD(P)+-dependent dehydrogenases (Figure S12). In addition to engineering thermophilic dehydrogenases on biomimetic nicotinamide analogues, this method can be modified to meet different goals. For example, starting with mesophilic enzymes, heat treatment-based screening49,50 can be applied before NESPA to identify thermostable mutants for coenzyme engineering (Figure S13A). Additionally, NESPA can be amended to screen for coenzyme-switched mutants or mNAD+-active mutants with desired specificity of the other substrate, by including an additional negative screening with unwanted coenzymes or substrates (Figure S13B,C). Because it minimizes endogenous metabolites and deactivates host proteins, NESPA is also a promising method for improving enzyme activities on other biomimetic coenzymes, such as riboflavin (FAD precursor51), FO-5′-phosphate (riboflavin analogue),52 and N-acetylcysteamine53 (coenzyme A analogue), via coupled enzyme assays.50,54,55 With the growing interest in enzyme catalysis using mNADs4,5,9,11,12,56 and the increasing number of natural/engineered thermostable enzymes,57 we believe that NESPA could be a widely used platform for altering the mNAD+ specificity of dehydrogenases. Its robustness for directed evolution would lay the foundation for developing biomimetic coenzyme-based biocatalysts in vitro and offers active templates for engineering biomimetic coenzyme-dependent bio-orthogonal systems in vivo.
EXPERIMENTAL SECTION
High-Throughput Screening Procedure of NESPA.
Transformed cells containing mutated plasmid libraries were first spread evenly on 10 cm LB-kanamycin agar plates (15 mL) and grown at 37 °C overnight with an expected colony number of 500 per Petri dish. After incubation at room temperature for an additional day for protein overexpression, the colonies were treated at 70 °C for 1 h to partially disrupt the cell membrane, deactivate mesophilic host proteins, and oxidize the endogenous reducing compounds. The heat-treated colonies were then transferred onto the surface of 7.5 cm filter paper, which was placed in a Buchner funnel. The colonies were then soaked with 50 mL of 50 mM sodium phosphate buffer (pH 7.5) for 3 min followed by buffer removal using vacuum filtration. The wash procedure was performed eight times to remove the endogenous NAD(P) in the colonies. For the first round of screening of 6PGDH and G6PDH variants, the washed colonies were placed in new Petri dishes containing 10 mL of 0.5% agarose gel with 150 μM WST-1, 0.13 μM GsDI (2.8 μg/mL), 2 mM substrate (e.g., 6PG or G6P), 1 mM NMN+, 50 mM tris-HCl (pH 7.5), 50 μg/mL chloramphenicol, and 0.1% sodium azide and were covered with another 10 mL of 0.5% melted agarose solution (60 °C) of the same composition. Color development was conducted at room temperature for 5 h for G6PDH screening and 12 h for 6PGDH screening. Subsequent rounds of evolution of 6PGDH used altered conditions (e.g., a shortened reaction time window and/or a lower NMN+ concentration) for higher screening stringency. To identify the positive mutants by image analysis, the colonies were photographed under white light with a consistent brightness level. The image files were analyzed using ImageJ. Up to 10 colonies exhibiting the highest color intensity (no lower than 90% compared to that of their parent) were regarded as positive mutants and were removed by using a sterile laboratory spatula, and the DNA was then isolated by following the standard minipreparation protocols. A more detailed protocol can be found in SI Materials and Methods.
Supplementary Material
ACKNOWLEDGMENTS
This study was mainly supported by the Department of Biological system Engineering, Virginia Tech, with additional support to J.C.L. from the NIH (R01 GM115665). Also, R.H. thanks Professor James Bowie, Professor Ryan Senger, Professor Caroline E. Paul, Professor Wei Tao, Doctor Wei Zhao, Doctor Chao Zhong, Harrison Snodgrass, and Natalie Chan David Scherr for project discussions and/or technique support and Professor Xueyang Feng for providing access to laboratory instruments.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.9b03840.
Detailed characterization of NESPA (e.g., work flow of screening, dye information collection, the retention efficiency of cytoplasmic 6PGDH and plasmid by the cell membrane, the scope of mNAD+, and detection limit of NESPA); a typical Petri dish screening result of NESPA; structural analysis of the final mutant; promising applications of NESPA; a detailed comparison of redox dyes; a detailed comparison of screening for coenzyme engineering; strains and plasmid/primer data as well as SI material and methods (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Cahn JK; Werlang CA; Baumschlager A; Brinkmann-Chen S; Mayo SL; Arnold FH A general tool for engineering the NAD/NADP cofactor preference of oxidoreductases. ACS Synth. Biol 2017, 6, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ji D; Wang L; Hou S; Liu W; Wang J; Wang Q; Zhao ZK Creation of bioorthogonal redox systems depending on nicotinamide flucytosine dinucleotide. J. Am. Chem. Soc 2011, 133, 20857–20862. [DOI] [PubMed] [Google Scholar]
- (3).You C; Huang R; Wei X; Zhu Z; Zhang Y-HP Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Syn. Syst. Biotechnol 2017, 2, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Paul CE; Hollmann F A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl. Microbiol. Biotechnol 2016, 100, 4773–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Paul CE; Arends IWCE; Hollmann F Is simpler better? synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar]
- (6).Zhang J-D; Yang X-X; Jia Q; Zhao J-W; Gao L-L; Gao W-C; Chang H-H; Wei W-L; Xu J-H Asymmetric ring opening of racemic epoxides for enantioselective synthesis of (S)-β-amino alcohols by a cofactor self-sufficient cascade biocatalysis system. Catal. Sci. Technol 2019, 9, 70–74. [Google Scholar]
- (7).You Z-N; Chen Q; Shi S-C; Zheng M-M; Pan J; Qian X-L; Li C-X; Xu J-H Switching cofactor dependence of 7β-hydroxysteroid dehydrogenase for cost-effective production of ursodeoxycholic acid. ACS Catal. 2019, 9, 466–473. [Google Scholar]
- (8).Chen F-F; Zheng G-W; Liu L; Li H; Chen Q; Li F-L; Li C-X; Xu J-H Reshaping the active pocket of amine dehydrogenases for asymmetric synthesis of bulky aliphatic amines. ACS Catal. 2018, 8, 2622–2628. [Google Scholar]
- (9).Knaus T; Paul CE; Levy CW; de Vries S; Mutti FG; Hollmann F; Scrutton NS Better than nature: nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc 2016, 138, 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang L; Liu B; Liu Y; Sun Y; Liu W; Yu D; Zhao ZK Escherichia coli strain designed for characterizing in vivo functions of nicotinamide adenine dinucleotide analogues. Org. Lett 2019, 21, 3218–3222. [DOI] [PubMed] [Google Scholar]
- (11).Nowak C; Pick A; Lommes P; Sieber V Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: providing a regeneration system for artificial cofactors. ACS Catal. 2017, 7, 5202–5208. [Google Scholar]
- (12).Zachos I; Nowak C; Sieber V Biomimetic cofactors and methods for their recycling. Curr. Opin. Chem. Biol 2018, 49, 59–66. [DOI] [PubMed] [Google Scholar]
- (13).Ryan JD; Fish RH; Clark DS Engineering cytochrome P450 enzymes for improved activity towards biomimetic 1,4-NADH cofactors. ChemBioChem 2008, 9, 2579–2582. [DOI] [PubMed] [Google Scholar]
- (14).Friedlos F; Jarman M; Davies LC; Boland MP; Knox RJ Identification of novel reduced pyridinium derivatives as synthetic co-factors for the enzyme DT diaphorase (NAD(P)H dehydrogenase (quinone), EC 1.6.99.2). Biochem. Pharmacol 1992, 44, 25–31. [DOI] [PubMed] [Google Scholar]
- (15).Nowak C; Beer B; Pick A; Roth T; Lommes P; Sieber V A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front. Microbiol 2015, 6, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang L; Ji D; Liu Y; Wang Q; Wang X; Zhou YJ; Zhang Y; Liu W; Zhao ZK Synthetic cofactor-linked metabolic circuits for selective energy transfer. ACS Catal. 2017, 7, 1977–1983. [Google Scholar]
- (17).Lutz J; Hollmann F; Ho TV; Schnyder A; Fish RH; Schmid A Bioorganometallic chemistry: biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp*Rh-(bpy)H]+ for selective organic synthesis. J. Organomet. Chem 2004, 689, 4783–4790. [Google Scholar]
- (18).Johannes TW; Woodyer RD; Zhao H Directed evolution of a thermostable phosphite dehydrogenase for NAD(P)H regeneration. Appl. Environ. Microbiol 2005, 71, 5728–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen H; Zhu Z; Huang R; Zhang Y-HP Coenzyme engineering of a hyperthermophilic 6-phosphogluconate dehydrogenase from NADP+ to NAD+ with its application to biobatteries. Sci. Rep 2016, 6, 36311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bornscheuer UT; Huisman GW; Kazlauskas RJ; Lutz S; Moore JC; Robins K Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- (21).Brinkmann-Chen S; Flock T; Cahn JKB; Snow CD; Brustad EM; McIntosh JA; Meinhold P; Zhang L; Arnold FH General approach to reversing ketol-acid reductoisomerase cofactor dependence from NADPH to NADH. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10946–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vázquez MJ; Ashman S; Ramón F; Calvo D; Bardera A; Martin JJ; Rudiger M; Tew D; Dominguez JM Utilization of substrate-induced quenching for screening targets promoting NADH and NADPH consumption. J. Biomol. Screening 2006, 11, 75–81. [DOI] [PubMed] [Google Scholar]
- (23).Mayer KM; Arnold FH A colorimetric assay to quantify dehydrogenase activity in crude cell lysates. J. Mol. Screening 2002, 7, 135–140. [DOI] [PubMed] [Google Scholar]
- (24).Johannes TW; Woodyer RD; Zhao H High-throughput screening methods developed for oxidoreductases. In Enzyme assays; Wiley-VCH: Weinheim, Germany, 2006; pp 77–93. [Google Scholar]
- (25).Liu Y; Feng Y; Wang L; Guo X; Liu W; Li Q; Wang X; Xue S; Zhao ZK Structural insights into phosphite dehydrogenase variants favoring a non-natural redox cofactor. ACS Catal. 2019, 9, 1883–1887. [Google Scholar]
- (26).El Hawrani AS; Sessions RB; Moreton KM; Holbrook JJ Guided evolution of enzymes with new substrate specificities. J. Mol. Biol 1996, 264, 97–110. [DOI] [PubMed] [Google Scholar]
- (27).Flores H; Ellington AD A modified consensus approach to mutagenesis inverts the cofactor specificity of Bacillus stearothermophilus lactate dehydrogenase. Protein Eng., Des. Sel 2005, 18, 369–377. [DOI] [PubMed] [Google Scholar]
- (28).Weiß MS; Pavlidis IV; Vickers C; Höhne M; Bornscheuer UT Glycine oxidase based high-throughput solid-phase assay for substrate profiling and directed evolution of (R)- and (S)-selective amine transaminases. Anal. Chem 2014, 86, 11847–11853. [DOI] [PubMed] [Google Scholar]
- (29).Huang R; Chen H; Zhong C; Kim J-E; Zhang Y-HP High-throughput screening of coenzyme preference change of thermophilic 6-phosphogluconate dehydrogenase from NADP+ to NAD+. Sci. Rep 2016, 6, 32644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gielen F; Hours R; Emond S; Fischlechner M; Schell U; Hollfelder F Ultrahigh-throughput–directed enzyme evolution by absorbance-activated droplet sorting (AADS). Proc. Natl. Acad. Sci. U. S. A 2016, 113, E7383–E7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Funk D; Schrenk H-H; Frei E Serum albumin leads to false-positive results in the XTT and the MTT assay. BioTechniques 2007, 43, 178–186. [DOI] [PubMed] [Google Scholar]
- (32).Chakrabarti R; Kundu S; Kumar S; Chakrabarti R Vitamin A as an enzyme that catalyzes the reduction of MTT to formazan by vitamin C. J. Cell. Biochem 2001, 80, 133–138. [DOI] [PubMed] [Google Scholar]
- (33).Wang Y; Zhang Y-HP Overexpression and simple purification of the Thermotoga maritima 6-phosphogluconate dehydrogenase in Escherichia coli and its application for NADPH regeneration. Microb. Cell Fact 2009, 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Joo H; Arisawa A; Lin Z; Arnold FH A high-throughput digital imaging screen for the discovery and directed evolution of oxygenases. Chem. Biol 1999, 6, 699–706. [DOI] [PubMed] [Google Scholar]
- (35).Kim E-J; Wu C-H; Adams MWW; Zhang Y-HP Exceptionally high rates of biological hydrogen production by biomimetic in vitro synthetic enzymatic pathways. Chem. – Eur. J 2016, 22, 16047–16051. [DOI] [PubMed] [Google Scholar]
- (36).Kim E-J; Kim J-E; Zhang Y-HP Ultra-rapid rates of water splitting for biohydrogen gas production through in vitro artificial enzymatic pathways. Energy Environ. Sci 2018, 11, 2064–2072. [Google Scholar]
- (37).Zhang J-H; Chung TD; Oldenburg KR A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- (38).Reetz MT; Carballeira JD; Vogel A Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew. Chem., Int. Ed 2006, 45, 7745–7751. [DOI] [PubMed] [Google Scholar]
- (39).Burks EA; Chen G; Georgiou G; Iverson BL In vitro scanning saturation mutagenesis of an antibody binding pocket. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ma C; Wu R; Huang R; Jiang W; You C; Zhu L; Zhu Z Directed evolution of a 6-phosphogluconate dehydrogenase for operating an enzymatic fuel cell at lowered anodic pHs. J. Electroanal. Chem 2019, 851, 113444. [Google Scholar]
- (41).Guarneri A; van Berkel WJ; Paul CE Alternative coenzymes for biocatalysis. Curr. Opin. Biotechnol 2019, 60, 63–71. [DOI] [PubMed] [Google Scholar]
- (42).Zhang YHP; Sun J; Zhong J-J Biofuel production by in vitro synthetic enzymatic pathway biotransformation. Curr. Opin. Biotechnol 2010, 21, 663–669. [DOI] [PubMed] [Google Scholar]
- (43).Sicsic S; Durand P; Langrené S; Goffic FL Activity of NMN+, nicotinamide ribose and analogs in alcohol oxidation promoted by horse-liver alcohol dehydrogenase. Eur. J. Biochem 1986, 155, 403–407. [DOI] [PubMed] [Google Scholar]
- (44).Campbell E; Meredith M; Minteer SD; Banta S Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun 2012, 48, 1898–1900. [DOI] [PubMed] [Google Scholar]
- (45).McLachlan MJ; Johannes TW; Zhao H Further improvement of phosphite dehydrogenase thermostability by saturation mutagenesis. Biotechnol. Bioeng 2008, 99, 268–274. [DOI] [PubMed] [Google Scholar]
- (46).Okamoto Y; Köhler V; Paul CE; Hollmann F; Ward TR Efficient in situ regeneration of NADH mimics by an artificial metalloenzyme. ACS Catal. 2016, 6, 3553–3557. [Google Scholar]
- (47).Josa-Culleré L; Lahdenperä AS; Ribaucourt A; Höfler GT; Gargiulo S; Liu Y-Y; Xu J-H; Cassidy J; Paradisi F; Opperman DJ; Hollmann F; Paul CE Synthetic biomimetic coenzymes and alcohol dehydrogenases for asymmetric catalysis. Catalysts 2019, 9, 207. [Google Scholar]
- (48).Matsumoto KI; Mukai Y; Ogata D; Shozui F; Nduko JM; Taguchi S; Ooi T Characterization of thermostable FMN-dependent NADH azoreductase from the moderate thermophile Geobacillus stearothermophilus. Appl. Microbiol. Biotechnol 2010, 86, 1431–1438. [DOI] [PubMed] [Google Scholar]
- (49).Huang R; Chen H; Zhou W; Ma C; Zhang Y-HP Engineering a thermostable highly active glucose 6-phosphate dehydrogenase and its application to hydrogen production in vitro. Appl. Microbiol. Biotechnol 2018, 102, 3203–3215. [DOI] [PubMed] [Google Scholar]
- (50).Zhou W; Huang R; Zhu Z; Zhang Y-HP Coevolution of both thermostability and activity of polyphosphate glucokinase from Thermobifida fusca YX. Appl. Environ. Microbiol 2018, 84, e01224–e01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Hou Y; Hossain GS; Li J; Shin H-D; Du G; Chen J; Liu L Metabolic engineering of cofactor flavin adenine dinucleotide (FAD) synthesis and regeneration in Escherichia coli for production of alpha-keto acids. Biotechnol. Bioeng 2017, 114, 1928–1936. [DOI] [PubMed] [Google Scholar]
- (52).Drenth J; Trajkovic M; Fraaije MW Chemoenzymatic synthesis of an unnatural deazaflavin cofactor that can fuel F420-dependent enzymes. ACS Catal. 2019, 9, 6435–6443. [Google Scholar]
- (53).Franke J; Hertweck C Biomimetic thioesters as probes for enzymatic assembly lines: synthesis, applications, and challenges. Cell Chem. Biol 2016, 23, 1179–1192. [DOI] [PubMed] [Google Scholar]
- (54).Rosini E; Caldinelli L; Piubelli L Assays of D-amino acid oxidase activity. Front Mol. 2018, 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leng P-Q; Zhao F-L; Yin B-C; Ye B-C A novel, colorimetric method for biogenic amine detection based on arylalkylamine N-acetyltransferase. Chem. Commun 2015, 51, 8712–8714. [DOI] [PubMed] [Google Scholar]
- (56).Nowak C; Pick A; Csepei LI; Sieber V Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus. ChemBioChem 2017, 18, 1944–1949. [DOI] [PubMed] [Google Scholar]
- (57).Vieille C; Zeikus GJ Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev 2001, 65, 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.