

Abstract
Background
Maple syrup urine disease (MSUD) is an autosomal recessive inborn error of amino acid metabolism, with unique clinico‐radiological findings. This study aims to show the benefit of using the clinico‐radiological findings for early diagnosis of children with MSUD, and confirming this diagnosis using the tandem mass spectrometry (MS/MS), in order to avoid deleterious outcome.
Methods
A prospective cohort study conducted in the period from August 2016 to December 2020. Twenty‐one children were included either by selective screening or by high‐risk screening. All children had clinical and neurodevelopmental evaluation, brain magnetic resonance imaging (MRI) assessment, and blood amino acids analysis at diagnosis. Patients were followed clinically.
Results
Most children had acute onsets neuro‐developmental symptoms, with wide range of brain parenchyma involvement on MRI (hyperintensity). Diagnosis of MSUD was confirmed by detecting high serum levels of leucine/isoleucine (mean value 2085.5 μmol/L) in all patients, and elevated levels of serum valine in (81%) of children. In addition, all children showed elevated leucine: alanine ratio, and leucine: phenylalanine ratio.
Conclusions
The characteristic clinico‐radiological features can help in the early diagnosis of MSUD children, thus preventing the delay in laboratory diagnosis and improving their outcomes.
Keywords: clinico‐radiological, diagnosis, Egypt, MSUD, outcome
This study aims to show the benefit of using the clinico‐radiological findings for early diagnosis of children with MSUD, and confirming this diagnosis using the tandem mass spectrometry (MS/MS), in order to avoid deleterious outcome.
1. INTRODUCTION
Maple syrup urine disease (MSUD) is a rare inborn error of amino acid metabolism. It has an autosomal recessive pattern of inheritance that runs in families (Patil et al., 2020). It occurs due to a deficiency in an enzyme complex [branched chain α‐keto acid dehydrogenase (BCKAD)] that results in defects in the catabolism of the branched chain amino acids (BCAAs): leucine, isoleucine, and valine. The accumulation of these BCAAs has a toxic effect on the brain cells (Chimbili et al., 2020), as they inhibit the activity of pyruvate dehydrogenase and α‐ketoglutarate, leading to interruption of the citric acid cycle and consequently affecting the synthesis of amino acids, causing cerebral edema and abnormal myelination (Li et al., 2021).
Clinical symptoms and signs of MSUD varies in severity according to the BCKAD residual activity (Blackburn et al., 2017). The onset of symptoms can occur within the first week of life, which can be non‐specific, variable and can be easily misdiagnosed. The clinical picture may present as poor feeding, poor activity, irritability, or vomiting. Without a proper management of this condition, these symptoms and signs would proceed to muscular rigidity, convulsions, coma, and even death (Kathait et al., 2018).
MSUD is classified according to age of onset, clinical manifestations and its severity. The subtypes include classic, intermediate (mild), intermittent, MSUD caused by a deficiency of E3 subunit, and thiamine‐responsive type. Classic subtype of MSUD is the commonest type (75%) and the most severe form (Blackburn et al., 2017).
Infants with MSUD can be identified early through newborn screening programs using the tandem mass spectrometry (MS/MS). Abnormal elevation of BCAAs in plasma and increased the urine organic acids are confirmatory. Tandem mass spectrometry (MS/MS) can provide the leucine–isoleucine concentration, in addition to its ratio with other amino acids including glutamate, glutamine, tryptophan, methionine, histidine, alanine, phenylalanine, and tyrosine. Organic acid analysis by gas chromatography (GC)–MS/MS also can provide qualitative assessment for the presence of branched chain α‐hydroxyacids and branched chain α‐ketoacids (BCKAs) in urine in patients with MSUD which is absent in normal individuals (Strauss & Morton, 2003; Strauss et al., 2010).
MRI brain findings in cases of classic MSUD are unique, in which characteristic patterns of edema occurs especially at diffusion‐weighted imaging, and so it provide a useful tool for early diagnosis (Li et al., 2021). The characteristic types of brain edema that can be seen with MSUD are intramyelinic edema (typical pattern) and vasogenic edema. The intramyelinic edema is an intense form that affects the myelinated white matter (cerebral peduncles, perirolandic cerebral white matter, cerebellar white matter, dorsal brain stem, posterior limb of the internal capsule), the thalami and globi pallidi, both of which have a high density of myelinated fibers. The intramyelinic edema has a high signal intensity on T2‐weighted & diffusion‐weighted MR images, low signal intensity on T1‐weighted MR images, and decreased ADC and fractional anisotropy values (Reddy et al., 2018).
Early diagnosis of infants with classic type MSUD and its proper management by lifelong diet restriction of BCAAs and supplementation with vitamin B1 (thiamine) could decrease the morbidity, mortality and provide better outcome.
The MSUD screening by tandem mass spectrometry (MS/MS) is currently incorporated in NBS programs throughout the United States, 5 Canadian provinces, 22 European countries, 2 Latin American countries (Costa Rica and Uruguay), and 8 countries in the Asia‐Pacific region; accordingly, the NBS for MSUD is not available in many regions worldwide (Therrell et al., 2015). Egypt like many developing countries, lacks mass newborn screening (NBS) program for inborn errors of metabolism, and the current Egyptian NBS is limited to hypothyroidism and phenylketonuria (Hassan et al., 2016; Zayed et al., 2019). Therefore, this study aims to show the benefit of using the clinico‐radiological findings for early diagnosis of children with MSUD, and confirming this diagnosis using the tandem mass spectrometry (MS/MS), in order to avoid deleterious outcome.
2. METHODS
2.1. Ethical compliance
The “Sohag University Medical Research Ethics Committee” approved this study with reference number (Soh‐Med‐21‐04‐34), Egypt.
2.2. Study population
From August 2016 to December 2020, we conducted a prospective cohort study in our pediatric department, at a tertiary level university hospital. The study included all consecutive children suspected to have symptoms and signs of MSUD, aged 1‐day to 2‐years old, admitted to the inpatient department and willing to participate in the study. Selective screening was done for: (a) any newborn presenting with neurological symptoms (e.g., coma, lethargy, and persistent of one or more of the following: convulsions, vomiting, diarrhea, metabolic acidosis, and hypoglycemia); (b) child with one or more of the following: delayed developmental milestones, altered level of consciousness, recurrent convulsions, metabolic acidosis with increased anion gap, recurrent attacks of hypoglycemia, or electrolyte imbalance (with no specific cause to explain this symptoms and signs). In addition, a high‐risk screening was performed to symptomatic family members of a known index child with MSUD or a family history of previous sibling death without a definite diagnosis.
Exclusion criteria were patients known to have definite history of perinatal brain injury, CNS infection, brain trauma or tumors, and intoxication. In addition, patients diagnosed with any chromosomal abnormalities.
2.3. Clinical and neuro‐developmental evaluation
All participants experienced thorough history taking, clinical examination, and neuro‐developmental assessment, focusing on patient's demographics, onset and details of symptoms, lag time between initial symptoms and establishing the diagnosis, manifestation severity and outcome related to MRI finding and management.
2.4. Neuroimaging assessment
Children underwent brain MRI using a 1.5‐T magnet system (different models and manufacturers) at various imaging centers. Available sequences were axial T2‐W fast spin echo (repetition/echo times: 3600–5540/100–110 ms) and T1‐W spin echo (480–565/14–15) with a slice thickness of 4–6 mm; sagittal T1‐W spin echo and axial fluid attenuation inversion recovery [FLAIR] (6000–11,000/120–146; inversion time 2000–2800 ms); coronal T1‐W spin echo or T2‐W fast spin echo; and diffusion‐weighted image. The MRI scan focused on the supratentorial area (including the frontal, temporal, parietal and occipital hemisphere regions, the centrum semiovale, and corona radiata), the internal capsule, corpus callosum, basal ganglia, thalamus, mesencephalon, pons, and cerebellum. Brain tissue manifesting with high signal on DWI can be identified as cytotoxic or intramyelinic edema. Affected areas with increased signal in T2‐FLAIR were recognized as dysmyelination or disturbed water content of the white matter.
An experienced pediatric neuro‐radiologist interpreted the MRI scans of the patients, while he was blinded to the clinical and laboratory data of them.
2.5. Biochemistry examinations
Arterial blood gas, serum glucose level, and ketonuria were assessed. In addition, the patient's blood levels of ammonia, lactic acid, and β‐hydroxybutyric acid were measured. Confirmatory testing of highly increased branched chain amino acid (BCAAs) levels in plasma via quantitative amino acid analysis was performed.
2.6. Blood amino acids and acylcarnitines analysis
Patients’ blood samples (2 ml) were obtained on a Guthrie card (Whatman 903 filter paper; GE Healthcare Ltd). Acylcarnitines and amino acids were analyzed by using ACQUITY TQD Tandem Quadrupole UPLC/MS/MS (Waters Corporation) with a positive electrospray ionization probe according to the manufacturer's instructions.
2.7. Statistical analysis
Data were expressed as frequency and percentage or means and standard deviations as appropriate. A p value ≤0.05 was considered significant for all statistical operations. Analyses were done using SPSS software 22.0 release, SPSS Inc., Chicago, USA.
3. RESULTS
The demographic and clinical characteristics of the studied population are shown in Table 1. There were 21 children (14 males and 7 females) diagnosed to have MSUD (the classic subtype). They belongs to 18 unrelated families (case 7 and 21 were sisters; case 4 and 11 were sister and brother; while case 12 and 13 were cousins).
TABLE 1.
Demographic, clinical characteristics, and outcome of 21 children with maple syrup urine disease
| Case | Sex | Screening | Onset of symptoms | Age of onset of symptoms | Age at MRI done | Age at diagnosis by MS/MS | Consanguinity | Family history | Seizures | DCL | Acidotic breathing | Ventilation support | The outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Selective | Acute | 6 days | 25 days | 45 days | + | + | + | + | − | + | Died |
| 2 | M | Selective | Acute | 75 days | 75 days | 105 days | + | − | − | + | + | + | Died |
| 3 | M | Selective | Acute | 9 days | 30 days | 45 days | + | + | + | + | − | + | Severe delay |
| 4 | F | Selective | Acute | 7 days | 15 days | 40 days | + | − | + | − | − | − | Died |
| 5 | F | Selective | Acute | 5 days | 12 days | 30 days | + | − | + | + | − | + | Died |
| 6 | M | Selective | Acute | 7 days | 15 days | 35 days | + | + | + | + | − | − | Died |
| 7 | F | Selective | Acute | 7 days | 8 days | 365 days | + | − | + | − | − | − | Severe delay |
| 8 | F | Selective | Acute | 3 days | 5 days | 8 days | + | + | + | + | − | − | Moderate delay |
| 9 | M | Selective | Acute | 6 days | 8 days | 10 days | + | − | − | + | − | + | Died |
| 10 | M | Selective | Acute | 15 day | 20 days | 35 days | + | + | + | − | − | + | Died |
| 11 | M | Selective | Acute | 3 days | 13 day | 25 days | + | + | + | + | − | + | Died |
| 12 | M | Selective | Acute | 8 days | 10 days | 30 days | + | + | + | + | + | + | Died |
| 13 | M | Selective | Acute | 5 days | 7 days | 20 days | + | + | + | + | − | + | Severe delay |
| 14 | M | Selective | Acute | 7 days | 13days | 30 days | + | + | + | − | + | + | Died |
| 15 | M | Selective | Acute | 6 days | 11 days | 25 days | + | + | + | + | − | + | Died |
| 16 | M | Selective | Acute | 8 days | 12 days | 30 days | − | − | − | + | − | + | Moderate delay |
| 17 | M | Selective | Acute | 9 days | 13 days | 35 days | − | − | − | + | − | − | Sever delay |
| 18 | M | Selective | Acute | 5 days | 10 days | 27 days | + | + | − | + | − | + | Moderate delay |
| 19 | M | Selective | Acute | 7 days | 13 days | 24 days | + | − | + | + | − | + | Died |
| 20 | F | Selective | Insidious | 35 days | 105 days | 135 days | − | + | − | − | − | − | Died |
| 21 | F | High risk | Asymptomatic | NA | 300 days | 360 days | + | + | − | − | − | − | Slight delay |
Abbreviation: DCL, disturbed conscious level.
All the symptomatic cases (20 child, 95.2%) were normal at birth; but the symptoms of MSUD appeared after a variable disease‐free interval (mean 8.3 ± 6.9 days; range: 3–75 days), and were included in the selective screening. Nineteen cases (90.4%) developed these symptoms acutely, one case (4.8%) had insidious onset, whereas one case (4.8%) was asymptomatic.
For the acute onset cases, they did the MRI at mean age of 16.5 ± 15.4 days (range 5–75 days), while their mean age at which the diagnosis was confirmed by MS/MS was 38.8 ± 22.7 days (range: 8–360 days).
Acute onset symptomatic children (total = 19 children) were presented with poor feeding, lethargy in 17 children (89.5%), irritability with excessive crying in 2 children (10.5%), disturbed conscious level in 15 children (78.9%), generalized seizures in 14 children (73.7%). Three cases (15.8%) presented by acidotic breathing, and 14 children (73.7%) developed generalized hypotonia and need mechanical ventilation support. The insidious onset symptomatic child (case no. = 20) presented with generalized developmental delay.
All the children with acute onset presentation received supportive management in the form of IV fluids, oxygen, and were fed on special formula with low‐branched chain amino acids.
The outcome of the acute symptomatic group showed 12 children (63.2%) who died before completing their first year, while the outcome of the insidious onset symptomatic child (case no. = 20) died at age of 10 months.
The brain parenchyma of all patients (n = 21) was analyzed by MRI and the findings are summarized in Table 2. All children with acute onset disease showed involvement of wide range of the brain parenchyma. Involvement of the basal ganglia occurred in 13 children (68.4%), the thalamus in 9 children (47.4%), and the internal capsule in 15 children (78.9%). The involvement of the optic chiasm, optic tracts, and optic radiations was reported in 5 children (26.3%), See Figures 1, 2, 3. The child (no. = 20) with the insidious onset presentation of the disease showed similar MRI findings like the acute onset children; with preservation of the basal ganglia and the thalamus. Asymptomatic child (no.= 21) which was diagnosed through high‐risk screening showed involvement of the dorsal brain stem and globus pallidus on both sides only, see (Figure 4).
TABLE 2.
MRI alteration in different anatomic brain regions
| Case | Supratentorial area | Corticospinal tract | Central portion of centrum semioval | Basal ganglia | Thalamus | Pones | Mesencephalon | Cerebellum |
|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | − | − | + | + | + |
| 2 | + | + | + | + | + | + | + | + |
| 3 | + | + | + | − | − | + | + | + |
| 4 | + | + | + | + | − | + | + | + |
| 5 | + | + | + | − | − | + | + | + |
| 6 | + | + | + | − | − | + | + | + |
| 7 | + | + | + | + | + | + | + | + |
| 8 | + | + | + | + | − | + | + | + |
| 9 | + | + | + | − | − | + | + | + |
| 10 | + | + | + | + | − | + | + | + |
| 11 | + | + | + | + | − | + | + | + |
| 12 | + | + | + | − | − | + | + | + |
| 13 | + | + | + | + | + | + | + | + |
| 14 | + | + | + | + | + | + | + | + |
| 15 | + | + | + | + | + | + | + | + |
| 16 | + | + | + | + | + | + | + | + |
| 17 | + | + | + | + | + | + | + | + |
| 18 | + | + | + | + | + | + | + | + |
| 19 | + | + | + | + | + | + | + | + |
| 20 | + | + | + | − | − | + | + | + |
| 21 | − | + | − | + | − | + | + | + |
FIGURE 1.
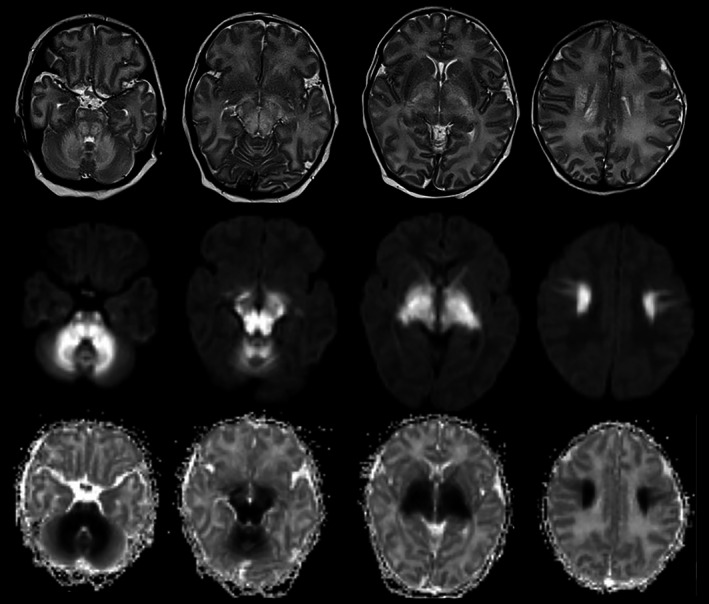
MRI of child 12 days with MSUD. Top row is axial T2, middle row is axial DWI and the bottom row is axial ADC map. There is bilateral symmetrical abnormal high T2 signal with related restriction in DWI and in ADC map involving cerebellar WM, brainstem tracts, optic tracts, cerebral peduncles, anterior, and posterior limbs of internal capsules, thalami, globus pallidus, and perirolandic WM
FIGURE 2.
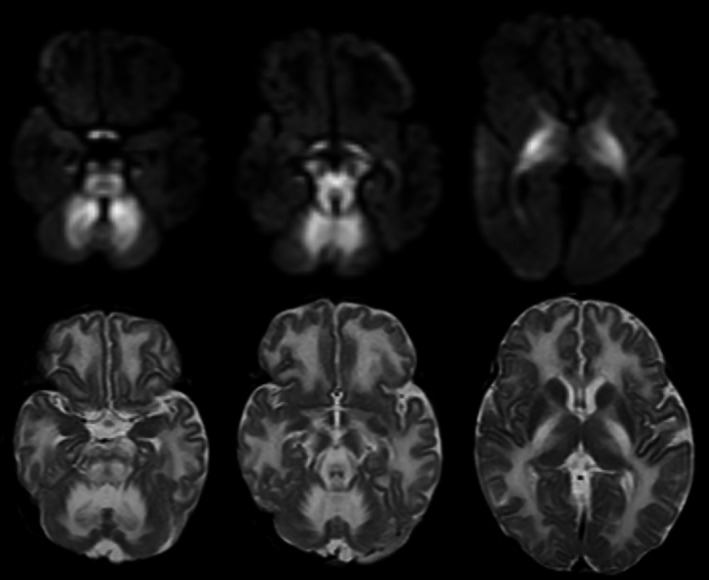
MRI of child 35 days with MSUD. The upper row is axial DWI and the lower row is axial T2. There is bilateral symmetrical abnormal high T2 signal with related restriction in DWI involving cerebellar WM, brainstem tracts, optic chiasm and tracts, cerebral peduncles, anterior, and posterior limbs of internal capsules, thalami, and globus pallidus
FIGURE 3.
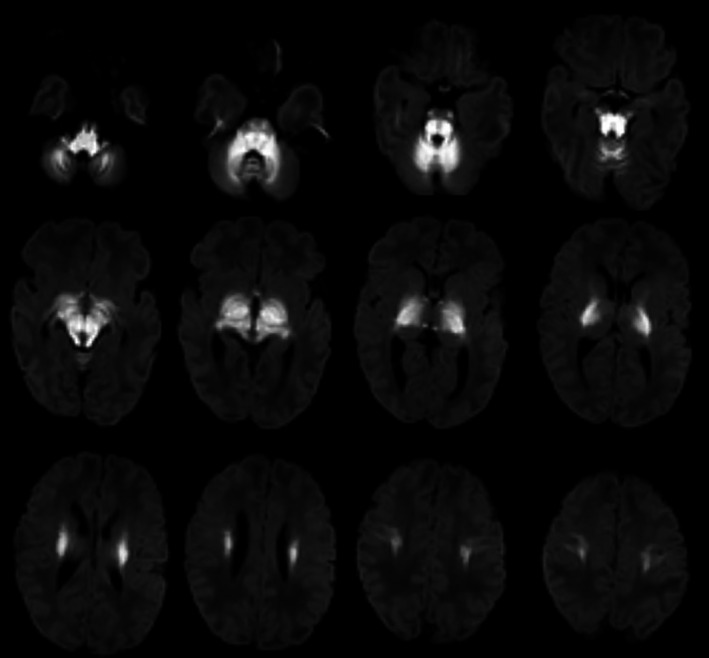
Axial diffusion‐weighted MRI of child 11 days with MSUD. There is bilateral symmetrical abnormal restriction involving cerebellar WM, brainstem tracts, cerebral peduncles, posterior and to less degree anterior limbs of internal capsules, thalami, globus pallidus, and perirolandic WM
FIGURE 4.
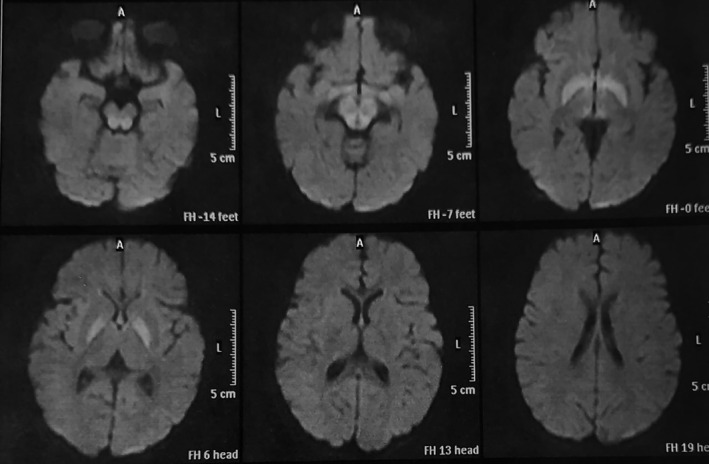
Axial diffusion‐weighted MRI of child 1 year with MSUD. There is bilateral symmetrical abnormal restriction involving the globus pallidus and dorsal brain stem. (Case 21 diagnosed by high‐risk screening)
Laboratory investigations of the cohort study are summarized in Table 3. An elevation of the serum ammonia level was reported in 14 children (66.7%) (Normal value = up to 85 μg/dl), 4 children out of them showed extremely high serum levels (>200 μg/dl). In addition, there was 6 children (28.6%) who showed high plasma lactate levels (Normal values =4.5–19.8 mg/dl), 14 children (66.7%) presented with metabolic acidosis, and 6 children (28.6%) with hypoglycemia. Ketonuria was reported in 14 children (66.7%).
TABLE 3.
Laboratory characteristics for diagnosis of children with MSUD
| Case no. | Leu/isoleu serum levels (normal up to 270 μmol/L) | Leu:alan ratio (normal up to 2.28) | Leu:phe ratio (normal up to 8) | Valine serum level (normal up to 198 μmol/L) | Ammonia serum level (normal up to 85 μg/dl) | Lactate serum level (normal up to 19.8 mg/dl) | Ketonuria | Hypo‐glycemia | Metabolic acidosis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2672 | 28 | 47 | 345 | 331 | 13 | ++ | + | + |
| 2 | 1146 | 5.4 | 15 | 261 | 80 | 70 | + | + | + |
| 3 | 1130 | 5.2 | 16 | 77 | 101 | 74 | + | − | + |
| 4 | 1068 | 3.7 | 24.6 | 368 | 125 | 21 | ++ | − | + |
| 5 | 2903 | 10.3 | 70.5 | 235 | 219 | 10 | − | + | + |
| 6 | 2943 | 21.8 | 51.8 | 424 | 85 | 18.9 | − | − | − |
| 7 | 2004 | 48.4 | 60.4 | 374 | 150 | 29 | − | − | − |
| 8 | 2730 | 21.9 | 75 | 188 | 104 | 10.2 | +++ | − | − |
| 9 | 3800 | 39.6 | 66.9 | 369 | 55 | 28 | + | + | − |
| 10 | 1133 | 5.2 | 16 | 76.7 | 110 | 15 | + | − | + |
| 11 | 1243 | 13.5 | 25.3 | 167.5 | 112 | 17.5 | + | − | + |
| 12 | 2672 | 27.7 | 47.1 | 354 | 138 | 9.5 | + | − | + |
| 13 | 2766 | 35.9 | 51.5 | 442 | 160 | 14 | + | + | + |
| 14 | 2800.0 | 28.0 | 46.0 | 325.0 | 230 | 23 | + | − | + |
| 15 | 2608.0 | 27.0 | 45.0 | 312.0 | 149 | 15.7 | + | − | + |
| 16 | 1234.0 | 5.8 | 17.0 | 273.0 | 63 | 53 | − | + | + |
| 17 | 1079.0 | 3.9 | 19.0 | 284.0 | 134 | 17 | + | − | + |
| 18 | 1006.0 | 4.2 | 23.3 | 296.0 | 72 | 12 | − | − | − |
| 19 | 2856.0 | 19.4 | 65.5 | 407.0 | 59 | 11.7 | + | − | + |
| 20 | 2900 | 28 | 42 | 319 | 215 | 16 | − | − | − |
| 21 | 1103 | 8.2 | 18 | 304 | 56 | 16 | − | − | − |
Abbreviations: Leu/isoleu, leucine/isoleucine; Leu:alan, leucine:alanine ratio; Leu:phe, leucine:phenylalanine ratio.
Diagnosis was confirmed by detection of elevated plasma levels of BCAAs using quantitative amino acid analysis and the tandem mass spectrometry. All children showed elevated serum levels of leucine/isoleucine with mean value 2085.5 μmol/L (ranging from 1006 to 3800 μmol/L) (Normal values = up to 270 μmol/L). Elevated serum valine was reported in 17 children (81%) (Normal values = up to 198 μmol/L); while the remaining 4 children (19%) had normal or low levels of serum valine as a result of starting protein restriction outside our center before testing with tandem mass spectrometry. In addition, all children showed elevated leucine: alanine ratio, and leucine: phenylalanine ratio.
4. DISCUSSION
MSUD is an inherited error of metabolism involving the BCAAs. The imaging features of this disease are very typical. The early clinico‐radiological correlations in MSUD could have an important role in the diagnosis, prevent the progress of neurological deficits, and help in appropriate management of this condition (Shah et al., 2018).
This study cohort included 21 MSUD patients (classic subtype). Other studies included nearly similar numbers of children with MSUD (classic subtype) ranging from 6 to 37 children (Cheng et al., 2017; Lee et al., 2008; Margutti et al., 2020; Yunus et al., 2012). The largest study for MUSD children was the one done by Strauss et al. who reported 184 MSUD children {including 176 children (95.7%) having the classic subtype} (Strauss et al., 2020).
In this study, the mean age of onset of symptoms to appear was mean 8.3 ± 6.9 days (range: 3–75 days). This consists with Lee et al. who reported that the mean age of onset of symptoms was 5 days (ranged from 2 to 14 days of life) (Lee et al., 2008), Cheng et al. who reported mean age of 10.5 days (Cheng et al., 2017), and Yunus et al. reported that all classic MSUD children developed initial symptoms within first week (Yunus et al., 2012).
In our study, we confirmed the diagnosis of MSUD by MS/MS at a mean age of 38.8 ± 22.7 days (range: 8–360 days). This delay in the diagnosis can be caused by: (1) the vague clinical manifestations at initial assessment resembling neonatal sepsis; especially in the absence of a positive family history for inborn errors of metabolism. Definitive laboratory diagnosis is needed to confirm clinical suspicion of MSUD, (2) Delay in referral from primary units, (3) time consumed on samples transfer and results receipt as there are limited number of tandem mass spectrometry in our locality. In other studies, there was variable delay (ranging from 3 to 12 months) in confirming the diagnosis of MSUD either by MS/MS or by genetic studies. The need for a rapid diagnostic test or implementation of MSUD newborn screening program to help pre‐symptomatic diagnosis was mandatory to avoid that delay (Cheng et al., 2017; Lee et al., 2008; Margutti et al., 2020; Strauss et al., 2020; Yunus et al., 2012). This time lag between age of symptoms and confirming the diagnosis of MSUD; allows MRI brain to be a great diagnostic tool that saves time and gives chance to start early a specific management for these children which may improves their outcome.
The main clinical findings in this study include poor feeding, poor activity, seizures, and disturbed conscious level. A common finding in almost children was positive consanguinity in 18 children (85.7%) and positive family history in 13 children (61.9%), which are consistent with the autosomal recessive mode of inherence that was reported in other studies (Blackburn et al., 2017; Chimbili et al., 2020; Jaradat et al., 2015; Li et al., 2021; O'Reilly et al., 2021; Patil et al., 2020; Yunus et al., 2012).
MRI brain imaging in MSUD patients (especially in classic subtype) has a characteristic hyperintensity particularly DW1 sequence in the first few weeks of life. It mainly involves the globus pallidus, thalamus, internal capsule, brainstem, and cerebellar white matter, which represent the typical myelinated areas in normal full‐term neonates (Li et al., 2021). In our study, all patients with acute onset showed involvement of wide range of brain parenchyma. This was inconsistent with other literatures that discussed the radiological finding of classic subtype of MSUD patients, and reported similar findings, see Table 4. These findings represent demyelination and disrupted water content in the white matter (Li et al., 2021).
TABLE 4.
Other studies showing brain parenchyma involvement in MRI of children with MSUD
| Similar studies |
Total no. of children |
Corticospinal Tract |
Central portion of centrum semioval |
Basal Ganglia |
Thalamus |
Pones |
Mesencephalon |
Cerebellum |
|---|---|---|---|---|---|---|---|---|
| Patil et al. (2020) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Chimbili et al. (2020) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Li et al. (2021) | 3 | 3 | 1 | 2 | 2 | 2 | 3 | 3 |
| Kathait et al. (2018) | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Shah et al. (2018) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Cheng et al. (2017) | 6 | 5 | 5 | 6 | 4 | 5 | 5 | 5 |
| O'Reilly et al. (2021) | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 |
| Jan et al. (2003) | 6 | 6 | 4 | 6 | 5 | 6 | 6 | 5 |
| Jain et al. (2013) | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Cornelius et al. (2014) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Sener (2007) | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Kilicarslan et al. (2012) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Ha et al. (2004) | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Indiran and Gunaseelan (2013) | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| Hou et al. (2016) | 7 | 5 | 3 | 5 | 4 | 5 | 5 | 5 |
The serum leucine/isoleucine at the time of diagnosis was elevated in all cases (mean 2085.5 μmol/L), and this was lower than the value reported from a study done by Yunus et al who reported the mean in Malaysian MSUD children to be 3037 μmol/L for the classic subtype (Yunus et al., 2012). However, in a study done by (Margutti et al., 2020), the mean plasma leucine level for 11 Brazilian children was 1976 μmol/L (range = 278.9–3252 μmol/L).
The outcome of children with MSUD (especially the classic subtype) is mainly affected by early diagnosis and long‐term control rather than leucine level at time of diagnosis (Margutti et al., 2020; O'Reilly et al., 2021). The MRI abnormalities that was reported in the first month of life were completely resolved by the age of 3 month after specific treatment as reported in many studies (Cheng et al., 2017; Margutti et al., 2020; O'Reilly et al., 2021). These data support the hypothesis that MSUD edema on DWI is intramedullary edema rather than cytotoxic.
Other studies using MR spectroscopy in MSUD children showed significant elevation of the methyl groups of BCAA and BCKA at 0.9–1 ppm; and these abnormalities resolved completely after recovery (Jan et al., 2003; Sato et al., 2014). So, MR spectroscopy can be considered as a specific diagnostic imaging study, which is also effective in follow‐up of stable periods.
4.1. Limitation of the study
The need of more studies with larger population samples are still required to confirm these clinico‐radiological observations in order to count on them on diagnosis and follow‐up till a local NBS program using MS/MS becomes well established. In addition, the need of specific studies reporting the use of MR spectroscopy at different intervals after MSUD diagnosis and its role as a follow‐up tool.
5. CONCLUSION
The unique clinico‐radiological features can help in early diagnosis of MSUD children, and thus decreasing the time lag until the confirmatory laboratory results (using tandem mass spectrometry) become available, which will result in improvement of their outcome.
CONFLICT OF INTEREST
The authors have declared that no competing interests exist.
AUTHOR CONTRIBUTIONS
MMM: conceived and designed the study. MAB: took part in database design, and data collection. RMM: assisted with the study design, data collection, and obtained ethical permission. HSE: undertook biochemical assay and interpretation. MHA: undertook MRI imaging and interpretation. HMA: assisted in the study design, design and interpretation of the statistical analysis, and drafted the initial manuscript. All authors revised and approved the final manuscript.
ETHICAL APPROVAL
All procedures performed in studies including human participants were as per the ethical standards of the institutional and/or national research committee.
INFORMED CONSENT
Informed consent was taken from all individual participants/parents included in the study.
ACKNOWLEDGMENTS
The authors acknowledge all the children and their parents who so willingly participated in this study.
Mohamed, M. M. , Bakheet, M. A. , Magdy, R. M. , El‐Abd, H. S. , Alam‐Eldeen, M. H. , & Abo‐Haded, H. M. (2021). The clinico‐radiological findings of MSUD in a group of Egyptian children: Contribution to early diagnosis and outcome. Molecular Genetics & Genomic Medicine, 9, e1790. 10.1002/mgg3.1790
DATA AVAILABILITY STATEMENT
Data available on request due to privacy/ethical restrictions.
REFERENCES
- Blackburn, P. R. , Gass, J. M. , Vairo, F. P. E. , Farnham, K. M. , Atwal, H. K. , Macklin, S. , Klee, E. W. , & Atwal, P. S. (2017). Maple syrup urine disease: Mechanisms and management. The Application of Clinical Genetics, 10, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, A. , Han, L. , Feng, Y. , Li, H. , Yao, R. , Wang, D. , & Jin, B. (2017). MRI and clinical features of maple syrup urine disease: Preliminary results in 10 cases. Diagnostic and Interventional Radiology, 23(5), 398–402. 10.5152/dir.2017.16466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimbili, S. S. , Shirbur, P. , & Agarwal, A. (2020). Imaging in a rare case of maple syrup urine disease. Journal of Evidence Based Medicine and Healthcare, 7(19), 963–966. 10.18410/jebmh/2020/210 [DOI] [Google Scholar]
- Cornelius, L. P. , Kannan, B. , Saravanan, V. , & Venkatesan, E. P. (2014). Intramyelinic edema in maple syrup urine disease. Annals of Indian Academy of Neurology, 17(2), 211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, J. S. , Kim, T. K. , Eun, B. L. , Lee, H. S. , Lee, K. Y. , Seol, H. Y. , & Cha, S. H. (2004). Maple syrup urine disease encephalopathy: A follow‐up study in the acute stage using diffusion‐weighted MRI. Pediatric Radiology, 34(2), 163–166. [DOI] [PubMed] [Google Scholar]
- Hassan, F. A. , El‐Mougy, F. , Sharaf, S. A. , Mandour, I. , Morgan, M. F. , Selim, L. A. , Hassan, S. A. , Salem, F. , Oraby, A. , Girgis, M. Y. , Mahmoud, I. G. , El‐Badawy, A. , El‐Nekhely, I. , Moharam, N. , Mehaney, D. A. , & Elmonem, M. A. (2016). Inborn errors of metabolism detectable by tandem mass spectrometry in Egypt: The first newborn screening pilot study. Journal of Medical Screening, 23(3), 124–129. 10.1177/0969141315618229 [DOI] [PubMed] [Google Scholar]
- Hou, L. , Han, L. S. , Chi, R. M. , & Wang, D. B. (2016). Brain MRI and clinical characteristics of maple syrup urine disease. Shiyong Fangshe Xue Zazhi, 10, 1582–1585. [Google Scholar]
- Indiran, V. , & Gunaseelan, R. E. (2013). Neuroradiological findings in maple syrup urine disease. Journal of Pediatric Neurosciences, 8(1), 31–33. 10.4103/1817-1745.111419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, A. , Jagdeesh, K. , Mane, R. , & Singla, S. (2013). Imaging in classic form of maple syrup urine disease: A rare metabolic central nervous system. Journal of Clinical Neonatology, 2(2), 98–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, W. , Zimmerman, R. A. , Wang, Z. J. , Berry, G. T. , Kaplan, P. B. , & Kaye, E. M. (2003). MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology, 45(6), 393–399. [DOI] [PubMed] [Google Scholar]
- Jaradat, S. A. , Al‐Qa'qa’, K. , Amayreh, W. , Backe, P. H. , Al‐Hawamdeh, A. , Karam, M. , Alzoubi, B. , Deebajah, H. , & Al Rababah, B. (2015). Molecular analysis of maple syrup urine disease in Jordanian families. Meta Gene, 10, 81–85. 10.1016/j.mgene.2015.10.002 [DOI] [Google Scholar]
- Kathait, A. S. , Puac, P. , & Castillo, M. (2018). Imaging findings in maple syrup urine disease: A case report. Journal of Pediatric Neurosciences, 13(1), 103–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilicarslan, R. , Alkan, A. , Demirkol, D. , Toprak, H. , & Sharifov, R. (2012). Maple syrup urine disease: Diffusion weighted MRI findings during acute metabolic encephalopathic crisis. Japanese Journal of Radiology, 30(6), 522–525. 10.1007/s11604-012-0079-2 [DOI] [PubMed] [Google Scholar]
- Lee, J. Y. , Chiong, M. A. , Estrada, S. C. , Cutiongco‐De la Paz, E. M. , Silao, C. L. T. , & Padilla, C. D. (2008). Maple syrup urine disease (MSUD)–Clinical profile of 47 Filipino patients. Journal of Inherited Metabolic Disease, 31(2), 281–285. 10.1007/s10545-008-0859-0 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Liu, X. , Duan, C. F. , Song, X. F. , & Zhuang, X. H. (2021). Brain magnetic resonance imaging findings and radiologic review of maple syrup urine disease: Report of three cases. World Journal of Clinical Cases, 9(8), 1844–1852. 10.12998/wjcc.v9.i8.1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margutti, A. V. B. , Silva, W. A. , Garcia, D. F. , de Molfetta, G. A. , Marques, A. A. , Amorim, T. , Prazeres, V. M. G. , Boy da Silva, R. T. , Miura, I. K. , Seda Neto, J. , Santos, E. D. S. , Santos, M. L. S. F. , Lourenço, C. M. , Tonon, T. , Sperb‐Ludwig, F. , de Souza, C. F. M. , Schwartz, I. V. D. , & Camelo, J. S. (2020). Maple syrup urine disease in Brazilian patients: Variants and clinical phenotype heterogeneity. Orphanet Journal of Rare Diseases, 15(1), 309–314. 10.1186/s13023-020-01590-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, D. , Crushell, E. , Hughes, J. , Ryan, S. , & Rogers, Y. (2021). Maple syrup urine disease: Clinical outcomes, metabolic control, and genotypes in a screened population after four decades of newborn bloodspot screening in the Republic of Ireland. Journal of Inherited Metabolic Disease, 44(3), 639–655. [DOI] [PubMed] [Google Scholar]
- Patil, R. , Giridhar, S. , Umadevi, L. , Rathinasamy, M. , & Antony, J. (2020). Maple syrup urine disease presenting as severe neonatal metabolic encephalopathy: A case report. International Journal of Contemporary Pediatrics, 7(10), 2072–2076. 10.18203/2349-3291.ijcp20204054 [DOI] [Google Scholar]
- Reddy, N. , Calloni, S. F. , Vernon, H. J. , Boltshauser, E. , Huisman, T. A. G. M. , & Soares, B. P. (2018). Neuroimaging findings of organic acidemias and aminoacidopathies. Radiographics, 38(3), 912–931. 10.1148/rg.2018170042 [DOI] [PubMed] [Google Scholar]
- Sato, T. , Muroya, K. , Hanakawa, J. , Asakura, Y. , Aida, N. , Tomiyasu, M. , Tajima, G. , Hasegawa, T. , & Adachi, M. (2014). Neonatal case of classic maple syrup urine disease: Usefulness of (1) H‐MRS in early diagnosis. Pediatrics International, 56(1), 112–115. [DOI] [PubMed] [Google Scholar]
- Sener, R. N. (2007). Maple syrup urine disease: diffusion MRI, and proton MR spectroscopy findings. Computerized Medical Imaging and Graphics, 31(2), 106–110. 10.1016/j.compmedimag.2006.11.005 [DOI] [PubMed] [Google Scholar]
- Shah, T. , Purohit, S. , & Raval, M. (2018). Imaging in maple syrup urine disease. Indian Journal of Pediatrics, 85(10), 927–928. 10.1007/s12098-018-2696-y [DOI] [PubMed] [Google Scholar]
- Strauss, K. A. , Carson, V. J. , Soltys, K. , Young, M. E. , Bowser, L. E. , Puffenberger, E. G. , Brigatti, K. W. , Williams, K. B. , Robinson, D. L. , Hendrickson, C. , Beiler, K. , Taylor, C. M. , Haas‐Givler, B. , Chopko, S. , Hailey, J. , Muelly, E. R. , Shellmer, D. A. , Radcliff, Z. , Rodrigues, A. , … Morton, D. H. (2020). Branched‐chain α‐ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. Molecular Genetics and Metabolism, 129(3), 193–206. 10.1016/j.ymgme.2020.01.006 [DOI] [PubMed] [Google Scholar]
- Strauss, K. A. , & Morton, D. H. (2003). Branched‐chain ketoacyl dehydrogenase deficiency: Maple syrup disease. Current Treatment Options in Neurology, 5(4), 329–341. [DOI] [PubMed] [Google Scholar]
- Strauss, K. A. , Wardley, B. , Robinson, D. , Hendrickson, C. , Rider, N. L. , Puffenberger, E. G. , Shelmer, D. , Moser, A. B. , & Morton, D. H. (2010). Classical maple syrup urine disease and brain development: principles of management and formula design. Molecular Genetics and Metabolism, 99(4), 333–345. 10.1016/j.ymgme.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrell, B. L. , Padilla, C. D. , Loeber, J. G. , Kneisser, I. , Saadallah, A. , Borrajo, G. J. C. , & Adams, J. (2015). Current status of newborn screening worldwide: 2015. Seminars in Perinatology, 39(3), 171–187. 10.1053/j.semperi.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Yunus, Z. M. , Kamaludin, D. A. , Mamat, M. , Choy, Y. S. , & Ngu, L. (2012). Clinical and biochemical profiles of maple syrup urine disease in Malaysian children. JIMD Reports, 5, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayed, H. , El Khayat, H. , Tomoum, H. , Khalifa, O. , Siddiq, E. , Mohammad, S. A. , Gamal, R. , Shi, Z. , Mosailhy, A. , & Zaki, O. K. (2019). Clinical, biochemical, neuroradiological and molecular characterization of Egyptian patients with glutaric acidemia type 1. Metabolic Brain Disease, 34(04), 1231–1241. 10.1007/s11011-019-00422-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request due to privacy/ethical restrictions.