

Abstract
Autosomal recessive non‐syndromic hearing loss (ARNSHL) is the most common inherited sensory impairment. It is particularly frequent in North African populations who have a high rate of consanguineous marriage. The c.242G>A homozygous variant in LRTOMT gene was previously established as pathogenic and is associated with NSHL in both humans and mice. The aim of this study is to determine the carrier frequency for the LRTOMT c.242G>A variant and also to estimate its age in addition to evaluating its diagnostic potential as a deafness biomarker among various populations and ethnicities in Northern African countries. A total of 179 Tunisian and 34 Libyan unrelated deafness patients were screened for this variant. The homozygous c.242G>A variant was found in 5.02% and 2.94% in Tunisian and Libyan families, respectively. Subsequent screening for this variant in 263 healthy controls of various ethnicities (136 Tunisian Berbers, 32 Andalusian and 95 Tunisian from undefined ethnic origin) revealed higher frequency for the heterozygous state among Tunisians of Berber origin only (19.11%). Genotyping 7 microsatellite markers nearby the variant location in ARNSHL patients who had the homozygous variant revealed the same haplotype suggesting a common founder origin for this variant. The age of this variant was estimated to be between 2025 and 3425 years (this corresponds to 3400 years when the variant rate was set at 10−3 or 2600 years when the variant rate is set at 10−2), spreading along with the Berber population who migrated to North Africa. In conclusion, the LRTOMT c.242G>A homozygous variant could be used as a useful deafness biomarker for North African ARNSHL patients meanwhile the heterozygous variant could be utilized in genealogical studies for tracing those of the Berber ethnic group.
Keywords: Arg81Gln, Berber and Biomarkers, DFNB63, LRTOMT
Genotyping 7 microsatellite markers nearby the variant location in ARNSHL patients who had the homozygous variant revealed the same haplotype suggesting a common founder origin for this variant. The LRTOMT c.242G>A homozygous variant could be used as a useful deafness biomarker for North African ARNSHL patients mean while the heterozygous variant could be utilized in genealogical studies for tracing those of Berber ethnic group.

1. INTRODUCTION
Hearing loss (HL) is the most common sensory disorder which affects nearly 1.8–2.7 out of every 1000 children globally (Morton & Nance, 2006). Genetic factors account for more than 50% of HL cases meanwhile the remaining percentage is due to environmental or age‐related deafness (Bouzid, Smeti, Chakroun, et al., 2018; Bouzid, Smeti, Dhouib, et al., 2018; Gates & Mills, 2015; Marazita et al., 1993). Almost 80% of genetic HL cases have impaired hearing without additional phenotypic disorders and are designated as non‐syndromic (NS). The latter is mainly inherited through autosomal recessive patterns with homogeneous and non‐progressive clinical manifestations (Morton & Nance, 2006). Up till now, 75 genes encoding key proteins for inner ear function and morphology were identified to be responsible for ARNSHL development (Camp & Smith, 2017).
Consanguineous marriage is very common in the Tunisian population, as part of their custom, and this increases the frequency and transmission of ARHL disorder among this population. Approximately 13 genes have been identified, so far, to cause ARNSHL in the Tunisian population. The LRTOMT [OMIM 612414] (Leucine‐Rich Transmembrane and O‐Methyl‐Transferase) gene variants which cause severe to profound pre‐lingual ARNSHL have been identified in few populations such as the Turkish, Pakistani and Tunisian population in 2008 (Ahmed et al., 2008). Recently, it was also confirmed in the Moroccan and Egyptian populations (Gibriel et al., 2019; Majida Charif et al., 2012). This gene includes a total of 10 exons comprising five different alternatively spliced transcripts that are widely expressed. Exons 5, 7 and 8 are included in transcripts encoding two different proteins: LRTOMT1 and LRTOMT2. These exons are predicted to be translated into two alternative reading frames and encode either the C terminus of LRTOMT1 or the N terminus of LRTOMT2 (Ahmed et al., 2008). When translation starts in exon 3, the LRTOMT1 protein presents two Leucine‐rich repeats. Beginning translation in exon 5 leads to the LRTOMT2 protein which has a catechol‐o‐methyltransferase (COMT) domain and hence it is named COMT2. Both proteins contain a predicted transmembrane domain located in the regions encoded by exon 5 for LRTOMT1 and by exon 8 of LRTOMT2 (Ahmed et al., 2008). The COMT protein catalyses the transfer of a methyl group donated by S‐adenosyl methionine to catecholamines and is involved in the inactivation of catecholamine neurotransmitters like dopamine, epinephrine and norepinephrine (Bonifácio et al., 2002; Tunbridge et al., 2006). Several studies have shown the involvement of the LRTOMT gene in the auditory function and many variants such as p.Arg81Gln (c.242G>A) and p.Glu110Lys (c.328G>A) in this gene led to loss of hearing function. So far, about 20 pathogenic mutations have been reported in the LRTOMT gene, 16 among them have been assigned to the COMT2 domain (Ahmed et al., 2008; Mojgan Babanejad et al., 2012; Sarmadi et al., 2020), whose the variant c.242G>A has been the most identified in several families with ARNSHL.
The purpose of this study is to screen for the c.242G>A variant in LRTOMT gene in 179 Tunisian and 34 Libyan families affected with ARNSHL in addition to 263 healthy controls of various ethnicities (136 Tunisian Berbers, 32 Andalusian and 95 Tunisian from undefined ethnic origin). We also genotyped seven microsatellite markers nearby this variant in order to determine haplotype frequency in our groups and also to see if there was a common founder origin for this variant. Moreover, the age of this variant among our Berber population was empirically estimated for genealogical purposes as well.
2. MATERIALS AND METHODS
2.1. Subject
This study was approved by the ethics committee of the University Hospital of Sfax (Tunisia).
We screened 179 Tunisian and 34 Libyan unrelated families with ARNSHL. These families had at least two affected members with an autosomal recessive pattern of inheritance. Informed consent was obtained from all study participants and from parents of subjects younger than 18 years of age. Clinical history interviews and physical examinations for families and their members ruled out involvement of environmental factors and other associated syndromes in hearing loss. In order to determine carrier frequency of the p.Arg81Gln variant among healthy controls with normal hearing capabilities and also to investigate the presence of common ancestors, we further recruited 263 individuals. This included 95 Tunisian individuals of unknown ethnicity, 32 Tunisian Andalusian, 136 Tunisian Berbers (51 Chennini‐Douiret subjects, 53 Sned and 32 Jradou Berbers). The sampling in the ‘Andalusian’ community was based on a patronymic criterion, whereas Berbers were Berber Chelha speakers born in one of the three villages mentioned above.
2.2. Methods
2.2.1. DNA extraction and PCR amplification
DNA was extracted from peripheral white blood cells using standard phenol–chloroform method. Quality and quantity of extracted DNA samples were determined by measuring the A260/A280 (Gibriel et al., 2013). The regions flanking the c.242G>A variant in LRTOMT (NG_021423.1 RefSeqGene hg19/GRCh37) were PCR amplified for all samples using PCR thermal cycler in a 30 µl reaction each using 20 ng DNA sample with 1 U TaqDNA polymerase (Biotools), 5 mM Taq Reaction Buffer and1 µM of each primer (Arg81Gln forward: 5′‐TGGTGGTAACATTGCTGGTG‐3′ and Arg81Gln reverse: 5′‐GCAACAGATCCCAAATATTCC‐3′). The following PCR conditions were implemented incubation at 95°C for 5 min, 38 cycles at 94°C for 30 s, 60°C for 30 s and 72°C for 1 min followed by final extension at 72°C for 10 min.
2.2.2. PCR‐RFLP analysis for the c.242G>A (p.Arg81Gln) variant
The generated PCR product (296 bp) was then digested with BsrB1 enzyme through overnight incubation at 37°C. RFLP reaction was conducted in 25 µl reaction volume using 0.5 µl (10 U/µl) BsrBI enzyme, 10 µl PCR amplicon and 2.5 µl stock buffer. The digested product was then visualized under UV after migration on a 3% agarose gel. BsrB1enzyme recognizes a restriction site in the normal wild‐type sequence and cut it into two fragments of 107 and 189 bp. The presence of LRTOMT c.242G>A variant abolishes such restriction site leading to no cut (296 bp) by the BsrB1enzyme.
2.2.3. Sanger sequencing
Following PCR‐RFLP gel electrophoresis, patients’ samples exhibiting anomalous run, compared to normal wild type, were Sanger sequenced in both directions to verify the presence of LRTOMT c.242G>A (p.Arg81Gln) variant. Briefly, PCR products were gel purified using the GeneJet gel extraction kit (Thermo Scientific) and then sequenced on the ABI 3500 Genetic Analyzer (Applied Biosystems) using either reverse or forward primer and 4 µl of BigDye terminator 3.1 ready reaction mix (Applied Biosystems). Sequencing chromatograms were analysed using Chromas software V2.6.4 followed by comparison to NCBI database.
2.2.4. Microsatellite genotyping
In order to get more evidences for the common origin and frequency of this variant in Tunisian population as well as in neighbouring area, patients confirmed to be carriers for the LRTOMT c.242G>A (p.Arg81Gln) variant were then genotyped for seven informative fluorescently labelled microsatellite markers. Those seven short tandem microsatellite repeat markers are located nearby the c.242G>A variant and three of them were recently generated. The seven microsatellite markers included D11S4139, D11S4162, D11S1314, D11S916, AP003783, AP002490 and AP000593. The True Allele PCR Premix was used for PCR reactions. Fluorescently labelled alleles were also analysed on ABI Prism 3100‐Avant DNA Analyser (Applied Biosystems).
2.2.5. Age of the c.242G>A LRTOMT variant
Calculation of the p.Arg81Gln variant age was performed using the C program developed by Genin et al. (2004). Compared with other methods using haplotype information, this approach was efficient with a very small number of affected individuals. Results are presented as the mean number of generations (with empirical 95% confidence interval) estimated over 5000 replicates.
2.2.6. Statistical analysis
Genotype and allele frequencies were determined using Hardy–Weinberg equation and were then compared with reported frequencies in other populations.
3. RESULTS
PCR‐RFLP digestion with the BsrB1enzyme revealed the presence of the p.Arg81Gln variant in homozygous state in 9 Tunisian families out of the 179 participating Tunisian families (5.02%). One of those nine carrier families was known to have a Berber origin. On the other hand, only one family out of the 34 unrelated Libyan families (2.94%) had the homozygous variant (Figure 1 and Table 1). Sanger sequencing confirmed the presence of this homozygous variant in those 10 cases (Figure 1). In a subsequent study, we screened for this variant in 263 healthy Tunisian individuals with normal hearing capability and of different ethnic origin to estimate the carrier frequency in those control subjects. This included 95 Tunisian individuals of unknown ethnicity, 32 Tunisian Andalusian, 136 Tunisian Berbers (51 Chennini‐Douiret subjects, 53 Sned and 32 Jradou Berbers). Interestingly, the frequency of heterozygous state for this variant in Berber controls was higher than that of the HL patients (19.11% vs. 5.02%). On the contrary, we did not find any heterozygous variants in neither the 32 Tunisian healthy Andalusians nor in the 95 Tunisians of unknown ethnic origin (Table 2). Moreover, genotyping analysis for the seven microsatellite markers in all the 10 positive HL patients revealed the same haplotype suggesting a common founder origin (Figure 2). We estimated the age of the p.Arg814Gln variant in the first haplotype to be about 136 generations (95% confidence interval, 81–239) when the variant rate was set at 10−3. Assuming that one generation is 25 years, this corresponds to 3400 years. When the variant rate is set at 10−2, the estimated age would be 104 generations corresponding to nearly 2600 years (95% confidence interval, 62–182).
FIGURE 1.
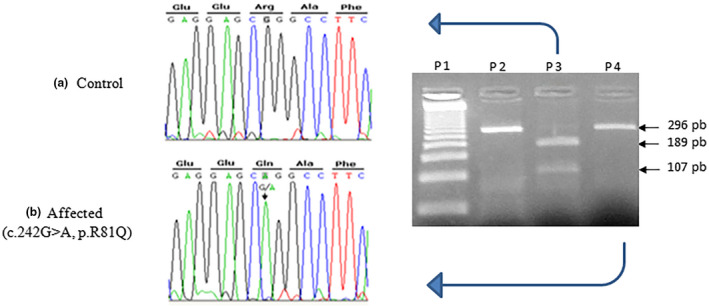
Sequencing chromatograms for the c.242 G>A variant in LRTOMT gene (NG_021423.1 RefSeqGene hg19/GRCh37) (a) wild‐type control with homozygous G base (Arg), (b) affected individual with homozygous A base (Gln), (c) PCR‐RFLP (BsrBI digestion) fragmentation pattern on the agarose gel stained with ethidium bromide for determination of the c.242G>A variant. DNA ladder (50 bp) is loaded into well 1 (p1), PCR product (296 bp) is loaded into well 2 (p2, PCR‐RFLP product for a patient with wild‐type c.242 G>A variant is loaded into well 3 (p3) with two digested fragments 189 and 107 bp, PCR‐RFLP product for a patient with homozygous c.242 G>A variant is loaded into well 4 (p4) with only one product 296 bp
TABLE 1.
Homozygous and heterozygous frequencies for the c.242 G>A variant (NG_021423.1 RefSeqGene hg19/GRCh37) in Tunisian and Libyan families
| Tunisian families (179) | Libyan families (34) | |||
|---|---|---|---|---|
| Homozygous variant (c.242G>A) | 9 | (5.02%) | 1 | 2.94% |
| Heterozygous variant (c.242G>A) | 0 | 0% | 0 | 0% |
| Non‐carrier of c.242G>A variant | 170 | (94.98%) | 33 | 97.05% |
TABLE 2.
Heterozygous and homozygous frequencies for the c.242 G>A variant (NG_021423.1 RefSeqGene hg19/GRCh37) in Tunisian Berber, Andalus and those of unknown ethnic groups
| Tunisian Berber ethnic | Other ethnic | ||||
|---|---|---|---|---|---|
| Chennini‐Douiret (51) | Sned (53) | Jradou (32) | Andalus (32) | Unknown (95) | |
|
Homozygous variant c.242G>A |
0 | 0 | 0 | 0 | 0 |
|
Heterozygous variant c.242G>A |
8 | 6 | 12 | 0 | 0 |
|
Heterozygous variant c.242G>A% |
15.68% | 11.32% | 37.5% | 0 | 0 |
| 19.11% | |||||
FIGURE 2.
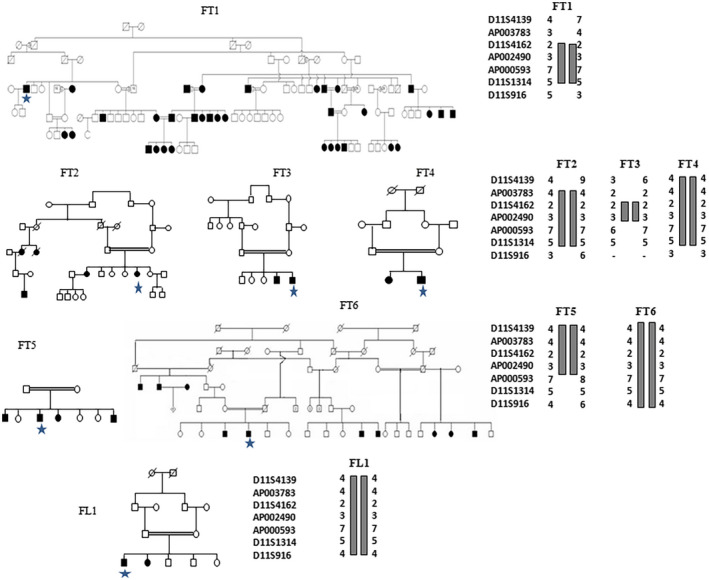
Pedigrees of six unpublished Tunisian families FT1–FT6 and Libyan family FL1 showing segregation of deafness consistent with linkage to genetic markers for the DFNB63 locus (RefSeqGene hg19/GRCh37). Haplotypes in family FT3 illustrate that D11S4162 is the centromeric flanking marker of the locus and Haplotypes in families FT3 and five illustrate that the telomeric marker for DFNB63 is AP000593. All families shared the same haplotype indicating a common ancestor. The stars represent tested patients
4. DISCUSSION
Several genetic variants were linked to various heterogenous diseases as in HL (Ahmed et al., 2008; Chakchouk et al., 2015). Various genotyping assays such as RFLP, single stranded conformational polymorphism, DNA microarrays and/or ligase chain reaction have been implemented to screen for genetic variants (Al‐Achkar et al., 2011; Chakchouk et al., 2015; Gibriel, 2012; Gibriel & Adel, 2017; Gibriel et al., 2013). The LRTOMT gene is located on chromosome 11 and contains 10 exons comprising five different alternatively spliced transcripts translated into LRTOMT1 and LRTOMT2 proteins (Ahmed et al., 2008). It is essential for auditory and vestibular function and is expressed in sensory hair cells of the inner ear of mice (Ahmed et al., 2008). Nearly 20 different variants affecting the LRTOMT gene have been identified as causing ARNSHL in different populations such as Iranian, Moroccan, Tunisian, Pakistan, Turkish and Japanese (Ichinose et al., 2015; Sarmadi et al., 2020). The substitution of guanine by adenine at position 242 on cDNA leads to missense variant with replacement of arginine by glutamine on position 81 of the amino acid sequence of the COMT domain of LRTOMT protein. The mutated residue p.Arg81Gln is in helix 1 and contributes to the formation of salt bridge between this helix and the loop following helix 2. The mutated Q81 residue could not form this bridge as it is not positively charged. In addition, the formation of hydrogen bonds is impaired because of the smaller size of glutamine compared to arginine (Ahmed et al., 2008). This particular variant was identified previously in Pakistani and Turkish populations in addition to 7 Tunisian families only (Ahmed et al., 2008). In this study, the p.Arg81Gln variant was found in 2.94% of unrelated Libyan families and 5.02% of Tunisian HL patients of whom 1 patient was of Berber origin. Given the frequency obtained for this variant in our population and its presence in the patient of Berber origin, we subsequently attempted to screen for this variant in 263 unrelated Tunisian families with ARNSHL after exclusion of the main recurrent variants such as the GJB2 [OMIM 121011] c.35delG (p.Gly12Valfs*2). Surprisingly this variant was not observed at all, even as a heterozygous state, neither in the 32 Tunisian healthy andalusians nor in the 95 Tunisians of unknown ethnic origin. It was rather observed with high frequency in a heterozygous state in 19.11% of Berber controls from Sned in South‐West, Jradou in the North and Chenini‐Douiret in the South of Tunisia. Interestingly, all the 10 ARNSHL Tunisian and Libyan ARNSHL families shared the same haplotype for the 7 investigated microsatellite markers present nearby this variant.
The p.Arg81Gln variant appeared 3400 years ago and this aligns well with the timing of the Berber descendent. Indeed it is described that the Berber culture was present in North Africa since Protohistoric times around 4700 years ago. The ancient and complex history of Berbers was marked by numerous events like conquests, invasions or migrations from different origins. Therefore several Berber communities were forced to settle in isolated areas which had resulted in geographic location of Berber populations mainly in heights. Moreover in previous studies it was shown via mitochondrial DNA analysis that pre‐historical events led to several contributions to the Berber gene pool corresponding especially to Western Eurasian, sub‐Saharian and North African haplogroups (Ben Arab et al., 2004; Coudray et al., 2009). The high consanguinity and endogamy rates that seem to characterize North African populations since ancient times could have led to counter selection of recessive variants which are lethal at homozygous state. This is not the case for HL patients who had the corresponding variant since ancient times. Other variants involved in hearing loss with founder effect have also been described.
Two old founder variants namely the c.35delG (p.Gly12Valfs*2) and the c.100C>T (p.R34*) variants in GJB2 [OMIM 121011] and TMC1 [OMIM 606706] genes, respectively, have been identified in Tunisian population. The high frequency of those variants was explained by the presence of a very old founder variant. Zied et al. showed that the c.35delG accounts for 85.4% of all GJB2 mutant alleles (Riahi et al., 2013); this result is in agreement with other data reported in different countries such as Portugal (64%; Nogueira et al., 2011), Italy (68%; Primignani et al., 2009) and Algeria (76%; Ammar‐Khodja et al., 2009) (Belguith et al., 2005; Rothrock et al., 2003). Lucotte tried to determine the origin of the c.35delG variant and to trace the path of its migration. They showed that variant originated from Greece where the rate of heterozygous carriers was 1/28 (Lucotte, 2001). Its spread would be made through the ancient Greek colonies. The age of the c.35delG variant was estimated at ~216 generations among the Lebanese population compared with 111 generations in the Tunisian and Moroccan populations. So the c.35delG variant has appeared 5500 years ago among the Lebanese compared with 2775 years among Tunisians and Moroccans (Belguith et al., 2005). On the other hand, the c.100C>T of the TMC1 gene represents more than 30% among of the 44 mutant alleles of this gene. This variant has been reported in hearing‐impaired persons originating from Iran, Iraq, Lebanon, Pakistan, Tunisia and Turkey (Ben Said et al., 2010; Hilgert et al., 2008; Kitajiri et al., 2007; Kurima et al., 2002; Sirmaci et al., 2009; Tlili et al., 2008). The highest frequency of this variant was reported in Tunisia and Pakistan (Kitajiri et al., 2007; Tlili et al., 2008). Analyses of the 21 markers flanking the TMC1 gene in 11 deaf (four Tunisians, two Algerians, one Iranian, one Iraqi, one Pakistani, one Lebanese and one Turkish) showed the presence of a common haplotype for all carriers, which is in favour of the presence of a common founder. The age of this variant was estimated between 1075 and 1900 years (Ben Said et al., 2010).
5. CONCLUSION
A homozygous state for the LRTOMT c.242G>A variant could be used as a useful deafness biomarker for North African ARNSHL patients. The heterozygous variant can also be used in genealogical studies for tracing individuals descendent from the Berber ethnic origin. This study indicates the importance of the LRTOMT c.242G>A variant not only in ARNSHL development but also in and also its role as an old founding variant for Berber population. So we believe that it would be interesting to add the p.Arg81Gln variant today with the 35delG variant in the routine diagnosis for deafness in the North Africa region notably to the Berber community.
PATIENT CONSENT
Informed consent was obtained from all participating individuals of this study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
ETHICS APPROVAL
This study was approved by the ethical committee of the University Hospital of Sfax, Tunisia.
ACKNOWLEDGEMENTS
We thank all persons and local communities that donated their DNA and made the present study feasible. We also thank Genin for the variant age calculation program.
Mosrati, M. A. , Fadhlaoui‐Zid, K. , Benammar‐Elgaaied, A. , Gibriel, A. A. , Ben Said, M. , & Masmoudi, S. (2021). Deep analysis of the LRTOMTc.242G>A variant in non‐syndromic hearing loss North African patients and the Berber population: Implications for genetic diagnosis and genealogical studies. Molecular Genetics & Genomic Medicine, 9, e1810. 10.1002/mgg3.1810
Funding information
This study was supported by the Tunisian Ministry of Higher Education and Scientific Research (LR15CBS07).
DATA AVAILABILITY STATEMENT
The data set(s) supporting the conclusions of this article is (are) included within the article.
REFERENCES
- Ahmed, Z. M. , Masmoudi, S. , Kalay, E. , Belyantseva, I. A. , Mosrati, M. A. , Collin, R. W. , Riazuddin, S. , Hmani‐Aifa, M. , Venselaar, H. , Kawar, M. N. , & Tlili, A. (2008). Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nature Genetics, 40, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Achkar, W. , Moassass, F. , Al‐Halabi, B. , & Al‐Ablog, A. (2011). Mutations of the connexin 26 gene in families with non‐syndromic hearing loss. Molecular Biology Reports, 4, 331–335. [DOI] [PubMed] [Google Scholar]
- Ammar‐Khodja, F. , Faugère, V. , Baux, D. , Giannesini, C. , Léonard, S. , Makrelouf, M. , Malek, R. , Djennaoui, D. , Zenati, A. , Claustres, M. , & Roux, A. F. (2009). Molecular screening of deafness in Algeria: high genetic heterogeneity involving DFNB1 and the Usher loci, DFNB2/USH1B, DFNB12/USH1D and DFNB23/USH1F. European Journal of Medical Genetics, 52(4), 174–179. 10.1016/j.ejmg.2009.03.018 [DOI] [PubMed] [Google Scholar]
- Belguith, H. , Hajji, S. , Salem, N. , Charfeddine, I. , Lahmar, I. , Amor, M. B. , Ouldim, K. , Chouery, E. , Driss, N. , Drira, M. , Megarbane, A. , Rebai, A. , Sefiani, A. , Masmoudi, S. , & Ayadi, H. (2005). Analysis of GJB2 mutation: Evidence for a Mediterranean ancestor for the 35delG mutation. Clinical Genetics, 68, 188–189. [DOI] [PubMed] [Google Scholar]
- Ben Arab, S. , Masmoudi, S. , Beltaief, N. , Hachicha, S. , & Ayadi, H. (2004). Consanguinity and endogamy in Northern Tunisia and its impact on non‐syndromic deafness. Genetic Epidemiology, 27(1), 74–79. 10.1002/gepi.10321 [DOI] [PubMed] [Google Scholar]
- Ben Said, M. , Hmani‐Aifa, M. , Amar, I. , Baig, S. M. , Mustapha, M. , Delmaghani, S. , Tlili, A. , Ghorbel, A. , Ayadi, H. , Van Camp, G. , Smith, R. J. , Tekin, M. , & Masmoudi, S. (2010). High frequency of the p. R34X mutation in the TMC1 gene associated with nonsyndromic hearing loss is due to founder effects. Genetic Testing and Molecular Biomarkers, 14, 307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifácio, M. J. , Archer, M. , Rodrigues, M. L. , Matias, P. M. , Learmonth, D. A. , Carrondo, M. A. , & Soares‐da‐Silva, P. (2002). Kinetics and crystal structure of catechol‐o‐methyltransferase complex with co‐substrate and a novel inhibitor with potential therapeutic application. Molecular Pharmacology, 62, 795–805. [DOI] [PubMed] [Google Scholar]
- Bouzid, A. , Smeti, I. , Chakroun, A. , Loukil, S. , Gibriel, A. A. , Grati, M. , Ghorbel, A. , & Masmoudi, S. (2018). CDH23 Methylation status and presbicusis risk in elderly women. Frontiers in Aging Neuroscience, 10, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzid, A. , Smeti, I. , Dhouib, L. , Roche, M. , Achour, I. , Khalfallah, A. , Gibriel, A. A. , Charfeddine, I. , Ayadi, H. , Lachuer, J. , & Ghorbel, A. (2018). Down‐expression of P2RX2, KCNQ5, ERBB3 and SOCS3 through DNA hypermethylation in elderly women with presbycusis. Biomarkers, 23(4), 347–356. [DOI] [PubMed] [Google Scholar]
- Chakchouk, I. , Said, M. B. , Jbeli, F. , Benmarzoug, R. , Loukil, S. , Smeti, I. , Chakroun, A. , Gibriel, A. A. , Ghorbel, A. , Hadjkacem, H. , & Masmoudi, S. (2015). NADf chip, a two color microarray for simultaneous screening of multigene mutations associated with hearing impairment in North African Mediterranean countries. The Journal of Molecular Diagnostics, 17, 155–161. [DOI] [PubMed] [Google Scholar]
- Coudray, C. , Olivieri, A. , Achilli, A. , Pala, M. , Melhaoui, M. , Cherkaoui, M. , El‐Chennawi, F. , Kossmann, M. , Torroni, A. , & Dugoujon, J. M. (2009). The complex and diversified mitochondrial gene pool of berber populations. Annals of Human Genetics, 73, 196–214. [DOI] [PubMed] [Google Scholar]
- Gates, G. A. , & Mills, J. H. (2015). Presbucusis. Lancet, 366, 1111–1120. [DOI] [PubMed] [Google Scholar]
- Genin, E. , Tullio‐Pelet, A. , Begeot, F. , Lyonnet, S. , & Abel, L. (2004). Estimating the age of rare disease mutations: The example of Triple‐A syndrome. Journal of Medical Genetics, 41(6), 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibriel, A. A. Y. (2012). Options available for labelling nucleic acid samples in DNA microarray‐based detection methods. Briefings in Functional Genomics, 11(4), 311–318. 10.1093/bfgp/els015 [DOI] [PubMed] [Google Scholar]
- Gibriel, A. A. , Abou‐Elew, M. H. , & Masmoudi, S. (2019). Analysis of p.Gly12Valfs*2, pTrp24* and p.Trp77Arg mutations in GJB2 and p.Arg81Gln Variant in LRTOMT among non syndromic hearing loss Egyptian patients: Implications for genetic diagnosis. Molecular Biology Reports, 46(2), 2139–2145. 10.1007/s11033-019-04667-0 [DOI] [PubMed] [Google Scholar]
- Gibriel, A. A. , & Adel, O. (2017). Advances in ligase chain reaction and ligation‐based amplifications for genotyping assays: Detection and applications. Mutation Research, 773, 66–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibriel, A. A. Y. , Tate, R. J. , Yu, Y. , Rawson‐Lax, E. , Hammer, H. M. , Tettey, J. N. A. , Pyne, N. J. , & Converse, C. A. (2013). The p.Arg86Gln change in GARP2 (glutamic acid‐rich protein‐2) is a common West African‐related polymorphism. Gene, 515(1), 155–158. 10.1016/j.gene.2012.11.005 [DOI] [PubMed] [Google Scholar]
- Hilgert, N. , Alasti, F. , Dieltjens, N. , Pawlik, B. , Wollnik, B. , Uyguner, O. , Delmaghani, S. , Weil, D. , Petit, C. , Danis, E. , Yang, T. , Pandelia, E. , Petersen, M. B. , Goossens, D. , Favero, J. D. , Sanati, M. H. , Smith, R. J. , & Van Camp, G. (2008). Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clinical Genetics, 74, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose, A. , Moteki, H. , Hattori, M. , Nishio, S. Y. , & Usami, S. I. (2015). Novel mutations in LRTOMT associated with moderate progressive hearing loss in autosomal recessive inheritance. The Annals of Otology, Rhinology, and Laryngology, 124, 142S–147S. [DOI] [PubMed] [Google Scholar]
- Kitajiri, S. I. , McNamara, R. , Makishima, T. , Husnain, T. , Zafar, A. U. , Kittles, R. A. , Ahmed, Z. M. , Friedman, T. B. , Riazuddin, S. , & Griffith, A. J. (2007). Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clinical Genetics, 72, 546–550. [DOI] [PubMed] [Google Scholar]
- Kurima, K. , Peters, L. M. , Yang, Y. , Riazuddin, S. , Ahmed, Z. M. , Naz, S. , Arnaud, D. , Drury, S. , Mo, J. , Makishima, T. , Ghosh, M. , Menon, P. S. , Deshmukh, D. , Oddoux, C. , Ostrer, H. , Khan, S. , Deininger, P. L. , Hampton, L. L. , Sullivan, S. L. , … Griffith, A. J. (2002). Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair‐cell function. Nature Genetics, 30, 277–284. [DOI] [PubMed] [Google Scholar]
- Lucotte, G. G. M. (2001). Meta‐analysis of GJB2 mutation 35delG frequencies in Europe. Genetic Testing, 5, 149–152. [DOI] [PubMed] [Google Scholar]
- Majida Charif, S. B. O. A. , Nahili, H. , Rouba, H. , Kandil, M. , Boulouiz, R. , & Barakat, A. (2012). The c.242G>A mutation in LRTOMT gene is responsible for a high prevalence of deafness in the Moroccan population. Molecular Biology Reports, 39, 11011–11016. [DOI] [PubMed] [Google Scholar]
- Marazita, M. L. , Ploughman, L. M. , Rawlings, B. , Remington, E. , Arnos, K. S. , & Nance, W. E. (1993). Genetic epidemiological studies of early‐onset deafness in the U.S. school‐age population. American Journal of Medical Genetics, 46(5), 486–491. 10.1002/ajmg.1320460504 [DOI] [PubMed] [Google Scholar]
- Mojgan Babanejad, Z. F. , Bazazzadegan, N. , Nishimura, C. , Meyer, N. , Nikzat, N. , Sohrabi, E. , Najmabadi, A. , Jamali, P. , Habibi, F. , Smith, R. J. H. , Kahrizi, K. , & Najmabadi, H. (2012). A comprehensive study to determine heterogeneity of autosomal recessive nonsyndromic hearing loss in Iran. American Journal of Medical Genetics, 158, 2485–2492. [DOI] [PubMed] [Google Scholar]
- Morton, C. C. , & Nance, W. E. (2006). Newborn hearing screening—A silent revolution. New England Journal of Medicine, 354(20), 2151–2164. 10.1056/NEJMra050700 [DOI] [PubMed] [Google Scholar]
- Nogueira, C. , Coutinho, M. , Pereira, C. , Tessa, A. , Santorelli, F. M. , & Vilarinho, L. (2011). Molecular investigation of pediatric portuguese patients with sensorineural hearing loss. Genetics Research International, 2011, 587602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primignani, P. , Trotta, L. , Castorina, P. , Lalatta, F. , Sironi, F. , Radaelli, C. , Degiorgio, D. , Curcio, C. , Travi, M. , Ambrosetti, U. , Cesarani, A. , Garavelli, L. , Formigoni, P. , Milani, D. , Murri, A. , Cuda, D. , & Coviello, D. A. (2009). Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: Frequencies, novel mutations, genotypes, and degree of hearing loss. Genetic Testing and Molecular Biomarkers, 13(2), 209–217. [DOI] [PubMed] [Google Scholar]
- Riahi, Z. , Hammami, H. , Ouragini, H. , Messai, H. , Zainine, R. , Bouyacoub, Y. , Romdhane, L. , Essaid, D. , Kefi, R. , Rhimi, M. , Bedoui, M. , Dhaouadi, A. , Feldmann, D. , Jonard, L. , Besbes, G. , & Abdelhak, S. (2013). Update of the spectrum of GJB2 gene mutations in Tunisian families with autosomal recessive nonsyndromic hearing loss. Gene, 525(1), 1–4. 10.1016/j.gene.2013.04.078 [DOI] [PubMed] [Google Scholar]
- Rothrock, C. R. , Murgia, A. , Sartorato, E. L. , Leonardi, E. , Wei, S. , Lebeis, S. L. , Yu, L. E. , Elfenbein, J. L. , Fisher, R. A. , & Friderici, K. H. (2003). Connexin 26 35delG does not represent a mutational hotspot. Human Genetics, 113, 18–23. [DOI] [PubMed] [Google Scholar]
- Sarmadi, A. , Nasrniya, S. , Soleimani Farsani, M. , Narrei, S. , Nouri, Z. , Sepehrnejad, M. , Nilforoush, M. H. , Abtahi, H. , & Tabatabaiefar, M. A. (2020). A novel pathogenic variant in the LRTOMT gene causes autosomal recessive non‐syndromic hearing loss in an Iranian family. BMC Medical Genetics, 21(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirmaci, A. , Duman, D. , Ozturkmen‐Akay, H. , Erbek, S. , Incesulu, A. , Ozturk‐Hismi, B. , Arici, Z. S. , Yuksel‐Konuk, E. B. , Tasir‐Yilmaz, S. , Tokgoz‐Yilmaz, S. , Cengiz, F. B. , Aslan, I. , Yildirim, M. , Hasanefendioglu‐Bayrak, A. , Aycicek, A. , Yilmaz, I. , Fitoz, S. , Altin, F. , Ozdag, H. , & Tekin, M. (2009). Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: A report of five novel mutations. International Journal of Pediatric Otorhinolaryngology, 86, 699–705. [DOI] [PubMed] [Google Scholar]
- Tlili, A. , Rebeh, I. B. , Aifa‐Hmani, M. , Dhouib, H. , Moalla, J. , Tlili‐Chouchene, J. , Said, M. B. , Lahmar, I. , Benzina, Z. , Charfedine, I. , Driss, N. , Ghorbel, A. , Ayadi, H. , & Masmoudi, S. (2008). TMC1 but not TMC2 is responsible for autosomal recessive nonsyndromic hearing impairment in Tunisian families. Audiology and Neurotology, 13, 213–218. [DOI] [PubMed] [Google Scholar]
- Tunbridge, E. M. , Harrison, P. J. , & Weinberger, D. R. (2006). Catechol‐omethyltransferase, cognition, and psychosis: Val158Met and beyond. Biological Psychiatry, 60, 141–151. [DOI] [PubMed] [Google Scholar]
- Van Camp, G. , & Smith, R. J. (2017). Hereditary hearing loss homepage. http://hereditaryhearingloss.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data set(s) supporting the conclusions of this article is (are) included within the article.